 Pablo Pérez-Ferreros1,2
Pablo Pérez-Ferreros1,2 Katharina Gaus
Katharina Gaus Jesse Goyette
Jesse Goyette- 1EMBL Australia Node in Single Molecule Science, University of New South Wales, Sydney, NSW, Australia
- 2ARC Centre of Excellence in Advanced Molecular imaging, University of New South Wales, Sydney, NSW, Australia
Leukocytes play critical roles in preventing pathogenic infection and controlling transformed cells, but must remain quiescent in response to healthy tissue. To execute this function, immune cells need to integrate signals from a host of activatory, co-activatory, and co-inhibitory immune receptors. When an immune cell interacts with another cell containing ligands for these receptors, an immunological synapse is formed at the contact interface that acts as a dynamic signaling hub into which cytoplasmic enzymes are recruited and tethered. Within this interface competing tethered enzymatic activities are integrated, ultimately leading to the cellular decision to respond or remain quiescent. Here, we review recent advances in our understanding of tethered signaling reactions, focusing on proximal signaling downstream of important T cell immune receptors. We discuss how a class of co-inhibitory receptors require co-localization with activatory receptors to function, how recent evidence that T cells use microvilli to probe antigen presenting cell surfaces may be important for immune receptor function, and how co-clustering between activatory and inhibitory receptors facilitates integration of tethered reactions.
Introduction
T cells play a critical role in the adaptive immune system, in which they must recognize and respond to foreign antigens whilst remaining quiescent to normal tissue antigens. Upon activation T cells undergo clonal expansion, differentiation, and mediate effector functions such as cytokine production and direct target cell killing [1].
Fundamentally, T cell function is dependent on the orchestration of signals generated by receptors found in their plasma membrane. T cells are activated when T cell receptors (TCRs) on their plasma membrane interact with cognate peptide antigen-major histocompatibility complexes (pMHC) presented on the surface of antigen presenting cells (APCs). These interactions occur at the contact interface between T cells and APCs that, upon antigen recognition, matures into a structure known as the immunological synapse [2]. In addition to the TCR there are numerous co-stimulatory and co-inhibitory receptors, the signals from which must be integrated to culminate in a specific output: the cell response [3]. Due to this integration of positive and negative regulation, APCs can prevent, dampen or enhance a potential response of the T cells by the expression of ligands for co-inhibitory or co-activatory receptors.
During the process of maturation, auto-reactive T cells are eliminated in the thymus [4], however, some autoreactive T cells escape thymic selection, and migrate to the periphery. Inhibitory receptors play an important role in restoring tolerance and preventing self-reactive T cells from initiating auto-immune reactions [4–10]. Inhibitory receptors that block the activation of T cells are also known as immune checkpoint receptors for their ability to prevent inappropriate autoreactive immune responses. Although this system is effective at protecting healthy cells from being attacked by T cells, some cancer cells and chronic viral infections take advantage of this protection mechanism, expressing high levels of the ligands for inhibitory receptors [11–13]. Due to the importance of immune checkpoint receptors, they have become increasingly important drug targets. Some of the most studied inhibitory receptors are Cytotoxic T lymphocyte-associated protein 4 (CTLA-4), Programmed death-1 (PD-1) and B and T lymphocyte attenuator (BTLA), with drugs targeting CTLA-4 and PD-1 already developed into frontline treatments for many cancers [14–17].
Here we review the mechanism of signal integration for a class of inhibitory receptors that function by recruiting cytosolic phosphatases. Specifically, we highlight how physical properties of the membrane and consequences of receptor-ligand interactions occurring at cell-cell interfaces may be important determinants of signal integration between these inhibitory receptors and the activatory signaling they modify.
Inhibitory and Activatory Receptors Need to be in Close Proximity to Integrate Their Signals
The TCR is a multi-chain complex consisting of alpha and beta subunits, responsible for binding pMHC, and CD3 chains (epsilon-delta, epsilon-gamma and zeta-zeta dimers), responsible for signaling. Upon TCR-pMHC binding, tyrosine residues within immunotyrosine-based activation motifs (ITAMs) present in the intracellular tails of the CD3 chains are phosphorylated by the Src-family kinase Lck. Phosphorylated ITAMs recruit the cytoplasmic kinase ZAP70 to the TCR complex, where the enzyme becomes activated and phosphorylates the membrane adaptor protein linker for activated T cells (LAT). Phosphorylated LAT in turn recruits other cytosolic adaptors that propagate the signal leading to full T cell activation [18, 19].
TCR mediated cellular activation can be modified by a number of co-stimulatory and co-inhibitory receptors [3]. Co-stimulatory receptors, such as CD28, can amplify T cell responses and in many contexts are essential for robust T cell mediated immune responses [20]. Similar to the TCR, when CD28 binds either of its ligands, CD80 and CD86, tyrosines on the intracellular tails are phosphorylated by Lck. These phosphorylated tyrosines recruit cytosolic mediators, such as phosphoinositide 3-kinase and Grb2, thereby augmenting T cell activation [21].
Conversely, co-inhibitory receptors, including PD-1, halt T cell activation [3, 22]. There are several mechanisms by which inhibitory receptors interfere with T cell activation. For example CTLA-4 competes with CD28 for CD80 and CD86 and thus limits co-activation via CD28. Alternatively, co-inhibitory receptors produce inhibitory intercellular signals that by recruiting cytosolic phosphatases that are subsequently integrated into the activating signals of the TCR and co-stimulatory receptors. In this category of co-inhibitory receptors, in which PD-1 belongs, the physical principles of signal integration are key. This subset of receptors has intracellular tails that harbor immunotyrosine-based inhibitory motifs (ITIM) or immunotyrosine-based switch motifs (ITSM), which are phosphorylated by Lck when these receptors bind their cognate ligand. Once phosphorylated, the endodomains of inhibitory receptors recruit phosphatases, such as Src homology region 2 domain-containing phosphatase-1 (SHP-1) or Src homology region 2 domain-containing phosphatase-2 (SHP-2) that dephosphorylate activatory receptors, or SH2 domain-containing inositol 5′-phosphatase (SHIP) that dephosphorylates phosphatidylinositol-3,4,5-trisphosphate [23].
Cellular studies have shown that co-ligation of PD-1 with TCR and/or CD28 results in decreased phosphorylation of CD3 chains, CD28 and proteins of the downstream cascade such as Vav1, protein kinase C (PKC)-θ and extracellular signal-regulated kinase (ERK)1/2 [24–26] and in vitro reconstitution experiments show that SHP-2 recruited to PD-1 can directly dephosphorylate CD3ζ and CD28 cytoplasmic tails [22]. At this point it is useful to make a distinction between receptor triggering and signal propagation. Van der Merwe and Dushek summarized it best when they defined TCR triggering as “[t]he process by which TCR binding to peptide–MHC molecules leads to biochemical changes in the cytoplasmic regions of the CD3 complex…” [27]. From this perspective, events such as ZAP70 recruitment or phosphorylation of LAT can be considered signal propagation downstream of TCR triggering. There are many competing models of TCR triggering [27, 28], but it is hard to imagine how a receptor like PD-1 might realistically interfere with any of the proposed triggering processes. Rather, the available data indicates that PD-1 intervenes post-triggering of individual TCR or CD28 receptors by decreasing the amount of phosphorylated receptor at any one time, or the lifetime of individual phosphotyrosines, on activatory receptors.
The integration of phosphatase activity tethered to co-inhibitory receptors with the kinase activity tethered to the TCR and co-inhibitory receptors can only occur if the spatial organization is such that the reach length of the phosphatase and kinase overlap. Since both signaling enzymes are tethered to the membrane via the recruitment to the respective receptors, it has become critical to examine the co-localization and co-clustering of activatory or co-activatory receptors in biological membranes. When ligands for PD-1, TCR, and CD28 are presented on the same surface the engaged receptors co-localize within clusters in the T cell [22, 26]. In an elegant series of experiments, Yokosuka et al. used PD-1 constructs with ectodomains elongated to different extents to demonstrate that the degree of mismatch between TCR/pMHC and PD-1/PD-L1 dimensions correlated inversely with co-localization and inhibitory function [26]. Elongating receptor-ligand interactions can have the additional effect of reducing the efficiency of receptor phosphorylation, which can be understood in the context of the kinetic segregation mechanism [29], and to separate this effect the authors used PD-1 constructs with SHP-2 fused to the intracellular tail, thus circumventing the need for triggering. These experiments build on earlier work by Köhler et al. and Eriksson et al. who demonstrated a similar principle with inhibitory and activatory receptors in NK cells [30, 31]. Accordingly, it was noted that most known inhibitory and stimulatory receptors that bind surface-associated ligands form similar ligand-receptor dimensions, which are smaller than the large surface phosphatases that regulate their triggering [32], suggesting that this is a common requirement for signal integration between activatory and co-inhibitory receptors. Taken together, it seems that the degree of co-localization of some receptors is an important determinant in the integration of their function.
Before we review the complex mechanisms that underlie receptor co-localization, we would like to point out that co-localization does not seem to be a requirement for CD28-mediated co-stimulation, and is thus not a universal requirement for T cell signal integration. If ligands for CD28 are presented on separate cells from agonistic pMHC molecules, T cells still have enhanced responses, although the effect appears to be less efficient than if both ligands are presented on the same cell surface [33].
Mechanics of Receptor-Ligand Interactions at Cell-Cell Interfaces
Bonds occurring between membrane-bound receptors and ligands are subject to forces that have consequences for the organization of proteins at the surface. In this section we will review the evidence for how these forces impact on the formation of initial bonds and how they result in the size-based segregation and clustering of surface proteins.
For bonds to occur between the TCR, co-inhibitory and co-activatory receptors and their respective membrane-bound ligands, the T cell and APC membranes must be the right distance apart. However, most immune receptor-ligand complexes span a short distance compared with the length of the glycocalyx. Given this size difference it is relevant to ask how bonds between smaller receptor-ligand pairs occur at all. To overcome the glycocalyx, which normally repulses the close apposition of two cell membranes, T cells actively probe APCs using thin (100–200 nm) actin-dependent finger-like membranous protrusions known as microvilli [34, 35] (Figure 1). These processes are highly dynamic and are continuously protruding and retracting [35]. Using a combination of super-resolution microscopy approaches Jung et al. suggested that these flexible projections are enriched with adhesion molecules and TCRs. However, it should be noted that all the molecules the authors investigated appeared to be enriched at the tip of microvilli, with the exception of CD45, which was modestly depleted [34]. The tips of microvilli were closer to the coverslip in their experiments and the authors may not have adequately corrected for the greater signal-to-noise at these points, which would be reflected in a greater probability of detection. Nevertheless, mounting evidence suggests that T cells probe the surface of APCs with these microvilli and that these are likely the sites of the initial bonds formed between the TCR, co-inhibitory and co-activatory receptors [34, 35].
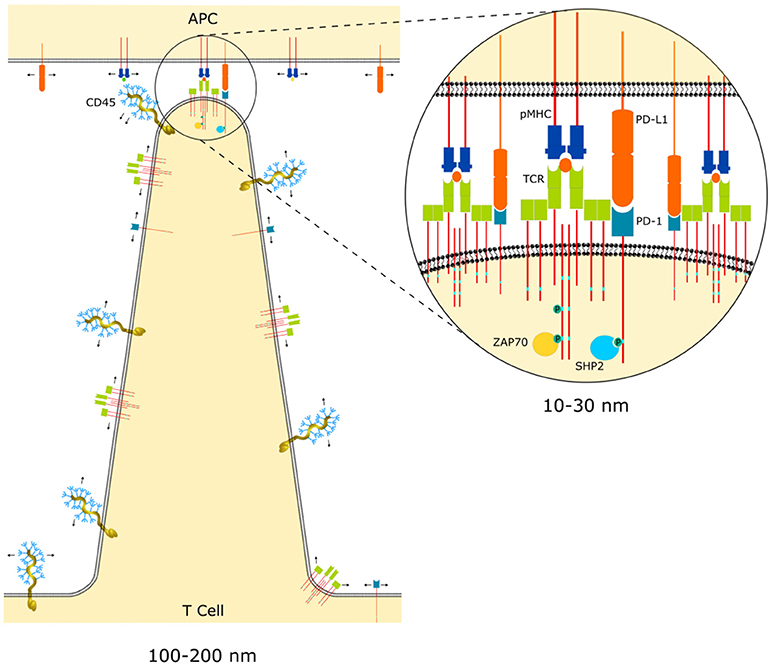
Figure 1. Microvilli of T cell probing an APC surface. The arrows indicate the free diffusion of unbound receptors on the membrane. This diffusion follows a random distribution of receptors except in the interface between the microvilli and the APC, where proteins larger than the size of the interface, such as CD45, are excluded. The zoomed in region illustrates how compatible receptor-ligand dimensions become co-clustered at points of close contact initiated at the tips of microvilli.
Once ligand-receptor interactions form across a cell-cell interface this has consequences for the organization of other proteins in the membrane. For example, the TCR-pMHC complex spans approximately 15 nm so that wherever these bonds exist the membranes of the two cells will be held apart at a distance equivalent to the complex dimensions (Figure 1). This gap will establish a size threshold that will segregate other surface proteins with an ectodomain larger than the interface height. CD45 is the best studied example of this and is excluded from points of TCR-pMHC bonds due to its large extracellular domain [36, 37]. This concept was also recently demonstrated in a reductionist system by Schmid et al. who used giant unilamellar vesicles coated with binding and non-binding fluorescent proteins of different dimensions [38]. Since this vesicular system lacked any cellular machinery the results demonstrate that segregation of proteins just 5 nm larger than the binding interface can occur purely as a result of the energy penalty caused by membrane bending to accommodate the larger protein.
Another important consequence of the uneven interface between the T cell and APC is that bonds formed between molecules on opposing membrane are much more likely to occur in regions where bonds of compatible dimensions already exist [39–41]. Thus, new ligand-receptor bonds gather around existing bonds, promoting the co-clustering of size-compatible ectodomain receptors, which is required for co-localization and signal integration between activatory and inhibitory receptors (Figure 1). As discussed earlier, initial interactions are likely formed at the tip of microvilli, but as the immune synapse evolves at later time points the contact interface flattens. Although, the nanoscale topology of the contact interface remains a topic of active investigation, receptor-ligand pairs with modestly different dimensions are known to segregate in the immunological synapse at late time points (several minutes after initial contact), with LFA1-ICAM-1, CD2-CD58, and TCR-pMHC forming a “bulls-eye” pattern of concentric circles in the synapse [42].
It should be noted here that TCR activation precedes full immunological synapse formation [43] and the typical “bulls-eye” pattern of the synapse (reviewed in detail elsewhere [2, 44]). It is easy to conflate events happening on the scale of the entire synapse (μm length scale, minutes time scale) with events happening at the scale of microvilli/clusters (100–200 nm length scale, 10 s of seconds time scale). This can be particularly confusing since the synapse-like patterns of integrin bonds surrounding TCR-pMHC bonds also occur in the form of micro-adhesion rings that surround TCR clusters at early timepoints [45]. However, as we will argue in the following section, signal integration between activatory and inhibitory receptors likely occurs at the scale of microvilli/clusters, and thus this distinction is important to keep in mind.
Physical Properties of Receptor Tails are an Important Determinant of Signal Integration
To understand why inhibitory receptors must co-localize with activatory receptors to function effectively we will conclude by more closely considering the molecular mechanisms of inhibitory signaling.
Proximal signaling downstream of the TCR and co-stimulatory receptor activation, initially occurs on the tails of the receptors, but eventually spreads to other proteins that are spatially separated from the receptors. In contrast, co-inhibitory receptors that recruit phosphatases do not seem to generate long-range inhibitory signals, but rather appear to function locally. The phosphatases that mediate inhibitory receptor function, SHP-1 and SHP-2, only become fully catalytically active when their SH2 domains are engaged by phosphorylated ITIM peptides [46, 47].
The concentration of substrate that SHP-1 or SHP-2 encounter when they are recruited to inhibitory receptors depends on the reach and flexibility of the cytoplasmic tail they are recruited to, the scale of co-localization with activatory receptors containing phosphorylated tyrosines, and the reach and flexibility of the cytoplasmic tail of the activatory receptors (Figure 2). The overall reaction rate is thus dictated by the binding kinetics between the recruited enzyme and the immune receptor tail, the intrinsic catalytic rate, and the reach and mechanical properties of the immune receptor tail. There are some similarities between these membrane-tethered reactions and signaling reactions on scaffolds [48]. However, since immune receptors diffuse in the membrane, cluster and co-cluster with other immune receptors the situation is analogous but more fluid than scaffold signaling and some authors have used the term “tethered signaling” to describe proximal signaling reactions occurring on the tails of immune receptors [49].
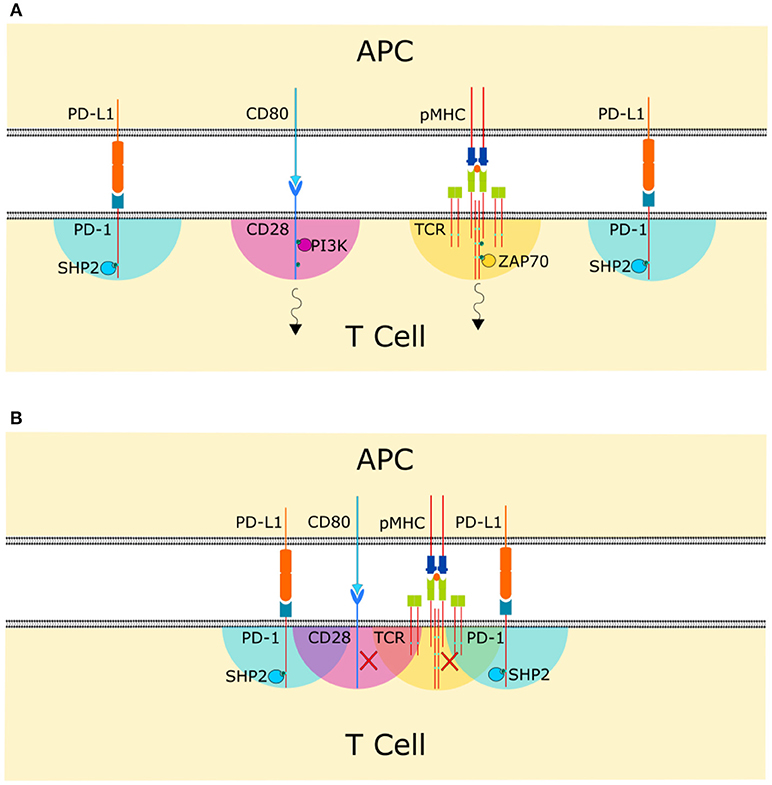
Figure 2. Co-clustering of compatible size receptor-ligand bonds in the cell-cell interphase allows signal integration. The colored areas indicate the span of action of each membrane tethered enzyme. (A) If receptor-ligand bonds are spaced too far apart at the cell-cell interface, inhibitory receptors are not able to carry out their function and activatory receptors can initiate their signaling cascade, indicated by the black arrows. (B) Co-clustering of receptor-ligand bonds allows the cytoplasmic domains to overlap and for phosphatases recruited to the cytoplasmic tails of co-inhibitory receptors to reach substrate phosphotyrosines, allowing signal integration.
Despite the importance of mechanics to tethered signaling reactions, the reach and flexibility of the cytoplasmic tails of activatory and inhibitory receptors remain largely unstudied. Local substrate concentration is an important consideration since tethering an enzyme to a poor substrate can override catalytic specificity and drive what would otherwise be an unfavorable reaction [50]. Mathematical modeling has been used to predict the local concentrations of enzymes tethered to substrates by unstructured linkers [51–53], which can dramatically enhance reaction rates. These studies often rely on modeling the unstructured regions of proteins the enzymes are tethered to, such as the tails of immune receptors, using the wormlike chain model. The worm-like chain model uses two parameters to describe the behavior of polymers: persistence length (quantifying the stiffness of the polymer) and contour length (the end-to-end length of the polymer). Although a good estimate, the specific sequence of the unstructured region can significantly affect the behavior [54, 55] and furthermore the size and orientation of the enzyme tethered to tail is often neglected. Windisch et al. used mesoscale modeling to address this second point finding that, for a tethered reaction in a bacterial chemotaxis receptor pathway, the physical size of the rigid enzyme impeded its ability to explore a significant portion of the tether-restricted space [51]. Given the difficulties in predicting the local enzyme concentrations on membrane tethers, we found it necessary to develop an experimental system that can capture the various parameters involved in tethered signaling independently.
To directly measure important parameters describing tethered signaling reactions, Goyette et al. recently described a novel biophysical assay utilizing surface plasmon resonance (SPR) [49]. The method can be used to measure the binding, catalytic and effective reach parameters for a phosphatase interacting with and dephosphorylating a phosphotyrosine-containing peptide immobilized on the SPR chip surface. The authors investigated the interaction of SHP-1 with an ITIM containing peptide derived from the inhibitory receptor leukocyte associated immunoglobulin like receptor 1 (LAIR1), finding that binding of the enzyme to the ITIM increased the catalytic rate 900-fold through a combination of allosteric activation and increased local substrate concentration [49]. Although the peptide used did not represent the full-length cytoplasmic tail of LAIR1 but was instead the ITIM portion with a PEG linker with shorter dimensions that the native tail, the results suggest that the enzyme itself contributed significantly to the effective reach length, which was measured to be 23 nm. Future studies using this technique with full length cytoplasmic tails of inhibitory receptors will hopefully shed some light on the effective reach and biophysical properties of tethered signaling reactions.
Based on the information with immobilized tethers, we know that the local concentration of recruited enzyme falls off rapidly with distance from the ligand-engaged receptor, highlighting the importance of close co-localization between receptors. Although we do not know the exact physical properties of immune receptor tails, for a reasonable estimate of receptor cytoplasmic tail length, a change in co-localization distance from 5 to 50 nm can cause a 1,000-fold decrease in effective enzyme concentration [49], assuming that no lateral diffusion takes place. Beyond the maximum reach of the tether, the recruited enzyme is unable to catalyze reactions. These considerations explain the observed requirement for co-localization and compatible receptor-ligand dimensions between activating and inhibitory receptors discussed in the previous section, but also suggest a relevant scale for the co-localization (<50 nm). This scale fits well with the dimensions of the tips of microvilli (Figure 1), suggesting that they could be the primary sites of signal integration. It also fits well with results from Cai et al. who patterned ligands for the TCR receptor at defined intermolecular distances and found a lateral spatial threshold of <50 nm to maintain robust signaling [56]. The authors note that this distance corresponds well the predicted reach length of intracellular tails of the TCR complex, suggesting that this allows the signaling to be reinforced. We are proposing a similar mechanism, with opposite consequences, for the co-clustering of activatory receptors such as TCR or CD28, with co-inhibitory receptors, such as PD-1.
While in the SPR assay and Cai et al's ligand patterning, proteins are immobilized on the surface, in the plasma membrane individual receptors can diffuse laterally. The diffusion of receptor-ligand bonds on opposing membranes at a cell-cell interface however, is less clear. While there are many imaging approaches to measure membrane diffusion [57] and we recently engineered a FRET sensor that measures TCR clustering dynamics [58], it is difficult to distinguish engaged from non-engaged receptors at cell interfaces with these technologies. The problem is confounded in that only ~23% of the TCRs signaled when stimulated with excess antigen or activating antibodies [20]. Thus, the diversity of receptors—engaged vs. non-engaged, signaling vs. non-signaling receptors—that are genetically and biochemically identical makes interpretation of bulk measurements difficult. In addition, we lack a detail understanding of the spatial organization when a T cell transitions from the first contact with an APC via microvilli to a mature synapse. The best indication we have is from single molecule imaging of live cells, which show that when the TCR binds pMHC on opposing membrane (in this case a supported lipid bilayer), the resulting ligand-receptor complex does not undergo free diffusion and is localized in the same diffraction-limited (>200 nm) spot on the timescale of seconds [59]. Ultimately, over the course of tens of seconds to minutes, the complex associates with the actin cytoskeleton and is transported to the center of the synapse where internalization and signal down-regulation occurs [42]. These experiments suggest that either complex formation drastically reduces mobility of the receptor and ligand, or the complex undergoes very confined diffusion within a small close-contact region, or both occur.
Finally, even the assumption that the tip of a microvillus or the mature synapse represents a 2-dimensional zone for ligand-receptor interactions is likely too simplistic. Even when opposing membranes are held very close to the optimal distance for binding, thermal fluctuations and cortical cytoskeletal stiffness can have large effects on binding kinetics on small scales (<100 × 100 nm) and receptor-ligand bonds can dampen thermal fluctuations, thus enhancing on-rates and reducing off-rates for compatible dimension receptors in their vicinity [60]. The theoretical upshot of this would again be distance-dependent synergy in compatible dimension receptor-ligand bond formation on a scale similar to the reach length of tethered reactions [49].
Concluding Remarks
A T cell must be able to integrate signals from inhibitory and activatory receptors. A possible mechanism is to use biophysical principles such as the spatial organization on the molecular scale to regulate biochemical reactions. This linkage occurs on two different levels: firstly, on the extracellular side, receptor engagement can lead to receptor clustering and co-localization and secondly, on the intracellular side, kinases and phosphatases can be tethered to the membrane by recruitment to phosophorylated receptor tails. The first is a result of the topology of the plasma membrane, including microvilli, together with passive mechanical forces acting to aggregate compatible dimension receptor-ligand complexes in close contact regions. Although some important specifics about the nanoscale topology of the contact interface and organization of receptors in the membrane remain uncertain, we propose a model in which these close contact points are the main site of co-inhibitory receptor signal integration. This model explains the requirement for compatible ectodomain dimensions between activatory and inhibitory ligand-receptor pairs and suggests that mechanical properties of receptor tails will have important consequences for the effectiveness of signal integration.
The relevant distances for these processes are on the molecular scale (tens of nanometres), which make them very difficult to study in situ. For this reason, modeling and reductionist systems have been important for highlighting the relevance of the biophysical properties of the cytoplasmic tails. With advances in microscopy techniques, and the development of complimentary biophysical methods, a clearer picture of how tethered signaling reactions occur within the complex environment of T cell-APC contact interfaces will emerge. As our understanding of signal integration processes becomes more nuanced it may become possible to manipulate and utilize them to construct more effective therapeutic strategies, such as synthetic chimeric antigen receptors [61].
Author Contributions
PP-F, JG, and KG made substantial intellectual contributions to writing and editing the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Australia Research Council (CE140100011 to KG) and National Health and Medical Research Council of Australia (APP1059278 to KG and APP1163814 to JG).
References
1. Wencker M, Turchinovich G, Di Marco Barros R, Deban L, Jandke A, Cope A, et al. Innate-like T cells straddle innate and adaptive immunity by altering antigen-receptor responsiveness. Nat Immunol. (2014) 15:80–7. doi: 10.1038/ni.2773
2. Alarcón B, Mestre D, Martínez-Martín N. The immunological synapse: a cause or consequence of T-cell receptor triggering? Immunology (2011) 133:420–5. doi: 10.1111/j.1365-2567.2011.03458.x
3. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibtion. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
4. Hogquist KA, Baldwin TA, Jameson SC. Central tolerance: learning self-control in the thymus. Nat Rev Immunol. (2005) 5:772–82. doi: 10.1038/nri1707
5. Schietinger A, Greenberg P. Tolerance and exhaustion: definin mechanisms of T cell dysfunction. Trends Immunol. (2014) 35:51–60. doi: 10.1016/j.it.2013.10.001
6. Hernandez J, Aung S, Redmond WL, Sherman LA. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J Exp Med. (2001) 194:707–17. doi: 10.1084/jem.194.6.707
7. Redmond WL, Marincek BC, Sherman LA. Distinct requirements for deletion versus anergy during CD8 T cell peripheral tolerance in vivo. J Immunol. (2005) 174:2046–53. doi: 10.4049/jimmunol.174.4.2046
8. Kurts C, Kosaka H, Carbone FR, Miller JFAP, Heath WR. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J Exp Med. (1997) 186:239–45. doi: 10.1084/jem.186.2.239
9. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. (2010) 11:7–13. doi: 10.1038/ni.1818
10. Schietinger A, Delrow JJ, Basom RS, Blattman JN, Greenberg PD. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science (2013) 335:723–7. doi: 10.1126/science.1214277
11. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. (2009) 229:114–25. doi: 10.1111/j.1600-065X.2009.00767.x
12. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443:350–4. doi: 10.1038/nature05115
13. Grakoui A, John Wherry E, Hanson HL, Walker C, Ahmed R. Turning on the off switch: regulation of anti-viral T cell responses in the liver by the PD-1/PD-L1 pathway. J Hepatol. (2006) 45:468–72. doi: 10.1016/j.jhep.2006.07.009
14. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
15. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
16. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
17. Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. A systematic review of immunotherapy in urologic cancer: evolving roles for targeting of CTLA-4, PD-1/PD-L1, and HLA-G. Eur Urol. (2015) 68:267–79. doi: 10.1016/j.eururo.2015.02.032
18. Malissen B, Grégoire C, Malissen M, Roncagalli R. Integrative biology of T cell activation. Nat Immunol. (2014) 15:790–7. doi: 10.1038/ni.2959
19. Roncagalli R, Hauri S, Fiore F, Liang Y, Chen Z, Sansoni A, et al. Quantitative proteomics analysis of signalosome dynamics in primary T cells identifies the surface receptor CD6 as a Lat adaptor–independent TCR signaling hub. Nat Immunol. (2014) 15:384–92. doi: 10.1038/ni.2843
20. Pageon SV, Tabarin T, Yamamoto Y, Ma Y, Bridgeman JS, Cohnen A, et al. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc Natl Acad Sci USA. (2016) 113:E5454–63. doi: 10.1073/pnas.1607436113
21. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. (2003) 3:939–51. doi: 10.1038/nri1248
22. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
23. Long EO, Sik Kim H, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. (2013) 31:227–58. doi: 10.1146/annurev-immunol-020711-075005
24. Chemnitz JM, Parry R V, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. (2004) 173:945–54. doi: 10.4049/jimmunol.173.2.945
25. Sheppard K-A, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
26. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
27. van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. (2011) 11:47–55. doi: 10.1038/nri2887
28. Malissen B, Bongrand P. Early T cell activation: integrating biochemical, structural, and biophysical cues. Annu Rev Immunol. (2015) 33:539–61. doi: 10.1146/annurev-immunol-032414-112158
29. Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol. (2006) 7:803–9. doi: 10.1038/ni1369
30. Köhler K, Xiong S, Brzostek J, Mehrabi M, Eissmann P, Harrison A, et al. Matched sizes of activating and inhibitory receptor/ligand pairs are required for optimal signal integration by human natural killer cells. PLoS ONE (2010) 5:e15374. doi: 10.1371/journal.pone.0015374
31. Eriksson M, Leitz G, Fällman E, Axner O, Ryan JC, Nakamura MC, et al. Inhibitory receptors alter natural killer cell interactions with target cells yet allow simultaneous killing of susceptible targets. J Exp Med. (1999) 190:1005–12.
32. Dushek O, Goyette J, Van der Merwe PA. Non-catalytic tyrosine- phosphorylated receptors. Immunol Rev. (2012) 250:258–76. doi: 10.1111/imr.12008
33. Lim H-S, Cordoba S-P, Dushek O, Goyette J, Taylor A, Rudd CE, et al. Costimulation of IL-2 production through CD28 is dependent on the size of its ligand. J Immunol. (2015) 195:5432–9. doi: 10.4049/jimmunol.1500707
34. Jung Y, Riven I, Feigelson SW, Kartvelishvily E, Tohya K, Miyasaka M, et al. Three-dimensional localization of T-cell receptors in relation to microvilli using a combination of superresolution microscopies. Proc Natl Acad Sci USA. (2016) 113:E5916–24. doi: 10.1073/pnas.1605399113
35. Cai E, Marchuk K, Beemiller P, Beppler C, Rubashkin MG, Weaver VM, et al. Visualizing dynamic microvillar search and stabilization during ligand detection by T cells. Science (2017) 356:eaal3118. doi: 10.1126/science.aal3118
36. Chang VT, Fernandes RA, Ganzinger KA, Lee SF, Siebold C, McColl J, et al. Initiation of T cell signaling by CD45 segregation at “close contacts.” Nat Immunol. (2016) 17:574–82. doi: 10.1038/ni.3392
37. Cordoba S-P, Choudhuri K, Zhang H, Bridge M, Basat AB, Dustin ML, et al. The large ectodomains of CD45 and CD148 regulate their segregation from and inhibition of ligated T-cell receptor. Blood (2013) 121:4295–302. doi: 10.1182/blood-2012-07-442251
38. Schmid EM, Bakalar MH, Choudhuri K, Weichsel J, Ann HS, Geissler PL, et al. Size-dependent protein segregation at membrane interfaces. Nat Phys. (2016) 12:704–11. doi: 10.1038/nphys3678
39. Taylor MJ, Husain K, Gartner ZJ, Mayor S, Vale RD. A DNA-based T cell receptor reveals a role for receptor clustering in ligand discrimination. Cell (2017) 169:108–19.e20. doi: 10.1016/j.cell.2017.03.006
40. Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature (2014) 511:319–25. doi: 10.1038/nature13535
41. Goyette J, Gaus K. Mechanisms of protein nanoscale clustering. Curr Opin Cell Biol. (2017) 44:86–92. doi: 10.1016/j.ceb.2016.09.004
42. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285:221–7. doi: 10.1126/science.285.5425.221
43. Lee K-H, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science (2002) 295:1539–42. doi: 10.1126/science.1067710
44. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse : a molecular machine controlling T cell activation. Science (1999) 285:221–8.
45. Tane AH, Sakuma M, Ike H, Yokosuka T, Kimura Y, Ohara O, et al. Micro – adhesion rings surrounding TCR microclusters are essential for T cell activation. J Exp Med. (2016) 213:1609–26. doi: 10.1084/jem.20151088
46. Hof P, Pluskey S, Dhe-paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. (1998) 92:441–50. doi: 10.1016/S0092-8674(00)80938-1
47. Pei D, Lorenz U, Klingmüller U, Neel BG, Walsh CT. Intramolecular regulation of protein tyrosine phosphatase SH-PTP1: a new function for Src homology 2 domains. Biochemistry (1994) 33:15483–93.
48. Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science (2011) 332:680–6. doi: 10.1126/science.1198701
49. Goyette J, Salas CS, Gordon NC, Bridge M, Isaacson S, Allard J, et al. Biophysical assay for tethered signalling reactions reveals tether-controlled activity for the phosphatase SHP-1. Sci Adv. (2017) 3:e1601692. doi: 10.1126/sciadv.1601692
50. Won AP, Garbarino JE, Lim WA. Recruitment interactions can override catalytic interactions in determining the functional identity of a protein kinase. Proc Natl Acad Sci USA. (2011) 108:9809–14. doi: 10.1073/pnas.1016337108
51. Windisch B, Bray D, Duke T. Balls and chains - a mesoscopic approach to tethered protein domains. Biophys J. (2006) 91:2383–92. doi: 10.1529/biophysj.105.078543
52. Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. Dependence of effective molarity on linker length for an intramolecular protein-ligand system. J Am Chem Soc. (2007) 129:1312–20. doi: 10.1021/ja066780e
53. Van Valen D, Haataja M, Phillips R. Biochemistry on a leash: the roles of tether length and geometry in signal integration proteins. Biophys J. (2009) 96:1275–92. doi: 10.1016/j.bpj.2008.10.052
54. Marsh JA, Forman-Kay JD. Sequence determinants of compaction in intrinsically disordered proteins. Biophys J. (2010) 98:2374–82. doi: 10.1016/j.bpj.2010.02.006
55. Moglich A, Joder K, Kiefhaber T. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc Natl Acad Sci USA. (2006) 103:12394–9. doi: 10.1073/pnas.0604748103
56. Cai H, Muller J, Depoil D, Mayya V, Sheetz MP, Dustin ML, et al. Full control of ligand positioning reveals spatial thresholds for T cell receptor triggering. Nat Nanotechnol. (2018) 13:610–7. doi: 10.1038/s41565-018-0113-3
57. Owen DM, Williamson D, Rentero C, Gaus K. Quantitative microscopy: Protein dynamics and membrane organisation. Traffic (2009) 10:962–71. doi: 10.1111/j.1600-0854.2009.00908.x
58. Ma Y, Pandzic E, Nicovich PR, Yamamoto Y, Kwiatek J, Pageon SV, et al. An intermolecular FRET sensor detects the dynamics of T cell receptor clustering. Nat Commun. (2017) 8:15100. doi: 10.1038/ncomms15100
59. O'Donoghue GP, Pielak RM, Smoligovets AA, Lin JJ, Groves JT. Direct single molecule measurement of TCR triggering by agonist pMHC in living primary T cells. Elife (2013) 2:e00778. doi: 10.7554/eLife.00778
60. Hu J, Lipowsky R, Weikl TR. Binding constants of membrane-anchored receptors and ligands depend strongly on the nanoscale roughness of membranes. Proc Natl Acad Sci USA. (2013) 110:15283–8. doi: 10.1073/pnas.1305766110
Keywords: microvilli, signal integration, PD-1, inhibitory receptors, inhibitory signaling
Citation: Pérez-Ferreros P, Gaus K and Goyette J (2019) Tethered Signaling in Inhibitory Immune Receptors. Front. Phys. 6:158. doi: 10.3389/fphy.2018.00158
Received: 30 September 2018; Accepted: 18 December 2018;
Published: 15 January 2019.
Edited by:
Jorge Bernardino De La Serna, United Kingdom Research and Innovation, United KingdomReviewed by:
Peter Jönsson, Lund University, SwedenSimon John Davis, University of Oxford, United Kingdom
Copyright © 2019 Pérez-Ferreros, Gaus and Goyette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katharina Gaus, ay5nYXVzQHVuc3cuZWR1LmF1
Jesse Goyette, ai5nb3lldHRlQHVuc3cuZWR1LmF1