 Mauricio Di Fulvio
Mauricio Di Fulvio Yakshkumar Dilipbhai Rathod
Yakshkumar Dilipbhai Rathod Shorooq Khader
Shorooq Khader
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 28 March 2025
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 16 - 2025 | https://doi.org/10.3389/fphar.2025.1513125
Thiazides, thiazide-like and loop diuretics are commonly prescribed to manage hypertension and heart failure. The main mechanism of action of these diuretics involve inhibition of Na+ reabsorption in the kidneys, leading to increased urine production. While effective, diuretics, particularly hydrochlorothiazide, have been linked to altered glucose metabolism and other metabolic issues. These disruptions in fuel homeostasis are not clearly related to their primary action of fluid management, raising concerns for patients with metabolic syndrome, in which high blood pressure coexists with obesity, insulin resistance, glucose intolerance and dyslipidemia. In this review, we conducted an extensive examination of existing literature on these classes of diuretics, covering publications from the late 1950s to the present. Our objective was to investigate the origins, development and current understanding of the widely recognized association between the use of diuretics in general and their potential negative impact on glucose homeostasis. We focused on the clinical and experimental evidence of the most commonly prescribed diuretics: hydrochlorothiazide, chlorthalidone, bumetanide and furosemide. On one hand, the clinical evidence supports the hypothesis that the metabolic effects on glucose homeostasis are primarily linked to hydrochlorothiazide, with little, if any impact observed in other diuretics. In addition, these metabolic effects do not appear to be related to their diuretic action or intended pharmacological targets, raising concerns about the long-term metabolic impact of specific diuretics, particularly in vulnerable populations, including those with metabolic syndrome. On the other hand, the experimental evidence using animal models suggest variable effects of diuretics in insulin secretion and general glucose metabolism. Although the mechanisms involved are not clearly understood, further research is needed to uncover the molecular mechanisms by which certain diuretics disrupt fuel metabolism and contribute to metabolic disturbances.
The common belief that thiazides (including thiazide-like diuretics) and loop diuretics impair glucose metabolism is viewed quite differently by clinicians and scientists. Some consider it a serious concern, while others see it as clinically insignificant (Zhang and Zhao, 2016; Hall et al., 2020), especially in patients with hypertension and coexisting conditions where blood pressure control is the primary, sometimes the only goal (Ramsay et al., 1994; Liang et al., 2017; Hall et al., 2020). In fact, many of the clinical trials from 1966 to 2004 focused solely on hypertension (Carter and Basile, 2005; Zillich et al., 2006) often overlooking coexisting metabolic complications such as those seen in metabolic syndrome (MetS). This condition is defined in hypertensive individuals with a constellation of interconnected metabolic abnormalities significantly increasing the risk of type 2 diabetes (T2D), heart disease and stroke (Samson and Garber, 2014). Indeed, MetS, prevalent among obese individuals, often manifests with glucose intolerance, insulin resistance and dyslipidemia (i.e., hypertriglyceridemia and hypercholesterolemia) (Cornier et al., 2008). As people age and gain weight, the prevalence of MetS also rises, exacerbating non-alcoholic fatty liver disease (NAFLD), recently renamed as metabolic dysfunction-associated fatty liver disease (MAFLD) (Eslam et al., 2020) and hypertension (Collaborators et al., 2017; Moore et al., 2017; Godoy-Matos et al., 2020). In fact, the relationship between hypertension and MetS is complex and bidirectional, with hypertension amplifying the risk of adverse health outcomes when combined with other MetS components (Haffner et al., 1992; Liese et al., 1997; Han et al., 2002). For example, abdominal obesity may contribute to insulin resistance and inflammation, aggravating hypertension, while insulin resistance may directly impact blood vessel function, further worsening hypertension (Kawai et al., 2021). In addition, hypertension worsens insulin resistance and disrupts glucose and lipid metabolism, which increases the risk of cardiovascular diseases and T2D in subjects with MetS (Arnlov et al., 2005; Hu and Stampfer, 2005). Consequently, these pathophysiological interconnections pose a significant clinical challenge when treating hypertensive patients with diuretics (Lassen and Jespersen, 2011).
Surprisingly, despite the extensive literature and the impressive research output over the last 75 years, our understanding of the physiopathological mechanisms underlying diuretic-induced metabolic abnormalities remains notably inadequate. Although some studies have proposed a link between hydrochlorothiazide-induced hypokalemia and elevated blood glucose levels (Carter and Basile, 2005), the causal mechanisms whereby some diuretics were associated with hyperglycemia or glucose intolerance (Zillich et al., 2006; Mukete and Rosendorff, 2013; Scheen, 2018) are unclear and hotly debated, in part due to inconsistent findings (Brown et al., 2015; Hall et al., 2020) and the main focus on hypokalemia as the primary electrolyte imbalance associated with diuretics. However, diuretic-induced sodium depletion may also play an underrecognized role in glucose homeostasis. Sodium is essential not only for the function of sodium-glucose cotransporters (SGLTs) in renal glucose reabsorption (Wright et al., 2007) but also for insulin secretion (Ernst et al., 2009; Nita et al., 2014) and insulin action, indirectly via activation of the renin-angiotensin-aldosterone system (Garg et al., 2011; Zhou et al., 2012). Therefore, both chronic and acute sodium depletion, whether induced by diuretics or other medications, such as antidiabetic SGLTs inhibitors (Ansary et al., 2019; Koh et al., 2023), may contribute to the worsening of metabolic disturbances. Moreover, certain metabolic effects of diuretics observed in animal models or humans, such as glucose intolerance (Zatuchni and Kordasz, 1961; Amery et al., 1978; Giugliano et al., 1980b;Sandstrom, 1988;Sandstrom and Sehlin, 1988d; Sandstrom and Sehlin, 1988b; Kempler et al., 1990;Sandstrom et al., 1993; Lopez et al., 1996; Brown et al., 2015;Brown et al., 2016) and insulin resistance (Bakris et al., 2006; Nathan et al., 2007; Sarafidis et al., 2007; Dronavalli and Bakris, 2008) suggest that diuretics exert effects beyond the kidneys. These findings challenge the assumption that diuretics act solely through renal mechanisms and highlight the need for further investigation into their systemic metabolic consequences.
Growing evidence suggest that thiazides, thiazide-like and loop diuretics may have clinically significant extra-renal effects. Advancements in next-generation sequencing and protein expression profiling have demonstrated that the renal targets of thiazides, i.e., Na+Cl− cotransporter (NCC, encoded by SLC12A3) and that of bumetanide/furosemide, i.e., Na+K+2Cl– cotransporter-2 (NKCC2, encoded by SLC12A1) are expressed in different tissues and cells, albeit at lower levels (Di Fulvio and Alvarez-Leefmans, 2009). For instance, NKCC2 has been found in insulin-secreting β-cells (Alshahrani et al., 2012), distal colonic epithelia (Zhu et al., 2011) or neurons of the hypothalamus (Konopacka et al., 2015), whereas NCC was detected in endothelial and smooth muscle cells, heart, lung and liver (Wang et al., 2015), adipocytes (Zhang et al., 2022a), and β-cells as well (Zhang et al., 2022b). In addition to that, diuretics may have “non-specific” yet metabolically relevant targets, a phenomenon that has been known from quite some time. For instance, furosemide can inhibit metabolic pathways modulated by several enzymes including UDP-glucuronyltransferases (Sorgel et al., 1980), 11β-hydroxysteroid dehydrogenases (Escher et al., 1995; Fuster et al., 1998), glucose-6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase and glutathione reductase (Adem and Ciftci, 2016). Similarly, hydrochlorothiazide and bumetanide can inhibit carbonic anhydrase Vb (Kucharczyk et al., 2023) and X (Malebari et al., 2020), respectively, whereas loop diuretics can interfere with signaling mediated by GABAA receptors (Korpi and Luddens, 1997; Thompson et al., 1999) and that of G protein-coupled receptor 35 (Yang et al., 2012). Moreover, bumetanide is well known to inhibit NKCC1, i.e., the ubiquitous Na+K+2Cl–cotransporter (Palfrey and Leung, 1993; Hannaert et al., 2002), while furosemide affects multiple K+Cl−transporters and NKCCs (at higher concentrations) (Haas and McManus, 1983;Popowicz and Simmons, 1988;Hegde and Palfrey, 1992; Lykke et al., 2015), which are unevenly distributed throughout tissues (Adragna et al., 2004; Zhang et al., 2023). Therefore, recognizing that diuretics have diverse pharmacodynamic properties (Wargo and Banta, 2009) and that their pharmacological effects can in turn vary based on many factors including age, gender or ethnicity (Andreasen et al., 1984; Chaudhry et al., 1984; Chun et al., 2008; Conde-Martel et al., 2024), along with the usually disregarded yet significant role of the kidneys in glucose production (Stumvoll et al., 1999; Mather and Pollock, 2011; Alsahli and Gerich, 2017; Legouis et al., 2022; Daza-Arnedo et al., 2023), may reduce bias when evaluating their metabolic effects.
In the next sections, we will briefly introduce thiazides, thiazide-like and loop diuretics from a historic perspective to illustrate how the success of one specific class of these diuretics in treating hypertension overshadowed their undesired metabolic effects and reduced our curiosity to study them to better understand their basic and clinical pharmacology. We will then revisit elemental concepts related to glucose homeostasis to refresh the intricacies of their regulation and identify potential points for future research while focusing on the available evidence relating the use of diuretics with altered fuel homeostasis within the context of the MetS.
Hydrochlorothiazide and chlorthalidone, a benzothiadiazide and a thiazide-like diuretic, respectively were introduced during 1957-59 and the former quickly became a mainstay in the treatment of hypertension. Its popularity at the time stemmed from its perceived clinical (anti-hypertensive) effectiveness, low cost and apparently better safety profiles compared to earlier diuretics (Au and Raisz, 1960; Conway and Lauwers, 1961; Laragh, 1962; Mizgala, 1965). Indeed, hydrochlorothiazide gained widespread popularity following a controlled trial for the management of hypertension published in 1970 (Author Anonymous, 1970). Importantly, over the period spanning the 1960s and 1970s, hydrochlorothiazide underwent continuous examination in many clinical trials, predominantly focused on controlling hypertension (Collins et al., 1990; Antonietta et al., 2022). Almost 20 years after its discovery, a pharmacokinetically dissimilar thiazide-like sulfonamide derivative of hydrochlorothiazide, i.e., chlorthalidone, emerged in the clinic (Riess et al., 1977; Chen and Chiou, 1992). Surprisingly, its efficacy in managing hypertension was first evaluated in 1979 (Author Anonymous, 1979) and several studies, decades later, consistently suggested that this and other thiazide-like diuretics may have a more favorable clinical profile than hydrochlorothiazide (Harrower et al., 1985; Ernst et al., 2006; Chalmers and Arima, 2010; Dorsch et al., 2011; Tziomalos et al., 2013; Liang et al., 2017; Khenhrani et al., 2023). Yet, hydrochlorothiazide prevailed, and still stands, as one of the most frequently prescribed medications in the United States, with a staggering ∼39 million prescriptions for this drug alone in 2021 (clincalc.com/DrugStats/Drugs/Hydrochlorothiazide).
Although there is little evidence that low doses of hydrochlorothiazide (12.5–25 mg daily) reduce the risk of heart attack, stroke or death (Messerli et al., 2011), higher doses have been proven effective in lowering blood pressure and improving cardiovascular outcomes in patients with hypertension. Early trials on small number of subjects during the late 1950s showed the benefits of higher doses, though they did not consider their metabolic effects (Beyer et al., 1957; Bayliss et al., 1958; Bunn, 1958; Freis et al., 1958; Laragh et al., 1958; Rochelle et al., 1958; Wilkins, 1958; Wilkins et al., 1958; Heinemann et al., 1959; Reinhardt, 1959; Laragh, 1967). As our understanding of hypertension grew in the 1980s and 1990s, hydrochlorothiazide remained a key treatment for hypertension (Wilhelmsen et al., 1981; Hebert et al., 1993; Moser and Hebert, 1996; Savage et al., 1998), even as newer drugs with fewer, if any, metabolic issues (e.g., ACE inhibitors, calcium channel blockers and β-blockers) began to replace it (Officers et al., 2002; Grossman and Messerli, 2006). Meanwhile, chlorthalidone was also effective in treating hypertension, as seen in the large ALLHAT trial (Elliott, 1996), which compared different blood pressure medications. Despite some criticisms of this trial (McInnes, 2003; Hebert et al., 2007), its findings heavily influenced future treatment guidelines, promoting the use of thiazide diuretics (Chobanian et al., 2003; Ernst and Moser, 2009). However, these guidelines largely focused on hydrochlorothiazide, not chlorthalidone or other thiazide-like drugs like indapamide (Stafford et al., 2010; Messerli and Bangalore, 2011). At this point, it is important to recognize that the term thiazide has often been used loosely to refer to hydrochlorothiazide, chlorthalidone and indapamide, despite their pharmacokinetic and pharmacodynamic differences between them (Kurtz, 2010). Over time, each of these diuretics has inherited the benefits and drawbacks of the most commonly prescribed and studied one, i.e., hydrochlorothiazide.
Indeed, early studies did suggest that (hydrochloro)thiazide diuretics might be linked to negative effects on glucose metabolism (Johnston and Cornish, 1959; Goldner et al., 1960; Zatuchni and Kordasz, 1961; Runyan, 1962; Author Anonymous, 1963; Anderson, 1966; Author Anonymous, 1971b; Hollenberg and Mickiewicz, 1989; Pollare et al., 1989; Lithell et al., 1990; Plavinik et al., 1992). However, not all research confirmed these findings, with some studies failing to show any such connections (Cornish et al., 1961; Runyan, 1962; Jackson and Nellen, 1966; Andersen and Persson, 1968; Chaudhury et al., 1968; Healy et al., 1970; Berglund et al., 1986; Grimm et al., 1996; Lakshman et al., 1999). The differences in results may be due to variations in study design, dosages, or the populations studied. At any rate, the metabolic effects of hydrochlorothiazide were considered mild or irrelevant from the standpoint of managing hypertension. More recently, however, research has focused on the use of hydrochlorothiazide, alone or in combination with other drugs, for treating hypertension in specific groups (Omboni et al., 2009), such as the elderly and those with obesity, MetS or T2D (Klauser et al., 1991; Middeke et al., 1997; Reisin et al., 1997; Maitland-van der Zee et al., 2005; Siegel et al., 2008; Cooper-DeHoff et al., 2010; Manrique et al., 2010; Gong et al., 2014; Brown et al., 2016; Huang et al., 2016; Georgianos and Agarwal, 2019). Interestingly, no substantial effects of hydrochlorothiazide or chlorthalidone on plasma insulin were reported in several of these and other trials (Klauser et al., 1991; Plavinik et al., 1992; Price et al., 2013). However, there has been less emphasis on thiazide-like diuretics, despite evidence that both chlorthalidone and indapamide may offer better metabolic outcomes compared to hydrochlorothiazide (Kostis et al., 1997; Black et al., 2008; Karnes et al., 2014; Singh et al., 2018). With apparently few exceptions (Jian-Liang et al., 2004), these drugs can reduce blood pressure with less impact on blood glucose and cholesterol levels (Liang et al., 2017). Despite this, many studies, especially those involving hydrochlorothiazide, have led to the widespread belief that diuretics invariably affect glucose metabolism, regardless of their class or specific characteristics.
The story of loop diuretics began in the early 1960s, when ethacrynic acid was found to increase urine production in both animals and humans (Beyer et al., 1962; Bernstein et al., 1965; Cannon et al., 1965). Ethacrynic acid became the first non-sulfonamide loop diuretic used in clinical settings, leading to the development of more powerful loop diuretics. In the mid-to-late 1960s, furosemide was synthetized and quickly gained popularity due to its strong diuretic effects, fast action and effectiveness, especially in treating heart failure, hypertension, edema and kidney failure (Ingram, 1964; Godwin and Gunton, 1965; Laragh et al., 1966; Stason et al., 1966; Davidov et al., 1967; Earley, 1967; Walker, 1967; Joynt and Morrin, 1968; Kirkendall and Stein, 1968; Cannon and Kilcoyne, 1969; Shanoff, 1969). By the 1970s, furosemide became one of the most commonly prescribed diuretics, with fewer undesired effects compared to earlier diuretics, including hydrochlorothiazide (Feit, 1971; Wertheimer et al., 1971; Cannon, 1972; Valmin and Hansen, 1975; Mahabir and Bacchus, 1976; Finnerty et al., 1977; Araoye et al., 1978; Coodley et al., 1979; Dettelbach and Bennett, 1979). Other loop diuretics with better bioavailability and longer-lasting effects, such as bumetanide, torsemide, azosemide and piretanide, were introduced around this time as well (Asbury et al., 1972; Murdoch and Auld, 1975; Hettiarachchi et al., 1977; Jayakumar and Puschett, 1977; Benet, 1979; Brater et al., 1979; Konecke, 1981; Whelton, 1981; Stroobandt et al., 1982; Halstenson and Matzke, 1983; McNabb et al., 1984; Ward and Heel, 1984; Clissold and Brogden, 1985; Car et al., 1988). Many of these diuretics are still in use today (Blose et al., 1995; Bagshaw et al., 2007; Carone et al., 2016; Mentz et al., 2016; Rahhal et al., 2019; Singh et al., 2023). Even after 40 years, loop diuretics remain a key treatment for conditions involving excess fluid retention, as supported by ongoing clinical trials (Blake, 1990; Kissling and Pickworth, 2014; Ozieranski et al., 2019; Eid et al., 2021; Verbrugge and Menon, 2022; Greene et al., 2023; Mentz et al., 2023; Cuthbert and Clark, 2024; Kapelios et al., 2024; Krim et al., 2024).
However, most clinical trials on loop diuretics over the past 50 years have primarily and understandably focused on how they affect edema and electrolyte balance, rather than their potential impact on glucose metabolism. As a result, there is limited evidence linking loop diuretics to metabolic issues, especially compared to hydrochlorothiazide. However, early on, loop diuretics seemed to inherit the perceived metabolic effects of hydrochlorothiazide (Toivonen and Mustala, 1966). This concern may have originated from a 1959 study that first raised the possibility of diuretics affecting glucose metabolism (Freis and Finnerty, 1959) based on the effects of hydrochlorothiazide. Although there are few direct studies connecting loop diuretics (such as furosemide) to metabolic problems (Lavender and McGill, 1974; Tasker and Mitchell-Heggs, 1976; Khaleeli and Wyman, 1978), isolated cases of glucose intolerance or diabetes in patients using furosemide have been reported. Nevertheless, one study in 1966 found that furosemide had little effect on glucose tolerance over 3 months in both healthy people and those with hypertension (Jackson and Nellen, 1966). Another study suggested that ethacrynic acid also had minimal effects on glucose levels in mildly hypertensive patients (Andersen and Persson, 1968). Yet, later reports documented some cases of glucose intolerance associated with furosemide (Coni et al., 1974; Cowley and Elkeles, 1978; Kobayakawa et al., 2003). On the other hand, short-term studies showed no significant impact on blood sugar levels from either furosemide or bumetanide in both healthy individuals and patients with T2D (Asbury et al., 1972; Kaldor et al., 1975). Similarly, studies in 1980 indicated that neither diuretic had a significant effect on insulin or glucagon secretion (Giugliano et al., 1980b; Luyckx et al., 1980), though furosemide did slightly alter insulin and glucagon responses without affecting glycemia (Giugliano et al., 1980a). In 1981, a study found that bumetanide even improved glucose tolerance, but furosemide did not (Robinson et al., 1981). Further research indicated that, unlike hydrochlorothiazide, bumetanide had no significant effect on insulin or other hormone levels in dog pancreas models (Hermansen et al., 1985). The introduction of piretanide in the 1980s also did not consistently affect glucose tolerance or insulin levels, though both piretanide and furosemide were linked to changes in cholesterol levels in hypertensive patients (Valimaki et al., 1983; Weidmann et al., 1983; Campbell et al., 1998). However, later studies did not confirm these findings consistently (Harno et al., 1985; Chaudhuri and Catania, 1988; Weidmann et al., 1993; Lind et al., 1995; van der Heijden et al., 1998). In fact, piretanide (Harno et al., 1985) and likely bumetanide (Harno et al., 1985) increased insulin secretion in humans.
Therefore, overall, it appears that the “diabetogenic” risks commonly associated with diuretics are more strongly linked to hydrochlorothiazide (Padwal and Laupacis, 2004; Stump et al., 2006) than other classes of diuretic or anti-hypertensive medications. Although “meta-analysis (97 comparisons across 95 trials) demonstrated a statistically significant but clinically unimportant increase in FPG [fasting plasma glucose]” (Hall et al., 2020), the impact of any diuretic on glucose homeostasis seems to depend on several factors, including the type of diuretic used and the specific metabolic context on which these diuretics are being studied (Grossman et al., 2011).
The common belief that diuretics negatively affect fuel balance in humans lacks strong experimental support, particularly for thiazide-like and loop diuretics. Nonetheless, we will focus on reviewing experimental evidence, mostly from animal studies, to better understand the potential effects of hydrochlorothiazide and loop diuretics on key processes involved in glucose regulation. This includes their impact on insulin secretion and the production and use of glucose in the liver and kidneys.
Insulin secreted from β-cells of the islets of Langerhans in the pancreas promotes the uptake of glucose from the blood into muscle and other insulin-sensitive tissues for immediate use (glycolysis) or fat storage (lipogenesis). In contrast, glucagon secreted by α-cells of the islet, has the opposite effect of insulin. When glycemia is low, such as during fasting or between meals, glucagon promotes the hepatic break-down of stored glycogen (glycogenolysis) into glucose for release into the bloodstream, or the renal synthesis of glucose from non-carbohydrate sources (Jiang and Zhang, 2003; Mutel et al., 2011; Bankir et al., 2016). Importantly, the liver and the kidneys, and to a much lesser extent the small intestine can produce glucose from amino acids and glycerol, through a process called de novo gluconeogenesis. This ensures a steady supply of glucose for organs and tissues, especially during long periods of fasting or prolonged exercise. In the case of insulin-sensitive tissues, such as the muscles and adipose tissue (Booth et al., 2016; Merz and Thurmond, 2020), when insulin binds to its receptors, glucose transporters (e.g., GLUT4) translocate to the cell membrane allowing glucose to enter the cell, where it can be used for energy during exercise or stored as glycogen (muscle) for future use. In adipose tissue, fat cells store energy in the form of triglycerides. On one hand, fatty acids produced from triglycerides by lipolysis can be used as an energy source by many tissues, including muscle cells (Hargreaves and Spriet, 2020). On the other hand, glycerol, also produced from triglycerides by lipolysis, can be converted into glucose through gluconeogenesis in the liver, providing an additional source of glucose during fasting or periods of increased energy demand (Han et al., 2016). These concepts, outlined in Figure 1, are relevant for our discussion; as insulin secretion, the glycolytic and/or lipolytic potential of tissues, gluconeogenesis and likely most aspects of glucose and energy homeostasis have been found defective and implicated in the pathogenesis and/or progression of hypertension and MetS (Katsimardou et al., 2020).
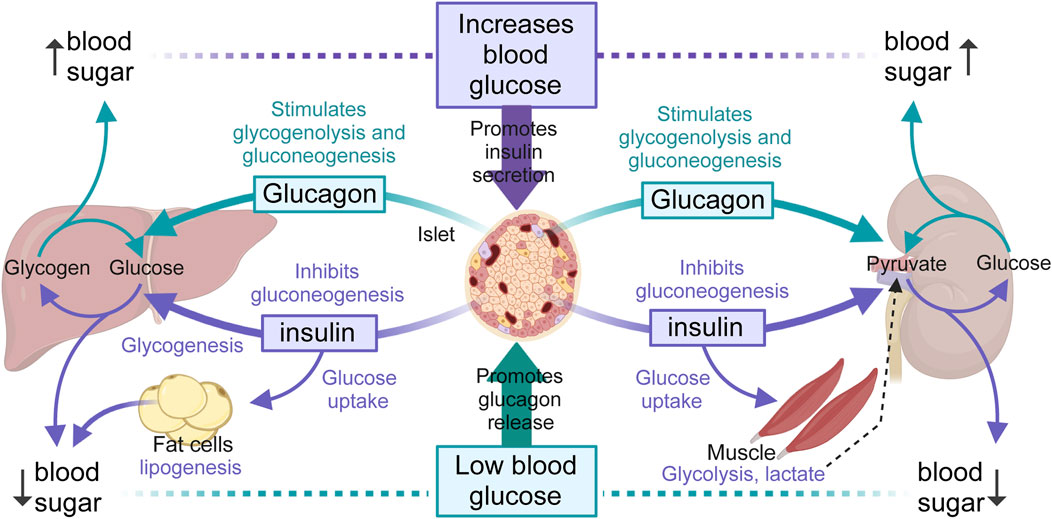
Figure 1. Overview of blood glucose regulation. The liver, muscles and kidneys are major modulators of blood glucose levels by releasing glucose through glycogenolysis (liver, muscle) and gluconeogenesis (liver and kidneys), in turn orchestrated by insulin (purple arrows) and glucagon (green arrows) secreted by β- and α-cells of the pancreatic islet, respectively. Glycogenolysis breaks down stored glycogen into glucose-6-phosphate, then free glucose after dephosphorylation, while gluconeogenesis forms glucose-6-phosphate from various non-hydrocarbon precursors (e.g., pyruvate, lactate, glycerol, glutamine). Only the liver, kidneys and small intestines (not represented) can release glucose from glucose-6-phosphate due to the presence of glucose-6-phosphatase activity. Hepatic glycogen breakdown releases glucose, while muscle glycogen breakdown releases lactate, a substrate that can be converted back into glucose by the liver and kidneys after conversion to pyruvate. The kidneys use glucose mainly in the renal medulla and release it from the renal cortex, due to enzyme differences alone the nephron. Renal medulla cells, like neurons, can accumulate glycogen but cannot release glucose. Renal cortex cells can produce and release glucose but cannot synthesize glycogen. In adipocytes, insulin promotes the uptake of glucose and its transformation into fat.
The process by which nutrients, particularly glucose, trigger insulin secretion from islet β-cells is complex and involves many signals (Di Fulvio et al., 2014). However, medical textbooks often oversimplify this process. Typically, the consensus mechanism is described as follows (see Figure 2): When glucose enters β-cells, it undergoes glycolysis, which raises intracellular ATP levels. This increase in ATP closes ATP-sensitive K+ channels (KATP channels), causing depolarization of the cell membrane. As a result, voltage-gated Ca2+ channels open, allowing Ca2+ to flow into the cell. The influx of Ca2+ triggers the release of insulin from the β-cells into the bloodstream. While this mechanism is important, it is incomplete (Henquin et al., 2009; Merrins and Kibbey, 2024). Indeed, Cl− channels and Cl− transporters also help regulate β-cell membrane potential and excitability, both crucial for insulin release (Best et al., 2010). In fact, recent studies have clearly defined the roles of some of these Cl− channels in islet physiology (Crutzen et al., 2016; Kang et al., 2018; Stuhlmann et al., 2018; Di Fulvio et al., 2020) and importantly, some Cl− transporters help maintain the intracellular Cl− concentration ([Cl−]i) above its predicted thermodynamic equilibrium, facilitating the movement of Cl−out of the cell and through Cl− channels in an electrogenic manner. Notably, some of these Cl− transporters in β-cells can be directly targeted by thiazide and loop diuretics (Di Fulvio and Aguilar-Bryan, 2019). In fact, hydrochlorothiazide (Hoskins and Jackson, 1978; Sandstrom et al., 1993; Kucharczyk et al., 2023), trichlormethiazide (Seltzer and Allen, 1969), hydroflumethiazide (Hermansen et al., 1985), bumetanide (Hermansen et al., 1985; Sandstrom, 1990), furosemide (Aynsley-Green and Alberti, 1973; Hermansen et al., 1986; Sandstrom and Sehlin, 1988c; Eberhardson et al., 2000) and indapamide (Hermansen et al., 1986) can all influence insulin secretory responses in vitro and in vivo in animal models. In addition, hydrochlorothiazide, bumetanide and furosemide were also consistently linked to altered blood glucose and impaired glucose tolerance in a variety of animal models (Foy, 1967; Weller and Borondy, 1967; Foy and Furman, 1969; Foy and Furman, 1971; Foy and Furman, 1972; Hoskins and Jackson, 1978;Papaccio and Esposito, 1987; Sandstrom, 1988; Sandstrom and Sehlin, 1988d; Sandstrom and Sehlin, 1988b; Ray et al., 1993; Sandstrom et al., 1993). Therefore, these data support the hypothesis that the metabolic effects associated with the use of thiazide, thiazide-like, loop-diuretics are related, at least in part, to direct or indirect effects on islet β-cell secretory function.
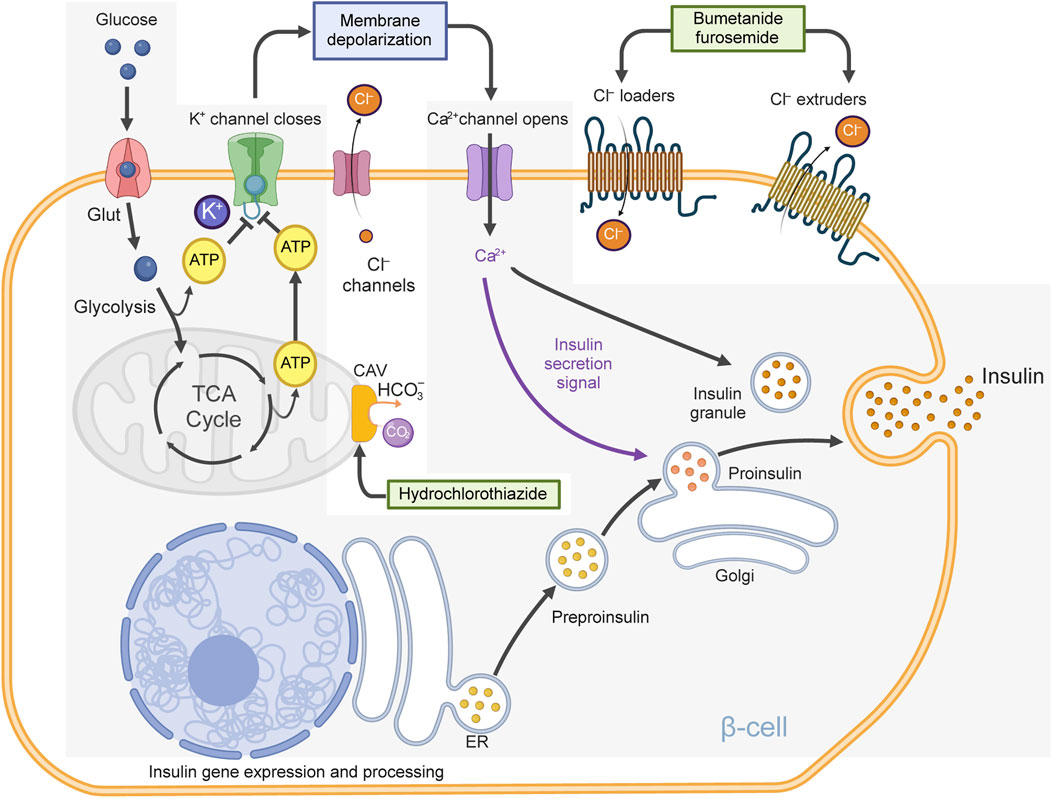
Figure 2. Oversimplified model of insulin secretion. Described is a β-cell containing glucose transporters (Glut), KATP-channels, voltage-gated Ca2+ channels, bumetanide-sensitive Cl− loaders (e.g., NKCC2, NKCC1), furosemide-sensitive Cl− extruders (e.g., KCC1, KCC2, KCC3, KCC4) and Cl− channels [e.g., volume-regulated anion channels, (VRAC), Ca2+ activated Cl− channels (ANO1) and others]. Note that Cl− loaders and extruders help maintain the intracellular Cl− concentration above thermodynamic equilibrium, making possible the electrogenic exiting of Cl− ions, when Cl− channels are opened, contributing to plasma membrane depolarization. When glucose is transported into the β-cell, it undergoes glycolysis, generating ATP and metabolites that affect cellular osmolarity and cell volume. ATP closes KATP-channels, reducing K+ permeability and causing plasma membrane depolarization. Metabolites and Ca2+ open Cl− channels triggering inward Cl− currents (Cl− exits the cell). Many Cl− channels likely contribute to these currents which together with reduced K+ permeability are responsible for the activation of voltage-gated Ca2+ channels, thus leading to Ca2+ influx, action potentials, electrical activity and insulin release. Note: hydrochlorothiazide can inhibit mitochondrial carbonic anhydrase Vb (CAV), which limits the supply of HCO3− to pyruvate carboxylase (and other carboxylases) reducing the biosynthesis of oxaloacetate, an intermediary of the tricarboxylic acid (TCA) cycle potentially reducing ATP and contributing to reduced KATP-channel closure. The consensus model of insulin secretion is greyed.
At first sight, the reported influence of hydrochlorothiazide on insulin secretion from rodent islets in vitro (Malaisse and Malaisse-Legae, 1968; Sandstrom et al., 1993) might now seem related to inhibition of NCC. However, the transcript levels of SLC12A3 were consistently very low or undetectable in both human and rodent islets, as determined by traditional methods (Zhang et al., 2022b) or advanced transcriptome profiling (Riahi et al., 2018; Jaafar et al., 2019; Chen et al., 2022). Moreover, hydrochlorothiazide have been shown to diminish insulin secretion from islets of obese mice by reducing Ca2+ influx rather than altering [Cl−]i, K+ or Cl−fluxes (Sandstrom et al., 1993). Therefore, the potential adverse effects possibly induced by hydrochlorothiazide on the islet secretory function might be influenced by targets other than NCC. Indeed, it is known that hydrochlorothiazide can target several ion transporters and enzymes including SLC4A8, a Na+-dependent Cl−/HCO3− exchanger (NDCBE), SLC26A4, a Na+-independent Cl−/HCO3− exchanger (Pendrin) and carbonic anhydrases (Pickkers et al., 1999; Leviel et al., 2010; Sinke et al., 2014), all of which were shown to play roles in insulin secretion (Parkkila et al., 1998; Sener et al., 2007). In fact, recent data suggest that hydrochlorothiazide may inhibit insulin secretion from normal mouse islets by blocking the activity of mitochondrial carbonic anhydrase Vb (Kucharczyk et al., 2023) (see Figure 2). Importantly, this enzyme provides HCO3− ions to different enzymes that participate in intermediary metabolism including pyruvate carboxylase (anaplerosis, gluconeogenesis), propionyl-CoA carboxylase, 3-methylcrotonyl-CoA carboxylase (branched chain amino acids catabolism) and carbamoylphosphate synthase 1 (urea cycle). Therefore, it is plausible that hydrochlorothiazide, by targeting carbonic anhydrases and other enzymes, may have wider metabolic effects than predicted, at least in animal models.
At any rate, the long-term effects of hydrochlorothiazide treatment on essential metabolic parameters such as body weight, body composition and dynamic evaluations of glucose homeostasis and metabolomics have not yet been conducted. Furthermore, the long-term role of renal NCC in the regulation and/or maintenance of glycemia also remains unknown. This is relevant, as thiazides in general have been proposed to promote metabolic dysregulation by inhibiting insulin secretory responses to nutrients through their hypokalemic effects (Zillich et al., 2006), and perhaps by direct effects on renal gluconeogenesis (Fulgraff et al., 1972). Moreover, the current hypothesis that hydrochlorothiazide may worsen glucose homeostasis through mechanisms related to insulin secretion has been recently challenged. Indeed, islets from young mice lacking NCC or NDCBE (NCCKO or NDCBEKO, respectively) exhibited normal secretory responses to glucose (Kucharczyk et al., 2023). Yet, hydrochlorothiazide triggered acute glucose intolerance in these mice. Hence, this diuretic can have metabolic effects independently of both transporters and by mechanisms unrelated to direct effects on islet NCC o NDCBE.
Nevertheless, a recent study has confirmed the presence of NCC in some but not all insulin-positive β-cells of both human and rodent islets (Zhang et al., 2022b). In these contexts, it was proposed that NCC may act as a receptor for interleukin 18 (IL-18), potentially collaborating with receptors for the incretin glucagon-like peptide 1 (GLP-1) to enhance β-cell mass and help maintain glucose homeostasis (Zhang et al., 2022b). Although it is unknown if the ion transport activity of NCC is required to interact with IL-18 (Wang et al., 2015), the potential functional/molecular interplay between NCC and GLP-1 receptors implies a permissive role for the former in the prandial islet secretory response to incretins. Notably, the insulinotropic effect of GLP-1 was lost in islets of NCCKO mice (Zhang et al., 2022b). Nevertheless, it remains unknown whether hydrochlorothiazide or thiazide-like diuretics reduce GLP-1 responses, glucose tolerance, energy intake behavior and feeding patterns in the long term. This constitutes an interesting hypothesis to test given that mice deficient in IL-18 signaling are insulin resistant, hyperphagic and obese (Netea et al., 2006; Zorrilla et al., 2007; Pazos et al., 2015). Further, some of IL-18 effects may be mediated by NCC (Wang et al., 2015; Zhang et al., 2022a; Zhang et al., 2022b) and potentially sensitive to thiazides and thiazide-like diuretics.
Like NCCKO islets, those from mice lacking NCC exclusively in β-cells (NCCβKO) showed preserved glucose-stimulated insulin secretion. However, these mice had reduced β-cell mass and enhanced islet inflammation under high fat diet (HFD) conditions (Zhang et al., 2022b). Therefore, it has become clear that β-cells can release insulin without relying on NCC, especially when triggered by glucose, although this may not be the case for other stimuli, including that elicited by GLP-1. Further, the data also imply a role for NCC in inflammatory processes, which may be of clinical relevance given the relationship that exists between low grade local tissue inflammation, obesity and the progression of MetS (Aronson et al., 2004; Grundy et al., 2005; Haffner, 2006). Although hydrochlorothiazide did impair glucose tolerance in normal mice through mechanisms related to β-cell insulin secretion, but independent of NCC (Kucharczyk et al., 2023), NCCβKO mice did not show reduced insulin responses to exogenous glucose. In fact, NCCβKO and NCCKO mice were normotolerant to glucose (Zhang et al., 2022b; Kucharczyk et al., 2023). Consequently, when considered collectively, these data suggest that hydrochlorothiazide could potentially induce glucose intolerance, particularly in mice models, through various mechanisms including those partially associated with β-cell function and mass, those related to intermediary metabolism, alongside others yet to be uncovered.
At any rate, the long-term role of NCC either as an ion transporter sensitive to thiazides, as an IL-18 receptor or as a potential partner for GLP-1 receptors in β-cells or in any capacity in metabolically active tissue awaits exploration, particularly within the context of obesity, the most prevalent component of MetS. Along these lines, HFD-fed NCCβKO mice showed exacerbated body weight gain, glucose intolerance and insulin resistance relative to chow fed mice (Zhang et al., 2022a; Zhang et al., 2022b). Therefore, it is possible that β-cell NCC may play a protective role against overnutrition and metabolic dysregulation. While it is uncertain whether these alterations also involve modified incretin responses, within the framework of obesity, MetS and the use of hydrochlorothiazide for treating hypertension associated with these conditions, these findings suggest an intriguing hypothesis: that overweight or overnutrition might amplify the metabolic effects of these diuretics by inhibiting NCC and/or other targets in β-cells and in metabolically active tissues but independently of insulin secretion.
When considering the potential metabolic effects of loop diuretics, a similar contextual line of thought as that conveyed for hydrochlorothiazide can be pragmatic. Certainly, several “extrarenal hypotheses” have been proposed over the years to better understand some observed metabolic effects of loop diuretics, mostly bumetanide and furosemide, in humans and animal models. For instance, it has been known for quite some time that these two diuretics may directly impair insulin secretion from islets in vitro and deteriorate glucose tolerance in mice (Sandstrom, 1988; Sandstrom and Sehlin, 1988d; Sandstrom and Sehlin, 1988b; Sandstrom, 1990; Sandstrom et al., 1993). Importantly, the demonstrated acute in vitro inhibitory effects of low concentrations of bumetanide on islet insulin secretion (Sandstrom, 1990) seem to stem mostly from inhibition of NKCC1, as its exclusive elimination from β-cells precluded the effects of bumetanide (Abdelgawad et al., 2022).
However, experiments using islets of null mice lacking NKCC1 (NKCC1KO) gave unexpected results. Contrary to initial expectations, pancreatic islets from 3-4w old NKCC1KO mice showed exaggerated insulin responses to glucose in vitro rather than a reduced response (Alshahrani and Di Fulvio, 2012). These data suggest that NKCC1 is dispensable for insulin secretion and that the dependence of insulin secretion on acute inhibition of NKCCs by bumetanide (Best, 2005) or furosemide (Sandstrom and Sehlin, 1988c;a) is rather complex. Along these lines, NKCC1KO mice exhibited exaggerated glucose tolerance (Alshahrani and Di Fulvio, 2012), which is also surprising given the well-known detrimental effects that bumetanide and furosemide have on glucose tolerance in mice (Sandstrom, 1988; Sandstrom and Sehlin, 1988d; Sandstrom and Sehlin, 1988b). Although these results are challenging to reconcile from a metabolic perspective, especially when considering that NKCC1KO null mice display a range of developmental and functional abnormalities (Delpire et al., 1999; Flagella et al., 1999; Evans et al., 2000; Meyer et al., 2002b; Walker et al., 2002; Bradford et al., 2016), recent data from patients harboring inactivating mutations in the SLC12A2 gene have suggested a potential implication for NKCC1 in intestinal function (Koumangoye et al., 2020) and energy metabolism (Omer et al., 2020). At any rate, the role of NKCC1 in insulin-secreting β-cell function is likely influenced by redundant mechanisms. In addition to NKCC1, islet β-cells express low levels of NKCC2A, a spliced variant of SLC12A1 (i.e., SLC12A1v1) (Alshahrani et al., 2012) exquisitely sensitive to bumetanide but functionally different than NKCC1 (Zeuthen and Macaulay, 2012). In fact, bumetanide did inhibit insulin secretion from NKCC1KO islets and impaired glucose tolerance in NKCC1KO mice (Alshahrani and Di Fulvio, 2012) whereas mice hemizygous for NKCC1 showed improved glucose tolerance associated to increased expression of NKCC2A in islet β-cells (Alshahrani et al., 2015). Therefore, it is plausible that NKCC2 may compensate, at least to some extent, the functional decrease or even absence of islet NKCC1 and play a minor, if any role per se in the secretory response. In line with this assumption, in vitro insulin responses to glucose from NKCC1-expressing islets but lacking NKCC2A were normal (Kelly et al., 2019). However, NKCC2AKO islets also showed increased expression of KCC2, i.e., a furosemide-sensitive and constitutively active K+Cl− cotransporter (Payne, 1997; Williams and Payne, 2004) recently implicated in facilitating insulin secretion (Kursan et al., 2017; Pae and Harper, 2021).
From the previous lines, it has become evident that β-cells possess overlapping, loop diuretic-sensitive mechanisms, which complicates the dissection of the specific role of each of them. Indeed, in addition to NKCC1, many K+Cl− cotransporter variants have been found at the mRNA levels in mammalian islets including KCC1, three and four splice variants of KCC2 and KCC3, respectively, and KCC4 (Davies et al., 2004; Kursan et al., 2017). Although these KCC variants are considered sensitive to loop diuretics, but not functionally equivalent (Adragna et al., 2004), our knowledge regarding the roles of these transporters in insulin secretory responses in vitro or glucose homeostasis in vivo is scant. Mammalian β-cells and islets do have furosemide-sensitive K+Cl−extrusion mechanisms, which become robust in response to cell swelling (Engstrom et al., 1991). As such, these transporters have been implicated in the quick inhibitory effect that furosemide has on islet insulin secretion in vitro (Aynsley-Green and Alberti, 1973;Hermansen et al., 1986;Sandstrom and Sehlin, 1988d;c;a;Eberhardson et al., 2000). Intriguingly, high doses of furosemide stimulated insulin secretion in vitro (Sandstrom and Sehlin, 1988c) producing a U-shaped dose-response like that observed with high doses of bumetanide (Sandstrom, 1990). Notably, these effects on islet insulin secretion were paralleled by changes in Cl− and Ca2+ fluxes (Sandstrom and Sehlin, 1987; 1988a; Sandstrom, 1990). However, while these experiments did not distinguish which KCC may be involved in the stimulatory effects of high doses of the diuretic, inhibition of β-cell KCC2 with highly selective drugs (Kursan et al., 2017) or its transient siRNA-mediated downregulation in islets (Pae and Harper, 2021) resulted in increased insulin secretion in response to glucose. Yet, the in vivo role of β-cell KCC2, or that of KCC1, KCC3 or KCC4 on glucose homeostasis, if any, remain to be explored.
The use of mice lacking NKCC1 specifically in insulin-secreting β-cells (NKCC1βKO) has provided some insight into the long-term metabolic effects of the bumetanide-sensitive NKCC1 in insulin secreting cells. For instance, NKCC1βKO mice gradually became overweight, hyperinsulinemic, hyperglycemic, hypertriglyceridemic, glucose intolerant and insulin resistant while developing mild non-alcoholic steatohepatitis and reduced β-cell mass and function, i.e., typical conditions found in MetS (Abdelgawad et al., 2022). Although the precise causal mechanisms underlying the initiation of this phenotype in NKCC1βKO mice remain unresolved, it is evident that fundamental deficiencies in β-cell function and/or mass are pivotal in the development/progression of age-dependent metabolic dysregulation (Hudish et al., 2019). Interestingly, NKCC1βKO mice also showed reduced satiation control to ad libitum feeding before developing overweight and a MetS-like phenotype (Rathod et al., 2023), consistent with the hypothesis that islets hormones participate in the control of food/energy intake (Woods et al., 2006). In that regard, it is known that chronic low doses of furosemide and potentially other diuretics can increase long-term energy intake in animal models (National Toxicology, 1989a; National Toxicology, 1989b; Bucher et al., 1990). Therefore, these data raise an intriguing possibility; in addition to provoke diuresis, loop diuretics may indirectly modulate feeding behavior and/or energy balance. However, as it is the case of many drugs in clinical use today, the role of diuretics in the behavioral control of food intake awaits further exploration.
The kidneys produce and release glucose primarily through gluconeogenesis (Weber, 1961; Schoolwerth et al., 1988) (Figure 3A). In fact, the kidneys contribute ∼50% of the total glucose released into the systemic circulation under fasting conditions (Gerich et al., 2001). Moreover, increased renal glucose production is a possible contributor to the development of hyperglycemia in patients with insulin resistance and MetS (Legouis et al., 2022). Indeed, insulin regulates renal gluconeogenesis by influencing enzyme production or activity associated with the availability of gluconeogenic precursors (Cano, 2001), an influence anticipated to be diminished or impaired in individuals with MetS or obesity-related insulin resistance (Rebelos et al., 2024). Yet, it remains uncertain whether any individual component of MetS, either alone or in combination, affects the gluconeogenic capacity of the kidneys. Much less certain is the potential effects that diuretics may have on renal glucose production.
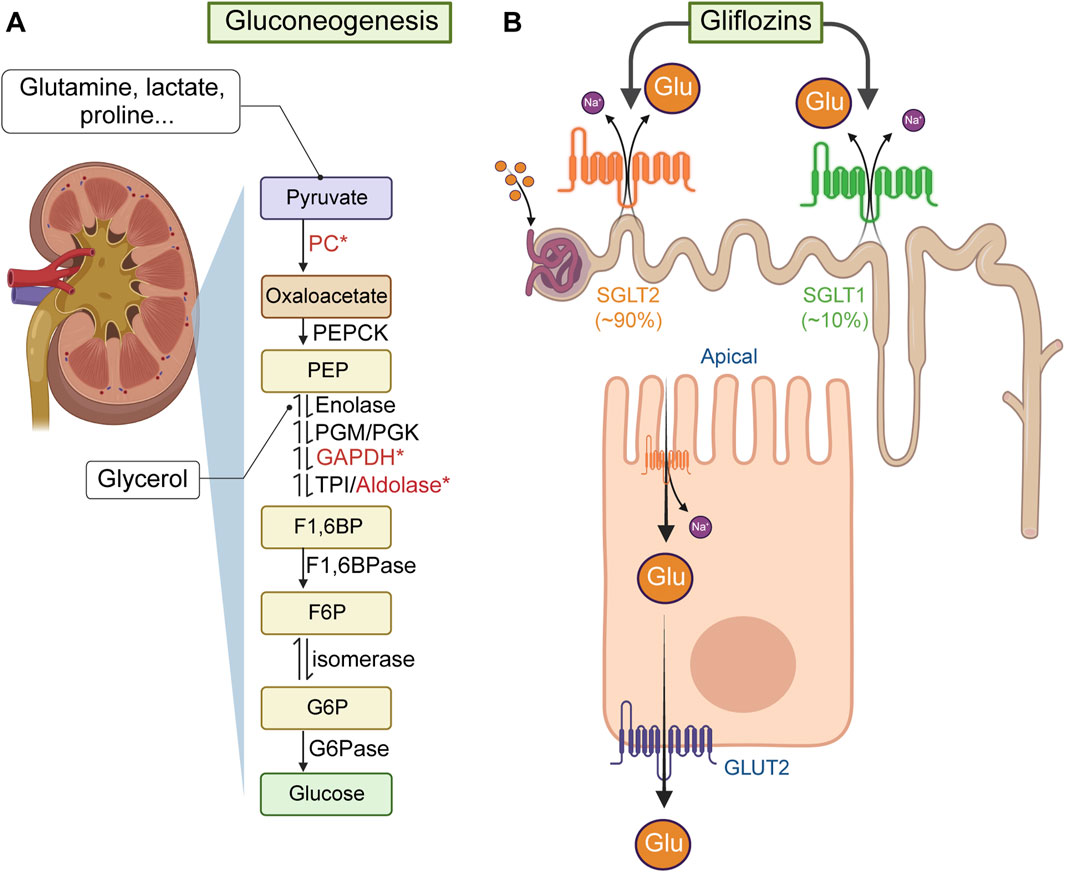
Figure 3. Renal de novo gluconeogenesis and glucose reabsorption. (A) Renal de novo gluconeogenesis is the process by which the kidneys produce glucose from non-carbohydrate sources (e.g., lactate, glycerol, amino acids). This process mainly occurs in the renal cortex and is particularly important during periods of fasting or intense exercise. Lactate or glutamine (from muscle) generate glucose in the kidneys after being transported into renal tubular cells, where they undergo enzymatic reactions to form pyruvate, which then is converted into oxaloacetate via pyruvate carboxylase (PC, which uses HCO3− provided by carbonic anhydrases, some of them potentially inhibited by hydrochlorothiazide). Oxaloacetate, through phosphoenolpyruvate carboxykinase (PEPCK) forms phosphonolpyruvate (PEP). Glycerol (from adipocytes), can enter the gluconeogenic process as a precursor of glyceraldehyde-3-phosphate (G3P) by the enzymes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and triose phosphate isomerase (TPI). G3P combined with dihydroxyacetone-phosphate, via aldolase B, forms fructose-1,6-bisphosphate (F1,6BP). Note: GAPDH was reported inhibited by furosemide and ethacrynic acid, and aldolase B can directly regulate NKCC2 functional expression. F1,6BP is then dephosphorylated to fructose-6-phosphate via fructose-1,6-bisphosphatase (F1,6BPase) and isomerized to form glucose-6-phosphate. (B) Renal glucose reabsorption primarily occurs in the proximal tubule of the nephron, ensuring that glucose is conserved and returned to the bloodstream rather than excreted in urine. This process involves two main types of glucose transporters: SGLTs and GLUTs. In particular, SGLT2, located in the apical side of epithelium of the proximal convoluted tubule uses the Na+ gradient to reabsorb ∼90% of glucose from the filtrate back into the cells lining the tubule. The remaining glucose filtered is absorbed by SGLT1 further down the proximal tubule. Once in the tubular cell, GLUT2, located in the basolateral side of the tubular epithelium, transports glucose into the bloodstream.
Nevertheless, studies performed ∼30 years ago have shown that furosemide and ethacrynic acid can inhibit mitochondrial electron transport in renal tissues (Manuel and Weiner, 1976; Orita et al., 1983) and that diuretics, in general, appear to have adverse effects on renal (and muscular) glycolysis and gluconeogenesis, at least in rodents (Jones and Landon, 1967; Yoshida et al., 1970; Klahr et al., 1971; Fulgraff et al., 1972; Cohen and Little, 1976; Vinay et al., 1987; Dimitriadis et al., 1988; Dimitriadis et al., 1993; Amores et al., 1994). More recently, a potential functional link between NKCC2 and renal glucose fate has been suggested. Indeed, fructose-bisphosphate aldolase B, an enzyme involved in both gluconeogenesis and glycolysis, and primarily located in the kidneys, liver and intestines, may bind to, sequester, and reduce the functional expression of NKCC2 (Benziane et al., 2007) in a manner dependent of fructose 1,6-bisphosphate (F1,6BP), the enzyme’s substrate. Moreover, fructose, once activated to fructose-1-phosphate, can also serve as a substrate for aldolase B and has been shown to increase NKCC2 functional expression in the kidney (Ares et al., 2019). Although it is unknown whether NKCC2 can modulate the enzymatic activity of aldolase B in tubular cells, or if loop diuretics in general directly influence this interaction, these findings suggest a complex regulatory relationship with potential clinical implications. On one hand, there appears to be a negative regulatory link between renal gluconeogenesis and NKCC2 function. On the other hand, dietary fructose consumption, a potential contributor to MetS (Reungjui et al., 2007), is linked to NKCC2 function.
Although the role of NKCC2 in renal handling of glucose remains poorly defined, NKCC2AKO mice developed several aspects of MetS including increased basal glycemia, glucose intolerance and insulin resistance (Kelly et al., 2019), but not hypertension, at least when mice were young (Oppermann et al., 2007). In addition, these mice showed enhanced glucose responses to alanine (Kelly et al., 2019), a substrate almost exclusively converted into glucose in the liver (Stumvoll et al., 1998; Meyer et al., 2002a; Mithieux et al., 2004; Mutel et al., 2011; Alsahli and Gerich, 2017; Sasaki et al., 2017). While these findings suggest increased hepatic de novo gluconeogenesis, the gluconeogenic response of NKCC2AKO mice to exogenous pyruvate, which is converted into glucose in the liver, kidneys and small intestines (Stumvoll et al., 1998; Meyer et al., 2002a; Mithieux et al., 2004) remained normal (Kelly et al., 2019). Therefore, these observations suggest that NKCC2AKO mice might have compromised renal gluconeogenesis. Moreover, aged NKCC2AKO male mice developed overweight and consumed excessive food and water indicating that, unsurprisingly, the kidneys and other organs may contribute to the impaired glucose homeostasis observed in NKCC2AKO mice (Kelly et al., 2019). In that regard, NKCC2 has been detected in other organs at much lower levels than those found in the kidneys, including small intestines (Xue et al., 2009) and hypothalamic regions of the brain (Konopacka et al., 2015). Even though the specific roles that NKCC2 in these organs may have in glucose homeostasis remain unexplored, the potential relevance of extrarenal NKCC2 is underlined by the following: i) the gluconeogenic capacity (Watford, 2005) of the small intestine supplies circulating glucose (Penhoat et al., 2014) and prevents obesity-related hepatic steatosis (Vily-Petit et al., 2020), ii) the hypothalamus plays a central role in endocrine integration of fuel homeostasis, control of water/energy intake and feeding behavior (Schwartz et al., 2000; Coll et al., 2007; Begg and Woods, 2013), and iii) as it has been known for a long time, diet and food intake affect renal gluconeogenesis and water balance (Author Anonymous, 1971a). Therefore, the metabolic phenotype of NKCC2AKO mice likely stems from complex, age-dependent and long-term functional interactions between the brain, pancreatic islets, kidneys and intestines as well as other tissues where NKCC2 may be expressed, even in minimal quantities relative to the kidneys.
The kidneys utilize ∼10% of the total glucose used by the body in a daily basis, filtering 180 g of glucose per day, which is then almost entirely brought back into circulation (Ross et al., 1986; Alsahli and Gerich, 2017). Glucose is actively reabsorbed in the proximal convoluted tubule via the Na+-glucose transporter 2 (SGLT2), which couples the transport of the sugar with that of Na+ following its electrochemical gradient created by the Na+/K+ ATPase on the basolateral membrane of the tubular cells (see Figure 3B). Once inside the tubular cell, glucose is transported across the basolateral membrane into the peritubular capillaries by GLUT2 to reach back the bloodstream (Kanai et al., 1994). Importantly, SGLT2 is targeted by a class of highly efficacious drugs known as gliflozins, which reduce renal glucose reabsorption, thereby aiding in the management of glycemia and improving cardiovascular and metabolic health (Teo et al., 2021; Matthews, 2024). Notably, there has long been awareness that at least two loop diuretics, i.e., furosemide and ethacrynic acid can moderately decrease glucose reabsorption in the proximal tubule (Bowman et al., 1973; Arruda et al., 1975; Boonjarern et al., 1977; Wen et al., 1978). However, the potential of loop diuretics (or thiazide and thiazide-like diuretics) to promote glycosuria through this or any mechanism remains uncertain. It is worth noting that SGLT2 inhibitors not only enhance glycemic control but also reduce hypertension and mitigate MetS in animal models co-administered with furosemide or hydrochlorothiazide (Rahman et al., 2016), as well as in clinical settings involving patients with chronic heart failure (Grodin and Tang, 2020; Ibrahim et al., 2020).
Hepatic gluconeogenesis is a highly regulated process that serves as a backup for synthesizing glucose and glycogen from non-sugar sources (Zhang et al., 2018). Like the liver, muscle cells store glucose as glycogen. However, muscle glycogen is used locally for energy rather than being released into the circulation. During muscle activity, for instance, glycogen is broken down into glucose-6-phosphate for ATP production through glycolysis. This process can occur either aerobically or anaerobically, the latter leading to lactate production and release. Muscle-derived lactic acid is converted into alanine, transported to the liver, converted back to lactic acid and then used in de novo gluconeogenesis to synthesize glucose (see Figure 1). Glucagon effectively stimulates gluconeogenesis from amino acids and other non-carbohydrate substrates in the liver, but not in muscle, while insulin has the opposite effect, i.e., it inhibits hepatic glucose production and release (Puigserver et al., 2003; Adeva-Andany et al., 2019). Importantly, hepatic gluconeogenesis produces glucose-6-phosphate, which together with that produced from glycogen degradation (glycogenolysis) must be hydrolyzed by glucose-6-phosphatase in the endoplasmic reticulum to be released as glucose into the circulation (Cahill et al., 1959). Therefore, tissue glucose-6-phosphatase plays a major role in the maintenance of glycemia, particularly under fasting conditions.
Very little is understood about the metabolic effects that thiazides, thiazide-like and loop diuretics may have in hepatic glucose production and/or degradation. Nonetheless, early evidence did suggest that mechanisms sensitive to loop diuretics, possibly involving NKCC1 and/or KCCs, may contribute to the phosphorylation of numerous protein substrates in the liver (Lang et al., 1998). Among these proteins, is the serum- and glucocorticoid-dependent kinase (Waldegger et al., 1997), which is now recognized for its role in promoting hepatic insulin resistance (Zhou et al., 2021). Although this kinase was shown to regulate plasma membrane trafficking of NKCC2 in vitro (Fillon et al., 2001), the specific role of loop diuretics in developing hepatic insulin resistance remains unclear. It has been suggested that loop diuretics might contribute to insulin resistance in the liver (Schliess et al., 2001) and as such contribute to increased hepatic gluconeogenesis, while thiazides may exacerbate insulin resistance in general (Ramsay et al., 1992; Eriksson et al., 2008). Despite these findings, our current knowledge about the overall impact of diuretics on hepatic gluconeogenesis related to insulin resistance remains very limited.
Also poorly understood is the potential relationship that may exist between hepatocyte swelling in response to amino acids, the obligatory KCC-dependent K+/Cl− extrusion, the resulting reduction in [Cl−]i and glycogen synthesis via activation of the Cl−-dependent enzyme glycogen synthase phosphatase (Meijer et al., 1992). As Cl−ions can directly inhibit this enzyme (Meijer et al., 1992) as well as glucose-6-phosphatase (Pederson et al., 1998) one would expect that changes in [Cl−]i may inversely correlate with glycogen biosynthesis or glucose production. However, like β-cells, the likely redundancy of diuretic-sensitive mechanisms involved in the regulation of hepatocyte [Cl−]i makes it challenging to study the role of loop diuretics on hepatic glucose metabolism. In addition, the potential of hydrochlorothiazide to indirectly impair the function of pyruvate carboxylase by inhibiting carbonic anhydrase Vb (Kucharczyk et al., 2023) may have widespread physiological implications; this enzyme is widely distributed and plays an essential role in de novo gluconeogenesis and lipogenesis (Jitrapakdee and Wallace, 1999).
In comparison, virtually nothing is known about the metabolic effects that diuretics may directly have on muscle glucose homeostasis.
Limited often conflicting evidence mostly involving hydrochlorothiazide is still taken as proof of increased risk of T2D in hypertensive patients treated with any thiazide, thiazide-like or loop diuretics. Indeed, early studies have found that hydrochlorothiazide has the potential to influence various facets of glucose homeostasis, spanning from insulin secretion in the islets to the production of glucose in the liver and kidneys under diverse physiopathological conditions in humans and animal models. In addition, some studies examining the effects of bumetanide or furosemide on carbohydrate metabolism in humans have produced inconsistent results. Further, despite the persistent notion that all diuretics might have “diabetogenic properties”, long-term studies in preclinical animal models are still missing and many questions remain unanswered regarding the mechanisms whereby these drugs may exert their metabolic effects under different chronic contexts. Untangling the potential effects of these diuretics on fuel homeostasis is additionally complicated by the intricate relationships among all components of the MetS, glucose intolerance (often confused with prediabetes), T2D, hypertension, the specific diuretic treatment and the functional redundancy that may exist among diuretic-sensitive targets. Whilst certain studies do hint at possible direct effects of thiazide, thiazide-like and loop diuretics on glucose homeostasis, and that its control is apparently beneficial for some aspects of the MetS, it is clear that additional research is necessary to fully understand the specific mechanisms involved and the potential clinical implications that they may have in hypertensive individuals with chronic metabolic conditions.
MD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. YR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Validation, Visualization, Writing–original draft, Writing–review and editing. SK: Conceptualization, Data curation, Formal Analysis, Investigation, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. The present investigation has been supported partly by funds from the American Diabetes Association and the National Institutes of Health (1-17-IBS-258 and R21DK113446-01 to MDiF).
We are grateful to Dr. Jeffrey Travers (Department of Pharmacology and Toxicology, WSU) who helped facilitate our research. The authors are thankful to Drs. Khalid Elased and Courtney Sulentic (WSU) for their valuable comments during the development of this and related projects. All figures were created by using BioRender.com and Adobe Illustrator.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelgawad, R., Rathod, Y. D., Alshammari, M., Kelly, L., Hubner, C. A., Aguilar-Bryan, L., et al. (2022). Loss of Slc12a2 specifically in pancreatic beta-cells drives metabolic syndrome in mice. PLoS One 17, e0279560. doi:10.1371/journal.pone.0279560
Adem, S., and Ciftci, M. (2016). Purification and characterization of glucose 6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, and glutathione reductase from rat heart and inhibition effects of furosemide, digoxin, and dopamine on the enzymes activities. J. Biochem. Mol. Toxicol. 30, 295–301. doi:10.1002/jbt.21793
Adeva-Andany, M. M., Funcasta-Calderon, R., Fernandez-Fernandez, C., Castro-Quintela, E., and Carneiro-Freire, N. (2019). Metabolic effects of glucagon in humans. J. Clin. Transl. Endocrinol. 15, 45–53. doi:10.1016/j.jcte.2018.12.005
Adragna, N. C., Di Fulvio, M., and Lauf, P. K. (2004). Regulation of K-Cl cotransport: from function to genes. J. Membr. Biol. 201, 109–137. doi:10.1007/s00232-004-0695-6
Alsahli, M., and Gerich, J. E. (2017). Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 133, 1–9. doi:10.1016/j.diabres.2017.07.033
Alshahrani, S., Almutairi, M. M., Kursan, S., Dias-Junior, E., Almiahuob, M. M., Aguilar-Bryan, L., et al. (2015). Increased Slc12a1 expression in beta-cells and improved glucose disposal in Slc12a2 heterozygous mice. J. Endocrinol. 227, 153–165. doi:10.1530/JOE-15-0327
Alshahrani, S., Alvarez-Leefmans, F., and Di Fulvio, M. (2012). Expression of the Slc12a1 gene in pancreatic β-cells: molecular characterization and in silico analysis. Cell. Physiol. Biochem. 30, 95–112. doi:10.1159/000339050
Alshahrani, S., and Di Fulvio, M. (2012). Enhanced insulin secretion and improved glucose tolerance in mice with homozygous inactivation of the Na(+)K(+)2Cl(-) co-transporter 1. J. Endocrinol. 215, 59–70. doi:10.1530/JOE-12-0244
Amery, A., Berthaux, P., Bulpitt, C., Deruyttere, M., De Schaepdryver, A., Dollery, C., et al. (1978). Glucose intolerance during diuretic therapy. Results of trial by the European working party on hypertension in the elderly. Lancet 1, 681–683. doi:10.1016/s0140-6736(78)90797-3
Amores, M. V., Hortelano, P., Garcia-Salguero, L., and Lupianez, J. A. (1994). Metabolic adaptation of renal carbohydrate metabolism. V. in vivo response of rat renal-tubule gluconeogenesis to different diuretics. Mol. Cell. Biochem. 137, 117–125. doi:10.1007/BF00944073
Andersen, O. O., and Persson, I. (1968). Carbohydrate metabolism during treatment with chlorthalidone and ethacrynic acid. Br. Med. J. 2, 798–801. doi:10.1136/bmj.2.5608.798
Andreasen, F., Hansen, U., Husted, S. E., Mogensen, C. E., and Pedersen, E. B. (1984). The influence of age on renal and extrarenal effects of frusemide. Br. J. Clin. Pharmacol. 18, 65–74. doi:10.1111/j.1365-2125.1984.tb05023.x
Ansary, T. M., Nakano, D., and Nishiyama, A. (2019). Diuretic effects of sodium glucose cotransporter 2 inhibitors and their influence on the renin-angiotensin system. Int. J. Mol. Sci. 20, 629. doi:10.3390/ijms20030629
Antonietta, C. M., Calvi, E., Faggiano, A., Maffeis, C., Bosisio, M., De Stefano, M., et al. (2022). Impact of loop diuretic on outcomes in patients with heart failure and reduced ejection fraction. Curr. Heart Fail Rep. 19, 15–25. doi:10.1007/s11897-021-00538-7
Araoye, M. A., Chang, M. Y., Khatri, I. M., and Freis, E. D. (1978). Furosemide compared with hydrochlorothiazide. Long-term treatment of hypertension. JAMA 240, 1863–1866. doi:10.1001/jama.1978.03290170045023
Ares, G. R., Kassem, K. M., and Ortiz, P. A. (2019). Fructose acutely stimulates NKCC2 activity in rat thick ascending limbs by increasing surface NKCC2 expression. Am. J. Physiol. Ren. Physiol. 316, F550–F557. doi:10.1152/ajprenal.00136.2018
Arnlov, J., Pencina, M. J., Nam, B. H., Meigs, J. B., Fox, C. S., Levy, D., et al. (2005). Relations of insulin sensitivity to longitudinal blood pressure tracking: variations with baseline age, body mass index, and blood pressure. Circulation 112, 1719–1727. doi:10.1161/CIRCULATIONAHA.105.535039
Aronson, D., Bartha, P., Zinder, O., Kerner, A., Markiewicz, W., Avizohar, O., et al. (2004). Obesity is the major determinant of elevated C-reactive protein in subjects with the metabolic syndrome. Int. J. Obes. Relat. Metab. Disord. 28, 674–679. doi:10.1038/sj.ijo.0802609
Arruda, J. A., Gutierrez, L. F., and Kurtzman, N. A. (1975). Diuretic agents and glucose reabsorption. Proc. Soc. Exp. Biol. Med. 149, 50–55. doi:10.3181/00379727-149-38741
Asbury, M. J., Gatenby, P. B., O'sullivan, S., and Bourke, E. (1972). Bumetanide: potent new “loop” diuretic. Br. Med. J. 1, 211–213. doi:10.1136/bmj.1.5794.211
Au, W. Y., and Raisz, L. G. (1960). Studies on the renal concentrating mechanism, 5. Effect of diuretic agents. J. Clin. Invest. 39, 1302–1311. doi:10.1172/JCI104147
Author Anonymous, (1970). Effects of treatment on morbidity in hypertension. II. Results in patients with diastolic blood pressure averaging 90 through 114 mm Hg. JAMA 213, 1143–1152. doi:10.1001/jama.1970.03170330025003
Author Anonymous, (1971a). Effects of dietary changes on kidney metabolism. Nutr. Rev. 29, 95–97. doi:10.1111/j.1753-4887.1971.tb07257.x
Author Anonymous, (1971b). Oral diuretics and carbohydrate metabolism. Report from the North-west England Faculty Research Committee of the Royal College of General Practitioners, J R Coll Gen Pract. 21, 535–542.
Author Anonymous, (1979). Five-year findings of the hypertension detection and follow-up program. I. Reduction in mortality of persons with high blood pressure, including mild hypertension. Hypertension Detection and Follow-up Program Cooperative Group. JAMA 242, 2562–2571. doi:10.1001/jama.1979.03300230018021
Aynsley-Green, A., and Alberti, K. G. (1973). Diuretics and carbohydrate metabolism: the effects of furosemide and amiloride on blood glucose, plasma insulin and cations in the rat. Diabetologia 9, 34–42. doi:10.1007/BF01225998
Bagshaw, S. M., Delaney, A., Jones, D., Ronco, C., and Bellomo, R. (2007). Diuretics in the management of acute kidney injury: a multinational survey. Contrib. Nephrol. 156, 236–249. doi:10.1159/000102089
Bakris, G., Molitch, M., Hewkin, A., Kipnes, M., Sarafidis, P., Fakouhi, K., et al. (2006). Differences in glucose tolerance between fixed-dose antihypertensive drug combinations in people with metabolic syndrome. Diabetes Care 29, 2592–2597. doi:10.2337/dc06-1373
Bankir, L., Bouby, N., Blondeau, B., and Crambert, G. (2016). Glucagon actions on the kidney revisited: possible role in potassium homeostasis. Am. J. Physiol. Ren. Physiol. 311, F469–F486. doi:10.1152/ajprenal.00560.2015
Bayliss, R. I., Marrack, D., Pirkis, J., Rees, J. R., and Zilva, J. F. (1958). Chlorothiazide: an oral diuretic. Lancet 1, 120–124. doi:10.1016/s0140-6736(58)90610-x
Begg, D. P., and Woods, S. C. (2013). The endocrinology of food intake. Nat. Rev. Endocrinol. 9, 584–597. doi:10.1038/nrendo.2013.136
Benet, L. Z. (1979). Pharmacokinetics/pharmacodynamics of furosemide in man: a review. J. Pharmacokinet. Biopharm. 7, 1–27. doi:10.1007/BF01059438
Benziane, B., Demaretz, S., Defontaine, N., Zaarour, N., Cheval, L., Bourgeois, S., et al. (2007). NKCC2 surface expression in mammalian cells: down-regulation by novel interaction with aldolase B. J. Biol. Chem. 282, 33817–33830. doi:10.1074/jbc.M700195200
Berglund, G., Andersson, O., and Widgren, B. (1986). Low-dose antihypertensive treatment with a thiazide diuretic is not diabetogenic. A 10-year controlled trial with bendroflumethiazide. Acta Med. Scand. 220, 419–424. doi:10.1111/j.0954-6820.1986.tb02790.x
Bernstein, A., Odze, M., Crews, A., and Simon, F. (1965). Ethacrynic acid: a new potent diuretic. Am. J. Med. Sci. 249, 551–560. doi:10.1097/00000441-196505000-00009
Best, L. (2005). Glucose-induced electrical activity in rat pancreatic beta-cells: dependence on intracellular chloride concentration. J. Physiol. 568, 137–144. doi:10.1113/jphysiol.2005.093740
Best, L., Brown, P. D., Sener, A., and Malaisse, W. J. (2010). Electrical activity in pancreatic islet cells: the VRAC hypothesis. Islets 2, 59–64. doi:10.4161/isl.2.2.11171
Beyer, K. H., Baer, J. E., and Hodes, M. E. (1962). The effect of ethacrynic acid (Merck) in edema. J. Am. Med. Assoc. 180, 1140–1143.
Beyer, K. H., Baer, J. E., Russo, H. F., and Haimbach, A. S. (1957). Chlorothiazide (6-chloro-7-sulfamyl-1,2,4-benzothiadiazine-1,1-dioxide): enhancement of sodium chloride excretion. Fed. Proc. 16, 282.
Black, H. R., Davis, B., Barzilay, J., Nwachuku, C., Baimbridge, C., Marginean, H., et al. (2008). Metabolic and clinical outcomes in nondiabetic individuals with the metabolic syndrome assigned to chlorthalidone, amlodipine, or lisinopril as initial treatment for hypertension: a report from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Diabetes Care 31, 353–360. doi:10.2337/dc07-1452
Blake, G. J. (1990). Furosemide for pulmonary edema. Nursing 20, 108. doi:10.1097/00152193-199008000-00038
Blose, J. S., Adams, K. F., and Patterson, J. H. (1995). Torsemide: a pyridine-sulfonylurea loop diuretic. Ann. Pharmacother. 29, 396–402. doi:10.1177/106002809502900411
Boonjarern, S., Mehta, P. K., Laski, M. E., Earnest, W. R., and Kurtzman, N. A. (1977). Effect of furosemide on renal handling of glucose in the rat. Am. J. Physiol. 232, F438–F442. doi:10.1152/ajprenal.1977.232.5.F438
Booth, A., Magnuson, A., Fouts, J., and Foster, M. T. (2016). Adipose tissue: an endocrine organ playing a role in metabolic regulation. Horm. Mol. Biol. Clin. Investig. 26, 25–42. doi:10.1515/hmbci-2015-0073
Bowman, R. H., Dolgin, J., and Coulson, R. (1973). Furosemide, ethacrynic acid, and iodoacetate on function and metabolism in perfused rat kidney. Am. J. Physiol. 224, 416–424. doi:10.1152/ajplegacy.1973.224.2.416
Bradford, E. M., Vairamani, K., and Shull, G. E. (2016). Differential expression of pancreatic protein and chemosensing receptor mRNAs in NKCC1-null intestine. World J. Gastrointest. Pathophysiol. 7, 138–149. doi:10.4291/wjgp.v7.i1.138
Brater, D. C., Anderson, S. A., and Strowig, S. (1979). Azosemide, a “loop” diuretic, and furosemide. Clin. Pharmacol. Ther. 25, 435–439. doi:10.1002/cpt1979254435
Brown, M. J., Williams, B., Macdonald, T. M., Caulfield, M., Cruickshank, J. K., Mcinnes, G., et al. (2015). Comparison of single and combination diuretics on glucose tolerance (PATHWAY-3): protocol for a randomised double-blind trial in patients with essential hypertension. BMJ Open 5, e008086. doi:10.1136/bmjopen-2015-008086
Brown, M. J., Williams, B., Morant, S. V., Webb, D. J., Caulfield, M. J., Cruickshank, J. K., et al. (2016). Effect of amiloride, or amiloride plus hydrochlorothiazide, versus hydrochlorothiazide on glucose tolerance and blood pressure (PATHWAY-3): a parallel-group, double-blind randomised phase 4 trial. Lancet Diabetes Endocrinol. 4, 136–147. doi:10.1016/S2213-8587(15)00377-0
Bucher, J. R., Huff, J., Haseman, J. K., Eustis, S. L., Davis, W. E., and Meierhenry, E. F. (1990). Toxicology and carcinogenicity studies of diuretics in F344 rats and B6C3F1 mice. 2. Furosemide. J. Appl. Toxicol. 10, 369–378. doi:10.1002/jat.2550100510
Bunn, W. H. (1958). A study of chlorothiazide (diuril) as an adjunctive antihypertensive agent. Ohio State Med. J. 54, 1168–1170.
Cahill, G. F., Ashmore, J., Renold, A. E., and Hastings, A. B. (1959). Blood glucose and the liver. Am. J. Med. 26, 264–282. doi:10.1016/0002-9343(59)90316-x
Campbell, N., Brant, R., Stalts, H., Stone, J., and Mahallati, H. (1998). Fluctuations in blood lipid levels during furosemide therapy: a randomized, double-blind, placebo-controlled crossover study. Arch. Intern Med. 158, 1461–1463. doi:10.1001/archinte.158.13.1461
Cannon, P. J. (1972). Diuretics in edema: how they work; how to pick the best one. Med. Times 100, 86–102.
Cannon, P. J., Heinemann, H. O., Stason, W. B., and Laragh, J. H. (1965). Ethacrynic acid: effectiveness and mode of diuretic action in man. Circulation 31, 5–18. doi:10.1161/01.cir.31.1.5
Cannon, P. J., and Kilcoyne, M. M. (1969). Ethacrynic acid and furosemide: renal pharmacology and clinical use. Prog. Cardiovasc Dis. 12, 99–118. doi:10.1016/0033-0620(69)90038-3
Cano, N. (2001). Inter-relationships between renal metabolism (both in physiology and renal dysfunction) and the liver. Curr. Opin. Clin. Nutr. Metab. Care 4, 279–285. doi:10.1097/00075197-200107000-00006
Car, N., Skrabalo, Z., and Verho, M. (1988). The effects of piretanide in patients with congestive heart failure and diabetes mellitus: a double-blind comparison with furosemide. Curr. Med. Res. Opin. 11, 133–141. doi:10.1185/03007998809110456
Carone, L., Oxberry, S. G., Twycross, R., Charlesworth, S., Mihalyo, M., and Wilcock, A. (2016). Furosemide. J. Pain Symptom Manage 52, 144–150. doi:10.1016/j.jpainsymman.2016.05.004
Carter, B. L., and Basile, J. (2005). Development of diabetes with thiazide diuretics: the potassium issue. J. Clin. Hypertens. (Greenwich) 7, 638–640. doi:10.1111/j.1524-6175.2005.04144.x
Chalmers, J., and Arima, H. (2010). Importance of blood pressure lowering in type 2 diabetes: focus on ADVANCE. J. Cardiovasc Pharmacol. 55, 340–347. doi:10.1097/fjc.0b013e3181d26469
Chaudhry, A. Y., Bing, R. F., Castleden, C. M., Swales, J. D., and Napier, C. J. (1984). The effect of ageing on the response to frusemide in normal subjects. Eur. J. Clin. Pharmacol. 27, 303–306. doi:10.1007/BF00542164
Chaudhuri, M. L., and Catania, J. (1988). A comparison of the effects of bumetanide (Burinex) and frusemide on carbohydrate metabolism in the elderly. Br. J. Clin. Pract. 42, 427–429. doi:10.1111/j.1742-1241.1988.tb08619.x
Chaudhury, R. R., Chugh, K. S., Gupta, G. S., Sodhi, P., and Gupta, K. K. (1968). A controlled clinical trial comparing the diuretic furosemide and hydrochlorothiazide. J. Assoc. Physicians India 16, 157–163.
Chen, C. W., Guan, B. J., Alzahrani, M. R., Gao, Z., Gao, L., Bracey, S., et al. (2022). Adaptation to chronic ER stress enforces pancreatic beta-cell plasticity. Nat. Commun. 13, 4621. doi:10.1038/s41467-022-32425-7
Chen, T. M., and Chiou, W. L. (1992). Large differences in the biological half-life and volume of distribution of hydrochlorothiazide in normal subjects from eleven studies. Correlation with their last blood sampling times. Int. J. Clin. Pharmacol. Ther. Toxicol. 30, 34–37.
Chobanian, A. V., Bakris, G. L., Black, H. R., Cushman, W. C., Green, L. A., Izzo, J. L., et al. (2003). The seventh report of the joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA 289, 2560–2572. doi:10.1001/jama.289.19.2560
Chun, T. Y., Bankir, L., Eckert, G. J., Bichet, D. G., Saha, C., Zaidi, S. A., et al. (2008). Ethnic differences in renal responses to furosemide. Hypertension 52, 241–248. doi:10.1161/HYPERTENSIONAHA.108.109801
Clissold, S. P., and Brogden, R. N. (1985). Piretanide. A preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 29, 489–530. doi:10.2165/00003495-198529060-00002
Cohen, J. J., and Little, J. R. (1976). Lactate metabolism in the isolated perfused rat kidney: relations to renal function and gluconeogenesis. J. Physiol. 255, 399–414. doi:10.1113/jphysiol.1976.sp011286
Coll, A. P., Farooqi, I. S., and O'rahilly, S. (2007). The hormonal control of food intake. Cell. 129, 251–262. doi:10.1016/j.cell.2007.04.001
Collaborators, G. B. D. O., Afshin, A., Forouzanfar, M. H., Reitsma, M. B., Sur, P., Estep, K., et al. (2017). Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 377, 13–27. doi:10.1056/NEJMoa1614362
Collins, R., Peto, R., Macmahon, S., Hebert, P., Fiebach, N. H., Eberlein, K. A., et al. (1990). Blood pressure, stroke, and coronary heart disease. Part 2, Short-term reductions in blood pressure: overview of randomised drug trials in their epidemiological context. Lancet 335, 827–838. doi:10.1016/0140-6736(90)90944-z
Conde-Martel, A., Trullas, J. C., Morales-Rull, J. L., Casado, J., Carrera-Izquierdo, M., Sanchez-Marteles, M., et al. (2024). Sex differences in clinical characteristics and outcomes in the CLOROTIC (combining loop with thiazide diuretics for decompensated heart failure) trial. Rev. Clin. Esp. (Barc) 224, 67–76. doi:10.1016/j.rceng.2023.11.003
Coni, N. K., Gordon, P. W., Mukherjee, A. P., and Read, P. R. (1974). The effect of frusemide and ethacrynic acid on carbohydrate metabolism. Age Ageing 3, 85–90. doi:10.1093/ageing/3.2.85
Conway, J., and Lauwers, P. (1961). Mode of action of chlorothiazide in the reduction of blood pressure in hypertension. Am. J. Cardiol. 8, 884–886. doi:10.1016/0002-9149(61)90256-9
Coodley, E. L., Nandi, P. S., and Chiotellis, P. (1979). Evaluation of a new diuretic, diapamide, in congestive heart failure. J. Clin. Pharmacol. 19, 127–136. doi:10.1002/j.1552-4604.1979.tb02470.x
Cooper-Dehoff, R. M., Wen, S., Beitelshees, A. L., Zineh, I., Gums, J. G., Turner, S. T., et al. (2010). Impact of abdominal obesity on incidence of adverse metabolic effects associated with antihypertensive medications. Hypertension 55, 61–68. doi:10.1161/HYPERTENSIONAHA.109.139592
Cornier, M. A., Dabelea, D., Hernandez, T. L., Lindstrom, R. C., Steig, A. J., Stob, N. R., et al. (2008). The metabolic syndrome. Endocr. Rev. 29, 777–822. doi:10.1210/er.2008-0024
Cornish, A. L., Mc, C. J., and Johnston, D. H. (1961). Effects of chlorothiazide on the pancreas. N. Engl. J. Med. 265, 673–675. doi:10.1056/NEJM196110052651403
Cowley, A. J., and Elkeles, R. S. (1978). Diabetes and therapy with potent diuretics. Lancet 1, 154. doi:10.1016/s0140-6736(78)90451-8
Crutzen, R., Virreira, M., Markadieu, N., Shlyonsky, V., Sener, A., Malaisse, W. J., et al. (2016). Anoctamin 1 (Ano1) is required for glucose-induced membrane potential oscillations and insulin secretion by murine beta-cells. Pflugers Arch. 468, 573–591. doi:10.1007/s00424-015-1758-5
Cuthbert, J. J., and Clark, A. L. (2024). Diuretic treatment in patients with heart failure: current evidence and future directions - Part I: loop diuretics. Curr. Heart Fail Rep. 21, 101–114. doi:10.1007/s11897-024-00643-3
Davidov, M., Kakaviatos, N., and Finnerty, F. A. (1967). Antihypertensive properties of furosemide. Circulation 36, 125–135. doi:10.1161/01.cir.36.1.125
Davies, S. L., Roussa, E., Le Rouzic, P., Thevenod, F., Alper, S. L., Best, L., et al. (2004). Expression of K+-Cl-cotransporters in the alpha-cells of rat endocrine pancreas. Biochim. Biophys. Acta 1667, 7–14. doi:10.1016/j.bbamem.2004.08.005
Daza-Arnedo, R., Rico-Fontalvo, J., Aroca-Martinez, G., Rodriguez-Yanez, T., Martinez-Avila, M. C., Almanza-Hurtado, A., et al. (2023). Insulin and the kidneys: a contemporary view on the molecular basis. Front. Nephrol. 3, 1133352. doi:10.3389/fneph.2023.1133352
Delpire, E., Lu, J., England, R., Dull, C., and Thorne, T. (1999). Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 22, 192–195. doi:10.1038/9713
Dettelbach, H. R., and Bennett, D. A. (1979). Furosemide compared with hydrochlorothiazide. JAMA 242, 712–713. doi:10.1001/jama.1979.03300080012012
Di Fulvio, M., and Aguilar-Bryan, L. (2019). Chloride transporters and channels in beta-cell physiology: revisiting a 40-year-old model. Biochem. Soc. Trans. 47, 1843–1855. doi:10.1042/BST20190513
Di Fulvio, M., and Alvarez-Leefmans, F. J. (2009). “The NKCC and NCC Genes: an in silico view,” in Physiology and pathology of chloride transporters and channels in the nervous system: from molecules to diseases. Editors F. J. Alvarez-Leefmans, and E. Delpire 1st ed (Incorporated: Academic Press), 169–208.
Di Fulvio, M., Bogdani, M., Velasco, M., Mcmillen, T. S., Ridaura, C., Kelly, L., et al. (2020). Heterogeneous expression of CFTR in insulin-secreting beta-cells of the normal human islet. PLoS One 15, e0242749. doi:10.1371/journal.pone.0242749
Di Fulvio, M., Brown, P. D., and Aguilar-Bryan, L. (2014). “Chloride channels and transporters in β-cell physiology,” in The islets of Langerhans. Editor M. S. Islam 2nd ed (Springer-Verlag), 401–451.
Dimitriadis, G., Leighton, B., Parry-Billings, M., and Newsholme, E. A. (1988). Effects of the diuretic furosemide on the sensitivity of glycolysis and glycogen synthesis to insulin in the soleus muscle of the rat. Diabetologia 31, 58–61. doi:10.1007/BF00279135
Dimitriadis, G., Tegos, C., Golfinopoulou, L., Roboti, C., and Raptis, S. (1993). Furosemide-induced hyperglycaemia: the implication of glycolytic kinases. Horm. Metab. Res. 25, 557–559. doi:10.1055/s-2007-1002176
Dorsch, M. P., Gillespie, B. W., Erickson, S. R., Bleske, B. E., and Weder, A. B. (2011). Chlorthalidone reduces cardiovascular events compared with hydrochlorothiazide: a retrospective cohort analysis. Hypertension 57, 689–694. doi:10.1161/HYPERTENSIONAHA.110.161505
Dronavalli, S., and Bakris, G. L. (2008). Mechanistic insights into diuretic-induced insulin resistance. Hypertension 52, 1009–1011. doi:10.1161/HYPERTENSIONAHA.108.120923
Eberhardson, M., Patterson, S., and Grapengiesser, E. (2000). Microfluorometric analysis of Cl-permeability and its relation to oscillatory Ca2+ signalling in glucose-stimulated pancreatic beta-cells. Cell. Signal 12, 781–786. doi:10.1016/s0898-6568(00)00122-4
Eid, P. S., Ibrahim, D. A., Zayan, A. H., Elrahman, M. M. A., Shehata, M. a.A., Kandil, H., et al. (2021). Comparative effects of furosemide and other diuretics in the treatment of heart failure: a systematic review and combined meta-analysis of randomized controlled trials. Heart Fail Rev. 26, 127–136. doi:10.1007/s10741-020-10003-7
Elliott, W. J. (1996). ALLHAT: the largest and most important clinical trial in hypertension ever done in the USA. Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial. Am. J. Hypertens. 9, 409–411. doi:10.1016/0895-7061(96)00093-3
Engstrom, K. G., Sandstrom, P. E., and Sehlin, J. (1991). Volume regulation in mouse pancreatic beta-cells is mediated by a furosemide-sensitive mechanism. Biochim. Biophys. Acta 1091, 145–150. doi:10.1016/0167-4889(91)90054-2
Eriksson, J. W., Jansson, P. A., Carlberg, B., Hagg, A., Kurland, L., Svensson, M. K., et al. (2008). Hydrochlorothiazide, but not Candesartan, aggravates insulin resistance and causes visceral and hepatic fat accumulation: the mechanisms for the diabetes preventing effect of Candesartan (MEDICA) Study. Hypertension 52, 1030–1037. doi:10.1161/HYPERTENSIONAHA.108.119404
Ernst, M. E., Carter, B. L., Goerdt, C. J., Steffensmeier, J. J., Phillips, B. B., Zimmerman, M. B., et al. (2006). Comparative antihypertensive effects of hydrochlorothiazide and chlorthalidone on ambulatory and office blood pressure. Hypertension 47, 352–358. doi:10.1161/01.HYP.0000203309.07140.d3
Ernst, M. E., and Moser, M. (2009). Use of diuretics in patients with hypertension. N. Engl. J. Med. 361, 2153–2164. doi:10.1056/NEJMra0907219
Ernst, S. J., Aguilar-Bryan, L., and Noebels, J. L. (2009). Sodium channel beta1 regulatory subunit deficiency reduces pancreatic islet glucose-stimulated insulin and glucagon secretion. Endocrinology 150, 1132–1139. doi:10.1210/en.2008-0991
Escher, G., Meyer, K. V., Vishwanath, B. S., Frey, B. M., and Frey, F. J. (1995). Furosemide inhibits 11 beta-hydroxysteroid dehydrogenase in vitro and in vivo. Endocrinology 136, 1759–1765. doi:10.1210/endo.136.4.7895688
Eslam, M., Sanyal, A. J., George, J., and International Consensus, P. (2020). MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014.e1. doi:10.1053/j.gastro.2019.11.312
Evans, R. L., Park, K., Turner, R. J., Watson, G. E., Nguyen, H. V., Dennett, M. R., et al. (2000). Severe impairment of salivation in Na+/K+/2Cl-cotransporter (NKCC1)-deficient mice. J. Biol. Chem. 275, 26720–26726. doi:10.1074/jbc.M003753200
Feit, P. W. (1971). Aminobenzoic acid diuretics. 2. 4-Substituted-3-amino-5-sulfamylbenzoic acid derivatives. J. Med. Chem. 14, 432–439. doi:10.1021/jm00287a014
Fillon, S., Warntges, S., Matskevitch, J., Moschen, I., Setiawan, I., Gamper, N., et al. (2001). Serum- and glucocorticoid-dependent kinase, cell volume, and the regulation of epithelial transport. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 130, 367–376. doi:10.1016/s1095-6433(01)00422-6
Finnerty, F. A., Maxwell, M. H., Lunn, J., and Moser, M. (1977). Long-term effects of furosemide and hydrochlorothiazide in patients with essential hypertension a two-year comparison of efficacy and safety. Angiology 28, 125–133. doi:10.1177/000331977702800209
Flagella, M., Clarke, L. L., Miller, M. L., Erway, L. C., Giannella, R. A., Andringa, A., et al. (1999). Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 274, 26946–26955. doi:10.1074/jbc.274.38.26946
Foy, J. M. (1967). Acute diuretic induced hyperglycaemia in rats. Life Sci. 6, 897–902. doi:10.1016/0024-3205(67)90074-4
Foy, J. M., and Furman, B. L. (1969). Diuretics and carbohydrate metabolism in the mouse. Br. J. Pharmacol. 36, 190P–191P.
Foy, J. M., and Furman, B. L. (1971). Effect of diuretics on mouse blood sugar following single dose administration. Br. J. Pharmacol. 42, 287–297. doi:10.1111/j.1476-5381.1971.tb07110.x
Foy, J. M., and Furman, B. L. (1972). The effect of fourteen day treatment with diuretics on mouse blood sugar and glucose tolerance. J. Pharm. Pharmacol. 24, 390–395. doi:10.1111/j.2042-7158.1972.tb09013.x
Freis, E., and Finnerty, F. A. (1959). Discussion of special problems in therapy of hypertension. Philadelphia: W.B. Saunders Company.
Freis, E. D., Wanko, A., Wilson, I. M., and Parrish, A. E. (1958). Treatment of essential hypertension with chlorothiazide (diuril); its use alone and combined with other antihypertensive agents. J. Am. Med. Assoc. 166, 137–140. doi:10.1001/jama.1958.02990020025004
Fulgraff, G., Nunemann, H., and Sudhoff, D. (1972). Effects of the diuretics furosemide, ethacrynic acid, and chlorothiazide on gluconeogenesis from various substrates in rat kidney cortex slices. Naunyn Schmiedeb. Arch. Pharmacol. 273, 86–98. doi:10.1007/BF00508082
Fuster, D., Escher, G., Vogt, B., Ackermann, D., Dick, B., Frey, B. M., et al. (1998). Furosemide inhibits 11beta-hydroxysteroid dehydrogenase type 2. Endocrinology 139, 3849–3854. doi:10.1210/endo.139.9.6175
Garg, R., Williams, G. H., Hurwitz, S., Brown, N. J., Hopkins, P. N., and Adler, G. K. (2011). Low-salt diet increases insulin resistance in healthy subjects. Metabolism 60, 965–968. doi:10.1016/j.metabol.2010.09.005
Georgianos, P. I., and Agarwal, R. (2019). Ambulatory blood pressure reduction with SGLT-2 inhibitors: dose-response meta-analysis and comparative evaluation with low-dose hydrochlorothiazide. Diabetes Care 42, 693–700. doi:10.2337/dc18-2207
Gerich, J. E., Meyer, C., Woerle, H. J., and Stumvoll, M. (2001). Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care 24, 382–391. doi:10.2337/diacare.24.2.382
Giugliano, D., Torella, R., Sgambato, S., and D'onofrio, F. (1980a). Effect of furosemide on insulin and glucagon responses to arginine in normal subjects. Diabetologia 18, 293–296. doi:10.1007/BF00251008
Giugliano, D., Varricchio, M., Cerciello, T., Varano, R., Saccomanno, F., and Giannetti, G. (1980b). Bumetanide and glucose tolerance in man. Farm. Prat. 35, 403–408.
Godoy-Matos, A. F., Silva Junior, W. S., and Valerio, C. M. (2020). NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 12, 60. doi:10.1186/s13098-020-00570-y
Godwin, T. F., and Gunton, R. W. (1965). Clinical trial of a new diuretic, furosemide: comparison with hydrochlorothiazide and mercaptomerin. Can. Med. Assoc. J. 93, 1296–1300.
Goldner, M. G., Zarowitz, H., and Akgun, S. (1960). Hyperglycemia and glycosuria due to thiazide derivatives administered in diabetes mellitus. N. Engl. J. Med. 262, 403–405. doi:10.1056/NEJM196002252620807
Gong, Y., Mcdonough, C. W., Beitelshees, A. L., Karnes, J. H., O'connell, J. R., Turner, S. T., et al. (2014). PROX1 gene variant is associated with fasting glucose change after antihypertensive treatment. Pharmacotherapy 34, 123–130. doi:10.1002/phar.1355
Greene, S. J., Velazquez, E. J., Anstrom, K. J., Clare, R. M., Dewald, T. A., Psotka, M. A., et al. (2023). Effect of torsemide versus furosemide on symptoms and quality of life among patients hospitalized for heart failure: the TRANSFORM-HF randomized clinical trial. Circulation 148, 124–134. doi:10.1161/CIRCULATIONAHA.123.064842
Grimm, R. H., Flack, J. M., Grandits, G. A., Elmer, P. J., Neaton, J. D., Cutler, J. A., et al. (1996). Long-term effects on plasma lipids of diet and drugs to treat hypertension. Treatment of Mild Hypertension Study (TOMHS) Research Group. JAMA 275, 1549–1556. doi:10.1001/jama.1996.03530440029033
Grodin, J. L., and Tang, W. H. W. (2020). Sodium-glucose cotransporter-2 inhibitors and loop diuretics for heart failure: priming the natriuretic and metabolic reserve of the kidney. Circulation 142, 1055–1058. doi:10.1161/CIRCULATIONAHA.120.048057
Grossman, E., and Messerli, F. H. (2006). Long-term safety of antihypertensive therapy. Prog. Cardiovasc Dis. 49, 16–25. doi:10.1016/j.pcad.2006.06.002
Grossman, E., Verdecchia, P., Shamiss, A., Angeli, F., and Reboldi, G. (2011). Diuretic treatment of hypertension. Diabetes Care 34 (Suppl. 2), S313–S319. doi:10.2337/dc11-s246
Grundy, S. M., Cleeman, J. I., Daniels, S. R., Donato, K. A., Eckel, R. H., Franklin, B. A., et al. (2005). Diagnosis and management of the metabolic syndrome: an American heart association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 112, 2735–2752. doi:10.1161/CIRCULATIONAHA.105.169404
Haas, M., and Mcmanus, T. J. (1983). Bumetanide inhibits (Na + K + 2Cl) co-transport at a chloride site. Am. J. Physiol. 245, C235–C240. doi:10.1152/ajpcell.1983.245.3.C235
Haffner, S. M. (2006). The metabolic syndrome: inflammation, diabetes mellitus, and cardiovascular disease. Am. J. Cardiol. 97, 3A–11A. doi:10.1016/j.amjcard.2005.11.010
Haffner, S. M., Valdez, R. A., Hazuda, H. P., Mitchell, B. D., Morales, P. A., and Stern, M. P. (1992). Prospective analysis of the insulin-resistance syndrome (syndrome X). Diabetes 41, 715–722. doi:10.2337/diab.41.6.715
Hall, J. J., Eurich, D. T., Nagy, D., Tjosvold, L., and Gamble, J. M. (2020). Thiazide diuretic-induced change in fasting plasma glucose: a meta-analysis of randomized clinical trials. J. Gen. Intern Med. 35, 1849–1860. doi:10.1007/s11606-020-05731-3
Halstenson, C. E., and Matzke, G. R. (1983). Bumetanide: a new loop diuretic (Bumex, Roche Laboratories). Drug Intell. Clin. Pharm. 17, 786–797. doi:10.1177/106002808301701101
Han, H. S., Kang, G., Kim, J. S., Choi, B. H., and Koo, S. H. (2016). Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 48, e218. doi:10.1038/emm.2015.122
Han, T. S., Williams, K., Sattar, N., Hunt, K. J., Lean, M. E., and Haffner, S. M. (2002). Analysis of obesity and hyperinsulinemia in the development of metabolic syndrome: San Antonio Heart Study. Obes. Res. 10, 923–931. doi:10.1038/oby.2002.126
Hannaert, P., Alvarez-Guerra, M., Pirot, D., Nazaret, C., and Garay, R. P. (2002). Rat NKCC2/NKCC1 cotransporter selectivity for loop diuretic drugs. Naunyn Schmiedeb. Arch. Pharmacol. 365, 193–199. doi:10.1007/s00210-001-0521-y
Hargreaves, M., and Spriet, L. L. (2020). Skeletal muscle energy metabolism during exercise. Nat. Metab. 2, 817–828. doi:10.1038/s42255-020-0251-4
Harno, K., Valimaki, M., and Verho, M. (1985). Effects of a new diuretic piretanide on glucose tolerance, insulin secretion and 125I-insulin binding. Eur. J. Clin. Pharmacol. 27, 697–700. doi:10.1007/BF00547052
Harrower, A. D., Mcfarlane, G., Donnelly, T., and Gray, C. E. (1985). Effect of indapamide on blood pressure and glucose tolerance in non-insulin-dependent diabetes. Hypertension 7, II161–163. doi:10.1161/01.hyp.7.6_pt_2.ii161
Healy, J. J., Mckenna, T. J., Canning, B. S., Brien, T. G., Duffy, G. J., and Muldowney, F. P. (1970). Body composition changes in hypertensive subjects on long-term oral diuretic therapy. Br. Med. J. 1, 716–719. doi:10.1136/bmj.1.5698.716
Hebert, L. A., Rovin, B. H., and Hebert, C. J. (2007). The design of ALLHAT may have biased the study's outcome in favor of the diuretic cohort. Nat. Clin. Pract. Nephrol. 3, 60–61. doi:10.1038/ncpneph0391
Hebert, P. R., Moser, M., Mayer, J., Glynn, R. J., and Hennekens, C. H. (1993). Recent evidence on drug therapy of mild to moderate hypertension and decreased risk of coronary heart disease. Arch. Intern Med. 153, 578–581. doi:10.1001/archinte.1993.00410050018004
Hegde, R. S., and Palfrey, H. C. (1992). Ionic effects on bumetanide binding to the activated Na/K/2Cl cotransporter: selectivity and kinetic properties of ion binding sites. J. Membr. Biol. 126, 27–37. doi:10.1007/BF00233458
Heinemann, H. O., Demartini, F. E., and Laragh, J. H. (1959). The effect of chlorothiazide on renal excretion of electrolytes and free water. Am. J. Med. 26, 853–861. doi:10.1016/0002-9343(59)90207-4
Henquin, J. C., Nenquin, M., Ravier, M. A., and Szollosi, A. (2009). Shortcomings of current models of glucose-induced insulin secretion. Diabetes Obes. Metab. 11 (Suppl. 4), 168–179. doi:10.1111/j.1463-1326.2009.01109.x
Hermansen, K., Schmitz, O., Arnfred, J., and Mogensen, C. E. (1986). Effects of furosemide and indapamide upon pancreatic insulin and somatostatin secretion in vitro. Diabetes Res. 3, 221–223.
Hermansen, K., Schmitz, O., and Mogensen, C. E. (1985). Effects of a thiazide diuretic (hydroflumethiazide) and a loop diuretic (bumetanide) on the endocrine pancreas: studies in vitro. Metabolism 34, 784–789. doi:10.1016/0026-0495(85)90031-9
Hettiarachchi, J., Mcinnes, G. T., Ramsay, L. E., Scott, P., and Shelton, J. (1977). Bumetanide and frusemide: qualitative differences (proceedings). Br. J. Clin. Pharmacol. 4, 644P–645P. doi:10.1111/j.1365-2125.1977.tb00816.x
Hollenberg, N. K., and Mickiewicz, C. (1989). Hyperkalemia in diabetes mellitus. Effect of a triamterene-hydrochlorothiazide combination. Arch. Intern Med. 149, 1327–1330. doi:10.1001/archinte.1989.00390060063013
Hoskins, B., and Jackson, C. M. (1978). The mechanism of chlorothiazide-induced carbohydrate intolerance. J. Pharmacol. Exp. Ther. 206, 423–430. doi:10.1016/s0022-3565(25)31343-1
Hu, F. B., and Stampfer, M. J. (2005). Insulin resistance and hypertension: the chicken-egg question revisited. Circulation 112, 1678–1680. doi:10.1161/CIRCULATIONAHA.105.568055
Huang, C. C., Leu, H. B., Huang, P. H., Lin, L. Y., Wu, T. C., Lin, S. J., et al. (2016). Hypertension subtypes modify metabolic response to thiazide diuretics. Eur. J. Clin. Invest. 46, 80–91. doi:10.1111/eci.12571
Hudish, L. I., Reusch, J. E., and Sussel, L. (2019). β Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J. Clin. Invest. 129, 4001–4008. doi:10.1172/JCI129188
Ibrahim, A., Ghaleb, R., Mansour, H., Hanafy, A., Mahmoud, N. M., Abdelfatah Elsharef, M., et al. (2020). Safety and efficacy of adding Dapagliflozin to furosemide in type 2 diabetic patients with decompensated heart failure and reduced ejection fraction. Front. Cardiovasc Med. 7, 602251. doi:10.3389/fcvm.2020.602251
Jaafar, R., Tran, S., Shah, A. N., Sun, G., Valdearcos, M., Marchetti, P., et al. (2019). mTORC1 to AMPK switching underlies beta-cell metabolic plasticity during maturation and diabetes. J. Clin. Invest. 129, 4124–4137. doi:10.1172/JCI127021
Jackson, W. P., and Nellen, M. (1966). Effect of frusemide on carbohydrate metabolism, blood-pressure, and other modalities. A comparison with chlorothiazide. Br. Med. J. 2, 333–336. doi:10.1136/bmj.2.5509.333
Jayakumar, S., and Puschett, J. B. (1977). Study of the sites and mechanisms of action of bumetanide in man. J. Pharmacol. Exp. Ther. 201, 251–258. doi:10.1016/s0022-3565(25)30849-9
Jiang, G., and Zhang, B. B. (2003). Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 284, E671–E678. doi:10.1152/ajpendo.00492.2002
Jian-Liang, Z., Yong-Wen, Q., Xing, Z., Jian-Li, Q., Jiang, C., and Rong-Liang, X. (2004). Possible induction of diabetes by treatment of hypertension with indapamide (with four case reports). Diabetes Res. Clin. Pract. 65, 243–246. doi:10.1016/j.diabres.2004.02.005
Jitrapakdee, S., and Wallace, J. C. (1999). Structure, function and regulation of pyruvate carboxylase. Biochem. J. 340 (Pt 1), 1–16. doi:10.1042/bj3400001
Johnston, D. H., and Cornish, A. L. (1959). Acute pancreatitis in patients receiving chlorothiazide. J. Am. Med. Assoc. 170, 2054–2056. doi:10.1001/jama.1959.03010170016003
Jones, V. D., and Landon, E. J. (1967). The effect of ouabain, meralluride and ethacrynic acid on respiration and glycolysis in kidney slices. Biochem. Pharmacol. 16, 2163–2169. doi:10.1016/0006-2952(67)90015-9
Joynt, M. S., and Morrin, P. A. (1968). Diuretic response of a severely diseased kidney to furosemide. Can. Med. Assoc. J. 99, 1256–1258.
Kaldor, A., Gachalyi, B., and Sebestyen, K. (1975). Diabetogenic effect of oral diuretics in asymptomatic diabetes. Int. J. Clin. Pharmacol. Biopharm. 11, 232–234.
Kanai, Y., Lee, W. S., You, G., Brown, D., and Hediger, M. A. (1994). The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J. Clin. Invest. 93, 397–404. doi:10.1172/JCI116972
Kang, C., Xie, L., Gunasekar, S. K., Mishra, A., Zhang, Y., Pai, S., et al. (2018). SWELL1 is a glucose sensor regulating beta-cell excitability and systemic glycaemia. Nat. Commun. 9, 367. doi:10.1038/s41467-017-02664-0
Kapelios, C. J., Greene, S. J., Mentz, R. J., Ikeaba, U., Wojdyla, D., Anstrom, K. J., et al. (2024). Torsemide versus furosemide after discharge in patients hospitalized with heart failure across the spectrum of ejection fraction: findings from TRANSFORM-HF. Circ. Heart Fail 17, e011246. doi:10.1161/CIRCHEARTFAILURE.123.011246
Karnes, J. H., Gong, Y., Arwood, M. J., Gums, J. G., Hall, K. L., Limacher, M. C., et al. (2014). Alteration in fasting glucose after prolonged treatment with a thiazide diuretic. Diabetes Res. Clin. Pract. 104, 363–369. doi:10.1016/j.diabres.2014.04.004
Katsimardou, A., Imprialos, K., Stavropoulos, K., Sachinidis, A., Doumas, M., and Athyros, V. (2020). Hypertension in metabolic syndrome: novel insights. Curr. Hypertens. Rev. 16, 12–18. doi:10.2174/1573402115666190415161813
Kawai, T., Autieri, M. V., and Scalia, R. (2021). Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell. Physiol. 320, C375–C391. doi:10.1152/ajpcell.00379.2020
Kelly, L., Almutairi, M. M., Kursan, S., Pacheco, R., Dias-Junior, E., Castrop, H., et al. (2019). Impaired glucose tolerance, glucagon, and insulin responses in mice lacking the loop diuretic-sensitive Nkcc2a transporter. Am. J. Physiol. Cell. Physiol. 317, C843–C856. doi:10.1152/ajpcell.00144.2019
Kempler, P., Jona, G., Somogyi, A., Varsanyi-Nagy, M., Rohla, M., and Balazsi, L. (1990). Late effect of tobanum and tobanum + furosemide therapy on the glucose tolerance of patients and on the insulin response to oral glucose doses. Ther. Hung 38, 22–25.
Khaleeli, A. A., and Wyman, A. L. (1978). Hyperosmolar non-ketotic diabetic coma induced by furosemide in modest dosage. Postgrad. Med. J. 54, 43–44. doi:10.1136/pgmj.54.627.43
Khenhrani, R. R., Nnodebe, I., Rawat, A., Adwani, R., Ghaffar, A., Devi, S., et al. (2023). Comparison of the effectiveness and safety of chlorthalidone and hydrochlorothiazide in patients with hypertension: a meta-analysis. Cureus 15, e38184. doi:10.7759/cureus.38184
Kirkendall, W. M., and Stein, J. H. (1968). Clinical pharmacology of furosemide and ethacrynic acid. Am. J. Cardiol. 22, 162–167. doi:10.1016/0002-9149(68)90221-x
Kissling, K. T., and Pickworth, K. K. (2014). Comparison of the effects of combination diuretic therapy with oral hydrochlorothiazide or intravenous chlorothiazide in patients receiving intravenous furosemide therapy for the treatment of heart failure. Pharmacotherapy 34, 882–887. doi:10.1002/phar.1456
Klahr, S., Yates, J., and Bourgoignie, J. (1971). Inhibition of glycolysis by ethacrynic acid and furosemide. Am. J. Physiol. 221, 1038–1043. doi:10.1152/ajplegacy.1971.221.4.1038
Klauser, R., Prager, R., Gaube, S., Gisinger, C., Schnack, C., Kuenburg, E., et al. (1991). Metabolic effects of isradipine versus hydrochlorothiazide in diabetes mellitus. Hypertension 17, 15–21. doi:10.1161/01.hyp.17.1.15
Kobayakawa, N., Sawaki, D., Otani, Y., Sekita, G., Fukushima, K., Takeuchi, H., et al. (2003). A case of severe diabetes mellitus occurred during management of heart failure with carvedilol and furosemide. Cardiovasc Drugs Ther. 17, 295. doi:10.1023/a:1026253013891
Koh, E. S., Kim, G. H., and Chung, S. (2023). Intrarenal mechanisms of sodium-glucose cotransporter-2 inhibitors on tubuloglomerular feedback and natriuresis. Endocrinol. Metab. (Seoul) 38, 359–372. doi:10.3803/EnM.2023.1764
Konecke, L. L. (1981). Clinical trial of bumetanide versus furosemide in patients with congestive heart failure. J. Clin. Pharmacol. 21, 688–690. doi:10.1002/j.1552-4604.1981.tb05684.x
Konopacka, A., Qiu, J., Yao, S. T., Greenwood, M. P., Greenwood, M., Lancaster, T., et al. (2015). Osmoregulation requires brain expression of the renal Na-K-2Cl cotransporter NKCC2. J. Neurosci. 35, 5144–5155. doi:10.1523/JNEUROSCI.4121-14.2015
Korpi, E. R., and Luddens, H. (1997). Furosemide interactions with brain GABAA receptors. Br. J. Pharmacol. 120, 741–748. doi:10.1038/sj.bjp.0700922
Kostis, J. B., Davis, B. R., Cutler, J., Grimm, R. H., Berge, K. G., Cohen, J. D., et al. (1997). Prevention of heart failure by antihypertensive drug treatment in older persons with isolated systolic hypertension. SHEP Cooperative Research Group. JAMA 278, 212–216. doi:10.1001/jama.1997.03550030052033
Koumangoye, R., Omer, S., Kabeer, M. H., and Delpire, E. (2020). Novel human NKCC1 mutations cause defects in goblet cell mucus secretion and chronic inflammation. Cell. Mol. Gastroenterol. Hepatol. 9, 239–255. doi:10.1016/j.jcmgh.2019.10.006
Krim, S. R., Anand, S., Greene, S. J., Chen, A., Wojdyla, D., Vilaro, J., et al. (2024). Torsemide vs furosemide among patients with new-onset vs worsening chronic heart failure: a substudy of the TRANSFORM-HF randomized clinical trial. JAMA Cardiol. 9, 182–188. doi:10.1001/jamacardio.2023.4776
Kucharczyk, P., Albano, G., Deisl, C., Ho, T. M., Bargagli, M., Anderegg, M., et al. (2023). Thiazides attenuate insulin secretion through inhibition of mitochondrial carbonic anhydrase 5b in beta -islet cells in mice. J. Am. Soc. Nephrol. 34, 1179–1190. doi:10.1681/ASN.0000000000000122
Kursan, S., Mcmillen, T. S., Beesetty, P., Dias-Junior, E., Almutairi, M. M., Sajib, A. A., et al. (2017). The neuronal K(+)Cl(-) co-transporter 2 (Slc12a5) modulates insulin secretion. Sci. Rep. 7, 1732. doi:10.1038/s41598-017-01814-0
Kurtz, T. W. (2010). Chlorthalidone: don't call it “thiazide-like” anymore. Hypertension 56, 335–337. doi:10.1161/HYPERTENSIONAHA.110.156166
Lakshman, M. R., Reda, D. J., Materson, B. J., Cushman, W. C., and Freis, E. D. (1999). Diuretics and beta-blockers do not have adverse effects at 1 year on plasma lipid and lipoprotein profiles in men with hypertension. Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. Arch. Intern Med. 159, 551–558. doi:10.1001/archinte.159.6.551
Lang, F., Busch, G. L., Ritter, M., Volkl, H., Waldegger, S., Gulbins, E., et al. (1998). Functional significance of cell volume regulatory mechanisms. Physiol. Rev. 78, 247–306. doi:10.1152/physrev.1998.78.1.247
Laragh, J. H. (1962). The mode of action and use of chlorothiazide and related compounds. Circulation 26, 121–132. doi:10.1161/01.cir.26.1.121
Laragh, J. H. (1967). The proper use of newer diuretics. Ann. Intern Med. 67, 606–613. doi:10.7326/0003-4819-67-3-606
Laragh, J. H., Cannon, P. J., Stason, W. B., and Heinemann, H. O. (1966). Physiologic and clincical observations on furosemide and ethacrynic acid. Ann. N. Y. Acad. Sci. 139, 453–465. doi:10.1111/j.1749-6632.1966.tb41219.x
Laragh, J. H., Heinemann, H. O., and Demartini, F. E. (1958). Effect of chlorothiazide on electrolyte transport in man; its use in the treatment of edema of congestive heart failure, nephrosis, and cirrhosis. J. Am. Med. Assoc. 166, 145–152. doi:10.1001/jama.1958.02990020033006
Lassen, C. K., and Jespersen, B. (2011). Management of diuretic treatment: a challenge in the obese patient. Scand. J. Urol. Nephrol. 45, 220–222. doi:10.3109/00365599.2011.552435
Lavender, S., and Mcgill, R. J. (1974). Nonketotic hyperosmolar coma and frusemide therapy. Diabetes 23, 247–248. doi:10.2337/diab.23.3.247
Legouis, D., Faivre, A., Cippa, P. E., and De Seigneux, S. (2022). Renal gluconeogenesis: an underestimated role of the kidney in systemic glucose metabolism. Nephrol. Dial. Transpl. 37, 1417–1425. doi:10.1093/ndt/gfaa302
Leviel, F., Hubner, C. A., Houillier, P., Morla, L., El Moghrabi, S., Brideau, G., et al. (2010). The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J. Clin. Invest. 120, 1627–1635. doi:10.1172/JCI40145
Liang, W., Ma, H., Cao, L., Yan, W., and Yang, J. (2017). Comparison of thiazide-like diuretics versus thiazide-type diuretics: a meta-analysis. J. Cell. Mol. Med. 21, 2634–2642. doi:10.1111/jcmm.13205
Liese, A. D., Mayer-Davis, E. J., Tyroler, H. A., Davis, C. E., Keil, U., Duncan, B. B., et al. (1997). Development of the multiple metabolic syndrome in the ARIC cohort: joint contribution of insulin, BMI, and WHR. Atherosclerosis risk in communities. Ann. Epidemiol. 7, 407–416. doi:10.1016/s1047-2797(97)00047-1
Lind, L., Berne, C., Pollare, T., and Lithell, H. (1995). Metabolic effects of anti-hypertensive treatment with nifedipine or furosemide: a double-blind, cross-over study. J. Hum. Hypertens. 9, 137–141.
Lithell, H. O., Pollare, T., and Berne, C. (1990). Insulin sensitivity in newly detected hypertensive patients: influence of captopril and other antihypertensive agents on insulin sensitivity and related biological parameters. J. Cardiovasc Pharmacol. 15 (Suppl. 5), S46–S52. doi:10.1097/00005344-199000005-00007
Lopez, R., Taboada, C., Rivas, C., and San Miguel, A. (1996). Metabolic effects of the combination of furosemide and captopril in rat. Rev. Esp. Fisiol. 52, 89–94.
Luyckx, A. S., Mendoza, E., and Lefebvre, P. (1980). Furosemide intravenous infusion in normal man: electrolytic, metabolic and hormonal effects. Lack of changes in basal insulin and glucagon plasma levels. Arch. Int. Pharmacodyn. Ther. 248, 305–313.
Lykke, K., Tollner, K., Romermann, K., Feit, P. W., Erker, T., Macaulay, N., et al. (2015). Structure-activity relationships of bumetanide derivatives: correlation between diuretic activity in dogs and inhibition of the human NKCC2A transporter. Br. J. Pharmacol. 172, 4469–4480. doi:10.1111/bph.13231
Mahabir, R. N., and Bacchus, R. (1976). Diuretic and clinical effects of low-dose furosemide in congestive heart failure patients. J. Clin. Pharmacol. 16, 510–517.
Maitland-Van Der Zee, A. H., Turner, S. T., Schwartz, G. L., Chapman, A. B., Klungel, O. H., and Boerwinkle, E. (2005). Demographic, environmental, and genetic predictors of metabolic side effects of hydrochlorothiazide treatment in hypertensive subjects. Am. J. Hypertens. 18, 1077–1083. doi:10.1016/j.amjhyper.2005.02.012
Malaisse, W., and Malaisse-Legae, F. (1968). Effect of thiazides upon insulin secretion in vitro. Arch. Int. Pharmacodyn. Ther. 171, 235–239.
Malebari, A. M., Ibrahim, T. S., Salem, I. M., Salama, I., Khayyat, A. N., Mostafa, S. M., et al. (2020). The anticancer activity for the bumetanide-based analogs via targeting the tumor-associated membrane-bound human carbonic anhydrase-IX enzyme. Pharm. (Basel) 13, 252. doi:10.3390/ph13090252
Manrique, C., Johnson, M., and Sowers, J. R. (2010). Thiazide diuretics alone or with beta-blockers impair glucose metabolism in hypertensive patients with abdominal obesity. Hypertension 55, 15–17. doi:10.1161/HYPERTENSIONAHA.109.142620
Manuel, M. A., and Weiner, M. W. (1976). Effects of ethacrynic acid and furosemide on isolated rat kidney mitochondria: inhibition of electron transport in the region of phosphorylation site II. J. Pharmacol. Exp. Ther. 198, 209–221. doi:10.1016/s0022-3565(25)30584-7
Mather, A., and Pollock, C. (2011). Glucose handling by the kidney. Kidney Int. Suppl. 79, S1–S6. doi:10.1038/ki.2010.509
Matthews, D. R. (2024). Sodium-glucose co-transporter-2 inhibitors: a paradigm shift in treatment for type 2 diabetes. Diabetes Obes. Metab. 26 (Suppl. 5), 3–4. doi:10.1111/dom.15963
Mcinnes, G. T. (2003). ALLHAT: a saga of missed opportunities. J. Hum. Hypertens. 17, 373–377. doi:10.1038/sj.jhh.1001564
Mcnabb, W. R., Noormohamed, F. H., Brooks, B. A., and Lant, A. F. (1984). Renal actions of piretanide and three other “loop” diuretics. Clin. Pharmacol. Ther. 35, 328–337. doi:10.1038/clpt.1984.38
Meijer, A. J., Baquet, A., Gustafson, L., Van Woerkom, G. M., and Hue, L. (1992). Mechanism of activation of liver glycogen synthase by swelling. J. Biol. Chem. 267, 5823–5828. doi:10.1016/s0021-9258(18)42627-0
Mentz, R. J., Anstrom, K. J., Eisenstein, E. L., Sapp, S., Greene, S. J., Morgan, S., et al. (2023). Effect of torsemide vs furosemide after discharge on all-cause mortality in patients hospitalized with heart failure: the TRANSFORM-HF randomized clinical trial. JAMA 329, 214–223. doi:10.1001/jama.2022.23924
Mentz, R. J., Hasselblad, V., Devore, A. D., Metra, M., Voors, A. A., Armstrong, P. W., et al. (2016). Torsemide versus furosemide in patients with acute heart failure (from the ASCEND-HF trial). Am. J. Cardiol. 117, 404–411. doi:10.1016/j.amjcard.2015.10.059
Merrins, M. J., and Kibbey, R. G. (2024). Glucose regulation of beta-cell KATP channels: it is time for a new model. Diabetes 73, 856–863. doi:10.2337/dbi23-0032
Merz, K. E., and Thurmond, D. C. (2020). Role of skeletal muscle in insulin resistance and glucose uptake. Compr. Physiol. 10, 785–809. doi:10.1002/cphy.c190029
Messerli, F. H., and Bangalore, S. (2011). Half a century of hydrochlorothiazide: facts, fads, fiction, and follies. Am. J. Med. 124, 896–899. doi:10.1016/j.amjmed.2011.05.009
Messerli, F. H., Makani, H., Benjo, A., Romero, J., Alviar, C., and Bangalore, S. (2011). Antihypertensive efficacy of hydrochlorothiazide as evaluated by ambulatory blood pressure monitoring: a meta-analysis of randomized trials. J. Am. Coll. Cardiol. 57, 590–600. doi:10.1016/j.jacc.2010.07.053
Meyer, C., Stumvoll, M., Dostou, J., Welle, S., Haymond, M., and Gerich, J. (2002a). Renal substrate exchange and gluconeogenesis in normal postabsorptive humans. Am. J. Physiol. Endocrinol. Metab. 282, E428–E434. doi:10.1152/ajpendo.00116.2001
Meyer, J. W., Flagella, M., Sutliff, R. L., Lorenz, J. N., Nieman, M. L., Weber, C. S., et al. (2002b). Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na(+)-K(+)-2Cl(-) cotransporter. Am. J. Physiol. Heart Circ. Physiol. 283, H1846–H1855. doi:10.1152/ajpheart.00083.2002
Middeke, M., Richter, W. O., Schwandt, P., and Holzgreve, H. (1997). The effects of antihypertensive combination therapy on lipid and glucose metabolism: hydrochlorothiazide plus sotalol vs. hydrochlorothiazide plus captopril. Int. J. Clin. Pharmacol. Ther. 35, 231–234.
Mithieux, G., Rajas, F., and Gautier-Stein, A. (2004). A novel role for glucose 6-phosphatase in the small intestine in the control of glucose homeostasis. J. Biol. Chem. 279, 44231–44234. doi:10.1074/jbc.R400011200
Mizgala, H. F. (1965). Newer drugs in the treatment of hypertension. Can. Med. Assoc. J. 92, 918–922.
Moore, J. X., Chaudhary, N., and Akinyemiju, T. (2017). Metabolic syndrome prevalence by race/ethnicity and sex in the United States, national health and nutrition examination survey, 1988-2012. Prev. Chronic Dis. 14, E24. doi:10.5888/pcd14.160287
Moser, M., and Hebert, P. R. (1996). Prevention of disease progression, left ventricular hypertrophy and congestive heart failure in hypertension treatment trials. J. Am. Coll. Cardiol. 27, 1214–1218. doi:10.1016/0735-1097(95)00606-0
Mukete, B. N., and Rosendorff, C. (2013). Effects of low-dose thiazide diuretics on fasting plasma glucose and serum potassium-a meta-analysis. J. Am. Soc. Hypertens. 7, 454–466. doi:10.1016/j.jash.2013.05.004
Murdoch, W. R., and Auld, W. H. (1975). Bumetanide--acute and long-term studies of a new high potency diuretic. Postgrad. Med. J. 51 (Suppl. 6), 10–14. doi:10.1136/pgmj.51.591.10
Mutel, E., Gautier-Stein, A., Abdul-Wahed, A., Amigo-Correig, M., Zitoun, C., Stefanutti, A., et al. (2011). Control of blood glucose in the absence of hepatic glucose production during prolonged fasting in mice: induction of renal and intestinal gluconeogenesis by glucagon. Diabetes 60, 3121–3131. doi:10.2337/db11-0571
Nathan, D. M., Davidson, M. B., Defronzo, R. A., Heine, R. J., Henry, R. R., Pratley, R., et al. (2007). Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care 30, 753–759. doi:10.2337/dc07-9920
National Toxicology, P. (1989a). Toxicology and carcinogenesis studies of furosemide (CAS No. 54-31-9) in F344/N rats and B6C3F1 mice (feed studies). Natl. Toxicol. Program Tech. Rep. Ser. 356, 1–190.
National Toxicology, P. (1989b). Toxicology and carcinogenesis studies of hydrochlorothiazide (CAS No. 58-93-5) in F344/N rats and B6C3F1 mice (feed studies). Natl. Toxicol. Program Tech. Rep. Ser. 357, 1–194.
Netea, M. G., Joosten, L. A., Lewis, E., Jensen, D. R., Voshol, P. J., Kullberg, B. J., et al. (2006). Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 12, 650–656. doi:10.1038/nm1415
Nita, Ii, Hershfinkel, M., Kantor, C., Rutter, G. A., Lewis, E. C., and Sekler, I. (2014). Pancreatic beta-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J. 28, 3301–3312. doi:10.1096/fj.13-248161
Officers, A.Coordinators for the A.C.R.G.T.A., and Lipid-Lowering Treatment to Prevent Heart Attack (2002). Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 288, 2981–2997. doi:10.1001/jama.288.23.2981
Omboni, S., Malacco, E., and Parati, G. (2009). Zofenopril plus hydrochlorothiazide fixed combination in the treatment of hypertension and associated clinical conditions. Cardiovasc Ther. 27, 275–288. doi:10.1111/j.1755-5922.2009.00102.x
Omer, S., Koumangoye, R., and Delpire, E. (2020). A mutation in the Na-K-2Cl cotransporter-1 leads to changes in cellular metabolism. J. Cell. Physiol. 235, 7239–7250. doi:10.1002/jcp.29623
Oppermann, M., Mizel, D., Kim, S. M., Chen, L., Faulhaber-Walter, R., Huang, Y., et al. (2007). Renal function in mice with targeted disruption of the A isoform of the Na-K-2Cl co-transporter. J. Am. Soc. Nephrol. 18, 440–448. doi:10.1681/ASN.2006091070
Orita, Y., Fukuhara, Y., Yanase, M., Ando, A., Okada, N., and Abe, H. (1983). Effect of furosemide on mitochondrial electron transport system and oxidative phosphorylation. Arzneimittelforschung 33, 1446–1450.
Ozieranski, K., Balsam, P., Kaplon-Cieslicka, A., Tyminska, A., Kowalik, R., Grabowski, M., et al. (2019). Comparative analysis of long-term outcomes of torasemide and furosemide in heart failure patients in heart failure registries of the European society of cardiology. Cardiovasc Drugs Ther. 33, 77–86. doi:10.1007/s10557-018-6843-5
Padwal, R., and Laupacis, A. (2004). Antihypertensive therapy and incidence of type 2 diabetes: a systematic review. Diabetes Care 27, 247–255. doi:10.2337/diacare.27.1.247
Pae, E. K., and Harper, R. M. (2021). Potential mechanisms underlying hypoxia-induced diabetes in a rodent model: implications for COVID-19. Child. (Basel) 8, 1178. doi:10.3390/children8121178
Palfrey, H. C., and Leung, S. (1993). Inhibition of Na-K-2Cl cotransport and bumetanide binding by ethacrynic acid, its analogues, and adducts. Am. J. Physiol. 264, C1270–C1277. doi:10.1152/ajpcell.1993.264.5.C1270
Papaccio, G., and Esposito, V. (1987). Hyperglycemic effects of hydrochlorothiazide and propranolol. A biochemical and ultrastructural study. Acta Diabetol. Lat. 24, 325–330. doi:10.1007/BF02742965
Parkkila, A. K., Scarim, A. L., Parkkila, S., Waheed, A., Corbett, J. A., and Sly, W. S. (1998). Expression of carbonic anhydrase V in pancreatic beta cells suggests role for mitochondrial carbonic anhydrase in insulin secretion. J. Biol. Chem. 273, 24620–24623. doi:10.1074/jbc.273.38.24620
Payne, J. A. (1997). Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am. J. Physiol. 273, C1516–C1525. doi:10.1152/ajpcell.1997.273.5.C1516
Pazos, P., Lima, L., Tovar, S., Gonzalez-Touceda, D., Dieguez, C., and Garcia, M. C. (2015). Divergent responses to thermogenic stimuli in BAT and subcutaneous adipose tissue from interleukin 18 and interleukin 18 receptor 1-deficient mice. Sci. Rep. 5, 17977. doi:10.1038/srep17977
Pederson, B. A., Nordlie, M. A., Foster, J. D., and Nordlie, R. C. (1998). Effects of ionic strength and chloride ion on activities of the glucose-6-phosphatase system: regulation of the biosynthetic activity of glucose-6-phosphatase by chloride ion inhibition/deinhibition. Arch. Biochem. Biophys. 353, 141–151. doi:10.1006/abbi.1998.0642
Penhoat, A., Fayard, L., Stefanutti, A., Mithieux, G., and Rajas, F. (2014). Intestinal gluconeogenesis is crucial to maintain a physiological fasting glycemia in the absence of hepatic glucose production in mice. Metabolism 63, 104–111. doi:10.1016/j.metabol.2013.09.005
Pickkers, P., Garcha, R. S., Schachter, M., Smits, P., and Hughes, A. D. (1999). Inhibition of carbonic anhydrase accounts for the direct vascular effects of hydrochlorothiazide. Hypertension 33, 1043–1048. doi:10.1161/01.hyp.33.4.1043
Plavinik, F. L., Rodrigues, C. I., Zanella, M. T., and Ribeiro, A. B. (1992). Hypokalemia, glucose intolerance, and hyperinsulinemia during diuretic therapy. Hypertension 19, II26–29. doi:10.1161/01.hyp.19.2_suppl.ii26
Pollare, T., Lithell, H., and Berne, C. (1989). A comparison of the effects of hydrochlorothiazide and captopril on glucose and lipid metabolism in patients with hypertension. N. Engl. J. Med. 321, 868–873. doi:10.1056/NEJM198909283211305
Popowicz, P., and Simmons, N. L. (1988). [3H]bumetanide binding and inhibition of Na+ + K+ + Cl-co-transport: demonstration of specificity by the use of MDCK cells deficient in co-transport activity. Q. J. Exp. Physiol. 73, 193–202. doi:10.1113/expphysiol.1988.sp003132
Price, A. L., Lingvay, I., Szczepaniak, E. W., Wiebel, J., Victor, R. G., and Szczepaniak, L. S. (2013). The metabolic cost of lowering blood pressure with hydrochlorothiazide. Diabetol. Metab. Syndr. 5, 35. doi:10.1186/1758-5996-5-35
Puigserver, P., Rhee, J., Donovan, J., Walkey, C. J., Yoon, J. C., Oriente, F., et al. (2003). Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 423, 550–555. doi:10.1038/nature01667
Rahhal, A., Saad, M. O., Tawengi, K., Assi, A. a.R., Habra, M., and Ahmed, D. (2019). Torsemide versus furosemide after acute decompensated heart failure: a retrospective observational study. BMC Cardiovasc Disord. 19, 127. doi:10.1186/s12872-019-1112-5
Rahman, A., Kittikulsuth, W., Fujisawa, Y., Sufiun, A., Rafiq, K., Hitomi, H., et al. (2016). Effects of diuretics on sodium-dependent glucose cotransporter 2 inhibitor-induced changes in blood pressure in obese rats suffering from the metabolic syndrome. J. Hypertens. 34, 893–906. doi:10.1097/HJH.0000000000000871
Ramsay, L. E., Yeo, W. W., and Jackson, P. R. (1992). Diabetes, impaired glucose tolerance and insulin resistance with diuretics. Eur. Heart J. 13 (Suppl. G), 68–71. doi:10.1093/eurheartj/13.suppl_g.68
Ramsay, L. E., Yeo, W. W., and Jackson, P. R. (1994). Metabolic effects of diuretics. Cardiology 84 (Suppl. 2), 48–56. doi:10.1159/000176457
Rathod, Y. D., Abdelgawad, R., Hubner, C. A., and Di Fulvio, M. (2023). Slc12a2 loss in insulin-secreting beta-cells links development of overweight and metabolic dysregulation to impaired satiation control of feeding. Am. J. Physiol. Endocrinol. Metab. 325, E581–E594. doi:10.1152/ajpendo.00197.2023
Ray, K., Sahana, C. C., Chaudhuri, S. B., Das, D. N., Mondal, S., Sarkar, D., et al. (1993). Frusemide-induced diabetes mellitus in insulin pretreated rabbits as an experimental model. Indian J. Med. Res. 98, 37–39.
Rebelos, E., Mari, A., Honka, M. J., Pekkarinen, L., Latva-Rasku, A., Laurila, S., et al. (2024). Renal cortical glucose uptake is decreased in insulin resistance and correlates inversely with serum free-fatty acids. J. Clin. Endocrinol. Metab. 109, 1033–1040. doi:10.1210/clinem/dgad663
Reinhardt, D. J. (1959). Experience with chlorothiazide (diuril) in hypertensive patients. Del State Med. J. 31, 44–52.
Reisin, E., Weir, M. R., Falkner, B., Hutchinson, H. G., Anzalone, D. A., and Tuck, M. L. (1997). Lisinopril versus hydrochlorothiazide in obese hypertensive patients: a multicenter placebo-controlled trial. Treatment in Obese Patients with Hypertension (TROPHY) Study Group. Hypertension 30, 140–145. doi:10.1161/01.hyp.30.1.140
Reungjui, S., Roncal, C. A., Mu, W., Srinivas, T. R., Sirivongs, D., Johnson, R. J., et al. (2007). Thiazide diuretics exacerbate fructose-induced metabolic syndrome. J. Am. Soc. Nephrol. 18, 2724–2731. doi:10.1681/ASN.2007040416
Riahi, Y., Israeli, T., Yeroslaviz, R., Chimenez, S., Avrahami, D., Stolovich-Rain, M., et al. (2018). Inhibition of mTORC1 by ER stress impairs neonatal beta-cell expansion and predisposes to diabetes in the Akita mouse. Elife 7, e38472. doi:10.7554/eLife.38472
Riess, W., Dubach, U. C., Burckhardt, D., Theobald, W., Vuillard, P., and Zimmerli, M. (1977). Pharmacokinetic studies with chlorthalidone (Hygroton) in man. Eur. J. Clin. Pharmacol. 12, 375–382. doi:10.1007/BF00562454
Robinson, D. S., Nilsson, C. M., Leonard, R. F., and Horton, E. S. (1981). Effects of loop diuretics on carbohydrate metabolism and electrolyte excretion. J. Clin. Pharmacol. 21, 637–646. doi:10.1002/j.1552-4604.1981.tb05676.x
Rochelle, J. B., Bullock, A. C., and Ford, R. V. (1958). Potentiation of antihypertensive therapy by use of chlorothiazide. J. Am. Med. Assoc. 168, 410. doi:10.1001/jama.1958.63000040002010a
Ross, B. D., Espinal, J., and Silva, P. (1986). Glucose metabolism in renal tubular function. Kidney Int. 29, 54–67. doi:10.1038/ki.1986.8
Runyan, J. W. (1962). Influence of thiazide diuretics on carbohydrate metabolism in patients with mild diabetes. N. Engl. J. Med. 267, 541–543. doi:10.1056/NEJM196209132671105
Samson, S. L., and Garber, A. J. (2014). Metabolic syndrome. Endocrinol. Metab. Clin. North Am. 43, 1–23. doi:10.1016/j.ecl.2013.09.009
Sandstrom, P. E. (1988). Evidence for diabetogenic action of bumetanide in mice. Eur. J. Pharmacol. 150, 35–41. doi:10.1016/0014-2999(88)90747-9
Sandstrom, P. E. (1990). Bumetanide reduces insulin release by a direct effect on the pancreatic beta-cells. Eur. J. Pharmacol. 187, 377–383. doi:10.1016/0014-2999(90)90365-d
Sandstrom, P. E., and Sehlin, J. (1987). Stereoselective inhibition of chloride transport by loop diuretics in pancreatic beta-cells. Eur. J. Pharmacol. 144, 389–392. doi:10.1016/0014-2999(87)90394-3
Sandstrom, P. E., and Sehlin, J. (1988a). Furosemide and Ca2+ affect 86Rb+ efflux from pancreatic beta-cells by different mechanisms. Biochim. Biophys. Acta 943, 28–34. doi:10.1016/0005-2736(88)90343-4
Sandstrom, P. E., and Sehlin, J. (1988b). Furosemide causes acute and long-term hyperglycaemia and reduces glucose tolerance in mice. Acta Physiol. Scand. 132, 75–81. doi:10.1111/j.1748-1716.1988.tb08300.x
Sandstrom, P. E., and Sehlin, J. (1988c). Furosemide reduces insulin release by inhibition of Cl- and Ca2+ fluxes in beta-cells. Am. J. Physiol. 255, E591–E596. doi:10.1152/ajpendo.1988.255.5.E591
Sandstrom, P. E., and Sehlin, J. (1988d). Furosemide-induced glucose intolerance in mice is associated with reduced insulin secretion. Eur. J. Pharmacol. 147, 403–409. doi:10.1016/0014-2999(88)90175-6
Sandstrom, P. E., Sehlin, J., and Amark, K. (1993). Furosemide treatment causes age-dependent glucose intolerance and islet damage in obese-hyperglycaemic mice. Pharmacol. Toxicol. 72, 304–309. doi:10.1111/j.1600-0773.1993.tb01655.x
Sarafidis, P. A., Mcfarlane, S. I., and Bakris, G. L. (2007). Antihypertensive agents, insulin sensitivity, and new-onset diabetes. Curr. Diab Rep. 7, 191–199. doi:10.1007/s11892-007-0031-5
Sasaki, M., Sasako, T., Kubota, N., Sakurai, Y., Takamoto, I., Kubota, T., et al. (2017). Dual regulation of gluconeogenesis by insulin and glucose in the proximal tubules of the kidney. Diabetes 66, 2339–2350. doi:10.2337/db16-1602
Savage, P. J., Pressel, S. L., Curb, J. D., Schron, E. B., Applegate, W. B., Black, H. R., et al. (1998). Influence of long-term, low-dose, diuretic-based, antihypertensive therapy on glucose, lipid, uric acid, and potassium levels in older men and women with isolated systolic hypertension: the Systolic Hypertension in the Elderly Program. SHEP Cooperative Research Group. Arch. Intern Med. 158, 741–751. doi:10.1001/archinte.158.7.741
Scheen, A. J. (2018). Type 2 diabetes and thiazide diuretics. Curr. Diab Rep. 18, 6. doi:10.1007/s11892-018-0976-6
Schliess, F., Von Dahl, S., and Haussinger, D. (2001). Insulin resistance induced by loop diuretics and hyperosmolarity in perfused rat liver. Biol. Chem. 382, 1063–1069. doi:10.1515/BC.2001.133
Schoolwerth, A. C., Smith, B. C., and Culpepper, R. M. (1988). Renal gluconeogenesis. Min. Electrolyte Metab. 14, 347–361.
Schwartz, M. W., Woods, S. C., Porte, D., Seeley, R. J., and Baskin, D. G. (2000). Central nervous system control of food intake. Nature 404, 661–671. doi:10.1038/35007534
Seltzer, H. S., and Allen, E. W. (1969). Hyperglycemia and inhibition of insulin secretion during administration of diazoxide and trichlormethiazide in man. Diabetes 18, 19–28. doi:10.2337/diab.18.1.19
Sener, A., Jijakli, H., Zahedi Asl, S., Courtois, P., Yates, A. P., Meuris, S., et al. (2007). Possible role of carbonic anhydrase in rat pancreatic islets: enzymatic, secretory, metabolic, ionic, and electrical aspects. Am. J. Physiol. Endocrinol. Metab. 292, E1624–E1630. doi:10.1152/ajpendo.00631.2006
Siegel, D., Meier, J., Maas, C., Lopez, J., and Swislocki, A. L. (2008). The effect of body mass index on fasting blood glucose after initiation of thiazide therapy in hypertensive patients. Am. J. Hypertens. 21, 438–442. doi:10.1038/ajh.2007.75
Singh, S., Goel, S., Duhan, S., Chaudhary, R., Garg, A., Tantry, U. S., et al. (2023). Effect of furosemide versus torsemide on hospitalizations and mortality in patients with heart failure: a meta-analysis of randomized controlled trials. Am. J. Cardiol. 206, 42–48. doi:10.1016/j.amjcard.2023.08.079
Singh, S., Mcdonough, C. W., Gong, Y., Alghamdi, W. A., Arwood, M. J., Bargal, S. A., et al. (2018). Genome wide association study identifies the HMGCS2 locus to be associated with chlorthalidone induced glucose increase in hypertensive patients. J. Am. Heart Assoc. 7, e007339. doi:10.1161/JAHA.117.007339
Sinke, A. P., Kortenoeven, M. L., De Groot, T., Baumgarten, R., Devuyst, O., Wetzels, J. F., et al. (2014). Hydrochlorothiazide attenuates lithium-induced nephrogenic diabetes insipidus independently of the sodium-chloride cotransporter. Am. J. Physiol. Ren. Physiol. 306, F525–F533. doi:10.1152/ajprenal.00617.2013
Sorgel, F., Beyhl, F. E., and Mutschler, E. (1980). Inhibition of uridinediphosphate glucuronyltransferase caused by furosemide. Experientia 36, 861–863. doi:10.1007/BF01978616
Stafford, R. S., Bartholomew, L. K., Cushman, W. C., Cutler, J. A., Davis, B. R., Dawson, G., et al. (2010). Impact of the ALLHAT/JNC7 Dissemination Project on thiazide-type diuretic use. Arch. Intern Med. 170, 851–858. doi:10.1001/archinternmed.2010.130
Stason, W. B., Cannon, P. J., Heinemann, H. O., and Laragh, J. H. (1966). Furosemide. A clinical evaluation of its diuretic action. Circulation 34, 910–920. doi:10.1161/01.cir.34.5.910
Stroobandt, R., Dodion, L., and Kesteloot, H. (1982). Clinical efficacy of torasemide, a new diuretic agent, in patients with acute heart failure: a double blind comparison with furosemide. Arch. Int. Pharmacodyn. Ther. 260, 151–158.
Stuhlmann, T., Planells-Cases, R., and Jentsch, T. J. (2018). LRRC8/VRAC anion channels enhance beta-cell glucose sensing and insulin secretion. Nat. Commun. 9, 1974. doi:10.1038/s41467-018-04353-y
Stump, C. S., Hamilton, M. T., and Sowers, J. R. (2006). Effect of antihypertensive agents on the development of type 2 diabetes mellitus. Mayo Clin. Proc. 81, 796–806. doi:10.4065/81.6.796
Stumvoll, M., Meyer, C., Mitrakou, A., and Gerich, J. E. (1999). Important role of the kidney in human carbohydrate metabolism. Med. Hypotheses 52, 363–366. doi:10.1054/mehy.1997.0655
Stumvoll, M., Meyer, C., Perriello, G., Kreider, M., Welle, S., and Gerich, J. (1998). Human kidney and liver gluconeogenesis: evidence for organ substrate selectivity. Am. J. Physiol. 274, E817–E826. doi:10.1152/ajpendo.1998.274.5.E817
Tasker, P. R., and Mitchell-Heggs, P. F. (1976). Non-ketotic diabetic precoma associated with high-dose frusemide therapy. Br. Med. J. 1, 626–627. doi:10.1136/bmj.1.6010.626-a
Teo, Y. H., Teo, Y. N., Syn, N. L., Kow, C. S., Yoong, C. S. Y., Tan, B. Y. Q., et al. (2021). Effects of sodium/glucose cotransporter 2 (SGLT2) inhibitors on cardiovascular and metabolic outcomes in patients without diabetes mellitus: a systematic review and meta-analysis of randomized-controlled trials. J. Am. Heart Assoc. 10, e019463. doi:10.1161/JAHA.120.019463
Thompson, S. A., Arden, S. A., Marshall, G., Wingrove, P. B., Whiting, P. J., and Wafford, K. A. (1999). Residues in transmembrane domains I and II determine gamma-aminobutyric acid type AA receptor subtype-selective antagonism by furosemide. Mol. Pharmacol. 55, 993–999. doi:10.1124/mol.55.6.993
Toivonen, S., and Mustala, O. (1966). Diabetogenic action of frusemide. Br. Med. J. 1, 920–921. doi:10.1136/bmj.1.5492.920-c
Tziomalos, K., Athyros, V. G., Mikhailidis, D. P., and Karagiannis, A. (2013). Hydrochlorothiazide vs. chlorthalidone as the optimal diuretic for the management of hypertension. Curr. Pharm. Des. 19, 3766–3772. doi:10.2174/13816128113199990315
Valimaki, M., Harno, K., and Nikkila, E. A. (1983). Serum lipoproteins and indices of glucose tolerance during diuretic therapy: a comparison between hydrochlorothiazide and piretanide. J. Cardiovasc Pharmacol. 5, 525–530. doi:10.1097/00005344-198307000-00003
Valmin, K., and Hansen, T. (1975). Treatment of benign essential hypertension: comparison of furosemide and hydrochlorothiazide. Eur. J. Clin. Pharmacol. 8, 393–401. doi:10.1007/BF00562312
Van Der Heijden, M., Donders, S. H., Cleophas, T. J., Niemeyer, M. G., Van Der Meulen, J., Bernink, P. J., et al. (1998). A randomized, placebo-controlled study of loop diuretics in patients with essential hypertension: the bumetanide and furosemide on lipid profile (BUFUL) clinical study report. J. Clin. Pharmacol. 38, 630–635. doi:10.1002/j.1552-4604.1998.tb04470.x
Verbrugge, F. H., and Menon, V. (2022). Torsemide comparison with furosemide for management of heart failure (TRANSFORM-HF) trial. Eur. Heart J. Acute Cardiovasc Care 11, 931–932. doi:10.1093/ehjacc/zuac144
Vily-Petit, J., Soty-Roca, M., Silva, M., Raffin, M., Gautier-Stein, A., Rajas, F., et al. (2020). Intestinal gluconeogenesis prevents obesity-linked liver steatosis and non-alcoholic fatty liver disease. Gut 69, 2193–2202. doi:10.1136/gutjnl-2019-319745
Vinay, P., Manillier, C., Lalonde, L., Thibault, G., Boulanger, Y., Gougoux, A., et al. (1987). Comparative effect of ANF and various diuretics on isolated nephron segments. Kidney Int. 31, 946–955. doi:10.1038/ki.1987.91
Waldegger, S., Barth, P., Raber, G., and Lang, F. (1997). Cloning and characterization of a putative human serine/threonine protein kinase transcriptionally modified during anisotonic and isotonic alterations of cell volume. Proc. Natl. Acad. Sci. U. S. A. 94, 4440–4445. doi:10.1073/pnas.94.9.4440
Walker, N. M., Flagella, M., Gawenis, L. R., Shull, G. E., and Clarke, L. L. (2002). An alternate pathway of cAMP-stimulated Cl secretion across the NKCC1-null murine duodenum. Gastroenterology 123, 531–541. doi:10.1053/gast.2002.34757
Walker, W. G. (1967). The clinical use of furosemide and ethacrynic acid. Med. Clin. North Am. 51, 1277–1283. doi:10.1016/s0025-7125(16)32994-7
Wang, J., Sun, C., Gerdes, N., Liu, C., Liao, M., Liu, J., et al. (2015). Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat. Med. 21, 820–826. doi:10.1038/nm.3890
Ward, A., and Heel, R. C. (1984). Bumetanide. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use. Drugs 28, 426–464. doi:10.2165/00003495-198428050-00003
Wargo, K. A., and Banta, W. M. (2009). A comprehensive review of the loop diuretics: should furosemide be first line? Ann. Pharmacother. 43, 1836–1847. doi:10.1345/aph.1M177
Watford, M. (2005). Is the small intestine a gluconeogenic organ. Nutr. Rev. 63, 356–360. doi:10.1111/j.1753-4887.2005.tb00114.x
Weber, G. (1961). Kidney enzymes of gluconeogenesis, glycogenesis, glycolysis and direct oxidation. Proc. Soc. Exp. Biol. Med. 108, 631–634. doi:10.3181/00379727-108-27018
Weidmann, P., De Courten, M., Ferrari, P., and Bohlen, L. (1993). Serum lipoproteins during treatment with antihypertensive drugs. J. Cardiovasc Pharmacol. 22 (Suppl. 6), S98–S105. doi:10.1097/00005344-199306226-00016
Weidmann, P., Gerber, A., and Mordasini, R. (1983). Effects of antihypertensive therapy on serum lipoproteins. Hypertension 5, III120–131. doi:10.1161/01.hyp.5.5_pt_2.iii120
Weller, J. M., and Borondy, M. (1967). Effect of furosemide on glucose metabolism. Metabolism 16, 532–536. doi:10.1016/0026-0495(67)90083-2
Wen, S. F., Boynar, J. W., and Stoll, R. W. (1978). Effects of diuretics on renal glucose transport in the dog. Clin. Sci. Mol. Med. 54, 481–488. doi:10.1042/cs0540481
Wertheimer, L., Finnerty, F. A., Bercu, B. A., and Hall, R. H. (1971). Furosemide in essential hypertension. A statistical analysis of three double-blind studies. Arch. Intern Med. 127, 934–938. doi:10.1001/archinte.127.5.934
Whelton, A. (1981). Long-term bumetanide treatment of renal edema. Comparison with furosemide. J. Clin. Pharmacol. 21, 591–598. doi:10.1002/j.1552-4604.1981.tb05669.x
Wilhelmsen, L., Berglund, G., Elmfeldt, D., and Wedel, H. (1981). Beta-blockers versus saluretics in hypertension. Comparison of total mortality, myocardial infarction, and sudden death: study design and early results on blood pressure reduction. Prev. Med. 10, 38–49. doi:10.1016/0091-7435(81)90004-9
Wilkins, R. W. (1958). Precautions in use of antihypertensive drugs, including chlorothiazide. J. Am. Med. Assoc. 167, 801–807. doi:10.1001/jama.1958.02990240001001
Wilkins, R. W., Hollander, W., and Chobanian, A. V. (1958). Chlorothiazide in hypertension: studies on its mode of action. Ann. N. Y. Acad. Sci. 71, 465–472. doi:10.1111/j.1749-6632.1958.tb46775.x
Williams, J. R., and Payne, J. A. (2004). Cation transport by the neuronal K(+)-Cl(-) cotransporter KCC2: thermodynamics and kinetics of alternate transport modes. Am. J. Physiol. Cell. Physiol. 287, C919–C931. doi:10.1152/ajpcell.00005.2004
Woods, S. C., Lutz, T. A., Geary, N., and Langhans, W. (2006). Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos. Trans. R. Soc. Lond B Biol. Sci. 361, 1219–1235. doi:10.1098/rstb.2006.1858
Wright, E. M., Hirayama, B. A., and Loo, D. F. (2007). Active sugar transport in health and disease. J. Intern Med. 261, 32–43. doi:10.1111/j.1365-2796.2006.01746.x
Xue, H., Liu, S., Ji, T., Ren, W., Zhang, X. H., Zheng, L. F., et al. (2009). Expression of NKCC2 in the rat gastrointestinal tract. Neurogastroenterol. Motil. 21, 1068–1e89. doi:10.1111/j.1365-2982.2009.01334.x
Yang, Y., Fu, A., Wu, X., and Reagan, J. D. (2012). GPR35 is a target of the loop diuretic drugs bumetanide and furosemide. Pharmacology 89, 13–17. doi:10.1159/000335127
Yoshida, T., Lewy, P. R., Voyer, L. E., Ward, A. B., Al-Ubaidi, Y. Y., and Metcoff, J. (1970). Effect of farosemide on renal glycolysis and oxygen uptake in rats. Pediatr. Res. 4, 448. doi:10.1203/00006450-197009000-00056
Zatuchni, J., and Kordasz, F. (1961). The diabetogenic effect of thiazide diuretics. Am. J. Cardiol. 7, 565–567. doi:10.1016/0002-9149(61)90513-6
Zeuthen, T., and Macaulay, N. (2012). Cotransport of water by Na⁺-K⁺-2Cl⁻ cotransporters expressed in Xenopus oocytes: NKCC1 versus NKCC2. J. Physiol. 590, 1139–1154. doi:10.1113/jphysiol.2011.226316
Zhang, S., Meor Azlan, N. F., Josiah, S. S., Zhou, J., Zhou, X., Jie, L., et al. (2023). The role of SLC12A family of cation-chloride cotransporters and drug discovery methodologies. J. Pharm. Anal. 13, 1471–1495. doi:10.1016/j.jpha.2023.09.002
Zhang, X., Luo, S., Wang, M., Cao, Q., Zhang, Z., Huang, Q., et al. (2022a). Differential IL18 signaling via IL18 receptor and Na-Cl co-transporter discriminating thermogenesis and glucose metabolism regulation. Nat. Commun. 13, 7582. doi:10.1038/s41467-022-35256-8
Zhang, X., Luo, S., Wang, M., Huang, Q., Fang, W., Li, J., et al. (2022b). IL18 signaling causes islet beta cell development and insulin secretion via different receptors on acinar and beta cells. Dev. Cell. 57, 1496–1511.e6. doi:10.1016/j.devcel.2022.05.013
Zhang, X., Yang, S., Chen, J., and Su, Z. (2018). Unraveling the regulation of hepatic gluconeogenesis. Front. Endocrinol. (Lausanne) 9, 802. doi:10.3389/fendo.2018.00802
Zhang, X., and Zhao, Q. (2016). Association of thiazide-type diuretics with glycemic changes in hypertensive patients: a systematic review and meta-analysis of randomized controlled clinical trials. J. Clin. Hypertens. (Greenwich) 18, 342–351. doi:10.1111/jch.12679
Zhou, B., Zhang, Y., Li, S., Wu, L., Fejes-Toth, G., Naray-Fejes-Toth, A., et al. (2021). Serum- and glucocorticoid-induced kinase drives hepatic insulin resistance by directly inhibiting AMP-activated protein kinase. Cell. Rep. 37, 109785. doi:10.1016/j.celrep.2021.109785
Zhou, M. S., Schulman, I. H., and Zeng, Q. (2012). Link between the renin-angiotensin system and insulin resistance: implications for cardiovascular disease. Vasc. Med. 17, 330–341. doi:10.1177/1358863X12450094
Zhu, J. X., Xue, H., Ji, T., and Xing, Y. (2011). Cellular localization of NKCC2 and its possible role in the Cl(-) absorption in the rat and human distal colonic epithelia. Transl. Res. 158, 146–154. doi:10.1016/j.trsl.2011.04.003
Zillich, A. J., Garg, J., Basu, S., Bakris, G. L., and Carter, B. L. (2006). Thiazide diuretics, potassium, and the development of diabetes: a quantitative review. Hypertension 48, 219–224. doi:10.1161/01.HYP.0000231552.10054.aa
Keywords: thiazides, hyperglycemia, metabolic syndrome, loop diuretics, insulin, overweight, hypertension, diabetes
Citation: Di Fulvio M, Rathod YD and Khader S (2025) Diuretics: a review of the pharmacology and effects on glucose homeostasis. Front. Pharmacol. 16:1513125. doi: 10.3389/fphar.2025.1513125
Received: 17 October 2024; Accepted: 07 March 2025;
Published: 28 March 2025.
Edited by:
Eliot Ohlstein, Drexel University School of Medicine, United StatesReviewed by:
Robertas Badaras, Vilnius University, LithuaniaCopyright © 2025 Di Fulvio, Rathod and Khader. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauricio Di Fulvio, bWF1cmljaW8uZGlmdWx2aW9Ad3JpZ2h0LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.