 Yuriel Núñez Fernández
Yuriel Núñez Fernández K. Hallberg
K. Hallberg- Centro Atómico Bariloche and Instituto Balseiro, CNEA, CONICET, Bariloche, Argentina
We implement an efficient numerical method to calculate response functions of complex impurities based on the Density Matrix Renormalization Group (DMRG) and use it as the impurity-solver of the Dynamical Mean Field Theory (DMFT). This method uses the correction vector to obtain precise Green's functions on the real frequency axis at zero temperature. By using a self-consistent bath configuration with very low entanglement, we take full advantage of the DMRG to calculate dynamical response functions paving the way to treat large effective impurities such as those corresponding to multi-orbital interacting models and multi-site or multi-momenta clusters. This method leads to reliable calculations of non-local self energies at arbitrary dopings and interactions and at any energy scale.
1. Introduction
Among the most intriguing problems in physics is the behavior of strongly correlated materials which present emergent behavior such as high temperature superconductivity, ferroelectricity, magnetism and metal-insulator transitions. These systems have triggered a great deal of research and are still far from being understood. However, a complete theoretical understanding is still lacking due to the presence of strongly interacting local orbitals in these materials. Methods to calculate electronic structure of weakly correlated materials, such as the Density Functional Theory (DFT) [1] which use the local density approximation (LDA) [2] and other generalizations, are unable to describe accurately the strong electronic correlation case. Non-perturbative numerical methods are, thus, the only reliable approach.
To include correlations, the Dynamical Mean Field Theory (DMFT) was developed more than 20 years ago. Together with its sucessive improvements [3–8], these methods have led to more reliable results. The combination of the DMFT with LDA has allowed for band structure calculations of a large variety of correlated materials (for reviews see [9, 10]), where the DMFT accounts mainly for local interactions [11, 12]. A recent proposal, the Density Matrix Embedding Theory (DMET) relies on the embedding of the wave functions of a local cluster fragment (instead of the local Green functions) in a self-consistent finite environment [13, 14].
The DMFT requires the calculation of an interacting quantum impurity for which the fermionic environment has to be determined self-consistently until convergence of the local Green functions and the local self-energies is reached. Therefore, the success and scope of the DMFT will depend on the existence of accurate methods to solve correlated and complex quantum impurities. This approach is exact for the infinitely coordinated system (infinite dimensions), the non-interacting model and in the atomic limit.
Several quantum impurity solvers have been proposed since the development of the DMFT, among which we can mention the iterative perturbation theory (IPT) [15, 16], exact diagonalization (ED) [17, 18], the Hirsch-Fye quantum Monte Carlo (HFQMC) [19], the continuous time quantum Monte Carlo (CTQMC) [20–24], non-crossing approximations (NCA) [25], the numerical renormalization group (NRG) [26–29], the rotationally invariant slave-boson mean-field theory (RISB) [30–32] and quantum chemistry-based techniques [33]. Although these methods allow for the calculation of relevant properties such as the metal-insulator transition and other low-lying energy properties, they present some problems. Among them, one can mention the sign problem and the difficulty in reaching low temperatures in the QMC-based algorithms, the difficulty of the NCA in obtaining a reliable solution for the metallic state, the limitation to few lattice sites of the ED, far from the thermodynamic limit, and the reduced high-energy resolution of the NRG technique.
To overcome some of these difficulties an impurity solver based on the Densit Matrix Renormalization Group (DMRG) technique [34–38] was proposed [39–42]. Subsequent improvements to this were introduced, such as those using the time evolution DMRG algorithm [43, 44], dynamical calculations using the Kernel Polynomial Method (Chebyshev expansion for Green functions) [45–48] and the application to non-equilibrium DMFT using MPS [49]. In a recent work [50], the authors converge the DMFT loop on the the imaginary-frequency axis rather than on the real-frequency one, reducing computational costs by orders of magnitude. This is because the bath can be represented in a controlled way with fewer bath sites and, most importantly, the imaginary-time evolution does not create quantum entanglement. This imaginary time algorithm is able to treat much more complex model Hamiltonians. However, the price to be paid is a reduced resolution on the real-frequency axis.
In spite of these developments, several difficulties still remain which hinder the calculation of reliable spectral densities for complex multi-band and multi-orbital correlated systems [51]. In this paper we present a novel technique based on the DMRG which includes important improvements and complements previous methods. It is based on an efficient selection of the relevant states due to low entanglement bath configurations and on the targetting of the correction vector for small real energy windows. This method, thus, provides detailed spectral functions for complex Hamiltonians at zero temperature and for any doping and correlations. In the following sections we describe the method and show some applications and potential uses.
2. General Formulation
In order to present a unified treatment of multi-site (or cluster) and multi-orbital Hamiltonians on the lattice, we start by interpreting the lattice as a superlattice such that:
1. The interaction is local and completely contained in the unit cell: , where i is the cell index.
2. The non-interacting Hamiltonian Ĥ0 is characterized by its local Green function matrix G0(ω1−T); being T = (tIJ) the coefficients of the local part of Ĥ0: , where creates an electron in cell i and local “orbital” I = 1, 2, .., Nc with spin σ = ↑, ↓.
These two points completely define our problem through the parameters , G0, T. Notice that G0 and T are typically well known one-particle quantities for a given lattice problem.
The key idea of the DMFT is to neglect the self-energy between different cells i and j in the lattice, that is, to consider only the local self-energy: Σij(ω) ≈ Σ(ω)δij. In this way, we are neglecting spatial correlations up to a certain degree while a good treatment of the local dynamical correlations is made. The relevant point is that the problem becomes tractable, as we will see in the following. Note that G0, T, and Σ are Nc × Nc matrices for the spin-symmetric solution, and 2Nc × 2Nc matrices in the general case. Spatial correlations or the momentum dependence of Σ can be obtain by periodization [52].
The local Green function is now given by DMFT [53]
which defines the self-consistency condition for the Nc × Nc matrices G and Σ. The lattice problem can now be mapped onto an auxiliar impurity problem that has the same local magnitudes G(ω) and Σ(ω). This impurity problem should be determined iteratively. The impurity Hamiltonian can be written:
where the non-interacting part Hb represents the bath:
corresponds to the creation operator for the bath-site q, associated to the “orbital” I and spin σ (see Figure 1), are real and symmetric and are symmetric coefficients.
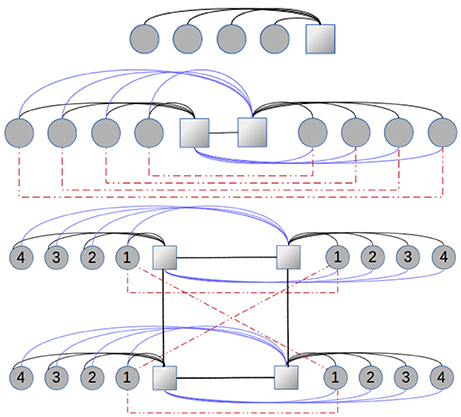
Figure 1. Graphic representation of Hamiltonian Equation (2) corresponding to the impurity problem for the one, two, and four-site cellular DMFT. The circles (squares) represent the non-interacting (interacting/impurity) sites. The red lines correspond to the parameters between bath sites q related to impurities I and J (they are the only hybridization between the baths related to different impurities). The blue lines are the with I≠J while the black lines are the . In the bottom scheme we omit some obvious connections for clarity.
The self-consistent iterations can be summarized as follows:
(i) Start with Σ(ω) = 0,
(ii) Calculate the Green's function:
(iii) Obtain the hybridization
(iv) Find a Hamiltonian representation Himp with hybridization to approximate Γ(ω). The hybridization is characterized by the parameters and of Hb through:
(v) Calculate the impurity Green's function matrix Gimp(ω) of the Hamiltonian Himp using DMRG. (vi) Obtain the self-energy
Return to (ii) until convergence. Step (iv) is a fitting problem for Υq and Λq, where we can use the general symmetries of the hybridization function matrix. If Γ can be diagonalized using the same unitary rotation R for all ω then we obtain (at most) Nc independent fittings. This can be seen from Equation (6) after applying R:
where the superscript D is used to stress that these matrices are diagonal, and MD = R† · M·R where M is an Nc × Nc matrix. In this new basis (the so-called molecular-orbital basis), we have to fit using the expression (9) for which depends on the parameters and , and similarly for , etc. Once these independent fittings are done, we bring the parameters back to our original basis through M = R · MD · R†.
In general, symmetries can be expoited for a better performance and stability. For example, at half-filling we could also have the electron-hole symmetry, giving a conection between G(−ω) and G(ω), implying the same structure for the hybridization Γ(ω).
The most resource-demanding part of the algorithm is carried out at step (v), where the dynamics of a complex many-body problem (see Figure 1) is calculated. Here we use the correction-vector method together with the DMRG essentially following [54, 55], although other methods to calculate dynamical response functions withing the DMRG can also be used [56, 57]. The one-dimensional representation of the problem (needed for a DMRG calculation) is shown in the figure, where we are also duplicating the graph when considering spin degress of freedom (not shown for clarity). In this configuration (star geometry), in spite of the high connectivity of the Hamiltonian, the DMRG shows a much better performance [50, 58].
The correction vector method is implemented in DMRG by targeting not only the ground state |E0〉 of the system but also the correction vector |CVr〉 associated to the applied operator at frequency ωr (and its neighborhood). For example, to obtain the single-particle density of states (DOS), the correction vector reads:
where a Lorentzian broadering η was introduced to deal with the poles of a finite-length impurity model. In this way a suitable renormalized representation of the operators is obtained to calculate the properties of the excitations around ωr, particularly the Green's function, for instance, and . Here ωr with r = 1, 2, …, Nω is a grid covering the frequencies of interest, typically Nω = 40–50 and are treated independently. Thus each DMFT iteration uses around 30 cores totalling less than 3 h for all cases considered in this work, considering system sizes of up to 36 sites.
3. One-Site DMFT
As we remark, only three parameters should be defined in order to apply the DMFT algorithm: , G0, T. We study the paramagnetic solution of the DMFT in the square (and Bethe) lattice using the following:
where , with k = (kx, ky) the Fourier space of the square lattice with N sites, N → ∞, and t (t′) denotes the (next-)nearest-neighbor hopping integral [59].
4. Two-Band Bethe Lattice
We consider the interaction:
where J > 0 is the Hund exchange, U (U2) is the intra (inter)-orbital Coulomb repulsion, and I = 1, 2 are the orbitals. The on-site non-interacting coefficients are
and the local Green's function:
where , and t1, t2 are the nearest-neighbor hoppings for each orbital.
Concerning step (iv), if t12 = 0 then all our 2 × 2 matrices are diagonal and we have only to calculate two Green's functions and do two independent fittings, one for each orbital. On the other hand, if t1 = t2 but t12 ≠ 0 then we can introduce the rotation to diagonalize the hybridization and we do again only two independent fittings. In the general case, a non-diagonal matrix fitting should be done to obtain a bath representation of the given hybridization Γ(ω), that is, to find the parameters Υq and Λq which minimize using, for instance, the matrix norm .
In Figure 2 we present the results for this model where, by analyzing the DOS for the different bandwidth case, the orbital-selective Mott transition can be clearly observed for a finite Hund's coupling J. This phase is robust for a certain range of interband hybridization, as is also shown in this figure. Previous calculations [60–62] either resorted to approximate analytic continuation methods to obtain the DOS or used exact diagonalization for small baths. The results shown here are calculated on the real energy axis directly (except for the small imaginary shift η). This is a main advantage over other methods and leads to much more precise and reliable results. It also has the potentiality of treating even larger clusters or more orbitals. For example, the advantages of the method presented here were crucial to find the in-gap holon-doublon quasiparticle peaks in the DOS when we appied it to calculate the asymmetric Hubbard model [63]. These quasiparticle peaks would have been either hindered using QMC or NRG solvers or they would have lacked a proper finite size analysis had an ED method been used.
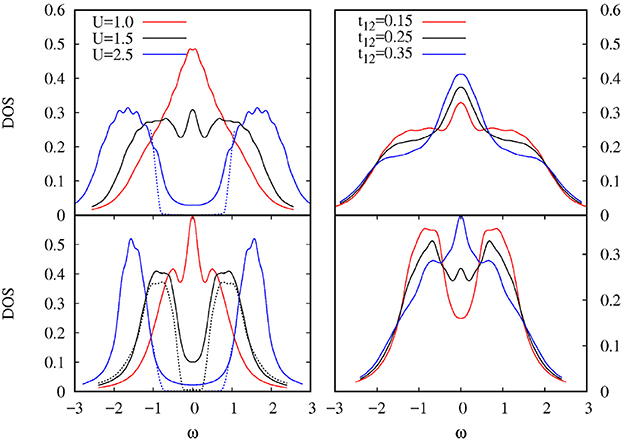
Figure 2. DOS for the half-filled two-band Kanamori-Hubbard model on the Bethe lattice showing the orbital-selective Mott transition for different bandwidths: t1 = 0.5 (Top panels) and t2 = 0.25 (Bottom panels). (Left panel) varying U for t12 = 0. (Right panel) Varying t12 for U = 1.5. We consider the rotationally invariant case U2 = U−2J and J = U/4. Two different values of the broadening η are depicted to emphasize the gapped region for the insulating phase.
5. Cellular DMFT on the Square Lattice
We consider the same physical problem of section 3 on the square lattice, but interpreted now in a superlattice of unit cell of size Nc = 2(or 4) corresponding to the two-site (c2) or four-site (c4) cellular DMFT [5, 64]. This case is illustrated in Figure 1. The next-nearest-neighbor-hoppings t′ for the c4-DMFT connect the opposite vertices of the 4 impurity square depicted at the bottom of this figure. Our three parameters , T, G0 are now:
respectively. Here, T is the non-interacting intracluster matrix and is the intercluster hopping on the superlattice Fourier space , which is connected to the one-site lattice through with K the intracluster Fourier-space vectors, see Equation (23) of Maier et al. [6].
for the c2-DMFT and
for the c4-DMFT. Finally, the hybridization matrix Γ has the following form, see Liebsch et al. [64]:
which can be diagonalized using the corresponding unitary rotation R, obtaining (at most) Nc independent fittings.
In Figure 3 we show the DOS for the Hubbard Hamiltonian on the square lattice with nearest (t = 0.25) and next nearest-neighbor hopping (t′ = 0) for two values of U. Larger clusters lead to a smaller critical U and to the appearence of pseudogaps [9]. The technique presented here enhances the scope and potentiality of the DMFT, for example, by considering larger systems (we considered here 32 bath sites compared to 8 in [65]).
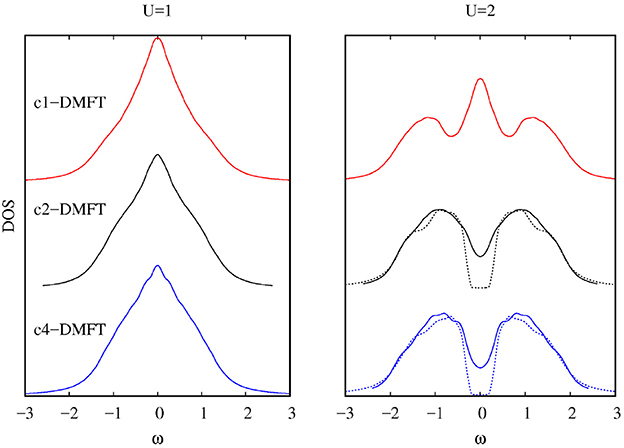
Figure 3. Comparison of the DOS calculated using different cluster sizes for the Hubbard Hamiltonian on the square lattice for two values of U. As in Figure 2, results for two different values of the broadening η are shown, emphasizing the gap for the insulating regime. The curves are shifted for clarity.
To illustrate the results with finite doping, in Figure 4 we show the DOS for the Hubbard Hamiltonian on the square lattice with nearest (t) and a finite next nearest-neighbor hopping (t′), together with the non-local Green's functions.
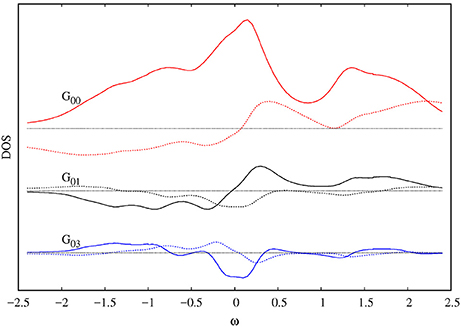
Figure 4. Imaginary (continuous lines) and real (dotted lines) Green's functions for the doped Hubbard model (μ = −0.3) on the square lattice with t′ = −0.05 and U = 2 calculated using c4-DMFT, arbitrary units. The red continuous curve corresponds to the density of states. The Fermi energy lies at ω = 0 and the horizontal lines are at zero.
6. Conclusions
We have presented here an efficient and reliable numerical method to calculate dynamical properties of complex impurities based on the DMRG. This technique uses the correction vector to obtain precise Green's functions on the real frequency axis directly thus avoiding ill-posed analytic continuation methods from the Matsubara frequencies and fermionic sign problems present in quantum Monte Carlo-based techniques, allowing also for zero temperature calculations. When used as the impurity-solver of the DMFT algorithm it leads to highly reliable spectral functions by using a self-consistent bath with low entanglement for which the density matrix renormalization works best.
To illustrate the versatility of the method, we have shown examples of densities of state and response functions within the DMFT framework for two paradigmatic models such as the Hubbard model at half filling on the square lattice on the one, two and four-site effective impurity models and at finite doping on the four-site case and also for the two-band Kanamori-Hubbard model on the Bethe lattice in the presence of Hund's coupling and interband hybridization.
This method leads to reliable results for non-local self energies at arbitrary dopings, hybridizations and interactions, at any energy scale. It also paves the way to treating large effective impurities not only within the framework of the DMFT to study multi-band interacting models and multi-site or multi-momenta clusters, but also for complex impurity problems such as adsorbed atoms, cold atoms and interacting nanoscopic systems like quantum dot arrays among others.
There is room to include additional improvements such as the consideration of symmetries, finite temperature, and more realistic systems by taking into account configurations given by ab-initio methods.
Author Contributions
YN implemented the numerical method, optimized it and adapted it for the models considered. KH did the tutoring, guiding, leading the research and providing the original DMRG and DMFT codes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge support from projects PICT 2012-1069 and PICT 2016-0402 from the Argentine ANPCyT and PIP 2015-2017 11220150100538CO (CONICET). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562 and is also funded in part by a QuantEmX grant from ICAM and the Gordon and Betty Moore Foundation through Grant GBMF5305 to KH. We thank G. Kotliar, D. García, P. Cornaglia, M. Imada, and S. Sakai for useful discussions.
References
1. Hohenberg P, Kohn W. Inhomogeneous electron gas. Phys Rev. (1964) 136:B864–71. doi: 10.1103/PhysRev.136.B864
2. Jones RO, Gunnarsson O. The density functional formalism, its applications and prospects. Rev Mod Phys. (1989) 61:689–746. doi: 10.1103/RevModPhys.61.689
3. Kotliar G, Vollhardt D. Strongly correlated materials: insights from dynamical mean-field theory. Phys Today (2004) 57:53–60. doi: 10.1063/1.1712502
4. Georges A, Kotliar G, Krauth W, Rozenberg MJ. Dynamical mean-field theory of strongly correlated fermion systems and the limit of infinite dimensions. Rev Mod Phys. (1996) 68:13. doi: 10.1103/RevModPhys.68.13
5. Kotliar G, Savrasov SY, Pálsson G, Biroli G. Cellular dynamical mean field approach to strongly correlated systems. Phys Rev Lett. (2001) 87:186401. doi: 10.1103/PhysRevLett.87.186401
6. Maier T, Jarrell M, Pruschke T, Hettler MH. Quantum cluster theories. Rev Mod Phys. (2005) 77:1027. doi: 10.1103/RevModPhys.77.1027
7. Hettler M, Tahvildar-Zadeh A, Jarrell M, Pruschke T, Krishnamurthy H. Nonlocal dynamical correlations of strongly interacting electron systems. Phys Rev B (1998) 58:R7475. doi: 10.1103/PhysRevB.58.R7475
8. Sénéchal D, Perez D, Pioro-Ladriere M. Spectral weight of the Hubbard model through cluster perturbation theory. Phys Rev Lett. (2000) 84:522. doi: 10.1103/PhysRevLett.84.522
9. Imada M, Miyake T. Electronic structure calculation by first principles for strongly correlated electron systems. J Phys Soc Jpn. (2010) 79:112001. doi: 10.1143/JPSJ.79.112001
10. Held K. Electronic structure calculations using dynamical mean field theory. Adv Phys. (2007) 56:829–926. doi: 10.1080/00018730701619647
11. Anisimov VI, Aryasetiawan F, Lichtenstein A. First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA+ U method. J Phys Condens Matt. (1997) 9:767. doi: 10.1088/0953-8984/9/4/002
12. Lichtenstein A, Katsnelson M. Ab initio calculations of quasiparticle band structure in correlated systems: LDA++ approach. Phys Rev B (1998) 57:6884. doi: 10.1103/PhysRevB.57.6884
13. Knizia G, Chan GKL. Density matrix embedding: a simple alternative to dynamical mean-field theory. Phys Rev Lett. (2012) 109:186404. doi: 10.1103/PhysRevLett.109.186404
14. Bulik IW, Chen W, Scuseria GE. Electron correlation in solids via density embedding theory. J Chem Phys. (2014) 141:054113. doi: 10.1063/1.4891861
15. Georges A, Kotliar G. Hubbard model in infinite dimensions. Phys Rev B (1992) 45:6479. doi: 10.1103/PhysRevB.45.6479
16. Rozenberg M, Kotliar G, Zhang X. Mott-Hubbard transition in infinite dimensions. II. Phys Rev B (1994) 49:10181. doi: 10.1103/PhysRevB.49.10181
17. Caffarel M, Krauth W. Exact diagonalization approach to correlated fermions in infinite dimensions: mott transition and superconductivity. Phys Rev Lett. (1994) 72:1545. doi: 10.1103/PhysRevLett.72.1545
18. Lu Y, Höppner M, Gunnarsson O, Haverkort MW. Efficient real-frequency solver for dynamical mean-field theory. Phys Rev B (2014) 90:085102. doi: 10.1103/PhysRevB.90.085102
19. Hirsch JE, Fye RM. Monte Carlo method for magnetic impurities in metals. Phys Rev Lett. (1986) 56:2521. doi: 10.1103/PhysRevLett.56.2521
20. Rubtsov AN, Savkin VV, Lichtenstein AI. Continuous-time quantum Monte Carlo method for fermions. Phys Rev B (2005) 72:035122. doi: 10.1103/PhysRevB.72.035122
21. Werner P, Comanac A, DeMedici L, Troyer M, Millis AJ. Continuous-time solver for quantum impurity models. Phys Rev Lett. (2006) 97:076405. doi: 10.1103/PhysRevLett.97.076405
22. Park H, Haule K, Kotliar G. Cluster dynamical mean field theory of the Mott transition. Phys Rev Lett. (2008) 101:186403. doi: 10.1103/PhysRevLett.101.186403
23. Gull E, Millis AJ, Lichtenstein AI, Rubtsov AN, Troyer M, Werner P. Continuous-time Monte Carlo methods for quantum impurity models. Rev Mod Phys. (2011) 83:349. doi: 10.1103/RevModPhys.83.349
24. Nomura Y, Sakai S, Arita R. Nonlocal correlations induced by Hund's coupling: a cluster DMFT study. Phys Rev B (2015) 91:235107. doi: 10.1103/PhysRevB.91.235107
25. Pruschke T, Cox DL, Jarrell M. Hubbard model at infinite dimensions: Thermodynamic and transport properties. Physical Review B. 1993;47(7):3553. doi: 10.1103/PhysRevB.47.3553
26. Wilson KG. The renormalization group: Critical phenomena and the Kondo problem. Reviews of Modern Physics. 1975;47(4):773. doi: 10.1103/RevModPhys.47.773
27. Bulla R. Zero temperature metal-insulator transition in the infinite-dimensional hubbard model. Phys Rev Lett. (1999) 83:136. doi: 10.1103/PhysRevLett.83.136
28. Stadler K, Yin Z, von Delft J, Kotliar G, Weichselbaum A. Dynamical mean-field theory plus numerical renormalization-group study of spin-orbital separation in a three-band hund metal. Phys Rev Lett. (2015) 115:136401. doi: 10.1103/PhysRevLett.115.136401
29. Stadler K, Mitchell A, von Delft J, Weichselbaum A. Interleaved numerical renormalization group as an efficient multiband impurity solver. Phys Rev B (2016) 93:235101. doi: 10.1103/PhysRevB.93.235101
30. Lechermann F, Georges A, Kotliar G, Parcollet O. Rotationally invariant slave-boson formalism and momentum dependence of the quasiparticle weight. Phys Rev B (2007) 76:155102. doi: 10.1103/PhysRevB.76.155102
31. Isidori A, Capone M. Rotationally invariant slave bosons for strongly correlated superconductors. Phys Rev B (2009) 80:115120. doi: 10.1103/PhysRevB.80.115120
32. Ferrero M, Cornaglia PS, De Leo L, Parcollet O, Kotliar G, Georges A. Valence bond dynamical mean-field theory of doped Mott insulators with nodal/antinodal differentiation. Europhys Lett. (2009) 85:57009. doi: 10.1209/0295-5075/85/57009
33. Zgid D, Chan GKL. Dynamical mean-field theory from a quantum chemical perspective. J Chem Phys. (2011) 134:094115. doi: 10.1063/1.3556707
34. White SR. Density matrix formulation for quantum renormalization groups. Phys Rev Lett. (1992) 69:2863. doi: 10.1103/PhysRevLett.69.2863
35. Peschel I, Wang X, Kaulke M, Hallberg K. Density Matrix Renormalization. Lecturer Notes in Physics (1999) 528. doi: 10.1007/BFb0106062
36. Schollwöck U. The density-matrix renormalization group. Rev Mod Phys. (2005) 77:259. doi: 10.1103/RevModPhys.77.259
37. Hallberg KA. New trends in density matrix renormalization. Adv Phys. (2006) 55:477–526. doi: 10.1080/00018730600766432
38. Nishimoto S, Gebhard F, Jeckelmann E. Dynamical density-matrix renormalization group for the Mott–Hubbard insulator in high dimensions. J Phys. (2004) 16:7063. doi: 10.1088/0953-8984/16/39/038
39. García DJ, Hallberg K, Rozenberg MJ. Dynamical mean field theory with the density matrix renormalization group. Phys Rev Lett. (2004) 93:246403. doi: 10.1103/PhysRevLett.93.246403
40. García D, Miranda E, Hallberg K, Rozenberg M. Mott transition in the Hubbard model away from particle-hole symmetry. Phys Rev B (2007) 75:121102. doi: 10.1103/PhysRevB.75.121102
41. Fernández YN, García D, Hallberg K. The two orbital Hubbard model in a square lattice: a DMFT+ DMRG approach. J Phys. 568:042009. doi: 10.1088/1742-6596/568/4/042009
42. Karski M, Raas C, Uhrig GS. Electron spectra close to a metal-to-insulator transition. Phys Rev B (2005) 72:113110. doi: 10.1103/PhysRevB.72.113110
43. Ganahl M, Aichhorn M, Evertz HG, Thunström P, Held K, Verstraete F. Efficient DMFT impurity solver using real-time dynamics with matrix product states. Phys Rev B (2015) 92:155132. doi: 10.1103/PhysRevB.92.155132
44. Bauernfeind D, Zingl M, Triebl R, Aichhorn M, Evertz HG. Fork tensor-product states: efficient multiorbital real-time DMFT solver. Phys Rev X (2017) 7:031013. doi: 10.1103/PhysRevX.7.031013
45. Weiße A, Wellein G, Alvermann A, Fehske H. The kernel polynomial method. Rev Mod Phys. (2006) 78:275. doi: 10.1103/RevModPhys.78.275
46. Holzner A, Weichselbaum A, McCulloch IP, Schollwöck U, von Delft J. Chebyshev matrix product state approach for spectral functions. Phys Rev B (2011) 83:195115. doi: 10.1103/PhysRevB.83.195115
47. Wolf FA, McCulloch IP, Parcollet O, Schollwöck U. Chebyshev matrix product state impurity solver for dynamical mean-field theory. Phys Rev B (2014) 90:115124. doi: 10.1103/PhysRevB.90.115124
48. Ganahl M, Thunström P, Verstraete F, Held K, Evertz HG. Chebyshev expansion for impurity models using matrix product states. Phys Rev B (2014) 90:045144. doi: 10.1103/PhysRevB.90.045144
49. Wolf FA, McCulloch IP, Schollwöck U. Solving nonequilibrium dynamical mean-field theory using matrix product states. Phys Rev B (2014) 90:235131. doi: 10.1103/PhysRevB.90.235131
50. Wolf FA, Go A, McCulloch IP, Millis AJ, Schollwöck U. Imaginary-time matrix product state impurity solver for dynamical mean-field theory. Phys Rev X (2015) 5:041032. doi: 10.1103/PhysRevX.5.041032
51. Hallberg K, García D, Cornaglia PS, Facio JI, Núñez-Fernández Y. State-of-the-art techniques for calculating spectral functions in models for correlated materials. Europhys Lett. (2015) 112:17001. doi: 10.1209/0295-5075/112/17001
52. Stanescu TD, Kotliar G. Fermi arcs and hidden zeros of the Green function in the pseudogap state. Phys Rev B (2006) 74:125110. doi: 10.1103/PhysRevB.74.125110
53. DMFT. DMFT at 25: Infinite Dimensions, Modeling and Simulation, vol. 4 Autumn School on Correlated Electrons. Jülich: Forschungszentrum Jülich Zentralbibliothek, Verlag (2014) Available online at: http://juser.fz-juelich.de/record/155829
54. Kühner TD, White SR. Dynamical correlation functions using the density matrix renormalization group. Phys Rev B (1999) 60:335. doi: 10.1103/PhysRevB.60.335
55. Ramasesha S, Pati S, Krishnamurthy HR, Shuai Z, Brédas JL. Low-Lying Electronic Excitations and Nonlinear Optic Properties of Polymers via Symmetrized Density Matrix Renormalization Group Method. Synth Metals (1997) 85:1019–22. doi: 10.1016/S0379-6779(97)80136-1
56. Hallberg KA. Density-matrix algorithm for the calculation of dynamical properties of low-dimensional systems. Phys Rev B (1995) 52:R9827–30. doi: 10.1103/PhysRevB.52.R9827
57. Jeckelmann E. Dynamical density-matrix renormalization-group method. Phys Rev B (2002) 66:045114. doi: 10.1103/PhysRevB.66.045114
58. Holzner A, Weichselbaum A, von Delft J. Matrix product state approach for a two-lead multilevel Anderson impurity model. Phys Rev B (2010) 81:125126. doi: 10.1103/PhysRevB.81.125126
59. Sakai S, Motome Y, Imada M. Doped high-T c cuprate superconductors elucidated in the light of zeros and poles of the electronic Greens function. Phys Rev B (2010) 82:134505. doi: 10.1103/PhysRevB.82.134505
60. Koga A, Kawakami N, Rice TM, Sigrist M. Orbital-selective mott transitions in the degenerate Hubbard Model. Phys Rev Lett. (2004) 92:216402. doi: 10.1103/PhysRevLett.92.216402
61. Koga A, Kawakami N, Rice TM, Sigrist M. Spin, charge, and orbital fluctuations in a multiorbital Mott insulator. Phys Rev B (2005) 72:045128. doi: 10.1103/PhysRevB.72.045128
62. Winograd EA, de', Medici L. Hybridizing localized and itinerant electrons: a recipe for pseudogaps. Phys Rev B (2014) 89:085127. doi: 10.1103/PhysRevB.89.085127
63. Núñez-Fernández Y, Kotliar G, Hallberg K. (2017). Emergent low-energy bound states in the two-orbital Hubbard model. arXiv:1710.08792.
64. Liebsch A, Ishida H, Merino J. Multisite versus multiorbital Coulomb correlations studied within finite-temperature exact diagonalization dynamical mean-field theory. Phys Rev B (2008) 78:165123. doi: 10.1103/PhysRevB.78.165123
Keywords: density matrix renormalization group, dynamical mean field theory, correlated electrons, density of states, multi-orbital models
Citation: Núñez Fernández Y and Hallberg K (2018) Solving the Multi-site and Multi-orbital Dynamical Mean Field Theory Using Density Matrix Renormalization. Front. Phys. 6:13. doi: 10.3389/fphy.2018.00013
Received: 20 November 2017; Accepted: 05 February 2018;
Published: 26 February 2018.
Edited by:
Gerardo Ortiz, Indiana University Bloomington, United StatesReviewed by:
Zohar Nussinov, Washington University in St. Louis, United StatesMarco Buongiorno Nardelli, University of North Texas, United States
Copyright © 2018 Núñez Fernández and Hallberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuriel Núñez Fernández, eXVyaWVsbmZAZ21haWwuY29t