
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Pharmacol., 21 March 2025
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 16 - 2025 | https://doi.org/10.3389/fphar.2025.1535259
 Zequn Zheng1,2*
Zequn Zheng1,2* Yongfei Song1*
Yongfei Song1*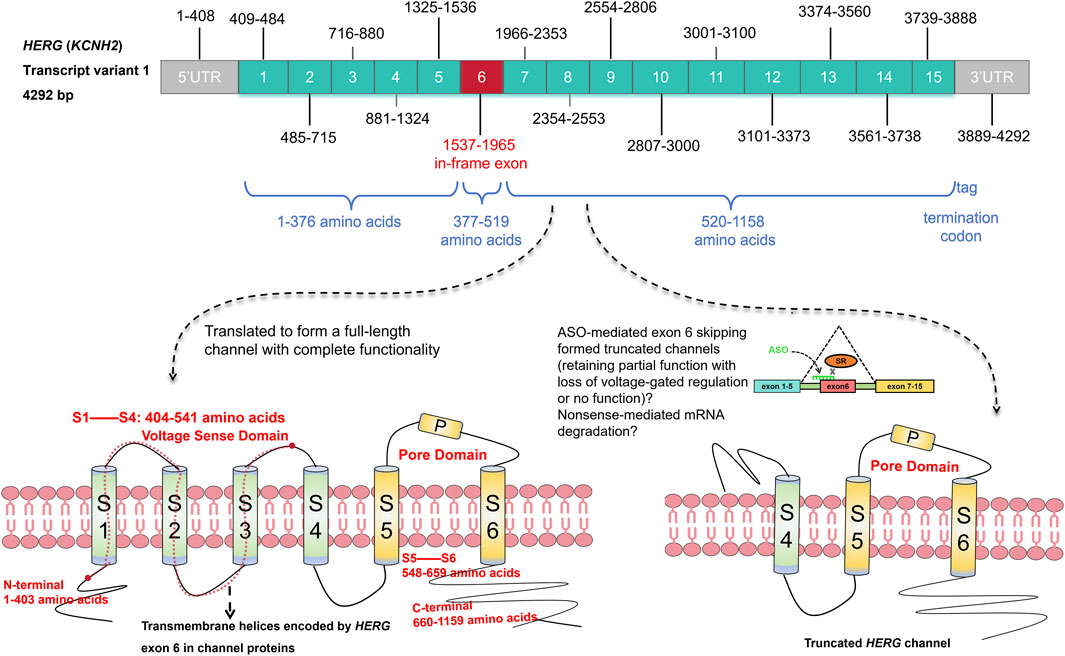
GRAPHICAL ABSTRACT | The unique in-frame exon 6 of the HERG gene as a potential target for antisense oligonucleotide-mediated exon skipping therapy.
Long QT Syndrome (LQTS) represents a paradigmatic genetic arrhythmia with profound impacts on affected individuals, manifesting in syncope, seizures, and sudden cardiac death (Wang et al., 2023; Schwartz et al., 2020). Predominantly triggered by mutations in the human ether-a-go-go-related gene (HERG, also known as KCNH2), LQTS Type 2 (LQT2) represents a considerable fraction of LQTS cases (Schwartz et al., 2020; Wilde et al., 2022). The HERG gene encodes the Kv11.1 channel, crucial for the cardiac action potential’s repolarization phase, with mutations typically leading to potassium channel dysfunctions, thereby extending the cardiac action potential duration and precipitating lethal arrhythmias such as Torsades de Pointes (TdP) (Curran et al., 1995). Over 30% of HERG mutations result in premature termination codons (PTCs), inducing nonsense-mediated mRNA decay (NMD) and subsequently diminishing functional Kv11.1 channel expression (Gong et al., 2007). Recent advancements have illuminated antisense oligonucleotide (ASO)-mediated exon skipping as a strategic approach to bypass mutant exons, offering learnable and promising therapeutic avenues for LQT2 (Michaels et al., 2022; Kim et al., 2022; Oren et al., 2022). Building on comprehensive review we previously published (Zheng et al., 2023), this commentary explores the potential of targeting exon 6 within the HERG gene for therapeutic intervention, highlighting the scientific rationale, associated challenges, and future directions.
ASOs are synthetic single-stranded oligonucleotides designed to hybridize with RNA transcripts, modulating pre-mRNA splicing. By masking splice sites, ASOs can effectively “skip” exons carrying pathogenic mutations, potentially restoring the open reading frame (ORF) and allowing for the production of partially functional proteins (Michaels et al., 2022). This innovative approach, previously applied in diseases like Duchenne Muscular Dystrophy, highlights its therapeutic possibilities (Young and Pyle, 2016). Recent foundational studies have applied exon skipping therapy to CFTR mutations causing cystic fibrosis, where ASO-mediated exon 23 skipping allowed the CFTR containing nonsense mutations to escape NMD, producing truncated amino acids with partial functionality (Michaels et al., 2022; Kim et al., 2022). For diseases caused by specific mutations, such as the exon 6 mutations in HERG associated with LQT2, the ideal ASO would specifically target transcripts from the mutant allele, allowing the wild-type allele to be expressed normally (Zheng et al., 2023).
Exon 6 of the HERG gene is uniquely characterized as an “in-frame exon,” being the only one among the fifteen exons in the longest transcript variant (4,292 bp) that maintains these properties. It precisely spans amino acids 377 to 519 without the risk of frame-shifting mutations since it is encoded by a multiple of three nucleotides and does not share codons with adjoining exons. This specific exon is critical in encoding the N-terminal tail to the transmembrane fragment S3 of the channel, areas frequently associated with mutations leading to NMD, which significantly reduces HERG protein levels (Zequn and Jiangfang, 2021) (Graphic abstract). The potential skipping of exon 6 could theoretically preserve the ORF, allowing for the production of a Kv11.1 channel variant with sufficient functionality to possibly ameliorate the LQT2 phenotype. This strategy hinges on the ability of ASOs targeting exon 6 to enable HERG mRNA with nonsense mutations to bypass NMD, potentially producing a functional protein that could compensate for the channel deficiency. While there are concerns that evasion from NMD might lead to dominant negative effects due to the presence of defective HERG protein within cells, the generation of assuredly functional channels is crucial to address the haploinsufficiency observed in these cases (Anderson et al., 2006; Aizawa et al., 2023).
The exon-skipping therapy, while innovative, creates uncertainty about the function of the resulting protein. As mentioned above, exon 6 of the HERG gene contains essential structural domains critical for channel activation and inactivation gating kinetics. The integrity of these structural domains (such as the N-terminal tail, S1-S2 connector, and S3 helix) is critical for the proper functioning of the Kv11.1 channel (Zequn and Jiangfang, 2021). Excision of exon 6 while preserving the ORF may result in the deletion of critical regions, generating truncated channels that may possess compromised electrophysiological properties. Even without direct evidence showing the impact of entirely removing transmembrane helices S1–S3 on channel function, the potential to trigger enhanced channel activity via specific nonsense mutations within HERG exon 6 remains plausible (Zheng et al., 2023). However, it is important to consider that truncated channels may still retain minimal ion permeability or exhibit unconventional functionality under certain conditions. Although the absence of S1–S3 helices theoretically destabilizes the S4 helix in the membrane environment, the lipid bilayer or other molecular partners may partially stabilize its position. Existing studies also suggest that even structurally defective channels can occasionally form ion leak pathways, contributing to cellular ion flux (Zhorov and Tikhonov, 2024). Such observations imply that the absence of traditional voltage-gating properties does not necessarily result in a complete loss of function. Moreover, alternative mechanisms of functional compensation cannot be ruled out. Truncated channels might interact with endogenous proteins or leverage the membrane environment to stabilize partial functionality, such as stochastic ion permeation. While this remains speculative, there is no definitive evidence to exclude the possibility of residual activity. Interestingly, observations of channels with severed loop regions connecting core transmembrane segments, such as the S4-S5 linker, the S2-S3 intracellular loop, and the S3-S4 extracellular loop, have demonstrated that truncated proteins can, in some cases, retain functionality (de la Peña et al., 2018). Notably, it is unclear whether nonsense mutations in the exon 6 sequence induce NMD or produce truncated proteins. Therefore, it is essential to determine the susceptibility of these variants to NMD in the laboratory before adopting the exon-skipping strategy.
The specificity of ASOs to target only the mutant allele, sparing the wild-type gene, poses a significant challenge, especially in the heterozygous context common in LQT2. Off-target effects could lead to unforeseen consequences, including potential toxicity (Li et al., 2018). In LQT2, the pathogenicity often arises from a heterozygous mutation where one allele carries the mutation while the other remains unaffected. Ideally, an ASO therapy would modulate only the mutant allele’s expression to restore normal function without disrupting the wild-type allele’s beneficial effects. This precise targeting requires identifying unique markers or sequences associated with the mutation that are absent in the wild-type allele, a task that can be complex depending on the nature of the mutation.
To improve delivery, innovations in ASO chemistry, such as locked nucleic acids (LNAs) or peptide nucleic acids (PNAs), improve binding affinity and specificity while reducing the risk of degradation and immunogenicity (Egli and Manoharan, 2023; Baker et al., 2022). These modifications can help achieve the delicate balance between efficacy and safety. In addition, the high specificity of not needing a cocktail but a single ASO sequence to target the heart is a worthy endeavor.
The prolonged impact of exon skipping remains largely unexplored. Continuous or repeated ASO application may cause cumulative toxicity, immune responses, or unforeseen genetic alterations (Egli and Manoharan, 2023). The potential for partially corrected channel function to induce substrates for arrhythmogenesis cannot be easily dismissed.
Beyond scientific and technological challenges, exon 6 skipping therapy raises ethical concerns over patient selection, consent, and treatment access. Moreover, the high costs associated with ASO therapy may limit accessibility, highlighting fairness issues that necessitate concurrent resolution with scientific advancement.
The development of novel strategies to address HERG mutations responsible for LQT2 highlights the necessity of comparing ASO-mediated exon skipping with existing therapeutic approaches. Given the complexity and heterogeneity of HERG mutations, various methods such as small molecule therapies, allele-specific downregulation, allosteric modulator and translational readthrough agents have been explored (Sala et al., 2016; Yu et al., 2014; Zheng et al., 2022; Schwartz et al., 2019; Mehta et al., 2018; Lu et al., 2013). Each of these strategies presents unique strengths and limitations in addressing distinct mutation classes. ASO-mediated exon skipping is a targeted approach that holds promise for addressing Class 1 HERG mutations, particularly nonsense or frameshift mutations in exon 6. Unlike allele-specific downregulation strategies, which reduce dominant-negative effects but risk off-target impacts, inefficient delivery, and potential haploinsufficiency (Lu et al., 2013; Qu et al., 2023), ASOs offer high specificity with reduced systemic toxicity. Compared to wild-type protein enhancement, which compensates for loss of function but risks cytotoxicity and aggregation (Zhang et al., 2022), exon skipping provides a safer alternative. Small molecule therapies, like Lumacaftor, effectively address Class 2 trafficking defects but lack the precision needed for Class 1 mutations, while translational readthrough agents have shown limited success due to toxicity and variability (Yu et al., 2014; Schwartz et al., 2019; Mehta et al., 2018). Although ASO-mediated exon skipping excels in specificity, challenges remain in confirming truncated protein functionality, minimizing off-target effects, and ensuring long-term safety. A personalized, integrative approach combining exon skipping with other methods tailored to specific mutation classes offers the most promise for effectively treating LQT2.
Constructing a minigene plasmid lacking exon 6 but including introns and testing it in tool cells like HEK293 using techniques such as patch-clamping is a straightforward and rapid approach to discern whether the resulting channel produces current. This method also allows for the investigation of phenotypes associated with any point of interest involving nonsense mutations in this transgenic model. Importantly, the functional assessment of HERG channel proteins without exon 6 can be more accurately evaluated in an in vitro model that retains and generalizes the genetic context of the individual. In contrast to using tool cells for transgenic mimicry, the derivation of human induced pluripotent stem cells (hiPSCs) from LQT2 patients facilitates personalized disease models that mirror the genetic variability inherent in the condition. Cardiomyocytes derived from these patient-specific hiPSCs (hiPSC-CMs) offer valuable insights into the functional efficacy and safety of strategies like exon 6 skipping, highlighting the potential for tailored therapeutic interventions in the treatment of LQT2 (Song et al., 2022; Yu et al., 2023). It is particularly valuable for evaluating the efficacy of ASOs designed to skip exon 6 and their impact on cardiomyocyte function, as well as identifying any unintended cardiotoxic effects. Furthermore, developing targeted delivery systems to enhance cardiac cells’ ASO uptake while minimizing systemic exposure presents a promising avenue. Finally, thoughtfully designed clinical trials assessing both safety and efficacy are crucial, incorporating endpoints that reflect meaningful clinical benefits for patients.
ASO-mediated exon 6 skipping in LQT2 marks a frontier in gene therapy, potentially offering targeted treatments for genetic arrhythmias caused by HERG mutations. However, this promising avenue is not without its significant challenges. Some of our colleagues in the field of HERG research would firmly argue that channels lacking the coding structural domain of exon 6 are inherently nonfunctional. We contend against such a definitive stance, as it aligns with Schrödinger’s cat principle: until experimentally confirmed, a critical yet cautiously optimistic perspective is essential—similar to the quantum mechanics thought experiment where a system exists in multiple states until observed, here too, the functionality of channels without exon 6 exists in a state of potential efficacy and inefficacy until definitive experimental evidence is available. We emphasize the need to balance optimism with rigor as we advance, ensuring that the pursuit of innovative therapies is grounded in ethical principles and a commitment to patient safety. This commentary also encourages researchers in the field of LQT2 to entertain the ideas presented here and push the boundaries of innovation further. We salute those contributing to research on LQT2 caused by HERG mutations and extend our gratitude to the reviewers of this manuscript.
ZZ: Conceptualization, Software, Visualization, Writing–original draft, Writing–review and editing. YS: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (grant number 82300347).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that Gen AI was used in the creation of this manuscript. ChatGPT for this manuscript language polish.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aizawa, T., Wada, Y., Hasegawa, K., Huang, H., Imamura, T., Gao, J., et al. (2023). Non-missense variants of KCNH2 show better outcomes in type 2 long QT syndrome. Eur. Eur. pacing, Arrhythm. cardiac Electrophysiol. J. Work. groups cardiac pacing, Arrhythm. cardiac Cell. Electrophysiol. Eur. Soc. Cardiol. 25, 1491–1499. doi:10.1093/europace/euac269
Anderson, C. L., Delisle, B. P., Anson, B. D., Kilby, J. A., Will, M. L., Tester, D. J., et al. (2006). Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 113, 365–373. doi:10.1161/circulationaha.105.570200
Baker, Y. R., Thorpe, C., Chen, J., Poller, L. M., Cox, L., Kumar, P., et al. (2022). An LNA-amide modification that enhances the cell uptake and activity of phosphorothioate exon-skipping oligonucleotides. Nat. Commun. 13, 4036. doi:10.1038/s41467-022-31636-2
Curran, M. E., Splawski, I., Timothy, K. W., Vincent, G. M., Green, E. D., and Keating, M. T. (1995). A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803. doi:10.1016/0092-8674(95)90358-5
de la Peña, P., Domínguez, P., and Barros, F. (2018). Functional characterization of Kv11.1 (hERG) potassium channels split in the voltage-sensing domain. Pflugers Archiv Eur. J. physiology 470, 1069–1085. doi:10.1007/s00424-018-2135-y
Egli, M., and Manoharan, M. (2023). Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic acids Res. 51, 2529–2573. doi:10.1093/nar/gkad067
Gong, Q., Zhang, L., Vincent, G. M., Horne, B. D., and Zhou, Z. (2007). Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation. 116, 17–24. doi:10.1161/circulationaha.107.708818
Kim, Y. J., Sivetz, N., Layne, J., Voss, D. M., Yang, L., Zhang, Q., et al. (2022). Exon-skipping antisense oligonucleotides for cystic fibrosis therapy. Proc. Natl. Acad. Sci. U. S. A. 119, e2114858118. doi:10.1073/pnas.2114858118
Li, D., Mastaglia, F. L., Fletcher, S., and Wilton, S. D. (2018). Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol. Sci. 39, 982–994. doi:10.1016/j.tips.2018.09.001
Lu, X., Yang, X., Huang, X., Huang, C., Sun, H. H., Jin, L., et al. (2013). RNA interference targeting E637K mutation rescues hERG channel currents and restores its kinetic properties. Heart rhythm. 10, 128–136. doi:10.1016/j.hrthm.2012.09.124
Mehta, A., Ramachandra, C. J. A., Singh, P., Chitre, A., Lua, C. H., Mura, M., et al. (2018). Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur. heart J. 39, 1446–1455. doi:10.1093/eurheartj/ehx394
Michaels, W. E., Pena-Rasgado, C., Kotaria, R., Bridges, R. J., and Hastings, M. L. (2022). Open reading frame correction using splice-switching antisense oligonucleotides for the treatment of cystic fibrosis. Proc. Natl. Acad. Sci. U. S. A. 119, e2114886119. doi:10.1073/pnas.2114886119
Oren, Y. S., Avizur-Barchad, O., Ozeri-Galai, E., Elgrabli, R., Schirelman, M. R., Blinder, T., et al. (2022). Antisense oligonucleotide splicing modulation as a novel Cystic Fibrosis therapeutic approach for the W1282X nonsense mutation. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21, 630–636. doi:10.1016/j.jcf.2021.12.012
Qu, Y., Kirby, R., Davies, R., Jinat, A., Stabilini, S., Wu, B., et al. (2023). Time is a critical factor when evaluating oligonucleotide therapeutics in hERG assays. Nucleic acid. Ther. 33, 132–140. doi:10.1089/nat.2022.0043
Sala, L., Yu, Z., Ward-van Oostwaard, D., van Veldhoven, J. P., Moretti, A., Laugwitz, K. L., et al. (2016). A new hERG allosteric modulator rescues genetic and drug-induced long-QT syndrome phenotypes in cardiomyocytes from isogenic pairs of patient induced pluripotent stem cells. EMBO Mol. Med. 8, 1065–1081. doi:10.15252/emmm.201606260
Schwartz, P. J., Ackerman, M. J., Antzelevitch, C., Bezzina, C. R., Borggrefe, M., Cuneo, B. F., et al. (2020). Inherited cardiac arrhythmias. Nat. Rev. Dis. Prim. 6, 58. doi:10.1038/s41572-020-0188-7
Schwartz, P. J., Gnecchi, M., Dagradi, F., Castelletti, S., Parati, G., Spazzolini, C., et al. (2019). From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome Type 2. Eur. heart J. 40, 1832–1836. doi:10.1093/eurheartj/ehz023
Song, Y. F., Zheng, Z. Q., and Lian, J. F. (2022). Deciphering common long QT syndrome using CRISPR/Cas9 in human-induced pluripotent stem cell-derived cardiomyocytes. Front. Cardiovasc. Med. 9, 889519. doi:10.3389/fcvm.2022.889519
Wang, G. Q., Chu, H. X., and Zhao, N. (2023). The clinical diagnosis and management of long QT syndrome: insights from the 2022 ESC guidelines. Rev. CARDIOVASC Med. 24, 170. doi:10.31083/j.rcm2406170
Wilde, A. A. M., Amin, A. S., and Postema, P. G. (2022). Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 108, 332–338. doi:10.1136/heartjnl-2020-318259
Young, C. S., and Pyle, A. D. (2016). Exon skipping therapy. Cell 167, 1144. doi:10.1016/j.cell.2016.10.050
Yu, H., Liu, X., Huang, J., Zhang, Y., Hu, R., and Pu, J. (2014). Comparison of read-through effects of aminoglycosides and PTC124 on rescuing nonsense mutations of HERG gene associated with long QT syndrome. Int. J. Mol. Med. 33, 729–735. doi:10.3892/ijmm.2013.1601
Yu, Y., Deschenes, I., and Zhao, M. T. (2023). Precision medicine for long QT syndrome: patient-specific iPSCs take the lead. Expert Rev. Mol. Med. 25, e5. doi:10.1017/erm.2022.43
Zequn, Z., and Jiangfang, L. (2021). Molecular insights into the gating kinetics of the cardiac hERG channel, illuminated by structure and molecular dynamics. Front. Pharmacol. 12, 687007. doi:10.3389/fphar.2021.687007
Zhang, S., Carlsen, L., Hernandez Borrero, L., Seyhan, A. A., Tian, X., and El-Deiry, W. S. (2022). Advanced strategies for therapeutic targeting of wild-type and mutant p53 in cancer. Cancer 12, 548. doi:10.3390/biom12040548
Zheng, Z., Song, Y., and Lian, J. (2022). What is the potential for lumacaftor as a chemical chaperone in promoting hERG trafficking? Front. Cardiovasc Med. 9, 801927. doi:10.3389/fcvm.2022.801927
Zheng, Z., Song, Y., and Tan, X. (2023). Deciphering HERG mutation in long QT syndrome type 2 using antisense oligonucleotide-mediated techniques: lessons from cystic fibrosis. Heart rhythm. 20, 1169–1177. doi:10.1016/j.hrthm.2023.04.021
Keywords: long QT syndrome type 2, hERG, nonsense-mediated mRNA decay, antisense oligonucleotide, exon skipping, therapy
Citation: Zheng Z and Song Y (2025) When HERG-caused LQT2 encounters antisense oligonucleotide: is exon 6 skipping therapy plausible?. Front. Pharmacol. 16:1535259. doi: 10.3389/fphar.2025.1535259
Received: 27 November 2024; Accepted: 13 March 2025;
Published: 21 March 2025.
Edited by:
Frederic Becq, University of Poitiers, FranceReviewed by:
Dario Melgari, IRCCS San Donato Polyclinic, ItalyCopyright © 2025 Zheng and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongfei Song, c29uZ3lvbmdmZWkxQGdtYWlsLmNvbQ==; Zequn Zheng, MTM0MTQwNTczODRAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.