 Signe Kjelstrup
Signe Kjelstrup Dick Bedeaux
Dick Bedeaux Alex Hansen
Alex Hansen Bjørn Hafskjold
Bjørn Hafskjold Olav Galteland1
Olav Galteland1- 1PoreLab, Department of Chemistry, Norwegian University of Science and Technology, Trondheim, Norway
- 2PoreLab, Department of Physics, Norwegian University of Science and Technology, Trondheim, Norway
We derive the entropy production for transport of multi-phase fluids in a non-deformable, porous medium exposed to differences in pressure, temperature, and chemical potentials. Thermodynamic extensive variables on the macro-scale are obtained by integrating over a representative elementary volume (REV). Contributions come from porous media specific properties, phase volumes, surface areas, and contact lines. Curvature effects are neglected. Using Euler homogeneity of the first order, we obtain the Gibbs equation for the REV. From this we define the intensive variables, the temperature, pressure, and chemical potentials, and, using the balance equations, we derive the entropy production for the REV. The entropy production defines sets of independent conjugate thermodynamic fluxes and forces in the standard way. The transport of two-phase flow of immiscible components is used to give a first illustration of the equations.
1. Introduction
The aim of this article is to develop the basis for a macro-scale description of multi-phase flow in porous media in terms of non-equilibrium thermodynamics. The system consists of several fluid phases in a medium of constant porosity. The aim is to describe the transport of these on the scale of measurements; i.e., on the macro-scale, using properties defined on this scale, which represent the underlying structure on the micro-scale. The effort is not new; it was pioneered more than 30 years ago [1–4], and we shall build heavily on these results, in particular those of Hassanizadeh and Gray [2, 3] and Gray and Miller [5].
The aim is also still the original one; to obtain a systematic description, which can avoid arbitrariness and capture the essential properties of multi-component multi-phase flow-systems. Not only bulk energies need be taken into account to achieve this for porous media. Also excess surface- and line-energies must be considered, see e.g., [6]. But, unlike what has been done before, we shall seek to reduce drastically the number of variables needed for the description, allowing us still to make use of the systematic theory of non-equilibrium thermodynamics. While the entropy production in the porous medium so far has been written as a combination of contributions from each phase, interface and contact line, we shall write the property for a more limited set of macro-scale variables. In this sense, we deviate widely from the Thermodynamically Constrained Averaging Theory [5]. Nevertheless, we will be able to describe experiments and connect variables within the classical scheme of non-equilibrium thermodynamics. The reduction of variables is possible as long as the system is Euler homogeneous of the first kind.
The theory of non-equilibrium thermodynamics was set up by Onsager [7, 8] and further developed for homogeneous systems during the middle of the last century [9]. It was the favored thermodynamic basis of Hassanizadeh and Gray for their description of porous media. These authors [2, 3] discussed also other approaches, e.g., the theory of mixtures in macroscopic continuum mechanics, cf. [1, 4]. Gray and Miller [5] argued that it is the simplest of the many approaches in non-equilibrium thermodynamics.
The theory of classical non-equilibrium thermodynamics has been extended to deal with a particular case of flow in heterogeneous systems, namely transport along [10] and perpendicular [11] to layered interfaces. A derivation of the entropy production for heterogeneous systems on the macro-scale has not been given, however, even if one can find several uses of this property [6]. Transport in porous media takes place, not only under pressure gradients. Temperature gradients will frequently follow from transport of mass, for instance in heterogeneous catalysis [12], in polymer electrolyte fuel cells, in batteries [11, 13], or in capillaries in frozen soils during frost heave [14]. The number of this type of phenomena is enormous. We have chosen to consider first the vectorial driving forces related to changes in pressure, chemical composition, and temperature, staying away for the time being from deformations, chemical reactions, or forces leading to stress [15]. The multi-phase flow problem is thus in focus.
The development of a general thermodynamic basis for multi-phase flow started by introduction of thermodynamic properties for each component in each phase, interface, and three-phase contact line [2, 3]. A representative volume element (REV) was introduced, consisting of bulk phases, interfaces, and three-phase contact lines. Balance equations were formulated for each phase in the REV, and the total REV entropy production was the sum of the separate contributions from each phase.
Hansen et al. [16] recognized recently that the motion of fluids at the coarse-grained level could be described by extensive variables. The properties of Euler homogeneous functions could then be used to create relations between the flow rates at this level of description. This work, however, did not address the coarse-graining problem itself. We shall take advantage of Euler homogeneity also here and use it in the coarse-graining process described above.
Like Gray et al. [2, 3, 5], we use the entropy production as the governing property. But rather than dealing with the total entropy production as a sum of several parts, we shall seek to define the total entropy production directly from a basis set of a few coarse-grained variables. This will be done here for the REV, see [17] for a preliminary version. Once the entropy production has been formulated, we shall set up the independent constitutive equations. This will be done in subsequent work, see the preliminary version [18]. There we highlight the consequences of the model, and show that new experimental relations can be found. We shall find that the description is able to add insight in already published experimental results and design new experiments.
The overall aim is thus to contribute toward solving the scaling problem; i.e., how a macro-level description can be obtained consistent with the micro-level one, by defining transport equations on the macro-level. The aim of the present work, seen in this context, is to present the basis for a description of central transport phenomena, namely those due to thermal, chemical, mechanical, and gravitational forces. We shall propose a systematic, course-grained procedure that will be simple in practical use.
2. System
Consider a heterogeneous system as illustrated by the (white) box in Figure 1. The system is a porous medium of fixed porosity filled with several immiscible fluids. There is net transport in one direction only, the x-direction. On the scale of measurement, the system is without structure. By zooming in, we see the pore scale. A collection of pores with two fluids is schematically shown in Figure 2.
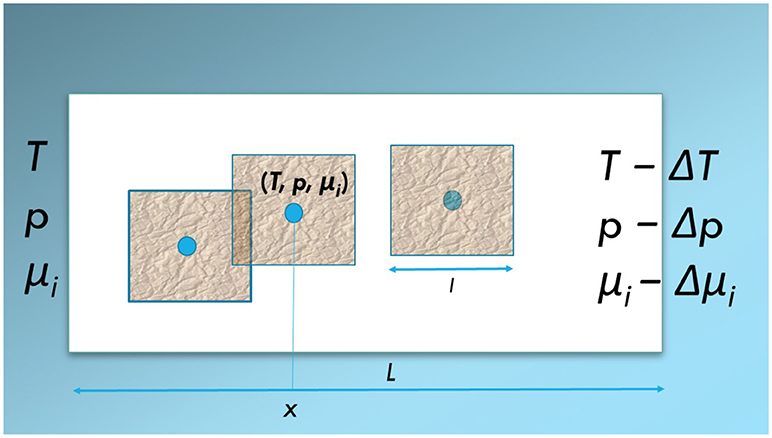
Figure 1. Schematic illustration of a heterogeneous system (white box, length L) exposed to a difference in temperature, ΔT, pressure, Δp, or chemical potential Δμi. The system is isolated in the y, z−directions. Net flows take place in the x−direction. Three representative elementary volumes, REVs (magenta squares, length l) are indicated. The REVs may overlap. Each is represented by a set of variables (p, T, μi) which defines a state (blue dot). Such states can be defined anywhere on the x-axis.
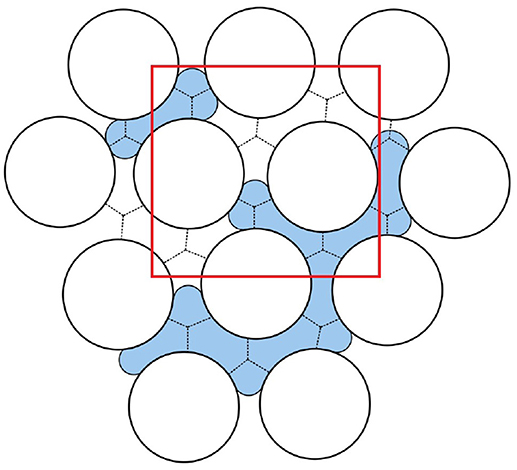
Figure 2. A schematic collection of pores filled with two immiscible liquids. In order to compute the system properties, we define a REV. A REV is indicated in the figure by the red square. Courtesy of M. Vassvik.
A temperature, pressure, and/or chemical potential difference is applied between the inlet and the outlet, and these differences can be measured. The pressure difference Δp between the outlet and the inlet was defined for steady state conditions by Tallakstad et al. [19], as the time average of the fluctuating difference Δp(t):
Here t is the time. Subscript “b” denotes beginning and “e” denotes the end of the measurement. We adopt similar definitions for ΔT and Δμi. It is possible, through application of separate inlet channels, to control the flow into and out of the system and find the flow of each component, to define the flow situation in Figure 1. In the presence of two immiscible phases, it is only possible to define the pressure difference between the inlet and the outlet for the phases, Δpw and Δpn, if there is continuity in the respective phases.
We will repeatedly use two-phase flow of single components as an example, where w indicates the most wetting and n the least wetting phase. We refer to them simply as the wetting and the non-wetting phase. In most of the paper we consider a multi-phase fluid. In the system pictured in Figure 2, there is flow within the REV in the direction of the pore. This is not necessarily the direction given by the overall pressure gradient. The flow on the macro-scale, however, is always in the direction of the pressure gradient. Net flow in other directions are zero due to isolation of the system in these directions. By flow on the macro-scale, we mean flow in the direction of the overall pressure gradient along the x-coordinate in Figure 1. The value of this average flow is of interest.
The representative volume element, REV, is constructed from a collection of pores like those contained in the red square in Figure 2. In Figure 1, three REVs are indicated (magenta structured squares). In a homogeneous system, statistical mechanical distributions of molecular properties lead to the macroscopic properties of a volume element. In a heterogeneous system like here, the statistical distributions are over the states within the REV. The collection of pores in the REV, cf. Figure 2, should be of a size that is large enough to provide meaningful values for the extensive variables, and therefore well defined intensive variables (see below, Equations 19 and 20), cf. section 3.2 below. Thermodynamic relations can be written for each REV.
State variables characterize the REV. They are represented by the (blue) dots in Figure 1. The size of the REV depends on its composition and other conditions. Typically, the extension of a REV, l, is large compared to the pore size of the medium, and small compared to the full system length L. This construction of a REV is similar to the procedure followed in smoothed particle hydrodynamics [20], cf. the discussion at the end of the work.
The REVs so constructed, can be used to make a path of states, over which we can integrate across the system. Each REV in the series of states, is characterized by variables T, p, μi, as indicated by the blue dots in Figure 1. Vice versa, each point in a porous medium can be seen as a center in a REV. The states are difficult to access directly, but can be accessed via systems in equilibrium with the states, as is normal in thermodynamics. This is discussed at the end of the work. We proceed to define the REV-variables.
3. Properties of the REV
3.1. Porosity and Saturation
Consider a solid matrix of constant porosity ϕ. We are dealing with a class of systems that are homogeneous in the sense that the typical pore diameter and pore surface area, on the average, are the same everywhere. There are m phases in the system. The pores are filled with a mixture of m − 1 fluid phases; the solid matrix is phase number m. Properties will depend on the time, but this will not be indicated explicitly in the equations.
In a simple case, the phases are immiscible single components. The chemical constituents are then synonymous with a phase, and the number of phases is the number of components. The state of the REV can be characterized by the volumes of the fluid phases Vα,REV, α = 1, .., m − 1 and of the solid medium Vm,REV. The total volume of the pores is
while the volume of the REV is
Superscript REV is used to indicate a property of the REV. The last term is the sum of the excess volumes of the three-phase contact lines. While the excess volume of the surfaces is zero by definition, this is not the case for the three-phase contact lines. The reason is that the dividing surfaces may cross each other at three lines which have a slightly different location. The corresponding excess volume is in general very small, and will from now on be neglected. This gives the simpler expression
All these volumes can be measured.
The porosity, ϕ, and the saturation, Ŝ, are given by
The porosity and the saturation are intensive variables. They do not depend on the size of the REV. They have therefore no superscript. It follows from these definitions that
In addition to the volumes of the different bulk phases (they are fluids or solids) m ≥ α ≥ 1, there are interfacial areas, Ω, between each two phases in the REV: Ωαβ,REV, m ≥ α > β ≥ 1. The total surface area of the pores is measurable. It can be split between various contributions
When the surface is not completely wetted, we can estimate the surface area between the solid m and the fluid phase α, from the total pore area available and the saturation of the component.
This estimate is not correct for strongly wetting components or dispersions. In those cases, films can form at the walls, and Ωmα,REV is not proportional to Ŝα. In the class of systems we consider, all fluids touch the wall, and there are no films of one fluid between the wall and another fluid.
3.2. Thermodynamic Properties of the REV
We proceed to define the thermodynamic properties of the REV within the volume VREV described above. In addition to the volume, there are other additive variables. They are the masses, the energy, and the entropy. We label the components (the chemical constituents) using italic subscripts. There are in total n components distributed over the phases, surfaces, and contact lines. The mass of component i, , in the REV is the sum of bulk masses, , m ≥ α ≥ 1, the excess interfacial masses, , m ≥ α > β ≥ 1, and the excess line masses, , m ≥ α > β > δ ≥ 1.
There is some freedom in how we allocate the mass to the various phases and interfaces [11, 21]. We are e.g., free to choose a dividing surface such that one equals zero. A zero excess mass will simplify the description, but will introduce a reference. The dividing surface with zero is the equimolar surface of component i. The total mass of a component in the REV is, however, independent of the location of the dividing surfaces. From the masses, we compute the various mass densities
where ρi and have dimension kg.m−3, has dimension kg.m−2 and has dimension kg.m−1.
All densities are for the REV. If we increase the size of the REV, by for instance doubling its size, VREV, and other extensive variables will all double. They will double, by doubling all contributions to these quantities. But this is not the case for the density ρi or the other densities. They remain the same, independent of the size of the REV. This is true also for the densities of the bulk phases, surfaces, and contact lines. Superscript REV is therefore not used for the densities.
Within one REV there are natural fluctuations in the densities. But the densities make it possible to give a description on the macro-scale independent of the precise size of the REV. The densities will thus be used in the balance equations on the macro-scale. The density may vary somewhat in Vα. We can then find as the integral of over Vα. Equation (10) then gives the volume-averaged densities.
The internal energy of the REV, UREV, is the sum of bulk internal energies, Uα,REV, m ≥ α ≥ 1, the excess interfacial internal energies, Uαβ,REV, m ≥ α > β ≥ 1, and the excess line internal energies, Uαβδ,REV, m ≥ α > β > δ ≥ 1:
The summation is taken over all phases, interfaces, and contact lines (if non-negligible). We shall see in a subsequent paper how these contributions may give specific contributions to the driving force. The internal energy densities are defined by
Their dimensions are J.m−3 (u, uα), J.m−2 (uαβ), and J.m−1 (uαβδ), respectively.
The entropy in the REV, SREV, is the sum of the bulk entropies, Sα,REV, m ≥ α ≥ 1, the excess entropies, Sαβ,REV, m ≥ α > β ≥ 1, the excess line entropies, Sαβδ,REV, m ≥ α > β > δ ≥ 1, and a configurational contribution, , from the geometrical distribution of the fluid phases within the pores:
The entropy densities are defined by
and have the dimensions J.K−1.m−3 (), J.K−1.m−2 (sαβ), and J.K−1.m−1 (sαβδ), respectively. To explain the configurational contribution in more detail; consider the example of stationary two-phase flow in a single tube of varying diameter described by Sinha et al. [22]. The tube contains one bubble of one fluid in the other. The bubble touches the wall; it can not form a film between the tube wall and the other fluid. The probability per unit of length of the tube to find the center of mass of the bubble at position xb, was Π(xb) [22]. Knowing this probability distribution, we can compute the entropy of an ensemble of single tubes (in this case a very long tube composed of the single ones). It is equal to
For a network of pores it is more appropriate to give the probability distribution for the fluid-fluid interfaces. This has not yet been done explicitly.
For the volume, Equations (2) and (4) apply when the contact lines give a negligible contribution. The dividing surfaces have by definition no excess volume. For all the other extensive thermodynamic variables, like the enthalpy, Helmholtz energy, Gibbs energy, and the grand potential, relations similar to Equations (11) and (13) apply. We shall later show how this affects the driving forces [18].
To summarize this section; we have defined a basis set of variables for a class of systems, where these variables are additive in the manner shown. From the set of REV-variables we obtain the densities, u, s, or ρi to describe the heterogeneous system on the macro-scale. A series of REVs of this type, is needed for integration across the system, see section 5.
3.3. REV Size Considerations
As an illustration of the REV construction, consider the internal energy of two isothermal, immiscible and incompressible fluids (water and decane) flowing in a Hele-Shaw type cell composed of silicone glass beads. The relevant properties of the fluids can be found in Table 1 The porous medium is a hexagonal network of 3,600 links, as illustrated in Figure 3. The network is periodic in the longitudinal and the transverse directions and a pressure difference of 1.8 × 104 Pa drives the flow in the longitudinal direction. The overall saturation of water is 0.4. The network flows were simulated using the method of Aker et al. [23], see [24] for details.
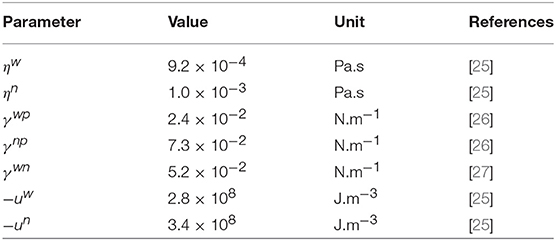
Table 1. Fluid properties used to compute the candidate REV internal energy, for a network containing water (n) and decane (w) within silica glass beads (p) at atmospheric pressure and 293 K.
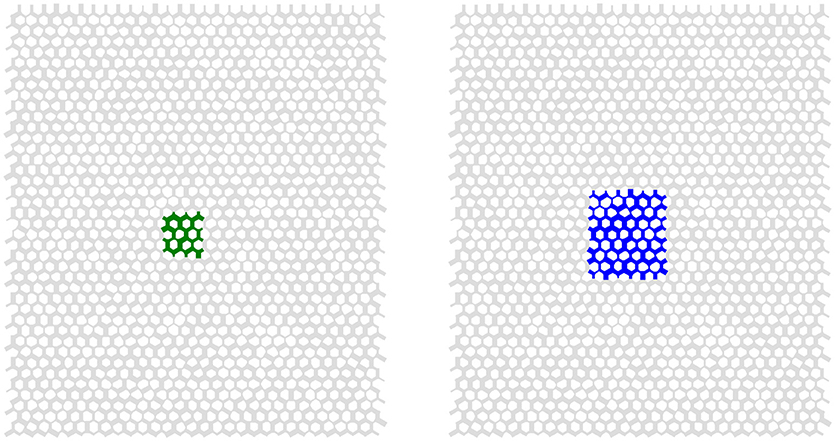
Figure 3. Illustration of the link network and two of the candidate REVs under consideration. The left candidate REV (green) is 5.2 × 6 mm and the right candidate REV (blue) is 10.4 × 12 mm.
The internal energy of the REV is, according to section 3.2, a sum over the two fluid bulk contributions and three interface contributions,
where, ui is the internal energy density of phase i and uij is the excess internal energy per interfacial area between phase i and phase j. We assume ui and uij to be constant. For simplicity, uij is approximated by interfacial tension, denoted γij. The internal energy of the porous matrix is constant in this example and is therefore set to zero.
Candidate REVs are of different sizes, see Table 2. The 5.4 × 6 mm (green), and 10.4 × 12 mm (blue) candidate REVs are shown in Figure 3. For all candidate REVs, UREV is calculated according to Equation (17) at each time step. Since the measured saturations and interfacial areas are fluctuating in time, so is the internal energy. A time-step weighted histogram of the internal energy presents the probability distribution.
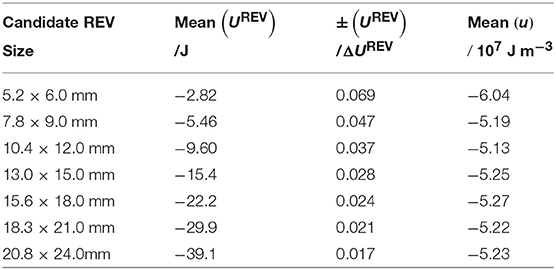
Table 2. Mean values of UREV and u for candidate REVs of different sizes, along with the standard deviation of UREV divided by ΔUREV. The latter quantity represents a measure of the relative size of the fluctuations in UREV.
The probability distributions of UREV are shown in Figure 4 for the 5.2 × 6 mm (green) and 10.4 × 12 mm (blue) candidate REVs. In both plots, the vertical lines represent the internal energy the REV would have if it were occupied by one of the fluids alone. We denote the difference in internal energy between these two single-phase states by ΔUREV.
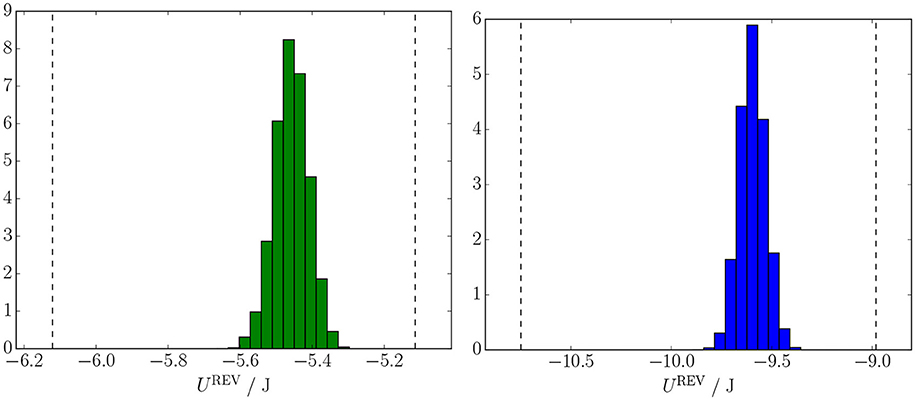
Figure 4. Probability distributions of internal energy in two the candidate REVs. The left candidate REV (green) is 5.2 × 6 mm and the right candidate REV (blue) is 10.4 × 12 mm. In each plot, the left dashed line represents the internal energy the candidate REV would have if it contained non-wetting fluid only and the right dashed line represents the internal energy the candidate REV would have if it contained wetting fluid only.
The mean value of the UREV for all candidate REVs are given in Table 2, along with mean density u = UREV/VREV and the standard deviation of UREV divided by ΔUREV. The latter quantity is a measure of the relative size of the fluctuations in UREV. Due to the additivity of UREV, the mean values of UREV increases roughly proportional to the candidate REV size. But this happens only after the REV has reached a minimum size, here 7.8 × 9.0 mm. For the larger candidate REVs, the mean value of u changes little as the size increases. The relative size of the fluctuations in UREV decreases in proportion to the linear size of the candidate REVs.
This example indicates that it makes sense to characterize the internal energy of a porous medium in terms of an internal energy density as defined by Equation (11), given that the size of the REV is appropriately large. About 100 links seem to be enough in this case. This will vary with the type of porous medium, cf. the 2D square network model of Savani et al. [28].
4. Homogeneity on the Macro-Scale
Before we address any transport problems, consider again the system pictured in Figure 1 (the white box). All REVs have variables and densities as explained above. By integrating to a somewhat larger volume V, using the densities defined, we obtain the set of basis variables, (U, S, Mi), in V. The internal energy U of the system is an Euler homogeneous function of first order in S, V, Mi:
where λ is a multiplication factor. The internal energy U, volume V, entropy S, and component mass Mi, obey therefore the Gibbs equation;
No special notation is used here to indicate that U, S, V, Mi are properties on the macro-scale. Given the heterogeneous nature on the micro-scale, the internal energy has contributions from all parts of the volume V, including from the excess surface and line energies. By writing Equation (18) we find that the normal thermodynamic relations apply for the heterogeneous system at equilibrium, for the additive properties U, S, V, Mi, obtained from sums of the bulk-, excess surface-, and excess line-contributions.
We can then move one more step and use Gibbs equation to define the temperature, the pressure, and chemical potentials on the macro-scale as partial derivatives of U:
The temperature, pressure, and chemical potentials on the macro-scale are, with these formulas, defined as partial derivatives of the internal energy. This is normal in thermodynamics, but the meaning is now extended. In a normal homogeneous, isotropic system at equilibrium, the temperature, pressure, and chemical equilibrium refer to a homogeneous volume element. The temperature of the REV is a temperature representing all phases, interfaces and lines combined, and the chemical potential of i is similarly obtained from the internal energy of all phases. Therefore, there are only one T, p, and μi for the REV. The state can be represented by the (blue) dots in Figure 1.
On the single pore level, the pressure and temperature in the REV will have a distribution. In the two immiscible-fluid-example the pressure, for instance, will vary between a wetting and a non-wetting phase because of the capillary pressure. One may also envision that small phase changes in one component (e.g., water) leads to temperature variations due to condensation or evaporation. Variations in temperature will follow changes in composition.
The intensive properties are not averages of the corresponding entities on the pore-scale over the REV. This was pointed out already by Gray and Hassanizadeh [3]. The definitions are derived from the total internal energy only, and this makes them uniquely defined. It is interesting that the intensive variables do not depend on how we split the energy into into bulk and surface terms inside the REV.
By substituting Equation (20) into Equation (19) we obtain the Gibbs equation for a change in total internal energy on the macro-scale
As a consequence of the condition of homogeneity of the first order, we also have
The partial derivatives T, p and μi are homogeneous functions of the zeroth order. This implies that
Choosing λ = 1/V it follows that
The temperature therefore depends only on the subset of variables s ≡ S/V, ρi ≡ Mi/V and not on the complete set of variables S, V, Mi. The same is true for the pressure, p, and the chemical potentials, μi. This implies that T, p and μi are not independent. We proceed to repeat the standard derivation of the Gibbs-Duhem equation which makes their interdependency explicit.
The Gibbs equation on the macro-scale in terms of the densities follows using Equations (21) and (22)
which can alternatively be written as
The Euler equation implies
By differentiating Equation (27) and subtracting the Gibbs equation (25), we obtain in the usual way the Gibbs-Duhem equation:
This equation makes it possible to calculate p as a function of T and μi and shows how these quantities depend on one another.
We have now described the heterogeneous porous medium by a limited set of coarse-grained thermodynamic variables. These average variables and their corresponding temperature, pressure, and chemical potentials, describe a coarse-grained homogeneous mixture with variables which reflect the properties of the class of porous media. In standard equilibrium thermodynamics, Gibbs' equation applies to a homogeneous phase. We have extended this use to be applicable for heterogeneous systems at the macro-scale. On this scale, the heterogeneous system (the REV) is then regarded as being in local equilibrium. Whether or not the chosen procedure is viable, remains to be tested. We refer to the section 7 of this paper for more discussion and to a paper to follow [18] for an experimental program.
5. Entropy Production in Porous Media
Gradients in mass- and energy densities produce changes in the variables on the macro-scale. These lead to transport of heat and mass. Our aim is to find the equations that govern this transport across the REV. We therefore expose the system to driving forces and return to Figure 1.
The balance equations for masses and internal energy of a REV are
The transport on this scale is in the x−direction only. The mass fluxes, Ji, and the flux of internal energy, Ju, are all macro-scale fluxes. The internal energy flux is the sum of the measurable (or sensible) heat flux, and the partial specific enthalpy (latent heat), Hi (in J.kg−1) times the component fluxes, Ji, see [3, 9, 11] for further explanations. Component m (the porous medium) is not moving and is the convenient frame of reference for the fluxes.
The entropy balance on the macro-scale is
Here Js is the entropy flux, and σ is the entropy production which is positive definite, σ ≥ 0 (the second law of thermodynamics). We can now derive the expression for σ in the standard way [9, 11], by combining the balance equations with Gibbs' equation. The entropy production is the sum of all contributions within the REV.
In the derivations, we assume that the Gibbs equation is valid for the REV also when transport takes place. Droplets can form at high flow rates, while ganglia may occur at low rates. We have seen above that there is a minimum size of the REV, for which the Gibbs equation can be written. When we assume that the Gibbs equation applies, we implicitly assume that there exists a uniquely defined state. The existence of such an ergodic state was postulated by Hansen and Ramstad [29]. Valavanides and Daras used it in their DeProF model for two-phase flow in pore networks [30]. Experimental evidence for the assumption was documented by Erpelding [31].
Under the conditions that we demand valid for the REV, the Gibbs Equation (26) keeps its form during a time interval dt, giving
We can now introduce the balance equations for mass and energy into this equation, see [11] for details. By comparing the result with the entropy balance, Equation (30), we identify first the entropy flux, Js,
The entropy flux is composed of the sensible heat flux over the temperature plus the sum of the specific entropies carried by the components. The form of the entropy production, σ, depends on our choice of the energy flux, Ju or . The choice of form is normally motivated by practical wishes; what is measurable or computable. We have
These expressions are equivalent formulations of the same physical phenomena. When we choose Ju as variable with the conjugate force ∂(1/T)/∂x, the mass fluxes are driven by minus the gradient in the Planck potential μi/T. When, on the other hand we choose as a variable with the conjugate force ∂(1/T)/∂x, the mass fluxes are driven by minus the gradient in the chemical potential at constant temperature over this temperature. The entropy production defines the independent thermodynamic driving forces and their conjugate fluxes. We have given two possible choices above to demonstrate the flexibility. The last expression is preferred for analysis of experiments.
In order to find the last line in Equation (33) from the first, we used the thermodynamic identities μi = Hi − TSi and ∂(μi/T)/∂(1/T) = Hi as well as the expression for the energy flux given in Equation (29). Here Si is the partial specific entropy (in J.kg−1.K−1).
5.1. The Chemical Potential at Constant Temperature
The derivative of the chemical potential at constant temperature is needed in the driving forces in the second line for σ in Equation (33). For convenience we repeat its relation to the full chemical potential [9]. The differential of the full chemical potential is:
where Si, Vi, and (∂μi/∂Mj)p,T,Mi are partial specific quantities. The partial specific entropy and volume are equal to:
and the last term of Equation (33) is denoted by
By reshuffling, we have the quantity of interest as the differential of the full chemical potential plus an entropic term;
The differential of the chemical potential at constant temperature is
With equilibrium in the gravitational field, the pressure gradient is dp/dx = −ρg, where ρ is the total mass density and g is the acceleration of free fall [32]. The well known separation of components in the gravitational field is obtained, with dμi,T = 0 and
where Wi is the molar mass (in kg.mol−1), Ŝi the saturation, and yi the activity coefficient of component i. The gas constant, R, has dimension J.K−1.mol−1. The gradient of the mole fraction of methane and decane in the geothermal gradient of the fractured carbonaceous Ekofisk oil field, was estimated to 5 × 10−4m−1 [33], in qualitative agreement with observations. We replace dμi,T below using these expressions.
It follows from Euler homogeneity that the chemical potentials in a (quasi-homogeneous) mixture are related by , which is Gibbs-Duhem's equation. By introducing Equation 37 into this equation we obtain an equivalent expression, to be used below:
6. Transport of Heat and Two-Phase Fluids
Consider again the case of two immiscible fluids of single components, one more wetting (w) and one more non-wetting (n). The entropy production in Equation (33) gives,
The solid matrix is the frame of reference for transport, Jr = 0 and does not contribute to the entropy production. The volume flux is frequently measured, and we wish to introduce this as new variable
Here JV has dimension (m3.m−2.s−1 = m.s−1), and the partial specific volumes have dimension m3.kg−1. The volume flows used by Hansen et al. [16] are related to ours by Jnυn = Ŝnυn, Jwυw = Ŝwυw and JV = υ = Ŝnυn + Ŝwυw.
The chemical potential of the solid matrix may not vary much if the composition of the solid is constant across the system. We assume that this is the case (), and use Equation (40) to obtain
The entropy production is invariant to the choice of variables. We can introduce the relations above and the explicit expression for dμi,T into Equation (41), and find the practical expression:
In the last line, the difference velocity vD is
This velocity (in m/s) describes the relative movement of the two components within the porous matrix on the macro-scale. In other words, it describes the ability of the medium to separate components. The main driving force for separation is the chemical driving force, related to the gradient of the saturation. The equation implies that also temperature and pressure gradients may play a role for the separation.
The entropy production has again three terms, one for each independent driving force. With a single fluid, the number of terms is two. The force conjugate to the heat flux is again the gradient of the inverse temperature. The entropy production, in the form we can obtain, Equations (41) or (44), dictates the constitutive equations of the system.
6.1. A Path of Sister Systems
As pointed out above, through the construction of the REV we were able to create a continuous path through the system, defined by the thermodynamic variables of the REVs. The path was illustrated by a sequence of dots in Figure 1. Such a path must exist, to make integration possible. Also continuum mixture theory hypothesizes such a path [4]: Hilfer introduced a series of mixture states, to define an integration path across the porous system, see e.g., [4].
The path created in section 2 is sufficient as a path of integration across the medium. The access to and measurement of properties in the REVs is another issue. It is difficult, if not impossible, to measure in situ as stated upfront. The measurement probe has a minimum extension (of some mm), and the measurement will represent an average over the surface of the probe. For a phase with constant density, the average is well-defined and measurable. A link between the state of the REV and a state where measurements are possible, is therefore needed. We call the state that provides this link a sister state.
Consider again the path of REVs in the direction of transport. To create the link between the REV and its sister state, consider the system divided into slices, see Figure 5. The slice (the sister system) contains homogeneous (pink) phases in equilibrium with the REV at the chosen location.
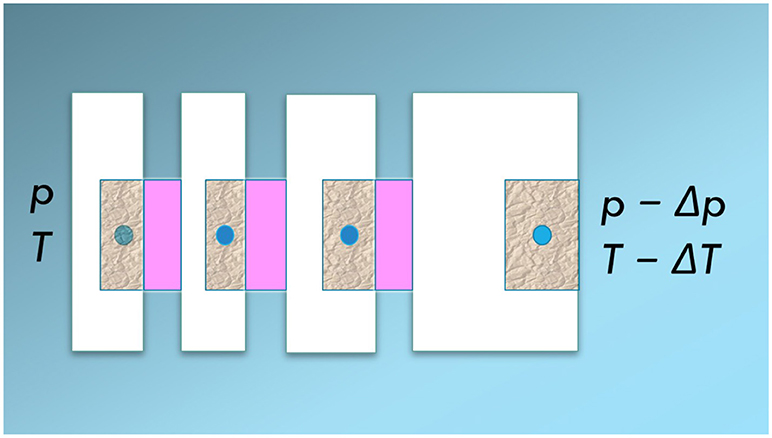
Figure 5. A one-dimensional heterogeneous system cut into slices. Each cut is brought in equilibrium with a homogeneous (pink) mixture at the same temperature and pressure as the REV.
We hypothesize that we can find such sister states; in the form of a multi-component mixture with temperature, pressure, and composition such that equilibrium can be obtained with the REV-variables at any slice position. The variables of the sister state can then be measured the normal way. The chemical potential of a component in the sister state can, for instance, be found by introducing a vapor phase above this state and measure the partial vapor pressure. We postulate thus that a sister state can be found, that obey the conditions
Here i = 1, …, n are the components in the REV, and superscript s denotes the sister state. With the sister states available, we obtain an experimental handle on the variables of the porous medium. The hypothesis must be checked, of course.
The series of sister states have the same boundary conditions as the REV-states, by construction, and the overall driving forces will be the same. Between the end states, we envision the non-equilibrium system as a staircase. Each step in the stair made up of a REV is in equilibrium with a step of the sister-state-stair. Unlike the states inside the porous medium, the sister states are accessible for measurements, or determination of T, p, and μi. The driving forces of transport can then be described by the sequence of the sister states.
7. Discussion
We have shown in this work how it is possible to extend the method of classical non-equilibrium thermodynamics [9] to describe transport in porous media. This was possible by
• constructing a REV in terms of a basis set of additive variables
• assuming that the REV is Euler homogeneous of degree one in the basis set.
The method is developed in the same manner as the classical theory is, but it extends the classical theory through the variable choice. The assumption about Euler homogeneity is the same for homogeneous (classical) as well as the heterogeneous porous media. The new variable set is necessary in order to account for the presence of the porous medium, i.e., the contributions from interfaces and contact line energies. Film formation in the pore is excluded. The properties of the porous medium will therefore enter in the definition of the variable set. The consequences of the choice will be elaborated in an article to come [18].
The classical equations have been written for single-phase systems, as these can be regarded as homogeneous on the molecular scale [34]. Equations (41) and (44), for instance, are well-established in theory of transport for polymer membranes, see e.g., [34]. The idea of the sister states to define the state of a porous media with larger pores and immiscible phases was inspired by this. The way of dealing with lack of knowledge of variables inside the system was for instance used in polymer membrane transport long ago, see [35, 36]. The procedure, to introduce a series of equilibrium states, each state in equilibrium with the membrane at some location between the external boundaries, was first used by Scatchard [35], and experimentally verified much later [36, 37].
With the condition of Euler homogeneity we can set up the Gibbs equation, which is essential in the derivation of the entropy production. The total entropy production follows directly from the new set of variables and Euler homogeneity. This procedure is new, when compared to the literature where focus was set on the single phases, interfaces and contact lines [2, 3, 5].
The REV obeys local equilibrium in the sense that it obeys Gibbs equation. Some support for this can be found in the literature. Prigogine and Mazur [38] investigated a mixture of two fluids using non-equilibrium thermodynamics. Their system consisted of superfluid - and normal helium. Two pressures were defined, one for each of the two fluids. The interaction between the two fluids was small, meaning that one phase flowed as if the other one (aside from a small frictional force) was not there. The situation here is similar, as we may have different liquid pressures inside the REV. But the interaction between the two immiscible components in our porous medium is large, not negligible as in the helium case.
We are adding the contributions from each phase, interface, and line to overall variables for the REV. But unlike Gray and Miller [5] and others [39], we do not need to require that thermodynamic equilibrium relations are obeyed within the REV. This may seem to be drastic, but the Gibbs-Duhem equation follows from Euler homogeneity alone, cf. section 5. The assumption of Euler homogeneity is sufficient to obtain the Gibbs-Duhem equation. In this aspect, we agree with those who use that equation for porous media, see e.g., [6].
The surface areas and the contact line lengths are not independent variables in our representation of the REV. These variables have been included through the assumption that the basic variables of the REV are additive. This means that a REV of a double size has double the energy, entropy, and mass, but also double the surface areas of various types and double the line lengths. The contraction to the small set of variables depend on this assumption. Otherwise, we need to expand the variable set. This can be done, however. A promising route seems to include Minkovski integrals [40]. Our approach can be compared to the up-scaling method used in Smoothed Particle Hydrodynamics [20]. Inspired by the idea behind smoothed particle hydrodynamics, we can also define a normalized weight function W(r), such that a microscopic variable a(r) may be represented by its average, defined as
For example, if a(r′) is the local void fraction in a porous material as determined from samples of the material, is the average porosity of the medium. The average is assigned to the point r and varies smoothly in space. The average porosity would then be suitable for e.g., a reservoir simulation at the macro-scale.
In general, the system is subject to external forces and its properties are non-uniform. The choice of W(r) is therefore crucial in that it defines the extent of the coarse-graining and the profile of the weighting. The illustration in Figure 1 alludes to a weight function that is constant inside a cubic box and zero outside, but other choices are possible. Popular choices used in mesoscale simulations are the Gaussian and spline functions (see [20] for details). A convenient feature of the coarse-graining is that the average of a gradient of a property a is equal to the gradient of the average.
Similarly the average of a divergence of a flux is equal to the divergence of the average. This implies that balance equations, which usually contain the divergence of a flux, remain valid after averaging. Time averages can also be introduced along the same lines.
Time scales relevant to porous media transport are usually large (minutes, hours); and much larger than times relevant for the molecular scale. Properties can change not only along the coordinate axis, but also on the time scale. In the present formulation, any change brought about in the REV must retain the validity of the Gibbs equation. As long as that is true, we can use the equations, also for transient phenomena.
The outcome of the derivations will enable us to deal with a wide range of non-isothermal phenomena in a systematic manner, from frost heave to heterogeneous catalysis, or multi-phase flow in porous media. We will elaborate on what this means in the next part of this work. In particular, we shall give more details on the meaning of the additive variables and the consequences for the REV pressure in a paper to come [18]. We will there return to the meaning of the REV variables and how they will contribute and help define new driving forces of transport.
8. Concluding Remarks
We have derived the entropy production for transport of heat and immiscible, single components (phases) in a porous medium. The derivations have followed standard non-equilibrium thermodynamics for heterogeneous systems [11]. The only, but essential, difference to current theories, has been the fact that we write all these equations for a porous medium on the macro-scale for the REV of a minimum size using its total entropy, energy and mass. These equations are mostly written for the separate contributions. Broadly speaking, we have been zooming out our view on the porous medium to first define some states that we take as thermodynamic states because they obey Euler homogeneity. The states are those illustrated by the dots in Figure 1. In order to define these states by experiments, we constructed the sister states of Figure 5.
The advantage of the present formulations is this; it is now possible to formulate the transport problem on the scale of a flow experiment in accordance with the second law of thermodynamics, with far less variables, see [18]. This opens up the possibility to test the thermodynamic models for consistency and compatibility with the second law. Such tests will be explicitly formulated together with the constitutive equations, in the next part of this work [18].
Author Contributions
SK and DB defined the variables of the REV and the sister states and wrote the first draft. AH, BH, and OG critically examined all proposals and contributed to revisions on the MS.
Funding
The authors are grateful to the Research Council of Norway through its Centers of Excellence funding scheme, project number 262644, PoreLab.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Per Arne Slotte is thanked for stimulating discussions.
References
2. Hassanizadeh SM, Gray WG. Mechanics and thermodynamics of multiphase flow in porous media including interphase boundaries. Adv Water Resour. (1990) 13:169–86.
3. Gray WG, Hassanizadeh SM. Macroscale continuum mechanics for multiphase porous-media flow including phases, interfaces, common lines and commpon points. Adv Water Resour. (1998) 21:261–81.
4. Hilfer R. Macroscopic equations of motion for two phase flow in porous media. Phys Rev E (1998) 58:2090.
5. Gray WG, Miller CT. Introduction to the Thermodynamically Constrained Averaging Theory for Porous Medium Systems. Cham: Springer (2014).
6. Revil A. Transport of water and ions in partially water-saturated porous media. Adv Water Resour. (2017) 103:119–38. doi: 10.1016/j.advwatres.2016.07.016
10. Bedeaux D, Albano AM, Mazur P. Boundary conditions and non-equilibrium thermodynamics. Phys A (1976) 82:438–62.
11. Kjelstrup S, Bedeaux D. Non-Equilibrium Thermodynamics of Heterogeneous Systems. Singapore: Wiley (2008).
12. Zhu L, Koper GJM, Bedeaux D. Heats of transfer in the diffusion layer before the surface and the surface temperature for a catalytic hydrogen oxidation reaction (H2+(1/2)O2 = H2O). J Phys Chem A (2006) 110:4080–8. doi: 10.1021/jp056301i
13. Richter F, Gunnarshaug AF, Burheim OS, Vie PJS, Kjelstrup S. Single electrode entropy change for LiCoO2 electrodes. ECS Trans. (2017) 80:219–38. doi: 10.1149/08010.0219ecst
14. Førland T, Ratkje SK. Irreversible thermodynamic treatment of frost heave. Eng Geol. (1981) 18:225–9.
15. Huyghe JM, Nikooee E, Hassanizadeh SM. Bridging effective stress and soil water retention equations in deforming unsaturated porous media: a thermodynamic approach. Transp Porous Med. (2017) 117:349–65. doi: 10.1007/s11242-017-0837-9
16. Hansen A, Sinha S, Bedeaux D, Kjelstrup S, Gjennestad MA, Vassvik M. Relations between seepage velocities in immiscible, incompressible two-phase flow in porous media. arXiv:1712.06823. Availbale online at: https://arxiv.org/abs/1712.06823
17. Kjelstrup S, Bedeaux D, Hansen A, Hafskjold B, Galteland O. Non-isothermal two-phase flow in porous media. The entropy production. (2018) arXiv.1805.03943.
18. Kjelstrup S, Bedeaux D, Hansen A, Hafskjold B, Galteland O. Non-isothermal two-phase flow in porous media. Constitutive equations. (2018) arXiv.1809.10378.
19. Tallakstad KT, Løvoll G, Knudsen HA, Ramstad T, Flekkøy EG, Måløy KJ. Steady-state, simultaneous two-phase flow in porous media: an experimental study. Phys Rev E (2009) 80:036308. doi: 10.1103/PhysRevE.80.036308
22. Sinha S, Hansen A, Bedeaux D, Kjelstrup S, Savani I, Vassvik M. Effective rheology of bubbles moving in a capillary tube. Phys Rev E (2013) 87:025001. doi: 10.1103/PhysRevE.87.025001
23. Aker E, Måløy KJ, Hansen A, Batrouni G. A two-dimensional network simulator for two-phase flow in porous media. Trans Porous Media (1998) 32:163–86.
24. Gjennestad MA, Vassvik M, Kjelstrup S, Hansen A. Stable and efficient time integration at low capillary numbers of a dynamic pore network model for immiscible two-phase flow in porous media. Front Phys. (2018) 6:56. doi: 10.3389/fphy.2018.00056
25. Linstrom PJ, Mallard WG, editors. NIST Chemistry WebBook, NIST Standard Reference Database Number 69. Gaithersburg MD: National Institute of Standards and Technology (2018).
26. Neumann A. Contact angles and their temperature dependence: thermodynamic status, measurement, interpretation and application. Adv Colloid Interfaces (1974) 4:105–91.
27. Zeppieri S, Rodríguez J, López de Ramos A. Interfacial tension of alkane + water systems. J Chem Eng Data (2001) 46:1086–8. doi: 10.1021/je000245r
28. Savani I, Sinha S, Hansen A, Kjelstrup S, Bedeaux D, Vassvik M. A Monte Carlo procedure for two-phase flow in porous media. Transp Porous Med. (2017) 116:869–88. doi: 10.1007/s11242-016-0804-x
29. Hansen A, Ramstad T. Towards a thermodynamics of immiscible two-phase steady-state flow in porous media. Comp Geosci. (2009) 13:227. doi: 10.1007/s10596-008-9109-7
30. Valavanides MS, Daras T. Definition and counting of configurational microstates in steady-state two-phase flows in pore networks. Entropy (2016) 18:1–28. doi: 10.3390/e18020054
31. Erpelding M, Sinha S, Tallakstad KT, Hansen A, Flekkøy EG, Måløy KJ. History independence of steady state in simultaneous two-phase flow through two-dimensional porous media. Phys Rev E (2013) 88:053004. doi: 10.1103/PhysRevE.88.053004
32. Førland KS, Førland T, Ratkje SK. Irreversible Thermodynamics. Theory and Applications. Chichester: Wiley (1988).
33. Holt T, Lindeberg E, Ratkje SK. The Effect of Gravity and Temperature Gradients on the Methane Distribution in Oil Reservoirs. Society of Petroleum Engineers, SPE-11761-MS (1983).
34. Katchalsky A, Curran PF. Nonequilibrium Thermodynamics in Biophysics. Harvard University Press (1965).
36. Lakshminarayanaiah N, Subrahmanyan V. Measurement of membrane potentials and test of theories. J Polym Sci Part A (1964) 2:4491–502.
37. Ratkje SK, Holt T, Skrede M. Cation membrane transport:evidence for local validity of nernst–planck equations. Berich Bunsen Gesell. (1988) 92:825–32.
38. Prigogine I, Mazur P. About two formulations of hydrodynamics and the problem of liquid helium II. Physica (1951) 17:661–79.
40. McClure JE, Armstrong RT, Berrill MA, Schluter S, Berg S, Gray WG, et al. A geometric state function for two-fluid flow in porous media. Phys Rev Fluids (2018) 3:084306. doi: 10.1103/PhysRevFluids.3.084306
Appendix
Mathematical symbols, superscripts, subscripts.
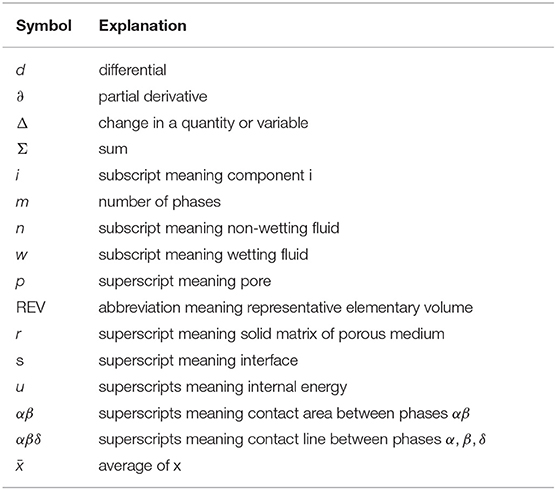
Greek symbols
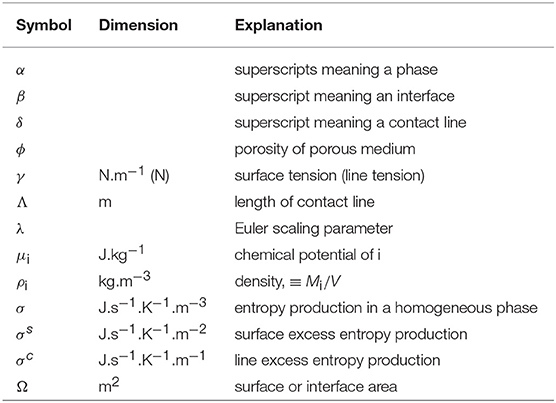
Latin symbols.

Keywords: porous media, energy dissipation, two-phase flow, excess surface- and line-energies, pore-scale, representative elementary volume, macro-scale, non-equilibrium thermodynamics
Citation: Kjelstrup S, Bedeaux D, Hansen A, Hafskjold B and Galteland O (2018) Non-isothermal Transport of Multi-phase Fluids in Porous Media. The Entropy Production. Front. Phys. 6:126. doi: 10.3389/fphy.2018.00126
Received: 05 September 2018; Accepted: 16 October 2018;
Published: 08 November 2018.
Edited by:
Antonio F. Miguel, Universidade de Évora, PortugalReviewed by:
Francisco J. Valdes-Parada, Universidad Autónoma Metropolitana, MexicoA. Murat, University of Ontario Institute of Technology, Canada
Copyright © 2018 Kjelstrup, Bedeaux, Hansen, Hafskjold and Galteland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Signe Kjelstrup, c2lnbmUua2plbHN0cnVwQG50bnUubm8=