 Ji Hye Kim
Ji Hye Kim In Jun Yeo
In Jun Yeo Dong Ju Son2
Dong Ju Son2 Do Young Yoon
Do Young Yoon Jin Tae Hong
Jin Tae Hong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 24 March 2025
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 16 - 2025 | https://doi.org/10.3389/fphar.2025.1548832
Chitinase-3-like protein 1 (Chi3l1) is a member of the mammalian Chitinase-like protein family, and several studies reported that Chi3l1 is associated with various inflammatory diseases as well as liver diseases. Acetaminophen (APAP) is usually used for antipyretic drug, but its overdose induces acute liver injury (ALI). Several studies reported that subsequent inflammatory responses of the immune system play a critical role in the severity and outcome of APAP-induced ALI. In the present study, we investigated the role of Chi3l1 and its mechanism during APAP-induced ALI using Chi3l1 knock-out (KO) mice. We explored the function of Chi3l1 using APAP-injected KO mice and sought proteins associated with Chi3l1 through biological research data program for investigating mechanism. Liver histological analysis revealed that APAP-induced ALI was attenuated in KO mice compared to wild-type (WT) mice. We observed that APAP-induced neutrophil infiltration was decreased in the liver of KO mice compared to WT mice. To investigate this mechanism, we sought proteins potentially associated with Chi3l1 by mRNA sequencing and protein correlation analysis data. We found lipocalin-2 (Lcn2) and examined Chi3l1, Lcn2, and their relationship in the APAP-induced ALI model using recombinant proteins and antibodies. Our results suggest that Chi3l1 deficiency ameliorates APAP-induced liver injury through abrogating Lcn2-mediated neutrophil infiltration in the liver.
Acetaminophen (APAP or paracetamol) is a frequently-used, antipyretic, and analgesic drug, but its overdose can cause severe acute liver injury (ALI) (Larsen and Wendon, 2014). The initial mechanism of ALI by APAP overdose is hepatocyte necrosis caused by increased generation of mitochondrial reactive oxygen species (ROS), mitochondrial permeabilization and dysfunction (Chambers and LoGrasso, 2011). However, several studies reported that subsequent immune responses critically play a role in the severity and outcome of APAP-induced liver injury (Antoniades et al., 2012; Krenkel et al., 2014). Several cytokines and chemokines are involved in the immune response, and various immune cells such as resident hepatic macrophages, also known as Kupffer cells (KC), infiltrating monocyte-derived macrophages, dendritic cells (DC), and lymphocytes are involved in APAP-induced liver injury (Li et al., 2022). KCs are early involved in mediating liver injury by sensing damage-associated molecular patterns (DAMPs) via Toll-like receptors (TLRs). This leads to the production of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β) and interleukin 6(IL-6) (Fisher et al., 2013), which are highly relevant for enhancing inflammation and infiltrating neutrophil and monocyte into the liver. Hepatic macrophages also attract other immune cells via the secretion of chemokines, such as C-C motif chemokine ligand 2(CCL2 or MCP-1) or C-X-C motif chemokine ligand 16(CXCL16) of which the first recruits monocytes into areas of necrosis (Holt et al., 2008) that, in turn, express high levels of TNF-α, IFN-γ, IL-1β, and IL-6 8. The specific role of DCs in APAP-induced liver injury is still ambiguous, but a recent study reported that DCs may play a protective role in APAP-induced liver injury (Connolly et al., 2011). Lymphocytes contribute to the pathogenesis of APAP-induced liver injury by producing interferon-γ(IFN-γ) and depleting stored glutathione (GSH) in hepatocytes through the Fas/FasL system (Tinel et al., 2004), but their role in APAP-induced liver injury is still not fully elucidated.
Among the immune cells, neutrophils play an important role in ALI such as ischemia-reperfusion injury (Jaeschke et al., 1990), endotoxemia (Jaeschke et al., 1991), alcoholic hepatitis (Bautista, 2002), obstructive cholestasis (Gujral et al., 2003), alpha naphthylisothiocyanate (ANIT) toxicity (Hill et al., 1999), and APAP toxicity (Liu et al., 2006). In ALI, various inflammatory mediators such as TNF-α, IL-1β, and chemokines mediate neutrophil infiltration into the liver (Essani et al., 1995; Bajt et al., 2001), subsequently, neutrophils induce liver toxicity through increasing oxidative stress by neutrophil-derived hydrogen peroxide (Jaeschke et al., 1999), aggravated inflammatory response by neutrophilic protease (Niehorster et al., 1990), activated stress signaling pathway by neutrophil-derived IFN-γ (Wu et al., 2023), and direct hepatocellular injury by neutrophil-derived proteases (Ho et al., 1996). In APAP-induced liver injury, neutrophils infiltrate into the periphery of necrotic areas by CXCL1, CXCL2 (MIP-2), and CXCL8 secreted by KC (Zimmermann and Tacke, 2011), and IL-17A secreted by γδ T cells (Wang et al., 2013). Wang et al. reported that Interleukin 17A (IL-17A) antibody administration attenuated APAP-induced liver injury by decreasing neutrophil infiltration and interleukin 23(IL-23) levels, which is a cytokine secreted by macrophages, and required for stimulating IL-17A production from γδ T Cells (Wang et al., 2013). These results suggest that chemokines and cytokines secreted from macrophages mediate neutrophil infiltration and damage-induced liver inflammation.
Chi3l1(YKL-40) is a member of the mammalian chitinase-like protein family (Roslind and Johansen, 2009). Chi3l1 plays a role in cell proliferation, differentiation, inflammation, and immune responses (Roslind and Johansen, 2009). Recently, studies reported that Chi3l1 levels were related to liver diseases such as fibrosis, chronic hepatitis C, and chronic hepatitis B (Tao et al., 2014; Kumagai et al., 2016). In addition, Chi3l1 is closely associated with the production of chemokines and cytokines in macrophages for immune cell infiltration. Recent studies have shown that alveolar macrophages exposed to recombinant Chi3l1 produce higher levels of the proinflammatory mediators matrix metalloproteinase-9 (MMP-9), CCL2, CCL3, and CXCL2 (Letuve et al., 2008), and silencing Chi3l1 decreases secretion of pro-inflammatory molecules by macrophages (Libreros et al., 2012). Moreover, Breyne et al. reported that Chi3l1 is required for neutrophil influx against Escherichia coli infection in the mouse pathogenic mastitis model (Breyne et al., 2018). These results suggest that Chi3l1 may play a role in chemokine and cytokine production in macrophages necessary for neutrophil infiltration. Recently, Shan et al. reported that chi3l1 is associated with APAP-induced liver injury by promoting platelet recruitment with the liver (Shan et al., 2021). Li et al. reported that chi3l1 blocking antibody attenuated liver damage caused by APAP (Li et al., 2023). These studies suggest that Chi3l1 is important role in ALI. However, the role of Chi3l1 in the infiltration of immune cells in APAP-induced liver damage is still unknown. In this present study, we investigated the effects and possible mechanisms of Chi3l1 on APAP -induced liver injury model.
Male and Female WT and Chi3l1 knock-out (KO) mice were obtained as described in the previous study (Im et al., 2020/01). In summary, maps depicting the wild-type Chi3l1 locus, the use of the CRISPR/Cas9 system for targeting and the predicted small deletion within exon 3 as a consequence of non-homologous end joining. WT and KO mice used had matched ages (about 3 months old). To generate APAP-induced ALI, mice were intraperitoneally (i.p.) injected with 500 mg/kg APAP, the dose used in previous stidies (Ruepp et al., 2002; Muhammad-Azam et al., 2019), and then sacrificed at 6 h. To examine the function of Chi3l1 or Lipocalin-2 (Lcn2 or NGAL) in the APAP-induced liver injury model, mice were first injected intravenously with recombinant Lcn2 (rLcn2, 10 μg/mouse, R&D, Minneapolis, MN, United States), recombinant Chi3l1 (rChi3l1, 10 μg/mouse, R&D, Minneapolis, MN, United States), with anti-Lcn2 (50 μg/mouse, Abcam, Cambridge, United Kingdom) or control antibody. After 3 h, mice were injected with APAP (300 mg/kg, i. p.), and sacrificed at 6 h. All studies received approval from and conducted in accordance with the ethical guidelines by the Chungbuk National University Animal Care Committee (CBNU-523–13–01).
Human serum samples from 20 healthy adult donors and 20 adult patients with hepatotoxicity were obtained from Chungbuk National University Hospital and Kyung Sang University Hospital in the Republic of Korea. The characteristics of these patients are described in the supplementary (Supplementary Table S1). All studies involving human serum samples were conducted in compliance with the Declaration of Helsinki and were approved by the Ethics Committee of Chungbuk National University Medical Centre (IRB No.: CBNU-201910-BR-937–01).
Mouse serum samples were acquired through the administration of an overdose of pentobarbital (100 mg/kg) to anesthetize the mice, followed by blood collection via cardiac puncture. The level of aspartate transaminase (AST) and alanine transaminase (ALT) in the serum of the liver of mice were determined using an automated analyzer (7,080, Hitachi Ltd., Japan) at Laboratory Animal Research Center in Chungbuk National University.
For histological processing, the livers were fixed in a phosphate buffer containing 10% formaldehyde and decalcified with EDTA. Fixed tissues were processed to paraffin blocks by routine methods. Specimens were sectioned at 4 μm, stained with hematoxylin and eosin (H&E) and examined for histopathological evidence of liver injury. Histopathology was scored for steatosis, inflammation, necrosis as follows: 0, normal; 1, mild changes; >2, mild to moderate severity; >3, moderate severity; 5, maximum severity.
To perform assay, liver tissues were homogenized and then normalized to protein concentration. Intracellular Hydrogen peroxides assay was performed as described in the manufacturer’s protocol (Cell biolabs, San Diego, CA). We measured the malondialdehyde (MDA) level using the TBARS assay kit (Cayman Chemical). The TBARS assay was performed as described in the manufacturer’s protocol.
To obtain cells from the liver, the liver was washed with cold PBS until it became pale, following the cutting of the inferior vena cava was cut above the diaphragm. After removing the connective tissue and gallbladder, the liver was minced into small pieces. Subesequently, it was gently forced through a 200 mm-gauge stainless steel mesh using a sterile syringe plunger. Finally, the minced liver was suspended in 50 mL RPMI-1640 medium containing 10% FCS (pH 7.4) and GlutaMAX™-1, 25 mM HEPES. The cells from the livers of CHI3L1 WT and KO mice were screened for CD45-APC (BD Bioscience, Franklin Lakes, NJ, United States), CD11b-FITC (BD Bioscience, Franklin Lakes, NJ, United States), and Ly6G-PE (BD Bioscience, Franklin Lakes, NJ, United States).
Homogenized livers were lysed by protein extraction solution (PRO-PREP, iNtRONBiotechnology, Korea), which included a phosphatase inhibitor cocktail (Roche, Germany) and a protease inhibitor cocktail (Calbiochem, Germany). 30 ug of total proteins were separated by SDS-PAGE and transferred onto a PVDF membrane (Millipore, Billerica, MA). After blocking overnight with 5% skim milk, the membrane was incubated with the primary antibodies (diluted 1:1000) for 1 h at room temperature. The membranes were immunoblotted with the following primary antibodies: anti-Lcn2 (Abcam, Cambridge, MA) and anti-β-actin (Santa Cruz Biotechnology, Dallas, TX). Following the washing step with Tris-buffered saline containing 0.05% Tween-20 (TBST), the PVDF membrane was incubated with horseradish peroxidase-conjugated secondary antibodies (diluted 1:3,000) for 1 h at room temperature. Detection of Antibody binding to the blot was conducted using enhanced chemiluminescence solution (Amersham Bioscience, United Kingdom) and X-ray film (AGFA, Belgium).
All specimens were fixed in formalin and embedded in paraffin for evaluation. Subsequently, Sections of 4 μm thickness were prepared, stained with hematoxylin and eosin (H&E) and conducted by immunohistochemistry analysis using primary rabbit anti-Lcn2 (Thermo Fisher, Waltham, MA), primary rat anti-Ly6G (Abcam, Cambridge, MA), and secondary horseradish peroxidase-conjugated anti-rabbit or anti-rat antibodies.
The primary mouse hepatic cells or Kupffer cells were isolated from the liver of 9-week-old, C57BL/6, male mice as described previously (Severgnini et al., 2012; Li et al., 2014). After filtering the isolated hepatocytes, they were placed into 100 mm2 dishes and grown in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) with 100 U/mL penicillin, and 100 mg/mL streptomycin (Gibco, Grand Island, NY, United States) at 37 °C in 5% CO2-humidified air. For the isolation of Kupffer cells, the filtered cells were washed Roswell Park Memorial Institute 1640 medium (RPMI 1640) and seeded into a 6-well plate at a density of 1–3 × 107/well in DMEM (Hyclone, United States) with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin. After then, the cells incubated for 2 h in a 5% CO2 atmosphere at 37 °C. After 2 h of incubation, KCs adhere to the plate and non-adherent cells can subsequently be removed by gently washing with PBS.
The relationship between liver injury and Chi3l1 was analyzed with the ArrayExpress web server (http://www.ebi.ac.uk/arrayexpress), which provided mRNA sequencing data of human patient or mouse disease model. We selected potential genes associated with liver injury that were either up- or downregulated in the human patient or mouse liver injury model and then analyzed the relationship between those expressed genes and Chi3l1 using the GENEMANIA web server (http://genemania.org) which analyzes gene-gene networks.
All experiment were conducted in triplicates and replicated at least three times. Analysis of data was performed utilizing GraphPad Prism four version 4.03 software (Graph-Pad Software, La Jolla, CA). The results are presented as mean ± standard error of the mean (SEM) and assessed by one-way analysis of variance followed by the Turkey’s test. Statistical differences were considered significant at P-value <0.05.
Several studies reported that the level of human chitinase three like protein 1(CHI3L1) increased in serum of patients with various liver diseases, including chronic liver disease and acute liver disease (Kumagai et al., 2016; Shan et al., 2021; Lee et al., 2011; Wang et al., 2020). Thus, we measured the level of serum CHI3L1 in patients with hepatotoxicity and found that it was significantly increased compared to normal donors (Figure 1A). Similarly, the levels of Chi3l1 in mouse serum and liver were dramatically increased by APAP administration (Figure 1B). Interestingly, we found that KO mice deficient in Chi3l1 were less susceptible to APAP-induced liver injury than WT mice. Histological analysis revealed massive damage in the livers of WT mice caused by APAP administration, compared to the livers of KO mice (Figures 1C,D). In accordance with the histological analysis, the AST and ALT levels induced APAP in serum were decreased in the KO mice (Figure 1E).
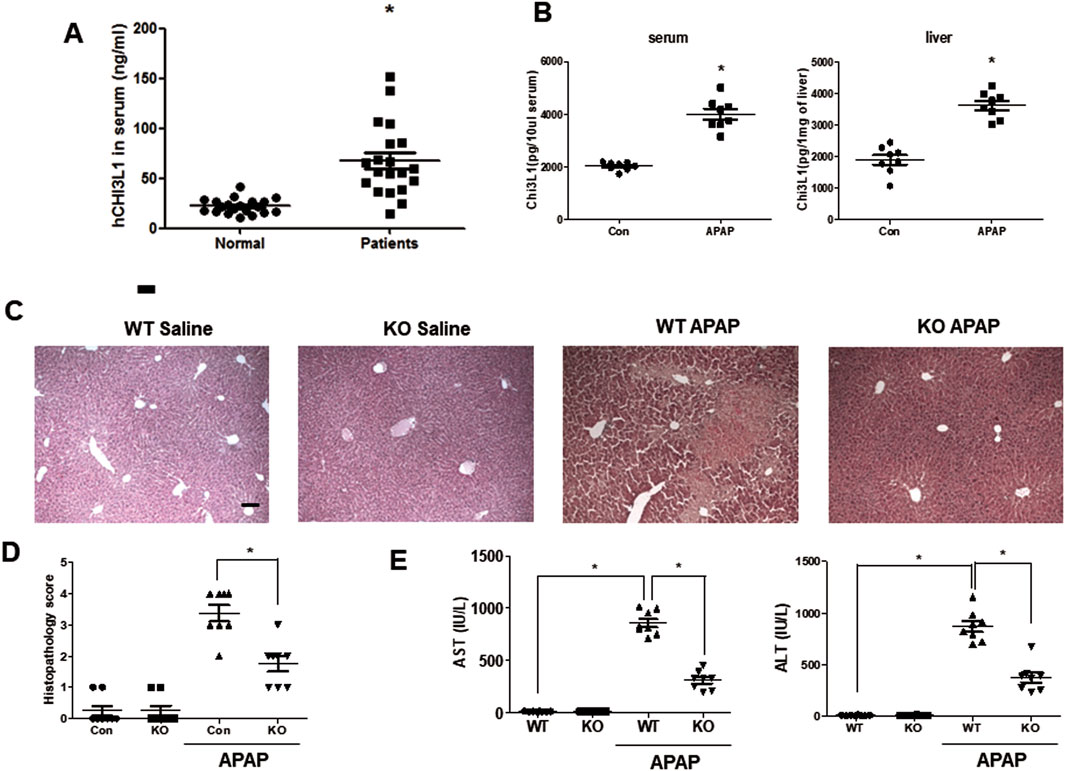
Figure 1. Chi3l1 deficiency shows protective effects in APAP-induced liver injury. (A) Serum analysis of CHI3L1 in patients with hepatotoxicity. N = 20 per group; means ± SEM, *P < 0.05. (B) The level of Chi3l1 was measured in the serum or liver of mice with or without APAP administration. N = 8 per group; means ± SEM, *P < 0.05. (C) Liver sections of WT and KO mice with or without APAP administration (500 mg/kg) were stained with hematoxylin and eosin (H&E) (Scale bars, 100 μm). (D) Histopathology score of liver H&E sections. (E) Serum AST and ALT levels in WT and KO mice with or without APAP administration (500 mg/kg). N = 8 per group; means ± SEM, *P < 0.05.
Neutrophils are the first responders to tissue injury (Graubardt et al., 2017), and the major constituent of leukocytes infiltrating the liver after APAP administration (Wang et al., 2013). Chi3l1 is closely associated with neutrophil infiltration (Breyne et al., 2018) thus, we measured the neutrophil population in the livers of WT and KO mice after APAP administration. Immunohistochemistry data reveal that, after APAP injection, neutrophils infiltrated the hepatic injury region in the livers of WT mice, whereas infiltrated neutrophils were almost completely mitigated in the livers of KO mice (Figure 2A). FACS analysis also showed that APAP-induced infiltrating neutrophils (CD11+Ly6G+) were decreased in the livers of KO mice compared to WT mice (Figures 2B,C). Myeloperoxydase (MPO), an indicator of neutrophil infiltration, is also decreased in the livers of KO mice compared to WT mice (Supplementary Figure S1). In contrast to neutrophils, macrophage migration was not affected by Chi3l1 deletion in the APAP-induced livers (Supplementary Figure S2). Neutrophils induce liver toxicity through oxidative stress by neutrophil-derived hydrogen peroxide, thus we investigated the oxidative stress in the livers of APAP-injected WT and KO mice. Our data reveal that the levels of hydrogen peroxide were elevated in the livers of APAP-injected WT mice whereas they were reduced in the livers of APAP-injected KO mice (Figure 2D). Accordingly, the level of MDA, a naturally occurring product of lipid peroxidation and an marker of oxidative stress, in the liver was also induced by APAP in WT mice; however, it was lower in the livers of APAP-injected KO mice (Figure 2E).
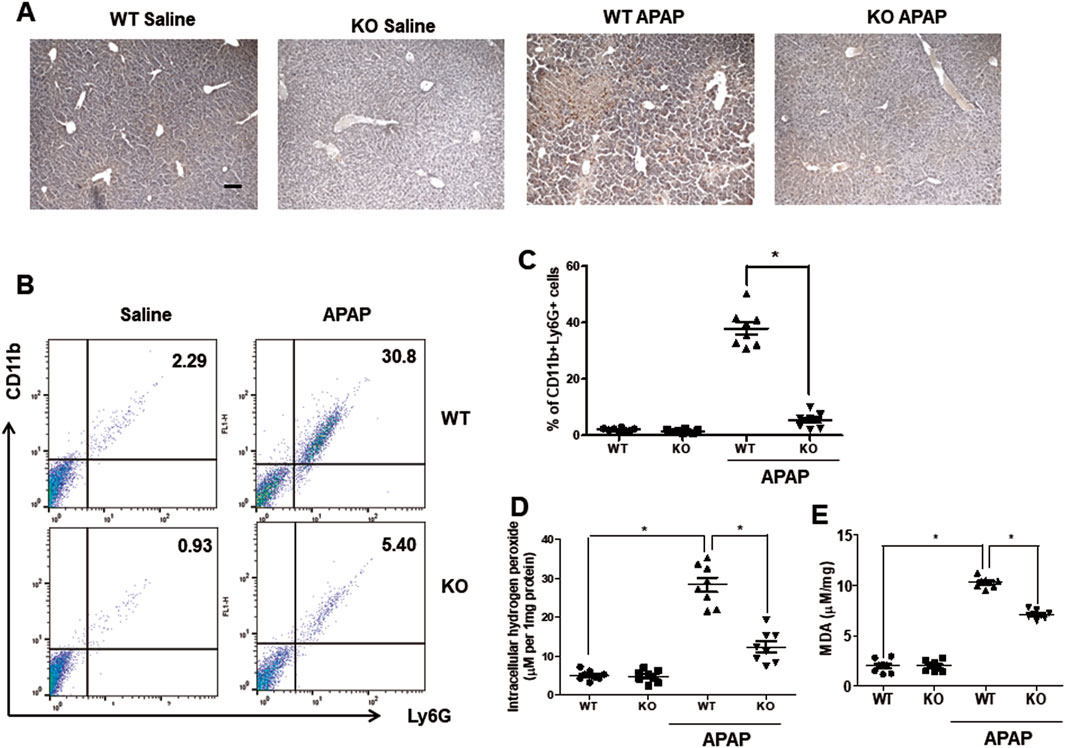
Figure 2. Chi3l1 deficiency abrogates recruitment of neutrophils into the liver by APAP overdose. (A) Immunohistochemistry of infiltrated neutrophils (Ly6G) in the livers of WT and KO mice with or without APAP administration (500 mg/kg) (Scale bars, 100 μm). Ly6G is marked with a brown dot. (B) Leukocytes were isolated from the livers of mice. The total CD11b+Ly6G + cells among all hepatic leukocytes were analyzed by flow cytometry. (C) Statistical analysis of the percentage of neutrophils in the hepatic leukocytes. N = 8 per group; means ± SEM, *P < 0.05. (D) Intracellular hydrogen peroxide levels and (E) MDA levels in the liver of WT and KO with or without APAP administration (500 mg/kg). N = 8 per group; means ± SEM, *P < 0.05.
Since previous studies reported that macrophages are the major cells expressing Chi3l1 (Zhao et al., 2023), we wondered which cells in the liver have the most Chi3l1 expression and production after APAP administration. To determine whether the expression of Chi3l1 could be induced by APAP in hepatocytes, we isolated mouse primary hepatocytes from WT mouse liver and determined the amount of Chi3l1 mRNA before and after APAP treatment. The levels of Chi3l1 mRNA were not changed by APAP treatment in mouse primary hepatocytes. However, Chi3l1 mRNA levels were significantly increased in Kupffer cells from WT mouse liver after treatment with the supernatant of APAP-treated mouse hepatocytes (Figures 3A,B). In addition, immunofluorescence data showed that the major site of APAP-induced Chi3l1 expression was in macrophages, labeled with mice macrophage maker, F4/80 (Figure 3C). These results suggest that Chi3l1 expression was not induced by APAP in hepatocytes, but its expression was induced in macrophage by secreted factors such as DAMPs from APAP-damaged hepatocytes. Next, we examined chemokines and cytokines required for neutrophil infiltration by Chi3l1 in APAP-induced liver injury. We found that the levels of APAP-induced IL-17 and IL-23, which are key factors for neutrophil infiltration, were abrogated in the livers of Chi3l1 KO mice (Supplementary Figure S3). The mRNA and protein levels of CCL2, CXCL2, and IL-23 were increased by APAP injection in the livers of WT mice, but these elevated mRNA levels were reduced in APAP-induced livers of KO mice (Figure 4A; Supplementary Figures S3, S4). Since Chi3l1 is expressed in macrophages and Kupffer cells, and resident macrophages are the main cells recruiting neutrophils to the liver, we isolated Kupffer cells from liver of WT mice then investigated the role of Chi3l1 in the expression of CCL2, CXCL2, and IL-23 which are all associated with neutrophils recruitment (Wang et al., 2013; Moles et al., 2014). The mRNA level of CCL2, CXCL2, and IL-23 were further increased by dose-dependent, rChi3l1 treatment in Kupffer cells from the livers of WT mice (Figure 4B). In contrast, after treatment of Kupffer cells with the supernatant of APAP-treated mouse hepatocytes, the mRNA levels of CCL2, CXCL2, and IL-23 were reduced in Kupffer cells from the livers of KO mice compared to WT mice, but these levels were restored by rChi3l1 treatment (Figure 4C). These results suggest that Chi3l1 plays a role in chemokine and cytokine expression in macrophages that is required for neutrophil infiltration.
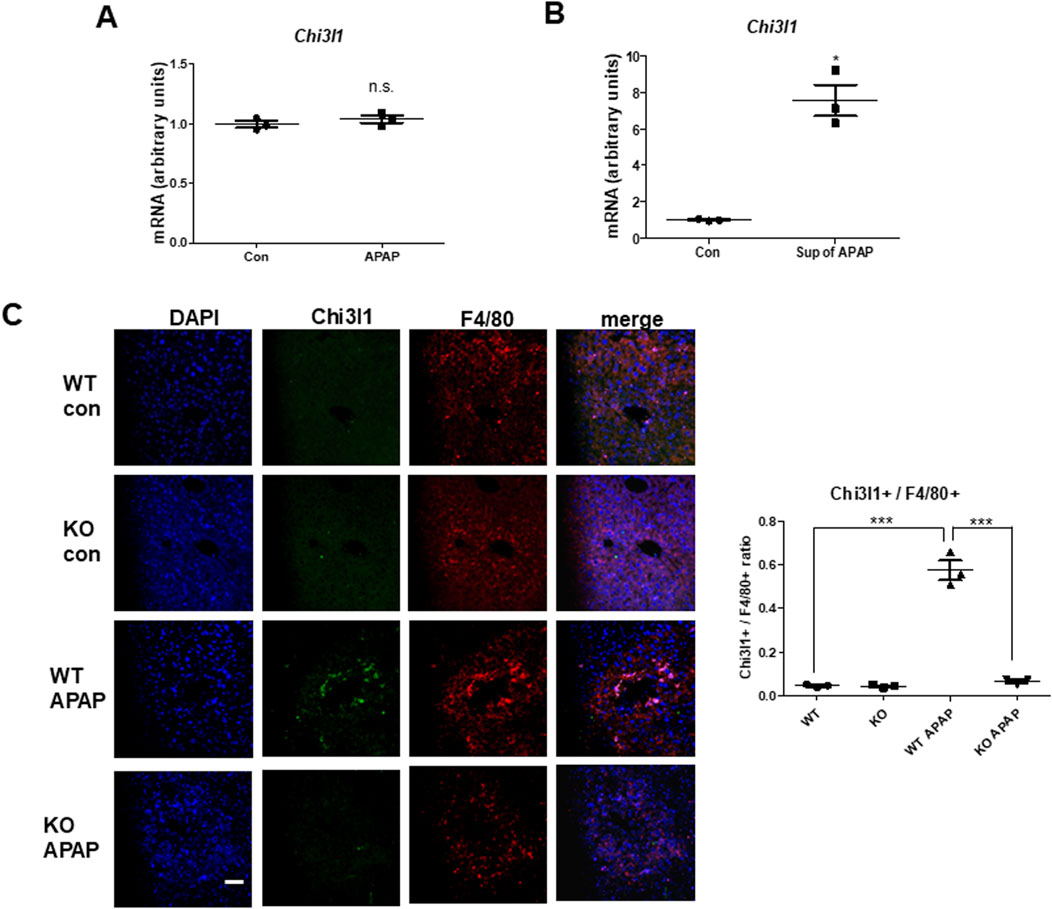
Figure 3. Chi3l1, which is induced by APAP administration, was expressed in macrophages, not hepatocytes. (A) Chi3l1 mRNA expression was measured in primary hepatocytes treated with or without APAP. (B) Kupffer cells were isolated from WT mice and then treated with the supernatant of APAP-treated hepatocytes (sup of APAP) or PBS-treated hepatocytes (Con). Then, Chi3l1 mRNA expression was measured. (C) Liver sections were triple-stained with Chi3l1 antibody (Alexa 488; green), F4/80 antibody (Alexa 568; red), and DAPI (nuclear counterstain; blue), and the images were analyzed by confocal laser-scanning microscopy (Scale bars, 50 μm) Values of chi3l1+/F4/80+ cells ratio are expressed as the means ± SEM. ***P < 0.001.
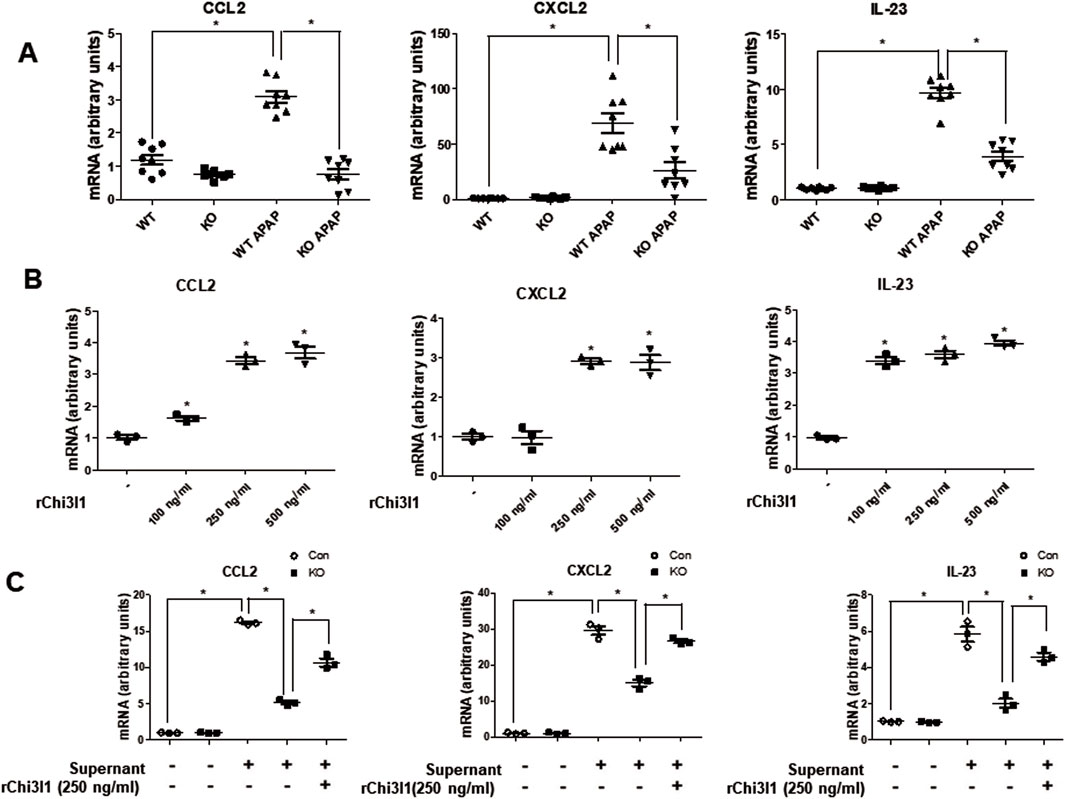
Figure 4. The gene expression of Ccl2, Cxcl2, and Il-23 which are involved in neutrophil recruitment. (A) The mRNA expression of Ccl2, Cxcl2, and Il-23 in the liver of WT and KO with or without APAP administration (500 mg/kg). N = 8 per group; means ± SEM, *P < 0.05. (B) The mRNA expression of Ccl2, Cxcl2, and Il-23 in Kupffer cells isolated from WT mice liver and dose-dependently treated with rChi3l1. Values are expressed as the mean ± SEM of three different experiments conducted in triplicates. *P < 0.05. (C) mRNA expression of Ccl2, Cxcl2, and Il-23 in Kupffer cells isolated from WT or KO mice liver pretreated with rCHI3L1 and then treated with or without supernatant of APAP-treated mouse hepatocytes. Values are expressed as the mean ± SEM of three different experiments conducted in triplicates. *P < 0.05.
Next, we investigated target proteins regulated by Chi3l1 in APAP-induced liver injury. Firstly, we analyzed RNA sequencing data, which are either data from the livers of mice orally treated with APAP (Accession No. E-GEOD-51969) or human blood samples of patients treated with APAP (Accession No. E-GEOD-70786), from ArrayExpress (www.ebi.ac.uk/arrayexpress) (Figure 5A). The analysis revealed that eight genes were upregulated and five genes were downregulated in both APAP-treated mouse liver and human blood samples. Then, we analyzed whether these genes are associated with Chi3l1 using GENEMANIA. We found that three upregulated genes (Lcn2, S100A8, Klf1) were correlated with Chi3l1 (Supplementary Figure S5). Thus, we validated the expression of these three genes using quantitative PCR in APAP-induced livers of WT or KO mice. The elevated mRNA expression of Lcn2 and S100A8 by APAP administration were abrogated in the livers of Chi3l1 KO mice, but the mRNA expression of Klf1 exhibited no changes due to APAP administration and loss of Chi3l1 (Figure 5B). We focused on the Lcn2 because STRING network analysis revealed a significant correlation between Lcn2 and Chi3l1 (Figure 5C). To validate if the expression of Lcn2 is affected by Chi3l1, we investigated the Lcn2 protein expressed in macrophages treated with rChi3l1. We show that Lcn2 expression was increased by dose-dependent, rChi3l1 treatment (Supplementary Figure S6). Moreover, expression of Lcn2 protein was significantly decreased in the APAP-treated livers of Chi3l1 KO mice compared to WT mice (Figure 5D). These data suggest that Lcn2 might be induced by Chi3l1.
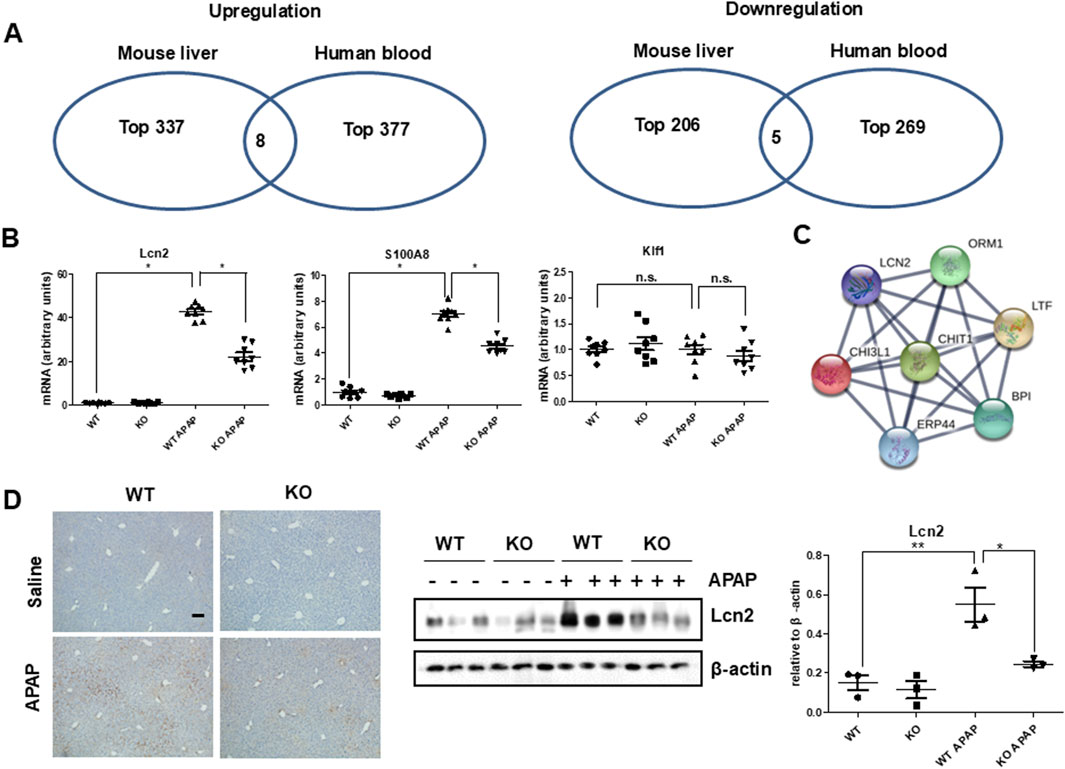
Figure 5. Strategy for target regulated by Chi3l1. (A) Select co-upregulated or co-downregulated genes from RNA sequencing analysis of mouse liver with or without APAP administration and human blood from a patient with APAP exposure or a healthy donor. (B) The mRNA expression of Lcn2, S100A8, and Klf1 in the liver of WT and KO with or without APAP administration (500 mg/kg). N = 8 per group; means ± SEM, *P < 0.05. (C) STRING network analysis with Chi3l1 and Lcn2. (D) Immunohistochemistry and immunoblot of Lcn2 in the liver of WT and KO mice with or without APAP administration (500 mg/kg) (Scale bars, 100 μm). Values are expressed as the means ± SEM. *P < 0.05, **P < 0.01.
Because APAP-induced Lcn2 expression was inhibited in Chi3l1 KO mice, we administered exogenous rLcn2 into Chi3l1 KO mice injected with or without APAP to investigate the effect of Lcn2 in APAP-treated Chi3l1 KO mice. The rLcn2 administration did not induce liver injury, but it weakly recruited neutrophils to the liver (Figure 6). However, attenuated APAP-induced liver injury in the Chi3l1 KO mice was aggravated by rLcn2 injection as well as increased neutrophil infiltration to the liver (Figures 6A,B). In accordance with the histological analysis, the APAP-induced CCL2, CXCL2, and IL-23 in the liver and AST, ALT serum levels were increased by rLcn2 in KO mice (Figures 6C,D). These results suggest that inhibited APAP-induced liver injury and neutrophil infiltration in Chi3l1 KO mice is mediated by Lcn2.
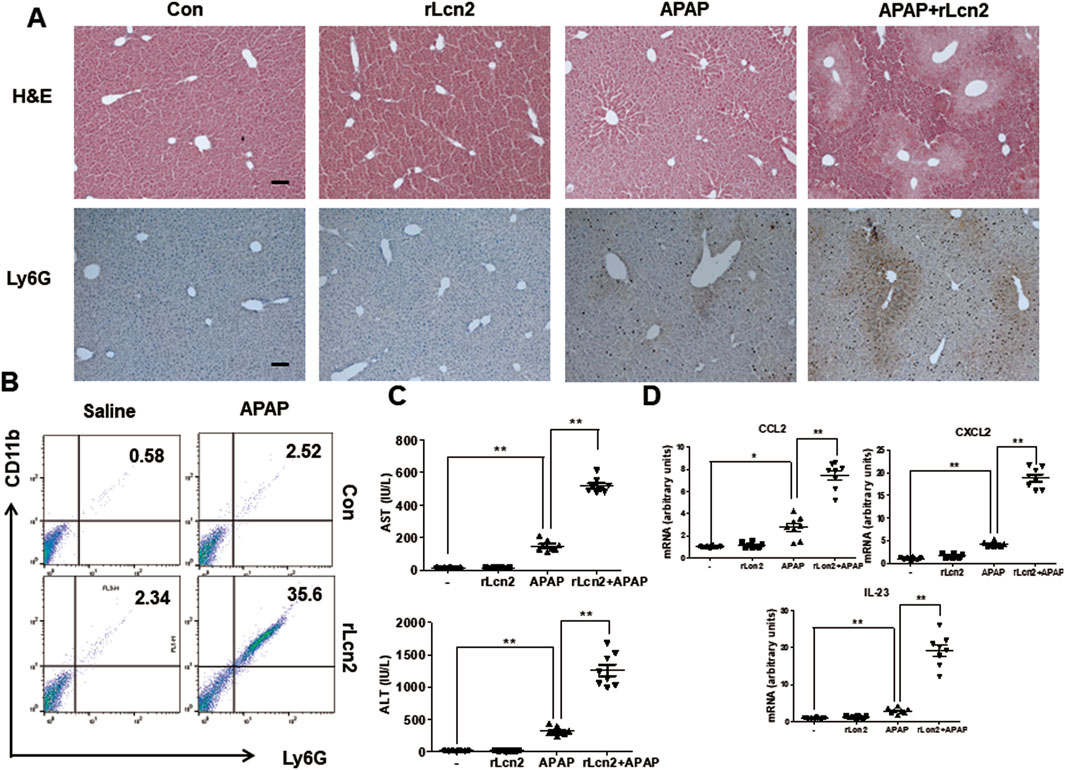
Figure 6. Lcn2 showed enhanced effects on APAP-induced liver injury in mice. (A) Hematoxylin and eosin (H&E) staining and immunohistochemistry of Ly6G of liver sections from KO mice injected with or without rLcn2 (10 μg/mouse) under APAP administration (300 mg/kg) (Scale bars, 100 μm). (B) Leukocytes were isolated from the liver. The total CD11b+Ly6G + cells among all hepatic leukocytes were analyzed by flow cytometry. (C) Serum AST and ALT levels in mice injected with rLcn2 (10 μg/mouse) in APAP administration (300 mg/kg) or not. N = 8 per group; means ± SEM, *P < 0.05. (D) The mRNA expression of Ccl2, Cxcl2, and Il-23 in the liver of KO mice injected with rLcn2 (10 μg/mouse) in APAP administration (300 mg/kg) or not. N = 8 per group; means ± SEM, *P < 0.05, **P < 0.01.
To further demonstrate that Chi3l1 can exacerbate APAP-induced liver injury through Lcn2, we treated Chi3l1 KO mice with rChi3l1 while neutralizing Lcn2. The data reveal that rChi3l1-induced deterioration of APAP-induced liver injury was abrogated by Lcn2 neutralization (Figures 7A,B). Moreover, APAP-induced neutrophil infiltration as well as CCL2, CXCL2, and IL-23 expression in the liver by rChi3l1 were inhibited by Lcn2 neutralization (Figures 7C,D). These results suggest that Lcn2, induced by Chi3l1, drives neutrophil infiltration in APAP-induced liver injury.
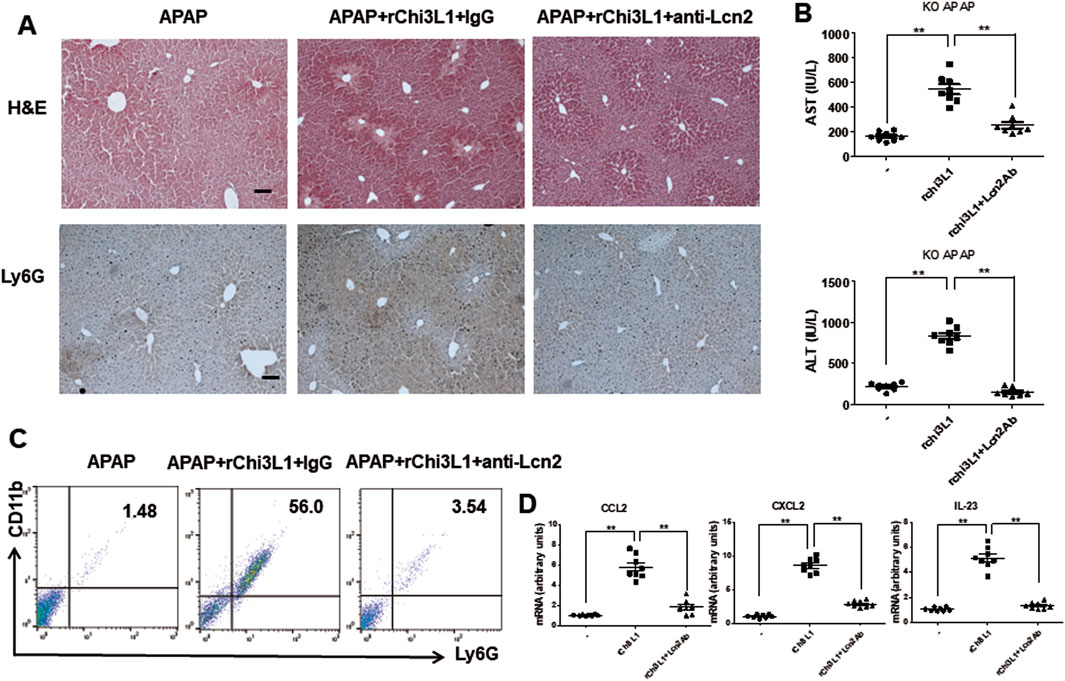
Figure 7. Lcn2 antibody recovered in exacerbated liver injury by rChi3l1 on APAP administration. (A) Hematoxylin and eosin (H&E) staining and immunohistochemistry of Ly6G of liver sections in rLcn2-and APAP-administrated KO mice pre-injected with anti-Lcn2 (50 μg/mouse) or anti-IgG (300 mg/kg) (Scale bars, 100 μm). (B) Serum AST and ALT levels in rLcn2-and APAP- administrated KO mice pre-injected with anti-Lcn2 (50 μg/mouse) or anti-IgG (300 mg/kg). N = 8 per group; means ± SEM, **P < 0.01. (C) Leukocytes were isolated from the liver. The total CD11b+Ly6G + cells among all hepatic leukocytes were analyzed by flow cytometry. (D) The mRNA expression of Ccl2, Cxcl2, and Il-23 in the liver of rLcn2-and APAP- administrated WT mice pre-injected with anti-Lcn2 (50 μg/mouse) or anti-IgG (300 mg/kg). N = 8 per group; means ± SEM, **P < 0.01.
CLPs (Chitinase like proteins) are proteins that are structurally similar to chitinase and bind to chitin but cannot be broken down (Yu et al., 2024). Studies of CLPs have revealed the types and various functions, for example, involved in immune response control and pathogen detection (Yu et al., 2024). Among them, chi3l1 is associated with cancer as well as non-neoplastic diseases characterized by inflammation such as arthritis, infectious disease, inflammatory bowel disease, kidney injury, and liver disease (Kumagai et al., 2016; Kjaergaard et al., 2014). In cancer, Chi3l1 plays a role in metastasis and is increased in serum; however, the function of Chi3l1 in various diseases remains unclear. Recently, a study was reported that CHI3L1 is involved in platelet recruitment and damage of the liver induced by APAP (Shan et al., 2021). However, many studies are still needed on the association between CHI3L1 and acute liver damage. To investigate this, we used Chi3l1 KO mice and employed an APAP-induced liver injury model which is frequently used for drug-induced hepatotoxicity.
APAP is a commonly-used drug, but its overdose leads to hepatocyte cell death by increasing oxidative stress and mitochondrial dysfunction during metabolism of APAP. Several recent studies reported the pathogenesis of APAP-induced liver injury. They revealed that not only acute hepatocytes necrosis but also subsequent inflammatory responses can critically affect the severity of the disease (Antoniades et al., 2012). Neutrophil-mediated liver injury has been reported in experimental drug/chemical-induced animal models such as ethanol toxicity, ANIT, and APAP (Ramaiah and Jaeschke, 2007; Smith et al., 1998). In drug/chemical-induced liver injury models, inflammatory mediators stimulated by oxidative stress and tissue injury lead to neutrophil invasion that often worsens the liver damage. Although it is still controversial whether neutrophils directly aggravate the course of APAP-induced liver injury (Jaeschke, 2008), several studies indicated that neutrophils can directly mediate hepatocyte necrosis in APAP-induced liver injury by neutrophil-derived ROS (Marques et al., 2012). In addition, it has been reported that decrease in neutrophil infiltration attenuates drug-induced acute liver damage (Matsuo et al., 2023). In our study, we found that Chi3l1-deficient mice had attenuated APAP-induced liver injury and abrogated neutrophil infiltration as well as decreased liver oxidative stress. In rodent experiments, a number of neutrophils are recruited into the liver at 4–24 h after treatment with a high dose of APAP (Lawson et al., 2000), and pretreatment with a neutrophil antiserum or anti-Gr-1 monoclonal antibody significantly attenuated hepatic neutrophil accumulation and liver injury after APAP administration (Liu et al., 2006; Smith et al., 1998). APAP-induced necrotic hepatocytes release DAMPs, such as high-mobility group box 1 (HMGB1), heat shock proteins, DNA, and cyclophilin A (Martin-Murphy et al., 2010; Dear et al., 2011). These DAMPs activate KCs, which are the resident hepatic macrophages, that in turn recruit neutrophils through various inflammatory mediators or γδ T cells (Wang et al., 2013). Our data show that Chi3l1 was not induced in hepatocytes and rChi3l1 treatment did not affect APAP-induced hepatic cell death. However, cell supernatant from APAP-treated hepatocytes induced Chi3l1 expression in macrophages, and immunofluorescence data revealed that Chi3l1 was mainly expressed in macrophages in the APAP-treated liver. These results suggest that Chi3l1 is not associated with APAP-induced cell death in hepatocytes, but the source of increasing Chi3l1 by APAP is maybe associated with macrophage-mediated liver injury. Furthermore, previous studies have reported the association of Chi3l1 with M1 macrophage and M2 macrophage (Zhao et al., 2023; Higashiyama et al., 2019). M2 secretes a large amount of CHI3L1 and CHI3L1 promotes M2 polarization in many diseases (Zhao et al., 2023). On the other hand, it has also been reported that CHI3L1 deficiency inhibit apoptosis of M1, but not apoptosis of M253. Accordingly, the association between CHI3L1 and macrophage still had an argument. In addition, it is known that the balance of M1/M2 is important in liver disease in many works (Wang et al., 2021). Therefore, further understanding of the association of Chi3l1 and M1/M2 is required in drug-induced acute liver disease.
Next, we investigated the function of Chi3l1 in APAP-induced liver injury. To study this, we first performed a cytokines/chemokines array and found that APAP-induced IL-17 and IL-23, which are key factors for neutrophil infiltration, were abrogated in the livers of Chi3l1-deficient mice. CHI3L1 is closely associated with the production of chemokines and cytokines in macrophages for immune cell infiltration (Letuve et al., 2008; Libreros et al., 2012; Breyne et al., 2018). Thus, we validated the levels of CCL2, CXCL2, and IL-23, which are involved in neutrophil infiltration, in the APAP-treated livers and macrophages. We found that they were not increased in the macrophages and livers of Chi3l1 KO mice. These results suggest that Chi3l1 may affect macrophage activation to produce chemokines and cytokines necessary for neutrophil infiltration.
Previous studies have shown that chi3l1 increased platelet recruitment by increasing podoplanin via receptor CD44 in macrophages, leading to platelet involvement in tissue damage in the early stages to AILI (Shan et al., 2021). Our data showed that neutrophil infiltration into the liver increased by chi3l1 derived from KC and was involved in ALI. These finding suggest that chi3l1 plays an important role in contributing to AILI exacerbation by inducing tissue damage through platelet and neutrophil recruitment. Platelet is involved in neutrophil recruitment to the liver (Morris and Chauhan, 2022), and platelet depletion within necrotizing foci in APAP-treated mice tends to reduce neutrophil accumulation (Miyakawa et al., 2015). Since these finding suggest that interactions between them are likely involved in promoting neutrophil accumulation in AILI, further studies are needed to reveal the mechanism by which platelet-neutrophils interactions causes APAP-induced liver damage.
Second, we investigated what protein is associated with Chi3l1 in APAP-induced liver injury. We thus downloaded RNA sequencing data of APAP-treated mice and patients from ArrayExpress site (www.ebi.ac.uk/arrayexpress). We found that Lcn2 was among the upregulated genes in APAP-treated mice and humans and was associated with Chi3l1. We then observed that Lcn2 was increased in the livers of APAP-treated mice but not in the APAP-treated Chi3l1 KO mice. Lcn2, also known as neutrophil gelatinase associated lipocalin (NGAL), may be an early biomarker of liver inflammation because it is highly upregulated in response to inflammation, injury, and metabolic stress in the liver (Xiao et al., 2017; Wieser et al., 2016). Several studies reported that Lcn2 may act to recruit neutrophils to the site of inflammation in various tissues (Xu et al., 2015). Moreover, Lcn2 is associated with neutrophilic inflammation and neutrophil-macrophage crosstalk in liver diseases (Wieser et al., 2016; Ye et al., 2016). Thus, we examined the relationship between Lcn2 and Chi3l1 in the livers of APAP-treated mice. In the present study, we found that Lcn2 was induced by Chi3l1, and the attenuated liver injury by Chi3l1 deficiency was exacerbated by Lcn2 administration in APAP-treated mouse liver. Moreover, exacerbated liver injury and elevated neutrophil infiltration by rChi3l1 were reduced through neutralizing Lcn2 by antibody treatment in APAP-treated Chi3l1 KO mouse liver. These results suggest that Lcn2 is induced by Chi3l1 and is associated with neutrophil infiltration in APAP-induced liver injury.
In summary, our results indicate that Chi3l1 induces Lcn2, which induced neutrophil infiltration leading to liver injury by APAP, and reveal a novel mechanism in which chi3l1 is involved in neutrophil infiltration. These studies support treatment strategies targeting chi3l1 in APAP-induced liver injury and suggest that Lcn2 inhibition has a protective effect on liver damage by APAP.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Ethics Committee of Chungbuk National University Medical Centre. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by the Chungbuk National University Animal Care Committee. The study was conducted in accordance with the local legislation and institutional requirements.
JK: Conceptualization, Formal Analysis, Investigation, Writing–original draft, Writing–review and editing. IY: Conceptualization, Formal Analysis, Investigation, Writing–original draft, Writing–review and editing. DS: Conceptualization, Formal Analysis, Resources, Writing–original draft, Writing–review and editing. SH: Conceptualization, Formal Analysis, Resources, Writing–original draft, Writing–review and editing. DY: Conceptualization, Formal Analysis, Resources, Writing–original draft, Writing–review and editing. DL: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Writing–original draft, Writing–review and editing. JH: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) MRC 2017R1A5A2015541 (to Jin Tae Hong), Chonnam National University 2024–1145–01 (to Dong Hun Lee), the National Research Foundation of Korea RS-2023–00251463 (to Dong Hun Lee) and the Global-Learning &Academic research institution for Master’s PhD students, and Postdocs (LAMP) Program of the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education RS-2024–00442775 (to Dong Hun Lee).
We are grateful to the patients, their families and referring physicians that have participated in these studies, and we also thank the RNA sequencing data program site administrators and its research data donors, and the fundings supporting this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1548832/full#supplementary-material
Antoniades, C. G., Quaglia, A., Taams, L. S., Mitry, R. R., Hussain, M., Abeles, R., et al. (2012). Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 56 (2), 735–746. doi:10.1002/hep.25657
Bajt, M. L., Farhood, A., and Jaeschke, H. (2001). Effects of CXC chemokines on neutrophil activation and sequestration in hepatic vasculature. Am. J. Physiol. Gastrointest. Liver Physiol. 281 (5), G1188–G1195. doi:10.1152/ajpgi.2001.281.5.G1188
Bautista, A. P. (2002). Neutrophilic infiltration in alcoholic hepatitis. Alcohol 27 (1), 17–21. doi:10.1016/s0741-8329(02)00206-9
Breyne, K., Steenbrugge, J., Demeyere, K., Lee, C. G., Elias, J. A., Petzl, W., et al. (2018). Immunomodulation of host chitinase 3-like 1 during a mammary pathogenic Escherichia coli infection. Front. Immunol. 9, 1143. doi:10.3389/fimmu.2018.01143
Chambers, J. W., and LoGrasso, P. V. (2011). Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 286 (18), 16052–16062. doi:10.1074/jbc.M111.223602
Connolly, M. K., Ayo, D., Malhotra, A., Hackman, M., Bedrosian, A. S., Ibrahim, J., et al. (2011). Dendritic cell depletion exacerbates acetaminophen hepatotoxicity. Hepatology 54 (3), 959–968. doi:10.1002/hep.24429
Dear, J. W., Simpson, K. J., Nicolai, M. P., Catterson, J. H., Street, J., Huizinga, T., et al. (2011). Cyclophilin A is a damage-associated molecular pattern molecule that mediates acetaminophen-induced liver injury. J. Immunol. 187 (6), 3347–3352. doi:10.4049/jimmunol.1100165
Essani, N. A., Fisher, M. A., Farhood, A., Manning, A. M., Smith, C. W., and Jaeschke, H. (1995). Cytokine-induced upregulation of hepatic intercellular adhesion molecule-1 messenger RNA expression and its role in the pathophysiology of murine endotoxin shock and acute liver failure. Hepatology 21 (6), 1632–1639. doi:10.1002/hep.1840210623
Fisher, J. E., McKenzie, T. J., Lillegard, J. B., Yu, Y., Juskewitch, J. E., Nedredal, G. I., et al. (2013). Role of Kupffer cells and toll-like receptor 4 in acetaminophen-induced acute liver failure. J. Surg. Res. 180 (1), 147–155. doi:10.1016/j.jss.2012.11.051
Graubardt, N., Vugman, M., Mouhadeb, O., Caliari, G., Pasmanik-Chor, M., Reuveni, D., et al. (2017). Ly6C(hi) monocytes and their macrophage descendants regulate neutrophil function and clearance in acetaminophen-induced liver injury. Front. Immunol. 8, 626. doi:10.3389/fimmu.2017.00626
Gujral, J. S., Farhood, A., Bajt, M. L., and Jaeschke, H. (2003). Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology 38 (2), 355–363. doi:10.1053/jhep.2003.50341
Higashiyama, M., Tomita, K., Sugihara, N., Nakashima, H., Furuhashi, H., Nishikawa, M., et al. (2019). Chitinase 3-like 1 deficiency ameliorates liver fibrosis by promoting hepatic macrophage apoptosis. Hepatol. Res. 49 (11), 1316–1328. doi:10.1111/hepr.13396
Hill, D. A., Jean, P. A., and Roth, R. A. (1999). Bile duct epithelial cells exposed to alpha-naphthylisothiocyanate produce a factor that causes neutrophil-dependent hepatocellular injury in vitro. Toxicol. Sci. 47 (1), 118–125. doi:10.1093/toxsci/47.1.118
Ho, J. S., Buchweitz, J. P., Roth, R. A., and Ganey, P. E. (1996). Identification of factors from rat neutrophils responsible for cytotoxicity to isolated hepatocytes. J. Leukoc. Biol. 59 (5), 716–724. doi:10.1002/jlb.59.5.716
Holt, M. P., Cheng, L., and Ju, C. (2008). Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 84 (6), 1410–1421. doi:10.1189/jlb.0308173
Im, J. H., Yeo, I. J., Park, P. H., Choi, D. Y., Han, S. B., Yun, J., et al. (2020/01/01/2020). Deletion of Chitinase-3-like 1 accelerates stroke development through enhancement of Neuroinflammation by STAT6-dependent M2 microglial inactivation in Chitinase-3-like 1 knockout mice. Exp. Neurol. 323, 113082. doi:10.1016/j.expneurol.2019.113082
Jaeschke, H., Farhood, A., and Smith, C. W. (1990). Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. 4 (15), 3355–3359. doi:10.1096/fasebj.4.15.2253850
Jaeschke, H., Farhood, A., and Smith, C. W. (1991). Neutrophil-induced liver cell injury in endotoxin shock is a CD11b/CD18-dependent mechanism. Am. J. Physiol. 261 (6 Pt 1), G1051–G1056. doi:10.1152/ajpgi.1991.261.6.G1051
Jaeschke, H., Ho, Y. S., Fisher, M. A., Lawson, J. A., and Farhood, A. (1999). Glutathione peroxidase-deficient mice are more susceptible to neutrophil-mediated hepatic parenchymal cell injury during endotoxemia: importance of an intracellular oxidant stress. Hepatology 29 (2), 443–450. doi:10.1002/hep.510290222
Jaeschke, H. (2008). Innate immunity and acetaminophen-induced liver injury: why so many controversies? Hepatology 48 (3), 699–701. doi:10.1002/hep.22556
Kjaergaard, A. D., Bojesen, S. E., Nordestgaard, B. G., and Johansen, J. S. (2014). YKL-40 and alcoholic liver and pancreas damage and disease in 86,258 individuals from the general population: cohort and mendelian randomization studies. Clin. Chem. 60 (11), 1429–1440. doi:10.1373/clinchem.2014.229096
Krenkel, O., Mossanen, J. C., and Tacke, F. (2014). Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg. Nutr. 3 (6), 331–343. doi:10.3978/j.issn.2304-3881.2014.11.01
Kumagai, E., Mano, Y., Yoshio, S., Shoji, H., Sugiyama, M., Korenaga, M., et al. (2016). Serum YKL-40 as a marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci. Rep. 6, 35282. doi:10.1038/srep35282
Larsen, F. S., and Wendon, J. (2014). Understanding paracetamol-induced liver failure. Intensive Care Med. 40 (6), 888–890. doi:10.1007/s00134-014-3293-9
Lawson, J. A., Farhood, A., Hopper, R. D., Bajt, M. L., and Jaeschke, H. (2000). The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol. Sci. 54 (2), 509–516. doi:10.1093/toxsci/54.2.509
Lee, C. G., Da Silva, C. A., Dela Cruz, C. S., Ahangari, F., Ma, B., Kang, M. J., et al. (2011). Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. physiology 73, 479–501. doi:10.1146/annurev-physiol-012110-142250
Letuve, S., Kozhich, A., Arouche, N., Grandsaigne, M., Reed, J., Dombret, M. C., et al. (2008). YKL-40 is elevated in patients with chronic obstructive pulmonary disease and activates alveolar macrophages. J. Immunol. 181 (7), 5167–5173. doi:10.4049/jimmunol.181.7.5167
Li, L., Wen, Y., Wrapp, D., Jeong, J., Zhao, P., Xiong, W., et al. (2023). A novel humanized Chi3l1 blocking antibody attenuates acetaminophen-induced liver injury in mice. Antib. Ther. 6 (1), 1–12. doi:10.1093/abt/tbac027
Li, P. Z., Li, J. Z., Li, M., Gong, J. P., and He, K. (2014). An efficient method to isolate and culture mouse Kupffer cells. Immunol. Lett. 158 (1-2), 52–56. doi:10.1016/j.imlet.2013.12.002
Li, Q., Chen, F., and Wang, F. (2022). The immunological mechanisms and therapeutic potential in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Cell Biosci. 12 (1), 187. doi:10.1186/s13578-022-00921-4
Libreros, S., Garcia-Areas, R., Shibata, Y., Carrio, R., Torroella-Kouri, M., and Iragavarapu-Charyulu, V. (2012). Induction of proinflammatory mediators by CHI3L1 is reduced by chitin treatment: decreased tumor metastasis in a breast cancer model. Int. J. Cancer 131 (2), 377–386. doi:10.1002/ijc.26379
Liu, Z. X., Han, D., Gunawan, B., and Kaplowitz, N. (2006). Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 43 (6), 1220–1230. doi:10.1002/hep.21175
Marques, P. E., Amaral, S. S., Pires, D. A., Nogueira, L. L., Soriani, F. M., Lima, B. H. F., et al. (2012). Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56 (5), 1971–1982. doi:10.1002/hep.25801
Martin-Murphy, B. V., Holt, M. P., and Ju, C. (2010). The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol. Lett. 192 (3), 387–394. doi:10.1016/j.toxlet.2009.11.016
Matsuo, S., Nabekura, T., Matsuda, K., Shibuya, K., and Shibuya, A. (2023). DNAM-1 immunoreceptor protects mice from concanavalin A-induced acute liver injury by reducing neutrophil infiltration. J. Immunol. 211 (6), 954–963. doi:10.4049/jimmunol.2200705
Miyakawa, K., Joshi, N., Sullivan, B. P., Albee, R., Brandenberger, C., Jaeschke, H., et al. (2015). Platelets and protease-activated receptor-4 contribute to acetaminophen-induced liver injury in mice. Blood 126 (15), 1835–1843. doi:10.1182/blood-2014-09-598656
Moles, A., Murphy, L., Wilson, C. L., Chakraborty, J. B., Fox, C., Park, E. J., et al. (2014). A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J. Hepatol. 60 (4), 782–791. doi:10.1016/j.jhep.2013.12.005
Morris, S. M., and Chauhan, A. (2022). The role of platelet mediated thromboinflammation in acute liver injury. Front. Immunol. 13, 1037645. doi:10.3389/fimmu.2022.1037645
Muhammad-Azam, F., Nur-Fazila, S. H., Ain-Fatin, R., Mustapha Noordin, M., and Yimer, N. (2019). Histopathological changes of acetaminophen-induced liver injury and subsequent liver regeneration in BALB/C and ICR mice. Vet. World 12 (11), 1682–1688. doi:10.14202/vetworld.2019.1682-1688
Niehorster, M., Tiegs, G., Schade, U. F., and Wendel, A. (1990). In vivo evidence for protease-catalysed mechanism providing bioactive tumor necrosis factor alpha. Biochem. Pharmacol. 40 (7), 1601–1603. doi:10.1016/0006-2952(90)90461-s
Ramaiah, S. K., and Jaeschke, H. (2007). Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol. Pathol. 35 (6), 757–766. doi:10.1080/01926230701584163
Roslind, A., and Johansen, J. S. (2009). YKL-40: a novel marker shared by chronic inflammation and oncogenic transformation. Methods Mol. Biol. 511, 159–184. doi:10.1007/978-1-59745-447-6_7
Ruepp, S. U., Tonge, R. P., Shaw, J., Wallis, N., and Pognan, F. (2002). Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol. Sci. 65 (1), 135–150. doi:10.1093/toxsci/65.1.135
Severgnini, M., Sherman, J., Sehgal, A., Jayaprakash, N. K., Aubin, J., Wang, G., et al. (2012). A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology 64 (2), 187–195. doi:10.1007/s10616-011-9407-0
Shan, Z., Li, L., Atkins, C. L., Wang, M., Wen, Y., Jeong, J., et al. (2021). Chitinase 3-like-1 contributes to acetaminophen-induced liver injury by promoting hepatic platelet recruitment. elife 10, e68571. doi:10.7554/eLife.68571
Smith, G. S., Nadig, D. E., Kokoska, E. R., Solomon, H., Tiniakos, D. G., and Miller, T. A. (1998). Role of neutrophils in hepatotoxicity induced by oral acetaminophen administration in rats. J. Surg. Res. 80 (2), 252–258. doi:10.1006/jsre.1998.5441
Tacke, F., and Zimmermann, H. W. (2014). Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 60 (5), 1090–1096. doi:10.1016/j.jhep.2013.12.025
Tao, H., Yang, J. J., Shi, K. H., Huang, C., Zhang, L., Lv, X. W., et al. (2014). The significance of YKL-40 protein in liver fibrosis. Inflamm. Res. 63 (4), 249–254. doi:10.1007/s00011-013-0698-9
Tinel, M., Berson, A., Vadrot, N., Descatoire, V., Grodet, A., Feldmann, G., et al. (2004). Subliminal Fas stimulation increases the hepatotoxicity of acetaminophen and bromobenzene in mice. Hepatology 39 (3), 655–666. doi:10.1002/hep.20094
Wang, C., Ma, C., Gong, L., Guo, Y., Fu, K., Zhang, Y., et al. (2021). Macrophage polarization and its role in liver disease. Front. Immunol. 12, 803037. doi:10.3389/fimmu.2021.803037
Wang, S., Hu, M., Qian, Y., Jiang, Z., Shen, L., Fu, L., et al. (2020). CHI3L1 in the pathophysiology and diagnosis of liver diseases. Biomed. and Pharmacother. 131, 110680. doi:10.1016/j.biopha.2020.110680
Wang, X., Sun, R., Wei, H., and Tian, Z. (2013). High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: interaction of γδ T cells with macrophages. Hepatology 57 (1), 373–384. doi:10.1002/hep.25982
Wieser, V., Tymoszuk, P., Adolph, T. E., Grander, C., Grabherr, F., Enrich, B., et al. (2016). Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J. Hepatol. 64 (4), 872–880. doi:10.1016/j.jhep.2015.11.037
Wu, H., Guo, C., Liu, Z., Cai, J., Wang, C., Yi, H., et al. (2023). Neutrophils exacerbate acetaminophen-induced liver injury by producing cytotoxic interferon-γ. Int. Immunopharmacol. 123, 110734. doi:10.1016/j.intimp.2023.110734
Xiao, X., Yeoh, B. S., and Vijay-Kumar, M. (2017). Lipocalin 2: an emerging player in iron homeostasis and inflammation. Annu. Rev. Nutr. 37, 103–130. doi:10.1146/annurev-nutr-071816-064559
Xu, M. J., Feng, D., Wu, H., Wang, H., Chan, Y., Kolls, J., et al. (2015). Liver is the major source of elevated serum lipocalin-2 levels after bacterial infection or partial hepatectomy: a critical role for IL-6/STAT3. Hepatology 61 (2), 692–702. doi:10.1002/hep.27447
Ye, D., Yang, K., Zang, S., Lin, Z., Chau, H. T., Wang, Y., et al. (2016). Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 65 (5), 988–997. doi:10.1016/j.jhep.2016.05.041
Yu, J. E., Yeo, I. J., Han, S. B., Yun, J., Kim, B., Yong, Y. J., et al. (2024). Significance of chitinase-3-like protein 1 in the pathogenesis of inflammatory diseases and cancer. Exp. Mol. Med. 56 (1), 1–18. doi:10.1038/s12276-023-01131-9
Zhao, H., Huang, M., and Jiang, L. (2023). Potential roles and future perspectives of chitinase 3-like 1 in macrophage polarization and the development of diseases. Int. J. Mol. Sci. 24 (22), 16149. doi:10.3390/ijms242216149
Zimmermann, H. W., and Tacke, F. (2011). Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm. Allergy Drug Targets 10 (6), 509–536. doi:10.2174/187152811798104890
ALT alanine transaminase
AST aspartate transaminase
ALI acute liver injury
ANIT alpha naphthylisothiocyanate
APAP Acetaminophen
Chi3l1 Chitinase-3-like protein 1
CHI3L1 human chitinase three like protein 1
CCL C-C motif chemokine ligand
CXCL C-X-C motif chemokine ligand CLPs: chitinase like proteins
DAMPs damage-associated molecular patterns
DC dendritic cells
DMEM Dulbecco’s Modified Eagle Medium
FBS fetal bovine serum
GSH glutathione
HMGB1 high-mobility group box one
I.p. intraperitoneally
IL Interleukin
IFN-γ Interferon-γ
KO Chi3l1 knock-out
KC Kupffer cells
Lcn2 lipocalin-2
MDA malondialdehyde
MMP-9 matrix metalloproteinase-9
MPO myeoloperoxydase
NGAL neutrophil gelatinase associated lipocalin
rLcn2 recombinant lipocalin-2
rChi3l1 recombinant Chitinase-3-like protein 1
ROS reactive oxygen species
RPMI 1640 Roswell Park Memorial Institute 1640 medium
TLRs Toll-like receptors
TNF-α Tumor necrosis factor alpha
TBST Tris-buffered saline containing 0.05% Tween-20
WT wild-type
Keywords: CHI3L1, acetaminophen, LCN2, leukocyte recruitment, oxidative stress
Citation: Kim JH, Yeo IJ, Son DJ, Han SB, Yoon DY, Lee DH and Hong JT (2025) Chitinase 3-like protein 1 deficiency ameliorates drug-induced acute liver injury by inhibition of neutrophil recruitment through lipocalin-2. Front. Pharmacol. 16:1548832. doi: 10.3389/fphar.2025.1548832
Received: 20 December 2024; Accepted: 06 March 2025;
Published: 24 March 2025.
Edited by:
Lin Yuan, Hunan University, ChinaReviewed by:
Longwei He, University of South China, ChinaCopyright © 2025 Kim, Yeo, Son, Han, Yoon, Lee and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Hun Lee, ZGh1bkBqbnUuYWMua3I=; Jin Tae Hong, amludGhvbmdAY2h1bmdidWsuYWMua3I=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.