 Wei Qin
Wei Qin Ping Nie
Ping Nie Xuejun Hui
Xuejun Hui Manyu Luo
Manyu Luo- Department of Nephropathy, The Second Hospital of Jilin University, Changchun, Jilin, China
Diabetic kidney disease is one of the common complications in diabetic patients and has gradually become an important pathogenic factor in chronic kidney disease. Therefore, studying the mechanisms of its occurrence and development is of great significance for the prevention and treatment of diabetic kidney disease. Some researchers have pointed out that there is a phenomenon of hypoxia in diabetic kidney tissue and believe that hypoxia-inducible factor-1α is closely related to the occurrence and progression of diabetic kidney disease. Additionally, the homeostasis of zinc plays a key role in the body’s adaptation to hypoxic environments. However, the specific relationship among these three factors remains unclear. This article provides a detailed review of the multiple roles of hypoxia-inducible factor-1α in the pathogenesis of diabetic kidney disease, including: regulating angiogenesis, increasing the expression of erythropoietin, modulating oxidative stress through the PI3K/AKT and HIF-1α/HO-1 pathways, promoting inflammatory cell infiltration and the release of inflammatory factors to induce inflammatory responses, facilitating epithelial-mesenchymal transition, pathological angiogenesis, and promoting the release of fibrotic factors, ultimately leading to renal fibrosis. Furthermore, HIF-1α also participates in the occurrence and development of diabetic kidney disease through mechanisms such as regulating apoptosis, inducing mitochondrial autophagy, and vascular calcification. At the same time, this article clarifies the regulatory role of the trace element zinc on hypoxia-inducible factor-1α in diabetic kidney disease. This article provides references and insights for further research on the pathogenesis and progression of diabetic kidney disease.
Introduction
With improvements in economic level and lifestyle changes, the prevalence of diabetes is increasing. The global prevalence of diabetes in 2021 was estimated at around 536 million individuals, representing 10.5% of the total world population. Projections indicate that by the year 2045, this figure is expected to rise to 783 million, resulting in a prevalence rate of 12.2% (Sun et al., 2022). China currently holds the distinction of having the largest population of diabetes patients globally, representing approximately 25% of the worldwide diabetic demographic. As reported by the International Diabetes Federation, projections indicate that by the year 2045, the number of individuals diagnosed with diabetes in China may rise to 170 million, resulting in a prevalence rate of up to 12.5%. Notably, over 90% of diabetes cases in the country are classified as type 2 diabetes mellitus (T2DM). In individuals with diabetes, alterations in glucose metabolism elevate the metabolic demands associated with glucose, resulting in disruptions in renal hemodynamics as well as the influence of local hormones and cytokines within the kidneys. These elements collectively play a significant role in the development of diabetic kidney disease within the diabetic population. According to Fenta et al. , the global prevalence of chronic kidney disease (CKD) among patients with T2DM is 27% (Fenta et al., 2023). In China, the prevalence of DKD among adult patients with T2DM is reported to be 21.8% (Zhang et al., 2020). The rising prevalence of diabetes may be stabilizing, yet the increasing number of diabetes patients will continue to expand the future population of those afflicted by DKD. A national sampling survey reveals that only 40% of diabetes patients in China are cognizant of their condition, while two-thirds have not received any form of treatment. Furthermore, among those who are undergoing treatment, approximately 50% exhibit inadequate disease control (Wang et al., 2021). The considerable prevalence, limited awareness, and low rates of treatment success for diabetes in China, along with the gradual progression of the disease, its various complications, and its chronic characteristics, render diabetes a critical public health concern within the nation.
The etiology of diabetes is intricate and remains incompletely elucidated. Among the principal types of diabetes, T2DM is characterized by insufficient insulin secretion from pancreatic β-cells or by insulin resistance, resulting in elevated blood glucose levels. This, in turn, leads to an increase in harmful factors such as advanced glycation end products (AGEs), lipid metabolites, peroxides, inflammatory mediators, and iron. These damaging agents exert their influence on multiple target organs, including the kidneys, heart, and retina, prompting pathological alterations such as apoptosis in the cells of these organs, ultimately culminating in functional impairment of the affected tissues. DKD represents a significant microvascular complication associated with T2DM. Clinically, it is characterized by persistent albuminuria, a gradual deterioration in glomerular filtration rate, and in advanced cases, progression to end-stage renal disease (ESRD). The pathogenesis of DKD is also complex. It is currently posited that the development of DKD is attributable to a combination of hemodynamic alterations, metabolic disturbances, and their interactions, which culminate in oxidative stress and inflammatory responses within the renal system (Tuttle et al., 2022). Hypoxia is detectable in the renal medulla during the initial phases of disease advancement in DKD. Using oxygen microelectronics, Palm found that PO2 in the renal tissue of diabetic rats was much lower than that in normal mice (Palm et al., 2003). Ries et al. used blood oxygenation level-dependent magnetic resonance imaging (BOLD-MRI) to determine that deoxyhemoglobin signals increased in the outer medullary region of diabetic animals (Ries et al., 2003). Researchers observed through BOLD-MRI that 2 days post-induction of diabetes, the oxygen partial pressure in the kidneys of diabetic rats significantly decreased, with worsening hypoxia over time (Dos et al., 2007). Concurrently, in patients with DKD, a continuous decline in cortical PO2 was noted as the disease progressed (Yin et al., 2012). A study in China indicated that the prevalence of obstructive sleep apnea syndrome among hospitalized patients with T2DM could be as high as 60.1% (Zhang P et al., 2016). As a primary pathophysiological mechanism of obstructive sleep apnea syndrome, intermittent hypoxia may be common among diabetic patients. However, due to the current limitations of technologies such as BOLD-MRI in clinical applications, there is still relatively little data regarding hypoxia in patients with DKD. Therefore, there is an urgent need for larger sample size studies and longer follow-up research to further explore the hypoxic conditions in patients with DKD. Beyond the kidneys, diabetes can also induce hypoxia in other organs of the body, such as the retina (Arden and Sivaprasad, 2011), islets (Sato et al., 2011), adipose (Lee et al., 2014) and other tissues.
A substantial body of research has established that hypoxia is associated with multiple factors, including hemodynamic alterations (Damiani et al., 2021), metabolic dysfunctions (Wheaton and Chandel, 2011), oxidative stress and inflammatory responses (McGarry et al., 2018). This indicates that renal hypoxia serves as a fundamental pathogenic mechanism in the onset and advancement of DKD. The human body utilizes a variety of mechanisms to adapt to hypoxic conditions. In instances of acute hypoxia, sodium ions function as second messengers, influencing the processes of mitochondrial oxidative phosphorylation and altering the fluidity of the mitochondrial inner membrane, as well as the transmission of redox signals. These changes subsequently impact cellular metabolism, facilitating adaptation to the acute hypoxic state (Hernansanz-Agustín et al., 2020). In contrast, prolonged and chronic hypoxia is primarily regulated by hypoxia-inducible factor-1 (HIF-1). HIF-1α, the principal regulatory component for hypoxic adaptation, plays a significant role in the pathogenesis and progression of DKD through several mechanisms, including the induction of angiogenesis, upregulation of erythropoietin (EPO) expression, suppression of oxidative stress and inflammatory responses, regulation of apoptosis, promotion of mitochondrial autophagy, and inhibition of fibrosis and vascular calcification. Given its potential protective effects on the kidneys, HIF-1α has emerged as a focal point of research in recent years.
The hypoxic conditions observed in the kidneys may arise from multiple factors (Figure 1). In the context of elevated glucose levels, the metabolism of glucose via the polyol pathway is enhanced, leading to an increased ratio of reduced to oxidized nicotinamide adenine dinucleotide. This alteration contributes to a state referred to as “pseudohypoxia” within the organism (Williamson et al., 1993). Elevated sugar levels can trigger hyperfiltration in healthy glomeruli by enhancing the reabsorption of glucose and sodium in renal tubular epithelial cells via the sodium-glucose co-transporter (SGLT) located in the proximal tubule, which results in significant oxygen consumption. In individuals with DKD, ongoing inflammatory responses can release a substantial quantity of inflammatory factors, thereby elevating the basal metabolic rate and contributing to increased oxygen consumption within the body (Hansell et al., 2013). In addition to the increased consumption of oxygen, the reduction in oxygen supply is also a contributing factor to renal hypoxia. In the initial phases of DKD, impairment of the primary renal microcirculation can result in diminished blood flow and reduced oxygen delivery to the kidneys (Eckardt et al., 2005). Furthermore, the activation of SGLT enhances sodium reabsorption, which subsequently decreases the sodium concentration in the macula densa of the distal renal tubule. This decrease triggers an increase in the release of renin and angiotensin II (Ang II) (Yin et al., 2012; Zhu et al., 2011). The resultant constriction of the afferent arterioles by Ang II further compromises oxygen supply within the renal tubular interstitium (Hesp et al., 2020). Prolonged hypoxia in the kidneys of DKD patients can lead to damage in the renal tubular interstitium, causing an imbalance in the synthesis and degradation of the extracellular matrix (ECM), ultimately resulting in renal fibrosis. This fibrosis restricts oxygen transport and exacerbates the reduction in oxygen supply. Mitochondrial damage or dysfunction can exacerbate hypoxic injury. Mitochondrial dysfunction and reduced adenosine triphosphate (ATP) production can be observed even before the histological changes of diabetic kidney disease manifest (Imasawa et al., 2017). In essence, the development of a hypoxic microenvironment within renal tissue is primarily attributed to increased oxygen consumption coupled with decreased oxygen availability (Friederich-Persson et al., 2013). To adapt to hypoxic conditions, cells have evolved intricate mechanisms, with HIF-1α being among the first to respond to low oxygen levels, mediating the expression and regulation of a variety of downstream genes (Maxwell, 2003).
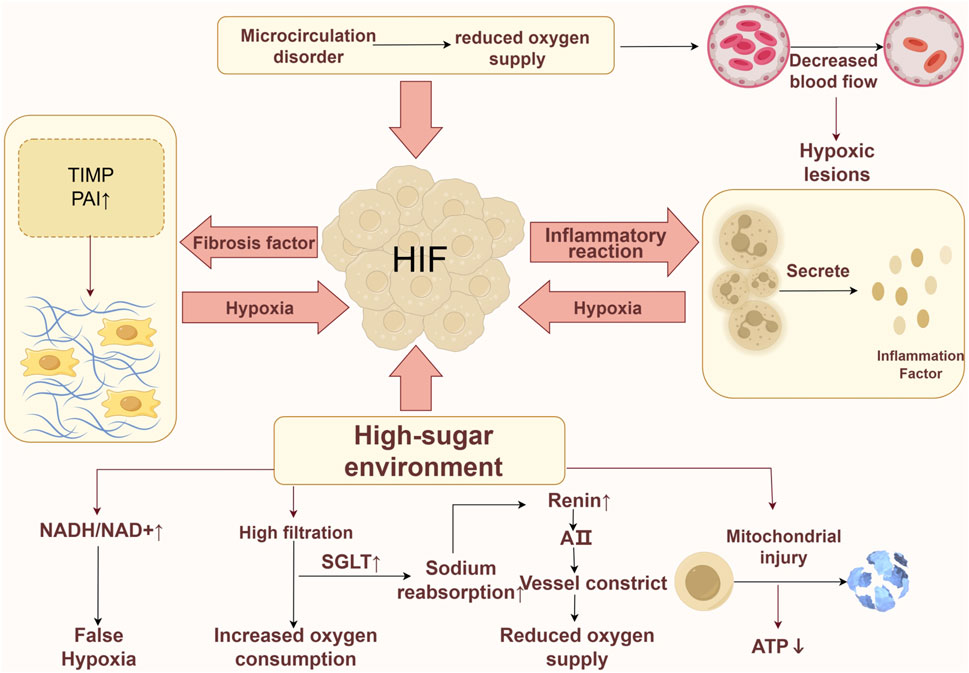
Figure 1. Hypoxia mechanism in DKD. The formation of a hypoxic environment can be induced by various factors. Firstly, in a high-glucose environment, the occurrence of pseudohypoxia is attributed to an imbalance in the NADH/NAD + ratio, while the excessive activation of SGLT leads to an increased consumption of oxygen and affects the release of Ang II, further impacting oxygen supply. Additionally, a high-glucose environment may impair mitochondria, resulting in reduced ATP production. Secondly, a vicious cycle is established among hypoxia, renal fibrosis, and inflammatory responses, exacerbating cellular hypoxia. Lastly, primary microcirculatory damage can lead to a decrease in microcirculatory blood flow, thereby reducing oxygen supply.
Numerous studies have demonstrated that trace elements may significantly influence the onset and progression of diseases associated with hypoxic conditions. Research suggests that the maintenance of zinc homeostasis is critical for the body’s ability to tolerate low oxygen environments (Guo et al., 2024b). Both animal and human subjects exposed to hypoxia have been observed to experience a reduction in plasma zinc levels (Guo et al., 2024a; Rawal et al., 1999; Vats et al., 2001), and appropriate zinc supplementation may mitigate symptoms related to hypoxia intolerance. Zinc is integral to the cellular regulation of HIF-1α. Evidence indicates that zinc can modulate the activity of HIF-1α through various mechanisms, underscoring the importance of intracellular zinc levels and homeostasis in preserving the normal function of HIF-1α (Nardinocchi et al., 2010). Consequently, zinc is pivotal in regulating the HIF-1α signaling pathway, which is crucial for cellular adaptation to hypoxic conditions. The pathogenesis of DKD is intricate, and the role of HIF-1α in mediating renal injury, along with its regulatory mechanisms, remains unclear. Further investigation is warranted to elucidate the regulatory effects of zinc on the HIF-1α signaling pathway in the context of DKD. This article reviews the research advancements surrounding HIF-1α and DKD, while exploring the clinical prospects of zinc as a therapeutic agent via the HIF-1α pathway for DKD, with the aim of providing reference and insight for future research.
Structure and biological characteristics of HIF-1α
HIF-1 mainly exists in the form of a heterogeneous dimer, consisting of HIF-1α subunit with sensitive oxygen concentration changes and HIF-1β subunit with stable expression (Maxwell, 2003). The primary physiological function of HIF-1 is regulated by HIF-1α. Under normal oxygen conditions, proline residues on the HIF-1α protein are hydroxylated by prolyl hydroxylase (PHD), and hydroxylated HIF-1α is connected by von Hippel-Lindau (VHL) ubiquitin. HIF-1α is identified, ubiquitinated, and finally hydrolyzed by the proteasome. Under normal oxygen conditions, HIF-1α could not be detected. In the hypoxic state, the activity of oxygen-dependent PHD is inhibited and the degradation of HIF-1α is blocked, resulting in the accumulation of HIF-1α. HIF-1α is transferred to the nucleus and HIF-1β dimerizes to form HIF-1. After combining with the hypoxia response element (HRE), the transcription of multiple downstream target genes can be regulated (Catrina and Zheng, 2021). Additionally, the expression of HIF-1α is not only affected by the hypoxic environment, but is also related to the regulation of insulin, insulin-like growth factors, AGEs, and blood sugar levels (Figure 2).
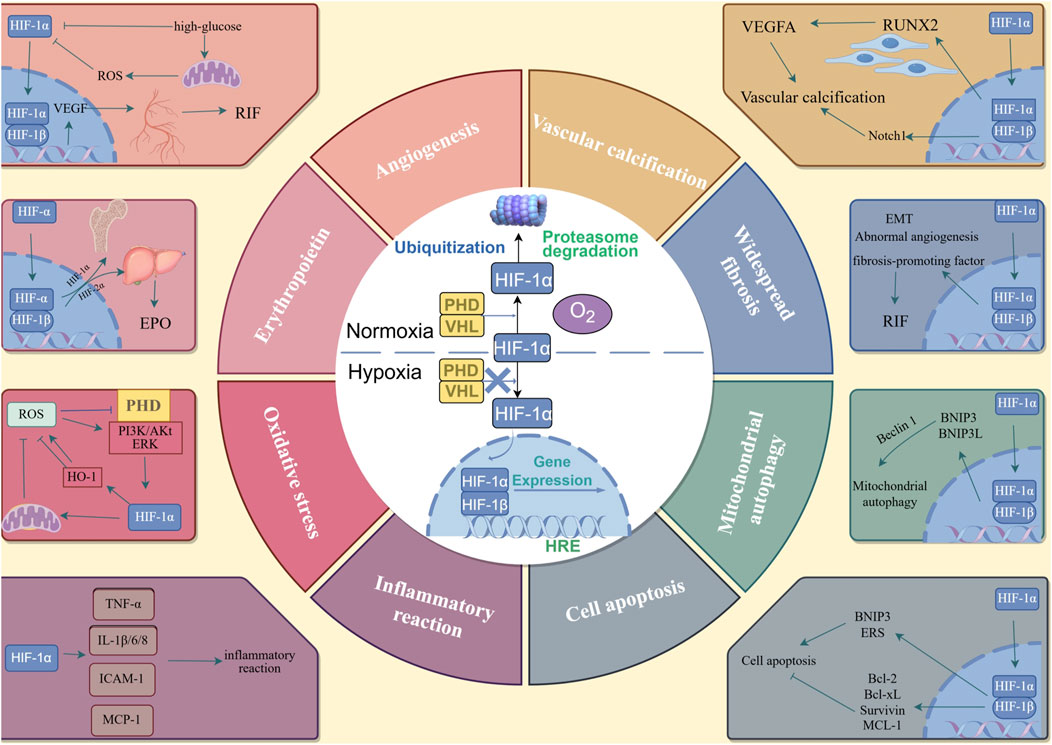
Figure 2. The metabolism of HIF-1α and the mechanism of HIF-1α in DKD. In the nomoxic conditions, proline residues on the HIF-1α protein are hydroxylated by PHD, and the hydroxylated HIF-1α is connected by VHL ubiquitin. HIF-1α is identified and ubiquitized, and finally hydrolyzed by protesome. In the hypoxia conditions, the activity of PHD is inhibited, and the degradation of the HIF-1α is blocked, HIF-1α is transferred to the nucleus and HIF-1β dimerized to form HIF-1. After combining with HRE, the transcription of multiple downstream target genes can be adjusted. The downstream genes can lead to angiogenesis; increased expression of EPO; oxidative stress; inflammatory reaction; cell apoptosis; mitochondrial autophagy; widespread fibrosis and vascular calcification.
The mechanism of HIF-1α in DKD
Angiogenesis
Under hypoxic conditions, HIF-1α can increase the formation of new blood vessels in renal tissue cells (Peng et al., 2023). Studies have found that the content of vascular endothelial growth factor (VEGF) in the serum of diabetic patients is significantly higher than that in normal individuals, and is positively correlated with the content of serum HIF-1α (Katavetin et al., 2006).
The VEGF family and its receptors are important cellular signaling molecules that play a significant role in the regulation of neovascularization. As one of the downstream target genes of HIF-1α, hypoxia can promote nuclear aggregation of HIF-1α in human microvascular endothelial cells (Hahne et al., 2018).
In many instances, the adaptation to hypoxia is not achievable, and as the condition advances, the phenomena of capillary rarefaction and hypoxia in DKD continue unabated. This persistence may be attributed to the combined effects of hypoxia and elevated glucose levels on renal function in the context of DKD. The hyperglycemic environment can influence the transcription of VEGF in renal tissue cells by altering the stability and activity of HIF-1α (Cerychova and Pavlinkova, 2018). Under conditions of high glucose and hypoxia, there is an overactivation of reactive oxygen species (ROS) radicals, which leads to a reduction in VEGF expression and vascularization. This, in turn, triggers a compensatory response characterized by a decrease in hypoxia-induced angiogenesis. Concurrently, the high-glucose environment diminishes cellular responsiveness to hypoxia, resulting in decreased expression of HIF-1α target genes and a significant reduction in VEGF levels within the glomeruli of diabetic patients (Chou et al., 2002). This decline in VEGF expression contributes to podocyte apoptosis, which serves as an intermediary link between thrombotic microvascular disease and DKD (Hernández-Arteaga et al., 2018).
It is crucial to recognize that inappropriate activation of HIF-1α can have detrimental effects. Research conducted by Hakroush et al. demonstrated that the upregulation of VEGF expression in renal tubules led to capillary proliferation and an increase in fibroblast activity surrounding these tubules, which subsequently resulted in the accumulation of ECM and the development of renal fibrosis (Hakroush et al., 2009). Additionally, investigations involving VHL knockout transgenic mice revealed that elevated levels of HIF-1α and increased expression of the downstream target gene VEGF were associated with significant endothelial cell proliferation around the renal tubules, as well as heightened interstitial fibrosis within the renal tubules (Theilig et al., 2011). These findings indicate that excessive activation of HIF-1α may facilitate capillary proliferation in the vicinity of renal tubules and stimulate fibroblast proliferation through the overexpression of VEGF, leading to the accumulation of newly formed ECM in the interstitial spaces between renal tubules and capillary loops. This process ultimately results in the formation of fibrous tissue characterized by a dense network of capillaries surrounding cysts, contributing to the progression of renal fibrosis.
Increased expression of EPO
EPO is a glycoprotein produced by endothelial cells in the kidneys and fibroblasts located in the peritubular interstitium. EPO interacts with EPO receptors on erythroid progenitor cells, facilitating their proliferation, differentiation, and maturation, while also enhancing the release of reticulocytes. The long-term hypoxic state of the kidneys in diabetic patients can induce the transformation of EPO-producing cells into α-smooth muscle actin (α-SMA) positive myofibroblasts, resulting in the loss of hypoxia-induced EPO expression. Patients suffering from DKD may experience anemia due to inadequate EPO production and a deficiency of hematopoietic components. In comparison to anemia associated with other forms of chronic kidney disease (CKD), the anemia linked to DKD manifests earlier, is more severe, and is associated with a poorer prognosis (Loutradis et al., 2016).
Research indicates that HIF-PHD inhibitors (HIF-PHI) can impede the hydroxylation of HIF-α by PHD, thereby diminishing the degradation of HIF-α. The resultant accumulation of HIF-α within cells facilitates the formation of the heterodimer HIF-1, which subsequently binds to HRE and stimulates the expression of downstream target genes, such as EPO and its receptor, thus enhancing erythropoiesis (Bernhardt et al., 2010). The HIF-β subunit is consistently expressed across various cell types, and its intracellular levels and biological activity remain unaffected by external hypoxic conditions, consequently, hypoxia primarily modulates EPO gene expression through HIF-α. There exist three isoforms of HIF-α: HIF-1α, HIF-2α, and HIF-3α (Holmquist-Mengelbier et al., 2006). Notably, HIF-1α is ubiquitously expressed in nearly all cell types, with a predominant presence in renal tubular cells. In contrast, HIF-2α is localized specifically to renal endothelial cells and fibroblasts adjacent to the renal tubules. Animal studies have revealed that the knockout of the HIF-2α gene in mice leads to a reduction in red blood cell count, hemoglobin levels, and hematocrit. Notably, PHD inhibitors did not exert any effects on mice deficient in HIF-2α (Gruber et al., 2007), indicating that HIF-2α serves as the principal regulator of EPO production under both physiological and stress-related conditions.
In comparison to HIF-2α, HIF-1α plays a more critical role in the preservation of hematopoietic stem cells within the hypoxic microenvironment of the bone marrow. Studies have demonstrated that hematopoietic stem cells cultured in conditions of extreme hypoxia can maintain the stable expression of HIF-1α (Takubo et al., 2010). Conversely, hematopoietic stem cells derived from HIF-1α deficient mice display notable functional impairments.
The above research indicates that HIF-1 can promote erythropoiesis by inducing the expression of endogenous EPO, thereby ameliorating renal anemia. Currently, HIF-PHI has entered the clinical research phase and is anticipated to become a novel therapeutic agent for the treatment of anemia.
Oxidative stress
The activation of HIF-1α is intricately linked to oxidative stress. ROS play a crucial role in maintaining the stability of HIF-1α under normoxic conditions, as physiological concentrations of ROS serve as signaling molecules that modulate various physiological processes within the organism. In conditions of physiological hypoxia, the mitochondrial electron transport chain may be impaired due to inadequate oxygen availability, resulting in the excessive generation of ROS. Specifically, hydrogen peroxide, a component of ROS, can inhibit the enzymatic activity of PHD by activating signaling pathways mediated by small GTPases such as Rac or Rho. This inhibition leads to the stabilization of HIF-1α and the subsequent activation of transcription for its downstream target genes (Rabbani et al., 2010). Furthermore, ROS can prevent the degradation of HIF-1α by inhibiting PHD activity and activating the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) signaling pathway, as well as the extracellular signal-regulated kinase pathway, thereby elevating HIF-1 levels (Iacobini et al., 2022).
HIF-1 exhibits a negative feedback mechanism on the production of ROS. Under typical conditions, HIF-1 mitigates ROS generation through several pathways. Primarily, HIF-1 modifies energy metabolism in hypoxic environments via metabolic reprogramming, which subsequently diminishes ROS production from the mitochondrial electron transport chain. Additionally, HIF-1 promotes selective autophagy of mitochondria, facilitating the removal of damaged and senescent mitochondria, thereby further reducing ROS output. Moreover, HIF-1α can inhibit ROS production by obstructing the activity of the electron transport chain during mitochondrial respiration.
Additionally, HIF-1 regulates ROS production through the HIF-1/heme oxygenase-1 (HO-1) pathway. HO-1 is a stress-inducible enzyme that catalyzes the breakdown of heme into carbon monoxide, iron, and biliverdin. As a downstream target gene of HIF-1, the expression of HIF-1α in skeletal muscle and fibroblasts cells is elevated under hypoxic conditions, which enhances the transcription of HO-1 (Dunn et al., 2021) and provides cytoprotection by facilitating the clearance of ROS (Rosenberger et al., 2008). However, excessive heme degradation by HO-1 may lead to the accumulation of its degradation products within the cell, resulting in iron overload, which can exacerbate oxidative stress and lipid peroxidation, ultimately increasing ROS production (Feng et al., 2021). Additionally, HIF-1 can influence downstream target genes to enhance the transcription of nicotinamide adenine dinucleotide phosphate oxidase 4, thereby further promoting ROS generation (Diebold et al., 2010).
Chronic oxidative stress may influence the generation of HIF-1α through epigenetic modifications. Epigenetic regulation encompasses DNA methylation, histone modifications, and the modulation of non-coding RNAs. Research has shown that ROS can affect DNA methylation (Wu and Ni, 2015). Methylation of superoxide dismutase (SOD) can activate HIF-1α, thus participating in the regulation and function of the HIF-1 signaling pathway. Additionally, under hypoxic conditions, HIF-1α can also regulate epigenetic modifications (Choudhry and Harris, 2018). This suggests that epigenetic regulation may be a crucial mechanism in the modulation and function of HIF-1 in the hypoxic response associated with diabetes (Pang et al., 2021). Therefore, considering it as a potential therapeutic target merits further investigation.
Inflammatory reaction
Under conditions of high-glucose and hypoxia, cells generate excessive ROS, which serve as a significant contributor to inflammation. Oxidative stress in glomerular mesangial cells stimulates the secretion of adhesion molecules, including intercellular cell adhesion molecule-1 and monocyte chemoattractant protein-1 (MCP-1), as well as inflammatory mediators such as nuclear factor-κB (NF-κB). Thioredoxin-interactingprotein (TXNIP) serves as a crucial regulatory factor in the activation of the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome within the organism. It facilitates the dissociation of thioredoxin and TXNIP through the action of ROS, thereby enhancing the activation of the NLRP3 inflammasome by TXNIP. This process ultimately results in the production and release of interleukin-1β (IL-1β), which initiates an inflammatory response (Yang et al., 2020). While the inflammatory response functions as a protective mechanism against external stimuli, chronic inflammation can result in tissue damage. The heightened metabolic demands of numerous infiltrating inflammatory cells within renal tissue can induce hypoxia in the renal microenvironment, which in turn can activate endogenous inflammatory responses. This interaction establishes a detrimental cycle between hypoxia and inflammation, ultimately resulting in renal injury.
In individuals with DKD or in relevant animal models, there is a marked increase in the expression of pro-inflammatory genes, which correlates with elevated levels of inflammatory cytokines in peripheral blood (Liu et al., 2018). Histological examination reveals a substantial presence of lymphocytes, macrophages, and mast cells in the kidneys of patients with DKD, these immune cells are capable of secreting significant quantities of inflammatory mediators and cytokines that contribute to renal damage, either directly or indirectly (Navarro-González et al., 2011). Prior research has identified key inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukins 1 (IL-1), interleukin-6 (IL-6), and interleukin-18 (IL-18), MCP-1, NF-κB, and NLRP3 inflammasome, as critical players in the inflammatory processes associated with DKD (Wada and Makino, 2013; Wu et al., 2024; Zhu et al., 2024). This body of evidence underscores the significance of the inflammatory response in the pathogenesis and progression of DKD.
HIF-1α is implicated in the inflammatory response associated with DKD by facilitating the production of various cytokines. In hyperglycemic conditions, HIF-1α expression is upregulated, which enhances the inflammatory response in mesangial cells and markedly increases the levels of pro-inflammatory factors such as TNF-α, IL-1β, and IL-6, thereby exacerbating inflammatory damage to vascular endothelial cells. Zhao et al. demonstrated that human umbilical vein endothelial cells exposed to high glucose in a hypoxic environment for 48 h exhibited an 8.84-fold increase in HIF-1α mRNA expression compared to cells exposed solely to high glucose. Additionally, the expression of inflammatory factors IL-6, IL-8, intercellular cell adhesion molecule-1, and MCP-1 mRNA was significantly elevated, an effect that could be attenuated by the HIF-1 inhibitor KC7F2, indicating that HIF-1 exacerbates renal damage by promoting the expression of inflammatory factors during DKD (Zhao et al., 2021). Furthermore, the application of YC-1, a specific HIF-1α inhibitor, has been shown to enhance the inflammatory response in tubular epithelial cells and increase the production of cytokines IL-1β and IL-18 (Lee et al., 2018).
In summary, HIF-1α exacerbates vascular endothelial damage by promoting the infiltration of inflammatory cells and the release of inflammatory factors, creating a vicious cycle of hypoxia and inflammation in the kidneys that contributes to the progression of DKD (Table 1). Therefore, inducing the degradation of HIF-1α may emerge as an effective strategy to disrupt this vicious cycle, warranting exploration as a potential therapeutic target for DKD.
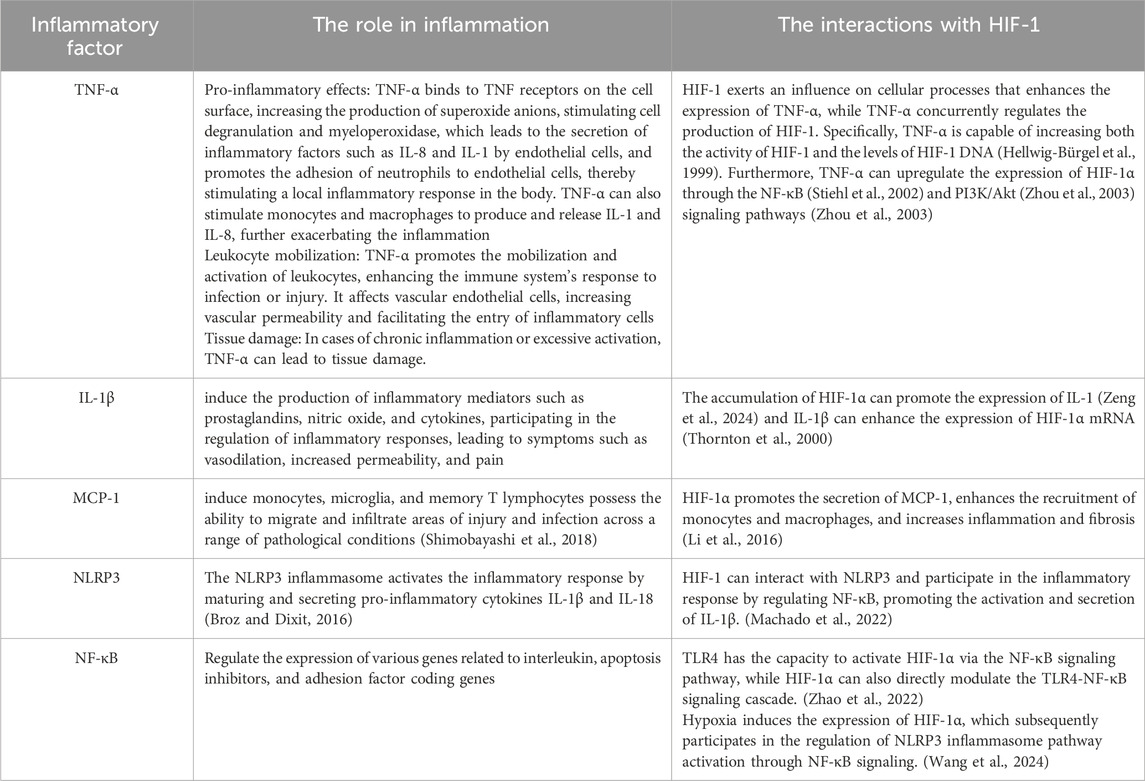
Table 1. The role of inflammatory factors in inflammation and their interactions with HIF-1.
Widespread fibrosis
A prolonged inflammatory response induces the infiltration of inflammatory cells into the kidneys, which in turn activates interstitial kidney cells, prompting them to release inflammatory molecules. This cascade incites vascular remodeling, ECM deposition, and mesangial proliferation, all of which contribute to the progression of renal fibrosis. Renal interstitial fibrosis (RIF) represents the primary pathological foundation and the ultimate common pathway for the progression of CKD to ESRD. This condition is characterized by the activation of fibroblasts and a substantial accumulation of ECM, which detrimentally impacts both the glomeruli and the overall kidney structure. Notably, the tubular basement membrane undergoes thickening, and there is an increase in the mesangial matrix, ultimately resulting in glomerulosclerosis and RIF. Research utilizing animal models has demonstrated that the expression of HIF-1α is elevated in the kidneys and ECM of diabetic model rats (Tang et al., 2012). In the context of DKD, hypoxic conditions can enhance the expression of HIF-1α. The sustained expression of HIF-1α may lead to glomerular hypertrophy and an increase in ECM, thereby facilitating the fibrotic process, generating significant scar tissue, disrupting the original tissue architecture of the kidneys, diminishing blood supply, and ultimately resulting in irreversible kidney dysfunction. In severe instances, this may culminate in renal failure.
The potential mechanisms through which HIF-1α contributes to RIF are as follows:
Epithelium-mesenchymal transition (Figure 3A).
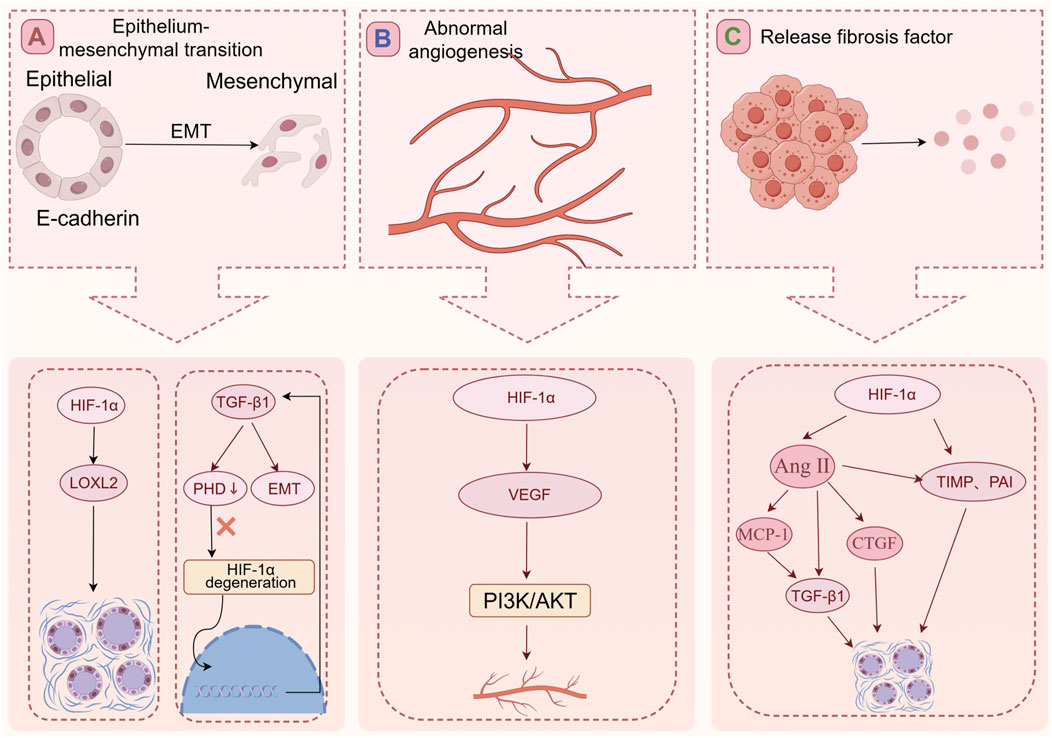
Figure 3. The mechanism of RIF. (A) HIF-1α can increase the expression LOXL2 to promote fibrosis. The TGF-β1 reduces the expression of PHD2 resulting in HIF-1α accumulation, and this PHD2/HIF-1α signaling pathway mediates TGF-β1-induced EMT in renal tubular cells. (B) The HIF-1α/VEGF signaling pathway of glomerular endothelial cells is activated, and the increased expression of VEGF can promote activation of signal path PI3K/AKT. The activation of the signal pathway leading to abnormal angiogenesis, exacerbating glomerular fibrosis, and ultimately the loss of glomerular function. (C) In a high-glucose and hypoxia environment, HIF-1α expression increases, leading to the activation of fibrosis-promoting factors such as TIMP and PAI. HIF-1α can also activate Ang II. Ang II further stimulates TGF-β1 expression and promotes pro-fibrotic factors, including CTGF, contributing to RIF by increasing ECM synthesis.
Epithelial-mesenchymal transition (EMT) is a biological process involving the dedifferentiation of epithelial cells, which is marked by the loss of cell polarity and specific epithelial markers, reorganization of the cytoskeleton, and the acquisition of a mesenchymal phenotype. This transformation enables epithelial cells to become fibroblast-like cells that are capable of secreting ECM components, which may accumulate in damaged renal tissues. A hyperglycemic and hypoxic environment can directly induce EMT in renal tubular epithelial cells and simultaneously enhance the expression of HIF-1α. The reduction of HIF-1α levels may mitigate EMT, indicating that HIF-1α is a critical factor in the EMT associated with DKD (Yang et al., 2021).
Research has shown that the activation of HIF-1α can enhance the expression of lysyl oxidase-like 2 (LOXL2), thereby facilitating the transition of epithelial cells to an interstitial phenotype, a process that contributes to fibrosis (Higgins et al., 2007). LOXL2, a member of the lysine oxidase family, is integral to the covalent crosslinking of collagen and elastin in the ECM, which is essential for the induction of EMT. Research has demonstrated that Rho-kinase inhibitors facilitate the degradation of HIF-1, inhibit the dimerization of HIF-1α with HIF-1β, and suppress the expression of their target genes (Matoba et al., 2013). Consequently, this leads to a decrease in the accumulation of glomerular mesangial matrix in diabetic patients and alleviates glomerular fibrosis. Silent information regulator 2 (Sirt1) has been shown to inhibit the activation of HIF-1α, resulting in a reduction of HIF-1α expression and a subsequent decrease in inflammation and fibrosis of glomerular cells under hyperglycemic conditions (Huang et al., 2015). Additionally, YC-1, a selective inhibitor of HIF-1α, has been found to significantly improve renal function and mitigate pathological injury in diabetic mice, as well as reduce the severity of renal fibrosis. Researchers have observed that the transfection of renal tubular cells with HIF-1α siRNA significantly inhibits transforming growth factor-β1 (TGF-β1) induced EMT. Furthermore, they have demonstrated that TGF-β1 can reduce PHD2 expression through a Smad-dependent mechanism, resulting in the accumulation of HIF-1α and consequently inducing EMT in renal tubular cells (Han et al., 2013).
The aforementioned study reveals that in DKD, HIF-1α induces EMT through pathways that promote the upregulation of LOXL2 and TGF-β1, leading to increased ECM generation and fostering RIF.
Abnormal angiogenesis (Figure 3B).
In DKD as we described above, abnormal neovascularization predominantly involves the formation of immature blood vessels, which can lead to renal fibrosis and, ultimately, a decline in glomerular function. The VEGF family and its associated receptors serve as critical signaling molecules that govern the process of neovascularization. Under conditions of elevated glucose, the HIF-1α/VEGF signaling pathway is activated in glomerular endothelial cells, resulting in an upregulation of VEGF expression. This increase in VEGF can subsequently activate the phosphorylation of endothelial nitric oxide synthase, which in turn leads to abnormal angiogenesis, exacerbates glomerular fibrosis, and ultimately contributes to the deterioration of glomerular function (Mahdy et al., 2010).
Activate the fibrosis-promoting factor (Figure 3C).
In environments characterized by elevated glucose levels and hypoxia, there is an upregulation of HIF-1α expression. HIF-1α has the capacity to activate downstream fibrosis-promoting factors, including tissue inhibitors of metalloproteinase (TIMP) and plasminogen activator inhibitor (PAI), which subsequently stimulate the synthesis of interstitial collagen and inhibit the degradation of the ECM. Furthermore, Ang II has the capacity to directly stimulate the expression of TGF-β1 molecules, which subsequently enhances TGF-β1 expression through the upregulation of cytokines such as MCP-1 (Zhou et al., 2006). This mechanism facilitates the accumulation of interstitial collagen, ultimately resulting in RIF. Furthermore, Ang II can elevate the expression of connective tissue growth factor (CTGF), a key mediator of fibrosis, through both direct and indirect pathways (Hussain et al., 2008). Additionally, Ang II may induce the expression of other pro-fibrotic factors, including a significant promotion of PAI transcription (Doller et al., 2009) and the activation of TIMP. These actions contribute to an increase in ECM synthesis while simultaneously decreasing its degradation, thereby culminating in the development of RIF. Additionally, both in vivo and in vitro studies have demonstrated that HIF-1α can also activate Ang II, thereby facilitating renal interstitial fibrosis (Tang et al., 2012). Krüppel-like factor 5 (KLF5) is expressed in the proximal tubular cells of fibrotic kidneys and may promote the expression of TGF-β1 through the transcriptional regulation of HIF-1α, thereby contributing to the onset and progression of kidney fibrosis. Studies indicate that, elevated concentrations of MK-8617, a HIF-PHI, have the potential to exacerbate renal tubular interstitial fibrosis through the activation of the HIF-1α/KLF5/TGF-β1 signaling pathway (Li et al., 2019).
In conclusion, HIF-1α exacerbates the RIF process in patients with DKD by inducing EMT, promoting pathological angiogenesis, and elevating the expression of profibrotic factors.
Cell apoptosis
HIF-1α exhibits a dual role in regulating apoptosis, functioning both to promote and inhibit this process. HIF-1α can activate the gene encoding induced nitric oxide synthase, which facilitates the induction of apoptosis in cells. Furthermore, HIF-1α interacts with HRE to enhance the expression of B-cell lymphoma-2 (Bcl-2) interacting protein 3 (BNIP3), a member of the Bcl-2 family, which is also implicated in the promotion of apoptosis. Additionally, endoplasmic reticulum stress (ERS) represents a significant pathogenic mechanism contributing to the onset and progression of DKD. Moderate ERS contributes to the restoration of cellular homeostasis. However, excessive activation of ERS results in apoptosis. In cardiomyocytes, HIF-1α has the capacity to activate ERS, thereby facilitating intermittent hypoxia and triggering apoptotic processes (Moulin et al., 2022). This intervention enhances the ERS response in diabetic mice, thereby mitigating renal pathological damage and delaying the deterioration of renal function. The mechanism underlying HIF-1α-induced ERS may involve the activation of ERS through its transcriptional activity, which promotes the expression of genes associated with apoptosis (Sun et al., 2020). It has also been shown that HIF-1α can directly participate in the protein kinase R-like endoplasmic reticulum kinase (PERK) pathway (Moulin et al., 2020).
As a significant regulator of apoptosis that operates downstream of HIF-1α, the anti-apoptotic protein Bcl-2 is subject to direct transcriptional regulation by HIF-1α. This includes members of the Bcl-2 family, such as B-cell lymphoma-extra large (Bcl-xL), as well as myeloid cell leukemia-1 and Survivin, which belongs to the inhibitor of apoptosis protein family (Chen et al., 2009). Dexmedetomidine can increase the level of HIF-1α after renal ischemia-reperfusion injury in diabetic rats, activate the anti-apoptotic protein Bcl-2, and inhibit the apoptotic protein, thus improving cell vitality, reducing apoptosis and protecting kidney function (Ji et al., 2019).
Mitochondrial autophagy
Mitochondria engage in selective self-removal through the process of autophagy to regulate their content and ensure quality control, thereby preserving mitochondrial homeostasis. A reduction in mitochondrial autophagy levels has been documented in mouse renal tubular epithelial cells subjected to high glucose conditions, as well as in kidney biopsy specimens from patients diagnosed with DKD (Zhan et al., 2015). This observation underscores the significant role of mitochondrial autophagy in the pathogenesis of DKD.
In experimental settings, researchers employed cobalt chloride to create a hypoxic environment in renal tubular epithelial cells. The application of a HIF-1α inhibitor resulted in decreased expression of HIF-1α and exacerbated mitochondrial damage within these cells (Jiang et al., 2020), suggesting that HIF-1α may confer protective effects on mitochondria under hypoxic conditions. Notably, hypoxia is associated with an increase in HIF-1α expression, which in turn enhances the expression of autophagy-related genes, including LC3, Atg5, Beclin 1, Atg7, and Bcl-2/BNIP3 (Chen et al., 2018). This indicates that HIF-1α may play a crucial role in maintaining mitochondrial homeostasis by promoting mitochondrial autophagy.
Chronic moderate hypoxia has been shown to elevate HIF-1α levels, which subsequently dimerizes with HIF-1β to form the HIF-1 complex. This complex enhances the expression of downstream target genes associated with cell death, such as BNIP3 and its homolog BNIP3L, by directly binding to the HRE located in their promoter regions, thereby inducing mitochondrial autophagy (Li et al., 2022). Furthermore, following the upregulation of BNIP3 expression by HIF-1α, there is also an increase in Beclin 1 levels, which occurs through the disruption of the interaction between Beclin 1 and Bcl-2, further promoting mitochondrial autophagy (Zhang et al., 2008). Under hypoxic conditions, HIF-1α facilitates mitochondrial autophagy by enhancing the expression of its downstream target genes, BNIP3 and BNIP3L, thereby sustaining mitochondrial homeostasis, improving mitochondrial morphology and function, and ultimately providing protection to renal tissues. Additionally, HIF-1α has the potential to upregulate genes encoding proteins involved in the mitochondrial tricarboxylic acid cycle, as well as proteins regulated by autophagy, thereby contributing to enhancements in mitochondrial morphology and functionality (Thomas et al., 2017).
Vascular calcification
Vascular calcification represents a pathological phenomenon akin to advanced arteriosclerosis characterized by ossification. This process affects the endothelium or media and is correlated with heightened cardiovascular morbidity and mortality among individuals with diabetes or ESRD (Dhakshinamoorthy et al., 2017). Vascular calcification is an active, complex, and chronic process characterized by inflammation, oxidative stress, and apoptosis. Vascular smooth muscle cells (VSMCs) are the primary cell types in the vascular interstitium, contributing to arterial medial calcification through the phenotypic conversion of osteotropters (McCarty and DiNicolantonio, 2014). Prolonged hypoxia in the kidneys induces an upregulation of HIF-1α expression in renal tubular and interstitial cells. This increase in HIF-1α levels may directly or indirectly interfere with mineral metabolism in the bloodstream, ultimately contributing to the development of vascular calcification.
Extensive research has established that HIF-1α is integral to the process of vascular calcification. An analysis of computed tomography scan data from 405 patients diagnosed with type 2 diabetes demonstrated a significant increase in serum HIF-1α concentrations correlating with the advancement of coronary calcification, as evidenced by a positive relationship with the coronary artery calcification score. Furthermore, serum HIF-1α levels were found to be independently associated with both the presence and severity of vascular calcification. In vitro studies utilizing human aortic smooth muscle cells cultured under hypoxic conditions revealed a time-dependent induction of ECM calcification mediated by HIF-1α (Zhu et al., 2018). Hypoxic conditions were shown to synergistically enhance vascular calcification in conjunction with inorganic phosphates, primarily through increased expression of HIF-1α (Mokas et al., 2016). These findings highlight the critical role of HIF-1α in the mechanisms underlying vascular calcification.
In CKD rats, there was a notable increase in nuclear staining for HIF-1α. Furthermore, two critical regulatory genes downstream of HIF-1α, namely, vascular endothelial growth factor A and Notch1 protein, also exhibited significant upregulation (Mokas et al., 2016). The inhibition of HIF-1α resulted in a decrease in the levels of vascular endothelial growth factor A and Notch1 proteins, indicating that these proteins may act as downstream mediators of vascular calcification associated with HIF-1α. Additionally, runt-related transcription factor 2 (RUNX2) is identified as a pivotal factor in the reprogramming of VSMCs during vascular calcification. Under both normoxic and hypoxic conditions, the expression of RUNX2 was markedly diminished in cells following the specific knockout of HIF-1α in VSMCs (Guo et al., 2024). This observation suggests that HIF-1α may regulate vascular calcification by enhancing RUNX2 expression in VSMCs, with RUNX2 also interacting with HIF-1α in osteoblasts to modulate its stability and transcriptional activity. Collectively, these findings indicate that HIF-1α can facilitate the osteogenic transformation and calcification of VSMCs, thereby contributing to the vascular calcification process in CKD. Moreover, the activation of HIF-1α may serve as an independent predictor of vascular calcification.
In the early stages of DKD, HIF-1α facilitates the adaptation of renal surrounding tissues and cells to hypoxic environments by regulating the expression of downstream target genes and promoting neovascularization, thereby alleviating ischemia and hypoxia’s detrimental effects on the kidneys and delaying disease progression. However, as DKD progressively worsens, prolonged hyperglycemia suppresses the expression of HIF-1α, leading to diminished protective effects. Additionally, an increasingly pronounced phenomenon of HIF-1α overinduction promoting renal cell apoptosis emerges, further fostering fibrosis, commencing the manifestations of renal fibrosis, exacerbating vascular calcification, and ultimately inflicting severe damage on the structure and function of the kidneys, thereby accelerating disease advancement.
Given the multifaceted roles of HIF-1α in DKD, developing targeted therapeutic strategies or drugs aimed at HIF-1α may represent a promising direction for future treatments of DKD. Currently, a growing number of researchers have commenced in-depth investigations into HIF-1α and its downstream pathways to identify effective approaches for treating DKD.
Li et al. conducted a study demonstrating that the administration of miRNA-122-5p analogs via tail vein injection in rat models of streptozotocin-induced DKD resulted in a notable decrease in renal tubular injury and interstitial fibrosis. This finding suggests that miRNA-122-5p may confer a protective effect on renal tissues in hyperglycemic conditions. In vitro investigations revealed that the transfection of cells exposed to high glucose levels with miRNA-122-5p led to a significant downregulation of factor-inhibiting HIF-1 (FIH-1) expression. Furthermore, in a hyperglycemic environment, the overexpression of FIH-1α was associated with increased cell apoptosis, whereas the introduction of miRNA-122-5p markedly reduced apoptotic rates. Additionally, miRNA-155-5p was found to enhance HIF-1α activity by inhibiting FIH-1 expression, thereby potentially delaying the progression of DKD (Cheng et al., 2022). Liu et al. explored the induction of kidney fibrosis in diabetic mice through intraperitoneal albumin injection, coupled with oral administration of the pyruvate kinase M2 activator TEPP-46. Their findings indicated that TEPP-46 mitigated HIF-1α accumulation and exerted anti-fibrotic effects by activating pyruvate kinase M2 and enhancing pyruvate kinase activity (Liu et al., 2021). Angelamellisy Revelian Ndibalema and colleagues reported that empagliflozin could alleviate damage to renal tubular epithelial cells in high-glucose environments by promoting HIF-1α production (Ndibalema et al., 2020). Pang et al. observed that administering hirudin to DKD mice over a 16-week period inhibited the HIF-1α/VEGF signaling pathway, resulting in reduced ECM deposition and improved renal fibrosis in the DKD model (Pang et al., 2020). Moreover, the HIF-PHI roxadustat was shown to prevent the hydroxylation of HIF-1α by PHD domain enzymes, leading to HIF-1α accumulation in the cytoplasm, followed by its translocation to the nucleus where it forms heterodimers with HIF-1β. This process promotes the expression of downstream target genes such as EPO, thereby ameliorating renal anemia. Current research on roxadustat has progressed to phase III clinical trials in various countries, indicating its efficacy in improving anemia and other effects of CKD patients. Other investigations have demonstrated that another HIF-PHI, Enarodustat, can inhibit the metabolic shift from the tricarboxylic acid cycle to glycolysis, thereby enhancing energy metabolism in the early stages of DKD (Hasegawa et al., 2020).
The aforementioned mechanism highlights the significant role of HIF-1α in the pathogenesis and progression of DKD (Figure 2), positioning HIF-1α as a promising target for therapeutic intervention in this condition. Recent investigations have identified various strategies and pharmacological agents capable of inhibiting HIF-1α activity and its associated pathways. The mechanisms of action of these agents predominantly involve multiple facets of the HIF-1α activation pathway, encompassing transcription, translation, stability, nuclear transport, heterodimerization, and the regulation of downstream target gene expression. Notably, anthracycline drugs and amifostine have been shown to suppress the expression of HIF-1α mRNA, while the active component of AFP-464, amino flavone, can completely inhibit the expression of HIF-1α protein. Furthermore, EZN-2968, mTOR inhibitors (rapamycin), COX-2 inhibitors (ibuprofen), topoisomerase inhibitors (mitoxantrone), and cardiac glycosides have all demonstrated the ability to obstruct the translation of HIF-1α mRNA (Qannita et al., 2024). Currently, research efforts aimed at inhibiting HIF-1α activity and its related pathways are predominantly concentrated in the oncology domain, with a notable deficiency in studies pertaining to renal diseases, particularly DKD.
Current research on HIF-1α in the context of renal diseases predominantly addresses the management of renal anemia, a topic that has been extensively reviewed in prior literature. Recent investigations have demonstrated that the administration of dapagliflozin in animal models of progressive DKD significantly ameliorates hyperglycemia and reduces urinary albumin excretion. Longitudinal studies indicate that this pharmacological agent can suppress the expression of HIF-1 in the nuclei of both glomerular and tubular cells, leading to improvements in glomerulosclerosis and RIF (Inada et al., 2022). Furthermore, the application of the HIF-1α inhibitor PX-478 in streptozotocin-induced diabetic murine models has yielded promising results, including the restoration of normal insulin secretion in response to glucose from the pancreatic islets, a reduction in blood glucose levels, and an enhancement of β-cell functionality. In vitro studies utilizing human pancreatic organoids have also shown that PX-478 can augment glucose-stimulated insulin secretion. These findings suggest that HIF-1α inhibitors may play a role in mitigating β-cell exhaustion by enhancing the activity of pancreatic β-cells under conditions of metabolic stress. Nonetheless, it is important to note that this research has primarily focused on the effects of HIF-1α inhibitors in diabetic animal models and human islets in vitro; thus, further investigation is necessary to ascertain their effects on pancreatic β-cells within the human physiological context. These inhibitors hold potential as effective therapeutic agents for the long-term management of diabetes in the future (Ilegems et al., 2022).
There are several critical areas within HIF-1α research that necessitate further investigation. Firstly, while HIF-PHI has demonstrated some effectiveness in the management of renal anemia, its inherent shortcomings and limitations warrant careful consideration. Given the variability in the distribution and function of HIF subtypes across renal cells, prolonged administration of HIF-PHI may result in the accumulation of HIF-1α, which could lead to adverse outcomes such as renal fibrosis, pathological angiogenesis, vascular calcification, and the promotion of tumorigenesis. Consequently, it is imperative to conduct clinical trials targeting specific patient populations to elucidate its therapeutic efficacy. Secondly, the mechanisms by which HIF-1α contributes to the onset and progression of DKD remain inadequately understood. In particular, there is a pressing need for in-depth exploration of the direct effects of various drug classes, including HIF-PHD inhibitors and SGLT-2 inhibitors, on HIF-1α signaling pathways.
Regulation of HIF-1α in DKD with zinc
Research has substantiated the regulatory function of trace elements on HIF-1α within the organism. Specifically, studies indicate that manganese ions are capable of downregulating PHD2, thereby activating the protein degradation pathway associated with HIF-1α, which results in a reduction of HIF-1α levels (Chen et al., 2024). Furthermore, iron is also critical in modulating HIF-1α expression, it not only facilitates the translation of HIF-1α transcripts but also activates PHD, which catalyzes the VHL-dependent degradation of HIF-1α (Linehan et al., 2019). Similarly, elevated concentrations of copper ions can inhibit the stabilizing effect of PHD on HIF-1α, thereby enhancing the transcriptional activity of HIF-1. In addition to the aforementioned trace elements, zinc can also regulate HIF-1α. Zinc is a vital micronutrient that plays a significant role in metabolic processes within the human body. It ranks as the second most prevalent trace element, following iron, and is predominantly located in skeletal muscle and bone (MacDonald, 2000). Zinc serves as a crucial cofactor for a variety of enzymes, thereby contributing significantly to numerous biochemical pathways. Furthermore, zinc is integral to processes such as cell proliferation, the synthesis of DNA and RNA, gene expression, the preservation of structural integrity, and the comprehensive regulation of the immune system (Li et al., 2013).
As DKD advances, there is a progressive decline in the glomerular filtration rate, accompanied by a reduction in plasma zinc levels (Nie et al., 2022). Furthermore, the diminished absorption of zinc in the intestines or the heightened urinary excretion of zinc in individuals with diabetes may contribute to a decrease in blood zinc levels among this population. A comparative analysis between patients with DKD and healthy individuals revealed that the plasma zinc ion concentration in DKD patients is significantly lower than that observed in healthy subjects (Nie et al., 2022). In patients undergoing hemodialysis, the prevalence of zinc deficiency ranges from 40% to 70%. Additionally, individuals with DKD frequently exhibit hypozincemia, particularly in developing nations. The elevated occurrence of hypozincemia may significantly contribute to the rising incidence of diabetes (Tang et al., 2010).
Numerous studies have suggested a possible interaction between zinc and diabetes, along with associated complications (Aschner et al., 2023; Nardinocchi et al., 2010). Physiological concentrations of zinc may confer advantages for individuals diagnosed with diabetes. Research, encompassing both in vitro and in vivo studies involving animal and human subjects, indicates that zinc has a variety of beneficial effects on both type 1 and type 2 diabetes (Takahashi, 2024; Yang et al., 2017). In a study, researchers administered zinc oxide nanoparticles at a dosage of 10 mg/kg/day orally to diabetic rats over a period of four consecutive weeks. Following the treatment, there was a significant enhancement in kidney function among the diabetic rats, accompanied by a notable reduction in blood glucose levels, blood urea nitrogen (BUN), serum creatinine, and urinary protein when compared to the control group (Abd El-Baset et al., 2023). In a separate investigation, researchers administered a daily dosage of 5 mg/kg of zinc chloride to mice with DKD. Histopathological examination of the renal tissue from the zinc-supplemented cohort revealed only mild congestion of the interstitial capillaries and minimal infiltration of mononuclear cells (Anton et al., 2022). Following a 6-week period of consistent supplementation with zinc carbonate in diabetic rats, a diet high in zinc has been shown to markedly decrease the concentrations of ROS and lipid peroxides in the renal tissues of these diabetic subjects (Barman et al., 2018). An investigation involving serum samples from 132 individuals diagnosed with T2DM, alongside in vitro experiments, revealed that zinc possesses the capacity to impede the propensity for calcification within the serum (Nakatani et al., 2020). Furthermore, zinc supplementation in diabetic patients has been shown to improve glycemic control and enhance lipid profiles (Takahashi, 2024). The significance of zinc in the prevention and mitigation of DKD progression has been thoroughly documented in numerous experimental investigations. The current management of DKD primarily aims to alleviate symptoms and postpone the decline in renal function. This is accomplished through the comprehensive regulation of various parameters, including blood pressure, blood glucose levels, and lipid profiles, in conjunction with symptomatic therapies designed to mitigate the progression of kidney disease. Frequently utilized pharmacological agents in clinical settings encompass RAS inhibitors, SGLT-2 inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, dipeptidyl peptidase-4 (DPP-4) inhibitors, and selective endothelin (ET) α receptor antagonists. A detailed examination of their specific mechanisms and clinical limitations is presented in Table 2. Nevertheless, the intricate pathogenesis of DKD, coupled with the absence of targeted therapeutic interventions, results in suboptimal treatment outcomes, contributing to an increasing prevalence of ESRD. Considering the significant role of HIF-1α in the pathogenesis and progression of DKD, the subsequent analysis will succinctly examine the interaction mechanisms between the trace element zinc and the targets of HIF-1α. This exploration aims to offer potential targeted therapeutic strategies for clinical application.
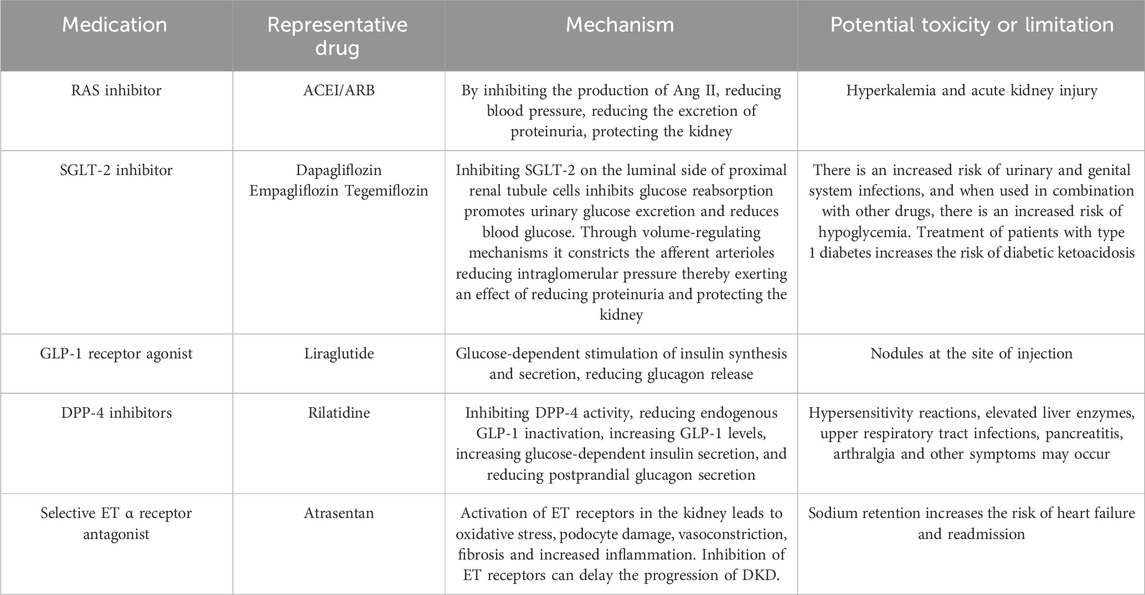
Table 2. In the clinical treatment of DKD commonly used medications, their mechanisms of action, and an analysis of their limitations.
The role of zinc in DKD through the regulation of HIF-1α
Inhibition of HIF-1α translocation (Figure 4A).
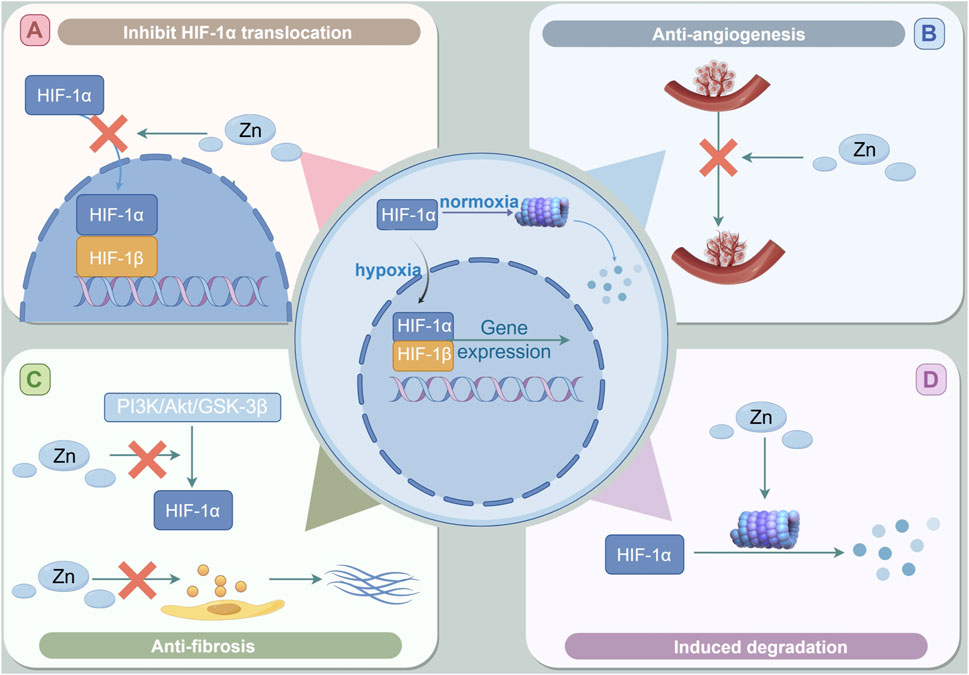
Figure 4. The role of Zinc in DKD through the regulation of HIF-1α. (A) Zinc supplementation may hinder the activation of HIF-1α by inhibiting the intranuclear shift of HIF-1α and destroying the heterodimerization of HIF-1. (B) Zinc can inhibit HIF-1-induced VEGF expression. (C) Zinc supplementation inhibits the expression of HIF-1α may involve the activation of zinc-induced PI3K/Akt/GSK-3β signal pathway.the anti-fibrosis effect of zinc may be lowered by inhibiting the activation of PI3K/Akt/GSK-3β signaling pathway to inhibit the hypoxia-induced HIF-1α accumulation and the change of EMT markers. (D) Zinc supplementation can reduce the expression decline of HIF-1α in cells by promoting the degradation of HIF-1α in the proteasome pathway, thus inhibiting the adverse effects of HIF-1α in the late stage of DKD.
Studies have shown that zinc inhibits the nuclear translocation of HIF-1α in astrocytes (Pan et al., 2013). In human microvascular endothelial cells, the cultivation of cells in a zinc-deficient environment, achieved through the application of the zinc-chelating agent TPEN, led to an increased intranuclear translocation of HIF-1α, heightened secretion of downstream ET, and augmented migration of endothelial cells (Morand et al., 2019). Zinc supplementation has the potential to hinder the activation of HIF-1 by obstructing the intranuclear translocation of HIF-1α and interfering with the heterodimerization of HIF-1 (Chun et al., 2001). Certain studies indicate that zinc may facilitate the accumulation and nuclear translocation of HIF-1α. However, it may concurrently inhibit the interaction of HIF-1 with DNA by impeding the nuclear translocation of HIF-1β, which subsequently leads to a decrease in the expression of the downstream target gene EPO (Guo et al., 2015).
Inhibit pathological angiogenesis (Figure 4B).
Investigations have demonstrated that the application of zinc chloride to hypoxic cancer cells results in a significant downregulation of HIF-1α protein levels and its downstream target gene, VEGF, both in vivo and in vitro. Notably, this reduction can be reversed by the proteasome inhibitor MG132. These findings indicate that zinc may facilitate the degradation of HIF-1α in hypoxic conditions via the proteasome pathway, thereby contributing to the observed decrease in HIF-1α levels (Nardinocchi et al., 2010). Researchers conducted an experiment in which primary human microvascular endothelial cells were cultured in a low zinc environment, induced by the chelator TPEN, for a duration of 5 hours. They observed a notable increase in the nuclear content of HIF-1α, as evidenced by immunofluorescence analysis. Furthermore, quantitative PCR analysis revealed that following TPEN treatment, the levels of the downstream target gene ET-1, associated with HIF-1α, were significantly elevated in the cell supernatant. This increase was found to be inhibited by the application of HIF-1α specific siRNA. These findings indicate that zinc plays a critical role in modulating the formation of the HIF-1 heterodimer and the expression of the downstream target gene ET-1 by influencing the translocation of HIF-1α into the nucleus of the cells (Morand et al., 2019). Homeodomain-interacting protein kinase 2 (HIPK2) has been identified as a potential biomarker for tumor proliferation. The research indicates that HIPK2 is capable of interacting with the promoter region of HIF-1, thereby inhibiting its activation. This interaction results in the downregulation of transcription of the downstream target genes of HIF-1 (Nardinocchi et al., 2009). Zinc supplementation has been shown to counteract the transcriptional repression of HIPK2 induced by hypoxic conditions in cancer cells, leading to a decrease in the expression of HIF-1α (Hatano et al., 2021).
Previous research has indicated that elevated levels of HIF-1α can enhance the expression of the downstream target gene VEGF, potentially resulting in pathological angiogenesis and subsequent renal fibrosis. Empirical studies have demonstrated that zinc supplementation can mitigate pathological angiogenesis and ameliorate renal fibrosis through the inhibition of the HIF-1α/VEGF signaling pathway.
Anti-fibrosis (Figure 4C).
A substantial body of research has indicated that zinc supplementation may effectively mitigate fibrosis across multiple tissue types (Malekahmadi et al., 2024). In previous investigations, we administered zinc supplementation and implemented zinc deficiency interventions in animal models of DKD, specifically in rats and mice, as well as in cellular models characterized by high glucose and high fat conditions. These studies corroborated the efficacy of zinc in mitigating fibrosis within diabetic renal tissues (Li et al., 2014; Wang et al., 2020). This aligns with findings from another study, which demonstrated that a 3-month regimen of zinc supplementation in diabetic rats led to a reduction in histological alterations, a significant decrease in the expression of fibrotic markers such as connective tissue growth factor, and an upregulation of metal-binding proteins in both cardiac and renal tissues (Tang et al., 2010).
The preceding discussion has elucidated the role of HIF-1α in facilitating the advancement of renal fibrosis through multiple mechanisms. Research indicates that zinc supplementation can mitigate renal tubular EMT and renal tubular interstitial fibrosis by downregulating HIF-1α in models of DKD (Zhang X et al., 2016). Specifically, Zhang et al. demonstrated that zinc supplementation in DKD rat models and in high glucose/hypoxia-induced cellular models significantly reduced the expression of fibrotic markers, including fibronectin, α-SMA, and E-cadherin. Han et al. found that both HIF-1α siRNA and the overexpression of PHD2 can effectively block the loss of epithelial markers and the increase in the expression of mesenchymal markers induced by TGF-β1 in renal tubular cells cultured in vitro (Han et al., 2013). In the cellular model, the antifibrotic effects of zinc were negated following treatment with PI3K inhibitors such as LY 294002 or wortmannin, which resulted in the restoration of E-cadherin levels and a reduction in α-SMA expression. This observation implies that zinc supplementation may downregulate HIF-1α expression and inhibit the activation of the PI3K/Akt/glycogen synthase kinase-3β (GSK-3β) signaling pathway, thereby diminishing the hypoxia-induced accumulation of HIF-1α and alterations in EMT markers, ultimately suppressing EMT in cells subjected to high glucose and hypoxia. The proposed mechanism of action may involve the mediation of zinc’s negative regulatory effects on the HIF-1α signaling pathway by disrupting the critical function of the zinc finger domain in PHD. Conversely, zinc deficiency, induced by the zinc chelator TPEN in primary human microvascular endothelial cells, has been shown to upregulate HIF-1α signaling, potentially through enhanced nuclear translocation of HIF-1α and increased secretion of ET-1 (Guo et al., 2015).
Zinc-induced proteasome degradation (Figure 4D).
In a standard oxygen environment, HIF-1α undergoes hydroxylation by PHD enzymes, which subsequently leads to its degradation via the proteasome, facilitated by the action of the VHL ubiquitin ligase. As a result, the detection of HIF-1α under normoxic conditions presents significant challenges. Some researchers have employed the proteasome inhibitor MG132 in cellular treatments, revealing that the impact of zinc supplementation on reducing HIF-1α levels can be negated by the presence of proteasome inhibitors. This finding suggests that zinc may modulate HIF-1α levels through the proteasomal degradation pathway (Nardinocchi et al., 2010). In a separate investigation, it was found that zinc supplementation inhibits the interaction between HIF-1α and the VEGF promoter by promoting the degradation of HIF-1α via the proteasome pathway. Furthermore, the introduction of a zinc chelating agent resulted in an increase in HIF-1α expression within cells, an effect that could be reversed by the addition of zinc. This indicates that zinc supplementation may mitigate the decline in HIF-1α expression in cells by enhancing its degradation through the proteasome pathway, thereby alleviating the adverse effects associated with HIF-1α during the later stages of DKD (Aschner et al., 2023; Nardinocchi et al., 2010).
The potential toxicity of zinc
The consumption of zinc is not without its drawbacks, as potential adverse effects may restrict its application. A study conducted in China identified a U-shaped correlation between dietary zinc intake and the risk of diabetes, with a critical threshold established at 9.1 mg/d (He et al., 2022). Excessive zinc consumption has been associated with an increased risk of developing diabetes. Furthermore, research indicates that prolonged hyperzincemia may elevate the risk of thrombosis (Hughes and Samman, 2006). An experimental study involving 11 healthy adult males demonstrated that continuous administration of 300 mg of zinc sulfate could result in reversible impairments in immune function (Chandra, 1984). In vitro investigations have revealed that elevated intracellular zinc levels can directly induce apoptosis in endothelial cells (Wiseman et al., 2010).
Common side effects associated with zinc supplementation include nausea, vomiting, and pruritus, with the symptoms of nausea and vomiting being mitigated when zinc is ingested postprandially. Additionally, increased zinc concentrations may lead to reduced serum copper levels (Koi et al., 2020), potentially resulting in copper deficiency-related anemia and leukopenia (Gembillo et al., 2022; Kodama et al., 2020). The proposed mechanism for this phenomenon suggests that excessive zinc intake enhances the binding of zinc to metallothionein in intestinal epithelial cells, which subsequently interferes with copper binding to metallothionein, thereby inducing copper deficiency (Duncan et al., 2015). Abnormalities in copper metabolism may compromise the activity of zinc-copper superoxide dismutase, consequently impairing antioxidant functions. Future research must delve deeper into the optimal intake levels of zinc and the potential mechanisms linking zinc to DKD.
Conclusion
DKD represents a significant complication associated with diabetes mellitus. The prevailing standard of care primarily emphasizes the management of glycemic levels, blood pressure, and lipid profiles. Nevertheless, a considerable number of patients continue to advance to ESRD, even while undergoing these interventions. Consequently, comprehensive investigations into the pathophysiology of DKD and the identification of potential therapeutic targets are crucial for the effective prevention and management of this condition.
The incidence of DKD is intricately linked to hemodynamic disturbances, metabolic irregularities, and the consequent inflammatory responses and oxidative stress, which collectively contribute to a hypoxic milieu within the renal system. HIF-1, a pivotal regulator of hypoxic responses, plays a significant role in angiogenesis, erythropoiesis, oxidative stress, inflammatory processes, mitochondrial autophagy, and extensive fibrosis by inducing aberrant expression of its downstream target genes, including VEGF, EPO, and HO-1. This involvement underscores the potential of HIF-1α as a therapeutic target for DKD. Currently, pharmacological agents aimed at HIF-1α are being utilized in the management of this condition. Roxadustat, a drug classified as a HIF-PHI, has progressed to clinical research phases, primarily for the treatment of renal anemia associated with CKD. HIF-1α mediates damage in DKD through various extensive signaling pathways, necessitating further investigation into targeted therapies for these alternative pathways. Although research on HIF-1α and DKD has gradually increased in recent years, many enigmas remain unresolved. For instance, does HIF-1α exert non-transcriptional effects while mediating hypoxic adaptation in a high-glucose and hypoxia environment? How can we effectively visualize the levels of PO2 within renal cells? Furthermore, the mechanisms, translational potential, and clinical exploration of treating DKD through the modulation of HIF-1α activity still warrant deeper investigation.
Patients suffering from DKD frequently exhibit dysregulated zinc metabolism. Existing literature suggests that zinc supplementation may exert beneficial effects on DKD. Zinc influences the expression of HIF-1α in cells subjected to hyperglycemic and hypoxic conditions through multiple mechanisms, including the inhibition of HIF-1α translocation, as well as exhibiting anti-inflammatory, antioxidant, anti-apoptotic, and anti-fibrotic properties. Consequently, zinc may represent a viable therapeutic strategy for modulating HIF-1α and decelerating the progression of DKD. However, current investigations into the efficacy of zinc predominantly focus on animal and cellular models, highlighting the necessity for further clinical research in human populations to elucidate the specific mechanisms by which zinc supplementation may affect DKD. While some studies have indicated a potential renal protective effect of zinc supplementation in DKD patients, there remains a pressing need for more randomized controlled trials with larger cohorts and extended follow-up durations to ascertain the long-term effects of zinc treatment. Given that zinc supplementation may negatively impact copper metabolism, additional clinical research is urgently required to establish the optimal zinc intake levels for DKD patients to maintain a balance between copper and zinc. Furthermore, under pathological conditions, the pharmacokinetics and pharmacodynamics of zinc supplementation may be altered by changes in the internal environment, necessitating comprehensive animal studies and clinical trials to identify the most effective zinc supplementation concentrations for patients with varying severities of DKD.
Zinc supplementation represents a promising area for further investigation. Currently, commonly utilized zinc supplements in clinical practice include zinc sulfate, zinc chloride, zinc nitrate, zinc gluconate, zinc acetate, and zinc lactate. Additionally, zinc oxide nanoparticles present a novel and effective treatment strategy for DKD, owing to their ability to address the limitations associated with the short duration of action of oral medications, low oral bioavailability, and significant side effects. However, current research on the application of zinc oxide nanoparticles in the treatment of DKD is relatively scarce, and the safety of this treatment process, along with associated adverse reactions, must be further validated through extensive animal experiments and clinical studies.
Author contributions
WQ: Data curation, Software, Writing–original draft. PN: Writing–original draft. XuH: Writing–original draft. FC: Writing–original draft. XiH: Writing–original draft. WS: Writing–original draft. ML: Writing–original draft. BL: Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by Department of Science and Technology of Jilin Province (YDZJ20241211ZYTS).
Acknowledgments
We thank our colleagues for critical advice on this manuscript. Figure by figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abd El-Baset, S. A., Mazen, N. F., Abdul-Maksoud, R. S., and Kattaia, A. A. A. (2023). The therapeutic prospect of zinc oxide nanoparticles in experimentally induced diabetic nephropathy. Tissue Barriers 11, 2069966. doi:10.1080/21688370.2022.2069966
Anton, I. C., Mititelu-Tartau, L., Popa, E. G., Poroch, M., Poroch, V., Pelin, A.-M., et al. (2022). Zinc chloride enhances the antioxidant status, improving the functional and structural organic disturbances in streptozotocin-induced diabetes in rats. Med. Kaunas. 58, 1620. doi:10.3390/medicina58111620
Arden, G. B., and Sivaprasad, S. (2011). Hypoxia and oxidative stress in the causation of diabetic retinopathy. Curr. Diabetes Rev. 7, 291–304. doi:10.2174/157339911797415620
Aschner, M., Skalny, A. V., Lu, R., Santamaria, A., Zhou, J.-C., Ke, T., et al. (2023). The role of hypoxia-inducible factor 1 alpha (HIF-1α) modulation in heavy metal toxicity. Arch. Toxicol. 97, 1299–1318. doi:10.1007/s00204-023-03483-7
Barman, S., Pradeep, S. R., and Srinivasan, K. (2018). Zinc supplementation alleviates the progression of diabetic nephropathy by inhibiting the overexpression of oxidative-stress-mediated molecular markers in streptozotocin-induced experimental rats. J. Nutr. Biochem. 54, 113–129. doi:10.1016/j.jnutbio.2017.11.008
Bernhardt, W. M., Wiesener, M. S., Scigalla, P., Chou, J., Schmieder, R. E., Günzler, V., et al. (2010). Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J. Am. Soc. Nephrol. 21, 2151–2156. doi:10.1681/ASN.2010010116
Broz, P., and Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi:10.1038/nri.2016.58
Catrina, S.-B., and Zheng, X. (2021). Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia 64, 709–716. doi:10.1007/s00125-021-05380-z
Cerychova, R., and Pavlinkova, G. (2018). HIF-1, metabolism, and diabetes in the embryonic and adult heart. Front. Endocrinol. (Lausanne) 9, 460. doi:10.3389/fendo.2018.00460
Chandra, R. K. (1984). Excessive intake of zinc impairs immune responses. JAMA 252, 1443–1446. doi:10.1001/jama.1984.03350110043027
Chen, J., Tao, Z., Zhang, X., Hu, J., Wang, S., Xing, G., et al. (2024). Manganese overexposure results in ferroptosis through the HIF-1α/p53/SLC7A11 pathway in ICR mouse brain and PC12 cells. Ecotoxicol. Environ. Saf. 279, 116481. doi:10.1016/j.ecoenv.2024.116481
Chen, K., Dai, H., Yuan, J., Chen, J., Lin, L., Zhang, W., et al. (2018). Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 9, 105–118. doi:10.1038/s41419-017-0127-z
Chen, N., Chen, X., Huang, R., Zeng, H., Gong, J., Meng, W., et al. (2009). BCL-xL is a target gene regulated by hypoxia-inducible factor-1{alpha}alpha. J. Biol. Chem. 284, 10004–10012. doi:10.1074/jbc.M805997200
Cheng, L., Qiu, X., He, L., and Liu, L. (2022). MicroRNA-122-5p ameliorates tubular injury in diabetic nephropathy via FIH-1/HIF-1α pathway. Ren. Fail. 44, 293–303. doi:10.1080/0886022X.2022.2039194
Chou, E., Suzuma, I., Way, K. J., Opland, D., Clermont, A. C., Naruse, K., et al. (2002). Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: a possible explanation for impaired collateral formation in cardiac tissue. Circulation 105, 373–379. doi:10.1161/hc0302.102143
Choudhry, H., and Harris, A. L. (2018). Advances in hypoxia-inducible factor biology. Cell Metab. 27, 281–298. doi:10.1016/j.cmet.2017.10.005
Chun, Y.-S., Choi, E., Yeo, E.-J., Lee, J. H., Kim, M.-S., and Park, J.-W. (2001). A new HIF-1 alpha variant induced by zinc ion suppresses HIF-1-mediated hypoxic responses. J. Cell Sci. 114, 4051–4061. doi:10.1242/jcs.114.22.4051
Damiani, E., Casarotta, E., Orlando, F., Carsetti, A., Scorcella, C., Domizi, R., et al. (2021). Effects of normoxia, hyperoxia, and mild hypoxia on macro-hemodynamics and the skeletal muscle microcirculation in anesthetised rats. Front. Med. 8, 672257. doi:10.3389/fmed.2021.672257
Dhakshinamoorthy, J., Elumalai, R. P., Dev, B., Hemamalini, A. J., Venkata Sai, P. M., and Periasamy, S. (2017). Assessment of abdominal aortic calcification in predialysis chronic kidney disease and maintenance hemodialysis patients. Saudi J. Kidney Dis. Transpl. 28, 1338–1348. doi:10.4103/1319-2442.220855
Diebold, I., Petry, A., Hess, J., and Görlach, A. (2010). The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 21, 2087–2096. doi:10.1091/mbc.e09-12-1003
Doller, A., Gauer, S., Sobkowiak, E., Geiger, H., Pfeilschifter, J., and Eberhardt, W. (2009). Angiotensin II induces renal plasminogen activator inhibitor-1 and cyclooxygenase-2 expression post-transcriptionally via activation of the mRNA-stabilizing factor human-antigen R. Am. J. Pathology 174, 1252–1263. doi:10.2353/ajpath.2009.080652
Dos, S., Elisabete, A., Li, L., Ji, L., and Prasad, P. (2007). Early changes with diabetes in renal medullary hemodynamics as evaluated by fiberoptic probes and BOLD magnetic resonance imaging. Investig. Radiol. 42 (3), 157–162. doi:10.1097/01.rli.0000252492.96709.36
Duncan, A., Yacoubian, C., Watson, N., and Morrison, I. (2015). The risk of copper deficiency in patients prescribed zinc supplements. J. Clin. Pathology 68, 723–725. doi:10.1136/jclinpath-2014-202837
Dunn, L. L., Kong, S. M. Y., Tumanov, S., Chen, W., Cantley, J., Ayer, A., et al. (2021). Hmox1 (heme oxygenase-1) protects against ischemia-mediated injury via stabilization of HIF-1α (Hypoxia-Inducible factor-1α). Arteriosclerosis, Thrombosis, Vasc. Biol. 41, 317–330. doi:10.1161/ATVBAHA.120.315393
Eckardt, K.-U., Bernhardt, W. W., Weidemann, A., Warnecke, C., Rosenberger, C., Wiesener, M. M., et al. (2005). Role of hypoxia in the pathogenesis of renal disease. Kidney Int. 68, S46–S51. doi:10.1111/j.1523-1755.2005.09909.x
Feng, X., Wang, S., Sun, Z., Dong, H., Yu, H., Huang, M., et al. (2021). Ferroptosis enhanced diabetic renal tubular injury via HIF-1α/HO-1 pathway in db/db mice. Front. Endocrinol. (Lausanne) 12, 626390. doi:10.3389/fendo.2021.626390
Fenta, E. T., Eshetu, H. B., Kebede, N., Bogale, E. K., Zewdie, A., Kassie, T. D., et al. (2023). Prevalence and predictors of chronic kidney disease among type 2 diabetic patients worldwide, systematic review and meta-analysis. Diabetology & Metabolic Syndrome 15, 245. doi:10.1186/s13098-023-01202-x
Friederich-Persson, M., Thörn, E., Hansell, P., Nangaku, M., Levin, M., and Palm, F. (2013). Kidney hypoxia, attributable to increased oxygen consumption, induces nephropathy independently of hyperglycemia and oxidative stress. Hypertension 62, 914–919. doi:10.1161/HYPERTENSIONAHA.113.01425
Gembillo, G., Visconti, L., Giuffrida, A. E., Labbozzetta, V., Peritore, L., Lipari, A., et al. (2022). Role of zinc in diabetic kidney disease. Nutrients 14, 1353. doi:10.3390/nu14071353
Gruber, M., Hu, C.-J., Johnson, R. S., Brown, E. J., Keith, B., and Simon, M. C. (2007). Acute postnatal ablation of Hif-2alpha results in anemia. Proc. Natl. Acad. Sci. 104, 2301–2306. doi:10.1073/pnas.0608382104
Guo, C., Quan, Z., Ke, J., Zang, H., Teng, Q., Li, X., et al. (2024). Hypoxia-inducible factor-1α regulates high phosphate-induced vascular calcification via type III sodium-dependent phosphate cotransporter 1. Cardiol. Res. Pract. 2024, 6346115. doi:10.1155/2024/6346115
Guo, L., Chen, Y., He, T., Qi, F., Liu, G., Fu, Y., et al. (2015). Nuclear-translocated endostatin downregulates hypoxia inducible factor-1α activation through interfering with Zn(II) homeostasis. Mol. Med. Rep. 11, 3473–3480. doi:10.3892/mmr.2015.3223
Guo, Y., Lu, Z.-S., Zhang, X.-C., Zhang, Q., Liu, X., Chen, J., et al. (2024a). Relationship between serum iron, zinc, calcium, and HIF-1a—comparative analysis of 2 regions and 4 ethnic groups in China. Front. Nutr. 11, 1433640. doi:10.3389/fnut.2024.1433640
Guo, Y., Yu, C., Lu, Z., Zhang, M., Zhang, Q., and Liu, X. (2024b). Zinc homeostasis plays important roles in hypoxia tolerance: a study conducted clinically and in vitro. High Alt. Med. & Biol. doi:10.1089/ham.2024.0036
Hahne, M., Schumann, P., Mursell, M., Strehl, C., Hoff, P., Buttgereit, F., et al. (2018). Unraveling the role of hypoxia-inducible factor (HIF)-1α and HIF-2α in the adaption process of human microvascular endothelial cells (HMEC-1) to hypoxia: redundant HIF-dependent regulation of macrophage migration inhibitory factor. Microvasc. Res. 116, 34–44. doi:10.1016/j.mvr.2017.09.004
Hakroush, S., Moeller, M. J., Theilig, F., Kaissling, B., Sijmonsma, T. P., Jugold, M., et al. (2009). Effects of increased renal tubular vascular endothelial growth factor (VEGF) on fibrosis, cyst formation, and glomerular disease. Am. J. Pathology 175, 1883–1895. doi:10.2353/ajpath.2009.080792
Han, W.-Q., Zhu, Q., Hu, J., Li, P.-L., Zhang, F., and Li, N. (2013). Hypoxia-inducible factor prolyl-hydroxylase-2 mediates transforming growth factor beta 1-induced epithelial–mesenchymal transition in renal tubular cells. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1833, 1454–1462. doi:10.1016/j.bbamcr.2013.02.029
Hansell, P., Welch, W. J., Blantz, R. C., and Palm, F. (2013). Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 40, 123–137. doi:10.1111/1440-1681.12034
Hasegawa, S., Tanaka, T., Saito, T., Fukui, K., Wakashima, T., Susaki, E. A., et al. (2020). The oral hypoxia-inducible factor prolyl hydroxylase inhibitor enarodustat counteracts alterations in renal energy metabolism in the early stages of diabetic kidney disease. Kidney Int. 97, 934–950. doi:10.1016/j.kint.2019.12.007
Hatano, N., Matsubara, M., Suzuki, H., Muraki, Y., and Muraki, K. (2021). HIF-1α dependent upregulation of ZIP8, ZIP14, and TRPA1 modify intracellular Zn2+ accumulation in inflammatory synoviocytes. IJMS 22, 6349. doi:10.3390/ijms22126349
He, P., Li, H., Liu, M., Zhang, Z., Zhang, Y., Zhou, C., et al. (2022). U-shaped association between dietary zinc intake and new-onset diabetes: a nationwide cohort study in China. J. Clin. Endocrinol. & Metabolism 107, e815–e824. doi:10.1210/clinem/dgab636
Hellwig-Bürgel, T., Rutkowski, K., Metzen, E., Fandrey, J., and Jelkmann, W. (1999). Interleukin-1β and tumor necrosis Factor-α stimulate DNA binding of hypoxia-inducible factor-1. Blood 94, 1561–1567. doi:10.1182/blood.v94.5.1561
Hernández-Arteaga, K., Soto-Abraham, V., Pérez-Navarro, M., de León-Garza, B., Rodríguez-Matías, A., Olvera-Soto, M. G., et al. (2018). Thrombotic microangiopathy in patients with diabetic nephropathy is associated with low VEGF expression and end-stage renal disease. Clin. Nephrol. 89, 429–437. doi:10.5414/CN109240
Hernansanz-Agustín, P., Choya-Foces, C., Carregal-Romero, S., Ramos, E., Oliva, T., Villa-Piña, T., et al. (2020). Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature 586, 287–291. doi:10.1038/s41586-020-2551-y
Hesp, A. C., Schaub, J. A., Prasad, P. V., Vallon, V., Laverman, G. D., Bjornstad, P., et al. (2020). The role of renal hypoxia in the pathogenesis of diabetic kidney disease: a promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 98, 579–589. doi:10.1016/j.kint.2020.02.041
Higgins, D. F., Kimura, K., Bernhardt, W. M., Shrimanker, N., Akai, Y., Hohenstein, B., et al. (2007). Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest 117, 3810–3820. doi:10.1172/JCI30487
Holmquist-Mengelbier, L., Fredlund, E., Löfstedt, T., Noguera, R., Navarro, S., Nilsson, H., et al. (2006). Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 10, 413–423. doi:10.1016/j.ccr.2006.08.026
Huang, K.-P., Chen, C., Hao, J., Huang, J.-Y., Liu, P.-Q., and Huang, H.-Q. (2015). AGEs-RAGE system down-regulates Sirt1 through the ubiquitin-proteasome pathway to promote FN and TGF-β1 expression in male rat glomerular mesangial cells. Endocrinology 156, 268–279. doi:10.1210/en.2014-1381
Hughes, S., and Samman, S. (2006). The effect of zinc supplementation in humans on plasma lipids, antioxidant status and thrombogenesis. J. Am. Coll. Nutr. 25, 285–291. doi:10.1080/07315724.2006.10719537
Hussain, A., Wyatt, A. W., Wang, K., Bhandaru, M., Biswas, R., Avram, D., et al. (2008). SGK1-dependent upregulation of connective tissue growth factor by angiotensin II. Kidney Blood Press. Res. 31, 80–86. doi:10.1159/000119703
Iacobini, C., Vitale, M., Haxhi, J., Pesce, C., Pugliese, G., and Menini, S. (2022). Mutual regulation between redox and hypoxia-inducible factors in cardiovascular and renal complications of diabetes. Antioxidants 11, 2183. doi:10.3390/antiox11112183
Ilegems, E., Bryzgalova, G., Correia, J., Yesildag, B., Berra, E., Ruas, J. L., et al. (2022). HIF-1α inhibitor PX-478 preserves pancreatic β cell function in diabetes. Sci. Transl. Med. 14, eaba9112. doi:10.1126/scitranslmed.aba9112
Imasawa, T., Obre, E., Bellance, N., Lavie, J., Imasawa, T., Rigothier, C., et al. (2017). High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB J. 31, 294–307. doi:10.1096/fj.201600293R
Inada, A., Inada, O., Yasunami, Y., Arakawa, K., Nabeshima, Y., and Fukatsu, A. (2022). Amelioration of murine diabetic nephropathy with a SGLT2 inhibitor is associated with suppressing abnormal expression of hypoxia-inducible Factors. Am. J. Pathology 192, 1028–1052. doi:10.1016/j.ajpath.2022.03.015
Ji, Z., Wang, L., Wang, S., and Liang, G. (2019). Dexmedetomidine hydrochloride up-regulates expression of hypoxia inducible factor-1α to alleviate renal ischemiareperfusion injury in diabetic rats. Nan Fang. Yi Ke Da Xue Xue Bao 39 (08.11), 944–949. doi:10.12122/j.issn.1673-4254.2019.08.11
Jiang, N., Zhao, H., Han, Y., Li, L., Xiong, S., Zeng, L., et al. (2020). HIF-1α ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif. 53, e12909. doi:10.1111/cpr.12909
Katavetin, P., Miyata, T., Inagi, R., Tanaka, T., Sassa, R., Ingelfinger, J. R., et al. (2006). High glucose blunts vascular endothelial growth factor response to hypoxia via the oxidative stress-regulated hypoxia-inducible factor/hypoxia-responsible element pathway. J. Am. Soc. Nephrol. 17, 1405–1413. doi:10.1681/ASN.2005090918
Kodama, H., Tanaka, M., Naito, Y., Katayama, K., and Moriyama, M. (2020). Japan’s practical guidelines for zinc deficiency with a particular focus on taste disorders, inflammatory bowel disease, and liver cirrhosis. Int. J. Mol. Sci. 21, 2941. doi:10.3390/ijms21082941
Koi, S., Arai, K., Nogami, A., Toma, H., Yamamoto, M., Miura, O., et al. (2020). Impaired hematopoiesis due to copper deficiency in a hemodialysis patient supplemented with zinc. Rinsho Ketsueki 61, 1487–1491. doi:10.11406/rinketsu.61.1487
Lee, W.-T., Tai, S.-H., Lin, Y.-W., Wu, T.-S., and Lee, E.-J. (2018). YC-1 reduces inflammatory responses by inhibiting nuclear factor-κB translocation in mice subjected to transient focal cerebral ischemia. Mol. Med. Rep. 18, 2043–2051. doi:10.3892/mmr.2018.9178
Lee, Y. S., Kim, J., Osborne, O., Oh, D. Y., Sasik, R., Schenk, S., et al. (2014). Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352. doi:10.1016/j.cell.2014.05.012
Li, B., Cui, W., Tan, Y., Luo, P., Chen, Q., Zhang, C., et al. (2014). Zinc is essential for the transcription function of Nrf2 in human renal tubule cells in vitro and mouse kidney in vivo under the diabetic condition. J. Cell. Mol. Med. 18, 895–906. doi:10.1111/jcmm.12239
Li, B., Tan, Y., Sun, W., Fu, Y., Miao, L., and Cai, L. (2013). The role of zinc in the prevention of diabetic cardiomyopathy and nephropathy. Toxicol. Mech. Methods 23, 27–33. doi:10.3109/15376516.2012.735277
Li, N., Li, Y., Li, Z., Huang, C., Yang, Y., Lang, M., et al. (2016). Hypoxia inducible factor 1 (HIF-1) recruits macrophage to activate pancreatic stellate cells in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 17, 799. doi:10.3390/ijms17060799
Li, W., Xiang, Z., Xing, Y., Li, S., and Shi, S. (2022). Mitochondria bridge HIF signaling and ferroptosis blockage in acute kidney injury. Cell Death Dis. 13, 308–312. doi:10.1038/s41419-022-04770-4
Li, Z.-L., Lv, L.-L., Wang, B., Tang, T.-T., Feng, Y., Cao, J.-Y., et al. (2019). The profibrotic effects of MK-8617 on tubulointerstitial fibrosis mediated by the KLF5 regulating pathway. FASEB J. 33, 12630–12643. doi:10.1096/fj.201901087RR
Linehan, W. M., Schmidt, L. S., Crooks, D. R., Wei, D., Srinivasan, R., Lang, M., et al. (2019). The metabolic basis of kidney cancer. Cancer Discov. 9, 1006–1021. doi:10.1158/2159-8290.CD-18-1354
Liu, H., Takagaki, Y., Kumagai, A., Kanasaki, K., and Koya, D. (2021). The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1α accumulation. J. Diabetes Investigation 12, 697–709. doi:10.1111/jdi.13478
Liu, S.-Y., Chen, J., and Li, Y.-F. (2018). Clinical significance of serum interleukin-8 and soluble tumor necrosis factor-like weak inducer of apoptosis levels in patients with diabetic nephropathy. J. Diabetes Investig. 9, 1182–1188. doi:10.1111/jdi.12828
Loutradis, C., Skodra, A., Georgianos, P., Tolika, P., Alexandrou, D., Avdelidou, A., et al. (2016). Diabetes mellitus increases the prevalence of anemia in patients with chronic kidney disease: a nested case-control study. World J. Nephrol. 5, 358–366. doi:10.5527/wjn.v5.i4.358
MacDonald, R. S. (2000). The role of zinc in growth and cell proliferation. J. Nutr. 130, 1500S-8S–8S. doi:10.1093/jn/130.5.1500S
Machado, M. G., Patente, T. A., Rouillé, Y., Heumel, S., Melo, E. M., Deruyter, L., et al. (2022). Acetate improves the killing of streptococcus pneumoniae by alveolar macrophages via NLRP3 inflammasome and glycolysis-HIF-1α axis. Front. Immunol. 13, 773261. doi:10.3389/fimmu.2022.773261
Mahdy, R. A., Nada, W. M., Hadhoud, K. M., and El-Tarhony, S. A. (2010). The role of vascular endothelial growth factor in the progression of diabetic vascular complications. Eye (Lond) 24, 1576–1584. doi:10.1038/eye.2010.86
Malekahmadi, M., Soltani, S., Pahlavani, N., Zahabi, E. S., Kazemizadeh, H., Hadavi, S., et al. (2024). Zinc status in cystic fibrosis patients; a systematic review and meta-analysis. Heliyon 10, e33686. doi:10.1016/j.heliyon.2024.e33686
Matoba, K., Kawanami, D., Okada, R., Tsukamoto, M., Kinoshita, J., Ito, T., et al. (2013). Rho-kinase inhibition prevents the progression of diabetic nephropathy by downregulating hypoxia-inducible factor 1α. Kidney Int. 84, 545–554. doi:10.1038/ki.2013.130
Maxwell, P. (2003). HIF-1: an oxygen response system with special relevance to the kidney. J. Am. Soc. Nephrol. 14, 2712–2722. doi:10.1097/01.asn.0000092792.97122.e0
McCarty, M. F., and DiNicolantonio, J. J. (2014). The molecular biology and pathophysiology of vascular calcification. Postgrad. Med. 126, 54–64. doi:10.3810/pgm.2014.03.2740
McGarry, T., Biniecka, M., Veale, D. J., and Fearon, U. (2018). Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 125, 15–24. doi:10.1016/j.freeradbiomed.2018.03.042
Mokas, S., Larivière, R., Lamalice, L., Gobeil, S., Cornfield, D. N., Agharazii, M., et al. (2016). Hypoxia-inducible factor-1 plays a role in phosphate-induced vascular smooth muscle cell calcification. Kidney Int. 90, 598–609. doi:10.1016/j.kint.2016.05.020
Morand, J., Briançon-Marjollet, A., Lemarie, E., Gonthier, B., Arnaud, J., Korichneva, I., et al. (2019). Zinc deficiency promotes endothelin secretion and endothelial cell migration through nuclear hypoxia-inducible factor-1 translocation. Am. J. Physiol. Cell Physiol. 317, C270-C276–C276. doi:10.1152/ajpcell.00460.2018
Moulin, S., Thomas, A., Arnaud, C., Arzt, M., Wagner, S., Maier, L. S., et al. (2020). Cooperation between hypoxia-inducible factor 1α and activating transcription factor 4 in sleep apnea-mediated myocardial injury. Can. J. Cardiol. 36, 936–940. doi:10.1016/j.cjca.2020.04.002
Moulin, S., Thomas, A., Wagner, S., Arzt, M., Dubouchaud, H., Lamarche, F., et al. (2022). Intermittent hypoxia-induced cardiomyocyte death is mediated by HIF-1 dependent MAM disruption. Antioxidants 11, 1462. doi:10.3390/antiox11081462
Nakatani, S., Mori, K., Sonoda, M., Nishide, K., Uedono, H., Tsuda, A., et al. (2020). Association between serum zinc and calcification propensity (T50) in patients with type 2 diabetes mellitus and in vitro effect of exogenous zinc on T50. Biomedicines 8, 337. doi:10.3390/biomedicines8090337
Nardinocchi, L., Pantisano, V., Puca, R., Porru, M., Aiello, A., Grasselli, A., et al. (2010). Zinc downregulates HIF-1α and inhibits its activity in tumor cells in vitro and in vivo. PLoS One 5, e15048. doi:10.1371/journal.pone.0015048
Nardinocchi, L., Puca, R., Sacchi, A., and D’Orazi, G. (2009). Inhibition of HIF-1alpha activity by homeodomain-interacting protein kinase-2 correlates with sensitization of chemoresistant cells to undergo apoptosis. Mol. Cancer 8, 1. doi:10.1186/1476-4598-8-1
Navarro-González, J. F., Mora-Fernández, C., de Fuentes, M. M., and García-Pérez, J. (2011). Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 7, 327–340. doi:10.1038/nrneph.2011.51
Ndibalema, A. R., Kabuye, D., Wen, S., Li, L., Li, X., and Fan, Q. (2020). Empagliflozin protects against proximal renal tubular cell injury induced by high glucose via regulation of hypoxia-inducible factor 1-alpha. DMSO 13, 1953–1967. doi:10.2147/DMSO.S243170
Nie, P., Lou, Y., Bai, X., Zhu, Y., Guo, Q., Luo, P., et al. (2022). Influence of zinc levels and Nrf2 expression in the clinical and pathological changes in patients with diabetic nephropathy. Nutr. Diabetes 12, 37–7. doi:10.1038/s41387-022-00212-4
Palm, F., Cederberg, J., Hansell, P., Liss, P., and Carlsson, P.-O. (2003). Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 46, 1153–1160. doi:10.1007/s00125-003-1155-z
Pan, R., Chen, C., Liu, W., and Liu, K. (2013). Zinc promotes the death of hypoxic astrocytes by upregulating hypoxia-induced hypoxia-inducible factor-1alpha expression via poly(ADP -ribose) polymerase-1. CNS Neurosci. Ther. 19, 511–520. doi:10.1111/cns.12098
Pang, M., Li, Y., Gu, W., Sun, Z., Wang, Z., and Li, L. (2021). Recent advances in epigenetics of macrovascular complications in diabetes mellitus. Heart, Lung Circulation 30, 186–196. doi:10.1016/j.hlc.2020.07.015
Pang, X., Zhang, Y., Shi, X., Peng, Z., Xing, Y., and Jiarui, H. (2020). Hirudin reduces the expression of markers of the extracellular matrix in renal tubular epithelial cells in a rat model of diabetic kidney disease through the hypoxia-inducible factor-1α (HIF-1α)/vascular endothelial growth factor (VEGF) signaling pathway. Med. Sci. Monit. 26, 9218944–e921900. doi:10.12659/MSM.921894
Peng, K., Xie, W., Wang, T., Li, Y., de Dieu Habimana, J., Amissah, O. B., et al. (2023). HIF-1α promotes kidney organoid vascularization and applications in disease modeling. Stem Cell Res. Ther. 14, 336. doi:10.1186/s13287-023-03528-9
Qannita, R. A., Alalami, A. I., Harb, A. A., Aleidi, S. M., Taneera, J., Abu-Gharbieh, E., et al. (2024). Targeting hypoxia-inducible factor-1 (HIF-1) in cancer: emerging therapeutic strategies and pathway regulation. Pharmaceuticals 17, 195. doi:10.3390/ph17020195
Rabbani, Z. N., Mi, J., Zhang, Y., Delong, M., Jackson, I. L., Fleckenstein, K., et al. (2010). Hypoxia inducible factor 1alpha signaling in fractionated radiation-induced lung injury: role of oxidative stress and tissue hypoxia. Radiat. Res. 173, 165–174. doi:10.1667/RR1816.1
Rawal, S. B., Singh, M. V., Tyagi, A. K., Roy, J., Dimri, G. P., and Selvamurthy, W. (1999). Effect of time exposure to high altitude on zinc and copper concentrations in human plasma. Aviat. Space Environ. Med. 70, 1161–1165.
Ries, M., Basseau, F., Tyndal, B., Jones, R., Deminière, C., Catargi, B., et al. (2003). Renal diffusion and BOLD MRI in experimental diabetic nephropathy. Blood oxygen level-dependent. J. Magn. Reson Imaging 17, 104–113. doi:10.1002/jmri.10224
Rosenberger, C., Khamaisi, M., Abassi, Z., Shilo, V., Weksler-Zangen, S., Goldfarb, M., et al. (2008). Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 73, 34–42. doi:10.1038/sj.ki.5002567
Sato, Y., Endo, H., Okuyama, H., Takeda, T., Iwahashi, H., Imagawa, A., et al. (2011). Cellular hypoxia of pancreatic β-cells due to high levels of oxygen consumption for insulin secretion in vitro. J. Biol. Chem. 286, 12524–12532. doi:10.1074/jbc.M110.194738
Shimobayashi, M., Albert, V., Woelnerhanssen, B., Frei, I. C., Weissenberger, D., Meyer-Gerspach, A. C., et al. (2018). Insulin resistance causes inflammation in adipose tissue. J. Clin. Invest 128, 1538–1550. doi:10.1172/JCI96139
Stiehl, D. P., Jelkmann, W., Wenger, R. H., and Hellwig-Bürgel, T. (2002). Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 512, 157–162. doi:10.1016/S0014-5793(02)02247-0
Sun, F., Du, J., Li, H., Hao, S., Zhao, G., and Lu, F. (2020). FABP4 inhibitor BMS309403 protects against hypoxia-induced H9c2 cardiomyocyte apoptosis through attenuating endoplasmic reticulum stress. J. Cell Mol. Med. 24, 11188–11197. doi:10.1111/jcmm.15666
Sun, H., Saeedi, P., Karuranga, S., Pinkepank, M., Ogurtsova, K., Duncan, B. B., et al. (2022). IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119. doi:10.1016/j.diabres.2021.109119
Takahashi, A. (2024). Zinc supplementation enhances the hematopoietic activity of erythropoiesis-stimulating agents but not hypoxia-inducible factor–prolyl hydroxylase inhibitors. Nutrients 16, 520. doi:10.3390/nu16040520
Takubo, K., Goda, N., Yamada, W., Iriuchishima, H., Ikeda, E., Kubota, Y., et al. (2010). Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 7, 391–402. doi:10.1016/j.stem.2010.06.020
Tang, L., Yi, R., Yang, B., Li, H., Chen, H., and Liu, Z. (2012). Valsartan inhibited HIF-1α pathway and attenuated renal interstitial fibrosis in streptozotocin-diabetic rats. Diabetes Res. Clin. Pract. 97, 125–131. doi:10.1016/j.diabres.2012.01.037
Tang, Y., Yang, Q., Lu, J., Zhang, X., Suen, D., Tan, Y., et al. (2010). Zinc supplementation partially prevents renal pathological changes in diabetic rats. J. Nutr. Biochem. 21, 237–246. doi:10.1016/j.jnutbio.2008.12.010
Theilig, F., Enke, A. K., Scolari, B., Polzin, D., Bachmann, S., and Koesters, R. (2011). Tubular deficiency of von hippel-lindau attenuates renal disease progression in anti-GBM glomerulonephritis. Am. J. Pathology 179, 2177–2188. doi:10.1016/j.ajpath.2011.07.012
Thomas, J. L., Pham, H., Li, Y., Hall, E., Perkins, G. A., Ali, S. S., et al. (2017). Hypoxia-inducible factor-1α activation improves renal oxygenation and mitochondrial function in early chronic kidney disease. Am. J. Physiol. Ren. Physiol. 313, F282-F290–F290. doi:10.1152/ajprenal.00579.2016
Thornton, R. D., Lane, P., Borghaei, R. C., Pease, E. A., Caro, J., and Mochan, E. (2000). Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem. J. 350 Pt 1, 307–312. doi:10.1042/0264-6021:3500307
Tuttle, K. R., Agarwal, R., Alpers, C. E., Bakris, G. L., Brosius, F. C., Kolkhof, P., et al. (2022). Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 102, 248–260. doi:10.1016/j.kint.2022.05.012
Vats, P., Singh, S. N., Kumria, M. M., Ranganathan, S., Arora, M. P., Jain, C. L., et al. (2001). Effect of hypoxia on the circulating levels of essential mineral elements in rats. J. Environ. Biol. 22, 277–282.
Wada, J., and Makino, H. (2013). Inflammation and the pathogenesis of diabetic nephropathy. Clin. Sci. (Lond) 124, 139–152. doi:10.1042/CS20120198
Wang, D., Wang, M., Sun, S., Zhang, C., Song, Y., Li, J., et al. (2024). Hypoxia-induced NLRP3 inflammasome activation via the HIF-1α/NF-κB signaling pathway in human dental pulp fibroblasts. BMC Oral Health 24, 1156. doi:10.1186/s12903-024-04936-w
Wang, L., Peng, W., Zhao, Z., Zhang, M., Shi, Z., Song, Z., et al. (2021). Prevalence and treatment of diabetes in China, 2013-2018. JAMA 326, 2498–2506. doi:10.1001/jama.2021.22208
Wang, S., Nie, P., Lu, X., Li, C., Dong, X., Yang, F., et al. (2020). Nrf2 participates in the anti-apoptotic role of zinc in Type 2 diabetic nephropathy through Wnt/β-catenin signaling pathway. J. Nutr. Biochem. 84, 108451. doi:10.1016/j.jnutbio.2020.108451
Wheaton, W. W., and Chandel, N. S. (2011). Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 300, C385–C393. doi:10.1152/ajpcell.00485.2010
Williamson, J. R., Chang, K., Frangos, M., Hasan, K. S., Ido, Y., Kawamura, T., et al. (1993). Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42, 801–813. doi:10.2337/diab.42.6.801
Wiseman, D. A., Sharma, S., and Black, S. M. (2010). Elevated zinc induces endothelial apoptosis via disruption of glutathione metabolism: role of the ADP translocator. Biometals 23, 19–30. doi:10.1007/s10534-009-9263-y
Wu, K., Zha, H., Wu, T., Liu, H., Peng, R., Lin, Z., et al. (2024). Cytosolic Hmgb1 accumulation in mesangial cells aggravates diabetic kidney disease progression via NFκB signaling pathway. Cell. Mol. Life Sci. 81, 408. doi:10.1007/s00018-024-05433-7
Wu, Q., and Ni, X. (2015). ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 16, 13–19. doi:10.2174/1389450116666150113121054
Yang, F., Li, B., Dong, X., Cui, W., and Luo, P. (2017). The beneficial effects of zinc on diabetes-induced kidney damage in murine rodent model of type 1 diabetes mellitus. J. Trace Elem. Med. Biol. 42, 1–10. doi:10.1016/j.jtemb.2017.03.006
Yang, X., Bao, M., Fang, Y., Yu, X., Ji, J., and Ding, X. (2021). STAT3/HIF-1α signaling activation mediates peritoneal fibrosis induced by high glucose. J. Transl. Med. 19, 283. doi:10.1186/s12967-021-02946-8
Yang, Y., Li, J., Han, T.-L., Zhou, X., Qi, H., Baker, P. N., et al. (2020). Endoplasmic reticulum stress may activate NLRP3 inflammasomes via TXNIP in preeclampsia. Cell Tissue Res. 379, 589–599. doi:10.1007/s00441-019-03104-9
Yin, W.-J., Liu, F., Li, X.-M., Yang, L., Zhao, S., Huang, Z.-X., et al. (2012). Noninvasive evaluation of renal oxygenation in diabetic nephropathy by BOLD-MRI. Eur. J. Radiology 81, 1426–1431. doi:10.1016/j.ejrad.2011.03.045
Zeng, J., Liu, J., Zhao, N., Wong, I. N., and Huang, R. (2024). Caulerpa chemnitzia polysaccharide exerts immunomodulatory activity in macrophages by mediating the succinate/PHD2/HIF-1α/IL-1β pathway. Int. J. Biol. Macromol. 277, 134450. doi:10.1016/j.ijbiomac.2024.134450
Zhan, M., Usman, I. M., Sun, L., and Kanwar, Y. S. (2015). Disruption of renal tubular mitochondrial quality control by myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 26, 1304–1321. doi:10.1681/ASN.2014050457
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903. doi:10.1074/jbc.M800102200
Zhang, X.-X., Kong, J., and Yun, K. (2020). Prevalence of diabetic nephropathy among patients with type 2 diabetes mellitus in China: a meta-analysis of observational studies. J. Diabetes Res. 2020, 2315607. doi:10.1155/2020/2315607
Zhang P, P., Zhang, R., Zhao, F., Heeley, E., Chai-Coetzer, C. L., Liu, J., et al. (2016). The prevalence and characteristics of obstructive sleep apnea in hospitalized patients with type 2 diabetes in China. J. Sleep Res. 25, 39–46. doi:10.1111/jsr.12334
Zhang X, X., Liang, D., Fan, J., Lian, X., Zhao, Y., Wang, X., et al. (2016). Zinc attenuates tubulointerstitial fibrosis in diabetic nephropathy via inhibition of HIF through PI-3K signaling. Biol. Trace Elem. Res. 173, 372–383. doi:10.1007/s12011-016-0661-z
Zhao, B., Niu, X., Huang, S., Yang, J., Wei, Y., Wang, X., et al. (2022). TLR4 agonist and hypoxia synergistically promote the formation of TLR4/NF-κB/HIF-1α loop in human epithelial ovarian cancer. Anal. Cell. Pathol. 2022, 4201262. doi:10.1155/2022/4201262
Zhao, M., Wang, S., Zuo, A., Zhang, J., Wen, W., Jiang, W., et al. (2021). HIF-1α/JMJD1A signaling regulates inflammation and oxidative stress following hyperglycemia and hypoxia-induced vascular cell injury. Cell. & Mol. Biol. Lett. 26, 40. doi:10.1186/s11658-021-00283-8
Zhou, J., Schmid, T., and Brüne, B. (2003). Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. MBoC 14, 2216–2225. doi:10.1091/mbc.e02-09-0598
Zhou, Y., Poczatek, M. H., Berecek, K. H., and Murphy-Ullrich, J. E. (2006). Thrombospondin 1 mediates angiotensin II induction of TGF-beta activation by cardiac and renal cells under both high and low glucose conditions. Biochem. Biophysical Res. Commun. 339, 633–641. doi:10.1016/j.bbrc.2005.11.060
Zhu, Q., Wang, Z., Xia, M., Li, P.-L., Van Tassell, B. W., Abbate, A., et al. (2011). Silencing of hypoxia-inducible factor-1α gene attenuated angiotensin II–induced renal injury in sprague-dawley rats. Hypertension 58, 657–664. doi:10.1161/HYPERTENSIONAHA.111.177626
Zhu, Y., Kang, D., Bai, X., Luo, P., Du, B., and Li, B. (2024). Plasma zinc levels in patients with diabetic nephropathy: is there a relationship with NLRP3 inflammasome activation and renal prognosis? Biol. Trace Elem. Res. doi:10.1007/s12011-024-04369-0
Zhu, Y., Ma, W.-Q., Han, X.-Q., Wang, Y., Wang, X., and Liu, N.-F. (2018). Advanced glycation end products accelerate calcification in VSMCs through HIF-1α/PDK4 activation and suppress glucose metabolism. Sci. Rep. 8, 13730. doi:10.1038/s41598-018-31877-6
Glossary
α-SMA α-smooth muscle actin
ACEI angiotensin-converting enzyme inhibitor
AGEs advanced glycation end products
AngⅡ angiotensin II
ARB angiotensin II receptor blocker
ATP adenosine triphosphate
Bcl-2 B-cell lymphoma-2
Bcl-xL B-cell lymphoma-extra large
BNIP3 Bcl-2 interacting protein 3
BOLD-MRI blood oxygenation level-dependent magnetic resonance imaging
BUN blood urea nitrogen
CKD chronic kidney disease
DKD diabetic kidney disease
DPP-4 dipeptidyl peptidase-4
ECM extracellular matrix
EMT epithelial-mesenchymal transition
EPO erythropoietin
ERS endoplasmic reticulum stress
ESRD end-stage renal disease
ET endothelin
FIH-1 factor-inhibiting HIF-1
GLP-1 glucagon-like peptide-1
GSK-3β glycogen synthase kinase-3β
HIF-1 hypoxia-inducible factor-1
HIF-PHI HIF-PHD inhibitors
HIPK2 homeodomain-interacting protein kinase 2
HO-1 heme oxygenase-1
HRE hypoxia response element
IL-1 interleukins 1
IL-1β interleukin-1β
IL-6 interleukin-6
IL-18 interleukin-18
KLF5 Krüppel-like factor 5
LOXL-2 lysyl oxidase-like 2
MCP-1 monocyte chemoattractant protein-1
NF-κB nuclear factor-κB
NLRP3 nucleotide-binding oligomerization domain-like receptor protein 3
PAI plasminogen activator inhibitor
PERK protein kinase R-like endoplasmic reticulum kinase
PHD prolyl hydroxylase
PI3K/AKT phosphatidylinositol 3-kinase/protein kinase B
RIF renal interstitial fibrosis
ROS reactive oxygen species
RUNX2 runt-related transcription factor 2
SGLT sodium-glucose co-transporter
SOD superoxide dismutase
TGF-β1 transforming growth factor-β1
TIMP tissue inhibitors of metalloproteinase
TNF-α tumor necrosis factor-α
TXNIP thioredoxin-interactingprotein
T2DM type 2 diabetes mellitus
VEGF vascular endothelial growth factor
VHL von Hippel-Lindau
VSMCs vascular smooth muscle cells
Keywords: diabetic kidney disease, hypoxia inducible factor-1α, zinc, HIF-1α, diabetic nephropathy
Citation: Qin W, Nie P, Hui X, Chen F, Hu X, Shi W, Luo M and Li B (2025) Research progress of hypoxia-inducible factor-1α and zinc in the mechanism of diabetic kidney disease. Front. Pharmacol. 16:1537749. doi: 10.3389/fphar.2025.1537749
Received: 01 December 2024; Accepted: 16 January 2025;
Published: 10 February 2025.
Edited by:
Abeda Jamadar, University of Kansas Medical Center, United StatesReviewed by:
Tripti Nair, University of Southern California, United StatesSwetha Thiyagarajan, Self-employed, Sunnyvale, United States
Amar Kumar, University of Illinois Chicago, United States
Purbasa Patnaik, Exelixis Inc., United States
Copyright © 2025 Qin, Nie, Hui, Chen, Hu, Shi, Luo and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manyu Luo, cm9iZXJ0bG15QGpsdS5lZHUuY24=; Bing Li, bGlfYmluZzIwMjBAamx1LmVkdS5jbg==
† These authors share first authorship