
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Photonics , 24 November 2022
Sec. Photovoltaic Materials and Devices
Volume 3 - 2022 | https://doi.org/10.3389/fphot.2022.1050189
This article is part of the Research Topic Bringing Together the Worlds of Photosynthesis and Photovoltaics: Mechanisms, Methods, and Applications View all 9 articles
 Jeroen Hustings1
Jeroen Hustings1 Robin Bonné2
Robin Bonné2 Rob Cornelissen1Filippo Morini1
Rob Cornelissen1Filippo Morini1 Roland Valcke3
Roland Valcke3 Koen Vandewal4
Koen Vandewal4 Jean V. Manca1*
Jean V. Manca1*Light-induced charge-transfer mechanisms are at the heart of both photosynthesis and photovoltaics. The underlying photophysical mechanisms occurring within photosynthesis and organic photovoltaics in particular show striking similarities. However, they are studied by distinct research communities, often using different terminology. This contribution aims to provide an introductory review and comparison of the light-induced charge-transfer mechanisms occurring in natural photosynthesis and synthetic organic photovoltaics, with a particular focus on the role of so-called charge-transfer complexes characterized by an excited state in which there is charge-transfer from an electron-donating to an electron-accepting molecular entity. From light absorption to fully separated charges, it is important to understand how a charge-transfer complex is excited, forming a charge-transfer state, which can decay to the ground state or provide free charge carries in the case of photovoltaics, or radicals for photochemistry in photosynthetic complexes. Our motivation originates from an ambiguity in the interpretation of charge-transfer states. This review attempts to standardize terminology between both research fields with the general aim of initiating a cross-fertilization between the insights and methodologies of these two worlds regarding the role of charge-transfer complexes, inspiring the cross-disciplinary development of next-generation solar cells. Likewise, we hope to encourage photosynthesis researchers to collaborate with the photovoltaics field, thereby gaining further knowledge of the charge-transfer process in natural light-harvesting systems.
One of the most critical global challenges nowadays is the development of low-cost, durable, and environmentally-compatible energy sources. Later this century, Earth’s population will reach 10.9 billion, according to estimates by the UN (UnitedNations, 2019). It is, therefore, more important than ever to look for a globally accessible energy source. Due to global challenges such as climate change, increased energy demand, and human overpopulation, there is growing pressure to improve both natural and artificial light-harvesting systems further. To answer global demand, we need crops that are more resilient to droughts and sudden changes in temperature and yield more for the same area (Lopes et al., 2015). Simultaneously, to decrease the emission of greenhouse gasses, renewable energy technology needs to be low-cost, durable, and highly efficient. Each hour, the Sun delivers enough light energy to cover the worldwide electricity need for an entire year (Armaroli and Balzani, 2007; IEA, 2021). Solar energy is, therefore, one of the most encouraging candidates as a renewable energy source for future society.
Nature has been using sunlight for energy production for a long time through the process of photosynthesis (PS); the first cyanobacteria able to split water and fixate carbon dioxide appeared around 2.4 billion years ago (Rasmussen et al., 2008; Gorka et al., 2021). The photosynthetic system can convert water and carbon dioxide into sugars and oxygen, driven by the absorption of sunlight. It uses pigments that absorb efficiently and in a broad range of the visible spectrum. However, not all energy from the Sun is converted to biomass. The theoretical maximum conversion efficiency of solar radiation into biomass lies around 6% (Zhu et al., 2008) (for plant species based on C4 photosynthesis), and average values in natural conditions typically reach only 0.1% (Chapin et al., 2002). The exact nature behind the loss mechanisms in energy conversion and energy transfer will be elaborated upon later when we clarify how a considerable portion of energy loss—often more than half of the incoming photon energy—occurs before charge separation at the reaction center of the photosynthetic apparatus (Golbeck and van der Est, 2014; Gorka et al., 2021). PS is therefore not a highly efficient process regarding energy conversion, but it is a robust one and serves the purpose of fueling the organism’s growth and survival.
Artificial light-energy conversion systems such as photovoltaics convert sunlight into electrical energy, which can be used to charge up a battery or power a device. Similar to PS, these systems cannot convert all the energy of incoming sunlight into usable electrical power. Single junction solar cells without complex light concentration systems have a theoretical maximum energy conversion efficiency around 33%, as described by Shockley and Queisser (Shockley and Queisser, 1961). More recent models that include recombination losses and non-perfect absorbance recently placed the upper limit at 31% for single-crystal silicon solar cells (Bhattacharya and John, 2019). Current advances have allowed conventional Si-based technology to record efficiencies of 26.7% (Yoshikawa et al., 2017; Green et al., 2022). Multijunction solar cells reach even higher efficiencies [38,8% (Green et al., 2022)] but at the expense of complexity and cost. As for organic photovoltaics (OPV), the highest reported efficiency has been increasing steadily in the past years, with the current certified record at 18.2% (Zhang et al., 2021; Green et al., 2022). The following section provides a detailed description of the working principle of these devices.
Photovoltaics and photosynthesis have many similarities but are studied by their respective research communities with little or no interaction, often using different terminology. Bridging these two worlds by a cross-fertilization of insights and methodologies would be beneficial for both fields. This contribution aims to bridge these two domains by providing a brief description of the underlying light-induced charge-transfer (CT) mechanisms in PS and OPV as an elemental introduction to OPV for PS researchers and vice versa. We, therefore, focus on the basic working principles of light absorption to charge separation in both OPV systems and PS organisms. CT complexes play an essential role in both systems, as their excited state consists of an electron-hole pair on adjacent molecules, the fate of which can be either to decay to the ground-state or to further dissociate (McNaught et al., 1997). The first part of this review focuses on OPV. Together with dye-sensitized solar cells (DSSC), it is arguably the PV technology closest related to PS. Organic solar cells outperform DSSCs, but their efficiency is still lower than that of Si solar cells. Their development and device architecture will be discussed, followed by a detailed explanation of the intricacies of charge generation, with particular emphasis on the definition of the CT state and its occurrence in OPV technologies. The second part addresses PS, introducing the capability of some biological systems to oxidize water and fixate carbon dioxide to synthesize carbohydrates. We will mainly discuss the multiple steps between light harvesting and charge separation to eventually clarify the role of CT states in PS. We conclude with an overview of the universal character of CT states and discuss the similarities and differences between OPV and PS with the goal of transferring knowledge between both fields.
Conventional solar cells, based on inorganic materials such as silicon (Si) or gallium-arsenide (GaAs), have high efficiencies, degrade little over time are resistant against elevated temperature and UV-radiation (Saga, 2010). Furthermore, thanks to continued development, the production cost of these devices has dropped dramatically. The levelized cost of efficiency (LCOE) of commercial PV has fallen tenfold over the past decade to 0.048 USD/kWh on a global scale (IRENA, 2022). As a downside, these devices need to be rather thick in order to absorb most of the incident light, owing to the low absorption coefficient of inorganic semiconductors devices, as is especially the case for Si with a typical absorption coefficient of 104 cm−1 (Green, 2008) in the visible spectrum, requiring device thickness in the order of 100 µm (Tiedje et al., 1984). The photoactive components in OPV are of molecular or polymeric nature and have an average absorption coefficient around 105 cm−1 (Lin and Kiang, 2014; Nyman et al., 2021), allowing for device thickness of about 100 nm. Further limitations of inorganic PV modules are their dull appearance and fragile nature, making them difficult to integrate into for instance buildings and unsuited for curved surfaces.
Organic solar cells (OSC) were developed in the 80’s when layers of porphyrin, which is related to the natural occurring pigment chlorophyll, were sandwiched between two electrodes. The resulting organic molecule-based device had a poor power conversion efficiency (<0.1%) (Chamberlain, 1983), but the photoactive layer had a high absorption coefficient which would make thin, semi-transparent, and even flexible devices possible. A unique property of organic semiconductors is their conjugated π-system, consisting of overlapping p-orbitals which allow for the delocalization of the electronic wavefunction along the backbone of the molecule. This is especially the case for semiconducting polymers, often displaying a band-like transport signature for the intrachain mobility. However, bulk transport is limited by interchain hopping (Fratini et al., 2020). The possibility to prepare single-junctions and tandem OSCs by continuous roll-to-roll processing using solution deposition or low pressure organic vapor phase deposition opens perspectives to low cost, large area production (Qu and Forrest, 2018; Ng et al., 2022). OPV became popular after the introduction of the bulk heterojunction configuration, allowing higher power-conversion efficiencies as described in the following section, reaching current lab record efficiencies of 18.2% (Zhang et al., 2021; Green et al., 2022) for single-junction OSCs.
In the most basic definition, an exciton is formed by excitation of an electron to the lowest unoccupied molecular orbital (LUMO) of an organic semiconductor, where it remains electrostatically bound to the remaining positive charge, or hole, residing on the highest occupied molecular orbital (HOMO). As opposed to inorganic materials, organic semiconductors typically possess a low relative permittivity (ϵr < 6 instead of ϵr > 10) (Liu et al., 2018), meaning that the attraction between opposite charges, as described by Coulomb’s law, is shielded to a lesser extend by polarization of the medium compared to inorganic materials. Consequently, the attraction between electron and hole remains strong, creating a collective state that defines an exciton. For inorganic semiconductors, the crystal structure allows the position of the wave function of the excited electron to delocalize quickly over a large area, assisting in the separation of the charges. Organic molecules tend to form non-covalent bonds with adjacent organic molecules preventing the formation of extended crystalline regions. As a result, the electron remains highly localized. However, it can diffuse throughout the material as a bound electron-hole pair, i.e., the exciton (Chen, 2019).
The rectifying behavior of the first, single component organic concept device—i.e., consisting of a thin film (a few µm) of copper phthalocyanine sandwiched between a metallic electrode and a transparent conductive oxide—was discovered by Delacote et al. (1964). However, it proved too much to ask for effective conduction of both electrons and holes by the same component (Spanggaard and Krebs, 2004). The alternative approach is a bilayer organic solar cell—i.e., devices where electron-donating and electron-accepting materials are cast in sequence as thin, stacked layers with a flat boundary (see Figure 1). The organic semiconductors have dissimilar bandgaps, creating a so-called heterojunction at the interface. Here, the difference in energy levels provides a driving force for exciton dissociation, resulting in an electron-hole pair at the materials interface. The donor (D) material has higher HOMO and LUMO levels as compared to the acceptor (A) material, resulting in a higher electron affinity of the acceptor layer (see Figure 1). At the D-A interface it is energetically favorable for the exciton to transfer its electron to the acceptor molecule while the hole remains behind on the donor, resulting in spontaneous charge separation. Tang et al. devised the first bilayer heterojunction in 1986 using copper phthalocyanine as D and a perylene tetracarboxylic derivative as A (Tang, 1986). After the introduction of the polymer/C60 junction by Sariciftci et al., in 1993, fullerene derivates, e.g., phenyl-C61-butyric acid methyl ester (PC60BM), were preferentially used as A in OPV because of their appropriate energy levels and conduction properties (Sariciftci et al., 1993; Facchetti, 2013).
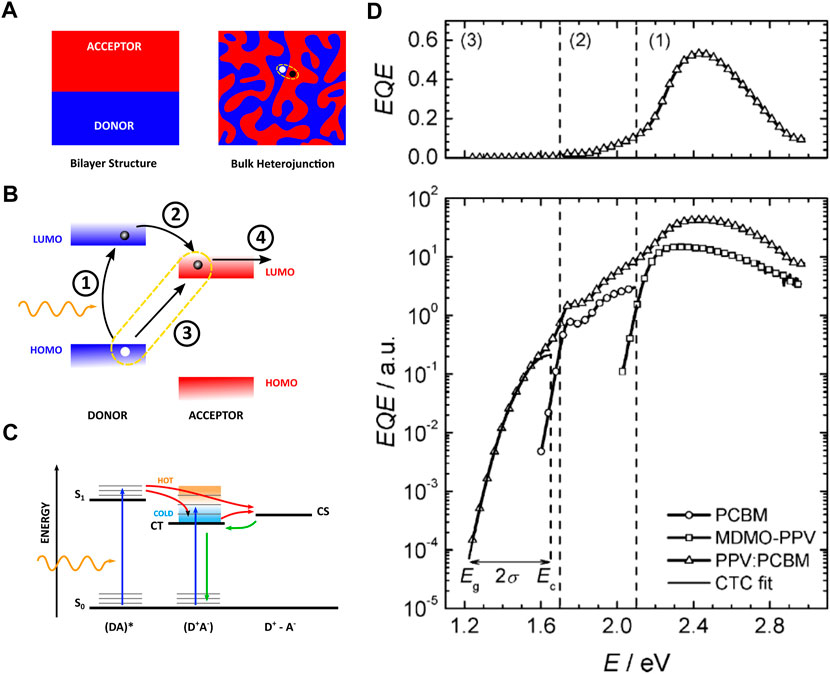
FIGURE 1. (A) (left) The architecture of a bilayer solar cell device with A (red) and D (blue) material cast in sequence as thin layers with a flat boundary. These devices suffer from the large diffusion lengths required for excitons before they reach the DA-interface. (right) The architecture of a BHJ solar cell device where acceptor and donor material are dispersed throughout the photoactive layer, resulting in a larger DA-interface and reducing the path length for excitons to reach the interface. The location of the CT complex is encircled. (B) Molecular orbital diagram of D and A near the interface. The arrows indicate the pathways for an electron from light absorption 1), excited state charge transfer 2) or ground state charge transfer 3), followed by charge separation 4). (C) Electronic state diagram showing the excitonic state, CT state, and CS state. (D) A typical spectrum of external quantum efficiency (conversion of photons to electrons) on a linear and logarithmic scale, indicating the influence of D (MDMO-PV) and A (PCBM) absorption. Direct CT state excitation is observed as an absorption band below excitation from D and (A) (Adapted from (Vandewal et al., 2008)).
Excitons created in the donor domain diffuse towards the donor-acceptor interface, the rate of diffusion can be predicted by the Förster mechanism (Tamai et al., 2015). Unfortunately, excitons only have a limited lifetime before they decay. They exist in the order of 1 ns, allowing the bound electron-hole pair to move only up to a distance of about 10 nm before recombination of the charges (Lunt et al., 2009). In order to absorb a good portion of the incoming light, the photoactive layer needs to have a thickness of around 100 nm (Bernède, 2008; Rafique et al., 2018). This is already much thinner compared to the layer thickness of Si-based devices (typically a few hundreds of µm), but a consequence for the bilayer device is that only a very small portion of the excitons will arrive at the D-A interface where it can be efficiently separated. To overcome the problems posed with a bilayer architecture, the D and A materials can be intimately mixed in the photoactive layer, creating localized regions of D and A material. This process extends the surface area of the interfaces and disperses them throughout the active layer, which ensures close proximity to any created exciton, yet maintaining the thickness needed for sufficient light absorption. This kind of device is termed a bulk-heterojunction (BHJ) solar cell, first conceived by Hiramoto et al. (1991) with the introduction of fullerene derivates occuring in 1994 (Lee et al., 1994). The design of such a device is displayed in Figure 1. One complication of the BHJ architecture is the need for electron- and hole-blocking layers at the contacts since both donor and acceptor material cross the entire photoactive layer. The electron-blocking layer maintains unfavorable energy levels for electrons to cross, preventing them from reaching the anode. As for the hole-blocking layer, it effectively inhibits the transport of holes at the cathode side (Bernède, 2008).
In PS organisms, the photosystems which include the reaction center, light-harvesting complexes and surrounding proteins, transfer the excitation energy absorbed by the antenna systems into an electron transport chain to produce reducing power (NADPH) and to store energy (ATP). These compounds drive biosynthesis of carbohydrates and proteins, cell division, and other energy-consuming processes all over the cell or organism.
We make the distinction between the PS process in purple bacteria on one hand, and plants, algae, and cyanobacteria on the other. Besides structural differences, purple bacteria perform anaerobic photosynthesis and use bacteriochlorophyll as main chromophore, while plants, algae and cyanobacteria have evolved to synthesize oxygen and utilize chlorophyll. In this review, we restrict ourselves to light absorption, excitation energy transfer (EET) and charge separation (CS). The focus will be on light-harvesting (LH) complexes and (type II) reaction centers in purple bacteria (PbRC) — more specifically Rhodobacter sphaeroides—as this model system is among the most studied. Algae and plants contain both the type I and type II reaction centers (RC), making them more complex, and so generally bacteria are chosen as a model system for study. However, some conclusions of this work will apply to both types of photosynthesis, especially concerning EET and CS.
Light harvesting in purple bacteria occurs in the cytoplasmic membrane. Here, specific complexes which overspan the membrane are responsible for the formation of ATP and NADPH, driven by respectively a proton gradient over the membrane and the supply of energetic electrons. Light-harvesting is initiated by absorption of sunlight by PS chromophores like bacteriochlorophylls (BChl) or carotenoids (Car) which are found in a complex of vast protein-pigment systems embedded in the membrane, known as light-harvesting (LH) complexes. These complexes are abundantly present in PS organisms, where α- and β-polypeptides function as a matrix to non-covalently bind the chromophores in ring-shaped structures. Having the molecules arranged in this way maximizes the chance of a match between the polarization direction of the incoming light and the molecular dipole moment of a chromophore, increasing the overall optical cross-section. The LH1 complex encloses the RC. Smaller LH2 complexes are arranged around the LH1-RC complex and function to gather light energy and transfer it to LH1-RC, where eventually CS takes place (Croce et al., 2018).
Although the local dielectric constant in the RCs and LH complexes varies greatly during electron transfer, its reported value is low (ϵr = 1–6) (Steffen et al., 1994; Guo et al., 2013; Aksu et al., 2019) — similar to the environment in OPV devices. The resulting Coulomb force between excited electron and hole is thus comparable to the attraction in OPV. Therefore, upon excitation of a chromophore in either RCs or LH complexes, bound electron-hole pairs or excitons are formed.
The environment in which the chromophores reside affects to a large extend the optical properties of the molecules. Excited energy is lowered by excitonically coupled assemblies of chromophores, like dimers or crystals. Being ring-shaped, the LH complex is considered to be a 1D array crystal and it forms excitonic bands (Reimers et al., 2016). The excitation energy levels of the (B)Chls are redshifted when nearing the RC, which will create a funnel-like system to guide excitonic energy to the RC, where subsequent CS will take place. The second function of the LH complex is regulation of the energy flow, as excessive light can damage the cell (e.g., reactive oxygen species can be sensitized which can break conjugated systems of chromophores). The LH complex can act as a light absorber but also as a light quencher, respectively transferring excitation energy to the RC or protecting the cell by non-radiative decay of the excitation energy (Drop et al., 2014; Magdaong and Blankenship, 2018; Cignoni et al., 2021).
With the use of two-dimensional electronic spectroscopy (2DES) in the early 2000s, indications for quantum-coherence was found in different PS systems (Brédas et al., 2016; Jumper et al., 2018). Long-lived oscillations in the Fenna-Mathews-Olson (FMO) complex of green sulfur bacteria was believed to originate from electronic coherence (Engel et al., 2007). Long-lived coherent mixing of electronic states leads to the delocalization of exciton energy over several chromophores, i.e., a superposition of excitonic states (Reimers et al., 2016). This mechanism has been used to explain the efficient energy transfer along the antenna system towards the RC. Excitons would essentially probe a large phase space for the most efficient downward path. This mechanism gets described by wave-like transport as opposed to the classical hopping transport, resulting in fast and efficient EET. (Romero et al., 2017; Jumper et al., 2018). However, more recent studies have refuted this claim by stating that the dephasing timescale of coherence exceeds the expected decoherence time of superpositon excitonic states (Jumper et al., 2018). The oscillations observed in the FMO complex excist on a timescale of ps, while theoretical models predict electronic decoherence over a range of only tens of fs (Duan et al., 2017; Jumper et al., 2018).
The controversy surrounding quantum-coherence in PS increased research efforts for alternative explanations. The delocalization of excitons over neighboring sites is now believed to be assisted by vibronic coupling (Duan et al., 2017), enabling fast EET (Higgins et al., 2021). This theory is an expansion of classical Förster resonance electron transfer (FRET), where the transfer rate is proportional to the Franck-Condon factor which incorporates the overlap of vibrational wavefunctions between donor and acceptor. In vibronic modified Förster theory, intermediate electronic coupling is included that redistributes the vibrational wavefunctions and modifies the Franck-Condon factor and thus the EET (Jumper et al., 2018). The coherent oscillations observed from 2DES in the FMO complex and other PS complexes is nowadays accepted to originate from this effect. Contemporary research has employed FRET to understand the coupling between bacteriorhodopsin and TiO2 in a biophotovoltaic device architecture (Barbiellini et al., 2018; Das et al., 2019).
The question remains whether biology intentionally exploits quantum mechanical principles to optimize photosynthesis. Higgins et al. has suggested this to be the case to steer the energy pathway of excitons in the FMO complex (Higgins et al., 2021).
The RC of both oxygenic PS organisms (plants, most algae, and cyanobacteria) and anoxygenic PS organisms (various types of bacteria) consists of two branches of molecules, which are nearly symmetrical.
In PbRCs, two closely placed BChl molecules form a dimer called the special pair P. From here, two branches, A and B, of cofactors spread out (see Figure 2). Both branches have a bacteriochlorophyll B and bacteriopheophytin H molecule (a bacteriochlorophyll lacking the Mg2+ ion), with a subscript corresponding to the specific branch (BA, BB, HA, HB). On the RC side lies one extra carotenoid molecule (spheroidene). Only the A-branch is active in CS. The function of the B-branch is still largely unknown, but it plays a role in photoprotection against the formation of triplet states which can sensitize harmful reactive oxygen species (ROS) (Croce et al., 2018).
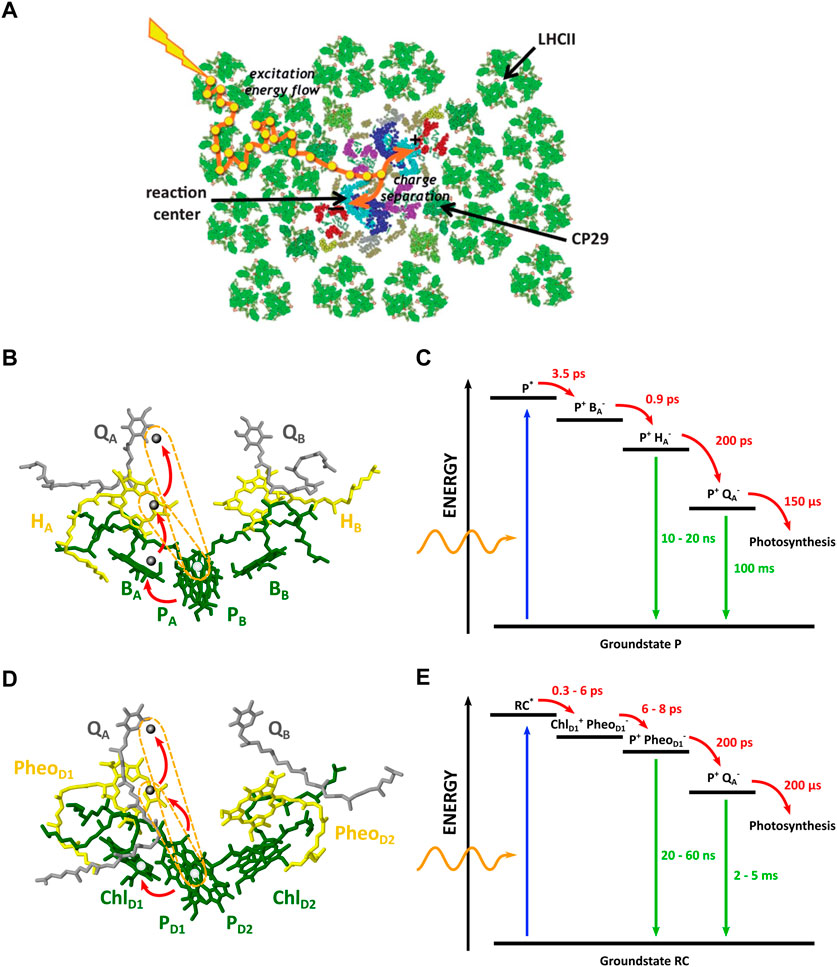
FIGURE 2. (A) Top view of a thylakoid membrane showing the architecture of the PSII supercomplex where antenna complexes surround two reaction centers. Excitation energy migrates from the antenna to the reaction center (Adapted from (Fleming et al., 2012)). (B) Structure of the PbRC with the main chromophores. Exciton injection from LH1 arrives at the special pair P. Subsequent CT occurs over the A branch, indicated by the red arrows. (C) Electronic state diagram in the PbRC, displaying the initial excitonic state and the subsequent free-energy levels of the electronic transfer chain, together with their time constants. The arrows indicate possible transitions. (D) Structure of the PSII RC with the main chromophores. Exciton injection from LH1 arrives at the special pair P. Subsequent CT occurs over the D1 branch. (E) Electronic state diagram of the PSII RC, displaying the initial excitonic state and the subsequent electronic states. (Adapted from (Allen et al., 1987; Umena et al., 2011; Croce et al., 2018)).
At excitation of the RC—most times by excited energy transfer from the antenna system—P will be populated by an exciton. The EET along the antenna complexes towards the RC is exceptionally efficient, reaching close to 100%. This is primarily due to the already mentioned fast decay in energy by tuning of the chromophore excitation levels by the protein environment, acting as an energy sink (Reimers et al., 2016; Croce et al., 2018).
The bacterial special pair absorbs light between 870 nm and 960 nm, generating the excited state P*. CS occurs over a timespan of 3.5 ps as the electron is transferred from P* to HA over a distance of about 9 Å. This is followed by electron transfer to quinone A (QA) in 200 ps over a similar distance of 9 Å. Ultimately, the excited electron is transferred to the exchangeable quinone B (QB), which can be rather easily detached from the protein by reducing it twice to plastoquinol QH2, which then further diffuses to the protein cytochrome bc1-complex. QH2 is now oxidized, releasing its protons and hereby initiating the creation of a proton gradient over the thylakoid membrane, which drives the formation of ATP by the membrane-crossing protein ATP synthase. Further steps will not be discussed here as we only focus on EET and CS (Romero et al., 2017). For an extensive review on CS in the PbRC and the RC of PSII, the reader is referred to (Croce et al., 2018).
In the context of organic materials, an exciton forms by exciting an electron from the HOMO to the LUMO. The resulting electron-hole pair resides on the same molecule and remains electrostatically bound. Nevertheless, diffusion of the exciton throughout the material remains often possible. The exciton can quickly decay back to the ground state. However, in the vicinity of another molecule with a greater electron affinity—referred to as the electron acceptor (A) — charge transfer might occur, in which case the electron jumps to A while the hole stays behind on the so-called electron-donor (D). This transition is energetically favorable by the difference in HOMO and LUMO levels between D and A (see Figure 1) (the terms donor and acceptor are used here as generic terminology and do not relate exclusively to the components of the photoactive layer in an OPV). The configuration where the charges are partly localized on different molecules is called a charge-transfer (CT) state (Pope, 1966; Pope et al., 1999). Population of the CT state can happen either after excitation of the donor component or after excitation of the acceptor component: D*A → (D+A−)* or DA* → (D+A−)*. A DA pair that can accommodate a CT state is called a CT complex over which the CT state is energetically spread. There is no necessity for a CT complex to consist of two different molecules. It can also comprise a single molecule with isolated D and A moieties. The CT state is an intermediate state between an exciton—where the charges remain closely bound—and complete charge separation (CS). Contrary to a CS state, the CT state represents the possibility of recombination, i.e., the electron falling back into the hole site. Recombination of separate charges, therefore, always has to pass through the CT state.
Taking these properties into account, we arrive at a simple definition that can be used in all systems, regardless of specific details. The following formal definitions apply:
• Exciton: excitation where the charges reside on the same molecule, and decay to the ground state is possible.
• Charge-transfer state: excitation where the charges reside on different molecular entities, and decay to the ground state remains possible.
• Charge-separated state: the charges reside on different molecular entities, and decay to the ground state is not possible without forming a CT state first.
In OPV, the creation of a CT state happens as follows: after formation of an exciton in the donor layer of the device, followed by diffusion to the donor-acceptor interface, it is energetically favorable for the exciton to transfer its electron to the acceptor molecule (LUMOD to LUMOA) while the hole remains localized on the donor, resulting in the creation of the CT state through excited state charge transfer (see Figure 1). The CT state is also observed to form by direct excitation of the CT complex from ground state charge transfer, promoting an electron from HOMOD to LUMOA (Goris et al., 2005; Vandewal et al., 2006). The energy associated to the CT state is expected to be less than required to produce a separate electron and hole: ECT < ECS (see Figure 1) (Pope, 1966).
An early overview stating the importance of CT states in polymer:fullerene solar cells was given by Benson-Smith et al. (2007), where the authors measured a small amount of absorption at excitation energies below the optical gap of both D and A material (see Figure 1). This sub-gap absorption indicated the presence of an extra electronic transition besides that of the pristine D or A HOMO-LUMO transition. In this study it was shown that the measurable presence of a CT state in the absorption spectrum had a large influence on the overall efficiency of the device. When a CT state is not detectable, its energy is too close to that of the D singlet energy level, which means that the energy difference
The performance of a solar cell in general can be described with the power conversion efficiency (PCE), which is the product of the current flowing at short-circuit (ISC), the voltage during open-circuit (VOC) and the Fill Factor (FF)—a measure for internal losses—divided by the power of the incident light (Pi). To generate current, an exciton (represented as (DA)*, where either D or A is excited) can dissociate into a CT state (D+A−), thereby populating the CT state, and this complex must further be separated into free charges (D+—A−) (see Figure 1). An observation of the CT state, as seen in Figure 1, is essential to the functionality of an OSC. In exceptional cases, sufficiently low ΔE—being material dependent—generates increased hybridization to so-called mixed excitonic-CT states. This phenomenon yields lower non-radiative recombination, which is beneficial in combating voltage losses, but also enables an overall increase in recombination. Hence, it is an interesting subject concerning the optimization of device performance through energy level tuning (Eisner et al., 2019).
For completeness, we briefly mention the observation of CT states in related PV technologies. Besides the classical polymer:fullerene OSC, CT states have also been detected in polymer:polymer OSCs (Clarke and Durrant, 2010). Even in DSSCs, CT behavior has been observed between the dye and the TiO2 anode. The presence of a dye-to-TiO2 CT band was detected at the low energy range in the incident-photon-to-current efficiency (IPCE) spectrum (Tae et al., 2005). Further reading on CT dynamics in DSSCs can be found in reference (Brauer et al., 2015).
To discuss the possible occurrence of CT states in the photosynthetic RC, we first return to the model system of purple bacteria. Considering the PbRC in Figure 2, CS occurs when the electron is localized on QB, and the hole remains behind on the special pair, P. To achieve this, several intermediate states form where the negative charge displaces over the A branch of the PbRC. The comlete CT pathway is summarized in Equation 1. The primary electron acceptor is HA, forming the state
It is worth comparing the dynamics of CS in PbRCs to the RC in photosystem II (PSII), found in plants and cyanobacteria. The structure of the PSII RC is displayed in Figure 2. PSII utilizes other chromophores: chlorophyll and pheophytin replace bacteriochlorophyll and bacteriopheophytin in purple bacteria. The general arrangement of cofactors is similar to PbRCs, however the designation of the cofactors differs, being a matter of convention. After collective excitation of the RC from the antenna complexes, the primary donor is briefly shifted to ChlD1 (Croce et al., 2018; Yoneda et al., 2022), the accessory chlorophyll on the D1 branch—an effect not observed in PbRCs. The negative charge switches back to P in the next stages of CT. At ambient temperatures, it is believed that multiple pathways exist with uncertainty surrounding ChlD1 or P as primary donor. Ambiguity over the identity of CT complexes also exists due to the observation of mixed excitonic-CT states in this stage of PSII CT. They are believed to be responsible for far-red absorption (Novoderezhkin et al., 2007; Wahadoszamen et al., 2014; Yoneda et al., 2022). The subsequent electron transfer to the CS state
All of the above mentioned intermediate states are called by a wide variety of names, including (transient) radical-pair (Novoderezhkin et al., 2005; Parson and Warshel, 2009), charge-separated state (Karlsson et al., 2010; Duan et al., 2017; Neumann et al., 2019; Sipka et al., 2021), yet also charge-transfer states (Novoderezhkin et al., 2007). To answer whether any of these intermediate states are truly CT states, we propose a nomenclature based on the definition of a CT state from earlier—differentiating excitons from CT states and CS states. To reiterate, a CT state is partly spread over two different molecular entities with the electron residing on the acceptor molecule and the hole residing on the donor molecule. In addition, a CT state remains coupled to the ground state, allowing for decay to occur, as opposed to a fully CS state. Taking this definition into account, the intermediate states between P* and

TABLE 1. Charge recombination rates and D-A center-to-center distances in the RCS of PSII (left) and purple bacteria (right).
From these values, all the above mentioned states, from
Further CS states beyond
To compare recombination and dissociation rates of CT states in PS to those in OPV, we briefly discuss the example by Hackl et al. (2022). They investigated a PCPDTBT:PCBM BHJ device and found a recombination of free charges via the CT state occurring on a timescale of krec = 109s−1. Dissociation of the CT state proceeded at a rate of kdis = 1012s−1. These values are typical for other OPV systems. The dynamics of CT dissociation and recombination in OPV occurs on much faster timescales than is the case for PS, which is important for efficient CS. The extend over which the charges are spread, i.e., the dimensions of a CT complex in OPV is related to the D-A distance. For a typical polymer:fullerene blends this distance is ∼7.3 Å (Wang et al., 2018). The smaller spacial separation compared to PS likely relates to the faster recombination dynamics observed in OPV.
Employing specialized optical techniques can assist in the designation of CT states in both fields of OPV and PS. One such technique is 2D-electronic spectroscopy (2DES) to study coherence and delocalization in electron-transfer processes (Oliver et al., 2014; Niedringhaus et al., 2018; Arsenault et al., 2021; Yoneda et al., 2022). In another way, Stark spectroscopy is used to study the CT character of excited states (Romero et al., 2017). Alongside calculations based on time-dependent density-functional theory (TDDFT) go hand-in-hand with these techniques and can improve our understanding of spectra (Sirohiwal and Pantazis, 2022).
This review presents a first comparison of the role of CT states between the worlds of PS and OPV. The formulation of definitions of the multiple stages between energy excitation and charge separation allows for concise terminology. We aim to standardize nomenclature to avoid confusion and lower the exploratory barrier between both fields.
We pointed out possible CT states in PS with their candidate CT complexes. However, caution should be taken surrounding the designation of CT complexes since these states are not always clearly localized—a phenomenon illustrated by the existence of mixed excitonic-CT states in both OPV and PS (Eisner et al., 2019; Yoneda et al., 2022). Also environmental factors can impact the recombination/dissociation rates of these states, possibly taking into question the CT character of these states. These factors include temperature, pH, OEC oxidation state, etc. (Croce et al., 2018). In addition, different organisms are affected in different ways, inhibiting us to provide a universal picture of CT in PS.
Considering the insights from the previous sections, what can we learn from comparing CT in both fields? There are similarities between PS and OPV, but they remain very different systems over the whole line. Most processes are uniquely assigned, but familiar concepts arise concerning the mechanisms behind EET and CT. The adaptations of the systems to these concepts should be studied across both worlds. Integrating these concepts from one field into the other might prove beneficial for research advancement. This review suggests different approaches toward future OPV design—to be further developed by researchers in the field. One such concept retrieved from the PS apparatus is the tuning of molecular band energies by proper orientation and interaction with their environment. The application of nanostructures and self-assembly can create systems favorable for EET and energy tunneling toward a CS center. Simulation-based research can assist in implementing FRET and quantum coherence—essential concepts in these processes. We continue by discussing two other concepts.
While the CT state in an OSC is very localized, CT in PS occurs over longer distances, populating multiple CT states and involving multiple CT complexes. This concept also exists in OPV research as so-called cascade solar cells (Schlenker et al., 2011). These devices contain an interlayer between D and A, typically a few nm thick. Other types of cascade solar cells have multiple D or A layers. The key behind this concept is a cascade of energy levels as charges cross the photoactive layer. As a result, the cascade architecture effectively reduces recombination losses since hole and electron are located further apart and have to cross a higher energy barrier during back transfer. Besides suppressing recombination, these devices can be tuned to cover a broader absorbance and facilitate efficient exciton transfer to the DA interface through an energy cascade.
With a vast antenna complex, PSII encapsulates roughly 300 pigment molecules per RC (Croce et al., 2018). One might expect this to prove disadvantageous. After all, the energy flow through the antenna system experiences severe losses before arriving at the RC. However, would it be beneficial to load the entire thylakoid membrane with RC complexes? Not necessarily, as one argument in favor of antenna systems is related to minimizing the number of recombination sites to increase the chemical potential (Ross and Calvin, 1967; Vandewal, 2016). This concept of reducing the number of recombination sites to increase VOC is also known in the world of OPV. As demonstrated by Vandewal et al., a reduction of the DA interface results in a higher electrical potential, or photovoltage (Vandewal et al., 2014).
Interdisciplinary research in biophotovoltaics can yield new insights into PS and developments in PV. One of society’s biggest challenges is expanding the use of clean, renewable energy sources in combating climate change. This research aspires to advance the field of OPV by transferring concepts from PS to OPV concerning device architecture, thereby improving efficiency and accessibility to society. At the same time, we face the issue of feeding all of humanity, challenging researchers to improve crop yields, which is directly related to PS efficiency. An understanding of the complex processes of PS from a PV perspective would in this respect certainly be of benefit.
Scientists involved in crossover research, like biophotovoltaics or bio-nano interfaces, will benefit from this review by gaining insight into the different charge-transfer processes in organics. The various approaches to charge separation by PS and OPV are highlighted and might aid researchers in approaching these systems from another perspective. Additionally, this basic introduction to the principles of PS and OPV may lower the barrier toward interdisciplinary cooperation.
We hope to extend the field of view of PV researchers to acquire further insight and inspiration, possibly aiding in developing future solar cell devices. Analogously, in the study of PS, this review might help researchers uncover the details on CS or even bring advances in the overall PS efficiency for the development of future crops. Lastly, we strongly encourage interdisciplinary cooperation between researchers in the worlds of PS and OPV.
JM conceived the design of the study. JH, RB, and RC gathered and analyzed literary material, and wrote the first draft of the manuscript. FM assisted in conceiving Figure 2. JH, RB, RC, RV, KV, and JM contributed to manuscript revision, read, and approved the submitted version.
This research was supported by the Research Foundation—Flanders (FWO) with research project G089918N (JH and JM).
The authors thank the colleagues from X-LAB from UHasselt for discussions and feedback.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aksu, H., Schubert, A., Geva, E., and Dunietz, B. D. (2019). Explaining spectral asymmetries and excitonic characters of the core pigment pairs in the bacterial reaction center using a screened range-separated hybrid functional. J. Phys. Chem. B 123, 8970–8975. doi:10.1021/acs.jpcb.9b07646
Allen, J. P., Feher, G., Yeates, T. O., Komiya, H., and Rees, D. C. (1987). Structure of the reaction center from rhodobacter sphaeroides r-26: The cofactors. Proc. Natl. Acad. Sci. U. S. A. 84, 5730–5734. doi:10.1073/pnas.84.16.5730
Armaroli, N., and Balzani, V. (2007). The future of energy supply: Challenges and opportunities. Angew. Chem. Int. Ed. 46, 52–66. doi:10.1002/anie.200602373
Arsenault, E. A., Bhattacharyya, P., Yoneda, Y., and Fleming, G. R. (2021). Two-dimensional electronic–vibrational spectroscopy: Exploring the interplay of electrons and nuclei in excited state molecular dynamics. J. Chem. Phys. 155, 020901. doi:10.1063/5.0053042
Barbiellini, B., Das, S., Renugopalakrishnan, V., and Somasundaran, P. (2018). Electromagnetic field in hybrid quantum plasmonic-photonic systems. Condens. Matter 3, 10. doi:10.3390/condmat3020010
Benson-Smith, J. J., Goris, L., Vandewal, K., Haenen, K., Manca, J. V., Vanderzande, D., et al. (2007). Formation of a ground-state charge-transfer complex in polyfluorene/[6, 6]- phenyl-C61 butyric acid methyl ester (PCBM) blend films and its role in the function of polymer/PCBM solar cells. Adv. Funct. Mat. 17, 451–457. doi:10.1002/adfm.200600484
Bernède, J. C. (2008). Organic photovoltaic cells: History, principle and techniques. J. Chil. Chem. Soc. 53. doi:10.4067/s0717-97072008000300001
Bhattacharya, S., and John, S. (2019). Beyond 30% conversion efficiency in silicon solar cells: A numerical demonstration. Sci. Rep. 9, 12482. doi:10.1038/s41598-019-48981-w
Brauer, J. C., Marchioro, A., Paraecattil, A. A., Oskouei, A. A., and Moser, J.-E. (2015). Dynamics of interfacial charge transfer states and carriers separation in dye-sensitized solar cells: A time-resolved terahertz spectroscopy study. J. Phys. Chem. C 119, 26266–26274. doi:10.1021/acs.jpcc.5b06911
Brédas, J.-L., Sargent, E. H., and Scholes, G. D. (2016). Photovoltaic concepts inspired by coherence effects in photosynthetic systems. Nat. Mat. 16, 35–44. doi:10.1038/nmat4767
Chamberlain, G. (1983). Organic solar cells: A review. Sol. Cells 8, 47–83. doi:10.1016/0379-6787(83)90039-X
Chapin, F. S., Matson, P. A., and Mooney, H. (2002). Principles of terrestrial ecosystem eology. Springer-Verlag New York. doi:10.1007/978-1-4419-9504-9
Chen, L. X. (2019). Organic solar cells: Recent progress and challenges. ACS Energy Lett. 4, 2537–2539. doi:10.1021/acsenergylett.9b02071
Cignoni, E., Lapillo, M., Cupellini, L., Acosta-Gutiérrez, S., Gervasio, F. L., and Mennucci, B. (2021). A different perspective for nonphotochemical quenching in plant antenna complexes. Nat. Commun. 12, 7152. doi:10.1038/s41467-021-27526-8
Clarke, T. M., and Durrant, J. R. (2010). Charge photogeneration in organic solar cells. Chem. Rev. 110, 6736–6767. doi:10.1021/cr900271s
Croce, R., van Grondelle, R., van Amerongen, H., and van Stokkum, I. (2018). Light harvesting in photosynthesis. Boca Raton, FL: CRC Press.
Das, S., Wu, C., Song, Z., Hou, Y., Koch, R., Somasundaran, P., et al. (2019). Bacteriorhodopsin enhances efficiency of perovskite solar cells. ACS Appl. Mat. Interfaces 11, 30728–30734. doi:10.1021/acsami.9b06372
De Causmaecker, S., Douglass, J. S., Fantuzzi, A., Nitschke, W., and Rutherford, A. W. (2019). Energetics of the exchangeable quinone, qb , in photosystem ii. Proc. Natl. Acad. Sci. U. S. A. 116, 19458–19463. doi:10.1073/pnas.1910675116
Delacote, G. M., Fillard, J. P., and Marco, F. J. (1964). Electron injection in thin films of copper phtalocyanine. Solid State Commun. 2, 373–376. doi:10.1016/0038-1098(64)90185-1
Drop, B., Webber-Birungi, M., Yadav, S. K. N., Filipowicz-Szymanska, A., Fusetti, F., Boekema, E. J., et al. (2014). Light-harvesting complex II (LHCII) and its supramolecular organization in Chlamydomonas reinhardtii. Biochimica Biophysica Acta (BBA) - Bioenergetics 1837, 63–72. doi:10.1016/j.bbabio.2013.07.012
Duan, H.-G., Prokhorenko, V. I., Cogdell, R. J., Ashraf, K., Stevens, A. L., Thorwart, M., et al. (2017). Nature does not rely on long-lived electronic quantum coherence for photosynthetic energy transfer. Proc. Natl. Acad. Sci. U. S. A. 114, 8493–8498. doi:10.1073/pnas.1702261114
Eisner, F. D., Azzouzi, M., Fei, Z., Hou, X., Anthopoulos, T. D., Dennis, T. J. S., et al. (2019). Hybridization of local exciton and charge-transfer states reduces nonradiative voltage losses in organic solar cells. J. Am. Chem. Soc. 141, 6362–6374. doi:10.1021/jacs.9b01465
Engel, G. S., Calhoun, T. R., Read, E. L., Ahn, T.-K., Mančal, T., Cheng, Y.-C., et al. (2007). Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 446, 782–786. doi:10.1038/nature05678
Facchetti, A. (2013). Polymer donor-polymer acceptor (all-polymer) solar cells. Mater. Today 16, 123–132. doi:10.1016/j.mattod.2013.04.005
Fleming, G. R., Schlau-Cohen, G. S., Amarnath, K., and Zaks, J. (2012). Design principles of photosynthetic light-harvesting. Faraday Discuss. 155, 27–41. doi:10.1039/c1fd00078k
Fratini, S., Nikolka, M., Salleo, A., Schweicher, G., and Sirringhaus, H. (2020). Charge transport in high-mobility conjugated polymers and molecular semiconductors. Nat. Mat. 19, 491–502. doi:10.1038/s41563-020-0647-2
Goris, L., Haenen, K., Nesládek, M., Wagner, P., Vanderzande, D., De Schepper, L., et al. (2005). Absorption phenomena in organic thin films for solar cell applications investigated by photothermal deflection spectroscopy. J. Mat. Sci. 40, 1413–1418. doi:10.1007/s10853-005-0576-0
Gorka, M., Baldansuren, A., Malnati, A., Gruszecki, E., Golbeck, J. H., and Lakshmi, K. V. (2021). Shedding light on primary donors in photosynthetic reaction centers. Front. Microbiol. 12, 735666. doi:10.3389/fmicb.2021.735666
Graige, M. S., Feher, G., and Okamura, M. Y. (1998). Conformational gating of the electron transfer reaction qa-.qb –> qaqb-. in bacterial reaction centers of rhodobacter sphaeroides determined by a driving force assay. Proc. Natl. Acad. Sci. U. S. A. 95, 11679–11684. doi:10.1073/pnas.95.20.11679
Green, M. A., Dunlop, E. D., Hohl-Ebinger, J., Yoshita, M., Kopidakis, N., Bothe, K., et al. (2022). Solar cell efficiency tables (version 60). Prog. Photovoltaics. 30, 687–701. doi:10.1002/pip.3595
Green, M. A. (2008). Self-consistent optical parameters of intrinsic silicon at 300k including temperature coefficients. Sol. Energy Mater. Sol. Cells 92, 1305–1310. doi:10.1016/j.solmat.2008.06.009
Guo, Z., Lin, S., and Woodbury, N. W. (2013). Utilizing the dynamic Stark shift as a probe for dielectric relaxation in photosynthetic reaction centers during charge separation. J. Phys. Chem. B 117, 11383–11390. doi:10.1021/jp4037843
Guskov, A., Kern, J., Gabdulkhakov, A., Broser, M., Zouni, A., and Saenger, W. (2009). Cyanobacterial photosystem ii at 2.9-Å resolution and the role of quinones, lipids, channels and chloride. Nat. Struct. Mol. Biol. 16, 334–342. doi:10.1038/nsmb.1559
Hackl, F., Fromherz, T., and Scharber, M. C. (2022). Radiative recombination in bulk-heterojunction solar cells. Israel J. Chem. 62, 134. doi:10.1002/ijch.202100134
Higgins, J. S., Lloyd, L. T., Sohail, S. H., Allodi, M. A., Otto, J. P., Saer, R. G., et al. (2021). Photosynthesis tunes quantum-mechanical mixing of electronic and vibrational states to steer exciton energy transfer. Proc. Natl. Acad. Sci. U. S. A. 118, e2018240118. doi:10.1073/pnas.2018240118
Hiramoto, M., Fujiwara, H., and Yokoyama, M. (1991). Three-layered organic solar cell with a photoactive interlayer of codeposited pigments. Appl. Phys. Lett. 58, 1062–1064. doi:10.1063/1.104423
Holten, D., Windsor, M. W., Parson, W. W., and Thornber, J. P. (1978). Primary photochemical processes in isolated reaction centers of rhodopseudomonas viridis. Biochimica Biophysica Acta (BBA) - Bioenergetics 501, 112–126. doi:10.1016/0005-2728(78)90100-7
IRENA (2022). Renewable power generation Costs in 2021. Abu Dhabi: Report, International Renewable Energy Agency.
Jumper, C. C., Rafiq, S., Wang, S., and Scholes, G. D. (2018). From coherent to vibronic light harvesting in photosynthesis. Curr. Opin. Chem. Biol. 47, 39–46. doi:10.1016/j.cbpa.2018.07.023
Karlsson, S., Boixel, J., Pellegrin, Y., Blart, E., Becker, H.-C., Odobel, F., et al. (2010). Accumulative charge separation inspired by photosynthesis. J. Am. Chem. Soc. 132, 17977–17979. doi:10.1021/ja104809x
Lee, K., Janssen, R. A. J., Sariciftci, N. S., and Heeger, A. J. (1994). Direct evidence of photoinduced electron transfer in conducting-polymer–c60 composites by infrared photoexcitation spectroscopy. Phys. Rev. B 49, 5781–5784. doi:10.1103/physrevb.49.5781
Lin, Y.-H., and Kiang, J.-F. (2014). Efficiency improvement of p-i-n solar cell by embedding quantum-dots. Prog. Electromagn. Res. 146, 167–180. doi:10.2528/PIER14032701
Liu, X., Xie, B., Duan, C., Wang, Z., Fan, B., Zhang, K., et al. (2018). A high dielectric constant non-fullerene acceptor for efficient bulk-heterojunction organic solar cells. J. Mat. Chem. A Mat. 6, 395–403. doi:10.1039/C7TA10136H
Lopes, M. S., El-Basyoni, I., Baenziger, P. S., Singh, S., Royo, C., Ozbek, K., et al. (2015). Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J. Exp. Bot. 66, 3477–3486. doi:10.1093/jxb/erv122
Lunt, R. R., Giebink, N. C., Belak, A. A., Benziger, J. B., and Forrest, S. R. (2009). Exciton diffusion lengths of organic semiconductor thin films measured by spectrally resolved photoluminescence quenching. J. Appl. Phys. 105, 053711. doi:10.1063/1.3079797
Magdaong, N. C. M., and Blankenship, R. E. (2018). Photoprotective, excited-state quenching mechanisms in diverse photosynthetic organisms. J. Biol. Chem. 293, 5018–5025. doi:10.1074/jbc.tm117.000233
Mattis, A., and Wraight, C. (2013). Bacterial photosynthetic reaction center from rhodobacter sphaeroides with ile m265 replaced with asn. [Dataset]. doi:10.2210/pdb4hbh/pdb
McNaught, A., Wilkinson, A., and Jenkins, A.IUPAC (1997). “Compendium of chemical terminology, 2nd ed,” in The ”gold book” (Oxford: Blackwell Scientific Publications).
Neumann, S., Kerzig, C., and Wenger, O. S. (2019). Quantitative insights into charge-separated states from one- and two-pulse laser experiments relevant for artificial photosynthesis. Chem. Sci. 10, 5624–5633. doi:10.1039/c9sc01381d
Ng, L. W. T., Lee, S. W., Chang, D. W., Hodgkiss, J. M., and Vak, D. (2022). Organic photovoltaics’ new renaissance: Advances toward roll-to-roll manufacturing of non-fullerene acceptor organic photovoltaics. Adv. Mat. Technol. 7, 2101556. doi:10.1002/admt.202101556
Niedringhaus, A., Policht, V. R., Sechrist, R., Konar, A., Laible, P. D., Bocian, D. F., et al. (2018). Primary processes in the bacterial reaction center probed by two-dimensional electronic spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 115, 3563–3568. doi:10.1073/pnas.1721927115
Novoderezhkin, V. I., Andrizhiyevskaya, E. G., Dekker, J. P., and van Grondelle, R. (2005). Pathways and timescales of primary charge separation in the photosystem II reaction center as revealed by a simultaneous fit of time-resolved fluorescence and transient absorption. Biophysical J. 89, 1464–1481. doi:10.1529/biophysj.105.060020
Novoderezhkin, V. I., Dekker, J. P., and Van Grondelle, R. (2007). Mixing of exciton and charge-transfer states in photosystem ii reaction centers: Modeling of Stark spectra with modified redfield theory. Biophysical J. 93, 1293–1311. doi:10.1529/biophysj.106.096867
Nyman, M., Sandberg, O. J., Li, W., Zeiske, S., Kerremans, R., Meredith, P., et al. (2021). Requirements for making thick junctions of organic solar cells based on nonfullerene acceptors. Sol. RRL 5, 2100018–2100028. doi:10.1002/solr.202100018
Oliver, T. A. A., Lewis, N. H. C., and Fleming, G. R. (2014). Correlating the motion of electrons and nuclei with two-dimensional electronic–vibrational spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 111, 10061–10066. doi:10.1073/pnas.1409207111
Orf, G. S., Gisriel, C., and Redding, K. E. (2018). Evolution of photosynthetic reaction centers: Insights from the structure of the heliobacterial reaction center. Photosynth. Res. 138, 11–37. doi:10.1007/s11120-018-0503-2
Parson, W. W., and Warshel, A. (2009). “Mechanism of Charge Separation in Purple Bacterial Reaction Centers,” in The Purple Phototrophic Bacteria. Editors C. N. Hunter, F. Daldal, M. C. Thurnauer, and J. T. Beatty (Dordrech: Springer Netherlands), 355–377. doi:10.1007/978-1-4020-8815-5_19
Pope, M. (1966). Charge-transfer exciton and ionic energy levels in organic crystals. Trans. N. Y. Acad. Sci. 29, 53–62. doi:10.1111/j.2164-0947.1966.tb02251.x
Pope, M., Swenberg, C., and Swenberg, P. (1999). Electronic processes in organic crystals and polymers. Oxford University Press.
Qu, B., and Forrest, S. R. (2018). Continuous roll-to-roll fabrication of organic photovoltaic cells via interconnected high-vacuum and low-pressure organic vapor phase deposition systems. Appl. Phys. Lett. 113, 053302. doi:10.1063/1.5039701
Rafique, S., Abdullah, S. M., Sulaiman, K., and Iwamoto, M. (2018). Fundamentals of bulk heterojunction organic solar cells: An overview of stability/degradation issues and strategies for improvement. Renew. Sustain. Energy Rev. 84, 43–53. doi:10.1016/j.rser.2017.12.008
Rappaport, F., Guergova-Kuras, M., Nixon, P. J., Diner, B. A., and Lavergne, J. (2002). Kinetics and pathways of charge recombination in photosystem ii. Biochemistry 41, 8518–8527. doi:10.1021/bi025725p
Rasmussen, B., Fletcher, I. R., Brocks, J. J., and Kilburn, M. R. (2008). Reassessing the first appearance of eukaryotes and cyanobacteria. Nature 455, 1101–1104. doi:10.1038/nature07381
Reimers, J. R., Biczysko, M., Bruce, D., Coker, D. F., Frankcombe, T. J., Hashimoto, H., et al. (2016). Challenges facing an understanding of the nature of low-energy excited states in photosynthesis. Biochimica Biophysica Acta - Bioenergetics 1857, 1627–1640. doi:10.1016/j.bbabio.2016.06.010
Romero, E., Novoderezhkin, V. I., and van Grondelle, R. (2017). Quantum design of photosynthesis for bio-inspired solar-energy conversion. Nature 543, 355–365. doi:10.1038/nature22012
Ross, R. T., and Calvin, M. (1967). Thermodynamics of light emission and free-energy storage in photosynthesis. Biophysical J. 7, 595–614. doi:10.1016/s0006-3495(67)86609-8
Saga, T. (2010). Advances in crystalline silicon solar cell technology for industrial mass production. NPG Asia Mat. 2, 96–102. doi:10.1038/asiamat.2010.82
Sariciftci, N. S., Braun, D., Zhang, C., Srdanov, V. I., Heeger, A. J., Stucky, G., et al. (1993). Semiconducting polymer-buckminsterfullerene heterojunctions: Diodes, photodiodes, and photovoltaic cells. Appl. Phys. Lett. 62, 585–587. doi:10.1063/1.108863
Schlenker, C. W., Barlier, V. S., Chin, S. W., Whited, M. T., McAnally, R. E., Forrest, S. R., et al. (2011). Cascade organic solar cells. Chem. Mat. 23, 4132–4140. doi:10.1021/cm200525h
Sebastian, E., and Hariharan, M. (2022). Symmetry-breaking charge separation in molecular constructs for efficient light energy conversion. ACS Energy Lett. 7, 696–711. doi:10.1021/acsenergylett.1c02412
Shockley, W., and Queisser, H. J. (1961). Detailed balance limit of efficiency of p-n junction solar cells. J. Appl. Phys. 32, 510–519. doi:10.1063/1.1736034
Sipka, G. B., Magyar, M., Mezzetti, A., Akhtar, P., Zhu, Q., Xiao, Y., et al. (2021). Light-adapted charge-separated state of photosystem ii: Structural and functional dynamics of the closed reaction center. Plant Cell 33, 1286–1302. doi:10.1093/plcell/koab008
Sirohiwal, A., and Pantazis, D. A. (2022). The electronic origin of far-red-light-driven oxygenic photosynthesis. Angew. Chem. Int. Ed. Engl. 61, e202200356. doi:10.1002/anie.202200356
Spanggaard, H., and Krebs, F. C. (2004). A brief history of the development of organic and polymeric photovoltaics. Sol. Energy Mater. Sol. Cells 83, 125–146. doi:10.1016/j.solmat.2004.02.021
Steffen, M. A., Lao, K., and Boxer, S. G. (1994). Dielectric asymmetry in the photosynthetic reaction center. Science 264, 810–816. doi:10.1126/science.264.5160.810
Suga, M., Akita, F., Hirata, K., Ueno, G., Murakami, H., Nakajima, Y., et al. (2014). Native structure of photosystem ii (dataset-1) by a femtosecond x-ray laser. [Dataset]. doi:10.1038/nature13991
Tae, E. L., Lee, S. H., Lee, J. K., Yoo, S. S., Kang, E. J., and Yoon, K. B. (2005). A strategy to increase the efficiency of the dye-sensitized tio2 solar cells operated by photoexcitation of dye-to-tio2 charge-transfer bands. J. Phys. Chem. B 109, 22513–22522. doi:10.1021/jp0537411
Tamai, Y., Ohkita, H., Benten, H., and Ito, S. (2015). Exciton diffusion in conjugated polymers: From fundamental understanding to improvement in photovoltaic conversion efficiency. J. Phys. Chem. Lett. 6, 3417–3428. doi:10.1021/acs.jpclett.5b01147
Tamura, H., Saito, K., and Ishikita, H. (2020). Acquirement of water-splitting ability and alteration of the charge-separation mechanism in photosynthetic reaction centers. Proc. Natl. Acad. Sci. U. S. A. 117, 16373–16382. doi:10.1073/pnas.2000895117
Tamura, H., Saito, K., and Ishikita, H. (2021). The origin of unidirectional charge separation in photosynthetic reaction centers: Nonadiabatic quantum dynamics of exciton and charge in pigment–protein complexes. Chem. Sci. 12, 8131–8140. doi:10.1039/d1sc01497h
Tang, C. W. (1986). Two-layer organic photovoltaic cell. Appl. Phys. Lett. 48, 183–185. doi:10.1063/1.96937
Tiedje, T., Yablonovitch, E., Cody, G. D., and Brooks, B. G. (1984). Limiting efficiency of silicon solar cells. IEEE Trans. Electron Devices 31, 711–716. doi:10.1109/t-ed.1984.21594
Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya, N. (2011). Crystal structure of oxygen-evolving photosystem ii at a resolution of 1.9Å. Nature 473, 55–60. doi:10.1038/nature09913
UnitedNations (2019). World population prospects 2019: Highlights. United Nations: Report, Population Division, Department of Economic and Social Affairs.
Vandewal, K., Goris, L., Haenen, K., Geerts, Y., and Manca, J. V. (2006). Highly sensitive spectroscopic characterization of inorganic and organic heterojunctions for solar cells. Eur. Phys. J. Appl. Phys. 36, 281–283. doi:10.1051/epjap:2006140
Vandewal, K., Gadisa, A., Oosterbaan, W. D., Bertho, S., Banishoeib, F., Van Severen, I., et al. (2008). The relation between open-circuit voltage and the onset of photocurrent generation by charge-transfer absorption in polymer: Fullerene bulk heterojunction solar cells. Adv. Funct. Mat. 18, 2064–2070. doi:10.1002/adfm.200800056
Vandewal, K. (2016). Interfacial charge transfer states in condensed phase systems. Annu. Rev. Phys. Chem. 67, 113–133. doi:10.1146/annurev-physchem-040215-112144
Vandewal, K., Widmer, J., Heumueller, T., Brabec, C. J., McGehee, M. D., Leo, K., et al. (2014). Increased open-circuit voltage of organic solar cells by reduced donor-acceptor interface area. Adv. Mat. 26, 3839–3843. doi:10.1002/adma.201400114
Vanmieghem, F., Brettel, K., Hillmann, B., Kamlowski, A., Rutherford, A. W., and Schlodder, E. (1995). Charge recombination reactions in photosystem-ii .1. yields, recombination pathways, and kinetics of the primary pair. Biochemistry 34, 4798–4813. doi:10.1021/bi00014a038
Vass, I., and Cser, K. (2009). Janus-faced charge recombinations in photosystem ii photoinhibition. Trends Plant Sci. 14, 200–205. doi:10.1016/j.tplants.2009.01.009
Vass, I. (2011). Role of charge recombination processes in photodamage and photoprotection of the photosystem ii complex. Physiol. Plant. 142, 6–16. doi:10.1111/j.1399-3054.2011.01454.x
Wahadoszamen, M., Margalit, I., Ara, A. M., Van Grondelle, R., and Noy, D. (2014). The role of charge-transfer states in energy transfer and dissipation within natural and artificial bacteriochlorophyll proteins. Nat. Commun. 5, 5287. doi:10.1038/ncomms6287
Wang, T., Chen, X., Ashokan, A., Zheng, Z., Ravva, M. K., and Brédas, J. (2018). Bulk heterojunction solar cells: Impact of minor structural modifications to the polymer backbone on the polymer–fullerene mixing and packing and on the fullerene–fullerene connecting network. Adv. Funct. Mat. 28, 1705868. doi:10.1002/adfm.201705868
Yoneda, Y., Arsenault, E. A., Yang, J., ShiunOrcutt, K., Iwai, M., and Fleming, G. R. (2022). The initial charge separation step in oxygenic photosynthesis. Nat. Commun. 13, 2275. doi:10.1038/s41467-022-29983-1
Yoshikawa, K., Kawasaki, H., Yoshida, W., Irie, T., Konishi, K., Nakano, K., et al. (2017). Silicon heterojunction solar cell with interdigitated back contacts for a photoconversion efficiency over 26%. Nat. Energy 2, 17032. doi:10.1038/nenergy.2017.32
Zhang, M., Zhu, L., Zhou, G., Hao, T., Qiu, C., Zhao, Z., et al. (2021). Single-layered organic photovoltaics with double cascading charge transport pathways: 18% efficiencies. Nat. Commun. 12, 309. doi:10.1038/s41467-020-20580-8
Keywords: charge-transfer state, photosynthesis, organic photovoltaics, reaction center, exciton
Citation: Hustings J, Bonné R, Cornelissen R, Morini F, Valcke R, Vandewal K and Manca JV (2022) Charge-transfer states in photosynthesis and organic solar cells. Front. Photonics 3:1050189. doi: 10.3389/fphot.2022.1050189
Received: 22 September 2022; Accepted: 14 November 2022;
Published: 24 November 2022.
Edited by:
Zhaoning Song, University of Toledo, United StatesReviewed by:
Dong Ryeol Whang, Hannam University, South KoreaCopyright © 2022 Hustings, Bonné, Cornelissen, Morini, Valcke, Vandewal and Manca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean V. Manca, amVhbi5tYW5jYUB1aGFzc2VsdC5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.