 Taras Bryk
Taras Bryk Taras Demchuk
Taras Demchuk Noël Jakse3
Noël Jakse3 Jean-François Wax
Jean-François Wax- 1Institute for Condensed Matter Physics of the National Academy of Sciences of Ukraine, Lviv, Ukraine
- 2Lviv Polytechnic National University, Lviv, Ukraine
- 3Université Grenoble Alpes, Centre National de la Recherche Scientifique, Grenoble INP, SIMaP, Grenoble, France
- 4Laboratoire de Chimie et de Physique A2MC, Université de Lorraine, Metz, France
Recent findings of pressure-induced emergence of unusual high-frequency contribution to transverse current spectral functions in several simple liquid metals at high pressures raised a question whether similar features can be observed in liquid metals at ambient conditions. We report here analysis of ab initio molecular dynamics-derived longitudinal and transverse current spectral functions and corresponding dispersions of collective excitations in liquid polyvalent metals Al, Tl, Ni. We have not found evidences of the second branch of high-frequency transverse modes in liquid Al and Ni, while in the case of liquid Tl they were clearly present in transverse dynamics. The vibrational density of states for liquid Tl has a pronounced high-frequency shoulder, which is located right in the frequency range of the second high-frequency transverse branch, while for liquid Al and Ni the vibrational density of states has only a weak indication of possible high-frequency shoulder. The origin of specific behavior of transverse excitations in liquid Tl is discussed.
1. Introduction
Collective dynamics in simple liquid metals is well understood on macroscopic spatial and temporal scales only, where all the contributions from relaxation and propagating collective modes to the measured in Brillouin light scattering experiments dynamic structure factors are well known. Hydrodynamic theory being in fact a set of local conservation laws provides perfect description of collective dynamics in fluids treated as continuum [1, 2]. On microscopic scale with atomic resolution, the collective dynamics in liquids can be studied by inelastic X-ray or neutron scattering experiments, as well as by molecular dynamics simulations (MD). Many new features appear on the atomic scales in dynamics of liquids and also in liquid metals, which are not described by hydrodynamic theory. They can be represented as effects of non-hydrodynamic collective modes like structural relaxation, shear waves, stress relaxation, heat waves, etc.[3].
Recent findings of pressure-induced emergence of unusual high-frequency contribution to transverse current spectral functions in liquid Li [4], Na [5], and Fe [6] at high pressures opened a new discussion on possible two types of transverse propagating modes coexisting on the spatial scale of the cage of nearest neighbors. The cage of nearest neighbors means the configuration of nearest atoms in the first coordination shell, which for dense liquids (especially in supercooled state) can be stable over some period of time, while diffusion of particles destroy the instantaneous cages. All these simulation results raise naturally the question whether similar features in transverse dynamics can be observed in liquid metals at ambient conditions. Another important and so far unclear issue is the range of wave numbers where the manifestation of the second branch of transverse excitations can be observed. As a matter of fact, analysis of inelastic X-ray scattering experiments indicated a possible coupling between longitudinal (L) and transverse (T) collective excitations and consequently two contributions of coupled propagating modes at relatively small wave numbers [7]. In contrast, ab initio MD (AIMD) simulations [4] clearly indicated the emergence of the second transverse branch only for wave numbers outside the first pseudo-Brillouin zone. Moreover, it has been reported earlier for polyvalent liquid metals such as Ga [7] and Sn [8] the existence of two contributions from propagating modes as measured by IXS experiments. Recent ab initio simulations of liquid Tl [9] near melting and Ni [10] at T = 1773 K showed a possibility to observe a small shoulder in the shape of dynamic structure factor S(k, ω) at wave numbers k within the first pseudo-Brillouin zone and frequencies ω below the L-acoustic mode, ascribed to a contribution from transverse excitations. Another liquid metal, liquid Zn at T=723 K, revealed a strongly smeared two-peak shape of L- and T- current spectral functions from ab initio simulations [11], and an attempt was made to explain this feature by mode-coupling effects. It will therefore be interesting to study collective dynamics in several other polyvalent and transition metals as well as different temperatures by means of computer simulations to clarify the issue with the possibility of emergence of two contributions from L- and T-collective excitations to different spectral functions.
In the present work we aim to perform ab initio simulations in order to derive L and T current spectral functions and well as dispersion of L- and T-collective excitations in liquid polyvalent metals Al, Tl as well as in transition liquid metal Ni at ambient pressure. The remaining part of paper has the following structure: next section contains information on the ab initio simulations that have been carried out for the studied liquid metals. In section 3 we report the spectra calculations and supply discussion on the obtained results. Finally, conclusions of this study are presented in the last section.
2. Simulation Background
The AIMD simulations for Al, Ni and Tl were performed in the present work within the framework of the density-functional theory (DFT) by the Vienna ab initio simulation package (VASP) [12]. A plane-wave basis set with a specific cut-off energy is used for each element, namely 241 eV for Al, 270 eV for Ni, and 90.14 eV for Tl. The cut-off energies were taken as VASP default values for corresponding electron-ion interactions represented by the projected augmented-wave potentials, which provide correct nodal structure of wave functions in the core regions [13, 14]. Specific exchange-correlation energy was used depending on the element considered, i.e., the local-density approximation (LDA) [15] for Al, and the generalized gradient approximation (GGA) in the Perdew-Burke-Ernzerhof (PBE) form [16, 17] for Ni and Tl. It appeared that for the case of liquid Tl at 577 K the agreement with experimental structure factor is worse, than from simulations with local density approximation. Therefore we additionally performed AIMD for liquid Tl at the same temperature using LDA.
All the simulations were carried out in the canonical ensemble (NVT), namely constant number of atoms, volume, and temperature, by applying the Nosé thermostat [18, 19]. Newton's equations of motion were integrated using Verlet's algorithm in the velocity form with a time step of 1 fs for Al, 1.5 fs for Ni, and 2 fs for Tl. The time steps were taken small enough in order to converge the DFT total energy within 4–5 iterations of wave functions. A cubic MD box with periodic boundary conditions containing 256 atoms for Al, 108 for Ni and 300 for Tl was used in the simulations, and only the Γ-point was considered to sample the supercell Brillouin zone.
The liquid systems for Al and Ni were initially prepared and equilibrated at some temperature well above the studied one. Then we cooled the samples to the lower desired temperatures for the given element with a rate of 1013 K/s followed by another equilibration for 3 ps. At the studied temperature, namely 1000 K for Al, 1735 K for Ni and 577 K for Tl, the volume V of cell was chosen to reproduce the experimental densities [20, 21]. The production runs were performed for 80 ps for Al and Ni. The initial configuration for liquid Tl was taken from classical MD simulations [22] and upon equilibration of 3 ps the production run of 126 ps was performed for liquid Tl.
All the time correlation functions and static wave-number dependent correlators were averaged over all possible directions of wave vectors corresponding to the same absolute value. The time correlation functions and static correlators were averaged over 80,000 configurations for Al and Ni, and for 40,000 ones in case of Tl. Note that the smallest accessible wave number for which the time correlation functions can be calculated in our simulations is 0.371Å−1 for Al, 0.569Å−1 for Ni and 0.301 Å−1 for Tl. It is worth mentioning that the AIMD simulations for Al and Ni used here were performed in our previous works [23–26].
3. Results and Discussion
For each of the three studied liquid polyvalent and transition metals we calculated directly the static structure factor via static density-density correlators
where n(k) are spatial-Fourier components of particle density and brackets denote ensemble average. The velocity autocorrelation functions ψ(t) were calculated with the purpose to analyze their time-Fourier transform , which outside the small-frequency region reflects the vibrational density of states in the simulated system. The collective dynamics was analyzed from the calculated L- and T-current spectral functions CL/T(k, ω), peaks of which are connected with the frequencies of corresponding collective propagating modes.
3.1. Liquid Al
Structure factor for liquid Al at temperature 1000 K is shown in Figure 1. A high number of configurations taken for calculations of the structure factor S(k) allowed to obtain very smooth k-dependence with a peak located at Kp = 2.69Å−1. The location of the first sharp diffraction peak in structure factors of liquid defines the region of de Gennes slowing down of density fluctuations, as well as the approximate location of the first pseudo-Brillouin zone boundary (Kp/2 ~ 1.345Å−1), where one usually for dense liquids observes the maximum of the dispersion curve for longitudinal collective excitations.
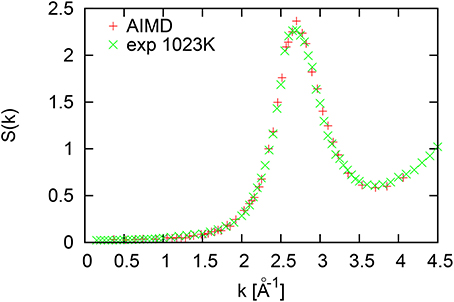
Figure 1. Structure factor of liquid Al at 1000 K.
The velocity autocorrelation function (VACF) characterizes single-particle motion of atoms in liquids, although it can be used also for extracting some information about the vibrational density of states (VDOS) for high-frequency excitations, because in the low-frequency region in liquids the vibrational density of states is hidden under the relaxing contribution which defines the diffusivity of particles. By date there is no unique theory-based scheme of how one can separate different contributions to the VACF in order to calculate the vibrational density of states, although several different approaches were suggested like 2PT-model [27]. Such a scheme for separation of relaxing and vibrational contributions to VACF should be based on the knowledge of the dispersion law of collective excitations and their damping - it is highly desirable because it would allow to study deviations from the Debye law (low frequency region of VDOS ~ ω2) due to existing “positive" or “negative" sound dispersion [28].
In Figure 2 the frequency spectrum of VACF is shown, and one can observe a single peak at ω ~ 20ps−1 and a tiny shoulder in the region of frequencies ~40–50ps−1.
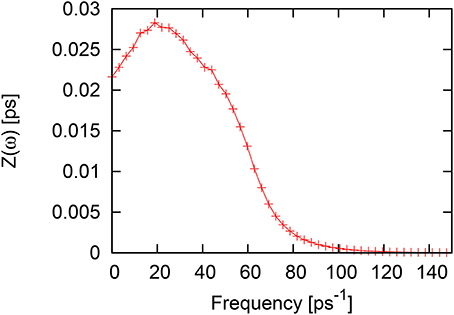
Figure 2. Fourier spectrum of velocity autocorrelation function for liquid Al.
Dispersion of L- and T-collective excitations can be roughly obtained from the location of peak position in corresponding L/T current spectral functions CL/T(k, ω), calculated for each available wave number as numerical time-Fourier transformation of the L/T current autocorrelation functions . In Figure 3 we show the shape of CL/T(k, ω) spectral functions for three wave numbers. One can see that for the wave numbers from the second pseudo-Brillouin zone only one peak is observed in the shape of CL(k, ω) and CT(k, ω), although a shoulder exists on the high-frequency side ω ~ 45ps−1 of CT(k, ω) close to wave number ~ 2Å−1. More precise schemes to analysis of different contributions to time correlation functions or to corresponding spectral functions is needed in order to find the possibility of emergence of another branch of collective excitations and estimate its origin.
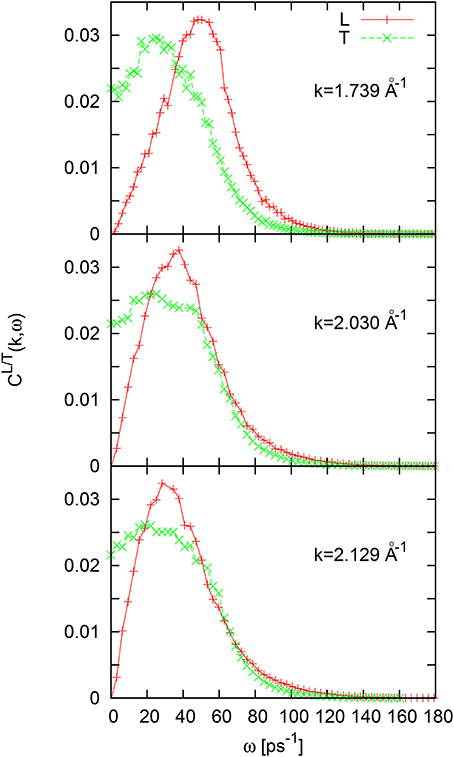
Figure 3. Longitudinal and transverse current spectral functions for liquid Al for three wave numbers in the second pseudo-Brillouin zone.
The peak position of L/T-current spectral functions as a function of wave number are shown in Figure 4. The linear dispersion by dashed line is just a guide for eye drawn through the frequency of longitudinal excitation at the smallest available wave number in order to show the existence of the “positive” sound dispersion in liquid Al, which is an evidence of strong viscoelastic crossover from hydrodynamic region with smaller adiabatic speed of sound to the elastic regime with the high-frequency apparent speed of sound. The transverse (T) branch of collective excitations has quite small propagation gap (the long-wavelength region where the transverse sound is absent), smaller than ~0.4Å−1. In the short-wavelength region the T-branch corresponds to very short-wavelength correlated oscillating motion of atoms in the cage of nearest neighbors. In the case of liquid Al the frequency of short-wavelength T-modes is located at 20–23 ps−1, that practically coincides with the peak location of the VACF frequency spectrum. We note, that the peak of the VACF frequency spectrum does not correspond to the maximum of frequencies of the L-branch of collective excitations which should be observed in case of the pure Debye model of the vibrational density of states. However, the roton-like minimum on the dispersion curve is located very close to the peak of the VACF frequency spectrum. Another finding is, that for liquid Al at ambient pressure the high-frequency branch of transverse excitations is not observed.
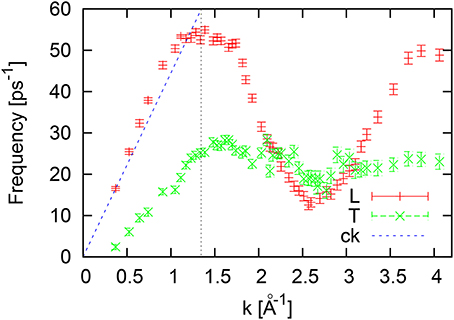
Figure 4. Dispersion of logitudinal (L) and transverse (T) collective excitations for liquid Al at 1000 K estimated from peak positions of the L/T current spectral functions. The vertical dashed line at k = 1.345Å−1 corresponds to the boundary of the first pseudo-Brillouin zone.
3.2. Liquid Ni
The structure factor of transition liquid metal Ni at 1735 K shown in Figure 5 does not show any specific features. Only the amplitude of the oscillations for wave numbers higher than the location of the first diffraction peak (Kp = 3.1Å−1) is smaller than for other studied here polyvalent metals.
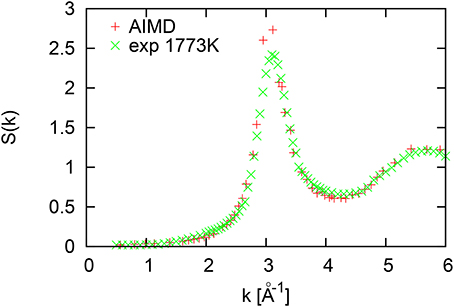
Figure 5. Structure factor of liquid Ni at 1735 K.
The spectrum of the VACF for liquid Ni (Figure 6) contains a single peak at ω ~ 25−27ps−1 and just a small change in the slope on the high-frequency side close to the frequency of ~ 45ps−1, which is much smaller than the small shoulder observed nearly at the same frequency for liquid Al (see Figure 2). We will check below at the analysis of the dispersion curves how the peak location of the VACF frequency spectrum correlates with the flat regions of L- or T-dispersions.
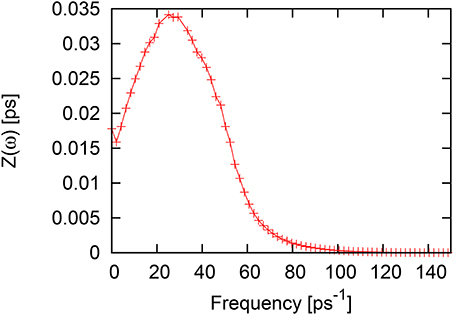
Figure 6. Fourier spectrum of velocity autocorrelation function for liquid Ni.
The current spectral functions CL/T(k, ω) for liquid Ni at 1735 K are shown in Figure 7. The L and T current spectral functions have just a single-peak form, although for the wave numbers k > 2Å−1 one may observe an emerging shoulder at frequency ~ 40ps−1.
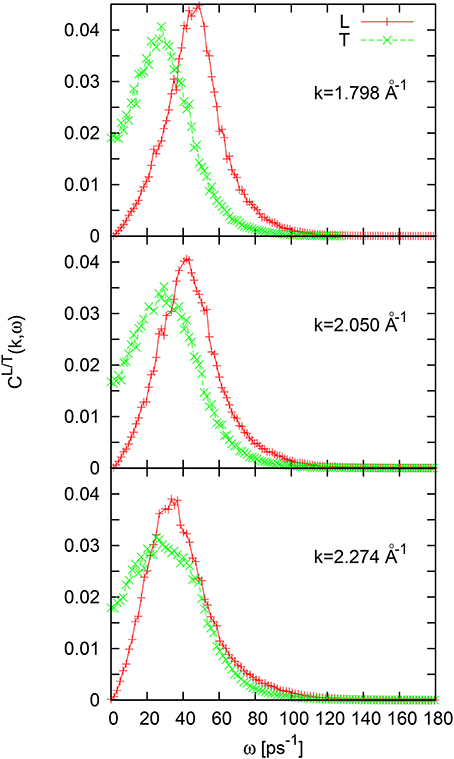
Figure 7. Longitudinal and transverse current spectral functions for liquid Ni for three wave numbers in the second pseudo-Brillouin zone.
The dispersion of L and T collective excitations for liquid Ni at 1735 K (Figure 8) have quite small error bars because of very long AIMD runs and well defined peaks of corresponding current spectral functions. Again, as for the case of liquid Al we do not see a correlation in the peak location of the VACF-spectrum and the highest longitudinal frequency, which is located at ω ~ 48ps−1, while the maximum of the Fourier-spectrum of VACF corresponds rather to the flat region of the short-wavelength region of T-dispersion at ω ~ 27−30ps−1. Again, for the liquid Ni we did not observe the second high-frequency branch of collective excitations, that correlates well with the absence of the high-frequency peak in the Fourier-spectrum of VACF.
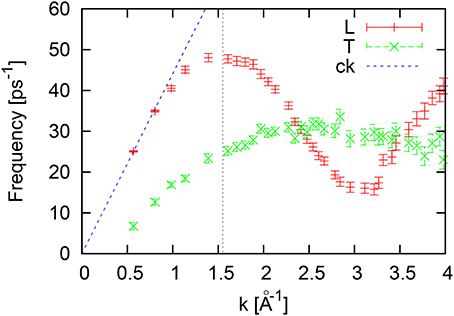
Figure 8. Dispersion of longitudinal (L) and transverse (T) collective excitations for liquid Ni at 1735 K estimated from peak positions of the L/T current spectral functions. The vertical dashed line at k = 1.55Å−1 corresponds to the boundary of the first pseudo-Brillouin zone.
3.3. Liquid Tl
The structure factors S(k) of liquid Tl at 577 K calculated with LDA and GGA are shown in Figures 9A,B. A comparison with the experimental S(k) gives evidence that LDA reproduces the experimental data much better than the GGA. The amplitide of the first sharp diffraction peak within LDA is Smax = 2.78 located at Kp ~ 2.25Å−1 which is in reasonable agreement with experimental S(k), while GGA results in much higher ampliture Smax = 3.3 at the same location Kp ~ 2.25Å−1. This is consistent with the fact that GGA generally gives rise to more compact and structured local environments, as it was shown in Jakse and Pasturel [23] for liquid Al.
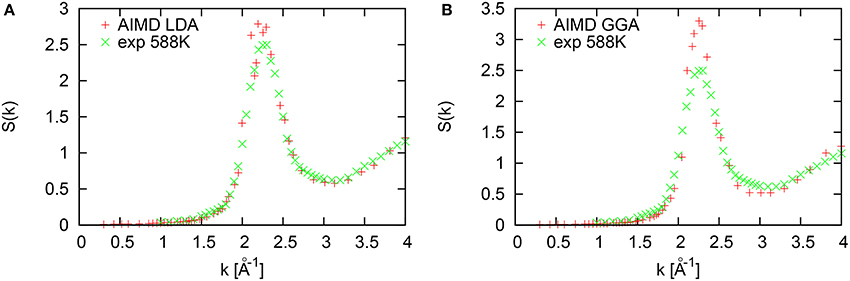
Figure 9. Structure factor of liquid Tl at 577 K from simulations in LDA (A) and GGA (B).
In contrast to the Fourier-spectrum of VACF of liquid Al and liquid Ni discussed above, the frequency spectrum of VACF for liquid Tl in LDA and GGA, shown in Figure 10, contains the well-pronounced high-frequency peak at frequency ~12ps−1 (LDA) and ~14ps−1 (GGA), while the main peak of the Fourier-spectrum of VACF is located at ω ~ 6ps−1 (LDA) and ω ~ 7ps−1 (GGA). Note, that the zero-frequency value of VACF is directly connected to diffusion coefficient, which is nearly twice higher in LDA simulations, than with the GGA. Again, the more compact local structures obtained with GGA lead to a more pronouneced cage effect and hence to a lower diffusion [23].
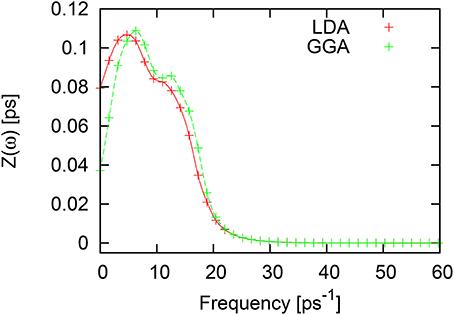
Figure 10. Fourier spectrum of velocity autocorrelation function for liquid Tl with LDA (red symbols and solid line) and GGA (green symbols and dashed line).
The L/T current spectral functions CL/T(k, ω) for the case of liquid Tl in contrast to liquid Al and liquid Ni contain well defined two-peak structure in T-spectral functions as shown in Figure 11. These features are observed only in the second pseudo-Brillouin zone, i.e., for wave numbers k > Kp/2. The second, high-frequency, peak of CT(k, ω) emerges at the frequency ~ 14ps−1, that coincides with the location of the high-frequency peak of the Fourier-spectrum of VACF.
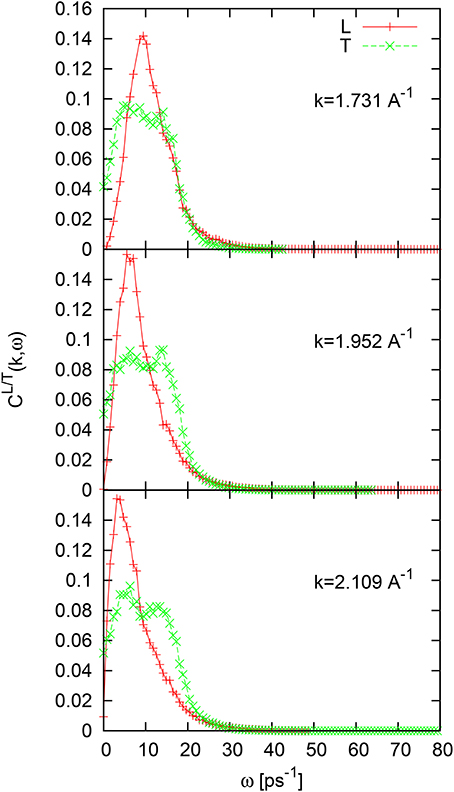
Figure 11. Longitudinal and transverse current spectral functions for liquid Tl for three wave numbers in the second pseudo-Brillouin zone from simulations with GGA.
In Figure 12 we report the longitudinal and transverse dispersions of collective excitations, with the L-branch reaching its maximum of ~16 ps−1 at Kp/2. The dispersion of T-excitations for the case of liquid Tl contains two transverse branches for wave numbers higher than the boundary of the first pseudo-Brillouin zone. The low-frequency T-branch in the short-wavelength region practically overlaps with the roton-like minimum of the L-dispersion, that is treated sometimes as a possible effect of emerging L-T coupling in atomic scale dynamics. On the other hand for liquid Al and Ni we did not observe the overlap of the short-wavelength regions of L- and T-dispersions. Hence, the dispersion of collective excitations in liquid Tl gives clear evidence of the correlation between the peaks of the Fourier-spectrum of VACF with the frequencies of the two types of T-excitations in the short-wavelength region (which practically corresponds to strong single-particle correlations in the collective quantities).
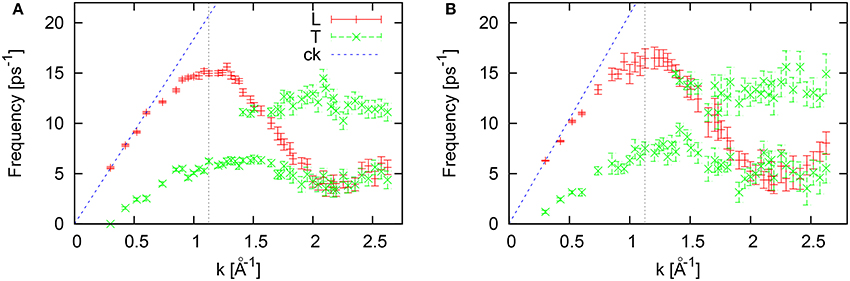
Figure 12. Dispersion of logitudinal (L) and transverse (T) collective excitations for liquid Tl at 577 K estimated from peak positions of the L/T current spectral functions in LDA (A) and GGA (B). The vertical dashed line at k = 1.125Å−1 corresponds to the boundary of the first pseudo-Brillouin zone.
4. Conclusion
We studied by ab initio molecular dynamics simulations collective dynamics of three liquid polyvalent metals at ambient pressure - Al, Tl, Ni, with a main focus on a possibility to observe directly from transverse current spectral functions CT(k, ω) unusually high-frequency short-wavelength transverse excitations previously found only in liquid metals at high pressures. The most original result is the existence of the two types of transverse excitations in liquid Tl close to the melting point from both LDA and GGA ab initio simulations. Our findings are as follows:
i. Comparing three different liquid polyvalent metals - only in the case of liquid Tl we observed two pronounced peaks of CT(k, ω) corresponding to two types of short-wavelength transverse excitations;
ii. The second high-frequency branch of transverse excitations in liquid Tl becomes visible only for wave numbers higher than the first pseudo-Brillouin zone boundary, i.e., for k > Kp/2, where Kp is the position of the main peak of static structure factor S(k). For smaller wave numbers the high-frequency transverse excitations were not found, in total agreement with the case of liquid Li [4] at high pressure;
iii. For the case of liquid Tl we observed a pronounced high-frequency shoulder and a small peak on the high-frequency side of the spectrum of velocity autocorrelation function. There exists a clear correlation between the existence of the high-frequency peak and emergence of the high-frequency transverse excitations with practically the same frequency as this is for liquid Tl. For liquid Al at 1000 K and liquid Ni at 1735 K only a tiny shoulder (and no peak) can be resolved in the high-frequency side of the spectrum of velocity autocorrelation functions in corresponding liquid metals;
iv. We observed shoulders on the high-frequency side CT(k, ω) for liquid Al close to k ~ 2Å−1. We refrain here to attribute this feature to the correlation between the high frequency-peak of the VDOS and the emergence of a high frequency transverse branch. However, this implies a possibility for liquid Al to have the short-wavelength high-frequency excitations that would be better revealed by applying relatively small pressure. This should be studied in more detail and will be reported elsewhere;
v. The flat dispersion of the high-frequency transverse branch implies localization of this mode. In general the existence of two transverse branches - low- and high-frequency ones on the spatial scale of the cage of nearest neighbors can be considered as some analogy of coexisting types of normal modes of stable over some period of time clusters of several atoms.
Author Contributions
TB and TD performed ab initio simulations of liquid Tl with different approximations of exchange-correlation functional (LDA and GGA), analyzed the spectral functions for Al, Ni, and Tl and calculated spectra of collective excitations for these liquid metals. NJ performed ab initio simulations for liquid Al and Ni. J-FW performed analysis of time correlation functions, calculated by the other authors. TB, NJ, and J-FW discussed the results and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
TB and TD gratefully acknowledge computing time allocation for the ab initio simulations in frames of the UA-GRID-project. The calculations have been performed using the ab initio total-energy and molecular dynamics program VASP (Vienna ab initio simulation program) developed at the Institute für Materialphysik of the Universität Wien [29–32]. We acknowledge the CINES and IDRIS under Project N INP2227/72914 as well as PHYNUM CIMENT for computational resources. This work was performed within the framework of the Centre of Excellence of Multifunctional Architectured Materials “CEMAM” nANR-10-LABX-44-01 funded by the “Investments for the Future” Program.
References
3. Bryk T. Non-hydrodynamic collective modes in liquid metals and alloys. Eur Phys J Special Top. (2011) 196:65–83. doi: 10.1140/epjst/e2011-01419-x
4. Bryk T, Ruocco G, Scopigno T, Seitsonen AP. Pressure-induced emergence of unusually high-frequency transverse excitations in a liquid alkali metal: evidence of two types of collective excitations contributing to the transverse dynamics at high pressures. J Chem Phys. (2015) 143:110204. doi: 10.1063/1.4928976
5. Marqués M, González DJ, González LE. Structure and dynamics of high-pressure Na close to the melting line: An ab initio molecular dynamics study. Phys Rev B (2016) 94:024204. doi: 10.1103/PhysRevB.94.024204
6. Marqués M, González LE, González DJ. Pressure-induced changes in structural and dynamic properties of liquid Fe close to the melting line. An ab initio study. J Phys Condens Matter (2016) 28:075101. doi: 10.1088/0953-8984/28/7/075101
7. Hosokawa S, Inui M, Kajihara Y, Matsuda K, Ichitsubo T, Pilgrim W-C, et al. Transverse acoustic excitations in liquid Ga. Phys Rev Lett. (2009) 102:105502. doi: 10.1103/PhysRevLett.102.105502
8. Hosokawa S, Munejiri S, Inui M, Kajihara Y, Pilgrim W-C, Ohmasa Y, et al. Transverse excitations in liquid Sn. J Phys Condens Matter (2013) 25:112101. doi: 10.1088/0953-8984/25/11/112101
9. del Rio BG, Rodriguez O, González LE, González DJ. First principles determination of static, dynamic and electronic properties of liquid Ti near melting. Comput Mater Sci. (2017) 139:243–51. doi: 10.1016/j.commatsci.2017.07.027
10. del Rio BG, González LE, González DJ. Ab initio study of several static and dynamic properties of bulk liquid Ni near melting. J Chem Phys. (2017) 146:034501. doi: 10.1063/1.4973803
11. del Rio BG, González LE. Longitudinal, transverse, and single-particle dynamics in liquid Zn: Ab initio study and theoretical analysis. Phys Rev B (2017) 95:224201. doi: 10.1103/PhysRevB.95.224201
12. Kresse G, Hafner J. Ab initio molecular dynamics for open-shell transition metals. Phys Rev B (1993) 48:13115. doi: 10.1103/PhysRevB.48.13115
13. Blöchl PE. Projector augmented-wave method. Phys Rev B (1994) 50:17953. doi: 10.1103/PhysRevB.50.17953
14. Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B (1999) 59:1758. doi: 10.1103/PhysRevB.59.1758
15. Ceperley DM, Alder BJ. Ground state of the electron gas by a stochastic method. Phys Rev Lett. (1980) 45:566. doi: 10.1103/PhysRevLett.45.566
16. Perdew JP, Zunger A. Self-interaction correction to density-functional approximations for many-electron systems. Phys Rev B (1981) 23:5048. doi: 10.1103/PhysRevB.23.5048
17. Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. (1996) 77, 3865–8. doi: 10.1103/PhysRevLett.77.3865%19
20. Assael MJ, Kakosimos K, Banish RM, Brillo J, Egry I, Brooks R. Reference data for the density and viscosity of liquid aluminum and liquid iron. J Phys Chem Ref Data (2006) 35:285. doi: 10.1063/1.2149380
22. Bryk T, Wax J-F. Collective dynamics in a liquid polyvalent metal: Liquid thallium at the melting point. J Chem Phys. (2010) 132:074504. doi: 10.1063/1.3319500
23. Jakse N, Pasturel A. Liquid Aluminum: Atomic diffusion and viscosity from ab initio molecular dynamics. Sci Rep. (2013) 3:3135. doi: 10.1038/srep03135
24. Jakse N. Pasturel A. Excess entropy scaling law for diffusivity in liquid metals. Sci Rep. (2016) 6:20689. doi: 10.1038/srep20689
25. Pasturel A, Jakse N. Validity of the stokes Einstein relation in liquids: simple rules from the excess entropy. J Phys Condens Matter (2016) 28:485101. doi: 10.1088/0953-8984/28/48/485101
26. Pasturel A, Jakse N. Transport properties and Stokes-Einstein relation in Al-rich liquid alloys. J Chem Phys (2016) 144:244502. doi: 10.1063/1.4954322
27. Lin S-T, Blanco M, Goddard III WA. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: validation for the phase diagram of Lennard-Jones fluids. J Chem Phys. (2003) 119:11792. doi: 10.1063/1.1624057
28. Bryk T, Mryglod I, Scopigno T, Ruocco G, Gorelli F, Santoro M. Collective excitations in supercritical fluids: analytical and molecular dynamics study of “positive” and “negative” dispersion. J Chem Phys. (2010) 133:024502. doi: 10.1063/1.3442412
29. Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys Rev B (1993) 47:558. doi: 10.1103/PhysRevB.47.558
30. Kresse G, Hafner J. Ab initio molecular-dynamics simulation of the liquid-metalamorphous-semiconductor transition in germanium. Phys Rev B (1994) 49:14251. doi: 10.1103/PhysRevB.49.14251
31. Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mat Sci. (1996) 6:15–50. doi: 10.1016/0927-0256(96)00008-0
Keywords: liquid metals, collective excitations, transverse dynamics, ab initio molecular dynamics, current spectral functions
Citation: Bryk T, Demchuk T, Jakse N and Wax J-F (2018) A Search for Two Types of Transverse Excitations in Liquid Polyvalent Metals at Ambient Pressure: An Ab Initio Molecular Dynamics Study of Collective Excitations in Liquid Al, Tl, and Ni. Front. Phys. 6:6. doi: 10.3389/fphy.2018.00006
Received: 27 October 2017; Accepted: 22 January 2018;
Published: 07 February 2018.
Edited by:
Gang Zhang, Institute of High Performance Computing (A*STAR), SingaporeReviewed by:
Arrigo Calzolari, Consiglio Nazionale Delle Ricerche (CNR), ItalyFlorent Calvayrac, University of Maine, France
Copyright © 2018 Bryk, Demchuk, Jakse and Wax. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taras Bryk, YnJ5a0BpY21wLmx2aXYudWE=