 Annalisa Contursi1,2†
Annalisa Contursi1,2† Stefania Tacconelli1,2†
Stefania Tacconelli1,2† Sara Di Berardino1,2Alessandra De Michele1,2
Sara Di Berardino1,2Alessandra De Michele1,2 Paola Patrignani1,2*
Paola Patrignani1,2*- 1Systems Pharmacology and Translational Therapeutics Laboratory, The Center for Advanced Studies and Technology (CAST), “G. d’Annunzio” University, Chieti, Italy
- 2Department of Neuroscience, Imaging and Clinical Science, “G. d’Annunzio” University Medical School, Chieti, Italy
Inflammation plays a critical role in the pathogenesis of various diseases by promoting the acquisition of new functional traits by different cell types. Shared risk factors between cardiovascular disease and cancer, including smoking, obesity, diabetes, high-fat diet, low physical activity, and alcohol consumption, contribute to inflammation linked to platelet activation. Platelets contribute to an inflammatory state by activating various normal cells, such as fibroblasts, immune cells, and vascular cells. This activation is achieved by releasing diverse molecules from platelets, including lipids (eicosanoids), growth and angiogenic factors, and extracellular vesicles (EVs) rich in various RNA species. Antiplatelet agents like low-dose aspirin can prevent cardiovascular disease and cancer by inhibiting platelet functions beyond the antithrombotic action. Throughout the initial phases of tumorigenesis, the activation of platelets induces the overexpression of cyclooxygenase (COX)-2 in stromal cells, leading to increased biosynthesis of prostaglandin (PG)E2. This prostanoid can contribute to tumor development by inhibiting apoptosis, promoting cancer cell proliferation and migration, and immune evasion. Notably, platelets induce the epithelial-mesenchymal transition (EMT) in tumor cells, enhancing their metastatic potential. Two platelet eicosanoids, PGE2 (generated as a minor product of COX-1) and 12S-hydroxyeicosatetraenoic acid (HETE) [derived from the platelet-type 12-lipoxygenase (LOX)], contribute to EMT. In addition to the pharmacological inhibition of eicosanoid biosynthesis, a potential strategy for mitigating platelet-induced metastasis might encompass the inhibition of direct interactions between platelets and cancer cells. For example, there is promise in utilizing revacept to inhibit the interaction between platelet collagen receptors (particularly GPVI) and galectin-3 in cancer cells. Identifying these novel platelet functions suggests the potential application of antiplatelet agents, such as low-dose aspirin, in mitigating cancer risk, particularly in the case of colorectal cancer. It is necessary to investigate the effectiveness of other antiplatelet drugs, such as ADP P2Y12 receptor antagonists, in cancer prevention. Other new antiplatelet drugs, such as revacept and selective 12-LOX inhibitors, currently under clinical development, are of interest due to their low risk of bleeding. Platelets and EVs carry important clinical information because they contain specific proteins and RNAs associated with disease conditions. Their analysis can improve the accuracy of liquid biopsies for early cancer detection, monitoring progression, and assessing drug response.
Introduction
The cellular components involved in the process of inflammation
Acute inflammation is a highly regulated physiological process essential in defending the body against various external and internal threats. Any disruption in tissue homeostasis triggers the activation of innate immune cells, which form the first line of defense intended to restore the affected tissue (Delves and Roitt, 2000). The primary inflammatory cells that mediate acute inflammation are the polymorphonuclear leukocytes (PMN). The production of chemotactic molecules induces the migration from the venous system to the damaged site of monocytes/macrophages, mast cells, dendritic cells, and natural killer (NK) cells. They amplify the inflammatory response, leading to pathogen elimination and tissue repair by releasing cytokines, chemokines, matrix-remodeling proteases, and reactive oxygen and nitrogen species (Coussens and Werb, 2002; Chitu and Stanley, 2006). Like that associated with wound healing, physiological inflammation is a strictly controlled and self-limiting process (Martin and Leibovich, 2005). However, losing control over immune components can lead to chronic inflammation.
Multiple factors can contribute to an excessive burden of inflammation, encompassing lifestyle factors such as smoking and dietary habits, along with conditions like obesity, diabetes, and exposure to environmental pollutants (Libby, 2012). The development of chronic inflammation is associated with a range of diseases, including fatty liver disease, inflammatory bowel disease (IBD), Alzheimer’s, Parkinson’s, atherosclerosis, and cancer (Chen et al., 2017). Current studies indicate that the trigger signaling of chronic inflammation is associated with enhanced platelet activation (Gawaz et al., 2005; Dovizio et al., 2014). It is now acknowledged that platelets are mediators of intercellular communication and propagation of the cellular activation response through the release and transfer of their molecular cargo to numerous cell types implicated in inflammation (Rondina et al., 2013; Varon and Shai, 2015). Consequently, this insight opens doors to innovative approaches to curbing chronic inflammation-related diseases (Gawaz et al., 2023; Patrignani and Patrono, 2015; 2018).
In atherosclerosis, the damage of vascular endothelial cells induces a rapid adhering to the vascular wall of activated platelets; this event contributes to the development of vascular inflammation through leukocyte chemoattraction and their subsequent infiltration into the vessel wall associated with the proliferation of smooth muscle cells (Ross et al., 1985; Gawaz et al., 2005). Interactions between platelets and leukocytes occur via PSGL-1-P-selectin (Evangelista et al., 1999; Yang et al., 1999), followed by the binding between Mac-1 (CD11b/CD18, αMb2) and GPIbα (Simon et al., 2000), JAM-3 (Santoso et al., 2002), or ICAM-2 (Diacovo et al., 1994). The binding facilitates the release of inflammatory molecules from platelets, triggering monocyte inflammatory cascades (Weyrich et al., 1996).
Platelets promote plaque formation by accelerating foam cell formation upon binding to oxidized LDL (Coenen et al., 2021). Platelet-derived matrix metalloproteinases (MMPs), a family of proteolytic enzymes that mediate physiological and pathophysiological extracellular matrix turnover (Gresele et al., 2021), such as MMP-1 and -2 can cleave protease-activated receptors (PARs). Among them, PAR-1 is a G protein-coupled receptor with important roles in hemostasis and inflammation (Willis Fox and Preston, 2020). Rana et al. (2018) demonstrated a role for MMP-1 in plaque formation in mice, mediated via interaction with endothelial PAR-1. Momi et al. (2022) discovered that platelet MMP-2 plays a role in mice’s early formation of arterial plaque. This occurs by activating endothelial PAR-1, which leads to endothelial activation and monocyte intravasation. They also found that in patients with coronary artery disease (CAD) and human immunodeficiency virus (HIV), MMP-2 is overexpressed on the surface of platelets compared to healthy individuals of similar age and sex. Furthermore, it was noted that platelet MMP-2 levels are positively associated with the severity of carotid artery stenosis in humans. These findings indicate that inhibiting PAR-1 activities could be a promising way to reduce atherothrombosis. New strategies may involve targeting the signaling downstream of PAR-1 activated by MMP-1 and -2 to specifically inhibit the proinflammatory activity of PAR-1 (Willis Fox and Preston, 2020).
The release of platelet-derived CD40 ligand (CD40L, CD154) fosters inflammation within the endothelium. The binding of CD40 on endothelial cells to CD40L on activated platelets boosts the release of IL-8 and monocyte chemoattractant protein-1 (MCP-1), major attractants for neutrophils and monocytes (Henn et al., 1998). The interaction also prompts smooth muscle cells and macrophages to release MMPs, facilitating the degradation and remodeling of inflamed tissues.
A persistent inflammatory microenvironment continuously provides various factors—such as cytokines, chemokines, and growth factors—that oppose cell death and repair mechanisms, causing genomic instability and predisposing tissues to cancer (Mantovani and Pierotti, 2008; Hussain and Harris, 2007). Consequently, mediators and cellular effectors of inflammation play a crucial role in the local tumor milieu. It was estimated that 20% of all cancers are linked to chronic infections and inflammation (Mantovani et al., 2008), highlighting inflammation as the seventh hallmark of cancer. The unrestrained nature of cancer-related inflammation promotes tumor growth, supports angiogenesis and metastasis, undermines adaptive immune responses, and modifies responses to chemotherapy (Balkwill et al., 2005; Mantovani et al., 2008). Although genetic studies in mouse models have shown that innate immune cells can get an adaptive immune response capable of eradicating nascent tumors (Negus et al., 1997), the genetic, epigenetic, and metabolic instability characteristic of neoplastic cells allows new variants to escape immune surveillance, leading to tumor establishment and progression (immunoediting process) (Dunn et al., 2002).
Notably, leukocyte infiltration observed in tumor tissues and the anticancer efficacy of antiinflammatory agents support the role of chronic inflammation in cancer development and progression (Menter et al., 2010; Wong et al., 2020; Li et al., 2020).
In the last decade, experimental evidence has supported platelets’ pivotal role in developing chronic inflammation beyond their roles in hemostasis and thrombosis (Davì and Patrono, 2007; Smyth et al., 2009; Patrignani and Patrono, 2015; 2018). Platelets transduce inflammation-related signals, contributing to the variable microenvironment facilitating cell plasticity and heterogeneity. Platelets interact directly with target cells and release numerous mediators, including eicosanoids [thromboxane (TX)A2 and prostaglandin (PG)E2], angiogenic and growth factors from α-granules, ADP from dense granules, and extracellular vesicles (EVs) containing genetic material such as mRNA and microRNAs (miRs) (Patrono et al., 2001; Patrignani and Patrono, 2015; 2018). These phenomena can contribute to developing the early events of tumorigenesis triggered by platelet activation (Figure 1).
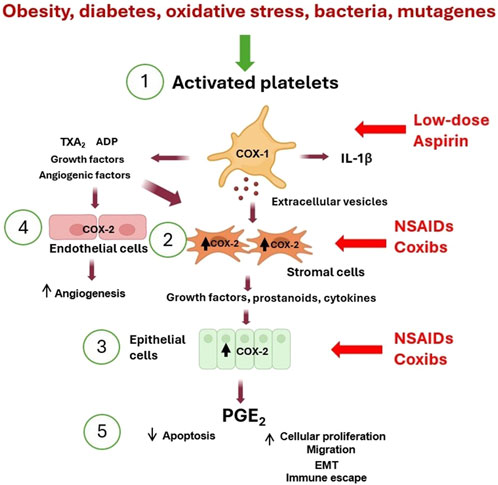
Figure 1. Platelet activation triggers the early phases of tumorigenesis. (1) Activated platelets release many mediators [including eicosanoids (such as TXA2), ADP, cytokines (such as IL-1β), growth and angiogenic factors, and extracellular vesicles (EVs) containing genetic material (such as mRNAs and microRNAs)] that contribute to (2) COX-2 overexpression in stromal cells (including macrophages and fibroblasts). (3) The crosstalk with epithelial cells leads to COX-2 induction. (4) Additionally, COX-2 is overexpressed in endothelial cells, leading to angiogenesis. (5) These events lead to the release of PGE2, which translates to inhibition of apoptosis, an increase in proliferation and migration, and induction of epithelial-mesenchymal transition (EMT). The inhibition of platelet COX-1 by low-dose aspirin prevents the downstream signaling pathways involved in the early events of tumorigenesis and indirectly inhibits COX-2 induction. In contrast, NSAIDs and coxibs have an antitumorigenic effect by directly inhibiting COX-2 activity and prostanoid generation in stromal cells, epithelial cells, and endothelial cells.
Platelet extravasation and the development of an inflammatory microenvironment
Platelets possess multifaceted functions beyond thrombosis (Figure 2), significantly contributing to various pathological conditions, including atherosclerosis, restenosis, cardiac fibrosis, airway hyperresponsiveness, airway wall remodeling, intestinal colitis, and tumor metastasis (Patrignani and Patrono, 2018; Rumbaut and Thiyagaraja, 2010; Golebiewska and Poole, 2015). An important function of platelets is the capacity to extravasate at sites of inflammation, thus interacting and activating other cells that constitute the cellular stromal compartment of different tissues. Platelets activate other cell types via direct interaction or through the release of many mediators with a wide range of activities and the capacity to release EVs that transfer platelet cargo far from the platelet. These events are associated with inflammation, tissue injury, and organ failure severity.
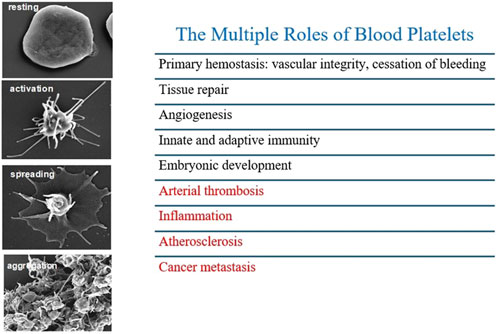
Figure 2. Different stages of platelet activation from resting to aggregation, by high resolution scanning electron microscopy. Platelets possess multifaceted functions beyond thrombosis contributing to inflammation, atherosclerosis and cancer metastasis.
Platelet recruitment to post-capillary venules at sites of acute inflammation has been shown in various experimental models, often associated with PMN-endothelial interactions. In a mouse model of corneal epithelial abrasion, an acute inflammatory response is necessary for effective wound healing (De La Cruz et al., 2021). In this model, PMNs and platelets are recruited to the small blood vessels surrounding the cornea in a mutually dependent manner. Depleting either cell type systemically inhibits the recruitment of the other. The mechanisms responsible for platelet extravasation in inflammation need to be clarified. However, the data suggest that in this model of corneal inflammation, platelet extravasation depends on CD18, mast cells, and PMNs, with a central role for mast cell degranulation in the responses. Platelet extravasation is accompanied by red blood cell (RBC) extravasation, with evidence of disruption of microvascular integrity (De La Cruz et al., 2021).
It has been reported that platelets influence leukocyte trafficking from blood vessels into lung tissue because platelets are necessary for the pulmonary recruitment of eosinophils and lymphocytes in murine allergic inflammation (Pitchford et al., 2003; Pitchford et al., 2005; Pitchford et al., 2008).
In hypertensive mice with prostacyclin (PGI2) receptor (IP) deletion (IPKO) fed with a high-salt diet associated with hypertension and cardiac fibrosis, enhanced systemic biosynthesis of TXA2 and left ventricular TXA2 receptor (TP) expression were detected (D’Agostino et al., 2021). Increased cardiac collagen deposition, profibrotic gene expression (including TGF-β), number of myofibroblasts at perivascular levels, and extravasated platelets were detected compared to WT mice treated with the same diet. The antiplatelet agent low-dose aspirin caused a selective inhibition of platelet TXA2 biosynthesis and mitigated enhanced blood pressure, cardiac fibrosis, and left ventricular profibrotic gene expression in IPKO but not WT mice. Moreover, the number of myofibroblasts and extravasated platelets in the heart was reduced (D’Agostino et al., 2021).
Platelets have been detected to extravasate and accumulate in the colonic lamina propria of chronic inflammation-associated fibrosis (Sacco et al., 2019) and intestinal adenomas in mice (Bruno et al., 2022), associated with an enhanced number of myofibroblasts.
Platelet-myofibroblast crosstalk: a key mechanism of intestinal inflammation-linked tumorigenesis
Under normal physiological conditions, the stroma comprises fibroblasts, smooth muscle cells, immune cells, endothelial cells, nerve cells, and the extracellular matrix (ECM) (Barron and Rowley, 2012). The stroma is a structural and functional support system for epithelial cells, regulating their behavior and enabling tissue repair in response to injury (Tuxhorn et al., 2001). Stromal activation during wound healing, characterized by myofibroblast activation, type I collagen deposition, and angiogenesis, is also observed in the tumor microenvironment, referred to as reactive stroma (Kalluri and Zeisberg, 2006; Tuxhorn et al., 2002).
In intestinal inflammation resulting from epithelial damage, such as experimentally induced colitis in mice or inflammatory bowel disease (IBD) in humans, platelets are observed to migrate from the bloodstream into the interstitial tissue of the colon (Danese et al., 2004; Petito et al., 2017). This extravasation may contribute to the persistence of chronic inflammation, leading to fibrosis and facilitating tumorigenesis through intercellular communication with myofibroblasts.
Sacco et al. (2019) conducted a study revealing that when human platelets are cocultured with intestinal myofibroblasts, the platelets undergo activation, resulting in increased production of TXA2, a major oxylipin derived from arachidonic acid (AA) through cyclooxygenase (COX)-1 activity. Subsequently, TXA2 induces phenotypic and functional alterations in myofibroblasts. In the presence of platelets, the characteristic spindle-shaped myofibroblasts turn towards a polarized phenotype, accompanied by elongation of the cellular body associated with enhanced proliferation and migratory) properties. The changes in myofibroblast morphology and functions induced by platelets were accompanied by reduced α-SMA, vimentin, fibronectin, and RhoA expression. These changes were prevented when platelets were exposed to aspirin, an irreversible inhibitor of platelet COX-1, and then washed away before incubation with myofibroblasts. Similar effects were observed when using an antagonist of the TP receptors, indicating that COX-1-dependent TXA2 is a crucial mediator released by platelets to activate myofibroblasts via TP receptors (Sacco et al., 2019). In the coculture of human platelets and myofibroblasts, platelet-derived TXA2 was involved in the induction of COX-2 in myofibroblasts since it was prevented by the selective inhibition of platelet COX-1 by aspirin or by a specific antagonist of TXA2 receptors (TP) (SQ 29,548) (Bruno et al., 2022). Enhanced COX-2-dependent PGE2 was detected under these experimental conditions. Interestingly, both TXA2 and PGE2 contribute to COX-2 induction in the coculture of platelet and myofibroblasts. The activation of TP by the TXA2 mimetic U46619 caused a rapid expression of COX-2 in myofibroblasts cultured alone, but PGE2 was required for the sustained expression of COX-2 (Bruno et al., 2022). Faour et al. (2001) reported that in IL-1β-treated human synovial fibroblasts, PGE2 stabilizes COX-2 mRNA and stimulates translation via EP4 receptor and the downstream kinases p38MAPK and cAMP-dependent protein kinase.
Enhanced COX-2-dependent PGE2 in the tumor microenvironment promotes tumor growth and inhibition of immunosurveillance and induces angiogenesis. Moreover, PGE2 can activate tumor epithelial cells, causing proliferation, survival, migration/invasion, and epigenetic changes (Wang and DuBois, 2013). However, it was found that TXA2 and PGE2 contribute to COX-2 induction in the coculture of platelet and myofibroblasts. The activation of TP by the TXA2 mimetic U46619 caused a rapid expression of COX-2 in myofibroblasts cultured alone, but PGE2 was required for the sustained expression of COX-2 (Bruno et al., 2022). Faour et al. (2001) reported that in IL-1β-treated human synovial fibroblasts, PGE2 stabilizes COX-2 mRNA and stimulates translation via EP4 receptor and the downstream kinases p38MAPK and cAMP-dependent protein kinase.
Proinflammatory stimuli, such as IL-1β, have been shown to upregulate COX-2 signaling in human colonic fibroblasts, modulating the proliferation and invasiveness of human colonic epithelial cancer cells (Zhu et al., 2012). Moreover, COX-2 expression has been detected in the stromal compartment of polyps from ApcMin/+ mice (Oshima et al., 1996).
These findings demonstrate the potential contribution of platelets to the development of an inflammatory microenvironment, thereby promoting the early stages of tumorigenesis through the upregulation of COX-2 and the increased production of PGE2 (Figure 1). Platelet-derived TXA2 is a pivotal trigger and sustainer of the cascade of events induced by platelet activation leading to intestinal tumorigenesis. Notably, antiplatelet agents can indirectly disrupt this cascade of downstream events (Patrignani and Patrono, 2015). Direct inhibition of COX-2 activity can achieve similar efficacy. Selective COX-2 inhibitors (coxibs) reduce tumor development by targeting COX-2 and PGE2. (Bertagnolli et al., 2006; Patrignani and Patrono, 2015; Wang and DuBois, 2013) (Figure 1). However, the heightened risk of cardiovascular side effects by COX-2 inhibitors hinders the utilization of these drugs for long-term cancer prevention regimens (Grosser et al., 2006).
The role of platelets in the development of intestinal tumorigenesis: a lesson from conditional COX-1 knockout mice in megakaryocytes/platelets
The role of platelet TXA2 in intestinal inflammation and tumorigenesis has been convincingly demonstrated by generating mice with the specific deletion of COX-1 in megakaryocytes/platelets (Sacco et al., 2019; Bruno et al., 2022).
We generated a mouse with floxed Ptgs1 (COX-1) (Sacco et al., 2019), in which exons 6 and 7 were flanked by loxP sites using transcription activator-like effector nucleases as a genome-editing tool, which significantly boosts genomic modification efficiency (Joung and Sander, 2013). These mice were bred with platelet factor 4 (Pf4)-Cre transgenic C57BL/6 mice [obtained from Jackson Laboratories (Bar Harbor, ME)] expressing a codon-improved Cre recombinase (iCre) under the control of the mouse Pf4 promoter, resulting in Cre recombinase expression in most megakaryocytes (Sacco et al., 2019). In these mice, the synthesis of TXA2 by platelet COX-1 was almost completely inhibited. The systemic biosynthesis of TXA2, as assessed by the enzymatic urinary metabolites of TXB2, was also profoundly reduced. However, COX-2-dependent markers of prostanoid biosynthesis (such as the urinary metabolite of PGE2 and PGI2) were not significantly affected (Sacco et al., 2019; Bruno et al., 2022). These effects observed in mice with the COX-1 specific deletion in platelets are similar to the impact of low-dose aspirin in humans, which is an antiplatelet agent that selectively inhibits COX-1 in platelets (Patrignani et al., 2014; Patrono et al., 2005).
In mice with experimental intestinal colitis treated with dextran sulfate sodium (DSS), there is increased biosynthesis of TXA2 from platelets (Sacco et al., 2019). TXA2 contributes to increased microvascular permeability (Turnage et al., 1985) and inflammatory, immune cells, and platelets’ movement, leading to colon accumulation (Vitiello et al., 2014). Deletion of COX-1 in platelets inhibits the chronic inflammatory response linked to the reversal of intestinal colitis symptoms in DSS-induced colitis. Furthermore, the specific deletion of COX-1 in platelets prevented intestinal inflammation-associated fibrosis (Sacco et al., 2019).
In ApcMin/+ mice that develop multiple intestinal neoplasia due to a nonsense mutation at codon 850 of the Apc gene, like humans with germline mutations in the APC gene (Moser et al., 1995), enhanced systemic TXA2 biosynthesis was detected deriving mainly from activated platelets (Bruno et al., 2022). The specific deletion of platelet COX-1 in ApcMin/+ mice was associated with a reduced systemic biosynthesis of TXA2 and a reduced number of intestinal adenomas (Figures 3A, B). ApcMin/+mice developed multiple sessile tubular adenomas with marked nuclear enlargement with hyperchromasia, denoting a lower grade of differentiation than ApcMin/+ mice with the specific deletion of platelet COX-1, as confirmed by the higher number of mitotic elements (Bruno et al., 2022). The adenomas of ApcMin/+mice showed higher staining of PCNA (proliferating cell nuclear antigen) than ApcMin/+mice with specific deletion of COX-1 in the platelet; PCNA is an auxiliary protein for DNA polymerase that reaches maximal expression during the S phase of the cell cycle (Celis et al., 1987). In polyps of ApcMin/+mice, enhanced expression of COX-2 and mPGES-1 (i.e., microsomal prostaglandin E synthase-1, the downstream enzyme responsible for PGE2 biosynthesis from COX-2 product PGH2) was detected in association with the downregulation of 15-prostaglandin dehydrogenase (15-PGDH, an enzyme oxidizing and degrading PGE2). These changes could contribute to enhanced biosynthesis of PGE2 in the tumor, which can reflect the increased systemic biosynthesis of PGE2 found in ApcMin/+ mice (Bruno et al., 2022). Deleting platelet COX-1 prevented the upregulation of COX-2 in the intestinal adenomas associated with reduced systemic biosynthesis of PGE2 (Figure 3B). The results of this study demonstrate that activated platelets can contribute to enhanced PGE2 generation in intestinal tumors, and inhibition of platelet function can indirectly prevent it, thus interfering with adenoma formation (Figures 3A, B).
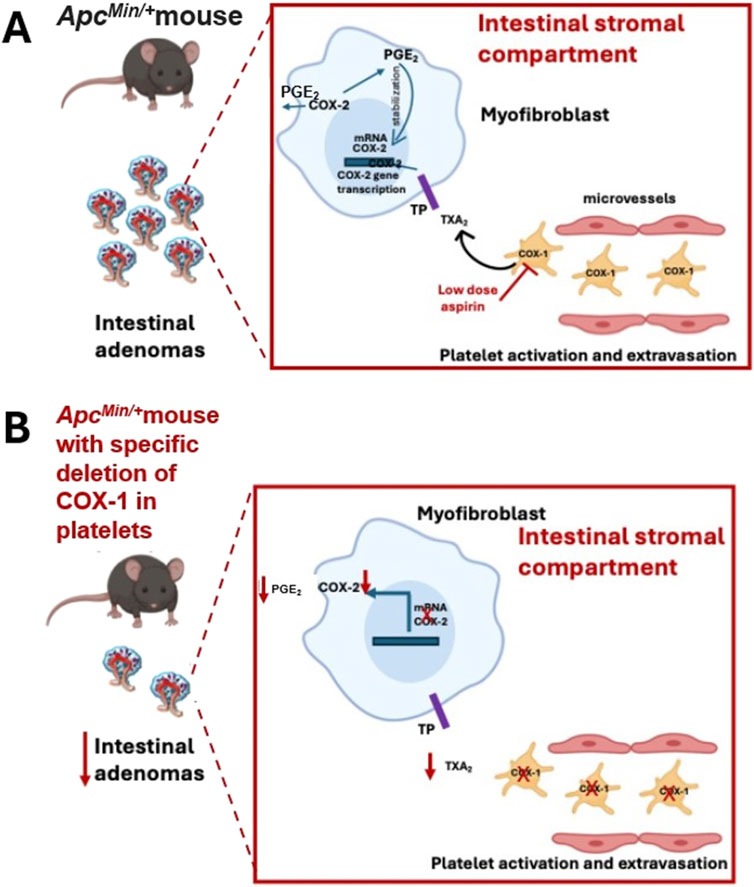
Figure 3. The role of platelet COX-1 in intestinal tumorigenesis of ApcMin/+ mice. (A) The platelet COX-1-dependent TXA2 (also from extravasated platelets) can contribute to intestinal neoplasia by triggering the expression of COX-2 in stromal cells, which is involved in the biosynthesis of PGE2; the selective inhibition of COX-1 in platelets by aspirin can prevent the biosynthesis of TXA2 and indirectly the upregulation of COX-2. (B) In ApcMin/+ mice, the specific deletion of platelet COX-1 caused a profound reduction in platelet TXA2 biosynthesis in vivo, which was associated with decreased COX-2 expression and PGE2 biosynthesis, and a reduced number and size of intestinal adenomas.
Platelets and the immune system
It is increasingly evident that platelets play a pivotal role in various immunological processes beyond their conventional function in hemostasis and thrombosis (Ali et al., 2015). Platelets can contribute to 1) defending against microbial threats, 2) recruiting and augmenting innate effector cell functions, 3) regulating antigen presentation, and 4) enhancing adaptive immune responses.
Platelets interact with immune complexes (ICs), which are associations of antibodies with their antigens. The presence of ICs in the bloodstream is documented in many pathological conditions characterized by chronic or acute inflammation (Cloutier et al., 2018). Platelets become activated when ICs bind to the IgG Fc receptor on human platelets, known as FcγRIIa (CD32) (Arman and Krauel, 2015). FcγRIIa is one of the three immunoreceptor-based activation motif (ITAM) family members of tyrosine kinase signaling receptors in human platelets (Boylan et al., 2008). The other two members are the collagen receptor glycoprotein (GP) VI and C-type lectin receptor 2. These receptors have a different signaling pathway from the G protein-coupled receptors (GPCRs) for other platelet agonists such as thrombin, adenosine 5′-diphosphate (ADP), and TXA2 (Boulaftali et al., 2014). Upon activation via ITAMs, platelets undergo aggregation and degranulation and are transformed into procoagulant platelets, which release procoagulant EVs (Puhm et al., 2021). Platelets express COX-1, crucial in converting AA released from membrane phospholipids following activation to the platelet agonist TXA2 (Patrignani and Patrono, 2015; 2018) that activates platelets through their TP receptors (Rovati et al., 2022). The selective and irreversible inhibition of COX-1 by aspirin is a fundamental aspect of antiplatelet therapy (Patrignani and Patrono, 2015). However, platelets also express 12-lipoxygenase (12S-LOX) (Contursi et al., 2022; Yoshimoto et al., 1992), and its specific role in platelet biology is not fully elucidated. AA released from cellular phospholipids in platelets can undergo a metabolic pathway involving 12-LOX (Figure 4), resulting in the generation of 12S-HpETE (hydroperoxyeicosatetraenoic acid), which subsequently undergoes rapid conversion to 12S-HETE (hydroxyeicosatetraenoic acid). The functional implications of 12-HETE in platelet physiology were hindered by the lack of specific 12-LOX inhibitors (Contursi et al., 2022). However, inhibitors that are selective for 12S-LOX over other LOXs and COXs have recently been developed (Luci et al., 2014). Their use showed the involvement of 12-LOX in the aggregation and degranulation of platelets activated by collagen, ADP, and thrombin, the latter via the PAR4 receptor, but interestingly, not via PAR1 (Tourdot and Holinstat, 2017). Using one of these new 12-LOX inhibitors (ML355), it was evidenced that 12-LOX also plays an important role in activating platelets through FcγRIIa (Yeung et al., 2014; Luci et al., 2014).
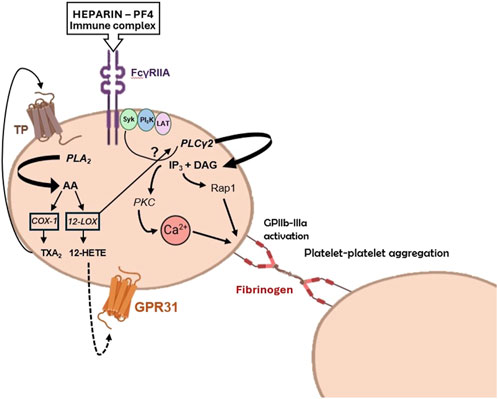
Figure 4. Potential roles of 12-lipoxygenase (LOX) in platelet activation by heparin-platelet factor 4 (PF4) immune complexes. PF4 is a chemokine released from platelet a-granules upon activation. It can form immune complexes with negatively charged substances, such as heparin. The formation of PF4-heparin complexes leads to the synthesis of antibodies, which activate platelets via FcγRIIa receptors. 12-LOX can amplify platelet activation through FcγRIIa via three different pathways: i) the arachidonic acid (AA)-derived compound 12S-HETE generated by 12-LOX, ii) the 12S-HETE receptor GPR31, and iii) the interaction with PLCγ2 or upstream signaling and adaptor molecules, such as Syk (spleen tyrosine kinase), PI3K (phosphoinositide 3-kinase), and LAT (T-cell activation linker). Modified from Alison H. Goodal et al. Blood 2014; 124 (14): 2166–2168 (comment on Yeung et al., Blood 2014; 124:2271-2279).
Heparin-induced thrombocytopenia (HIT) is a condition where platelet count decreases due to the disintegration and release of platelet-derived EVs into the bloodstream, promoting thrombin generation (Ahmed et al., 2007). It can be life-threatening and is caused by heparin binding to platelet factor 4 (PF4) released from platelets, which in some patients can result in immune recognition of the structurally modified heparin-PF4 complexes (Arepally and Padmanabhan, 2021). In HIT, 12-LOX is important in stimulating platelets through FcγRIIa leading to GPIIb-IIIa (the platelet fibrinogen receptor) activation (Goodall, 2014) (Figure 4). Thus, ML355 attenuates aggregation and degranulation via a mechanism that also affects phosphorylation of phospholipase Cγ2 (PLCγ2), calcium mobilization, and phosphorylation of protein kinase C (PKC) and Ras-proximate-1 or Ras-related protein 1 (Rap1involved in activation of GPIIb-IIIa) (Figure 4), while having no direct effect on phosphorylation of FcγRIIa itself (Yeung et al., 2014). The decreased phosphorylation of Rap1 was confirmed in platelets from 12-LOX−/− mice, indicating that the effects were specific to 12-LOX. The data suggests that targeting 12-LOX could be a promising treatment for patients with HIT. Preclinical studies have shown that ML355 has a good safety profile (Adili et al., 2017). As 12-LOX enhances platelet activation by other agonists like TXA2 and ADP, selectively inhibiting 12-LOX may reduce the platelet response without completely blocking it. This indicates a favorable balance between the risk of clotting and bleeding. ML355 (also known as VLX-1005) is currently in phase 2 clinical trials for treating HIT and thrombosis (Stanger and Holinstat, 2023).
Due to immune surveillance, tumor cells face significant challenges surviving in the bloodstream. Consequently, metastasis and subsequent extravasation involve intricate mechanisms facilitating cancer cell survival in the systemic circulation. Fundamental to this process is the recruitment of monocytes, neutrophils, and platelets, which protect tumor cells from immune surveillance and facilitate the formation of metastases (Kitamura et al., 2015). In addition to these mechanisms, platelets have been found to impair the immune response through various other avenues. They play a pivotal role in regulating innate and adaptive immunity, particularly T cells. Furthermore, platelets can serve as antigen-presenting cells (APCs), as demonstrated by the expression of MHC class I proteins on their surface, initiating the adaptive immune response (Ali et al., 2015). It has been reported that platelets can transfer MHC class I proteins to tumor cells, resulting in a tumor cell phenotype termed the “phenotype of false pretenses.” This allows platelets to disrupt the self from non-self-recognition by NK cells, ultimately failing to protect the host by producing IFN-γ (Placke et al., 2012).
Emerging evidence indicates that platelets express the immune checkpoint molecule PD-L1 (Programmed Death-Ligand 1), which interacts with PD-1 (Programmed Cell Death Protein 1) on T cells, leading to their exhaustion (Figure 5). Notably, platelets from cancer patients with non-small cell lung cancer (NSCLC) showed significant levels of PD-L1 (Hinterleitner et al., 2021). Using specific antibodies, such as pembrolizumab, to block the PD-L1/PD-1 interaction has resulted in long-term responses in patients with metastatic NSCLC (Dang et al., 2016). Some tumor cells also express high levels of PD-L1, aiding in immune system evasion (Juneja et al., 2017).
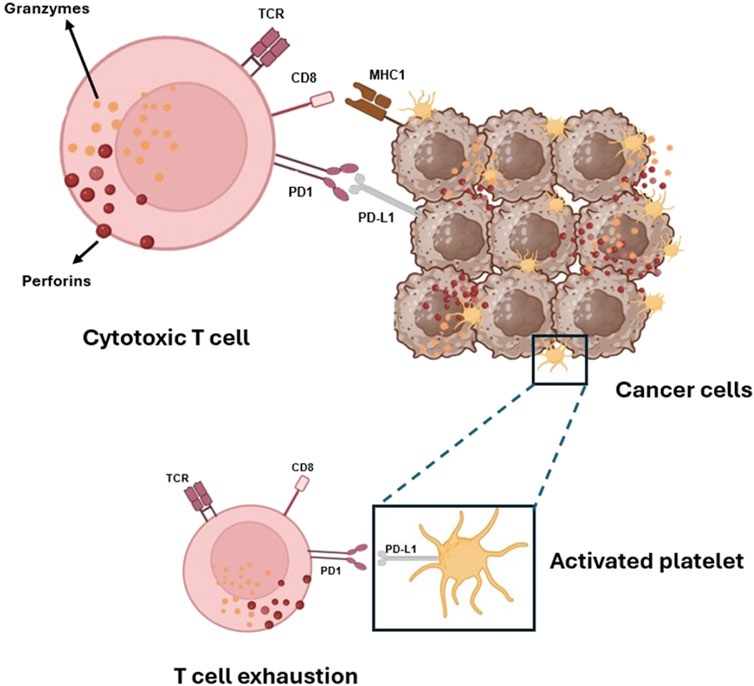
Figure 5. Platelets and tumor cells’ immune evasion via activation of PD-1 on T-cells by its ligands PD-L1. Platelets, like cancer cells, can activate the immune checkpoint protein PD-1, which controls the immune response of cytotoxic CD8+ T cells that carry out their killing function by releasing two types of preformed cytotoxic proteins: the granzymes, which seem able to induce apoptosis in any target cell, and the pore-forming protein perforin, which punches holes in the target cell membrane through which the granzymes can enter.
Interestingly, Rachidi and colleagues (2017) identified a potential role for the non-signaling TGF-β-docking receptor glycoprotein A repetitions predominant (GARP), constitutively expressed on the platelet surface. Platelets are the primary source of TGF-β in systemic circulation and the tumor microenvironment. Platelet TGF-β production and secretion occur through GARP expression and the secretion of TGF-β itself (Tran et al., 2009). It has been shown that platelet-derived TGF-β has an immunosuppressive effect, primarily affecting T cells, and that the deletion of Lrrc32 (the gene encoding GARP) enhances the protective effect of the immune system against both melanoma and colon cancer. Combining immunotherapy with antiplatelet agents is an effective therapeutic strategy in mouse models. This effectiveness is attributed to the interruption of the GARP-TGF-β axis (Rachidi et al., 2017). Thus, antiplatelet agents may interfere with cancer cells’ capacity to evade immune surveillance (Patrignani and Patrono, 2018).
Platelets appear to be involved in the pathogenesis of hepatocellular carcinoma (HCC), a potential complication of chronic HBV infection (Sitia et al., 2012). During chronic hepatitis B virus (HBV) infection, HBV-specific CD8+ T cells fail to eradicate the infection, leading to necrosis, regeneration, inflammation, and ultimately HCC development (Guidotti and Chisari, 2006). It was reported that administering aspirin and clopidogrel (two antiplatelet agents) reduces the number of HBV-specific CD8+ T cells and secondary nonspecific infiltrates, preventing liver injury and fibrosis (Sitia et al., 2012). The pathophysiology of HBV infection and the role of platelet/T cell crosstalk are not fully understood. It has been proposed that interaction through platelet-CD40L and lymphocyte-CD40 (Elzey et al., 2003) could trigger an inflammatory endothelial response (Henn et al., 1998). Studies have shown an increased number of thrombotic events in patients with liver diseases (Tripodi and Mannucci, 2011), suggesting the potential for investigating antiplatelet drugs in further randomized clinical trials (RCTs) to assess their effects on HCC development in patients with chronic HBV infection.
Platelets fuel cancer metastasis
Cancer metastasis, which involves the spread of cancer cells from a primary lesion to distant organs, is the main cause of cancer-related deaths (Massagué and Obenauf, 2016). Numerous studies have highlighted the crucial role platelets play in this process. Clinical observations have underlined the potential link between cancer cell diffusion via the bloodstream and platelet involvement in coagulation (Gay and Felding-Habermann, 2011; Contursi et al., 2017). Once cancer cells enter systemic circulation, they interact with platelets. Platelets can surround cancer cells, forming aggregates that promote survival, protect them from immune responses, and enhance their capacity to adhere to the endothelium, arrest, and exit blood vessels (Gay and Felding-Habermann, 2011).
Recent findings have shown that platelets induce a more malignant phenotype in cancer cells by enhancing their migratory properties (Labelle et al., 2011; Dovizio et al., 2013; Guillem-Llobat et al., 2016). Platelets release mediators such as PDGF, TGF-β, and PGE2, which promote EMT, that endows cancer cells with mesenchymal markers like vimentin and fibronectin and transcription factors such as Twist, Snail, and Zeb while downregulating epithelial markers like E-cadherin, thereby enabling these cells to acquire migratory properties and colonize distant organs (Kalluri and Weinberg, 2009). Labelle et al. (2011) demonstrated that EMT is induced in colon and breast cancer cells via platelet-derived TGF-β1, which activates the TGF-β/Smad pathway, thus promoting the metastatic potential of tumor cells. Dovizio et al. (2013) demonstrated that platelets trigger EMT and COX-2 overexpression in human adenocarcinoma cells (such as HT29 cells) through direct contact and PDGF release. The direct interaction involves the platelet collagen receptor GPVI and cancer cell galectin-3, unique among galectins due to its collagen-like domain. Inhibition of galectin-3 function [using β-lactose, a dominant-negative form of galectin-3, Gal-3C (Yang et al., 2008), or anti-galectin-3 antibody M3/38] or the inhibition of platelet adhesion to collagen-like binding sites on cancer cells by revacept, the soluble dimeric GPVI receptor fusion protein (GPVI-Fc) (Ungerer et al., 2011), prevented abnormal COX-2 expression. Revacept, which prevents the interaction between platelets and cancer cells, and the inhibition of COX-2 by rofecoxib, a highly selective COX-2 inhibitor, avoided platelet-induced changes in mRNA levels of EMT markers. This suggests that the direct platelet-cancer cell interaction and abnormal COX-2 expression contribute to the modifications in gene expression associated with EMT. Mammadova-Bach et al. (2020) found that genetic deficiency of platelet GPVI in mice reduced both experimental and spontaneous colon and breast cancer cell metastasis. Similar results were observed in mice lacking the spleen-tyrosine kinase Syk in platelets, a crucial component of the ITAM-signaling cascade. Both in vitro and in vivo analyses confirmed that mouse and human GPVI facilitated platelet adhesion to colon and breast cancer cells. Through a CRISPR/Cas9-based gene knockout approach, galectin-3 was identified as the primary counterreceptor of GPVI on tumor cells. Further, in vivo studies revealed that the interaction between platelet GPVI and tumor cell–expressed galectin-3 involves ITAM-signaling components in platelets and promotes the extravasation of tumor cells. Additionally, it was demonstrated that inhibiting GPVI by JAQ1 F (abʹ) 2 antibody efficiently impairs platelet–tumor cell interaction and tumor metastasis (Mammadova-Bach et al., 2020).
Guillem-Llobat et al. (2016) studied the impact of platelets on the spread of colorectal cancer cells (HT29 cells) to the lungs and the effect of the antiplatelet agent low-dose aspirin. When HT-29 cells were exposed to platelets in vitro, they were more likely to cause lung metastasis once injected in humanized immunodeficient mice than untreated HT29 cells. This effect was linked to the development of mesenchymal-like cancer cells by platelets associated with acquiring migratory properties and EMT. Mesenchymal-like cancer cells had an enhanced capacity to activate platelets, thus promoting the formation of platelet aggregates surrounding tumor cells, and this event is central in the development of cancer metastases. Administering a low dose of aspirin to the mice, prevented the increased rate of lung metastasis associated with the prevention of platelet activation in vivo in response to the injection of mesenchymal-like cancer cells. These results provide a mechanistic understanding of the reported antimetastatic properties of low-dose aspirin in posthoc analyses of randomized trials for cardiovascular prevention (Patrignani and Patrono, 2016) and reinforce the rationale for performing adjuvant trials of low-dose aspirin and possibly other antiplatelet agents, in colorectal cancer patients. In vitro studies of cocultures of platelets and HT29 cells showed that platelet-derived PGE2 activated the EP4 receptor on cancer cells, promoting EMT (Guillem-Llobat et al., 2016). Different antiplatelet agents, such as aspirin DG-041 (an EP3 receptor antagonist), and ticagrelor (a P2Y12 receptor antagonist), prevented EMT of cancer cells by inhibiting platelet activation and platelet-derived PGE2 release involved in cancer cell EP4-dependent migration (Guillem-Llobat et al., 2016).
Further studies investigated the potential of anticoagulant agents to disrupt communication between platelets and cancer cells. Battinelli et al. (2014) discovered that the interaction between breast cancer cells (MCF-7) and platelets stimulated the secretion of VEGF from platelets, a process that was prevented by the administration of low molecular weight heparin (LMWH) or fondaparinux. This suggests a thrombin-dependent release of angiogenic molecules by platelets after interaction with tumor cells. Human adenocarcinoma cell lines (such as HCT-8 and LoVo) and anaplastic murine tumor cells (such as Hut-20) can generate thrombin, which in turn activates platelets (Nierodzik and Karpatkin, 2006). Mitrugno et al. (2014) found that the colorectal cancer cell line Caco-2 and the prostate carcinoma cell line PC3M-luc induce platelet activation and granule secretion. They showed that the platelet immune receptor FcγRIIa plays a crucial role in this process by activating the FcγRIIa-spleen tyrosine kinase (Syk)-PLCγ signaling pathway, resulting in further platelet activation and granule secretion. This enhanced platelet recruitment and protection of circulating tumor cells from the immune system promote tumor cell survival and metastasis (Mitrugno et al., 2014), suggesting FcγRIIa as a target for antimetastatic therapy. Mannori et al. (1995) highlighted interactions between platelets and colon cancer cells (LS 180, T84, COLO 205, COLO 320, and HT-29) involving P-selectin on platelets and mucin-type glycoproteins on the cancer cell surface. Other studies reported the role of the platelet GPIIb/IIIa receptor in platelet/cancer cell crosstalk. In the melanoma cell line M3Dau, the interaction between the platelet receptor GPIIb/IIIa and a similar complex on tumor cells led to larger platelet-tumor aggregates (Boukerche et al., 1989). Mammadova-Bach et al. (2016) reported interactions between platelets and breast or colon tumor cells via platelet integrin α6β1 and ADAM-9 on tumor cells. This interaction promotes platelet activation, granule secretion, and endothelial transmigration, thus promoting metastasis. Genetic deletion of platelet α6β1 or an integrin α6-blocking antibody (GoH3) reduced lung metastasis by preventing platelet-tumor cell crosstalk (Mammadova-Bach et al., 2016). Lysophosphatidic acid (LPA), a platelet-derived mediator, plays a significant role in cancer development (Mills and Moolenaar, 2003). LPA acts through GPCRs, promoting platelet shape change and aggregation (Williams et al., 2009). Platelet-derived LPA promotes bone metastasis in ovarian and breast cancers by binding to LPA1 on tumor cells, and a specific LPA1 antagonist can inhibit this interaction without affecting normal platelet function (Boucharaba et al., 2004; 2006). Platelets also contribute to LPA biosynthesis via the enzyme autotaxin (ATX), which hydrolyzes LPA precursors to form LPA, binding to its receptors on cancer cells to promote invasion and metastasis (Leblanc et al., 2014). LPA can induce EMT in ovarian cancer cells by promoting nuclear translocation of β-catenin, activating Wnt/β-catenin target genes and hypoxia-induced factor-1α (HIF1α), leading to mesenchymal marker expression (Burkhalter et al., 2015; Ha et al., 2016).
The metabolite 12S-HETE, generated by platelet 12S-LOX, plays a crucial role in cancer development and metastasis through various mechanisms (Contursi et al., 2022). The involvement of 12S-LOX in cancer development has been demonstrated in human gastric cancer cells with ALOX12 overexpression, and it is suggested as a marker of cancer progression in melanoma (Timár et al., 1999). 12-HETE is generated as a free acyl and is found in esterified form in membrane phospholipids, particularly in platelets where it is involved in coagulation (Thomas et al., 2010; Slatter et al., 2018; Lauder et al., 2017). HT29 cells do not express 12-LOX or synthesize 12-HETE when cultured alone, but in coculture with platelets, they express the protein and synthesize 12-HETE (Contursi et al., 2021). The interaction with cancer cells activates platelets that release EVs containing catalytically active 12S-LOX, quickly transferred to cancer cells. The cancer cells then acquire the capacity to generate 12-HETE, which is esterified rapidly, mainly in plasmalogen phospholipids. In cancer cells exposed to platelets, endogenous but not exogenous 12S-HETE contributed to changes in EMT gene expression by modifying cancer cell phospholipids by 12-HETE (Contursi et al., 2021). Modifying cancer cell phospholipids by 12S-HETE may functionally impact cancer cell biology, and the pharmacological inhibition of 12-LOX represents a novel anticancer strategy.
Platelet-derived extracellular vesicles in cancer
Platelets secrete medium-size EVs (mEVs) (Gasecka et al., 2019; Kailashiya, 2018) ranging from 100 to 1,000 nm, released under various physiological and pathological conditions (Kailashiya, 2018). They are characterized by the inversion of membrane phospholipids, exposure of phosphatidylserine (PS) on the outer membrane—rendering them 50 to 100 times more procoagulant than platelets—and the expression of various integrins and enzymes (Janowska-Wieczorek et al., 2005; Italiano et al., 2010). These mEVs promote the communication between platelets and various cell types (Italiano et al., 2010). They act by (1) stimulating target cells through their surface ligands, (2) transferring surface receptors, and (3) delivering their cargo, i.e., proteins, bioactive lipids, mRNAs, transcription factors, and miRs, which can significantly alter cellular phenotypes. mEVs can also transfer infectious particles (e.g., HIV, prions) and intact organelles (e.g., mitochondria) (Ratajczak et al., 2006). Due to their carrier role, mEVs are involved in various biological processes, including hemostasis, thrombosis, inflammation, tumorigenesis, angiogenesis, and immunity (Italiano et al., 2010). For instance, platelet-derived EVs transfer receptors to both normal and cancer cells, enhancing adhesion, proliferation, and survival (e.g., CD41, CD61, CD62, CXCR4, PAR-1) (Baj-Krzyworzeka et al., 2002). mEVs also carry bioactive lipids like sphingosine-1-phosphate (S1P) and AA. Barry and colleagues demonstrated the transfer of platelet AA via mEVs to monocytes and endothelial cells, inducing COX-2 expression and prostanoid synthesis (Barry et al., 1997; 1999). Platelet mEVs transfer mRNA to monocytic THP-1 cells, increasing gamma-globin transcripts and hemoglobin subunits (Risitano et al., 2012). Additionally, platelet-derived mEVs contribute to tumorigenesis through miR transfer. For instance, the transfer of miR-223 to endothelial cells downregulates tumor-suppressor genes (Laffont et al., 2013), while the transfer of miR-939 to ovarian cancer cells promotes EMT (Tang et al., 2017). Conversely, mEVs carrying miR-24 induce apoptosis and suppress tumor growth (Michael et al., 2017). Platelet mEVs interact with monocytes/macrophages in inflammatory contexts, influencing proinflammatory cytokine release (Linke et al., 2017; Vasina et al., 2011).
Platelets contain important RNA biomarkers [mRNAs, miRs, circular (circ) RNAs, long non-coding (lnc) RNAs, mitochondrial-RNAs] and have a protein translation machinery for processing the RNA transcripts (Harrison and Goodall, 2008). CircRNAs, generated from exons of protein-coding genes through back-splicing, are abundant in platelets. They have received increasing attention for their potential role as cancer biomarkers due to their high stability and spatiotemporal-specific expression (Bach et al., 2019). In addition, lncRNAs (Dahariya et al., 2019) and miRs (Nagalla et al., 2011) represent important cancer biomarkers. Boerrigter et al. (2020) reported that the measurement of these molecules in prostate cancer (PCa) offers promising prospects because they are highly tumor-specific and relatively easy to detect. Through their crosstalk with other cells, including cancer cells, platelets can uptake these RNAs. These molecules can be found in isolated platelets. Analyzing RNAs from patients’ platelets offers significant promise for accurately distinguishing between metastatic and non-metastatic tumors. Additionally, RNA sequencing from platelets could identify the primary tumor location of six distinct tumor types with an accuracy of 71% (Hu et al., 2024).
Liquid biopsy involves analyzing biomarkers found in non-solid biological tissues, primarily blood. It offers significant advantages over traditional methods: it is risk-free, non-invasive, painless, does not necessitate surgery, and lowers costs and diagnostic time. The most extensively studied non-invasive cancer biomarkers include circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), and EV cargo (Marrugo-Ramírez et al., 2018). They offer several advantages, including the ability for early detection, assessment of an individual patient’s prognosis, such as cancer stage and spread, identification of new targets for personalized treatments, and predict therapy responses. Currently, blood-based biopsy measurements focus on the evaluation of biomarker biosources, including circulating tumor DNA (ctDNA), circulating tumor cells (CTCs), EVs (i.e., exosomes, mEVs and oncosomes), and tumor-educated platelets (TEPs) (Marrugo-Ramírez et al., 2018; Best et al., 2015; Nilsson et al., 2011; 2016; Skog et al., 2008).
Contursi et al. (2023) studied platelet-derived mEVs collected from non-metastatic CRC patients at initial diagnosis versus healthy controls (HCs). The aim was to address how these mEVs can change the expression of genes related to the process of EMT in four different human colorectal cancer cell lines: HCA7, HCT116, HT29, and Caco2. These cell lines have different genetic features and metastatic potentials. The study found that platelet-derived mEVs from CRC patients affect colorectal cancer cells differently depending on their genetic makeup and behavior. Specifically, the mEVs from CRC patients caused an increase in the expression of TWIST1 and VIM (protein name: vimentin) in all the studied cancer cell lines, while those from healthy subjects did not. In HCA7 cells, the expression of CDH1 (protein name, E-cadherin) was not affected by the mEVs from either CRC patients or HCs. However, the HCT116, HT29, and Caco2 cells showed decreased CDH1 expression when cultured with mEVs from CRC patients, with the HCT116 cells (which have high metastatic potential) showing decreased expression after just 4 hours of exposure. Moreover, the mEVs from CRC patients and HCs induced the expression of COX-2 in colorectal cancer cell lines at 24 but not at 4 hours, suggesting that this effect may be due to post-transcriptional regulation (Contursi et al., 2023).
Cancer-associated thrombosis is a leading cause of death in cancer patients (Mahajan A. et al., 2022). Contursi et al. (2023) explored whether platelet-derived mEVs collected from CRC patients affected the TXB2 biosynthesis of 4 CRC cell lines in vitro. Proteomic analysis revealed that COX-1 and TXA2 synthase were expressed similarly in mEVs collected from CRC patients and healthy controls (HC). HCA7 cells can generate endogenously TXB2, and when exposed to mEVs, the TXB2 levels were unchanged. Other CRC cell lines have very limited capacity to generate TXB2, but when cocultured with mEVs from both CRC patients and HCs, TXB2 was detectable. This may suggest that platelet-derived mEVs could provide AA to cancer cells, leading to TXB2 generation. Alternatively, platelet-derived mEVs might transfer COX-1 and/or TXA2 synthase to cancer cells, allowing them to produce TXB2.
A comparison of the proteomic profiles of mEVs from CRC patients and HCs revealed 208 significantly altered proteins. High expression of HLA-B class I and PSMD2 in CRC patient mEVs was detected. The human leukocyte antigen (HLA) system or complex is a group of related proteins encoded by the major histocompatibility complex (MHC) gene; it has been reported that cell surface expression of HLA I/MHC I molecules is a marker of young, reactive platelets (Angénieux et al., 2019). PSMD2 is part of the ubiquitin-proteasome system and plays a role in immune modulation (Colberg et al., 2020). Platelets contain proteasome and immunoproteasome components, allowing them to process foreign proteins into peptide fragments. These fragments are then loaded onto MHC I molecules and presented on the surface of the platelets as peptide-MHC I complexes. Immunoproteasomes may regulate platelets’ involvement in innate and adaptive immunity, highlighting the connection between hemostasis and inflammation. The increased protein levels of HLA-B class I and PSMD2 found in platelet-derived mEVs from CRC patients by Contursi et al. (2023) suggest that platelet-derived mEVs are modulators of the immune response in CRC. A comprehensive examination of the protein composition and its alterations associated with cancer underscores the potential of platelet-derived mEV proteomics to facilitate early diagnosis, provide continuous monitoring, and support personalized treatment strategies, ultimately enhancing patient outcomes.
Conclusion
The complex and intricate interplay between inflammation and platelets in the development of tumors presents exciting opportunities for pioneering new avenues in cancer prevention and treatment strategies. A wealth of clinical and experimental evidence underscores platelets’ pivotal and multifaceted role in fostering an inflammatory microenvironment that fuels tumor growth and progression. Moreover, platelets play a key role in the development of cancer metastasis. This huge amount of knowledge opens the way to using antiplatelet agents, such as low-dose aspirin, which exhibit considerable potential in dampening platelet activation and consequent inflammatory processes, thereby lowering cancer risk, particularly in cases of CRC (Patrignani and Patrono, 2016; 2018). It is necessary to investigate further the effectiveness of alternative antiplatelet medications, such as ADP P2Y12 receptor antagonists (Ballerini et al., 2018). Other potential new antiplatelet drugs, such as revacept (Ungerer et al., 2011; Mayer et al., 2021) and selective 12-LOX inhibitors (Tourdot and Holinstat, 2017), currently under clinical development, are of interest due to their lower risk of bleeding (Sim et al., 2023).
The role of platelets in promoting the initial stages of cancer development is supported by various evidence, including studies using knockout mouse models and clinical research (Bruno et al., 2022; Patrignani and Patrono, 2016). Platelets contribute to inflammation by releasing various molecules and EVs. The molecules delivered by platelets activate different signaling pathways in the cells involved in inflammation and immunity (Patrignani and Patrono, 2018). Platelets are essential for cell-cell communication and significantly influence the phenotype of cellular components within the stromal compartment of tissues. An emerging and continuously evolving paradigm underscores the synergistic connection between cardiovascular disease (CVD) and cancer, elucidated by their shared modifiable risk factors, which encompass tobacco use, obesity, diabetes mellitus, dietary patterns, physical activity, and alcohol consumption. It is important to note that all these factors contribute to the activation of platelets, making this response a key event in both cancer and cardiovascular disease (Patrignani and Patrono, 2016; Davì and Patrono, 2007). This helps explain why using antiplatelet agents like low-dose aspirin effectively prevents cardiovascular disease and possibly cancer (Patrignani and Patrono, 2016). Platelet activation results in an increased generation of TXA2 and PGE2, subsequently triggering the activation of immune cells, fibroblasts, and endothelial cells. Thus, the inhibition of platelet function effectively impedes the release of molecules from platelets involved in initial events linked to tumorigenesis (Patrignani and Patrono, 2016). In cases where cancer has already developed, antiplatelet agents are likely less effective because they have limited capacity to directly affect the cellular component of a cancerous lesion (Patrignani et al., 2024). However, platelets can infiltrate cancerous tissues (Bruno et al., 2022), which can lead to tumor immune evasion, inflammation, and EMT (Patrignani and Patrono, 2016; 2018). This suggests that antiplatelet agents could indirectly cause antitumor effects by reducing platelet accumulation in tumors.
Genetic alterations may affect an individual’s response to aspirin. However, Frouws et al. (2017) reported that the mutation status of the BRAF and KRAS genes should not be regarded as reliable indicators for personalized aspirin therapy. In contrast, Liao et al. (2012) showed that tumors with mutations in the PIK3CA gene (which codes for the protein phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform) exhibit increased sensitivity to the effects of aspirin. Nonetheless, the underlying mechanisms explaining this association remain to be elucidated.
Ongoing clinical trials investigate aspirin’s potential as an adjuvant cancer therapy in various doses and durations in CRC patients (Table 1). Some of these trials are targeting patients based on specific biomarkers. These trials are part of the Prospective Aspirin Meta-analysis, registered on PROSPERO in October 2023. The main objective of this meta-analysis is to ascertain the potential moderate effects of aspirin, both overall and within different subgroups. These subgroups include those defined by tumor location (right-sided versus left-sided) and specific biomarkers.
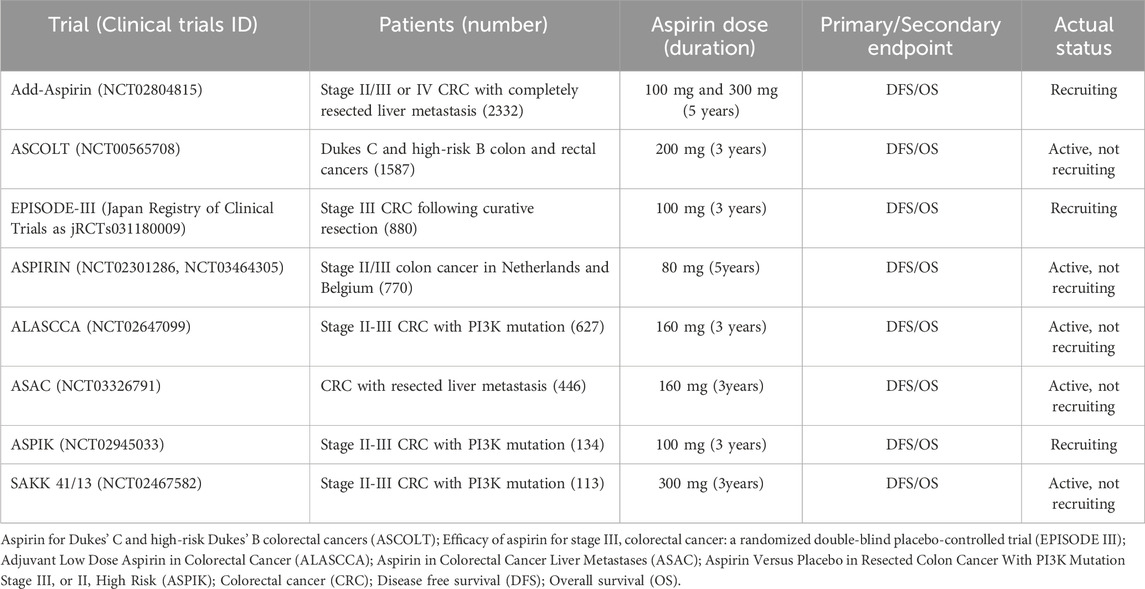
Table 1. Ongoing Phase III placebo-controlled trials for aspirin in adjuvant colorectal cancer.
At long follow-up, aspirin (300 mg BID) has been shown to prevent Lynch Syndrome (LS) cancers, including CRC (Burn et al., 2008; 2011; 2020). LS is a dominantly inherited cancer predisposition syndrome characterized by an increased risk of numerous cancers. LS-associated tumors are mismatch repair deficient (Engel et al., 2020). Aspirin is now recommended in NICE guidelines to prevent CRC in LS (National Institute for Health and Care, 2020; Serrano et al., 2022). CaPP3 trial (https://www.capp3.org/) is ongoing to define the most appropriate aspirin dose in LS. The trial is a non-inferiority study comparing daily aspirin of 600, 300, and 100 mg (100 mg/d is the dose recommended for preventing CVD). The finding of comparable efficacy of the higher doses versus the low dose of aspirin (100 mg/d) would support the platelet hypothesis in tumorigenesis (Patrignani and Patrono, 2016; 2018), thus opening the way to its use for cancer prevention in LS.
However, chronic aspirin use, even at low doses, can be associated with increased bleeding (Patrono and Baigent, 2019), especially in older individuals. Therefore, the decision to treat individual patients with low-dose aspirin should consider the bleeding risk weighed against CVD prevention and cancer benefits.
In the current era of precision medicine, developing treatment protocols involving the safe utilization of antiplatelet agents to prevent cancer and cardiovascular disease necessitates adopting a systems biology approach. This strategy entails a comprehensive analysis of heterogeneous datasets, including genomics, epigenomics, proteomics, lipidomics, and clinical data, at the level of the individual patient. This procedure involves dynamic systems modeling to identify candidate pathways that contribute to the benefits and harms of aspirin and possibly other antiplatelet agents. Additionally, this strategy will help identify susceptibility profiles for CRC and possibly other types of cancer, verifying whether platelet- and EV-based liquid biopsy can predict the onset and recurrence of cancer.
Author contributions
AC: Funding acquisition, Writing–review and editing, Writing–original draft. ST: Funding acquisition, Writing–review and editing, Writing–original draft. SDB: Writing–review and editing. ADM: Writing–review and editing. PP: Writing–review and editing, Conceptualization, Funding acquisition, Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This review was funded by Associazione Italiana per la Ricerca sul Cancro (AIRC) [under IG 2017- ID. 20365 Project; Principal Investigator PP], and by Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR) [Fondi per la Ricerca Scientifica di Ateneo, (ex 60%)] to PP and ST. In addition, this work was conducted on behalf of the Aspirin for Cancer Prevention Group, Wolfson Institute of Preventive Medicine, Queen Mary School of Medicine and Dentistry, University of London (United Kingdom). Moreover, it was funded by the European Union - European Social Fund - PON Research and Innovation 2014–2020 to AC.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adili, B. E. T., Mast, K., Yeung, J., Freedman, J. C., Green, A., Luci, D. K., et al. (2017). First selective 12-LOX inhibitor, ML355, impairs thrombus formation and vessel occlusion in vivo with minimal effects on hemostasis. Arterioscler. Thromb. Vasc. Biol. 37, 1828–1839. doi:10.1161/ATVBAHA.117.309868
Ahmed, I., Majeed, A., and Powell, R. (2007). Heparin induced thrombocytopenia: diagnosis and management update. Postgrad. Med. J. 83 (983), 575–582. doi:10.1136/pgmj.2007.059188
Ali, R. A., Wuescher, L. M., and Worth, R. G. (2015). Platelets: essential components of the immune system. Curr. Trends Immunol. 16, 65–78.
Angénieux, C., Dupuis, A., Gachet, C., de la Salle, H., and Maître, B. (2019). Cell surface expression of HLA I molecules as a marker of young platelets. J. Thromb. Haemost. 17 (9), 1511–1521. doi:10.1111/jth.14537
Arepally, G. M., and Padmanabhan, A. (2021). Heparin-induced thrombocytopenia: a focus on thrombosis. Arterioscler. Thromb. Vasc. Biol. 41 (1), 141–152. doi:10.1161/ATVBAHA.120.315445
Arman, M., and Krauel, K. (2015). Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J. Thromb. Haemost. 13 (6), 893–908. doi:10.1111/jth.12905
Bach, D. H., Lee, S. K., and Sood, A. K. (2019). Circular RNAs in cancer. Mol. Ther. Nucleic Acids 16, 118–129. doi:10.1016/j.omtn.2019.02.005
Baj-Krzyworzeka, M., Majka, M., Pratico, D., Ratajczak, J., Vilaire, G., Kijowski, J., et al. (2002). Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp. Hematol. 30, 450–459. doi:10.1016/s0301-472x(02)00791-9
Balkwill, F., Charles, K. A., and Mantovani, A. (2005). Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7 (3), 211–217. doi:10.1016/j.ccr.2005.02.013
Ballerini, P., Dovizio, M., Bruno, A., Tacconelli, S., and Patrignani, P. (2018). P2Y12 receptors in tumorigenesis and metastasis. Front. Pharmacol. 9, 66. doi:10.3389/fphar.2018.00066
Barron, D. A., and Rowley, D. R. (2012). The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 19 (6), R187–R204. doi:10.1530/ERC-12-0085
Barry, O. P., Kazanietz, M. G., Praticò, D., and FitzGerald, G. A. (1999). Arachidonic acid in platelet microparticles up-regulates cyclooxygenase-2-dependent prostaglandin formation via a protein kinase C/mitogen-activated protein kinase-dependent pathway. J. Biol. Chem. 274, 7545–7556. doi:10.1074/jbc.274.11.7545
Barry, O. P., Pratico, D., Lawson, J. A., and FitzGerald, G. A. (1997). Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Investigation 99, 2118–2127. doi:10.1172/JCI119385
Battinelli, E. M., Markens, B. A., Kulenthirarajan, R. A., Machlus, K. R., Flaumenhaft, R., and Italiano, J. E. (2014). Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood 123 (1), 101–112. doi:10.1182/blood-2013-02-485011
Bertagnolli, M. M., Eagle, C. J., Zauber, A. G., Redston, M., Solomon, S. D., Kim, K., et al. (2006). Celecoxib for the prevention of sporadic colorectal adenomas. N. Engl. J. Med. 355 (9), 873–884. doi:10.1056/NEJMoa061355
Best, M. G., Sol, N., Kooi, I., Tannous, J., Westerman, B. A., Rustenburg, F., et al. (2015). RNA-seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell 28 (5), 666–676. doi:10.1016/j.ccell.2015.09.018
Boerrigter, E., Groen, L. N., Van Erp, N. P., Verhaegh, G. W., and Schalken, J. A. (2020). Clinical utility of emerging biomarkers in prostate cancer liquid biopsies. Expert Rev. Mol. Diagn 20 (2), 219–230. doi:10.1080/14737159.2019.1675515
Boucharaba, A., Serre, C. M., Grès, S., Saulnier-Blache, J. S., Bordet, J. C., Guglielmi, J., et al. (2004). Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Invest 114, 1714–1725. doi:10.1172/JCI22123
Boucharaba, A., Serre, C. M., Guglielmi, J., Bordet, J. C., Clézardin, P., and Peyruchaud, O. (2006). The type 1 lysophosphatidic acid receptor is a target for therapy in bone metastases. Proc. Natl. Acad. Sci. U. S. A. 103, 9643–9648. doi:10.1073/pnas.0600979103
Boukerche, H., Berthier-Vergnes, O., Tabone, E., Doré, J. F., Leung, L. L., and McGregor, J. L. (1989). Platelet-melanoma cell interaction is mediated by the glycoprotein IIb-IIIa complex. Blood 74, 658–663. doi:10.1182/blood.v74.2.658.bloodjournal742658
Boulaftali, Y., Hess, P. R., Kahn, M. L., and Bergmeier, W. (2014). Platelet immunoreceptor tyrosine-based activation motif (ITAM) signaling and vascular integrity. Circ. Res. 114 (7), 1174–1184. doi:10.1161/CIRCRESAHA.114.301611
Boylan, B., Gao, C., Rathore, V., Gill, J. C., Newman, D. K., and Newman, P. J. (2008). Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood 112 (7), 2780–2786. doi:10.1182/blood-2008-02-142125
Bruno, A., Contursi, A., Tacconelli, S., Sacco, A., Hofling, U., Mucci, M., et al. (2022). The specific deletion of cyclooxygenase-1 in megakaryocytes/platelets reduces intestinal polyposis in ApcMin/+ mice. Pharmacol. Res. 185, 106506. doi:10.1016/j.phrs.2022.106506
Burkhalter, R. J., Westfall, S. D., Liu, Y., and Stack, M. S. (2015). Lysophosphatidic acid initiates epithelial to mesenchymal transition and induces β-Catenin-mediated transcription in epithelial ovarian carcinoma. J. Biol. Chem. 290, 22143–22154. doi:10.1074/jbc.M115.641092
Burn, J., Bishop, D. T., Mecklin, J. P., Macrae, F., Möslein, G., Olschwang, S., et al. (2008). Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N. Engl. J. Med. 359 (24), 2567–2578. doi:10.1056/NEJMoa0801297
Burn, J., Gerdes, A. M., Macrae, F., Mecklin, J. P., Moeslein, G., Olschwang, S., et al. (2011). Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 378 (9809), 2081–2087. doi:10.1016/S0140-6736(11)61049-0
Burn, J., Sheth, H., Elliott, F., Reed, L., Macrae, F., Mecklin, J. P., et al. (2020). Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet 395 (10240), 1855–1863. doi:10.1016/S0140-6736(20)30366-4
Celis, P. M., Celis, A., Nielsen, H. V., and Gesser, B. (1987). Cyclin (PCNA, auxiliary protein of DNA polymerase delta) is a central component of the pathway(s) leading to DNA replication and cell division. FEBS Lett. 220, 1–7. doi:10.1016/0014-5793(87)80865-7
Chen, L., Deng, H., Cui, H., Fang, J., Zuo, Z., Deng, J., et al. (2017). Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9 (6), 7204–7218. doi:10.18632/oncotarget.23208
Chitu, V., and Stanley, E. R. (2006). Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 18 (1), 39–48. doi:10.1016/j.coi.2005.11.006
Cloutier, N., Allaeys, I., Marcoux, G., Machlus, K. R., Mailhot, B., Zufferey, A., et al. (2018). Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. PNAS 115, E1550-E1559–E1559. doi:10.1073/pnas.1720553115
Coenen, D. M., Heinzmann, A. C. A., Karel, M. F. A., Cosemans, JMEM, and Koenen, R. R. (2021). The multifaceted contribution of platelets in the emergence and aftermath of acute cardiovascular events. Atherosclerosis 319, 132–141. doi:10.1016/j.atherosclerosis.2020.12.017
Colberg, L., Cammann, C., Greinacher, A., and Seifert, U. (2020). Structure and function of the ubiquitin-proteasome system in platelets. J. Thromb. Haemost. 18, 771–780. doi:10.1111/jth.14730
Contursi, A., Fullone, R., Szklanna-Koszalinska, P., Marcone, S., Lanuti, P., Taus, F., et al. (2023). Tumor-educated platelet extracellular vesicles: proteomic profiling and crosstalk with colorectal cancer cells. Cancers (Basel) 15 (2), 350. doi:10.3390/cancers15020350
Contursi, A., Sacco, A., Grande, R., Dovizio, M., and Patrignani, P. (2017). Platelets as crucial partners for tumor metastasis: from mechanistic aspects to pharmacological targeting. Cell. Mol. Life Sci. 74, 3491–3507. doi:10.1007/s00018-017-2536-7
Contursi, A., Schiavone, S., Dovizio, M., Hinz, C., Fullone, R., Tacconelli, S., et al. (2021). Platelets induce free and phospholipid-esterified 12-hydroxyeicosatetraenoic acid generation in colon cancer cells by delivering 12-lipoxygenase. J. Lipid Res. 62, 100109. doi:10.1016/j.jlr.2021.100109
Contursi, A., Tacconelli, S., Hofling, U., Bruno, A., Dovizio, M., Ballerini, P., et al. (2022). Biology and pharmacology of platelet-type 12-lipoxygenase in platelets, cancer cells, and their crosstalk. Biochem. Pharmacol. 205, 115252. doi:10.1016/j.bcp.2022.115252
Coussens, L. M., and Werb, Z. (2002). Inflammation and cancer. Nature 420 (6917), 860–867. doi:10.1038/nature01322
D’Agostino, I., Tacconelli, S., Bruno, A., Contursi, A., Mucci, L., Hu, X., et al. (2021). Low-dose Aspirin prevents hypertension and cardiac fibrosis when thromboxane A2 is unrestrained. Pharmacol. Res. 170, 105744. doi:10.1016/j.phrs.2021.105744
Dahariya, S., Paddibhatla, I., Kumar, S., Raghuwanshi, S., Pallepati, A., and Gutti, R. K. (2019). Long non-coding RNA: classification, biogenesis and functions in blood cells. Mol. Immunol. 112, 82–92. doi:10.1016/j.molimm.2019.04.011
Danese, S., Motte, Cd C., and Fiocchi, C. (2004). Platelets in inflammatory bowel disease:clinical, pathogenic, and therapeutic implications. Am. J. Gastroenterol. 99, 938–945. doi:10.1111/j.1572-0241.2004.04129.x
Dang, T. O., Ogunniyi, A., Barbee, M. S., and Drilon, A. (2016). Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev. Anticancer Ther. 16 (1), 13–20. doi:10.1586/14737140.2016.1123626
Davì, G., and Patrono, C. (2007). Platelet activation and atherothrombosis. N. Engl. J. Med. 357 (24), 2482–2494. doi:10.1056/NEJMra071014
De La Cruz, A., Hargrave, A., Magadi, S., Courson, J. A., Landry, P. T., Zhang, W., et al. (2021). Platelet and erythrocyte extravasation across inflamed corneal venules depend on CD18, neutrophils, and mast cell degranulation. Int. J. Mol. Sci. 22 (14), 7360. doi:10.3390/ijms22147360
Delves, P. J., and Roitt, I. M. (2000). The immune system First of two parts. N. Engl. J. Med. 343 (1), 37–49. doi:10.1056/NEJM200007063430107
Diacovo, T. G., deFougerolles, A. R., Bainton, D. F., and Springer, T. A. (1994). A functional integrin ligand on the surface of platelets: intercellular adhesion molecule-2. J. Clin. Invest 94 (3), 1243–1251. doi:10.1172/JCI117442
Dovizio, M., Alberti, S., Guillem-Llobat, P., and Patrignani, P. (2014). Role of platelets in inflammation and cancer: novel therapeutic strategies. Basic Clin. Pharmacol. Toxicol. 114 (1), 118–127. doi:10.1111/bcpt.12156
Dovizio, M., Maier, T. J., Alberti, S., Di Francesco, L., Marcantoni, E., Munch, G., et al. (2013). Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 84, 25–40. doi:10.1124/mol.113.084988
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3 (11), 991–998. doi:10.1038/ni1102-991
Elzey, B. D., Tian, J., Jensen, R. J., Swanson, A. K., Lees, J. R., Lentz, S. R., et al. (2003). Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity 19, 9–19. doi:10.1016/s1074-7613(03)00177-8
Engel, C., Ahadova, A., Seppälä, T. T., Aretz, S., Bigirwamungu-Bargeman, M., and Bläker, H.German HNPCC Consortium, the Dutch Lynch Syndrome Collaborative Group (2020). Associations of pathogenic variants in MLH1, MSH2, and MSH6 with risk of colorectal adenomas and tumors and with somatic mutations in patients with Lynch syndrome. Gastroenterology 158 (5), 1326–1333. doi:10.1053/j.gastro.2019.12.032
Evangelista, V., Manarini, S., Sideri, R., Rotondo, S., Martelli, N., Piccoli, A., et al. (1999). Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood 93 (3), 876–885. doi:10.1182/blood.v93.3.876.403k25_876_885
Faour, W. H., He, Y., He, Q. W., de Ladurantaye, M., Quintero, M., Mancini, A., et al. (2001). Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J. Biol. Chem. 276 (34), 31720–31731. doi:10.1074/jbc.M104036200
Frouws, M. A., Reimers, M. S., Swets, M., Bastiaannet, E., Prinse, B., van Eijk, R., et al. (2017). The influence of BRAF and KRAS mutation status on the association between aspirin use and survival after colon cancer diagnosis. PLoS One 12 (1), e0170775. doi:10.1371/journal.pone.0170775
Gasecka, A., Nieuwland, R., van der Pol, E., Hajji, N., Ćwiek, A., Pluta, K., et al. (2019). P2Y12 antagonist ticagrelor inhibits the release of procoagulant extracellular vesicles from activated platelets. Cardiol. J. 26 (6), 782–789. doi:10.5603/CJ.a2018.0045
Gawaz, M., Geisler, T., and Borst, O. (2023). Current concepts and novel targets for antiplatelet therapy. Nat. Rev. Cardiol. 20 (9), 583–599. doi:10.1038/s41569-023-00854-6
Gawaz, M., Langer, H., and May, A. E. (2005). Platelets in inflammation and atherogenesis. J. Clin. Invest 115, 3378–3384. doi:10.1172/JCI27196
Gay, L. J., and Felding-Habermann, B. (2011). Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 11, 123–134. doi:10.1038/nrc3004
Golebiewska, , and Poole, A. W. (2015). Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. 29, 153–162. doi:10.1016/j.blre.2014.10.003
Goodall, A. H. (2014). Platelet 12-LOX scores a HIT. Blood 124 (14), 2166–2168. doi:10.1182/blood-2014-08-595652
Gresele, P., Falcinelli, E., Momi, S., Petito, E., and Sebastiano, M. (2021). Platelets and matrix metalloproteinases: a bidirectional interaction with multiple pathophysiologic implications. Hamostaseologie 41 (2), 136–145. doi:10.1055/a-1393-8339
Grosser, T., Fries, S., and FitzGerald, G. A. (2006). Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest 116 (1), 4–15. doi:10.1172/JCI27291
Guidotti, L. G., and Chisari, F. V. (2006). Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathology 1, 23–61. doi:10.1146/annurev.pathol.1.110304.100230
Guillem-Llobat, P., Dovizio, M., Bruno, A., Ricciotti, E., Cufino, V., Sacco, A., et al. (2016). Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 7, 32462–32477. doi:10.18632/oncotarget.8655
Ha, J. H., Ward, J. D., Radhakrishnan, R., Jayaraman, M., Song, Y. S., and Dhanasekaran, D. N. (2016). Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Gαi2, Src, and HIF1α signaling nexus. Oncotarget 7, 37664–37679. doi:10.18632/oncotarget.9224
Harrison, P., and Goodall, A. H. (2008). Message in the platelet-more than just vestigial mRNA. Platelets 19 (6), 395–404. doi:10.1080/09537100801990582
Henn, V., Slupsky, J. R., Gräfe, M., Anagnostopoulos, I., Förster, R., Müller-Berghaus, G., et al. (1998). CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 391, 591–594. doi:10.1038/35393
Hinterleitner, C., Strähle, J., Malenke, E., Hinterleitner, M., Henning, M., Seehawer, M., et al. (2021). Platelet PD-L1 reflects collective intratumoral PD-L1 expression and predicts immunotherapy response in non-small cell lung cancer. Nat. Commun. 12 (1), 7005. doi:10.1038/s41467-021-27303-7
Hu, H., Song, H., Han, B., Zhao, H., and He, J. (2024). Tumor-educated platelet RNA and circulating free RNA: emerging liquid biopsy markers for different tumor types. Front. Biosci. Landmark Ed. 29 (2), 80. doi:10.31083/j.fbl2902080
Hussain, S. P., and Harris, C. C. (2007). Inflammation and cancer: an ancient link with novel potentials. Int. J. Cancer 121 (11), 2373–2380. doi:10.1002/ijc.23173
Italiano, J. E., Mairuhu, A. T., and Flaumenhaft, R. (2010). Clinical relevance of microparticles from platelets and megakaryocytes. Curr. Opin. Hematol. 17, 578–584. doi:10.1097/MOH.0b013e32833e77ee
Janowska-Wieczorek, A., Wysoczynski, M., Kijowski, J., Marquez-Curtis, L., Machalinski, B., Ratajczak, J., et al. (2005). Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 113, 752–760. doi:10.1002/ijc.20657
Joung, J. K., and Sander, J. D. (2013). TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 14, 49–55. doi:10.1038/nrm3486
Juneja, V. R., McGuire, K. A., Manguso, R. T., LaFleur, M. W., Collins, N., Haining, W. N., et al. (2017). PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 214, 895–904. doi:10.1084/jem.20160801
Kailashiya, J. (2018). Platelet-derived microparticles analysis: techniques, challenges and recommendations. Anal. Biochem. 546, 78–85. doi:10.1016/j.ab.2018.01.030
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Investigation 119, 1420–1428. doi:10.1172/JCI39104
Kalluri, R., and Zeisberg, M. (2006). Fibroblasts in cancer. Nat. Rev. Cancer 6 (5), 392–401. doi:10.1038/nrc1877
Kitamura, T., Qian, B. Z., and Pollard, J. W. (2015). Immune cell promotion of metastasis. Nat. Rev. Immunol. 15, 73–86. doi:10.1038/nri3789
Labelle, M., Begum, S., and Hynes, R. O. (2011). Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20, 576–590. doi:10.1016/j.ccr.2011.09.009
Laffont, B., Corduan, A., Plé, H., Duchez, A. C., Cloutier, N., Boilard, E., et al. (2013). Activated platelets can deliver mRNA regulatory Ago2⋅microRNA complexes to endothelial cells via microparticles. Blood 122, 253–261. doi:10.1182/blood-2013-03-492801
Lauder, S. N., Allen-Redpath, K., Slatter, D. A., Aldrovandi, M., O'Connor, A., Farewell, D., et al. (2017). Networks of enzymatically oxidized membranelipids support calcium-dependent coagulation factor binding to maintain hemostasis. Sci. Signal. 10, eaan2787. doi:10.1126/scisignal.aan2787
Leblanc, R., Lee, S. C., David, M., Bordet, J. C., Norman, D. D., Patil, R., et al. (2014). Interaction of platelet-derived autotaxin with tumor integrin αVβ3 controls metastasis of breast cancer cells to bone. Blood 124, 3141–3150. doi:10.1182/blood-2014-04-568683
Li, S., Jiang, M., Wang, L., and Yu, S. (2020). Combined chemotherapy with cyclooxygenase-2 (COX-2) inhibitors in treating human cancers: recent advancement. Biomed. Pharmacother. 129, 110389. doi:10.1016/j.biopha.2020.110389
Liao, X., Lochhead, P., Nishihara, R., Morikawa, T., Kuchiba, A., Yamauchi, M., et al. (2012). Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N. Engl. J. Med. 367 (17), 1596–1606. doi:10.1056/NEJMoa1207756
Libby, P. (2012). Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32 (9), 2045–2051. doi:10.1161/ATVBAHA.108.179705
Linke, B., Schreiber, Y., Picard-Willems, B., Slattery, P., Nüsing, R. M., Harder, S., et al. (2017). Activated platelets induce an anti-inflammatory response of monocytes/macrophages through cross-regulation of PGE2 and cytokines. Mediat. Inflamm. 2017, 1463216. doi:10.1155/2017/1463216
Luci, D. K., Jameson, J. B. 2nd, Yasgar, A., Diaz, G., Joshi, N., Kantz, A., et al. (2014). Synthesis and structure-activity relationship studies of 4-((2-hydroxy-3-methoxybenzyl)amino)benzenesulfonamide derivatives as potent and selective inhibitors of 12-lipoxygenase. J. Med. Chem. 57 (2), 495–506. doi:10.1021/jm4016476
Mahajan, A., Brunson, A., Adesina, O., Keegan, T. H. M., and Wun, T. (2022). The incidence of cancer-associated thrombosis is increasing over time. Blood Adv. 6, 307–320. doi:10.1182/bloodadvances.2021005590
Mammadova-Bach, E., Gil-Pulido, J., Sarukhanyan, E., Burkard, P., Shityakov, S., Schonhart, C., et al. (2020). Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood 135 (14), 1146–1160. doi:10.1182/blood.2019002649
Mammadova-Bach, E., Zigrino, P., Brucker, C., Bourdon, C., Freund, M., De Arcangelis, A., et al. (2016). Platelet integrin α6β1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight 1, e88245. doi:10.1172/jci.insight.88245
Mannori, G., Crottet, P., Cecconi, O., Hanasaki, K., Aruffo, A., Nelson, R. M., et al. (1995). Differential colon cancer cell adhesion to E-P-and L-selectin: role of mucin-type glycoproteins. Cancer Res. 55 (19), 4425–4431.
Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. (2008). Cancer-related inflammation. Nature 454 (7203), 436–444. doi:10.1038/nature07205
Mantovani, A., and Pierotti, M. A. (2008). Cancer and inflammation: a complex relationship. Cancer Lett. 267 (2), 180–181. doi:10.1016/j.canlet.2008.05.003
Marrugo-Ramírez, J., Mir, M., and Samitier, J. (2018). Blood-based cancer biomarkers in liquid biopsy: a promising non-invasive alternative to tissue biopsy. Int. J. Mol. Sci. 19 (10), 2877. doi:10.3390/ijms19102877
Martin, P., and Leibovich, S. J. (2005). Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 15 (11), 599–607. doi:10.1016/j.tcb.2005.09.002
Massagué, J., and Obenauf, A. C. (2016). Metastatic colonization by circulating tumour cells. Nature 529 (7586), 298–306. doi:10.1038/nature17038
Mayer, K., Hein-Rothweiler, R., Schüpke, S., Janisch, M., Bernlochner, I., Ndrepepa, G., et al. (2021). Efficacy and safety of revacept, a novel lesion-directed competitive antagonist to platelet glycoprotein VI, in patients undergoing elective percutaneous coronary intervention for stable ischemic heart disease: the randomized, double-blind, placebo-controlled ISAR-PLASTER phase 2 trial. JAMA Cardiol. 6 (7), 753–761. doi:10.1001/jamacardio.2021.0475
Menter, D. G., Schilsky, R. L., and DuBois, R. N. (2010). Cyclooxygenase-2 and cancer treatment: understanding the risk should be worth the reward. Clin. Cancer Res. 16 (5), 1384–1390. doi:10.1158/1078-0432.CCR-09-0788
Michael, J. V., Wurtzel, J. G. T., Mao, G. F., Rao, A. K., Kolpakov, M. A., Sabri, A., et al. (2017). Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood 130 (5), 567–580. doi:10.1182/blood-2016-11-751099
Mills, G. B., and Moolenaar, W. H. (2003). The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 3, 582–591. doi:10.1038/nrc1143
Mitrugno, A., Williams, D., Kerrigan, S. W., and Moran, N. (2014). A novel and essential role for FcγRIIa in cancer cell-induced platelet activation. Blood 123, 249–260. doi:10.1182/blood-2013-03-492447
Momi, S., Falcinelli, E., Petito, E., Ciarrocca Taranta, G., Ossoli, A., and Gresele, P. (2022). Matrix metalloproteinase-2 on activated platelets triggers endothelial PAR-1 initiating atherosclerosis. Eur. Heart J. 43 (6), 504–514. doi:10.1093/eurheartj/ehab631
Moser, C. L., Gould, K. A., McNeley, M. K., Shoemaker, A. R., and Dove, W. F. (1995). ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur. J. Cancer 31A, 1061–1064. doi:10.1016/0959-8049(95)00181-h
Nagalla, S., Shaw, C., Kong, X., Kondkar, A. A., Edelstein, L. C., Ma, L., et al. (2011). Platelet microRNA-mRNA coexpression profiles correlate with platelet reactivity. Blood 117 (19), 5189–5197. doi:10.1182/blood-2010-09-299719
National Institute for Health and Care (2020). Excellence NICE-effectiveness of aspirin in the prevention of colorectal cancer in people with Lynch syndrome. London, UK: National Institute for Health and Care.
Negus, R. P., Stamp, G. W., Hadley, J., and Balkwill, F. R. (1997). Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am. J. Pathol. 150 (5), 1723–1734.
Nierodzik, M. L., and Karpatkin, S. (2006). Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 10 (5), 355–362. doi:10.1016/j.ccr.2006.10.002
Nilsson, R. J., Balaj, L., Hulleman, E., van Rijn, S., Pegtel, D. M., Walraven, M., et al. (2011). Blood platelets contain tumor-derived RNA biomarkers. Blood 118 (13), 3680–3683. doi:10.1182/blood-2011-03-344408
Nilsson, R. J., Karachaliou, N., Berenguer, J., Gimenez-Capitan, A., Schellen, P., Teixido, C., et al. (2016). Rearranged EML4-ALK fusion transcripts sequester in circulating blood platelets and enable blood-based crizotinib response monitoring in non-small-cell lung cancer. Oncotarget 7 (1), 1066–1075. doi:10.18632/oncotarget.6279
Oshima, M., Dinchuk, J. E., Kargman, S. L., Oshima, H., Hancock, B., Kwong, E., et al. (1996). Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87 (5), 803–809. doi:10.1016/s0092-8674(00)81988-1
Patrignani, P., and Patrono, C. (2015). Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochim. Biophys. Acta 1851, 422–432. doi:10.1016/j.bbalip.2014.09.016
Patrignani, P., and Patrono, C. (2016). Aspirin and cancer. J. Am. Coll. Cardiol. 68, 967–976. doi:10.1016/j.jacc.2016.05.083
Patrignani, P., and Patrono, C. (2018). Aspirin, platelet inhibition and cancer prevention. Platelets 29 (8), 779–785. doi:10.1080/09537104.2018.1492105
Patrignani, P., Tacconelli, S., Piazuelo, E., Di Francesco, L., Dovizio, M., Sostres, C., et al. (2014). Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J. Thromb. Haemost. 12 (8), 1320–1330. doi:10.1111/jth.12637
Patrignani, P., Tacconelli, S., Contursi, A., Piazuelo, E., Bruno, A., Nobili, S., et al. (2024). Optimizing aspirin dose for colorectal cancer patients through deep phenotyping using novel biomarkers of drug action. Front. Pharmacol. 29;15, 1362217. doi:10.3389/fphar.2024.1362217
Patrono, C., and Baigent, C. (2019). Role of aspirin in primary prevention of cardiovascular disease. Nat. Rev. Cardiol. 16 (11), 675–686. doi:10.1038/s41569-019-0225-y
Patrono, C., García Rodríguez, L. A., Landolfi, R., and Baigent, C. (2005). Low-dose aspirin for the prevention of atherothrombosis. N. Engl. J. Med. 353 (22), 2373–2383. doi:10.1056/NEJMra052717
Patrono, C., Patrignani, P., and García Rodríguez, L. A. (2001). Cyclooxygenase selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J. Clin. Invest 108, 7–13. doi:10.1172/JCI13418
Petito, E., Momi, S., and Gresele, P. (2017). “The migration of platelets and their interaction with other migrating cells,” in Platelets in thrombotic and non-thrombotic disorders. Editors P. Gresele, N. Kleiman, J. Lopez, and C. Page (Cham, Switzerland: Springer), 337–351. Ray MK, Fagan SP, and Brunicardi.
Pitchford, S. C., Momi, S., Baglioni, S., Casali, L., Giannini, S., Rossi, R., et al. (2008). Allergen induces the migration of platelets to lung tissue in allergic asthma. Am. J. Respir. Crit. Care Med. 177 (6), 604–612. doi:10.1164/rccm.200702-214OC
Pitchford, S. C., Momi, S., Giannini, S., Casali, L., Spina, D., Page, C. P., et al. (2005). Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood 105 (5), 2074–2081. doi:10.1182/blood-2004-06-2282
Pitchford, S. C., Yano, H., Lever, R., Riffo-Vasquez, Y., Ciferri, S., Rose, M. J., et al. (2003). Platelets are essential for leukocyte recruitment in allergic inflammation. J. Allergy Clin. Immunol. 112 (1), 109–118. doi:10.1067/mai.2003.1514
Placke, T., Örgel, M., Schaller, M., Jung, G., Rammensee, H. G., Kopp, H. G., et al. (2012). Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 72, 440–448. doi:10.1158/0008-5472.CAN-11-1872
Puhm, F., Boilard, E., and Machlus, K. R. (2021). Platelet extracellular vesicles: beyond the blood. Arterioscler. Thromb. Vasc. Biol. 41 (1), 87–96. doi:10.1161/ATVBAHA.120.314644
Rachidi, S., Metelli, A., Riesenberg, B., Wu, B. X., Nelson, M. H., Wallace, C., et al. (2017). Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci. Immunol. 2 (11), eaai7911. doi:10.1126/sciimmunol.aai7911
Rana, R., Huang, T., Koukos, G., Fletcher, E. K., Turner, S. E., Shearer, A., et al. (2018). Noncanonical matrix metalloprotease 1–protease-activated receptor 1 signaling drives progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 38, 1368–1380. doi:10.1161/ATVBAHA.118.310967
Ratajczak, J., Wysoczynski, M., Hayek, F., Janowska-Wieczorek, A., and Ratajczak, M. Z. (2006). Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 20, 1487–1495. doi:10.1038/sj.leu.2404296
Risitano, A., Beaulieu, L. M., Vitseva, O., and Freedman, J. E. (2012). Platelets and platelet-like particles mediate intercellular RNA transfer. Blood 119, 6288–6295. doi:10.1182/blood-2011-12-396440
Rondina, M. T., Weyrich, A. S., and Zimmerman, G. A. (2013). Platelets as cellular effectors of inflammation in vascular diseases. Circ. Res. 112 (11), 1506–1519. doi:10.1161/CIRCRESAHA.113.300512
Ross, R., Bowen-Pope, D. F., and Raines, E. W. (1985). Platelets, macrophages, endothelium, and growth factors. Their effects upon cells and their possible roles in atherogenesis. Ann. N. Y. Acad. Sci. 454, 254–260. doi:10.1111/j.1749-6632.1985.tb11865.x
Rovati, G., Contursi, A., Bruno, A., Tacconelli, S., Ballerini, P., and Patrignani, P. (2022). Antiplatelet agents affecting GPCR signaling implicated in tumor metastasis. Cells 11 (4), 725. doi:10.3390/cells11040725
Rumbaut, R. E., and Thiagarajan, P. (2010). Platelet-vessel wall interactions in hemostasis and thrombosis. San. Rafael Morgan Claypool Life Sci. 2, 1–75. doi:10.4199/C00007ED1V01Y201002ISP004
Sacco, A., Bruno, A., Contursi, A., Dovizio, M., Tacconelli, S., Ricciotti, E., et al. (2019). Platelet-specific deletion of cyclooxygenase-1 ameliorates dextran sulfate sodium-induced colitis in mice. J. Pharmacol. Exp. Ther. 370 (3), 416–426. doi:10.1124/jpet.119.259382
Santoso, S., Sachs, U. J., Kroll, H., Linder, M., Ruf, A., Preissner, K. T., et al. (2002). The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J. Exp. Med. 196 (5), 679–691. doi:10.1084/jem.20020267
Serrano, D., Patrignani, P., Stigliano, V., Turchetti, D., Sciallero, S., Roviello, F., et al. (2022). Aspirin colorectal cancer prevention in Lynch syndrome: recommendations in the era of precision medicine. Genes (Basel) 13 (3), 460. doi:10.3390/genes13030460
Sim, M. M. S., Shiferawe, S., and Wood, J. P. (2023). Novel strategies in antithrombotic therapy: targeting thrombosis while preserving hemostasis. Front. Cardiovasc Med. 10, 1272971. doi:10.3389/fcvm.2023.1272971
Simon, D. I., Chen, Z., Xu, H., Li, C. Q., Dong, Jf, McIntire, L. V., et al. (2000). Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 192 (2), 193–204. doi:10.1084/jem.192.2.193
Sitia, G., Aiolfi, R., Di Lucia, P., Mainetti, M., Fiocchi, A., Mingozzi, F., et al. (2012). Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. U. S. A. 109, E2165–E2172. doi:10.1073/pnas.1209182109
Skog, J., Würdinger, T., van Rijn, S., Meijer, D. H., Gainche, L., Sena-Esteves, M., et al. (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10 (12), 1470–1476. doi:10.1038/ncb1800
Slatter, D. A., Percy, C. L., Allen-Redpath, K., Gajsiewicz, J. M., Brooks, N. J., Clayton, A., et al. (2018). Enzymatically oxidized phospholipids restore thrombin generation in coagulation factor deficiencies. JCI Insight 3 (6), e98459. doi:10.1172/jci.insight.98459
Smyth, S. S., McEver, R. P., Weyrich, A. S., Morrell, C. N., Hoffman, M. R., Arepally, G. M., et al. (2009). Platelet functions beyond hemostasis. J. Thromb. Haemost. 7 (11), 1759–1766. doi:10.1111/j.1538-7836.2009.03586.x
Stanger, L., and Holinstat, M. (2023). Bioactive lipid regulation of platelet function, hemostasis, and thrombosis. Pharmacol. Ther. 246, 108420. doi:10.1016/j.pharmthera.2023.108420
Tang, M., Jiang, L., Lin, Y., Wu, X., Wang, K., He, Q., et al. (2017). Platelet microparticle-mediated transfer of miR-939 to epithelial ovarian cancer cells promotes epithelial to mesenchymal transition. Oncotarget 8, 97464–97475. doi:10.18632/oncotarget.22136
Thomas, C. P., Morgan, L. T., Maskrey, B. H., Murphy, R. C., Kühn, H., Hazen, S. L., et al. (2010). Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J. Biol. Chem. 285 (10), 6891–6903. doi:10.1074/jbc.M109.078428
Timár, J., Rásó, E., Honn, K. V., and Hagmann, W. (1999). 12-lipoxygenase expression in human melanoma cell lines. Adv. Exp. Med. Biol. 469, 617–622. doi:10.1007/978-1-4615-4793-8_89
Tourdot, B. E., and Holinstat, M. (2017). Targeting 12-lipoxygenase as a potential novel antiplatelet therapy. Trends Pharmacol. Sci. 38 (11), 1006–1015. doi:10.1016/j.tips.2017.08.001
Tran, D. Q., Andersson, J., Wang, R., Ramsey, H., Unutmaz, D., and Shevach, E. M. (2009). GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. U. S. A. 106 (32), 13445–13450. doi:10.1073/pnas.0901944106
Tripodi, A., and Mannucci, P. M. (2011). The coagulopathy of chronic liver disease. N. Engl. J. Med. 365, 147–156. doi:10.1056/NEJMra1011170
Turnage, R. H., LaNoue, J. L., Kadesky, K. M., Meng, Y., and Myers, S. I. (1985). Thromboxane A2 mediates increased pulmonary microvascular permeability after intestinal reperfusion. J. Appl. Physiol. 82 (2), 592–598. doi:10.1152/jappl.1997.82.2.592
Tuxhorn, J. A., Ayala, G. E., and Rowley, D. R. (2001). Reactive stroma in prostate cancer progression. J. Urol. 166 (6), 2472–2483. doi:10.1016/s0022-5347(05)65620-0
Tuxhorn, J. A., Ayala, G. E., Smith, M. J., Smith, V. C., Dang, T. D., and Rowley, D. R. (2002). Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 8 (9), 2912–2923.
Ungerer, M., Rosport, K., Bültmann, A., Piechatzek, R., Uhland, K., Schlieper, P., et al. (2011). Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation 123, 1891–1899. doi:10.1161/CIRCULATIONAHA.110.980623
Varon, D., and Shai, E. (2015). Platelets and their microparticles as key players in pathophysiological responses. J. Thromb. Haemost. 13 (Suppl. 1), S40–S46. doi:10.1111/jth.12976
Vasina, E. M., Cauwenberghs, S., Feijge, M. A., Heemskerk, J. W., Weber, C., and Koenen, R. R. (2011). Microparticles from apoptotic platelets promote resident macrophage differentiation. Cell Death and Dis. 2, e211. doi:10.1038/cddis.2011.94
Vitiello, L., Spoletini, I., Gorini, S., Pontecorvo, L., Ferrari, D., Ferraro, E., et al. (2014). Microvascular inflammation in atherosclerosis. IJC Metab. Endocr. 3, 1–7. doi:10.1016/j.ijcme.2014.03.002
Wang, D., and DuBois, R. N. (2013). An inflammatory mediator, prostaglandin E2, in colorectal cancer. Cancer J. 19 (6), 502–510. doi:10.1097/PPO.0000000000000003
Weyrich, A. S., Elstad, M. R., McEver, R. P., McIntyre, T. M., Moore, K. L., Morrissey, J. H., et al. (1996). Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Invest 97 (6), 1525–1534. doi:10.1172/JCI118575
Williams, J. R., Khandoga, A. L., Goyal, P., Fells, J. I., Perygin, D. H., Siess, W., et al. (2009). Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J. Biol. Chem. 284, 17304–17319. doi:10.1074/jbc.M109.003194
Willis Fox, O., and Preston, R. J. S. (2020). Molecular basis of protease-activated receptor 1 signaling diversity. J. Thromb. Haemost. 18, 6–16. doi:10.1111/jth.14643
Wong, C. C., Baum, J., Silvestro, A., Beste, M. T., Bharani-Dharan, B., Xu, S., et al. (2020). Inhibition of IL-1β by canakinumab may be effective against diverse molecular subtypes of lung cancer: an exploratory analysis of the CANTOS trial. Cancer Res. 80 (24), 5597–5605. doi:10.1158/0008-5472.CAN-19-3176
Yang, J., Hirata, T., Croce, K., Merrill-Skoloff, G., Tchernychev, B., Williams, E., et al. (1999). Targeted gene disruption demonstrates that P-selectin glycoprotein ligand 1 (PSGL-1) is required for P-selectin-mediated but not E-selectin-mediated neutrophil rolling and migration. J. Exp. Med. 190 (12), 1769–1782. doi:10.1084/jem.190.12.1769
Yang, R. Y., Rabinovich, G. A., and Liu, F. T. (2008). Galectins: structure, function and therapeutic potential. Expert Rev. Mol. Med. 10, e17. doi:10.1017/S1462399408000719
Yeung, J., Tourdot, B. E., Fernandez-Perez, P., Vesci, J., Ren, J., Smyrniotis, C. J., et al. (2014). Platelet 12-LOX is essential for FcγRIIa-mediated platelet activation. Blood 124 (14), 2271–2279. doi:10.1182/blood-2014-05-575878
Yoshimoto, T., Arakawa, T., Hada, T., Yamamoto, S., and Takahashi, E. (1992). Structure and chromosomal localization of human arachidonate 12-lipoxygenase gene. J. Biol. Chem. 267, 24805–24809. doi:10.1016/s0021-9258(18)35835-6
Keywords: platelets, cancer, aspirin, antiplatelet agents, inflammation, revacept, 12-lipoxygenase, extracellular vesicles
Citation: Contursi A, Tacconelli S, Di Berardino S, De Michele A and Patrignani P (2024) Platelets as crucial players in the dynamic interplay of inflammation, immunity, and cancer: unveiling new strategies for cancer prevention. Front. Pharmacol. 15:1520488. doi: 10.3389/fphar.2024.1520488
Received: 31 October 2024; Accepted: 04 December 2024;
Published: 23 December 2024.
Edited by:
Galina Sud’ina, Lomonosov Moscow State University, RussiaReviewed by:
Luciana Mucci, Italian Medicines Agency (AIFA), ItalyNadji Hannachi, University Ferhat Abbas of Setif, Algeria
Copyright © 2024 Contursi, Tacconelli, Di Berardino, De Michele and Patrignani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Patrignani, cHBhdHJpZ25hbmlAdW5pY2guaXQ=
†These authors have contributed equally to this work and share first authorship