 Mykhailo Vladymyrov
Mykhailo Vladymyrov Neda Haghayegh Jahromi
Neda Haghayegh Jahromi Elisa Kaba
Elisa Kaba Britta Engelhardt
Britta Engelhardt Akitaka Ariga
Akitaka Ariga- 1Laboratory for High Energy Physics (LHEP), Albert Einstein Center for Fundamental Physics, University of Bern, Bern, Switzerland
- 2Theodor Kocher Institute, University of Bern, Bern, Switzerland
- 3Science IT Support, Mathematical Institute, University of Bern, Bern, Switzerland
Intravital multiphoton microscopy has become one of the central tools used in the investigation of dynamic cellular activity and function in living animals under nearly physiological conditions and is particularly important for studying the dynamic immune system. During intravital imaging in mice, periodic motion of tissue caused by respiration, induces significant shifts of the imaged region. In slow laser scanning imaging modalities, such as multiphoton microscopy, this movement can lead to considerable distortion and discontinuity in three dimensions of the acquired images. Here, we introduce VivoFollow 2, a toolkit that concurrent with image acquisition performs a precise measurement of the respective image distortion, enabling subsequent automated correction of the imaging data. Recovery of one three-dimensional image stack, corresponding to the tomographic tissue sectioning by the optical plane from each single raw image stack, preserves the time continuity within each image stack. Implementation of VivoFollow 2 thus enables a minimization of motion artifacts in tissues exposed to periodic movements and allows for long-term time-lapse imaging and subsequent precise image analysis of the dynamics of cellular and humoral factors in vivo.
Introduction
In the last two decades, intravital multiphoton microscopy has become one of the central tools in basic and preclinical biological research. It allows for the investigation of dynamic cellular activity and function in living animals under nearly physiological conditions (reviewed in [1] and [2]). Due to the high tissue penetration depth, low phototoxicity, and the possibility of imaging cellular behavior over an extended period of time in living animals, 2-photon intravital microscopy (2P-IVM) has become a sophisticated technique in the field of neurosciences and immunology [3]. For instance, 2P-IVM has been used to visualize a wide variety of biological processes, such as the homing and interaction of dendritic cells with T cells in lymph nodes [4, 5], the migration of T cells across the blood-brain barrier into the central nervous system (CNS) [6–8], the processes of tumor angiogenesis and metastasis (reviewed in [9] and [10]), and the post-arrest behavior of immune cells on endothelial cells lining blood vessels during their extravasation. A common hurdle is to get optical access to the region of interest, for example, when aiming to image immune cell migration in the CNS, which is shielded by bone, the dura mater, and the leptomeningeal layers [11]. Nevertheless, different surgical preparations have been developed allowing for imaging of the rodent brain and spinal cord, such as cranial windows, thinned skull preparations, and lumbar and thoracic spinal cord windows [12–17]. Recently, we have developed a novel window preparation that allows visualization of immune cell migration across microvessels in the cervical spinal cord of mice using 2P-IVM [18].
A major challenge of 2P-IVM is to minimize motion artifacts in the region of interest that are present during the imaging. In the conventional intravital imaging setting, drift of the tissue and movement caused by the vital functions of the experimental animal can compromise the precise analysis of, for example, motility parameters of individual cells, or even make the acquired data completely unanalyzable. In recent years, several approaches have been developed to overcome this problem. When tissue motion is minor, offline processing tools such as StackReg [19] can be used following image acquisition to correct for the shift caused by the tissue drift. Yet, when the tissue shift over the course of time-lapse acquisition becomes comparable or greater than the size of the imaged volume, these tools are not applicable.
Previously, we have developed the VivoFollow feedback microscopy system that performs tracking of the sample drift in real time and adjusts the imaging position directly during the acquisition [20]. VivoFollow has thus solved the issue of severe tissue drift. While originally developed for 2P-IVM, this system can be applied to a wide range of bioimaging settings.
However, the periodic tissue motion caused by respiration (usual frequency 2–4 Hz) poses a very different problem. This issue is especially present while imaging the cervical spinal cord region in a living animal due to its anatomical position. In this case, the shift of the region of interest becomes significant already within a single image stack of a three-dimensional (3-D) tomographic time-lapse imaging stack for the fast imaging modalities (when image stack acquisition speed is >20 frames/s). In the commonly used 2P-IVM employing the galvanometric scanning mirrors, the frame rate is 0.5–2 frames per second, which corresponds to 1–8 periods of tissue motion within a single image frame. Effectively, this causes a significant distortion in each of the acquired images of a 3-D stack. Moreover, uncoherent distortion of each Z frame leads to the discontinuity of the acquired 3-D volume.
For some studies, tissue motion can be reduced by physical restraints by employing coverslips, suction holders, or other stabilization holders [21], but their application presents other significant issues. They can cause obstruction of the physiological blood flow in the tissue or induce tissue damage and inflammation and are thus a trade-off between minimizing the impact on physiological tissue condition and stabilization efficacy. On the other hand, a number of motion stabilization approaches have been developed from the image analysis side. Several Image-J [22] plugins have been introduced for the correction of motion artifacts in time-lapse videos, for the separation of cellular movement from slow tissue movement [23], or for removing distorted and out-of-focus images caused by tissue motion [24]. In another study [25], the authors proposed a method of recovering an undistorted image by composing fragments of the original dataset. Another technique employing time gating of the image acquisition was proposed [26] that enables acquiring image data only during the time when the sample is stationary. Unfortunately, these tools are not easily applicable to 3-D time-lapse imaging of dynamic processes, such as cell migration in vivo.
As it will be shown, a simple synchronization of image acquisition with the respiration of the animal generally solves the issue of 3-D discontinuity, yet the acquired image stacks remain significantly distorted, compromising precise measurements of, for example, cell motility parameters. Moreover, in this case, one cannot directly estimate the value of distortion and corresponding systematic error of the position measurements.
Here, we present a system named VivoFollow 2, which allows precise distortion measurement and subsequent distortion correction by employing a real-time processing pipeline and a novel approach for image acquisition. This approach relies on the synchronization of the image acquisition with the periodic movement of the tissue of anesthetized mice. For the particular examples reported in the present study, the movement was caused by the mouse respiration, and the synchronization was achieved by monitoring the periodic motion of the inhalation anesthesia ventilation unit used for the endotracheal intubation. VivoFollow 2 enables imaging at a close-to-nominal frame rate, requires minimal changes to the 2P-IVM setup, and recovers the images corresponding to the tomographic tissue sectioning by the optical plane, enabling high-precision 3-D time-lapse imaging.
Methods
VivoFollow 2 System
When a static sample is imaged, the acquired images correspond to the tomographic tissue sectioning by the optical plane (Figure 1A). But if the sample is subjected to periodic movement with a frequency comparable to or higher than image acquisition frame rate, each image does not correspond to a plane, but instead to a complicated sheet sourced from a range of depths in the sample. Furthermore, when acquisition of sequential images in an image stack is not synchronized, these surfaces interleave, leading to the discontinuity of the acquired 3-D volume, as shown in Figure 1B.
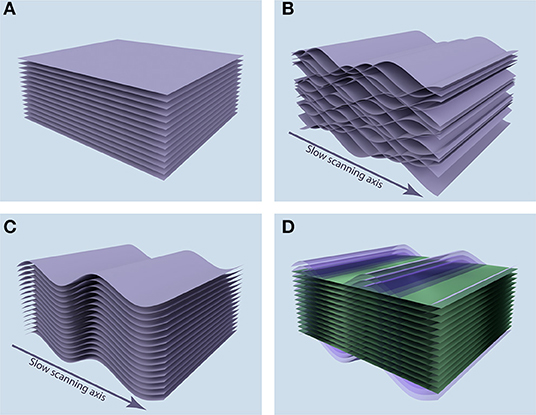
Figure 1. Position of acquired image stacks in the tissue. (A) When the sample is static, the acquired images (purple sheets) correspond to the tomographic tissue sectioning by the optical plane. (B) When the sample is subjected to the periodic motion along the depth axis, acquired images correspond to complicated surfaces in the sample, leading to discontinuity in the 3-D volume of the acquired image stack. (C) Synchronization allows for the recovery of 3-D continuity, but the image stack corresponds to a distorted volume in the sample. (D) After measuring the periodic distortion profile, we can reconstruct the plane tomographic tissue sectioning (green planes) by resampling the acquired image data (purple sheets).
The VivoFollow 2 system recovers the 3-D image stack corresponding to the optical plane tomographic tissue sectioning in three steps. First, image acquisition is synchronized with the respiration of the mouse, ensuring recovery of the 3-D continuity in the acquired volume (Figure 1C). To this end, we have developed a specialized Triggering VivoFollow device, named “TrigViFo,” which enables synchronization of individual image frame acquisition with respiration by performing a readout from the piston position of the mouse ventilator, that provides anesthetic gas mixture to the animal, employing a slotted optical switch (see Hardware Synchronization for details). This device allows for a remote automated control of the phase of the respiration at which the image acquisition is triggered.
Second, to recover the optical sectioning of the tissue, we perform a short calibration measurement of the periodic tissue movement at the beginning of the image acquisition. This calibration is done by acquiring 32 “calibration” image stacks at eight different phases uniformly distributed over the respiration period, employing the TrigViFo that can trigger acquisition at any phase of the periodical movement. The tissue motion profile is then built based on the anatomical landmark position in the sequence of the calibration image stacks. This motion profile and the corresponding image distortion map are computed in real time during the image acquisition using these calibration stacks. Finally, by correcting the image stacks for distortion, the tomographic tissue sectioning by the optical plane is reconstructed (Figure 1D; see Distortion Mapping and Correction).
After the calibration measurement is completed, all of the subsequently acquired image stacks are corrected for the distortion in real time. Building upon the original VivoFollow [20], the present system employs the distortion corrected images to additionally perform tissue drift correction during the course of image acquisition.
Following the experiment, the distortion correction is iteratively refined, and the final corrected image stack sequence is generated. This reduces the displacement of the tissue on the images from typical value of 15 μm to 0.4–0.8 μm, which is comparable with the error of measuring the coordinates of individual cells. The accuracy achieved by VivoFollow 2 allows also the first 32 calibration image stacks to be used for further biological analysis, for example, analysis of the immune cell motility.
Hardware Synchronization
To perform triggering at different phases of the respiration cycle, a dedicated TrigViFo device, allowing for synchronization signal readout, period measurement, triggering pulse generation at adjustable phase, and remote control, was designed. Our setup relies on the use of the Raspberry Pi 3 B+.
Schematic representation of the circuit is given in Figure 2A. The IR LED–PIN photodiode pair is used as a slotted optical sensor (Figure 2B) to read the position of the piston of the MiniVent (Harvard Apparatus) ventilator that supplies the isoflurane anesthesia mixed with the oxygen to the anesthetized mouse. The sensor is attached to the transparent tube of the ventilator pump and fixed with a plastic screw (Figure 2C). The analog signal from the photodiode is amplified and then digitized using the Adafruit ADS115 Analog-to-Digital Converter (ADC). Pulses on one of the microcontroller's GPIO pins are generated to trigger the image acquisition on the microscope (Figure 2D).

Figure 2. Tissue movement profile measurement procedure in the VivoFollow 2 system. (A) Schematic representation of the TrigViFo triggering device circuit. (B) A slotted sensor composed of an infrared LED and a PIN photodiode is used for synchronization of the image acquisition with the piston movement of the ventilator. (C) The sensor is attached to the pump of the MiniVent ventilator and connected to TrigViFo. (D) Imaging setup. (E–H). Distortion mapping procedure. (E) Image volume is split into overlapping crops along the slow scanning axis. (F) Pairwise offsets are found between all time points of calibration stacks for each crop. (G) Relative position of crops at all time points is found by global solving. (H) Spline fit of the periodic tissue motion after pooling all crops together.
The custom-designed software running on the TrigViFo performs a continuous readout of the signal from the ADC, so that signal sampling corresponds to the ADC frequency, 860 Hz. This signal is then filtered for readout duplicates and smoothened by averaging with a sliding window of size 5. Afterward, the rising edges are detected at the level of a manually selected threshold. An example of the filtered waveform is shown in Supplementary Figure 1. We estimate the time resolution of the device as , where σV is the signal voltage standard deviation at the linear part of the rising edge and is the voltage slope at the rising edge. The breathing period τ is measured using a sliding window of the last n = 10 periods, corresponding to the period measurement resolution of . For usual acquisition conditions, according to the motion profiles reconstructed with our system, the maximal tissue motion speed due to animal respiration can be estimated as 200 μm/s. Consequently, the period measurement resolution corresponds to an error of 0.02 μm in tissue position estimation, which is below the resolution of 2-photon microscopy imaging modality.
The rising edge detection threshold can be manually adjusted to the approximate level of the steepest part of the waveform using the graphical user interface (GUI) on the touch screen. The GUI allows the operator to see the current period and the waveform profile to facilitate sensor installation.
The triggering pulse is then generated at time tt = tre + f τ + Δt where tre is the time point of the rising edge, the factor f defines the phase of the breathing cycle at which the triggering occurs, and delay Δt = 20 ms is introduced to ensure stable operation. In practice, it can be reduced for applications where the motion period τ is significantly smaller than in our case.
TrigViFo can be controlled remotely using the Remote Procedure Call (RPC) interface operating over the TCP network layer. This allows setting the value of the phase factor f as well as switching the triggering on and off. Acquisition of each image frame is triggered by the pulse from this device and thus is synchronized to the respiration cycle. Diagram of the algorithm running on the device is shown in Supplementary Figure 2. It is implemented in C++ and is running on a standard Raspbian build.
Image Acquisition
Image acquisition was performed using a TrimScope-II 2-Photon microscope (2PM) system equipped with an Olympus BX51WI fluorescence microscope (LaVision BioTec) using a Nikon CFI Apo LWD 25× water immersion objective. Images were acquired using excitation wavelength of 920 and 1,045 nm. The laser beam scanner was running at 800 Hz, and acquisition was performed in a bidirectional mode.
The computational setup for the real-time correction consists of two PC and the TrigViFo device interconnected via 1 Gbit local network. One PC runs the commercial 2PM acquisition program ImSpector (LaVision BioTec), which controls the microscope hardware and image acquisition. This program allows for access to the acquired images, and customization of the acquisition pipeline by a Python script. We have designed a script defining an image acquisition pipeline that controls the triggering phase and the scanning position. The acquisition consists of two sequential measurements. First, we acquired a time-lapse image stack sequence for mapping the distortion and then continued the normal image acquisition with the synchronization as well as with the tissue drift correction. A flowchart of the pipeline is shown in Supplementary Figure 3.
Distortion Mapping and Correction
To perform the distortion mapping, we acquired a sequence of the calibration 3-D image stacks at nph different phases along the respiration cycle. This is achieved by setting the triggering phase factor f for each of the image stacks sequentially to . The total number of acquired image stacks is nst = nph·nr, where nr is the number of repetitions. In our experiments, we set nph = 8 and nr = 4. These numbers were chosen empirically to balance robust operation and keep the time of the calibration stacks acquisition short.
Acquired image stacks It are then filtered for the out-of-range noise, smoothed, and downscaled by a factor of 2 in XY plane. Next, they are split into overlapping image crops Ic, t of width 32 pixels with step 8 pixels along the slow scanning axis (Figure 2E).
For each crop c, we then find the relative position of all nst image stacks. This is performed in two steps. We denote the offset in 3-D between crops Ic, i and Ic, j at time points i, j as . At first, we find relative offsets between crops at all time points pairwise (Figure 2F) using fine pattern matching in 3-D as previously described [20]. For this, we rely on the image channel containing the anatomical landmarks. The measurement error is defined by the standard deviation of the peak center measurement on the Pearson correlation map σp; c, i→j and the maximum correlation value Cpeak as
where Cmin = 0.6 is the minimum accepted correlation value and ϵ = 10−4. This approach allows us to keep track of the alignment error when the distortion causes the appearance of the two image crops to differ significantly.
Next, we perform global solving to find relative positions of the crops c at all time points i > 0 with respect to the position of the corresponding crop at the first time point . This is performed by the weighted least square minimization of with respect to free parameters rc, 1, …, rc,nst−1. Also, the errors σc, i of each position are evaluated. This operation is performed for each crop c and each axis independently.
The tissue can be subjected to the slow drift during the calibration stacks acquisition often caused by subtle changes in tissue tone. The offset during each repetition k ∈ {0, …, nr − 1} due to the slow drift can be estimated as Δrk = 𝔼c(rc, (k+1)·nph−rc, k·nph), where 𝔼c denotes weighted average with weight 1/σc, ij, to account for measurement error of each crop's absolute position. Such weighting is mandatory since errors σc,i can significantly vary when the anatomical landmarks cover the imaging region non-uniformly, which is often the case in practice. The corresponding drift speed is then , where Δt is the time step size between consecutive time frames. We then correct the crop positions to account for the linear motion component occurring due to the slow tissue drift: rc,k·nph+i → rc,k·nph+i−vk · Δt · i, ∀k ∈ [0, nr), ∀i ∈ [0, nph). Afterward, we shift all crop positions to make average positions across all time points equal to zero: , so that we will obtain rc,i oscillating around zero (Figure 2G).
Finally, all crops c are pulled together to obtain the sample motion profile (Figure 2H). For this, we fit the motion profile with a periodic third-order spline with ns nodes uniformly distributed along the period , where is the motion phase, kph–phase index of the image stack, y is the coordinate on the image along the slow scanning axis (axis Y in our experiments), λy is the effective “wavelength” of the motion along the slow scanning axis, and rs is the set of all the spline nodes positions , i ∈ {0, …, ns − 1}. Please note that while the wavelength λy is in fact defined by the breathing period τ and line scanning speed vscan as λy = τ vscan, at this point, it is more convenient to treat it as an independent variable. To find the wavelength λy, we first fit the Z motion profile with a simple sine function: .
Distortion correction is then performed by resampling the image taken at phase kph by shifting each pixel at pixel coordinates to the new coordinates . This image resampling is performed employing trilinear interpolation.
The most resource-intensive operation is finding the relative offsets between pairs of image crops. To enable performing distortion correction in real time, in addition to relying on the GPU processing, we have developed a parallel processing scheme. We perform pairwise offset evaluation before all image stacks are available, namely, the offsets and corresponding errors are calculated right after the time frame j is acquired and during the acquisition of the next time frame j + 1. This allowed us to perform the global solving and fitting of the motion profile right after the acquisition of the last calibration image stack Inst−1, so that the parameters for distortion correction are ready before the acquisition of the first stack of normal data taking Inst is completed.
Finding the distortion map and performing the distortion correction in real time allow us to apply the tissue drift correction procedure during the acquisition on the images corrected for the distortion.
Finally, we perform additional post-processing after the image acquisition to achieve image quality suitable for further biological data analysis. For this, we iteratively repeat the distortion mapping and correction to find the residual distortion until convergence. During the first iteration, we correct only the Z offset because significant shifts in Z can impair the accuracy of the distortion mapping in XY. Iteration continues until the change in the distortion map remains above 0.1 μm but not more than five iterations to prevent potential divergence in poor landmark signal or landmark coverage conditions. This iterative procedure improves the distortion measurement by 10% (Supplementary Figure 4) and enables performing the distortion correction with subpixel resolution.
The image processing server and the post-processing tool are implemented in C++ and can be run on the Windows or Linux platforms with CUDA-enabled GPU.
Results
Evaluation on Synthetic Data
To assess performance of the distortion measurement and correction procedure, we have first evaluated it on a synthetic dataset. For this, we have generated three image stacks of resolution 512 × 512 × 22 pixels with randomly distributed Gaussian spots (width 10 pixels in XY, 2 pixels in Z) of density 240, 30, and 10 spots/field of view (FoV), corresponding to good, average, and poor landmark coverage in the sample (Figure 3A). We then distorted them by applying image resampling according to a simulated motion profile , where ϕ is the phase of periodic motion. In case of laser scanning imaging, the phase depends on the position on the image: , where kph is the phase index of the image stack, nph is the number of used phases, y is the coordinate on the image along the slow scanning Y axis, and λy is the effective wavelength of the motion along the slow scanning axis (see Distortion Mapping and Correction for details). We have chosen an amplitude along all of the three axes of A = 4 pixels that approximately corresponds to the usual experimental conditions. We generated 32 image stacks for each density sample corresponding to four repetitions of eight image stacks at different phases (nph = 8) and added random Poisson pixel noise to the images. The periodic motion wavelength λy was chosen to be 343.3 pixels, so that the image height is 1.5 λy.
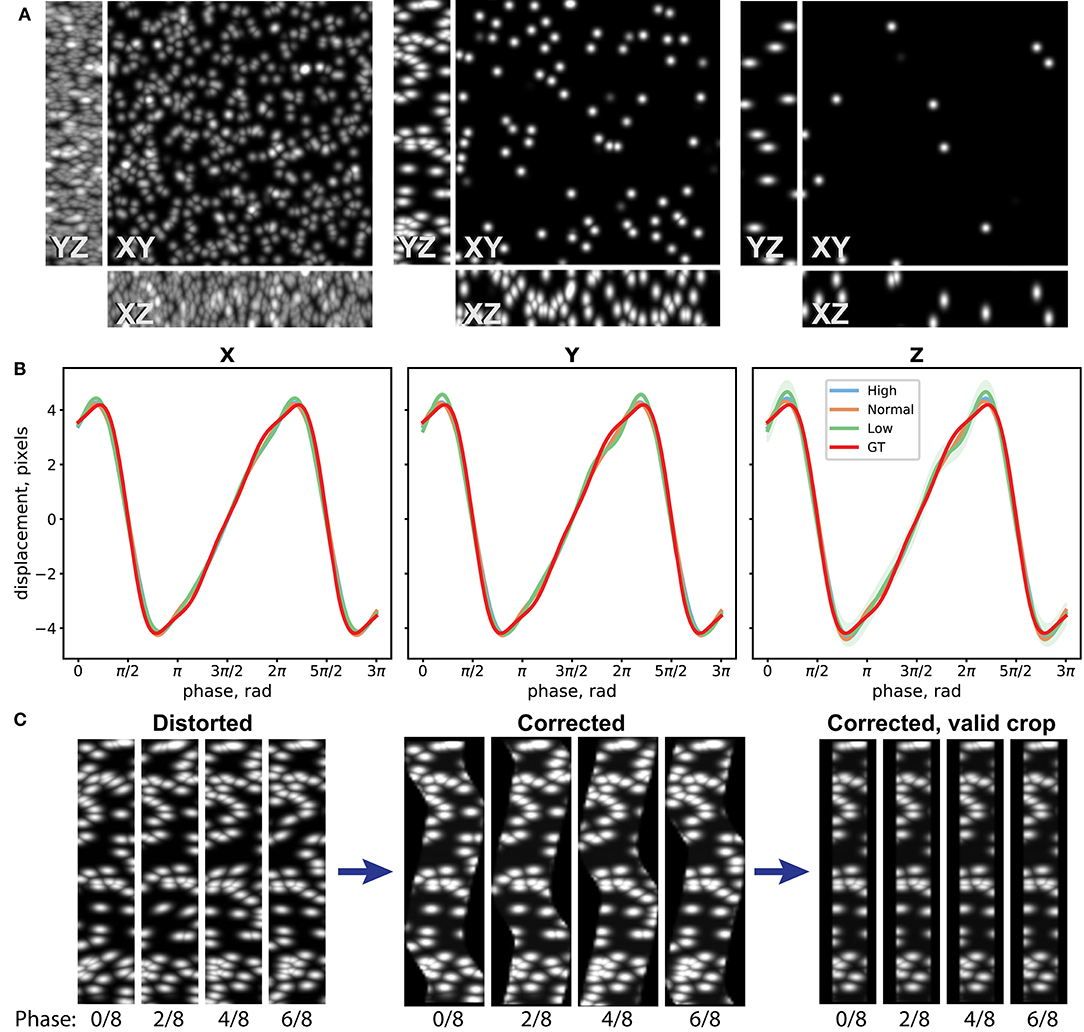
Figure 3. Evaluation of the distortion measurement and correction procedure on synthetic datasets. (A) Maximum intensity projections (MIP) of one Z stack of three synthetic datasets with high, normal, and low densities of Gaussian spots, used as landmarks for distortion measurement. (B) Reconstruction of distortion profile for the three datasets and the ground truth (GT) profile. Eight-node spline fit is performed. (C) YZ MIPs for the normal-density distorted data, reconstructed data according to the measured distortion, and crop of the valid data volume are shown for four different phases.
The synthetic dataset was then processed, and the image distortion was iteratively reconstructed as described in the Methods section. In Figure 3B, the 8-node spline fit of the reconstructed periodic motion profiles for the three datasets and the ground truth (GT) used to distort the images are shown. Comparison between different landmark coverage for spline fit with 16, 8, and 4 nodes is shown in Supplementary Figure 5. One can see that the developed method can reconstruct the distortion pattern with accuracy up to 0.1 pixel for a wide range of experimental conditions. In Figure 3C, the YZ maximum intensity projections (MIPs) for the distorted data with average landmark coverage, reconstructed volume according to the measured distortion, and crop of the valid volume are shown for four different phases. The time-lapse video of the MIPs and the 3-D view of the distorted dataset, reconstructed, and the crop of the valid volume of the reconstructed dataset can be seen in the Supplementary Videos 1, 2.
Evaluation on the 2P-IVM Data
We demonstrate the performance of the system in 2P-IVM imaging of the cervical spinal cord vasculature of mice [18]. To this end, we used CX3CR1-GFP//CCR2-RFP reporter mice systemically injected with a non-function-blocking anti-PECAM-1-Alexa Fluor 555 antibody. This allowed to visualize tissue resident myeloid cells via the GFP signal and infiltrating monocytes by the RFP signal. Systemic injection of the anti-PECAM-1 antibody readily labeled circulating immune cells. In Figure 4A, we show the MIPs of an image stack of size 395μm × 395μm × 84μm acquired without image acquisition synchronization. One can clearly see the duplicates of the fine GFP+ cellular processes of the CNS myeloid cells in XY projection caused by the motion of the sample, as shown in Figure 4C. Due to significant motion in Z direction, the 3-D structural information is lost in this case, as seen on the XZ and YZ projections in Figures 4A,C. In Figures 4B,D, we show the MIPs of the image stack in the same region acquired with triggering by TrigViFo, synchronizing the start of each Z frame acquisition with the respiration cycle. One can see that the consecutive Z planes are aligned, and the 3-D structure is recovered. Since the period of the motion (τ≈ 0.54s) is, however, comparable with the acquisition time of a single Z frame (0.69 s), the acquired volume is effectively distorted. To measure the distortion pattern, we performed acquisition of several Z stacks, with triggering the start of each Z frame acquisition at different phases of the respiration cycle, as described in the Methods section. In Figure 4E, the examples of the YZ MIPs acquired with triggering phases , , , and are presented. The difference in the position of the tissue on the respective images is clearly visible. The peak-to-peak amplitude amounts in this case to about 15 μm. We performed the first iteration of the distortion measurement using the myeloid cell CX3CR1-GFP signal as landmark in real time during the acquisition. The corresponding YZ MIPs after the final iterative offline distortion correction of the same dataset are presented in Figure 4F. The tissue position after the distortion correction is the same, confirming that the correction quality is sufficient to include the original calibration image stacks, used for distortion measurement, into the biological analysis. Hence, the first 16 min of acquisition are not lost and remain accessible for subsequent image analysis. The XY, XZ, and YZ corrected MIPs are shown in Figure 5A. We compared the distortion measurement performance using 4-, 8-, and 16-node spline fit in Figure 5B. The four-node fit only slightly (<1 μm) affects the precision of the distortion measurement. Performing the distortion correction in real time enables subsequent accurate real-time tracking of the region of interest in the tissue. This is done by performing continuous offset measurement using fine pattern matching of the acquired image stacks in 3-D and adjusting the stage position where the next time frame is to be scanned in real time during the course of the imaging [20]. The tissue drift profile for a 60-min acquisition following the distortion measurement (16 min) is shown in Figure 5C. The 3-D time-lapse of the acquired raw image sequence without synchronization, with our triggering scheme, distortion-corrected image stacks, and the crop of valid volume after distortion correction for the two datasets, as well as time-lapse of the corresponding MIPs are presented in Supplementary Videos 3–6.
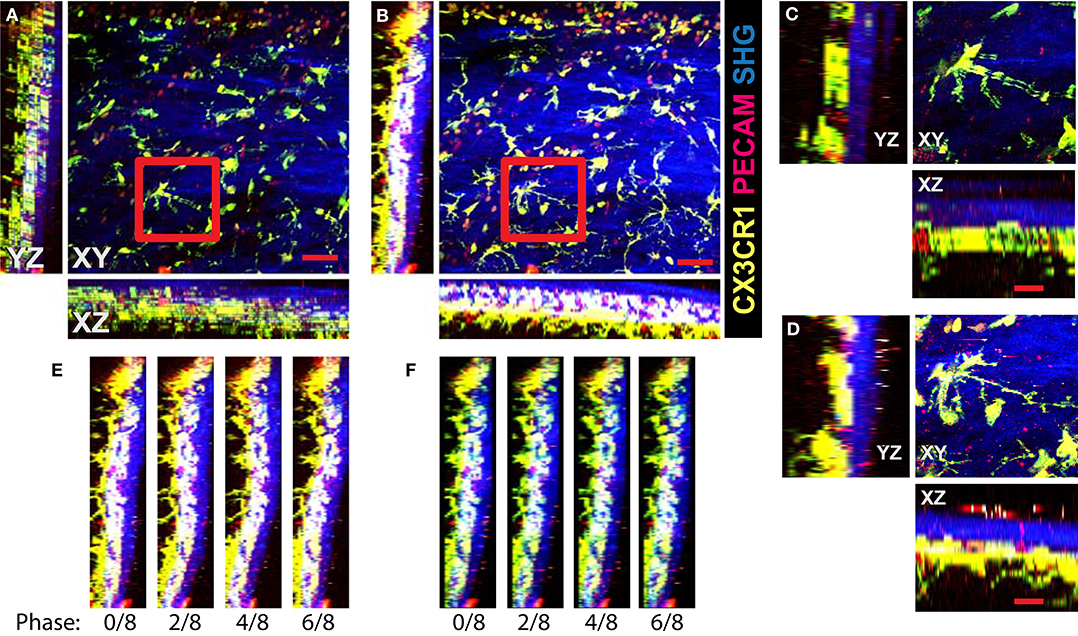
Figure 4. Image acquisition with the VivoFollow 2 system in the 2P-IVM imaging of the cervical spinal cord (SC) vasculature of mice. Maximum intensity projection (MIP) of the SC vasculature images in CX3CR1-GFP//CCR2-RFP reporter mouse. (A) Usual acquisition without triggering. Duplicates of the fine processes of GFP+ microglial cells are visible in XY projection (red frame). Due to severe motion, the depth information is incoherent and practically lost. Scale bar is 50 μm. (B) Acquisition of each Z frame is synchronized to respiration using TrigViFo. Three-dimensional volume continuity is recovered. (C,D) Zoom on the highlighted regions from (A,B) correspondingly. Scale bar is 20 μm. (E) YZ MIPs of the same region acquired at four different phases of respiration. Significant “wavy” distortion is present. (F) Same YZ MIPs after distortion correction. Smooth lining of the tissue is recovered.
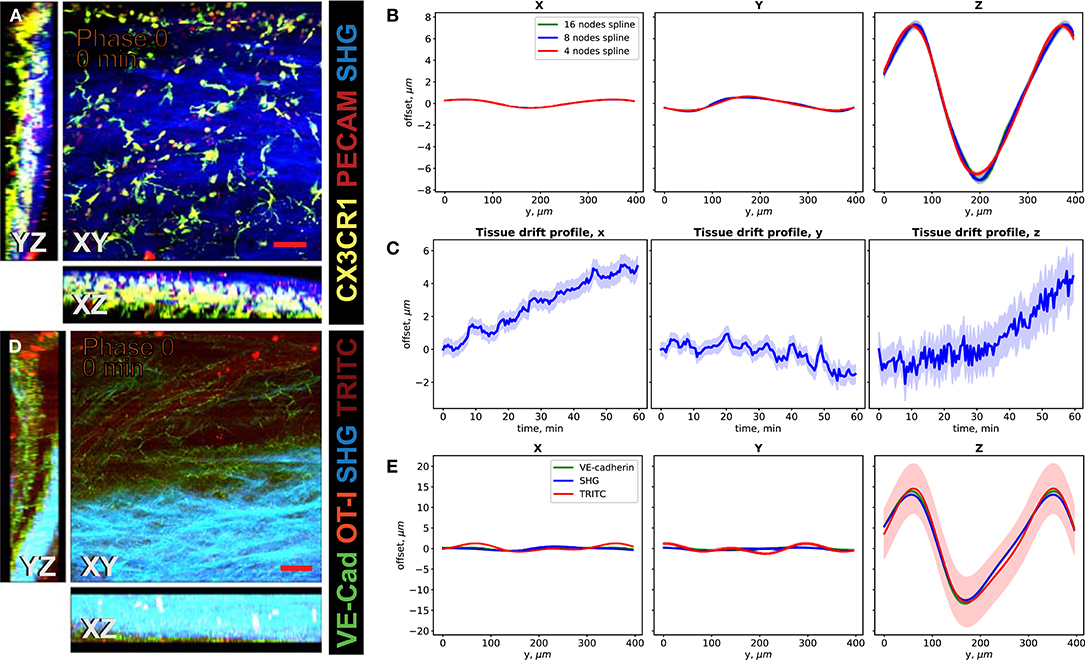
Figure 5. Performance of the VivoFollow 2 system in the 2P-IVM imaging of the cervical spinal cord (SC) vasculature of mice. (A) Maximum intensity projection (MIP) of an image stack of the SC vasculature images in a CX3CR1-GFP//CCR2-RFP reporter mouse after iterative distortion correction. (B) Comparison of periodic motion profile reconstructed by the CX3CR1-GFP signal as reference landmark with 4-, 8-, and 16-node spline fit. The offset is shown as a function of position on the image for phase 0. (C) Tissue drift profile over 60 min following the calibration stack acquisition (16 min). (D) MIP of the SC vasculature images in VE-cadherin-GFP knock-in mice after distortion correction. (E) Comparison of periodic motion profile reconstructed by the VE-cadherin-GFP, Second Harmonic Generation (SHG), and low concentration of TRITC dextran vascular tracer signals. The significant Z motion is well-reconstructed in all cases. Color bands in (B,C,E) show standard error.
We then used a dataset with three types of anatomical landmarks (Figure 5D), namely, the endogenous VE-Cadherin-GFP reporter, allowing to see adherens cellular junctions of endothelial cells lining the vascular walls, meningeal collagen layers (second harmonic generation, SHG), and an intravascular dextran tracer (TRITC) to assess the distortion mapping performance. These landmarks have different quality and coverage in the imaging volume. For example, we deliberately used a low concentration of the TRITC tracer to assess the performance on poor landmarks. In Figure 5E, the periodic motion profiles along the X, Y, and Z axes of the tissue motion reconstructed using the VE-Cadherin-GFP, SHG, and TRITC signals are shown. We used the four-node spline fit for this dataset. Using an eight-node fit led to divergence of the fit curve because not enough spatial information is available. This is consistent with the synthetic dataset results (Supplementary Figure 4). Flexibility in the number of the spline fit nodes allows us to apply this method to a wide variety of imaging settings. The motion pattern spline fit allows the method to perform robustly also in cases when anatomical landmark coverage is sparse. For example, the SHG signal covers <50% of the FoV. One can also see that the reconstructed Z motion profile, which has the highest impact on the final motility parameter estimation, is reconstructed well in all cases, even for the low signal-to-noise ratio TRITC signal (Figure 5E, red line). The reconstructed distortion corrected image stacks enabled subsequent tracking of the motility of immune cells in the bloodstream, as visualized by tdTomato+ CD8 T cells (Figure 5D and Supplementary Videos 7, 8).
Discussion and Outlook
In this study, we have developed the VivoFollow 2 system, which allows for the measurement and correction of distortion caused by periodic motion, such as animal breathing activity during relatively slow (i.e., acquisition frame rate is comparable with breathing rate) 2P-IVM image acquisition. This system enables further precise localization of objects of interest over time, which is, for example, a prerequisite for the precise measurement of cell motility parameters, movement of humoral factors, or sites of cellular extravasation from blood vessels. While previously described methods discard distorted frames [24] or recover undistorted images by combining tiles of multiple image stacks using pattern matching [25] or gating schemes [26], our method recovers one image stack corresponding to the optical plane tomographic tissue sectioning from each single raw image stack. Preserving the time continuity within each 3-D image stack allows this method to be used for time-lapse imaging of dynamic processes in vivo, such as cell interactions and cell migration. Adopting this method requires minimal changes to the usual 2P-IVM setup, namely, triggering the single frames acquisition and access to the pump of a suitable animal anesthesia ventilation system to perform readout of its motion. In addition, by relying mainly on image processing, the method does not require a piezo actuator for image acquisition. The image acquisition speed is slightly reduced by the increased delay between image frames due to the necessity of synchronization and an increased imaging depth. However, since the method does not involve a gating scheme, the effect on the overall image acquisition speed remains minimal (less than one respiration cycle per frame).
We have demonstrated the applicability of the developed VivoFollow 2 system to spinal cord window preparations of mice that feature different types of anatomical landmarks that we have successfully used for distortion mapping and correction. Most importantly, the implemented spline fit of the motion pattern allows the system to be applicable even in cases when the anatomical landmark coverage in the region of interest is sparse. Finally, by measuring and applying the distortion correction in real time, we were able to perform the tissue drift correction in regions subjected to significant periodic motion.
While in this work we have focused on a single imaging region, the presented method is directly applicable to any imaging of a periodically moving sample with the motion frequency comparable to the image acquisition frame rate. For example, abdominal organs or even the lung are subjected to periodic motion caused by the animal breathing, and the acquired images exhibit the same motion aberrations. Furthermore, this technique can be applied to faster imaging modalities, such as spinning disc confocal microscopy or 2P-IVM employing the resonant laser scanning, when motion is negligible within a single Z frame but remains significant in a Z stack. In this case, one could perform synchronization on the whole Z stack acquisition instead of synchronizing each single frame. By applying the same procedure, the distortion map would be found as a function of the depth Z. Alternatively, fast imaging modalities can benefit from this method when imaging tissue regions subjected to fast periodic motion, such as the heartbeat. The only difference in this case would be the synchronization to the electrocardiogram of the animal, instead of respiration.
One current limitation of the method is the requirement of inhalation anesthesia with intubation, allowing for external breathing control, and consecutively stable periodic tissue motion. While many experiments employ isoflurane anesthesia with intubation, a significant amount of experiments relies on injection anesthesia and recently also perform imaging of awake mice. Demanding the use of intubation would put additional hurdles on the often already complex experimental setups. We plan to address this issue in our further work by performing synchronization for experimental setups, which do not employ ventilators. Also, as previously mentioned, the employed triggering scheme can affect acquisition speed. The increase of acquisition time depends on the imaging settings, ranging between zero and one respiration period per image frame. Researchers should bear this effect in mind when choosing imaging parameters. Finally, since the excitation laser power is adjusted with depth for each image frame during deep tissue imaging, the fluorescent signal brightness will exhibit some degree of inconsistency due to sample movement, the same way as in usual triggered acquisition. While currently we did not address the compensation of the fluorescent signal intensity, with our approach, this becomes feasible due to knowledge of the absolute position of the tissue at each location. This will open the possibility to perform quantitative analysis of the fluorescent signals and, for example, study the T-cell activation dynamics employing fluorescent reporter mouse lines in a moving tissue.
Materials and Equipment
Mice
C57BL/6J mice were obtained from Janvier Labs (Genest Saint Isle, France). VE-cadherin-GFP knock-in mice express a C-terminal GFP fusion protein of VE-cadherin in the endogenous VE-cadherin locus [27, 28]. VE-cadherin-GFP knock-in mice were used for visualizing and imaging of the endothelial adherens junctions [18]. CX3CR1GFP/+::CCR2RFP/+ mice were kindly provided by Dr. Israel Charo and Dr. Richard Ransohoff [29] and were generated by crossbreeding CX3CR1GFP/+ mice (B6.129P-Cx3cr1tm1Litt/J) with CCR2RFP/+ mice [B6.129(Cg)-Ccr2tm2.1Ifc/J] [30]. OT-I tdTomato mice were kindly provided by Dr. Jens Stein (University of Fribourg, Switzerland) and were generated by crossing OT-I mice [C57BL/6-Tg(TcraTcrb)1100Mjb/J] [31] with Ai14 mice [B6.Cg-Gt (ROSA)26Sortm14(CAG−tdTomato)Hze/J]. Before crossing to OT-I mice, the stop cassette was removed from the Ai14 mice by breeding to Tg(ZP3-cre)93Knw. Prior to the experiment, the Ai14.1 allele was bred to homozygosity. All transgenic mice were backcrossed to a C57BL/6J background for at least eight generations. Mice were housed in individually ventilated cages under specific pathogen-free conditions. Adult female and male animals, 8–10 weeks old were used in the experiments. All animal experiments were performed in accordance with the Swiss legislation on the protection of animals and were approved by the veterinary office of the Canton of Bern (permission BE31/17).
Cervical Spinal Cord Window Preparation
Preparation of the cervical spinal cord window was performed as described previously [18]. Briefly, a carotid catheter pointing toward the aortic arch was placed in the right carotid artery of an anesthetized with fentanyl/midazolam/medetomidine mouse for systemic injection of exogenously labeled cells, fluorochrome-labeled plasma markers, or antibodies via the arterial blood supply prior to the imaging. Tracheotomy was done to place the tracheal cannula and connect it via a tube to a mouse ventilator (MiniVent, Harvard Apparatus) providing the humidified gas mixture to the mouse (1% isoflurane in a mixture of 40% O2 and 60% air, tidal volume of 175 μl, inspiration rate of 120 strokes/min). Laminectomy was then performed to expose the mouse cervical spinal cord at the level of C2–C6. The dura mater was kept intact to protect the spinal cord tissue. The physiological parameters (ECG and temperature) of the anesthetized mouse were monitored during the preparative surgery and imaging.
Imaging of Immune Cell Interaction With the Cervical Spinal Cord Postcapillary Venules in Mice
Fluorescent Labeling of Endogenous Blood Cells
To image the interaction of fluorescently labeled endogenous blood cells with the spinal cord microvessels, a non-function-blocking rat anti-mouse PECAM-1 antibody (mAb 390) [32] conjugated to Alexa Fluor 555 was systemically injected via the carotid artery catheter into the surgically prepared mouse (30 μg/mouse). mAb 390 was kindly provided by Dr. Steven Albelda (Philadelphia, USA). The systemic injection of the PECAM-1 antibody allows for direct labeling of endogenous blood cells, including platelets, monocytes, neutrophils, and subsets of T cells that are circulating in the cervical spinal cord vasculature.
Adoptive Transfer of CD8+ T Cells Expressing a Fluorescent Protein (tdTomato Reporter)
The interaction of adoptively transferred tdTomato CD8+ T cells with the postcapillary venules of the mouse spinal cord was imaged. To provide the in vitro activated CD8+ T cells for the adoptive transfer, the single cell suspension was prepared from the spleen and lymph nodes of T-cell receptor (TCR) transgenic OT-I tdTomato mice. The purification and in vitro activation of CD8+ T cells were performed as previously described [31]. Prior to the imaging, 4–5 × 106 in vitro activated OT-I CD8+ T cells were suspended in pre-warmed (37°C) 0.9% isotonic NaCl and then injected via the carotid catheter into the surgically prepared VE-cadherin GFP mice. In addition to visible endothelial adherens junctions of the blood vessels in VE-cadherin GFP mice, 1% TRITC-conjugated dextran (MW = 155,000, Sigma-Aldrich, Switzerland) was applied as a plasma marker via intra-carotid artery injection [18].
Image Processing
Image and video preparation for this publication was performed with ImageJ2 and Imaris 9.2 software packages.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The animal study was reviewed and approved by Veterinary office of the Canton of Bern (permission BE31/17).
Author Contributions
MV implemented the hardware and software for the described system, helped with experimental design and imaging, and wrote the manuscript. NH performed experiments and contributed to manuscript writing. EK performed the experiments. AA and BE provided guidance, coordinated this study, and wrote the manuscript.
Funding
This work was supported by the Swiss National Science Foundation interdisciplinary grant CR23I3_156234 to AA, a University of Bern ID grant to AA and BE, and a Fidelity Bermuda Foundation research grant to BE.
Licensing
A license is required for the nonacademic and/or for-profit use of the VivoFollow 2 system.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Urban Deutsch for managing our mouse breeding, Dr. Giuseppe Locatelli for the support during the imaging sessions, and Dr. Steven Albelda for kindly providing the PECAM-1 antibodies.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2019.00222/full#supplementary-material
References
1. Mempel TR, Scimone ML, Mora JR, von Andrian UH. In vivo imaging of leukocyte trafficking in blood vessels and tissues. Curr Opin Immunol. (2004) 16:406–17. doi: 10.1016/j.coi.2004.05.018
2. Germain RN, Miller MJ, Dustin ML, Nussenzweig MC. Dynamic imaging of the immune system: progress, pitfalls and promise. Nat Rev Immunol. (2006) 6:497–507. doi: 10.1038/nri1884
3. Kawakami N, Bartholomäus I, Pesic M, Kyratsous NI. Intravital imaging of autoreactive T cells in living animals. Methods Cell Biol. (2013) 113:149–68. doi: 10.1016/B978-0-12-407239-8.00008-2
4. Miller MJ, Wei SH, Cahalan MD, Parker I. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc Natl Acad Sci USA. (2003) 100:2604–9. doi: 10.1073/pnas.2628040100
5. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. (2004) 427:154–9. doi: 10.1038/nature02238
6. Pai S, Danne KJ, Qin J, Cavanagh LL, Smith A, Hickey MJ, et al. Visualizing leukocyte trafficking in the living brain with 2-photon intravital microscopy. Front Cell Neurosci. (2013) 6:67. doi: 10.3389/fncel.2012.00067
7. Flügel A, Odoardi F, Nosov M, Kawakami N. Autoaggressive effector T cells in the course of experimental autoimmune encephalomyelitis visualized in the light of two-photon microscopy. J Neuroimmunol. (2007) 191:86–97. doi: 10.1016/j.jneuroim.2007.09.017
8. Zenaro E, Rossi B, Angiari S, Constantin G. Use of imaging to study leukocyte trafficking in the central nervous system. Immunol Cell Biol. (2013) 91:271–80. doi: 10.1038/icb.2012.81
9. Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer. (2002) 2:266–76. doi: 10.1038/nrc778
10. Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. (2003) 3:921–30. doi: 10.1038/nrc1231
11. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. (2017) 18:123–31. doi: 10.1038/ni.3666
12. Piccio L, Rossi B, Scarpini E, Laudanna C, Giagulli C, Issekutz AC, et al. Molecular mechanisms involved in lymphocyte recruitment in inflamed brain microvessels: critical roles for P-selectin glycoprotein ligand-1 and heterotrimeric Gi-linked receptors. J. Immunol. (2014) 168:1940–9. doi: 10.4049/jimmunol.168.4.1940
13. Battistini L, Piccio L, Rossi B, Bach S, Galgani S, Gasperini C, et al. CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: a critical role for P-selectin glycoprotein ligand-1. Blood. (2003) 101:4775–82. doi: 10.1182/blood-2002-10-3309
14. Siffrin V, Brandt AU, Radbruch H, Herz J, Boldakowa N, Leuenberger T, et al. Differential immune cell dynamics in the CNS cause CD4 T cell compartmentalization. Brain. (2009) 132:1247–58. doi: 10.1093/brain/awn354
15. Jolivel V, Bicker F, Binamé F, Ploen R, Keller S, Gollan R, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. (2015) 129:279–95. doi: 10.1007/s00401-014-1372-1
16. Niesner R, Siffrin V, Zipp F. Two-photon imaging of immune cells in neural tissue. Cold Spring Harb Protoc. (2013) 8:224–30. doi: 10.1101/pdb.prot073528
17. Schläger C, Litke T, Flügel A, Odoardi F. In Vivo Visualization of (Auto)Immune Processes in the Central Nervous System of Rodents. New York, NY: Humana Press (2014). p. 117–29.
18. Haghayegh Jahromi N, Tardent H, Enzmann G, Deutsch U, Kawakami N, Bittner S, et al. A novel cervical spinal cord window preparation allows for two-photon imaging of T-cell interactions with the cervical spinal cord microvasculature during experimental autoimmune encephalomyelitis. Front Immunol. (2017) 8:406. doi: 10.3389/fimmu.2017.00406
19. Thévenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process. (1998) 7:27–41. doi: 10.1109/83.650848
20. Vladymyrov M, Abe J, Moalli F, Stein JV, Ariga A. Real-time tissue offset correction system for intravital multiphoton microscopy. J Immunol Methods. (2016) 438:35–41. doi: 10.1016/j.jim.2016.08.004
21. Vinegoni C, Lee S, Weissleder R. Image processing technologies for motion compensation. In: Fong Y, Giulianotti PC, Lewis J, Koerkamp BG, and Reiner T, editors. Imaging and Visualization in The Modern Operating Room. New York, NY: Springer (2015). p. 181–191.
22. Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, et al. ImageJ2: imageJ for the next generation of scientific image data. BMC Bioinformatics. (2017) 18:529. doi: 10.1186/s12859-017-1934-z
23. Hecker D, Kappler J, Glassmann A, Schilling K, Alt W. Image analysis of time-lapse movies—a precision control guided approach to correct motion artefacts. J Neurosci Methods. (2008) 172:67–73. doi: 10.1016/j.jneumeth.2008.04.010
24. Soulet D, Paré A, Coste J, Lacroix S. Automated filtering of intrinsic movement artifacts during two-photon intravital microscopy. PLoS ONE. (2013) 8:e53942. doi: 10.1371/journal.pone.0053942
25. Lee S, Vinegoni C, Sebas M, Weissleder R. Automated motion artifact removal for intravital microscopy, without a priori information. Sci Rep. (2015) 4:4507. doi: 10.1038/srep04507
26. Vinegoni C, Aguirre AD, Lee S, Weissleder R. Imaging the beating heart in the mouse using intravital microscopy techniques. Nat Protoc. (2015) 10:1802–19. doi: 10.1038/nprot.2015.119
27. Abadier M, Haghayegh Jahromi N, Cardoso Alves L, Boscacci R, Vestweber D, Barnum S, et al. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur J Immunol. (2015) 45:1043–58. doi: 10.1002/eji.201445125
28. Winderlich M, Keller L, Cagna G, Broermann A, Kamenyeva O, Kiefer F, et al. VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol. (2009) 185:657–71. doi: 10.1083/jcb.200811159
29. Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE. (2010) 5:e13693. doi: 10.1371/journal.pone.0013693
30. Locatelli G, Theodorou D, Kendirli A, Jordão MJC, Staszewski O, Phulphagar K, et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat Neurosci. (2018) 21:1196–208. doi: 10.1038/s41593-018-0212-3
31. Rudolph H, Klopstein A, Gruber I, Blatti C, Lyck R, Engelhardt B. Postarrest stalling rather than crawling favors CD8 + over CD4 + T-cell migration across the blood-brain barrier under flow in vitro. Eur J Immunol. (2016) 46:2187–203. doi: 10.1002/eji.201546251
Keywords: intravital microscopy, multiphoton microscopy, tissue motion, GPU-accelerated data processing, real-time image processing, immune cell trafficking, cell migration
Citation: Vladymyrov M, Haghayegh Jahromi N, Kaba E, Engelhardt B and Ariga A (2020) VivoFollow 2: Distortion-Free Multiphoton Intravital Imaging. Front. Phys. 7:222. doi: 10.3389/fphy.2019.00222
Received: 02 September 2019; Accepted: 02 December 2019;
Published: 17 January 2020.
Edited by:
Zhen Cheng, Stanford University, United StatesReviewed by:
Xin Liu, University of Queensland, AustraliaHongguang Liu, Northeastern University, China
Copyright © 2020 Vladymyrov, Haghayegh Jahromi, Kaba, Engelhardt and Ariga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mykhailo Vladymyrov, bXlraGFpbG8udmxhZHlteXJvdkB0a2kudW5pYmUuY2g=
†Present address: Neda Haghayegh Jahromi, Institute of Pharmaceutical Sciences, ETH Zürich, Zurich, Switzerland