 Lorenzo Petrolli
Lorenzo Petrolli Francesco Tommasino
Francesco Tommasino Emanuele Scifoni
Emanuele Scifoni Gianluca Lattanzi
Gianluca Lattanzi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Phys. , 30 October 2020
Sec. Medical Physics and Imaging
Volume 8 - 2020 | https://doi.org/10.3389/fphy.2020.576284
This article is part of the Research Topic Applied Nuclear Physics at Accelerators View all 57 articles
Advanced track structure codes excel as state-of-the-art tools to low-scale dosimetric models: the rational evolution for a cell-like scenario is detailed within a microsecond of an ion collision, that is the standard timescale for critical DNA modifications. The in vitro DNA double-strand breaks (DSB) yield is matched indirectly by nanodosimetric track structure assessments; however, the score to specific DSB motifs (i.e., the yield associated to each DSB distance between DNA cuts) is mostly overlooked. In this work, we extend the PDB4DNA example of the Geant4-DNA toolkit, to briefly assess the hit and DSB scores over a nucleosome tetramer framework (Protein Data Bank entry: 1zbb). We describe a critical scenario that biases the statistical significance for an event-by-event track structure assessment at the nanometric scale, based on a Shannon's entropy estimate of the volumetric hit score; finally, we draw a tentative correlation between the mean DSB quality and a proton track, and conclude that short-distanced DSBs by direct effect are favored within a Bragg peak-relevant energy range.
Mechanistic estimates of cell radiosensitivity benefit from the sharply detailed information on the local density of radiation-induced, DNA-lethal events in cell nuclei, down to a nanometric scale [1, 2].
The description of a rational frame for the overall outcome associated to a specific radiation quality (which leads to the early cell reaction) involves several research fields. Particularly, this is critical to ion beam irradiation, which is associated with a highly complex energy deposition cascade. The event-by-event reconstruction of the cell-like scenario, however, forces a choice in scale and time; indeed, advanced Monte Carlo radiation track structure toolkits depict the microsecond timescale of radiolytic radical diffusion and reactivity [3–6], while molecular dynamics (MD) may achieve a few microseconds of non-reactive, all-atom DNA dynamics [7, 8]. Beside actual technical limitations, however, there is a multitude of choices as to which chromatin scenarios are worth the simulation effort, to what extent, and what to look for.
As nuclear DNA suffers all carriers of molecular toxicity, cells enable substrate-selective recovery mechanisms [9]; each mechanism is associated to one activation factor, that senses a defined DNA aberration class (i.e., base excisions, DNA strands crosslinks, strand breaks, etc.)—still, we lack a rational scenario to describe the subsequent scale of mechanistic evolution for the DNA lesion [10]. All aforementioned DNA modification examples relate, in fact, to a wide variety of biochemical scenarios; it was remarked that the early dynamics for a short double-stranded DNA chain, as it suffers a double-strand break (DSB) event—that is, the close cuts in the covalent DNA backbone over the two strands—is defined markedly by the distance between strand cuts [11]. Furthermore, broken DNA chains are resilient to thermal fluctuations in the nucleosome, where the core histone tails hold the DNA ends, within a few microseconds [12].
Track structure toolkits benefit from coarse criteria that qualify as a DSB any closely associated, local energy deposition event over the DNA backbone (either beyond or within an arbitrary dose threshold) to a certain likelihood [6, 13, 14]. We assume, however, that different DSB motifs are distinctly harmful, and short-distanced DSBs alone would unfold nucleosomal DNA within a few microseconds of an ion collision, while it seems unlikely that a DSB frame distanced by 10 bases would crack by thermal fluctuations. The mechanistic assessment of DSB distances meets a biochemical intuition; in fact, the explicit scenario that unfolds after a DSB event, and the kinetic implications distinct DSBs have on cell activation factors, are not fully known. Furthermore, while track structure mechanistic assessments are meant to focus the overall radiation field effect on a local scale, to indirectly match absolute strand break outcomes, the score to specific DSB distances is mostly overlooked.
We hereby show the results of a brief assessment for a proton track structure within a Bragg peak-related energy range, at a nanometric scale. As a fit environment, we exploited and extended the basic PDB4DNA example classes [15] of the Geant4-DNA toolkit [16–19], to keep track of the hit and DSB distance scores (by early direct effect), over a nanometric water simulation frame. The natural framework to such analysis are the atomic crystal coordinates of a nucleosome tetramer [20] (hereafter tetranucleosome, Protein Data Bank entry 1ZBB [21]). Nucleosomes define the elemental units of chromatin in the eukaryotic cell [22, 23], where a 147 bp double-stranded DNA helix wraps a core histone octamer over 1.67 turns [24]; furthermore, nucleosomes are threaded as “beads” into 30-nm-wide chromatin fibers [25], thus the tetrameric framework involves linear, linker DNA chains and wrapped core DNA, and fairly depicts a chromatin-like scenario.
Initially, our first aim was to track the likelihood of each nucleotide over the nucleosome tetramer framework to be involved by a DSB event. However, our assessment revealed a critical scenario, where the hit collection over the tetranucleosome DNA backbone is statistically biased by a “truncated” track structure artifact; we thus involve a statistical estimator based on Shannon's entropy to assess the level of bias for a collection of hit events, within the nanometric volume. As the hit artifact is trivially fixed by an expansion of the water box, we finally draw a tentative correlation between the mean DSB quality and a proton track, and conclude that short-distanced DSBs by direct effect are favored within a Bragg peak-related energy range.
The PDB4DNA example of the Geant4-DNA toolkit includes a set of C++ libraries that let users create nanometric simulation volumes, tailored over the atomic coordinates of DNA biomolecular structures (available within the RCSB Protein Data Bank, RRID:SCR_012820 [26]). Protein Data Bank (PDB) files collect exhaustive information of a biomolecule's framework as, for instance, a detailed list of atom coordinates in a readable ASCII format; DNA nucleotides are further classified and labeled by a serial index that codifies their location over the DNA chain (1–694 for the overall nucleosome tetramer framework referred to in this work).
The 1ZBB PDB entry describes the elemental symmetrical unit for the nucleosome tetramer (i.e., a dinucleosome). This was further transformed via a dedicated PDB file editor (VMD, RRID:SCR_001820 [27]) and added with its complementary element, to achieve a full tetrameric framework (694 bp) that was associated to the PDB4DNA example; the edited PDB file records were fixed to comply with the PDBlib reader format criteria (these are detailed in [15]).
We will hereafter refer to the tetranucleosome-tailored water box extracted by PDB4DNA (13.0 × 15.2 × 25.4 nm) as the reference volume; all framework-related information, that is, the DNA atomic coordinates and nucleotides' center of mass, is implicitly extracted and cached within the Geant4 environment. Such default reference volume (a G4Box instance) is made of G4_WATER (a NIST database material) and lies within a void environment of “Galactic,” vacuum material.
The basic PDB4DNA analysis tools keep track of the overall energy deposition, single- and DSB scores within the water box. We extended the default classes and involved a further set of ROOT [28] histograms to keep track of1 (i) the energy deposition events (hits) score over the reference volume coordinates; (ii) the hits and DNA strand breaks score over the DNA backbone; (iii) the DNA DSBs distance score, as the absolute distance between individual strand breaks over complementary DNA strands, within a 10 bp threshold. A DNA strand break is scored as an overall 8.22-eV dose deposition (lower threshold) to a nucleotide backbone moiety, that is, the ribose-phosphate residue. Several criteria that yield reliable estimates of DNA strand breaks by early direct effect are detailed in the literature [29]; however, we remark that our analysis, in this concern, is merely qualitative.
The overall dataset was collected within the Geant4 (version 10.02-P03) toolkit environment [30–32]; “raw” data were analyzed via ROOT scripts as detailed in the “Results” section. Each of several runs covered 107 tracks and diverse volume choices; however, the default G4EmDNAPhysics list constructor2 to low-energy electron cross-sections3 was set. The default PDB4DNA layout involves an isotropic, outer spherical source, that is defined over the vertex coordinates of the reference volume; thus, particles (500 keV to 5 MeV protons, in this work) are randomly shot by the edges toward the water box. The source is bound to the active box, therefore it stretches as the water volume is expanded (vide infra). Such scenario let us factor varied, random nucleosome layouts in the overall assessment, that is representative of a chromatin fiber scenario.
We started by the default PDB4DNA extension layout, where particles (500 keV protons) randomly strike the reference volume by a perfectly isotropic, spherical source. Figure 1A shows how the hit counter (the score to each energy deposition event over the DNA backbone) is explicitly non-homogenous, as well as the strand break and DSB counters (not shown here). We expected an explicit dependence for the hit score on local DNA morphology, hence the likelihood for a nucleotide to be involved by a DSB event would vary between the linker and core DNA chains; however, no such correlation seems to arise, as the hit spikes in Figure 1A involve nucleotides over linker and wrapped DNA likewise. Remarkably, such spikes describe a cluster of DNA nucleotides over the “core” of the tetranucleosome framework (highlighted in red in Figure 1D), which we show to be an artifact (vide infra); indeed, the hit counter to all energy deposition events within the reference volume (Figure 2A) shows that the central core is oversampled effectively over the z-axis (as defined in the nucleosome tetramer PDB atom coordinates file). The same holds true for the x-and y-axes (not shown here).
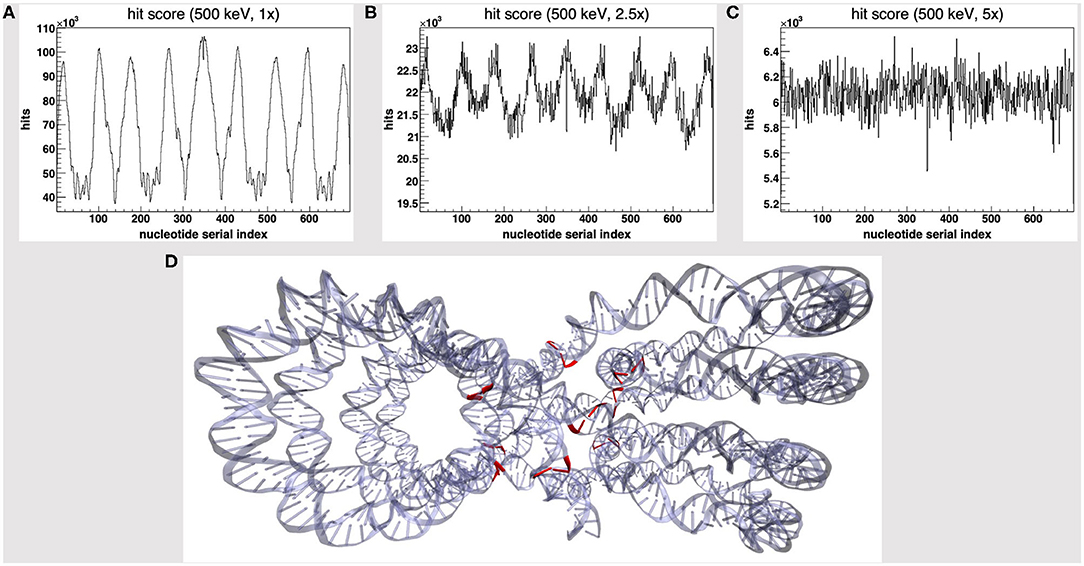
Figure 1. Hit score over the nucleotides serial index, for a set of 500 keV-protons PDB4DNA runs at a (A) 1-fold, (B) 2.5-fold, and (C) 5-fold linear expansion of the reference volume sizes; the spiked behavior shown by the hit counter in (A) vanishes off as the water box is expanded. (D) The nucleotide cluster at the core of the tetranucleosome framework (in red) involves the hit counter spikes in (A).
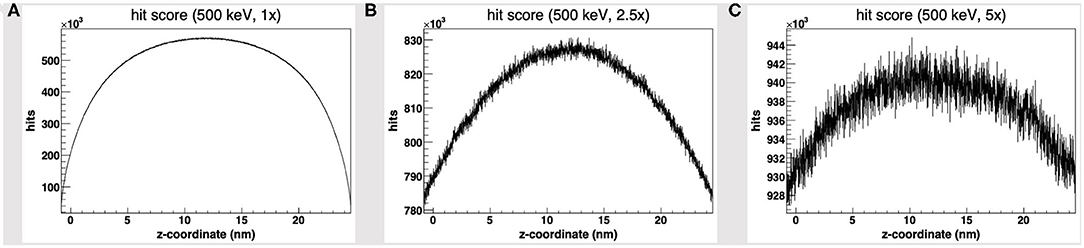
Figure 2. Hit score over the z-axis [as defined in the nucleosome tetramer Protein Data Bank (PDB) atom coordinates file] to all dose deposition events within the reference volume frame, for a set of 500 keV-protons PDB4DNA runs at a (A) 1-fold, (B) 2.5-fold, and (C) 5-fold linear expansion of the reference volume sizes; the reference volume is oversampled at its core, whereas the disparity to the outlying compartments is explicit to smaller systems (hit artifact).
We will hereafter refer to such effect as a hit artifact. As particles leak off the water box (to a vacuum environment), their tracks are cut off. Hence, no further collisions/events are detailed by the reference volume outer shell, and we lack track information at the system boundary, where there is a local unbalance. As a consequence, a minor dose fraction is deposited over outlying DNA nucleotides, and the dosimetric information we draw out of the tetranucleosome framework is an effective oversample of the reference volume central core (that is incidentally taken up by linker DNA chains), while we overlook the outlying nucleosome compartments. Such hit artifact remarks how we likely misestimate the early effect of an ion traversal over a small DNA framework, as we lack either boundary conditions or track information at its outer solvation shell.
To avoid the hit artifact, we expanded the water box further off the default size of the reference volume, i.e., we symmetrically applied a multiplicative linear expansion factor to each box size (1–5-fold). Figures 1A–C show the hit counter spikes effectively vanish off the DNA backbone, as well as the DNA strand break counter (not shown here), where the water volume is expanded; hence, such trivial symmetrical expansion ensures all nucleotides are sampled evenly over the tetranucleosome framework. So, while we expect the hit artifact to apply to all nanometric systems likewise, a substantially thick “solvation shell” makes sure the track is detailed over the reference volume (and the DNA backbone), which now lies within a wider water box. This raises a further issue, that is, to achieve a convenient tradeoff in track structure details and the least effort, i.e., to establish where the water box is overexpanded.
We will hereafter refer to the volume hit score (VHS) as the overall score of energy deposition events within the reference volume frame, while the DNA hit score (DHS) is the subset of all VHS events that fall over the DNA backbone; such scorers effectively estimate the overall amount of information we collect (and lose) over the reference volume frame, while the water box is expanded.
The VHS increases with the volume expansion, as shown in Figure 3A; this is expected, as a thick solvation shell ensures an increase in the overall information we collect over the reference (and its outer) volume, as shown explicitly in Figures 2A–C. However, Figure 3B shows that the DHS coincidently decreases, thus we scored fewer hits over the DNA backbone, while the hit counter had increased within the reference volume.
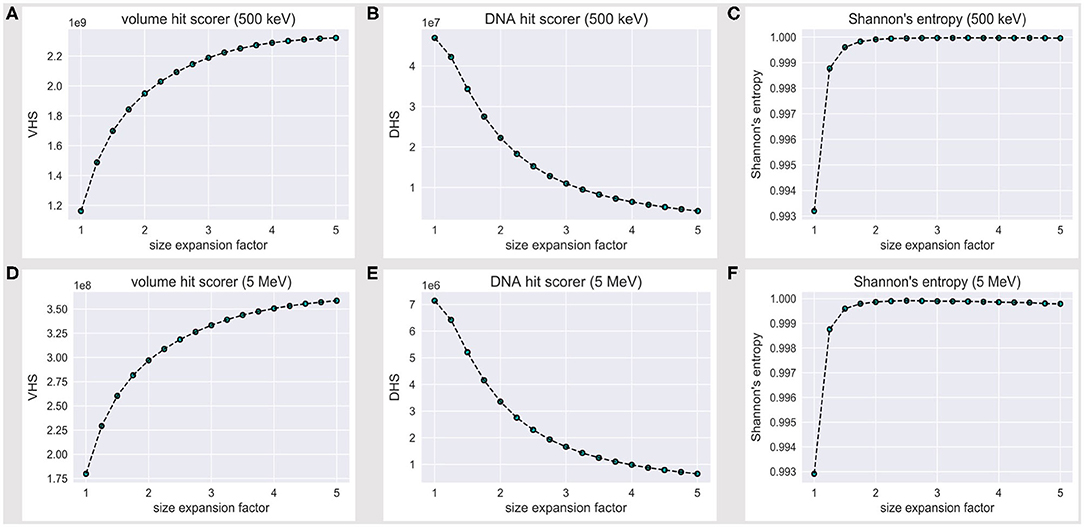
Figure 3. Volume hit score (VHS) at (A) 500 keV and (D) 5 MeV; DNA hit score (DHS) at (B) 500 keV and (E) 5 MeV; the VHS increase with the expansion of the water box is associated with a decrease in the DHS. The level of flatness for the DNA hit counter is established via a normalized Shannon's entropy formula (Eq. 1) at (C) 500 keV and (F) 5 MeV; no significant increase in S is achieved beyond a 2.5-fold linear expansion of the reference volume sizes.
The latter outcome looks counterintuitive; however, nucleotides effectively take up a minor fraction of a nucleosome volume. By the PDB4DNA default scenario (1-fold expansion factor), we overstrike the tetranucleosome “crowded” core, where tracks are extremely effective; they turn less effective over the DNA backbone, however, as we shoot over a wider water system (lower DHS). This likely reflects an uneven nucleotide framework, where a major hit fraction strikes an outlying volume associated to a low nucleotide concentration.
To establish and quantify the level of flatness for the DNA hit counter (and estimate a VHS–DHS tradeoff), we referred to a normalized Shannon's entropy formula, defined as:
where the index i runs over the N = 694 nucleotide pairs, and pi is defined as the hit score over the i-th nucleotide pair, divided by the overall DNA hit score; therefore, S varies between 0 (maximally biased distribution) and 1 (unbiased distribution).
Figure 3C shows that Shannon's entropy increases steeply with the linear expansion factor; however, no significant increase in S is achieved beyond a 2.5-fold expansion of the default reference volume sizes, which we therefore established as a minimum threshold to achieve an unbiased sample of the tetranucleosome DNA backbone.
Such a threshold is, however, strictly bound to the 500 keV scenario. We thus extended the assessment to a 5-MeV particle case; Figures 3D,E show the volume and DNA hit score to behave exactly alike at 500 keV and 5 MeV, within an order of magnitude decrease for all values in the latter case, as expected by an effective difference in LET, i.e., with fewer energy deposition events overall. Remarkably, the 500-keV steep increase in Shannon's entropy (Figure 3C) is matched at 5 MeV (Figure 3F)—likewise, S is maximum at a 2.5-fold linear expansion factor of the default reference volume sizes. We therefore established a 2.5 expansion factor to be a minimum threshold to achieve an unbiased and statistically significant sample of the tetranucleosome framework, within the 500 keV−5 MeV energy range.
In view of the latter outcome, we extended our assessment of the 500-keV to 5-MeV energy range scenario at fixed 2.5-fold volume expansion and involved a further estimator we will hereafter refer to as double strand break distance mean score (DMS).
It is widely shared that the definition of the DNA double strand break is based over a threshold distance criterion between individual strand breaks over complementary DNA strands [33]. Such criterion is rational to a microdosimetry level of theory; each DSB motif (that is associated to a strand breaks distance), however, implies a local unique chemical aberration and mechanical behavior, where the (virtual) timescales for a broken DNA framework to crack by thermal fluctuations vary in each DSB scenario [11]. We thus added a ROOT histogram that kept track of each DSB distance.
The DSB distance scores at 500 keV, 1.5 MeV, and 5 MeV seemingly share a Poisson fit, as shown by Figures 4A–C, and are biased towards short-distanced DSBs; Figure 4D further shows how the DSB DMS decreases slightly with the particle energy, within the 500 keV to 5 MeV range. While we lack a yet significant dataset (that extends over a wider energy range), we would speculate the existence of an effective correlation between a particle track structure and a “mean DSB event” quality, over a unique DNA framework. Rather remarkably, short-distanced DSB events by direct effect (one- to five-nucleotide distance) would thus look favored by a proton source within a Bragg peak-related energy range.
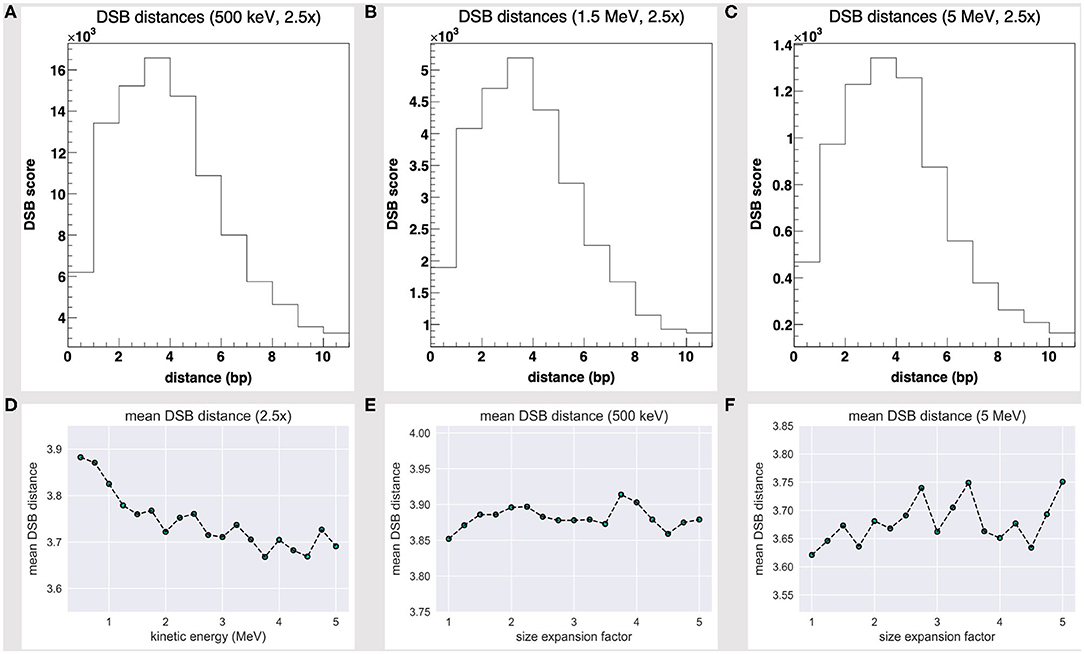
Figure 4. DSB distance scores at fixed 2.5-fold linear size expansion, at (A) 500 keV, (B) 1.5 MeV, and (C) 5 MeV; all share a Poisson fit and are biased towards short-distanced DSBs. The DSB distance mean score at a fixed 2.5-fold size expansion (D) shows a slight decrease with the particle energy and fluctuates as the water box is expanded at (E) 500 keV and (F) 5 MeV.
We further realized that the DSB DMS value slightly fluctuates as the volume is expanded—Figures 4E,F show the DMS within a 1- to 5-fold linear size expansion, at 0.5 and 5 MeV. As we overexpand the water box sizes beyond a 2.5-fold factor (that is, the threshold where we achieve a substantially thick solvation shell and moderate the DHS loss), we shall collect no further track structure information and collaterally oversample the water volume far off the tetranucleosome framework; thus, we expect the DSB DMS to converge eventually.
We detailed a brief assessment of the early, direct DNA lesions associated to the energy deposition track by a proton beam isotropic source over a Bragg peak-related energy range. We extended the default PDB4DNA (a Geant4-DNA example) C++ classes and involved a set of ROOT histograms to keep track of the hit and DNA strand break scores over a nanometric-sized water box, tailored over the atomic coordinates of a nucleosome tetramer (Protein Data Bank entry: 1ZBB).
By the default PDB4DNA extension layout, we achieved a non-homogeneous, spiked hit score over the nucleosome tetramer DNA backbone, which describes a nucleotides cluster over the reference volume central core and where the outlying nucleosome compartments are undersampled; this leads to a local unbalance in the hit counts. We thus symmetrically expanded each of the reference volume box sizes up to 5-fold and allowed a thick solvation shell about the nucleosome tetramer, where an ion track is not broken off; we eventually established (via a normalized Shannon's entropy formula) that a 2.5 linear size expansion threshold achieves an unbiased sample of the nucleosome tetramer DNA backbone, within a 500 keV to 5 MeV energy range.
Clinical treatments (108-109 ions/cm2 fluence) strike cell nuclei by a few hundred projectiles; in silico track structure assessments of a nucleosome (that is a frame size smaller by a factor 108 than a cell nucleus) shall thus infer mean dosimetric information at the nanometric level, which we achieved by an exhaustive and unbiased collection of events over the DNA backbone. A bias estimation by a Shannon's entropy algorithm is, however, strictly framework dependent: in fact, we shall expect it to be not as effective where DNA is highly symmetrical over the volume.
In conclusion, we remarked that a DSB coarse nanodosimetric description based over a distance threshold (i.e., that is inclusive of all double strand break motifs, within an arbitrary distance) is weak by molecular dynamics (MD) criteria, where we are not allowed chemical ambivalence. To such aim, we noticed that the DSB distance scores share a Poisson fit and are biased towards short-distanced DSBs (one- to five-nucleotide distance), within a 500 keV to 5 MeV proton energy range. As a further biophysical estimator, the DSB DMS slightly fluctuates as the volume is expanded and decreases with the particle energy. While we lack a yet significant dataset and a careful assessment of DSB criteria, we speculated a correlation between a particle quality and energy and a “mean break event” assumption (by direct effect), whereby particles are associated with a DSB distance likelihood based over a track structure description. We acknowledge, however, that a further, updated analysis, where indirect effects are taken into account will be needed.
As done initially in Landuzzi et al. [11] and Cleri et al. [12], MD shall be exploited to further assess the early evolution of chromatin-like DNA frameworks, despite within limited timescales, as a clear scenario of early DNA lesions is collected. To create a cross framework, where to meet a nanodosimetric and biochemical intuition, further structural feedback shall be collected, whereas, to date, we lack atomistic datasets to irradiated, chromatin-like frameworks. An isolated attempt that unifies DNA lesions by ion irradiation and classic MD was carried out in the context of a multiscale approach [34], although explicitly focused on the channels of shockwave induction by local heat spikes in high-LET regimes. As exhaustive datasets on the local features of clustered DNA lesions by different radiation sources will become accessible, multiscale approaches shall become straightforward; however, such issues are yet a matter for further debate.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
LP carried out the numerical simulations and analysis and wrote the draft. All authors conceived the current ideas, discussed the assessment, reviewed, and contributed to the draft.
This work was partially supported by the INFN CSN5 Call MoVe IT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank Drs. Yann Perrot, Emmanuel Delage, and Sebastien Incerti for useful intellectual discussions.
1. ^Data are collected at runtime, on a track basis, and analyzed offline.
2. ^It involves low-energy electromagnetic interactions by electrons (excitation, ionization, vibrational excitation, attachment, elastic scattering) and light ions (excitation, ionization, charge increase/decrease).
3. ^To a quantitative assessment, the G4EmDNAPhysics_option4 and G4EmDNAPhysics_option6 list constructors shall be favored—see Incerti et al. [19].
1. Friedrich T, Scholz U, Elsässer T, Durante M, Scholz M. Calculation of the biological effects of ion beams based on the microscopic spatial damage distribution pattern. Int J Radiat Biol. (2012) 88:103–7. doi: 10.3109/09553002.2011.611213
2. Hawkins RB. A microdosimetric-kinetic model for the effect of non-poisson distribution of lethal lesions on the variation of RBE with LET. Radiat Res. (2003) 160:6169. doi: 10.1667/rr3010
3. Karamitros M, Luan S, Bernal MA, Allison J, Baldacchino G, Davidkova M, et al. Diffusion-controlled reactions modeling in Geant4-DNA. J Comput Phys. (2014) 274:841–82. doi: 10.1016/j.jcp.2014.06.011
4. Boscolo D, Krämer M, Durante M, Fuss MC, Scifoni E. TRAX-CHEM: A pre-chemical and chemical stage extension of the particle track structure code TRAX in water targets. Chem Phys Lett. (2018) 698:11–8. doi: 10.1016/j.cplett.2018.02.051
5. Ramos-Méndez J, Perl J, Schuemann J, McNamara A, Paganetti H, Faddegon B. Monte Carlo simulation of chemistry following radiolysis with TOPAS-nBio. Phys Med Biol. (2018) 63:105014. doi: 10.1088/1361-6560/aac04c
6. Friedland W, Dingfelder M, Kundrát P, Jacob P. Track structures, DNA targets and radiation effects in the biophysical Monte Carlo simulation code PARTRAC. Mutat Res. (2011) 711:28–40. doi: 10.1016/j.mrfmmm.2011.01.003
7. Dans PD, Walther J, Gómez H, Orozco M. Multiscale simulation of DNA. Curr Opin Struct Biol. (2016) 37:29–45. doi: 10.1016/j.sbi.2015.11.011
8. Shaytan AK, Armeev GA, Goncearenco A, Zhurkin VB, Landsman D, Panchenko AR. Coupling between histone conformations and DNA geometry in nucleosomes on a microsecond timescale: atomistic insights into nucleosome functions. J Mol Biol. (2016) 428:221–37. doi: 10.1016/j.jmb.2015.12.004
9. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071–8. doi: 10.1038/nature08467
10. McMahon SJ, Prise KM. Mechanistic modelling of radiation responses. Cancers. (2019) 11:e205. doi: 10.3390/cancers11020205
11. Landuzzi F, Palla PL, Cleri F. Stability of radiation-damaged DNA after multiple strand breaks. Phys Chem Chem Phys. (2017) 19:14641–51. doi: 10.1039/c7cp02266b
12. Cleri F, Landuzzi F, Blossey R. Mechanical evolution of DNA double-strand breaks in the nucleosome. PLoS Comput Biol. (2018) 14:e1006224. doi: 10.1371/journal.pcbi.1006224
13. Nikjoo H, O'Neill P, Goodhead DT, Terrissol M. Computational modelling of low-energy electron-induced DNA damage by early physical and chemical events. Int J Radiat Biol. (1997) 71:467–83. doi: 10.1080/095530097143798
14. Lampe N, Karamitros M, Breton V, Brown JMC, Kyriakou I, Sakata D, et al. Mechanistic DNA damage simulations in Geant4-DNA part 1: a parameter study in a simplified geometry. Phys Medi. (2018) 48:135–45. doi: 10.1016/j.ejmp.2018.02.011
15. Delage E, Pham QT, Karamitros M, Payno H, Stepan V, Incerti S, et al. PDB4DNA: implementation of DNA geometry from the protein data bank (PDB) description for Geant4-DNA Monte-Carlo simulations. Comput Phys Commun. (2015) 192:282–8. doi: 10.1016/j.cpc.2015.02.026
16. Incerti S, Baldacchino G, Bernal M, Capra R, Champion C, Francis Z, et al. The Geant4-DNA project. Int J Model Simul Sci Comput. (2010) 1:157–78. doi: 10.1142/S1793962310000122
17. Incerti S, Ivanchenko A, Karamitros M, Mantero A, Moretto P, Tran HN, et al. Comparison of GEANT4 very low energy cross section models with experimental data in water. Med Phys. (2010) 37:4692–708. doi: 10.1118/1.3476457
18. Bernal MA, Bordage MC, Brown JMC, Davídková M, Delage E, El Bitar Z, et al. Track structure modeling in liquid water: a review of the Geant4-DNA very low energy extension of the Geant4 Monte Carlo simulation toolkit. Phys Med. (2015) 31:861–74. doi: 10.1016/j.ejmp.2015.10.087
19. Incerti S, Kyriakou I, Bernal MA, Bordage MC, Francis Z, Guatelli S, et al. Geant4-DNA Example applications for track structure simulations in liquid water: a report from the Geant4-DNA project. Med Phys. (2018) 45:e722–39. doi: 10.1002/mp.13048
20. Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. (2005) 436:138–41. doi: 10.1038/nature03686
21. Schalch T, Duda S, Sargent DF, Richmond TJ. Structure of the 4_601_167 Tetranucleosome, RCSB Protein Data Bank ID “1ZBB” (2005). doi: 10.2210/pdb1zbb/pdb
22. Olins AL, Olins DE. Spheroid chromatin units (ν bodies). Science. (1974) 183:330–2. doi: 10.1126/science.183.4122.330
23. Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. (1999) 98:285–94. doi: 10.1016/s0092-8674(00)81958-3
24. Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol. (2002) 319:1097–113. doi: 10.1016/S0022-2836(02)00386-8
25. Boulé JB, Mozziconacci J, Lavelle C. The polymorphisms of the chromatin fiber. J Physi Condensed Matter. (2014) 27:033101. doi: 10.1088/0953-8984/27/3/033101
26. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. (2000) 28:235–42. doi: 10.1093/nar/28.1.235
27. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. (1996) 14:33–8. doi: 10.1016/0263-7855(96)00018-5
28. Brun R, Rademakers F. ROOT - an object oriented data analysis framework. Nucl Instrum Methods Phys Res A. (1997) 389:81–6. doi: 10.1016/S0168-9002(97)00048-X
29. Schuemann J, McNamara AL, Warmenhoven JW, Henthorn NT, Kirkby KJ, Merchant MJ, et al. A new standard DNA damage (SDD) data format. Radiat Res. (2019) 191:76–92. doi: 10.1667/RR15209.1
30. Agostinelli S, Allison J, Amako K, Apostolakis J, Araujo H, Arce Dubois P, et al. Geant4 - a simulation toolkit. Nucl Instrum Methods Phys Res A. (2003) 506:250–303. doi: 10.1016/S0168-9002(03)01368-8
31. Allison J, Amako K, Apostolakis J, Araujo H, Arce Dubois P, Asai M, et al. Geant4 developments and applications. IEEE Trans Nucl Sci. (2006) 53:270–8. doi: 10.1109/TNS.2006.869826
32. Allison J, Amako K, Apostolakis J, Arce Dubois P, Asai M, Aso T, et al. Recent developments in Geant4. Nucl Instrum Methods Phys Res A. (2016) 835:186–225. doi: 10.1016/j.nima.2016.06.125
33. Elsässer T, Scholz M. Cluster effects within the local effect model. Radiat Res. (2007) 167:319–29. doi: 10.1667/RR0467.1
Keywords: tetranucleosome, PDB4DNA, DNA double strand breaks, Geant4-DNA, track structure, chromatin, Monte Carlo simulations, ion beam irradiation
Citation: Petrolli L, Tommasino F, Scifoni E and Lattanzi G (2020) Can We Assess Early DNA Damage at the Molecular Scale by Radiation Track Structure Simulations? A Tetranucleosome Scenario in Geant4-DNA. Front. Phys. 8:576284. doi: 10.3389/fphy.2020.576284
Received: 25 June 2020; Accepted: 29 September 2020;
Published: 30 October 2020.
Edited by:
Marco Durante, GSI Helmholtz Center for Heavy Ion Research, GermanyReviewed by:
Ziad Francis, Saint Joseph University, LebanonCopyright © 2020 Petrolli, Tommasino, Scifoni and Lattanzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emanuele Scifoni, ZW1hbnVlbGUuc2NpZm9uaUB0aWZwYS5pbmZuLml0; Gianluca Lattanzi, Z2lhbmx1Y2EubGF0dGFuemlAdW5pdG4uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.