 Haipeng Jie
Haipeng Jie Jingjing Zhang
Jingjing Zhang Shuzhen Wu
Shuzhen Wu Luyao Yu1
Luyao Yu1 Shengnan Li
Shengnan Li Bo Dong
Bo Dong Feng Yan
Feng Yan- 1Department of Cardiology, Shandong Provincial Hospital, Cheeloo College of Medicine, Shandong University, Jinan, China
- 2Department of Emergency Medicine, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, China
Sustained production of reactive oxygen species (ROS) and an imbalance in the antioxidant system have been implicated in the development of cardiovascular diseases (CVD), especially when combined with diabetes, hypercholesterolemia, and other metabolic disorders. Among them, NADPH oxidases (NOX), including NOX1-5, are major sources of ROS that mediate redox signaling in both physiological and pathological processes, including fibrosis, hypertrophy, and remodeling. Recent studies have demonstrated that mitochondria produce more proteins and energy in response to adverse stress, corresponding with an increase in superoxide radical anions. Novel NOX4-mediated modulatory mechanisms are considered crucial for maintaining energy metabolism homeostasis during pathological states. In this review, we integrate the latest data to elaborate on the interactions between oxidative stress and energy metabolism in various CVD, aiming to elucidate the higher incidence of CVD in individuals with metabolic disorders. Furthermore, the correlations between NOX and ferroptosis, based on energy metabolism, are preliminarily discussed. Further discoveries of these mechanisms might promote the development of novel therapeutic drugs targeting NOX and their crosstalk with energy metabolism, potentially offering efficient management strategies for CVD.
1 Introduction
Cardiovascular diseases (CVD) constitute a group of preventable conditions, including coronary heart disease, hypertension, heart failure, myocardial infarction, hypertrophic cardiomyopathy, and others. Since 1990, CVD has been the leading cause of death worldwide (Thomas et al., 2018). These diseases are associated with risk factors such as unhealthy diet, obesity, age, genetics, smoking, and diabetes (Schloss et al., 2020). The heart functions as the body’s pumping organ, driving blood circulation and requiring balanced energy metabolism to maintain its proper function (Bertero and Maack, 2018).
Cardiac energy metabolism is intricately linked with various metabolic substrates used by the heart. In a healthy heart, ATP is primarily produced through the oxidation of fatty acids, glucose, and lactate, whereas the fetal and newborn heart predominantly relies on glycolysis and lactic acid metabolism. As the heart matures, fatty acids become the preferred substrate (Kolwicz and Tian, 2011; Li et al., 2023). After birth, with changes in the levels of these substrates in the blood, the pattern of energy supplementation shifts, and the proportion of fatty acid oxidation (FAO) increases in healthy hearts (Li et al., 2023). Additionally, glucose, lactate, pyruvate, ketone bodies, and amino acids are also utilized as energy substrates. However, under various pathological conditions such as hypoxia, dyslipidemia, and diabetes, disturbances in energy metabolism and the development of CVD are interrelated (Li et al., 2023; Lopaschuk et al., 2021; Olkowicz et al., 2021), potentially mediated by NADPH oxidase (NOX) (Nabeebaccus et al., 2023; van der Pol et al., 2019; Zhang et al., 2020).
The NOX family comprises seven members in the human body, including NOX1-5 and DUOX1-2, all of which are multi-transmembrane proteins (NOX1-5 are six-pass transmembrane proteins, while DUOX1-2 are seven-pass transmembrane proteins) (Lambeth, 2004). All NOX enzymes share structural properties, including an NH2-terminal hydrophobic transmembrane region and a COOH-terminal flavin adenine dinucleotide binding domain. NOX1-4 include the catalytic subunit gp91phox (known as NOX2 in its most studied form), along with regulatory subunits p22phox, p47phox, p40phox, p67phox, and the small GTPase RAC. NOX5, based on gp91phox, has an amino-terminal calmodulin-like domain bound with calcium. DUOX1-2, based on NOX5, extend an amino-terminal peroxidase-homology domain (Begum et al., 2022; Gu et al., 2021). NOX enzymes are ubiquitously expressed in various subcellular localizations within cardiovascular tissues, regulating their physiological and pathological functions (Begum et al., 2022; Zhang et al., 2020).
Reactive oxygen species (ROS) are a chemically defined group that includes superoxide (O2•¯), hydrogen peroxide (H2O2), hydroxyl radical (OH•), and their reaction products, which can trigger cellular dysfunction. In the cardiovascular system, ROS originate from the mitochondrial electron transport chain, NOX, xanthine oxidase, lipoxygenase, and cyclooxygenase (Begum et al., 2022; Lambeth, 2004). Among these, NOX, as membrane protein donors, can be selectively activated to participate in physiological and pathological processes such as proliferation, migration, angiogenesis, and cell death (Begum et al., 2022). Based on NOX-mediated oxidative stress, this paper primarily focuses on the role of NOX-mediated energy metabolism in various CVD.
2 Cardiac energy metabolism
2.1 Fatty acid metabolism
The heart predominantly utilizes non-esterified fatty acids, chylomicrons, and very low-density lipoproteins for myocardial energy production (Hahn et al., 2023). Cardiomyocytes acquire fatty acids from plasma albumin (circulating) and lipoproteins for esterification. Fatty acid-binding protein on cardiomyocyte surfaces facilitates the uptake of free fatty acids (FFAs), which are transported into cardiomyocytes and recognized as substrates for fatty acid acetyl-coenzyme A synthetase, promoting lipid synthesis. These fatty acids also generate acetyl-CoA, which participates in the tricarboxylic acid (TCA) cycle to supply energy for the heart (Da Dalt et al., 2023; Gibb and Hill, 2018). High-fat diets, such as those rich in palmitic acid, can alter the heart’s fatty acid composition and slightly affect heart function in mice (Pakiet et al., 2020). Excessive accumulation of lipids in the heart can lead to oxidative and mitochondrial stress and apoptosis, partially modulated by NOX (Li et al., 2023; Nabeebaccus et al., 2023; Yamamoto and Sano, 2022).
2.2 Glucose metabolism
Glucose, another vital energy substrate for the heart, also provides metabolic substrates for various cell physiological activities (Kolwicz and Tian, 2011). Cardiomyocytes primarily take up circulating glucose via glucose transport proteins (GLUT), with GLUT1 (insulin-independent) and GLUT4 (insulin-dependent) as the main subtypes expressed in the heart (Aerni-Flessner et al., 2012). Glucose is then phosphorylated to glucose-6-phosphate (G-6-P) by hexokinase, producing pyruvate through glycolysis, which is transferred to the mitochondria to enter the TCA cycle for energy production. Additionally, glucose is utilized in the pentose phosphate pathway (PPP), glycogen synthesis, and the hexosamine biosynthetic pathway (HBP) (Aerni-Flessner et al., 2012; Gibb and Hill, 2018). The oxidative PPP is recognized for maintaining reduced glutathione levels to combat ROS by producing NADPH.
2.3 Lactate metabolism
Lactate is one of the sources of energy metabolism for the myocardium (Gibb and Hill, 2018). Before and after birth, the myocardium preferentially selects fatty acids over carbohydrates as energy substrates, a process accompanied by changes in lactate metabolism. Specifically, cardiomyocytes can take up and release lactate simultaneously, where lactate oxidation is inhibited by FAO (Bartelds et al., 1999). Typically, lactate provides about 10% of the energy supply required for daily consumption in the heart (Ouyang et al., 2023). Lactate and pyruvate are mutually converted to one another and can be produced or eliminated through a reversible redox reaction catalyzed by lactate dehydrogenase (Bonen, 2000). Monocarboxylate transporters (MCT) facilitate the transport of lactate from the blood into cardiomyocytes for lactate removal. Lactate is then oxidized to pyruvate, which subsequently enters the TCA cycle to provide energy for the heart. Interestingly, lactate has been demonstrated to enable the lactylation of lysine residues on histones or other proteins, playing protective roles in myocardial infarction (MI) and heart failure (Ouyang et al., 2023; Wang N. et al., 2022; Zhang et al., 2023).
2.4 Ketone body metabolism
Circulating ketone bodies, including acetoacetic acid, acetone, and β-hydroxybutyrate, serve as additional energy sources for the heart (de Koning et al., 2021; Manolis et al., 2023). These ketone body levels are increased in CVD such as heart failure, MI, and atherosclerosis, contributing to compensatory energy mechanisms (de Koning et al., 2021). Specifically, ketone bodies are produced by FAO in mitochondria and transported across cell membranes. Monocarboxylate transporters 1 and 2 (MCT1 and MCT2) facilitate their transport into cardiomyocyte mitochondria for ketolysis, thereby producing acetyl-CoA to enter the TCA cycle and produce ATP.
2.5 Branched-chain amino acid metabolism
Amino acids, including branched-chain amino acids (BCAAs: valine, leucine, and isoleucine), glutamic acid, cystine, histidine, and lysine, serve as a fuel source for the heart (Lopaschuk and Ussher, 2016). The metabolism of amino acids is closely related to obesity, ischemic cardiomyopathy, heart failure, and other diseases, and may predict the risk of cardiovascular events (McGarrah and White, 2023; Shah et al., 2012). Specifically, BCAAs are first transaminated by branched-chain amino acid transferases (BCATs) to form branched-chain α-keto acids (BCKAs) and glutamate (Dimou et al., 2022). Subsequently, BCKAs are oxidized and decarboxylated to their branched-chain acyl-CoA esters, with α-ketoglutarate dehydrogenase playing a pivotal role (Patrick et al., 2022). Finally, various branched-chain acyl-CoA compounds promote cardiac energy production through different pathways.
3 The role of ROS in physiological processes
Most studies indicate that excessive ROS accumulation leads to DNA damage, protein modification, and lipid peroxidation, ultimately inducing irreparable cellular injury (Checa and Aran, 2020). However, it is noteworthy that localized, low levels of ROS are critical for redox signaling. Current research predominantly focuses on H₂O₂, which functions as a second messenger in signaling pathways. H₂O₂ mediates protein post-translational modifications (e.g., cysteine and tyrosine residues), transcription factor activity, and epigenetic modifications of DNA and histones (Lennicke and Cochemé, 2021a). These processes regulate downstream signaling pathways involved in autophagy, cell proliferation, apoptosis, extracellular matrix repair, and immune defense (Checa and Aran, 2020; Holmström and Finkel, 2014). While the precise mechanisms are not detailed here, we emphasize the relationships between ROS and energy metabolism under physiological conditions. Generally, ROS modulates cellular energy metabolism by activating or inhibiting protein kinases and suppressing phosphatases. This is evident in signaling pathways such as insulin, AMPK, and mTOR (Checa and Aran, 2020; Lennicke and Cochemé, 2021a; Zmijewski et al., 2010). In the insulin signaling pathway, ligand-receptor interactions induce ROS production via NOX4, which is critical for proper cascade reaction and glucose metabolism (Lennicke and Cochemé, 2021b). Interestingly, NOX4 has been shown to improve high-fat diet-induced adipose accumulation, insulin resistance, and liver steatosis (Li et al., 2012). Notably, NOX4 produces H₂O₂ instead of O2•¯, which may play vital roles through redox signaling (Schürmann et al., 2015). AMPK is activated when energy metabolism demand increases, and studies have shown that H₂O₂ can activate AMPK by oxidizing its cysteine residues (Zmijewski et al., 2010). In mitochondria, ROS is the byproduct of the respiratory chain, linking them closely to cellular metabolism (Shadel and Horvath, 2015). Recent findings suggest that elevated hypothalamic ROS suppresses food intake and increases energy expenditure, indicating the physiological roles of ROS in maintaining energy homeostasis (Benani et al., 2007). While most studies focus on the pathological overproduction of ROS driven by NOX, their roles in physiological processes, particularly in relation to cellular metabolism, remain underexplored and warrant further investigation.
4 The roles of NOX in CVD
4.1 Atherosclerosis
Atherosclerosis is a pathological condition characterized as a chronic multifocal immune-inflammatory disease driven by lipids, mainly occurring in large and medium-sized arteries (Tedgui and Mallat, 2006). NOX1, NOX2, NOX4, NOX5 are the main source of ROS in vasculature (Langbein et al., 2016). Upregulated NOX1 in atherosclerosis has been demonstrated to promote vascular smooth muscle cell (VSMC) proliferation and extracellular matrix (ECM) production, inducing the formation of vascular neointima (Lee et al., 2009; Valente et al., 2012). Knockout NOX1 has been shown to delay the progression of atherosclerosis by reducing ROS production, suppressing inflammation, improving mitochondrial apoptosis, and alleviating endothelial cell dysfunction (Liu et al., 2020; Sorescu et al., 2004). NOXO1 (NOX organizer 1) and NOXA1 (NOX activator 1) are necessary for the activation of NOX1 and their inhibition produces similar effects (Buchmann et al., 2020; Siu etal., 2016). In atherosclerosis, NOX1 is elevated early but declines later, while NOX4 increases in advanced stages (Xu et al., 2014). Unlike the harmful effects of other NOX isoforms, the roles of NOX4 in atherosclerosis remain under discussion. Recent studies have revealed that NOX4-derived H₂O₂ has vascular protective effects (Langbein et al., 2016; Schürmann et al., 2015). At the same time, overexpression of NOX4 in endothelial cells reduces the expression of interferon-gamma and increases the proportion of T regulatory cells (Craige et al., 2015). However, the proinflammatory phenotype of VSMCs mediated by NOX4 is also recognized to cause plaque instability and rupture (Xu et al., 2014). Recent studies have revealed that the overexpression of NOX4 in mitochondria can accelerate the formation of aortic sclerosis. This effect can be partially reversed by mitochondrial oxidative stress inhibitors (Canugovi et al., 2019; Vendrov et al., 2015).
The unique role of NOX4 in atherosclerosis may be linked to its production of H₂O₂, which functions as a second messenger widely involved in regulating cellular signaling pathways. However, the precise mechanisms remain unclear. Beyond its role in modulating inflammation infiltration and VSMC phenotype switching, the metabolic effects of NOX4 warrant further investigation. The current study has shown that silencing NOX4 in hepatocytes reduces insulin and fatty acid utilization, but its metabolic role in atherosclerosis remains to be elucidated (Wu and Williams, 2012).
4.2 Hypertension
Essential hypertension, a major contributor to the global disease burden, is closely associated with a high-salt diet, obesity, dyslipidemia, and diabetes (Di Raimondo et al., 2021; Munir and du Toit, 2024; Poulter et al., 2015). NOX1 and NOX4 are significantly elevated in VSMC of spontaneously hypertensive rats (SHR), particularly in the endoplasmic reticulum (ER) and nucleus, promoting ROS production and ER stress through protein sulfenylation and hyperoxidation (Camargo et al., 2018). Moreover, associations between NOX and hypertension have been demonstrated in animal models induced by L-NAME, Ang-II, and DOCA-salt, affecting both the vascular system and extravascular systems, including the renal system, central nervous system, and immune system (Griendling et al., 2021; Zhang et al., 2020). Several studies have revealed the harmful roles of NOX1/2-mediated oxidative stress in hypertension, including but not limited to the inactivation of NO and the production of peroxynitrite (Drummond et al., 2011; Drummond and Sobey, 2014; Matsuno et al., 2005; Murdoch et al., 2011). However, NOX4 remains controversial in the pathologic progression of hypertension. Some studies have demonstrated that NOX4 has similar effects to other NOX, but recent studies have revealed that NOX4 plays a protective role in vasculature by producing H₂O₂ and NO (Drummond et al., 2011; Ray et al., 2011; Schröder et al., 2012). Specially, this includes reducing Ang-II induced vascular dysfunction and increasing ischemia induced angiogenesis (Schröder et al., 2012).
Recently, NOX4-mediated metabolism remodeling has gradually come into view (Nabeebaccus et al., 2023). In Dahl salt-sensitive (SS) hypertensive rats, kidney metabolomics reveal that the PPP and glycolysis are increased, accompanied by reduced glutathione reductase activity and TCA cycle activity, promoting ROS mediated by NOX (Wang Y. et al., 2018). Additionally, fatty acid and amino acid metabolism also change (Wang Y. et al., 2018). Recent clinical studies reveal that high salt intake in daily life increases serum sodium and osmotic pressure, which are considered risks for hypertension (Kuwabara et al., 2020). It is also demonstrated that sodium and fructose intake contribute to metabolic syndrome, consisting of insulin resistance, obesity, dyslipidemia, and hypertension in children (Genovesi et al., 2021). Fructose, whether produced by the liver on a high-salt diet or ingested externally, can increase intracellular uric acid levels. This, in turn, recruits NOX into mitochondria, leading to oxidative phosphorylation uncoupling and oxidative stress, ultimately contributing to hypertension (Sánchez-Lozada et al., 2023). Interestingly, knocking out NOX4 reduces the utilization of glucose and lipids, inducing insulin signaling disturbances (Lennicke and Cochemé, 2021b; Li et al., 2012; Wu and Williams, 2012). Therefore, NOX4-mediated metabolic remodeling may also be involved in hypertension.
Given the predominant influence of diet on hypertension, the gut microbiome in hypertension patients has attracted significant interest. Studies have revealed that Prevotella is a main contributor to prehypertension and hypertension, and mice that receive stool from hypertensive patients experience elevated blood pressure (Li et al., 2017). Fecal microbiota transplantation from losartan-treated SHR improves endothelial function and reduces NOX activity in untreated SHR, leading to lower blood pressure (Robles-Vera et al., 2020). Dysregulation of the gut microbiota and barrier impairment during hypertension induces chronic translocation of lipopolysaccharide (LPS) into circulation, causing an imbalance of NOX and antioxidant enzymes through toll-like receptor 4 (TLR4). This finding provides a theoretical basis for the use of probiotics in treating hypertension (Grylls et al., 2021; Kim et al., 2018; Robles-Vera et al., 2020). In response to high-fat diets, TLR4 knockout prevents obesity-induced endothelial dysfunction and hypertension by reducing NOX1/4 and ROS content, lowering inflammatory factors, and increasing eNOS activity (Liang et al., 2013). Blocking the TLR4-MD2 complex effectively improves blood pressure by reducing oxidative stress (de Oliveira et al., 2020). Additionally, apocynin, a selective NOX inhibitor, reduces the stability and expression of LPS-induced TLR4, indicating the crosstalk between oxidative stress produced by NOX p47phox and TLR4 or its subsequent pathways (Lin et al., 2006). Similarly, decreased expression of NOX1 and NOX4 in the vascular tissue of TLR4-mutated mice protects against arterial endothelial dysfunction in diabetic mice (Liang et al., 2013) (Figure 1).
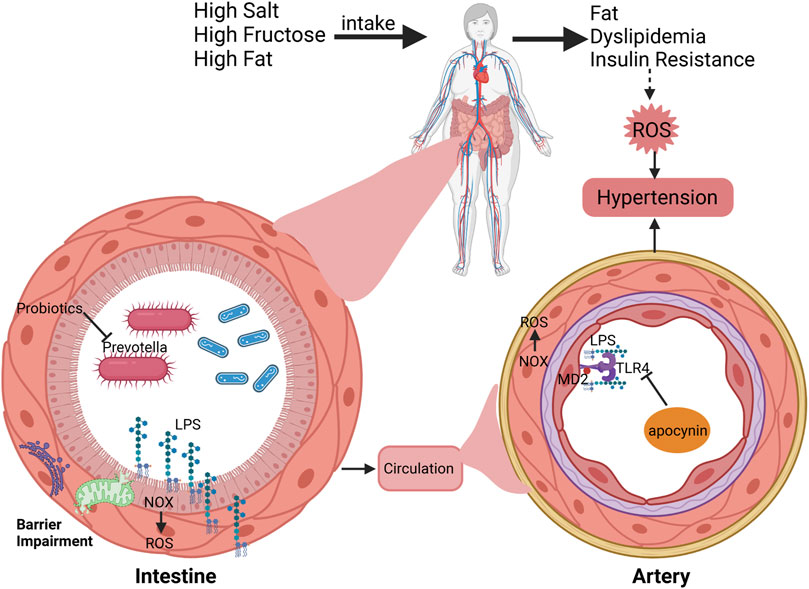
Figure 1. Interactions between hypertension, dietary factors, gut microbiome, and NOX pathways. Daily intake of high levels of salt, fructose, and fat could cause obesity, dyslipidemia, insulin resistance and hypertension. In particular, changes in daily diet induce dysfunction of gut microbiome and NOX-mediated ROS production, resulting in barrier damage and chronic LPS translocation to the circulation. And LPS also acts on the arterial endothelium through the TLR4/MD2 complex, promoting oxidative stress to cause hypertension.
Long-term intake of a high-fat, high-sugar diet increases vascular wall oxidative stress through enteral (gut microbiota) and extrenteral pathways, causing vascular remodeling and hypertension. NOX4 may play a vascular protective role due to the production of H2O2 and NO. At the same time, H2O2, as a second messenger, may also participate in downstream signaling pathways, but further confirmation is still needed.
4.3 Heart failure
Heart failure (HF) is a decompensated manifestation of various CVD, seriously threatening the health and quality of life of patients (Ziaeian and Fonarow, 2016). The imbalance between oxidative stress and endogenous antioxidant systems accelerates the pathogenesis of heart failure (van der Pol et al., 2019; Zhang et al., 2020). A failing heart exhibits significant changes in energy metabolism, accompanied by reduced ATP synthesis, and shows a preference for ketone body and lactate metabolism over FAO (Lopaschuk et al., 2021; Montaigne et al., 2021). Myocardial metabolomics reveal that amino acids (especially BCAAs), pyruvate, and ketone bodies are increased in patients with heart failure, acting as supplementary substrates in the TCA cycle to maintain energy homeostasis (Hahn et al., 2023). Mitochondrial oxidative phosphorylation is essential for ATP production, and functional impairment in this process is frequently accompanied by the production of superoxide anions, especially in the energy-intensive heart. Therefore, exploring the crosstalk between oxidative stress mediated by NOX and metabolism remodeling in heart failure is crucial. Multiple studies have revealed that NOX2, NOX4, and NOX5 increase ROS production, promoting cardiac pathological remodeling and dysfunction (Lozhkin et al., 2022; Parajuli et al., 2014; Zhao et al., 2020). Specifically, knockdown of NOX2 improves oxidative stress, myocardial fibrosis, and remodeling in the transverse aortic constriction (TAC) model, while also inhibiting MAPK activation (Parajuli et al., 2014). ROS have been demonstrated to activate MAPK, inducing the differentiation of fibroblasts into myofibroblasts and the production of inflammatory factors (Romero-Becerra et al., 2020). Moreover, NOX4 promotes the differentiation of fibroblasts into myofibroblasts through the TGF-β/SMAD pathway (Cucoranu et al., 2005). Overexpression of NOX4 in mitochondria leads to mitochondrial dysfunction and fission, causing cardiac dysfunction mediated by oxidative stress. The possible mechanism involves a significant decrease in the activity of mitochondrial citrate synthase and complex I, leading to oxidative phosphorylation decoupling (Lozhkin et al., 2022). Interestingly, the increased expression of NOX4 in heart failure may be a compensatory effect. Recent studies have shown that NOX4 induces the expression and release of Hypoxia-inducible factor 1 alpha (HIF-1α) and vascular endothelial growth factor (VEGF), increasing myocardial capillary density to improve cardiac function (Zhang et al., 2010). NOX5, a calcium-sensitive subtype, has also been demonstrated to promote myocardial hypertrophy and systolic dysfunction (Zhao et al., 2020).
Glucose-6-phosphate dehydrogenase (G6PD), the first regulatory enzyme in the PPP, is significantly upregulated in patients with HF, resulting in increased superoxide production, which can be suppressed by inhibitors of NOX and G6PD (Gupte et al., 2007). This phenomenon has been further demonstrated in HF models (Gupte et al., 2006). Previous studies have shown that glycolysis is upregulated in HF, potentially increasing the utilization of glycolytic intermediates and glucose to enhance PPP activity and induce ROS production (Bertero and Maack, 2018). Insulin resistance in ischemic heart failure activates NOX, which can be alleviated by enhancing Akt phosphorylation and GLUT4 translocation using apocynin (Fukushima et al., 2016; Ohta et al., 2011). However, the above studies did not identify specific types of NOX. Most studies have shown that NOX2 can produce oxidative stress that mediates insulin resistance (Souto Padron de Figueiredo et al., 2015; Zhang et al., 2014). Interestingly, NOX2 deficiency reduced the improvement of exercise training on diet-induced insulin resistance and obesity (Henriquez-Olguin et al., 2023). To some extent, NOX4 is universally recognized to improve insulin resistance due to producing H₂O₂, which activates insulin receptor kinase and inhibits phosphatase (Lennicke and Cochemé, 2021b; Li et al., 2012; Plecitá-Hlavatá et al., 2020). However, NOX4 has also been demonstrated to cause insulin resistance and inflammation in adipose tissue (Den Hartigh et al., 2017). Although there is substantial evidence that NOX4 can increase insulin signaling, further validation is needed. Increased glycolysis is not sufficient to compensate for inadequate energy metabolism in heart failure, whereas NOX4 may improve metabolic remodeling in pathological states to improve heart function (Lopaschuk et al., 2021). AMP-activated protein kinase (AMPK), a key energy sensor regulating glucose and lipid metabolism, is influenced by oxidative stress primarily derived from NOX to maintain cellular homeostasis (Song and Zou, 2012). Trimetazidine improves insulin resistance and mitochondrial function through AMPK activation, protecting against TAC-induced HF (Shu et al., 2021). Further studies reveal that the AMPKα2 subtype restores impaired mitophagy by phosphorylating PINK1 at Ser495, thereby enhancing mitochondrial function and reducing oxidative stress to mitigate HF progression (Wang B. et al., 2018). Therefore, AMPK activators such as metformin and statins can improve impaired heart function by reducing NOX activity and maintaining mitochondrial and energy metabolism homeostasis (Song and Zou, 2012). Mineralocorticoid receptor (MR) antagonists, including spironolactone and eplerenone, are fundamental drugs in HF management, as they partly inhibit NOX activity to reduce oxidative stress (Nagata et al., 2006). MR activation by small GTPase Rac1 increases NOX4 expression in TAC-induced HF (Ayuzawa et al., 2016). Sodium-glucose cotransporter 2 (SGLT2) inhibitors, which are new therapeutic drugs for HF, enhance the AMPK/Rac1 pathway to inhibit NOX activity and increase tetrahydrobiopterin bioavailability through additional SGLT1 inhibition effects (Heidenreich et al., 2022; Kondo et al., 2021). Moreover, ATF4 targets several enzymes in the PPP and one-carbon metabolic pathways to maintain redox homeostasis in the heart, findings validated by metabolomic and transcriptomic analyses (Wang X. et al., 2022) (Figure 2).
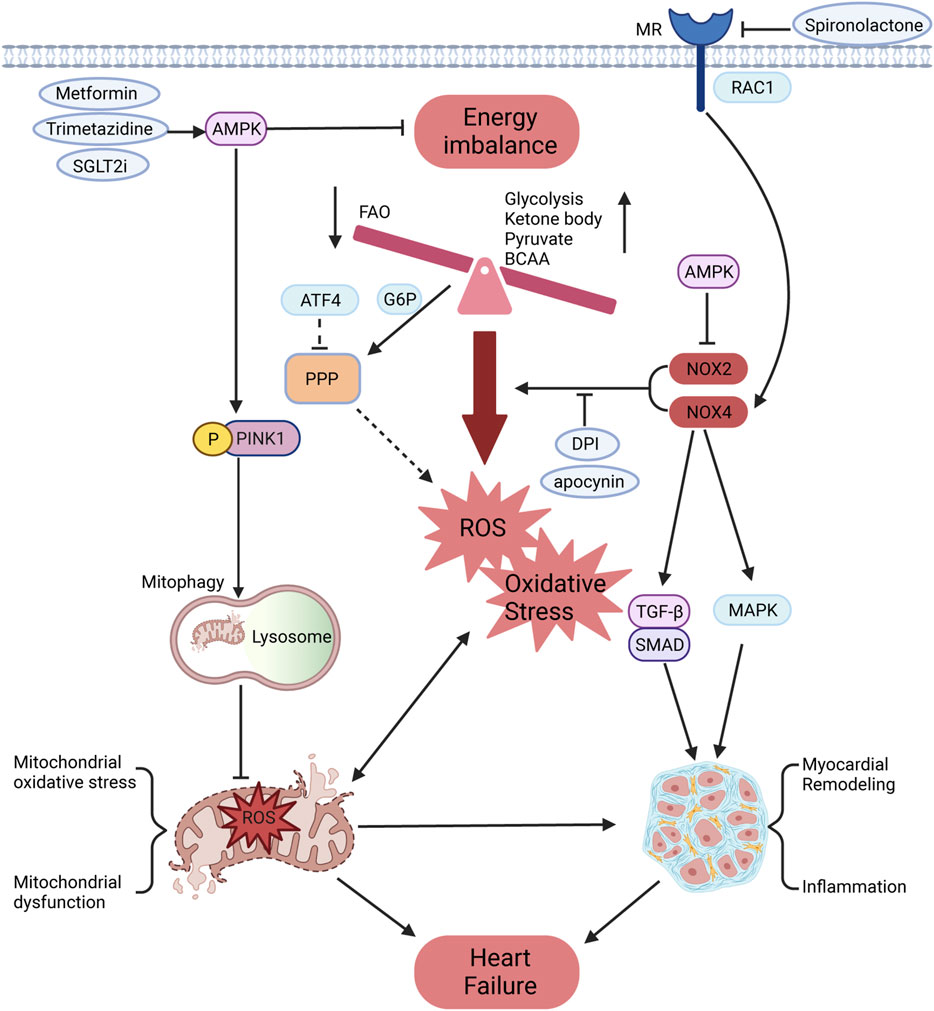
Figure 2. Effects of NOX on energy metabolism and oxidative stress in HF. The cardiac energy metabolism of HF is manifested as reduced FAO, increased metabolism of glucose, ketone bodies, pyruvate, BCAAs, and other substrates, which is accompanied by oxidative phosphorylation imbalance, leading to ROS production and oxidative stress. The PPP, a glycometabolic bypass, is also enhanced in HF and might provide more oxidizing substrates for NOX to promote ROS production. Meanwhile, NOX could promote the conversion of fibroblast into myofibroblast and increase the expression of inflammatory factors through TGF-β/SMAD and MAPK pathways, leading to myocardial remodeling. The mineralocorticoid receptor can be activated by RAC1 to promote NOX4 expression, which is partially inhibited by mineralocorticoid receptor antagonists. As the core regulator of energy homeostasis, AMPK can inhibit NOX activity and phosphorylate PINK1 to restore damaged mitophagy. This process improves mitochondrial function and reduces oxidative stress, thereby preventing the progression of HF.
4.4 Myocardial infarction
Myocardial infarction (MI) is a severe event characterized by a sudden decrease in blood supply and energy deprivation, often accompanied by metabolic remodeling and oxidative stress (Li et al., 2023; Liu et al., 2022; Zhang et al., 2020). Metabolomic studies have shown significant changes in glucose, amino acid, and ketone metabolism in peripheral blood and tissues during MI (Bai et al., 2020; Liu et al., 2022). The role of NOX1 in accelerating vascular disease progression has been extensively studied, but its specific role in the heart remains a topic of ongoing investigation. In MI mice subjected to ischemia-reperfusion, knockdown of NOX1/NOX2 decreases the size of MI, although oxidative stress levels remain unchanged in NOX1-deficient mice (Braunersreuther et al., 2013). However, inhibition of NOX1 does not improve heart function in MI mice, and late ischemic preconditioning actually increases MI size and apoptosis, potentially by blocking protective effects mediated by NF-kB activation (Jiang et al., 2014). Knockdown of NOX2 or p47phox improves systolic and diastolic function in MI models, accompanied by reduced fibrosis and apoptosis (Doerries et al., 2007; Looi et al., 2008). Conversely, specific overexpression of NOX2 in cardiomyocytes or endothelium does not deteriorate systolic and diastolic function, although cardiomyocyte overexpression led to increased fibrosis and myocardial hypertrophy (Sirker et al., 2016). NOX2-specific siRNA and microRNA oligonucleotide particles containing miR-106b, miR-148b, and miR-204 have been designed to reduce oxidative stress and improve heart function through intramyocardial injection (Somasuntharam et al., 2013; Yang et al., 2017). In contrast to NOX2, cardiomyocyte overexpression of NOX4 promotes M2 macrophage polarization, improving myocardial remodeling and survival in MI mice, while also increasing autophagy to cope with energy stress (Mongue-Din et al., 2017; Sciarretta et al., 2013). However, inhibition of NOX4 reduces MI size, accompanied by decreased oxidative stress and inflammatory infiltration (Stevenson et al., 2019). Increased NOX4 expression in the paraventricular nucleus impairs heart function through oxidative stress-mediated activation of sympathetic nerves in MI mice, an effect that is mitigated by NOX4 inhibition in the paraventricular nucleus (Infanger et al., 2010). The roles of NOX1 and NOX4 in MI demonstrate contradictory effects that warrant further investigation (Hahn et al., 2012). There are fewer studies showing the associations between NOX5 and MI, which could cause endothelium dysfunction by regulating calcium or oxidase stress to exacerbate disease progression (Marqués et al., 2022; Zhao et al., 2020).
Previous studies have revealed that metabolic dysfunctions in glycolysis, FAO, ketone bodies, and BCAAs promote the progression of MI (Li et al., 2023). Under hypoxic conditions, cardiomyocytes preferentially utilize glycolysis for ATP production, increasing GLUT1/4 expression and enhancing the affinity of phosphofructokinase 1 for substrates, a process promoted by AMPK (Li et al., 2023; Sosa et al., 2007). Multiple studies have demonstrated that H₂O₂ produced by NOX4 promotes insulin signaling, which probably promotes metabolic balance (Lennicke and Cochemé, 2021b; Plecitá-Hlavatá et al., 2020). Metformin, an AMPK activator, has been shown to reduce oxidative stress mediated by NOX4 (Sosa et al., 2007). Overexpression of NOX4 in cardiomyocytes reduces glycolysis and promotes FAO by increasing O-linked N-acetylglucosamine (O-GlcNAcylation) and CD36 combination, regulated by ATF4 via the hexosamine biosynthetic pathway (Nabeebaccus et al., 2017). NOX4 also protects cell survival during MI through the eIF2α/ATF4 pathway, driving pro-survival and metabolic transcriptional programs including autophagy and amino acid metabolism (Nabeebaccus et al., 2023). HIF-1α translocates into the nucleus under hypoxic conditions to induce protective pathways such as increased glycolysis, enhanced lactate transportation, and stabilization of cellular homeostasis, which includes autophagy, mitophagy, and management of oxidative stress (Ong and Hausenloy, 2012; Sciarretta et al., 2013). Notably, NOX2/4 may serve as upstream and downstream regulators of HIF-1α, playing pivotal roles in oxidative stress during myocardial energy metabolism (Nabeebaccus et al., 2023). Exogenous fat supplementation can exacerbate oxidative stress during MI (Liu and Lloyd, 2013). The peroxisome proliferator-activated receptor (PPAR) family, including PPARα, PPARγ, and PPARδ, are crucial regulators of lipid metabolism in CVD (Montaigne et al., 2021). The role of PPARα in MI is still controversial. PPARα possibly improves insulin resistant and oxidative stress, but overexpression of PPARα in cardiomyocytes causes glycogen deposition, macrophage infiltration, antioxidant system imbalance, and heart function deterioration (Duerr et al., 2014; Ibarra-Lara et al., 2016; Wagner and Wagner, 2020). Despite multicenter randomized controlled trials showing that the PPARα agonist pemafibrate does not reduce cardiovascular events in patients with type 2 diabetes or dyslipidemia, its efficacy remains uncertain (Das Pradhan et al., 2022). Several studies have demonstrated that PPARγ is upregulated in MI and exerts protective effects. Increased myocardial inflammatory infiltration and NOX2/4 exacerbate heart function following PPARγ knockdown in myeloid cells (Shen et al., 2018; Wagner and Wagner, 2020). Similar to NOX4, the PPARγ agonist pioglitazone reduces macrophage infiltration and promotes macrophage conversion to the M2 phenotype, improving myocardial remodeling (Mongue-Din et al., 2017; Tokutome et al., 2019) (Figure 3).
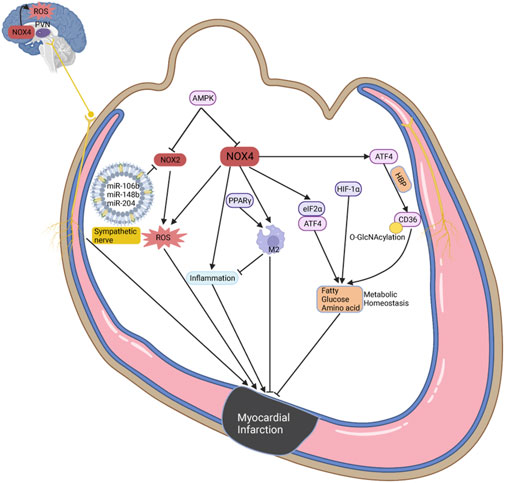
Figure 3. The potential roles of NOX in oxidative stress and metabolic homeostasis during MI. The most common function of NOX2/4 is to produce ROS and oxidative stress, which can be inhibited by AMPK. In the paraventricular nucleus, NOX promotes ROS production and activates sympathetic nerve to deteriorate cardiac function. And NOX4 can increase inflammatory infiltration, but is also recognized to promote macrophage conversion to M2 phenotype, and improves myocardial remodeling. Interestingly, NOX4 maintains metabolic homeostasis during MI through ATF4, eIF2α, and HIF-1α, including but not limited to fatty acid, glucose, and amino acid metabolism.
4.5 Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized by ventricular hypertrophy and abnormal load with unknown etiology, often presenting with myocardial fibrosis, cardiomyocyte hypertrophy, and disordered arrangement (Previs et al., 2022; Teekakirikul et al., 2019). Several proteomics and metabolomics studies reveal that the imbalance of energy metabolism in HCM not only involves decreased FAO, glycolysis and intermediates of TCA, but also increases the utilization of ketone body, lactate and BCAAs, eventually causing impaired ATP production. Oxidative stress, mitochondrial dysfunction and impaired mitophagy are also demonstrated in HCM, which mutually exacerbate metabolic disorder (Coats et al., 2018; Previs et al., 2022; Ranjbarvaziri et al., 2021). NOX inhibitor apocynin, mitochondria-targeted antioxidant SS-31, and mitoquidone are also proven to alleviate myocardial dysfunction, remodeling and fibrosis mediated by oxidative stress and mitochondrial dysfunction (Dai et al., 2011; Goh et al., 2019; Saleem et al., 2018). Mice with specific overexpression of NOX1 in VSMC develop vascular hypertrophy, which is associated with oxidative stress and can be prevented by the antioxidant tempol (Dikalova et al., 2005). In TAC-induced HCM model, NOX2 has been shown to promote oxidative stress, cardiac dysfunction, and fibrosis (Grieve et al., 2006; Parajuli et al., 2014). The role of NOX4 in HCM remains controversial, as studies have suggested both protective and detrimental effects. Similar to NOX2, NOX4 is generally recognized to enhance oxidative stress and contribute to cardiac dysfunction in HCM, primarily through pathways involving mTOR and NF-kB (Ago et al., 2010; Zhao et al., 2015). NOX4 siRNA delivered via small extracellular vesicles targeting the heart has shown potential in mitigating cardiac dysfunction and fibrosis in Ang-II-induced HCM models (Kang et al., 2023). Conversely, previous studies indicate that increased cardiac capillary density in NOX4-overexpressing HCM may provide protection against heart dysfunction and facilitate adaptation to chronic stress, possibly regulated by the HIF-1α/VEGF axis (Zhang et al., 2010). Specific knockdown of NOX4 in cardiomyocytes and endothelial cells has been shown to exacerbate myocardial dysfunction and remodeling in TAC models (Zhang et al., 2018). Additionally, NOX5 has been implicated in inducing heart dysfunction and fibrosis in HCM through oxidative stress mechanisms (Zhao et al., 2020).
As described above, stress-overloaded hearts exhibit decreased FAO and glycolysis, accompanied by mitochondrial dysfunction and oxidative stress, leading to disturbances in energy supply. The balance between oxidative phosphorylation and oxidative stress is critical for maintaining cellular homeostasis. The contradictory role of NOX4 in HCM may be linked to simultaneous increases in FAO and oxidative stress, which can respectively be protective and harmful. Recent studies have shown that in hearts overexpressing NOX4, FAO is upregulated, whereas glucose oxidation is reduced, despite no significant change in glucose uptake. This metabolic shift is facilitated through the activation of the HBP, which enhances FAO by increasing CD36 expression (Nabeebaccus et al., 2017). PPP is also recognized as a glucose utilization branch and is enhanced by increased G6PD activity, leading to elavated superoxide production, which may have adverse effects in NOX4 overexpression (Gupte et al., 2007). Key regulators such as the PPAR family, NRF2, and AMPK play pivotal roles in energy metabolism and redox homeostasis under various stress conditions, including in HCM (Hayes and Dinkova-Kostova, 2014; Li et al., 2023; Nabeebaccus et al., 2023). Activators of PPARα and PPARγ have been shown to reduce the levels of p22phox and p47phox in endothelial cells, thereby aiding in redox balance (Inoue et al., 2001). Conversely, NOX2-mediated oxidative stress downregulates PPARα expression, exacerbating heart dysfunction in HCM (Harvey et al., 2020). The interplay between NOX enzymes and the PPAR family involves complex redox and metabolic crosstalk, which requires further investigation. Chronic oxidative stress mediated by NRF2 can induce myocardial remodeling, mitochondrial dysfunction, and caspase 3-independent cell death, ultimately leading to HCM and heart failure (Shanmugam et al., 2020; Smyrnias et al., 2015). Interestingly, NRF2 activation by endogenous NOX4 has shown a protective role in HCM, independent of increased capillary density (Smyrnias et al., 2015) (Figure 4).
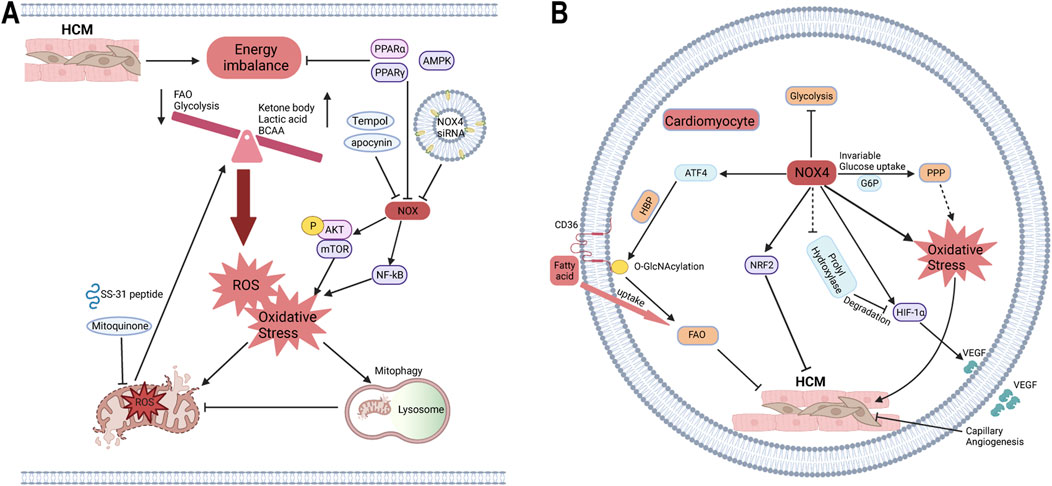
Figure 4. The roles of NOX in HCM in the context of energy metabolism and oxidative stress. (A) The crosstalk of NOX on energy metabolism and oxidative stress. The cardiac energy metabolism of HCM is manifested as decreased FAO and glycolysis, increased metabolism of ketone bodies, pyruvate, BCAAs, etc., accompanied by mitochondrial dysfunction and oxidative phosphorylation imbalance leading to ROS production and oxidative stress. Meanwhile, NOX can promote oxidative stress through p-AKT/mTOR and NF-kB pathways, and further induce mitochondrial oxidative stress and dysfunction, which are inhibited by apocynin, DPI, mitoquinone, mitochondria-targeted antioxidant SS-31, and NOX4 siRNA. PPARα, PPARγ, and AMPK can maintain energy homeostasis and inhibit NOX activity, which may be a bridge between energy metabolism and oxidative stress. (B) The paradoxical role of NOX4 in HCM. Traditionally, the most classic role of NOX4 is to produce oxidative stress. Overexpression of NOX4 does not change glucose uptake but inhibits glycosis, possibly providing oxidizing substrates via glycometabolic bypass PPP. And HBP, as a glycometabolic branch, increases the O-GlcNAcylation of CD36 to promote FAO. Meanwhile, NOX4 promotes myocardial capillary angiogenesis via HIF-1α/VEGF, possibly by inhibiting prolyl hydroxylase activity to reduce HIF-1α degradation. NOX4 also activates NRF2 to protect against HCM without capillary angiogenesis. FAO, fatty acid oxidation; PPP, pentose phosphate pathway; HBP, hexosamine biosynthesis pathway.
4.6 Diabetic cardiomyopathy
Diabetic cardiomyopathy (DCM) is a form of heart dysfunction that occurs in the context of diabetes, without the presence of coronary artery disease (CAD), hypertension, or valvular diseases. It is characterized by myocardial insulin resistance, mitochondrial dysfunction, and abnormal coronary microcirculation (Jia et al., 2016). Unlike MI and HCM, DCM is marked by increased FAO and decreased glycolysis, leading to lipid toxicity and oxidative stress. These processes disrupt cellular metabolism and contribute to mitochondrial dysfunction (Jia et al., 2016; Kenny and Abel, 2019; Tong et al., 2019). Both redox imbalance and metabolic disorders play roles in the pathogenesis of DCM, although the crosstalk between these factors remains enigmatic (Kenny and Abel, 2019). Specifically, oxidative stress mediated by NOX1 exacerbates heart dysfunction and fibrosis through the TLR2/NF-κB pathway, a process that can be ameliorated by the NOX1 inhibitor ML171 (Zhang et al., 2022). Diabetic patients often exhibit increased blood glucose and oxidative stress, which enhance O-GlcNAcylation of multiple proteins through HBP. This contributes to mitochondrial dysfunction, endoplasmic reticulum stress, and metabolic disturbances, including those related to fatty acids and amino acids (Zhang et al., 2022). Recent studies have revealed that hyperglycemia increases O-GlcNAcylation of CaMKII, activating NOX2 and inducing oxidative stress, which can be suppressed by specific inhibitors (Lu et al., 2020). Additionally, Rac1/NOX2-mediated oxidative stress has been shown to induce apoptosis and exacerbate dysfunction in DCM (Kowluru and Kowluru, 2014). In various DCM models, NOX4 is overactivated, promoting oxidative stress and fibrosis through the TGF-β pathway and matrix metalloproteinases (Wang D. et al., 2023). Cardiac fibrosis and dysfunction are improved with the application of NOX4 antisense oligonucleotides in DCM (Maalouf et al., 2012). Furthermore, FOXO1 has been found to bind to the promoter of KLF5, thereby activating NOX4 and inducing oxidative stress, mitochondrial dysfunction, and impaired heart function (Kyriazis et al., 2021).
The development of DCM is closely linked to chronic hyperglycemia and insulin resistance, accompanied by systemic metabolic disorders, lipid accumulation, and advanced glycation end products. It is recognized that PPARα and PGC-1α are upregulated in early DCM but downregulated in advanced DCM, leading to reduced myocardial metabolic efficiency (Jia et al., 2016). Interestingly, the metabolic and pathological phenotypes observed in hearts with specific overexpression of PPARα are similar to those seen in DCM (Finck et al., 2002). Silencing PPARα in DCM has been shown to reduce NOX1 expression and oxidative stress, which in turn improves heart function (Wang L. et al., 2020). However, NOX-mediated oxidative stress increases when a PPARα activator is applied to macrophages, ultimately leading to ox-LDL production (Teissier et al., 2004). This suggests that PPARα-induced oxidative stress may exacerbate heart function in DCM by increasing FAO and decreasing energy efficiency. In contrast, recent studies have highlighted the cardioprotective role of PPARγ, leading to the use of several agonists to treat diabetes and its complications. Obesity and diabetes are accompanied by elevated inflammatory factors, such as TNF-α, which promote oxidative stress and reduce PPARγ expression (Su et al., 2013). The cardioprotective effects of the PPARγ activator rosiglitazone are likely due to its ability to reduce NOX4 expression (Guo et al., 2012). Additionally, rosiglitazone has been shown to activate AMPK, reducing oxidative stress mediated by NOX in endothelial cells exposed to high glucose levels (Ceolotto et al., 2007). Further research has revealed that silencing AMPK increases NOX2 expression, promoting apoptosis, pyroptosis, and ferroptosis in ischemia-reperfusion models under high glucose conditions (Wang C. et al., 2020). The glucagon-like peptide 1 (GLP-1) receptor agonist also protects the heart from high glucose toxicity by inhibiting p47phox translocation to the plasma membrane (Balteau et al., 2014).
The PPAR family and AMPK are key regulators of glycolipid and energy metabolism in DCM and play key roles in mitigating NOX-mediated oxidative stress. In HF, MI, and HCM, NOX4 has been recognized to maintain metabolic balance including glucose and lipid metabolism. However, NOX4 is currently only known to worsen DCM. Considering the metabolic characteristics of DCM, the role of NOX4 should not be summarized by producing oxidative stress alone, and further research is needed to demonstrate its regulation of metabolic function.
5 The crosstalk between NOX, mitochondrial dysfunction, and ferroptosis
Iron, an essential trace element, is present in the human body at an average amount of 3–4 g and participates in numerous processes, such as energy metabolism, essential for sustaining cell function. The redox properties of iron, including electron transfer, are crucial in forming Fe-sulfur clusters, hemoglobin, and other functional subunits (Ganz, 2013). Cardiomyocytes acquire iron through ferroportin (FPN), transferrin-bound iron via transferrin receptor 1 (TfR1), or non-transferrin-bound iron (NTBI) through various channels like L-type and T-type calcium channels, ZIP14 and DMT1 (Lakhal-Littleton et al., 2015). Both iron deficiency (ferritin <100 ng/mL or transferrin saturation <20%) and iron overload can damage cell function, underscoring the critical importance of maintaining intracellular iron homeostasis for cardiovascular health (Chung et al., 2023; Jankowska et al., 2013).
Mitochondrial dysfunction is regarded as a major contributor to intracellular oxidative stress, which significantly accelerates the progression of CVD (Ranjbarvaziri et al., 2021; Zhang et al., 2020). NOX-mediated oxidative stress induces mitochondrial dysfunction, myocardial remodeling, and cardiac dysfunction, which can be alleviated by NOX inhibitors and targeted mitochondrial antioxidants (Ago et al., 2010; Daiet al., 2011; Goh et al., 2019; Lozhkin et al., 2022). Ferroptosis, a form of programmed cell death regulated by lipid peroxidation and iron accumulation, is linked to disturbances in iron, lipid, and glutathione metabolism. Mitochondria, being iron-enriched organelles responsible for producing heme, Fe-S clusters, and ROS, exhibit morphological abnormalities and dysfunction, which contribute to ferroptosis (Chen et al., 2021; Fang et al., 2023). Additionally, NOX activation in CVD impedes the conversion of oxidized glutathione back to glutathione, reducing protection against lipid peroxidation damage (Fang et al., 2023). Excess ROS leads to mitochondrial DNA damage and a decrease in membrane potential, triggering lipid peroxidation, which enhances mitophagy in an effort to restore mitochondrial function and counteract ferroptosis (Su et al., 2023). However, excessive mitophagy releases abundant Fe2⁺ into the labile iron pool (LIP), inducing ferroptosis while increasing oxidative stress via the Fenton reaction and reducing ATP production (Yu et al., 2022). Previous studies have also shown that FAO decreases in CVD, leading to lipid accumulation and facilitating lipid peroxidation (Bertero and Maack, 2018; Chen et al., 2021; Previs et al., 2022; Ranjbarvaziri et al., 2021). Therefore, oxidative stress mediated by NOX not only diminishes protection against ferroptosis but also facilitates ferroptosis by inducing mitochondrial dysfunction and lipid peroxidation. Specifically, NOX2 has been shown to reduce the expression of the ferroptosis suppressor GPX4 in H9C2 cells exposed to high glucose and hypoxia/reoxygenation (Wang et al., 2020a). NOX4 impairs mitochondrial respiratory chain complexes I-V, leading to increased oxidative stress, mitochondrial fragmentation, and dysfunction, which in turn promotes iron accumulation and lipid peroxidation (Park et al., 2021). A recent study demonstrates that inhibiting O-GlcNAcylation leads to Fe2⁺ accumulation in mitochondria and increases mitophagy, while the loss of O-GlcNAcylation on ferritin heavy chain (FTH) increases interaction with nuclear receptor coactivator 4 (NCOA4), expanding the LIP and accelerating ferroptosis (Yu et al., 2022). Our research group has previously demonstrated that NCOA4-mediated ferritinophagy promote ferroptosis and myocardial fibrosis in DCM (Zhang et al., 2024). Interestingly, NOX4 has been shown to promote O-GlcNAcylation of CD36, increasing fatty acid uptake and providing novel insights into the regulation of mitochondrial dysfunction and ferroptosis (Nabeebaccus et al., 2017) (Figure 5).
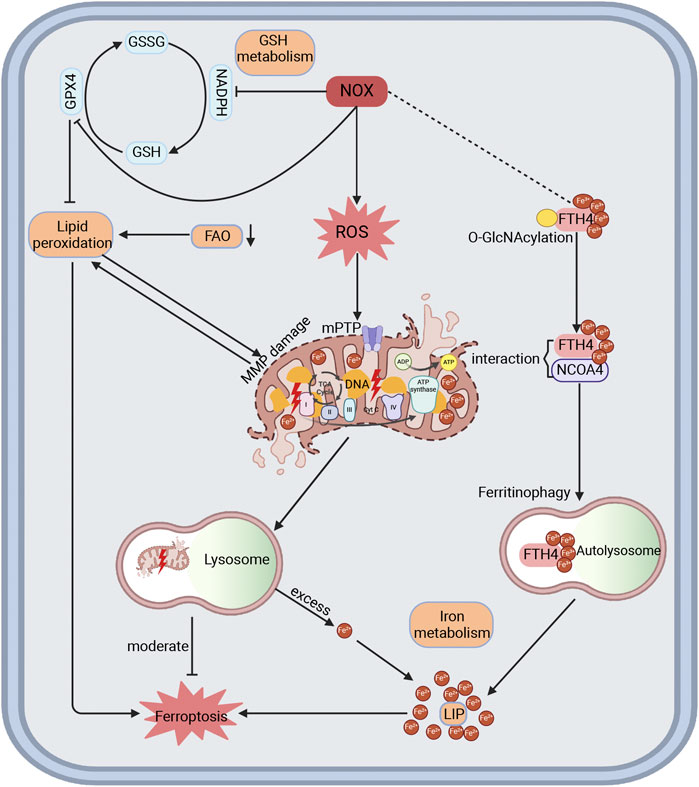
Figure 5. The interactions among NOX, mitochondria dysfunction, and ferroptosis in CVD. NOX is recognized to produce ROS and oxidative stress, leading to impaired membrane potential, DNA damage, electron transport chain damage, and lipid peroxidation of mitochondria, thereby increasing mitophagy in an attempt to restore mitochondrial function and mitigate ferroptosis in CVD. Excessive mitophagy can release Fe2+ into cytoplasm to form a labile iron pool and promote ferroptosis. Loss of O-GlcNAcylation in FTH could enhance its interaction with NCOA4, which induces ferritinophagy and expands the labile iron pool, thereby increasing ferroptosis. Here, NOX4 might affect the O-GlcNAcylation of target proteins to modulate ferroptosis. Moreover, NOX could also block GSH metabolism by consuming NADPH and inhibiting GPX4, thereby promoting lipid peroxidation and mitochondrial damage, which reduces protection against ferroptosis. Reduced FAO is demonstrated in HF, HCM, and ICM in proteomics, promoting lipid peroxidation and ferroptosis. FAO, fatty acid oxidation.
6 NOX inhibitor
Current research on NOX inhibitors encompasses non-selective NOX inhibitors, selective NOX inhibitors, and specific peptide-based inhibitors. Among them, apocynin and diphenyleneiodonium chloride (DPI) are the most widely used non-selective NOX inhibitors, with off-target effects that broadly eliminated the production of ROS (Savla et al., 2021; Zhang et al., 2020). They are demonstrated to reduce oxidative stress mediated by NOX in MI, HCM, and DCM models (Cheng et al., 2021; Li et al., 2010; Saleem et al., 2018). Further advancements include non-selective NOX inhibitors such as VAS2870, VAS3947, ML171, and APX-115, which have shown efficacy in reducing oxidative stress mediated by NOX enzymes (Cha et al., 2017; Zhang et al., 2020). Apocynin, for example, has been shown to inhibit ROS production, potentially altering the protective effects of exercise during cardiac ischemia-reperfusion (Frasier et al., 2013). DPI, on the other hand, inhibits NOX4, which plays a role in promoting neovascularization through VEGF, impacting cardiac microcirculation and energy metabolism (Vogel et al., 2015). The dual nature of NOX4 in both protective and harmful roles in heart function underscores the need for selective NOX inhibitors. GKT137831, a selective NOX1/4 inhibitor, has demonstrated efficacy in reducing inflammatory macrophage infiltration, mitochondrial dysfunction, and cardiac remodeling in stress-induced cardiomyopathy and HCM models (Lozhkin et al., 2022; Vendrov et al., 2023; Zhao et al., 2015). Another dual inhibitor, GKT136901 (NOX1/4), has shown promise in cardiovascular diseases (Zhang et al., 2020). GSK2795039, the first NOX2 inhibitor, enhances macrophage efferocytosis and reduces apoptosis and 4-HNE production, potentially preventing plaque rupture in atherosclerosis (Wang Y. et al., 2023; Zhang et al., 2020). Similar to heart, the brain is an energy-sensitive organ that easily sufferes from ROS. NCATS-SM7270 is designed according to GSK2795039 to improve neuronal survival by suppressing NOX2. Interestingly, NOX4 knockdown in mild traumatic brain injury exacerbates cell death, which is partly reversed by NCATS-SM7270 (Mason et al., 2023). Selective NOX4 inhibitors such as GLX7013114 and GLX351322 have been identified for their protective effects against high glucose and palmitate-induced damage to islet cells (Anvari et al., 2015; Wang X. et al., 2018). Given the enigmatic role of NOX4 in CVD, the application of selective NOX4 inhibitors requires further validation. Meanwhile, peptide inhibitors targeting NOX1/2 have been designed and confirmed their effects (Altenhöfer et al., 2015) or instance, NoxA1ds has been shown to effectively reduce ROS production by suppressing NOX1 in models such as SHR (Camargo et al., 2018). Additionally, intranasal administration of NOX2ds-tat over 7 days has demonstrated improvement in trauma-associated olfactory deficits by inhibiting NOX2, a major contributor to oxidative stress in cardiovascular diseases (Liu et al., 2023).
7 Conclusion
During prolonged dietary alterations, systemic metabolic diseases, and pathological cardiac conditions, the limited metabolic reserve of the heart necessitates a metabolic substrate shift. This shift is often accompanied by increased oxidative stress, energy metabolism disorders, and the progression of CVD. This paper investigates the potential mechanisms by which NOX regulates energy metabolism, with a particular focus on the role of NOX4. In addition to its functions similar to the other NOX, NOX4 is also believed to regulate glucose, fatty acid and amino acid metabolism, thereby improving myocardial remodeling under stress challenges. Moreover, this study also preliminarily explores the mechanisms mediated by NOX to promote ferroptosis. NOX4 may regulate ferroptosis progression through the O-GlcNAcylation of proteins, potentially linking NOX4-mediated metabolic disorders with ferroptosis. Further investigation is warranted to substantiate these mechanisms.
Author contributions
HJ: Conceptualization, Data curation, Investigation, Methodology, Visualization, Writing–original draft. JZ: Conceptualization, Data curation, Investigation, Methodology, Visualization, Writing–original draft. SW: Conceptualization, Data curation, Investigation, Visualization, Writing–original draft. LY: Conceptualization, Investigation, Writing–original draft. SL: Conceptualization, Investigation, Writing–original draft. BD: Conceptualization, Funding acquisition, Investigation, Methodology, Supervision, Visualization, Writing–original draft, Writing–review and editing. FY: Conceptualization, Funding acquisition, Investigation, Methodology, Resources, Software, Supervision, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. 81601721, 82070382, and 82371574); Natural Science Foundation of Shandong Province (No. ZR2023MH306) and the Program of Taishan Scholars Programme (No. ts 20190979).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aerni-Flessner, L., Abi-Jaoude, M., Koenig, A., Payne, M., and Hruz, P. W. (2012). GLUT4, GLUT1, and GLUT8 are the dominant GLUT transcripts expressed in the murine left ventricle. Cardiovasc Diabetol. 11, 63. doi:10.1186/1475-2840-11-63
Ago, T., Kuroda, J., Pain, J., Fu, C., Li, H., and Sadoshima, J. (2010). Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 106 (7), 1253–1264. doi:10.1161/CIRCRESAHA.109.213116
Altenhöfer, S., Radermacher, K. A., Kleikers, P. W., Wingler, K., and Schmidt, H. H. (2015). Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid. Redox Signal 23 (5), 406–427. doi:10.1089/ars.2013.5814
Anvari, E., Wikström, P., Walum, E., and Welsh, N. (2015). The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice. Free Radic. Res. 49 (11), 1308–1318. doi:10.3109/10715762.2015.1067697
Ayuzawa, N., Nagase, M., Ueda, K., Nishimoto, M., Kawarazaki, W., Marumo, T., et al. (2016). Rac1-Mediated activation of mineralocorticoid receptor in pressure overload-induced cardiac injury. Hypertension 67 (1), 99–106. doi:10.1161/HYPERTENSIONAHA.115.06054
Bai, H., Sun, K., Wu, J. H., Zhong, Z. H., Xu, S. L., Zhang, H. R., et al. (2020). Proteomic and metabolomic characterization of cardiac tissue in acute myocardial ischemia injury rats. PLoS One 15 (5), e0231797. doi:10.1371/journal.pone.0231797
Balteau, M., Van Steenbergen, A., Timmermans, A. D., Dessy, C., Behets-Wydemans, G., Tajeddine, N., et al. (2014). AMPK activation by glucagon-like peptide-1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 307 (8), H1120–H1133. doi:10.1152/ajpheart.00210.2014
Bartelds, B., Knoester, H., Beaufort-Krol, G. C., Smid, G. B., Takens, J., Zijlstra, W. G., et al. (1999). Myocardial lactate metabolism in fetal and newborn lambs. Circulation 99 (14), 1892–1897. doi:10.1161/01.cir.99.14.1892
Begum, R., Thota, S., Abdulkadir, A., Kaur, G., Bagam, P., and Batra, S. (2022). NADPH oxidase family proteins: signaling dynamics to disease management. Cell Mol. Immunol. 19 (6), 660–686. doi:10.1038/s41423-022-00858-1
Benani, A., Troy, S., Carmona, M. C., Fioramonti, X., Lorsignol, A., Leloup, C., et al. (2007). Role for mitochondrial reactive oxygen species in brain lipid sensing: redox regulation of food intake. Diabetes 56 (1), 152–160. doi:10.2337/db06-0440
Bertero, E., and Maack, C. (2018). Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 15 (8), 457–470. doi:10.1038/s41569-018-0044-6
Bonen, A. (2000). Lactate transporters (MCT proteins) in heart and skeletal muscles. Med. Sci. Sports Exerc 32 (4), 778–789. doi:10.1097/00005768-200004000-00010
Braunersreuther, V., Montecucco, F., Asrih, M., Pelli, G., Galan, K., Frias, M., et al. (2013). Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J. Mol. Cell Cardiol. 64, 99–107. doi:10.1016/j.yjmcc.2013.09.007
Buchmann, G. K., Schürmann, C., Warwick, T., Schulz, M. H., Spaeth, M., Müller, O. J., et al. (2020). Deletion of NoxO1 limits atherosclerosis development in female mice. Redox Biol. 37, 101713. doi:10.1016/j.redox.2020.101713
Camargo, L. L., Harvey, A. P., Rios, F. J., Tsiropoulou, S., Da Silva, R. N. O., Cao, Z., et al. (2018). Vascular nox (NADPH oxidase) compartmentalization, protein hyperoxidation, and endoplasmic reticulum stress response in hypertension. Hypertension 72 (1), 235–246. doi:10.1161/HYPERTENSIONAHA.118.10824
Canugovi, C., Stevenson, M. D., Vendrov, A. E., Hayami, T., Robidoux, J., Xiao, H., et al. (2019). Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 26, 101288. doi:10.1016/j.redox.2019.101288
Ceolotto, G., Gallo, A., Papparella, I., Franco, L., Murphy, E., Iori, E., et al. (2007). Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 27 (12), 2627–2633. doi:10.1161/ATVBAHA.107.155762
Cha, J. J., Min, H. S., Kim, K. T., Kim, J. E., Ghee, J. Y., Kim, H. W., et al. (2017). APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Invest 97 (4), 419–431. doi:10.1038/labinvest.2017.2
Checa, J., and Aran, J. M. (2020). Reactive oxygen species: drivers of physiological and pathological processes. J. Inflamm. Res. 13, 1057–1073. doi:10.2147/JIR.S275595
Chen, X., Li, J., Kang, R., Klionsky, D. J., and Tang, D. (2021). Ferroptosis: machinery and regulation. Autophagy 17 (9), 2054–2081. doi:10.1080/15548627.2020.1810918
Cheng, W., Sun, Y., Wu, Q., Ooi, K., Feng, Y., Xia, C., et al. (2021). Paraventricular nucleus P2X7 receptors aggravate acute myocardial infarction injury via ROS-induced vasopressin-V1b activation in rats. Neurosci. Bull. 37 (5), 641–656. doi:10.1007/s12264-021-00641-8
Chung, B., Wang, Y., Thiel, M., Rostami, F., Rogoll, A., Hirsch, V. G., et al. (2023). Pre-emptive iron supplementation prevents myocardial iron deficiency and attenuates adverse remodelling after myocardial infarction. Cardiovasc Res. 119 (10), 1969–1980. doi:10.1093/cvr/cvad092
Coats, C. J., Heywood, W. E., Virasami, A., Ashrafi, N., Syrris, P., Dos Remedios, C., et al. (2018). Proteomic analysis of the myocardium in hypertrophic obstructive cardiomyopathy. Circ. Genom Precis. Med. 11 (12), e001974. doi:10.1161/CIRCGEN.117.001974
Craige, S. M., Kant, S., Reif, M., Chen, K., Pei, Y., Angoff, R., et al. (2015). Endothelial NADPH oxidase 4 protects ApoE-/- mice from atherosclerotic lesions. Free Radic. Biol. Med. 89, 1–7. doi:10.1016/j.freeradbiomed.2015.07.004
Cucoranu, I., Clempus, R., Dikalova, A., Phelan, P. J., Ariyan, S., Dikalov, S., et al. (2005). NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 97 (9), 900–907. doi:10.1161/01.RES.0000187457.24338.3D
Da Dalt, L., Cabodevilla, A. G., Goldberg, I. J., and Norata, G. D. (2023). Cardiac lipid metabolism, mitochondrial function, and heart failure. Cardiovasc Res. 119 (10), 1905–1914. doi:10.1093/cvr/cvad100
Dai, D. F., Chen, T., Szeto, H., Nieves-Cintrón, M., Kutyavin, V., Santana, L. F., et al. (2011). Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 58 (1), 73–82. doi:10.1016/j.jacc.2010.12.044
Das Pradhan, A., Glynn, R. J., Fruchart, J. C., MacFadyen, J. G., Zaharris, E. S., Everett, B. M., et al. (2022). Triglyceride lowering with pemafibrate to reduce cardiovascular risk. N. Engl. J. Med. 387 (21), 1923–1934. doi:10.1056/nejmoa2210645
de Koning, M. L. Y., Westenbrink, B. D., Assa, S., Garcia, E., Connelly, M. A., van Veldhuisen, D. J., et al. (2021). Association of circulating ketone bodies with functional outcomes after ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 78 (14), 1421–1432. doi:10.1016/j.jacc.2021.07.054
Den Hartigh, L. J., Omer, M., Goodspeed, L., Wang, S., Wietecha, T., O'Brien, K. D., et al. (2017). Adipocyte-specific deficiency of NADPH oxidase 4 delays the onset of insulin resistance and attenuates adipose tissue inflammation in obesity. Arterioscler. Thromb. Vasc. Biol. 37 (3), 466–475. doi:10.1161/ATVBAHA.116.308749
de Oliveira, A. A., Faustino, J., Webb, R. C., and Nunes, K. P. (2020). Blockade of the TLR4-MD2 complex lowers blood pressure and improves vascular function in a murine model of type 1 diabetes. Sci. Rep. 10 (1), 12032. doi:10.1038/s41598-020-68919-x
Dikalova, A., Clempus, R., Lassègue, B., Cheng, G., McCoy, J., Dikalov, S., et al. (2005). Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112 (17), 2668–2676. doi:10.1161/CIRCULATIONAHA.105.538934
Dimou, A., Tsimihodimos, V., and Bairaktari, E. (2022). The critical role of the branched chain amino acids (BCAAs) catabolism-regulating enzymes, branched-chain aminotransferase (BCAT) and branched-chain α-keto acid dehydrogenase (BCKD), in human pathophysiology. Int. J. Mol. Sci. 23 (7), 4022. doi:10.3390/ijms23074022
Di Raimondo, D., Buscemi, S., Musiari, G., Rizzo, G., Pirera, E., Corleo, D., et al. (2021). Ketogenic diet, physical activity, and hypertension-A narrative review. Nutrients 13 (8), 2567. doi:10.3390/nu13082567
Doerries, C., Grote, K., Hilfiker-Kleiner, D., Luchtefeld, M., Schaefer, A., Holland, S. M., et al. (2007). Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ. Res. 100 (6), 894–903. doi:10.1161/01.RES.0000261657.76299.ff
Drummond, G. R., Selemidis, S., Griendling, K. K., and Sobey, C. G. (2011). Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 10 (6), 453–471. doi:10.1038/nrd3403
Drummond, G. R., and Sobey, C. G. (2014). Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol. Metab. 25 (9), 452–463. doi:10.1016/j.tem.2014.06.012
Duerr, G. D., Heinemann, J. C., Arnoldi, V., Feisst, A., Kley, J., Ghanem, A., et al. (2014). Cardiomyocyte specific peroxisome proliferator-activated receptor-α overexpression leads to irreversible damage in ischemic murine heart. Life Sci. 102 (2), 88–97. doi:10.1016/j.lfs.2014.03.019
Fang, X., Ardehali, H., Min, J., and Wang, F. (2023). The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 20 (1), 7–23. doi:10.1038/s41569-022-00735-4
Finck, B. N., Lehman, J. J., Leone, T. C., Welch, M. J., Bennett, M. J., Kovacs, A., et al. (2002). The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Invest 109 (1), 121–130. doi:10.1172/JCI14080
Frasier, C. R., Moukdar, F., Patel, H. D., Sloan, R. C., Stewart, L. M., Alleman, R. J., et al. (2013). Redox-dependent increases in glutathione reductase and exercise preconditioning: role of NADPH oxidase and mitochondria. Cardiovasc Res. 98 (1), 47–55. doi:10.1093/cvr/cvt009
Fukushima, A., Kinugawa, S., Takada, S., Matsumoto, J., Furihata, T., Mizushima, W., et al. (2016). Direct renin inhibitor ameliorates insulin resistance by improving insulin signaling and oxidative stress in the skeletal muscle from post-infarct heart failure in mice. Eur. J. Pharmacol. 779, 147–156. doi:10.1016/j.ejphar.2016.03.022
Ganz, T. (2013). Systemic iron homeostasis. Physiol. Rev. 93 (4), 1721–1741. doi:10.1152/physrev.00008.2013
Genovesi, S., Giussani, M., Orlando, A., Orgiu, F., and Parati, G. (2021). Salt and sugar: two enemies of healthy blood pressure in children. Nutrients 13 (2), 697. doi:10.3390/nu13020697
Gibb, A. A., and Hill, B. G. (2018). Metabolic coordination of physiological and pathological cardiac remodeling. Circ. Res. 123 (1), 107–128. doi:10.1161/CIRCRESAHA.118.312017
Goh, K. Y., He, L., Song, J., Jinno, M., Rogers, A. J., Sethu, P., et al. (2019). Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biol. 21, 101100. doi:10.1016/j.redox.2019.101100
Griendling, K. K., Camargo, L. L., Rios, F. J., Alves-Lopes, R., Montezano, A. C., and Touyz, R. M. (2021). Oxidative stress and hypertension. Circ. Res. 128 (7), 993–1020. doi:10.1161/CIRCRESAHA.121.318063
Grieve, D. J., Byrne, J. A., Siva, A., Layland, J., Johar, S., Cave, A. C., et al. (2006). Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J. Am. Coll. Cardiol. 47 (4), 817–826. doi:10.1016/j.jacc.2005.09.051
Grylls, A., Seidler, K., and Neil, J. (2021). Link between microbiota and hypertension: focus on LPS/TLR4 pathway in endothelial dysfunction and vascular inflammation, and therapeutic implication of probiotics. Biomed. Pharmacother. 137, 111334. doi:10.1016/j.biopha.2021.111334
Gu, F., Krüger, A., Roggenkamp, H. G., Alpers, R., Lodygin, D., Jaquet, V., et al. (2021). Dual NADPH oxidases DUOX1 and DUOX2 synthesize NAADP and are necessary for Ca(2+) signaling during T cell activation. Sci. Signal 14 (709), eabe3800. doi:10.1126/scisignal.abe3800
Guo, Z., Qin, Z., Zhang, R., Li, J., and Yin, Y. (2012). Effect of rosiglitazone on the expression of cardiac adiponectin receptors and NADPH oxidase in type 2 diabetic rats. Eur. J. Pharmacol. 685 (1-3), 116–125. doi:10.1016/j.ejphar.2012.04.010
Gupte, R. S., Vijay, V., Marks, B., Levine, R. J., Sabbah, H. N., Wolin, M. S., et al. (2007). Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J. Card. Fail 13 (6), 497–506. doi:10.1016/j.cardfail.2007.04.003
Gupte, S. A., Levine, R. J., Gupte, R. S., Young, M. E., Lionetti, V., Labinskyy, V., et al. (2006). Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J. Mol. Cell Cardiol. 41 (2), 340–349. doi:10.1016/j.yjmcc.2006.05.003
Hahn, N. E., Meischl, C., Kawahara, T., Musters, R. J., Verhoef, V. M., van der Velden, J., et al. (2012). NOX5 expression is increased in intramyocardial blood vessels and cardiomyocytes after acute myocardial infarction in humans. Am. J. Pathol. 180 (6), 2222–2229. doi:10.1016/j.ajpath.2012.02.018
Hahn, V. S., Petucci, C., Kim, M. S., Bedi, K. C., Wang, H., Mishra, S., et al. (2023). Myocardial metabolomics of human heart failure with preserved ejection fraction. Circulation 147 (15), 1147–1161. doi:10.1161/CIRCULATIONAHA.122.061846
Harvey, A. P., Robinson, E., Edgar, K. S., McMullan, R., O'Neill, K. M., Alderdice, M., et al. (2020). Downregulation of PPARα during experimental left ventricular hypertrophy is critically dependent on Nox2 NADPH oxidase signalling. Int. J. Mol. Sci. 21 (12), 4406. doi:10.3390/ijms21124406
Hayes, J. D., and Dinkova-Kostova, A. T. (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39 (4), 199–218. doi:10.1016/j.tibs.2014.02.002
Heidenreich, P. A., Bozkurt, B., Aguilar, D., Allen, L. A., Byun, J. J., Colvin, M. M., et al. (2022). 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation 145 (18), e895–e1032. doi:10.1161/CIR.0000000000001063
Henriquez-Olguin, C., Meneses-Valdes, R., Raun, S. H., Gallero, S., Knudsen, J. R., Li, Z., et al. (2023). NOX2 deficiency exacerbates diet-induced obesity and impairs molecular training adaptations in skeletal muscle. Redox Biol. 65, 102842. doi:10.1016/j.redox.2023.102842
Holmström, K. M., and Finkel, T. (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15 (6), 411–421. doi:10.1038/nrm3801
Ibarra-Lara, L., Sánchez-Aguilar, M., Sánchez-Mendoza, A., Del Valle-Mondragón, L., Soria-Castro, E., Carreón-Torres, E., et al. (2016). Fenofibrate therapy restores antioxidant protection and improves myocardial insulin resistance in a rat model of metabolic syndrome and myocardial ischemia: the role of angiotensin II. Molecules 22 (1), 31. doi:10.3390/molecules22010031
Infanger, D. W., Cao, X., Butler, S. D., Burmeister, M. A., Zhou, Y., Stupinski, J. A., et al. (2010). Silencing nox4 in the paraventricular nucleus improves myocardial infarction-induced cardiac dysfunction by attenuating sympathoexcitation and periinfarct apoptosis. Circ. Res. 106 (11), 1763–1774. doi:10.1161/CIRCRESAHA.109.213025
Inoue, I., Goto, S., Matsunaga, T., Nakajima, T., Awata, T., Hokari, S., et al. (2001). The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+,Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism 50 (1), 3–11. doi:10.1053/meta.2001.19415
Jankowska, E. A., Malyszko, J., Ardehali, H., Koc-Zorawska, E., Banasiak, W., von Haehling, S., et al. (2013). Iron status in patients with chronic heart failure. Eur. Heart J. 34 (11), 827–834. doi:10.1093/eurheartj/ehs377
Jia, G., DeMarco, V. G., and Sowers, J. R. (2016). Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 12 (3), 144–153. doi:10.1038/nrendo.2015.216
Jiang, S., Streeter, J., Schickling, B. M., Zimmerman, K., Weiss, R. M., and Miller, F. J. (2014). Nox1 NADPH oxidase is necessary for late but not early myocardial ischaemic preconditioning. Cardiovasc Res. 102 (1), 79–87. doi:10.1093/cvr/cvu027
Kang, J. Y., Mun, D., Chun, Y., Park, D. S., Kim, H., Yun, N., et al. (2023). Engineered small extracellular vesicle-mediated NOX4 siRNA delivery for targeted therapy of cardiac hypertrophy. J. Extracell. Vesicles 12 (10), e12371. doi:10.1002/jev2.12371
Kenny, H. C., and Abel, E. D. (2019). Heart failure in type 2 diabetes mellitus. Circ. Res. 124 (1), 121–141. doi:10.1161/CIRCRESAHA.118.311371
Kim, S., Goel, R., Kumar, A., Qi, Y., Lobaton, G., Hosaka, K., et al. (2018). Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. (Lond) 132 (6), 701–718. doi:10.1042/CS20180087
Kolwicz, S. C., and Tian, R. (2011). Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 90 (2), 194–201. doi:10.1093/cvr/cvr071
Kondo, H., Akoumianakis, I., Badi, I., Akawi, N., Kotanidis, C. P., Polkinghorne, M., et al. (2021). Effects of canagliflozin on human myocardial redox signalling: clinical implications. Eur. Heart J. 42 (48), 4947–4960. doi:10.1093/eurheartj/ehab420
Kowluru, A., and Kowluru, R. A. (2014). Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem. Pharmacol. 88 (3), 275–283. doi:10.1016/j.bcp.2014.01.017
Kuwabara, M., Kanbay, M., Niwa, K., Ae, R., Andres-Hernando, A., Roncal-Jimenez, C. A., et al. (2020). Hyperosmolarity and increased serum sodium concentration are risks for developing hypertension regardless of salt intake: a five-year cohort study in Japan. Nutrients 12 (5), 1422. doi:10.3390/nu12051422
Kyriazis, I. D., Hoffman, M., Gaignebet, L., Lucchese, A. M., Markopoulou, E., Palioura, D., et al. (2021). KLF5 is induced by FOXO1 and causes oxidative stress and diabetic cardiomyopathy. Circ. Res. 128 (3), 335–357. doi:10.1161/CIRCRESAHA.120.316738
Lakhal-Littleton, S., Wolna, M., Carr, C. A., Miller, J. J., Christian, H. C., Ball, V., et al. (2015). Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. U. S. A. 112 (10), 3164–3169. doi:10.1073/pnas.1422373112
Lambeth, J. D. (2004). NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4 (3), 181–189. doi:10.1038/nri1312
Langbein, H., Brunssen, C., Hofmann, A., Cimalla, P., Brux, M., Bornstein, S. R., et al. (2016). NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur. Heart J. 37 (22), 1753–1761. doi:10.1093/eurheartj/ehv564
Lee, M. Y., San Martin, A., Mehta, P. K., Dikalova, A. E., Garrido, A. M., Datla, S. R., et al. (2009). Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 29 (4), 480–487. doi:10.1161/ATVBAHA.108.181925
Lennicke, C., and Cochemé, H. M. (2021a). Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 81 (18), 3691–3707. doi:10.1016/j.molcel.2021.08.018
Lennicke, C., and Cochemé, H. M. (2021b). Redox regulation of the insulin signalling pathway. Redox Biol. 42, 101964. doi:10.1016/j.redox.2021.101964
Li, J., Zhao, F., Wang, Y., Chen, J., Tao, J., Tian, G., et al. (2017). Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 5 (1), 14. doi:10.1186/s40168-016-0222-x
Li, J., Zhu, H., Shen, E., Wan, L., Arnold, J. M., and Peng, T. (2010). Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 59 (8), 2033–2042. doi:10.2337/db09-1800
Li, Q., Zhang, S., Yang, G., Wang, X., Liu, F., Li, Y., et al. (2023). Energy metabolism: a critical target of cardiovascular injury. Biomed. Pharmacother. 165, 115271. doi:10.1016/j.biopha.2023.115271
Li, Y., Mouche, S., Sajic, T., Veyrat-Durebex, C., Supale, R., Pierroz, D., et al. (2012). Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int. J. Obes. (Lond) 36 (12), 1503–1513. doi:10.1038/ijo.2011.279
Liang, C. F., Liu, J. T., Wang, Y., Xu, A., and Vanhoutte, P. M. (2013). Toll-like receptor 4 mutation protects obese mice against endothelial dysfunction by decreasing NADPH oxidase isoforms 1 and 4. Arterioscler. Thromb. Vasc. Biol. 33 (4), 777–784. doi:10.1161/ATVBAHA.112.301087
Lin, F. Y., Chen, Y. H., Tasi, J. S., Chen, J. W., Yang, T. L., Wang, H. J., et al. (2006). Endotoxin induces toll-like receptor 4 expression in vascular smooth muscle cells via NADPH oxidase activation and mitogen-activated protein kinase signaling pathways. Arterioscler. Thromb. Vasc. Biol. 26 (12), 2630–2637. doi:10.1161/01.ATV.0000247259.01257.b3
Liu, J., and Lloyd, S. G. (2013). High-fat, low-carbohydrate diet alters myocardial oxidative stress and impairs recovery of cardiac function after ischemia and reperfusion in obese rats. Nutr. Res. 33 (4), 311–321. doi:10.1016/j.nutres.2013.02.005
Liu, J., Meng, Z., Gan, L., Guo, R., Gao, J., Liu, C., et al. (2020). C1q/TNF-related protein 5 contributes to diabetic vascular endothelium dysfunction through promoting Nox-1 signaling. Redox Biol. 34, 101476. doi:10.1016/j.redox.2020.101476
Liu, W., Zhang, L., Shi, X., Shen, G., and Feng, J. (2022). Cross-comparative metabolomics reveal sex-age specific metabolic fingerprints and metabolic interactions in acute myocardial infarction. Free Radic. Biol. Med. 183, 25–34. doi:10.1016/j.freeradbiomed.2022.03.008
Liu, X., Lei, Z., Gilhooly, D., He, J., Li, Y., Ritzel, R. M., et al. (2023). Traumatic brain injury-induced inflammatory changes in the olfactory bulb disrupt neuronal networks leading to olfactory dysfunction. Brain Behav. Immun. 114, 22–45. doi:10.1016/j.bbi.2023.08.004
Looi, Y. H., Grieve, D. J., Siva, A., Walker, S. J., Anilkumar, N., Cave, A. C., et al. (2008). Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 51 (2), 319–325. doi:10.1161/HYPERTENSIONAHA.107.101980
Lopaschuk, G. D., Karwi, Q. G., Tian, R., Wende, A. R., and Abel, E. D. (2021). Cardiac energy metabolism in heart failure. Circ. Res. 128 (10), 1487–1513. doi:10.1161/CIRCRESAHA.121.318241
Lopaschuk, G. D., and Ussher, J. R. (2016). Evolving concepts of myocardial energy metabolism: more than just fats and carbohydrates. Circ. Res. 119 (11), 1173–1176. doi:10.1161/CIRCRESAHA.116.310078
Lozhkin, A., Vendrov, A. E., Ramos-Mondragón, R., Canugovi, C., Stevenson, M. D., Herron, T. J., et al. (2022). Mitochondrial oxidative stress contributes to diastolic dysfunction through impaired mitochondrial dynamics. Redox Biol. 57, 102474. doi:10.1016/j.redox.2022.102474
Lu, S., Liao, Z., Lu, X., Katschinski, D. M., Mercola, M., Chen, J., et al. (2020). Hyperglycemia acutely increases cytosolic reactive oxygen species via O-linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circ. Res. 126 (10), e80–e96. doi:10.1161/CIRCRESAHA.119.316288
Maalouf, R. M., Eid, A. A., Gorin, Y. C., Block, K., Escobar, G. P., Bailey, S., et al. (2012). Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am. J. Physiol. Cell Physiol. 302 (3), C597–C604. doi:10.1152/ajpcell.00331.2011
Manolis, A. S., Manolis, T. A., and Manolis, A. A. (2023). Ketone bodies and cardiovascular disease: an alternate fuel source to the rescue. Int. J. Mol. Sci. 24 (4), 15472. doi:10.3390/ijms242015472
Marqués, J., Fernández-Irigoyen, J., Ainzúa, E., Martínez-Azcona, M., Cortés, A., Roncal, C., et al. (2022). NADPH oxidase 5 (NOX5) overexpression promotes endothelial dysfunction via cell apoptosis, migration, and metabolic alterations in human brain microvascular endothelial cells (hCMEC/D3). Antioxidants (Basel) 11 (11), 2147. doi:10.3390/antiox11112147
Mason, H., Rai, G., Kozyr, A., De Jonge, N., Gliniewicz, E., Berg, L. J., et al. (2023). Development of an improved and specific inhibitor of NADPH oxidase 2 to treat traumatic brain injury. Redox Biol. 60, 102611. doi:10.1016/j.redox.2023.102611
Matsuno, K., Yamada, H., Iwata, K., Jin, D., Katsuyama, M., Matsuki, M., et al. (2005). Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112 (17), 2677–2685. doi:10.1161/CIRCULATIONAHA.105.573709
McGarrah, R. W., and White, P. J. (2023). Branched-chain amino acids in cardiovascular disease. Nat. Rev. Cardiol. 20 (2), 77–89. doi:10.1038/s41569-022-00760-3
Mongue-Din, H., Patel, A. S., Looi, Y. H., Grieve, D. J., Anilkumar, N., Sirker, A., et al. (2017). NADPH oxidase-4 driven cardiac macrophage polarization protects against myocardial infarction-induced remodeling. JACC Basic Transl. Sci. 2 (6), 688–698. doi:10.1016/j.jacbts.2017.06.006
Montaigne, D., Butruille, L., and Staels, B. (2021). PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 18 (12), 809–823. doi:10.1038/s41569-021-00569-6
Munir, L. Z., and du Toit, E. F. (2024). Impact of chronic psychological stress on cardiovascular disease risk: a narrative review. A Narrat. Rev. 8 (4), 268–278. doi:10.4103/hm.hm-d-24-00040
Murdoch, C. E., Alom-Ruiz, S. P., Wang, M., Zhang, M., Walker, S., Yu, B., et al. (2011). Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res. Cardiol. 106 (4), 527–538. doi:10.1007/s00395-011-0179-7
Nabeebaccus, A. A., Reumiller, C. M., Shen, J., Zoccarato, A., Santos, C. X. C., and Shah, A. M. (2023). The regulation of cardiac intermediary metabolism by NADPH oxidases. Cardiovasc Res. 118 (17), 3305–3319. doi:10.1093/cvr/cvac030
Nabeebaccus, A. A., Zoccarato, A., Hafstad, A. D., Santos, C. X., Aasum, E., Brewer, A. C., et al. (2017). Nox4 reprograms cardiac substrate metabolism via protein O-GlcNAcylation to enhance stress adaptation. JCI Insight 2 (24), e96184. doi:10.1172/jci.insight.96184
Nagata, K., Obata, K., Xu, J., Ichihara, S., Noda, A., Kimata, H., et al. (2006). Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low-aldosterone hypertensive rats. Hypertension 47 (4), 656–664. doi:10.1161/01.HYP.0000203772.78696.67
Ohta, Y., Kinugawa, S., Matsushima, S., Ono, T., Sobirin, M. A., Inoue, N., et al. (2011). Oxidative stress impairs insulin signal in skeletal muscle and causes insulin resistance in postinfarct heart failure. Am. J. Physiol. Heart Circ. Physiol. 300 (5), H1637–H1644. doi:10.1152/ajpheart.01185.2009
Olkowicz, M., Tomczyk, M., Debski, J., Tyrankiewicz, U., Przyborowski, K., Borkowski, T., et al. (2021). Enhanced cardiac hypoxic injury in atherogenic dyslipidaemia results from alterations in the energy metabolism pattern. Metabolism 114, 154400. doi:10.1016/j.metabol.2020.154400
Ong, S. G., and Hausenloy, D. J. (2012). Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol. Ther. 136 (1), 69–81. doi:10.1016/j.pharmthera.2012.07.005
Ouyang, J., Wang, H., and Huang, J. (2023). The role of lactate in cardiovascular diseases. Cell Commun. Signal 21 (1), 317. doi:10.1186/s12964-023-01350-7
Pakiet, A., Jakubiak, A., Mierzejewska, P., Zwara, A., Liakh, I., Sledzinski, T., et al. (2020). The effect of a high-fat diet on the fatty acid composition in the hearts of mice. Nutrients 12 (3), 824. doi:10.3390/nu12030824
Parajuli, N., Patel, V. B., Wang, W., Basu, R., and Oudit, G. Y. (2014). Loss of NOX2 (gp91phox) prevents oxidative stress and progression to advanced heart failure. Clin. Sci. (Lond) 127 (5), 331–340. doi:10.1042/CS20130787
Park, M. W., Cha, H. W., Kim, J., Kim, J. H., Yang, H., Yoon, S., et al. (2021). NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer's diseases. Redox Biol. 41, 101947. doi:10.1016/j.redox.2021.101947
Patrick, M., Gu, Z., Zhang, G., Wynn, R. M., Kaphle, P., Cao, H., et al. (2022). Metabolon formation regulates branched-chain amino acid oxidation and homeostasis. Nat. Metab. 4 (12), 1775–1791. doi:10.1038/s42255-022-00689-4
Plecitá-Hlavatá, L., Jabůrek, M., Holendová, B., Tauber, J., Pavluch, V., Berková, Z., et al. (2020). Glucose-stimulated insulin secretion fundamentally requires H(2)O(2) signaling by NADPH oxidase 4. Diabetes 69 (7), 1341–1354. doi:10.2337/db19-1130
Poulter, N. R., Prabhakaran, D., and Caulfield, M. (2015). Hypertension. Lancet 386 (9995), 801–812. doi:10.1016/S0140-6736(14)61468-9
Previs, M. J., O'Leary, T. S., Morley, M. P., Palmer, B. M., LeWinter, M., Yob, J. M., et al. (2022). Defects in the proteome and metabolome in human hypertrophic cardiomyopathy. Circ. Heart Fail 15 (6), e009521. doi:10.1161/CIRCHEARTFAILURE.121.009521
Ranjbarvaziri, S., Kooiker, K. B., Ellenberger, M., Fajardo, G., Zhao, M., Vander Roest, A. S., et al. (2021). Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 144 (21), 1714–1731. doi:10.1161/CIRCULATIONAHA.121.053575
Ray, R., Murdoch, C. E., Wang, M., Santos, C. X., Zhang, M., Alom-Ruiz, S., et al. (2011). Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 31 (6), 1368–1376. doi:10.1161/ATVBAHA.110.219238
Robles-Vera, I., Toral, M., de la Visitación, N., Sánchez, M., Gómez-Guzmán, M., Muñoz, R., et al. (2020). Changes to the gut microbiota induced by losartan contributes to its antihypertensive effects. Br. J. Pharmacol. 177 (9), 2006–2023. doi:10.1111/bph.14965
Romero-Becerra, R., Santamans, A. M., Folgueira, C., and Sabio, G. (2020). p38 MAPK pathway in the heart: new insights in health and disease. Int. J. Mol. Sci. 21 (19), 7412. doi:10.3390/ijms21197412
Saleem, N., Prasad, A., and Goswami, S. K. (2018). Apocynin prevents isoproterenol-induced cardiac hypertrophy in rat. Mol. Cell Biochem. 445 (1-2), 79–88. doi:10.1007/s11010-017-3253-0
Sánchez-Lozada, L. G., Madero, M., Mazzali, M., Feig, D. I., Nakagawa, T., Lanaspa, M. A., et al. (2023). Sugar, salt, immunity and the cause of primary hypertension. Clin. Kidney J. 16 (8), 1239–1248. doi:10.1093/ckj/sfad058
Savla, S. R., Laddha, A. P., and Kulkarni, Y. A. (2021). Pharmacology of apocynin: a natural acetophenone. Drug Metab. Rev. 53 (4), 542–562. doi:10.1080/03602532.2021.1895203
Schloss, M. J., Swirski, F. K., and Nahrendorf, M. (2020). Modifiable cardiovascular risk, hematopoiesis, and innate immunity. Circ. Res. 126 (9), 1242–1259. doi:10.1161/CIRCRESAHA.120.315936
Schröder, K., Zhang, M., Benkhoff, S., Mieth, A., Pliquett, R., Kosowski, J., et al. (2012). Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 110 (9), 1217–1225. doi:10.1161/CIRCRESAHA.112.267054
Schürmann, C., Rezende, F., Kruse, C., Yasar, Y., Löwe, O., Fork, C., et al. (2015). The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur. Heart J. 36 (48), 3447–3456. doi:10.1093/eurheartj/ehv460
Sciarretta, S., Zhai, P., Shao, D., Zablocki, D., Nagarajan, N., Terada, L. S., et al. (2013). Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α/activating transcription factor 4 pathway. Circ. Res. 113 (11), 1253–1264. doi:10.1161/CIRCRESAHA.113.301787
Shadel, G. S., and Horvath, T. L. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163 (3), 560–569. doi:10.1016/j.cell.2015.10.001
Shah, S. H., Sun, J. L., Stevens, R. D., Bain, J. R., Muehlbauer, M. J., Pieper, K. S., et al. (2012). Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am. Heart J. 163 (5), 844–850. doi:10.1016/j.ahj.2012.02.005
Shanmugam, G., Wang, D., Gounder, S. S., Fernandes, J., Litovsky, S. H., Whitehead, K., et al. (2020). Reductive stress causes pathological cardiac remodeling and diastolic dysfunction. Antioxid. Redox Signal 32 (18), 1293–1312. doi:10.1089/ars.2019.7808
Shen, Z. X., Yang, Q. Z., Li, C., Du, L. J., Sun, X. N., Liu, Y., et al. (2018). Myeloid peroxisome proliferator-activated receptor gamma deficiency aggravates myocardial infarction in mice. Atherosclerosis 274, 199–205. doi:10.1016/j.atherosclerosis.2018.05.005
Shu, H., Hang, W., Peng, Y., Nie, J., Wu, L., Zhang, W., et al. (2021). Trimetazidine attenuates heart failure by improving myocardial metabolism via AMPK. Front. Pharmacol. 12, 707399. doi:10.3389/fphar.2021.707399
Sirker, A., Murdoch, C. E., Protti, A., Sawyer, G. J., Santos, C. X., Martin, D., et al. (2016). Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J. Mol. Cell Cardiol. 98, 11–17. doi:10.1016/j.yjmcc.2016.07.003
Siu, K. L., Gao, L., and Cai, H. (2016). Differential roles of protein complexes NOX1-NOXO1 and NOX2-p47phox in mediating endothelial redox responses to oscillatory and unidirectional laminar shear stress. J. Biol. Chem. 291 (16), 8653–8662. doi:10.1074/jbc.M115.713149
Smyrnias, I., Zhang, X., Zhang, M., Murray, T. V., Brandes, R. P., Schröder, K., et al. (2015). Nicotinamide adenine dinucleotide phosphate oxidase-4-dependent upregulation of nuclear factor erythroid-derived 2-like 2 protects the heart during chronic pressure overload. Hypertension 65 (3), 547–553. doi:10.1161/HYPERTENSIONAHA.114.04208
Somasuntharam, I., Boopathy, A. V., Khan, R. S., Martinez, M. D., Brown, M. E., Murthy, N., et al. (2013). Delivery of Nox2-NADPH oxidase siRNA with polyketal nanoparticles for improving cardiac function following myocardial infarction. Biomaterials 34 (31), 7790–7798. doi:10.1016/j.biomaterials.2013.06.051
Song, P., and Zou, M. H. (2012). Regulation of NAD(P)H oxidases by AMPK in cardiovascular systems. Free Radic. Biol. Med. 52 (9), 1607–1619. doi:10.1016/j.freeradbiomed.2012.01.025
Sorescu, G. P., Song, H., Tressel, S. L., Hwang, J., Dikalov, S., Smith, D. A., et al. (2004). Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ. Res. 95 (8), 773–779. doi:10.1161/01.RES.0000145728.22878.45
Sosa, V., Carbó, R., and Guarner, V. (2007). Participation of glucose transporters on atrial natriuretic peptide-induced glucose uptake by adult and neonatal cardiomyocytes under oxygenation and hypoxia. Eur. J. Pharmacol. 568 (1-3), 83–88. doi:10.1016/j.ejphar.2007.04.040
Souto Padron de Figueiredo, A., Salmon, A. B., Bruno, F., Jimenez, F., Martinez, H. G., Halade, G. V., et al. (2015). Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. J. Biol. Chem. 290 (21), 13427–13439. doi:10.1074/jbc.M114.626077
Stevenson, M. D., Canugovi, C., Vendrov, A. E., Hayami, T., Bowles, D. E., Krause, K. H., et al. (2019). NADPH oxidase 4 regulates inflammation in ischemic heart failure: role of soluble epoxide hydrolase. Antioxid. Redox Signal 31 (1), 39–58. doi:10.1089/ars.2018.7548
Su, H., Yuan, Y., Wang, X. M., Lau, W. B., Wang, Y., Wang, X., et al. (2013). Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFα-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Res. Cardiol. 108 (1), 315. doi:10.1007/s00395-012-0315-z
Su, L., Zhang, J., Gomez, H., Kellum, J. A., and Peng, Z. (2023). Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 19 (2), 401–414. doi:10.1080/15548627.2022.2084862
Tedgui, A., and Mallat, Z. (2006). Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol. Rev. 86 (2), 515–581. doi:10.1152/physrev.00024.2005
Teekakirikul, P., Zhu, W., Huang, H. C., and Fung, E. (2019). Hypertrophic cardiomyopathy: an overview of genetics and management. Biomolecules 9 (12), 878. doi:10.3390/biom9120878
Teissier, E., Nohara, A., Chinetti, G., Paumelle, R., Cariou, B., Fruchart, J. C., et al. (2004). Peroxisome proliferator-activated receptor alpha induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-alpha activation properties. Circ. Res. 95 (12), 1174–1182. doi:10.1161/01.RES.0000150594.95988.45
Thomas, H., Diamond, J., Vieco, A., Chaudhuri, S., Shinnar, E., Cromer, S., et al. (2018). Global atlas of cardiovascular disease 2000-2016: the path to prevention and control. Glob. Heart 13 (3), 143–163. doi:10.1016/j.gheart.2018.09.511
Tokutome, M., Matoba, T., Nakano, Y., Okahara, A., Fujiwara, M., Koga, J. I., et al. (2019). Peroxisome proliferator-activated receptor-gamma targeting nanomedicine promotes cardiac healing after acute myocardial infarction by skewing monocyte/macrophage polarization in preclinical animal models. Cardiovasc Res. 115 (2), 419–431. doi:10.1093/cvr/cvy200
Tong, M., Saito, T., Zhai, P., Oka, S. I., Mizushima, W., Nakamura, M., et al. (2019). Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ. Res. 124 (9), 1360–1371. doi:10.1161/CIRCRESAHA.118.314607
Valente, A. J., Yoshida, T., Murthy, S. N., Sakamuri, S. S., Katsuyama, M., Clark, R. A., et al. (2012). Angiotensin II enhances AT1-Nox1 binding and stimulates arterial smooth muscle cell migration and proliferation through AT1, Nox1, and interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 303 (3), H282–H296. doi:10.1152/ajpheart.00231.2012
van der Pol, A., van Gilst, W. H., Voors, A. A., and van der Meer, P. (2019). Treating oxidative stress in heart failure: past, present and future. Eur. J. Heart Fail 21 (4), 425–435. doi:10.1002/ejhf.1320
Vendrov, A. E., Vendrov, K. C., Smith, A., Yuan, J., Sumida, A., Robidoux, J., et al. (2015). NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid. Redox Signal 23 (18), 1389–1409. doi:10.1089/ars.2014.6221
Vendrov, A. E., Xiao, H., Lozhkin, A., Hayami, T., Hu, G., Brody, M. J., et al. (2023). Cardiomyocyte NOX4 regulates resident macrophage-mediated inflammation and diastolic dysfunction in stress cardiomyopathy. Redox Biol. 67, 102937. doi:10.1016/j.redox.2023.102937
Vogel, J., Kruse, C., Zhang, M., and Schröder, K. (2015). Nox4 supports proper capillary growth in exercise and retina neo-vascularization. J. Physiol. 593 (9), 2145–2154. doi:10.1113/jphysiol.2014.284901
Wagner, K. D., and Wagner, N. (2020). PPARs and myocardial infarction. Int. J. Mol. Sci. 21 (24), 9436. doi:10.3390/ijms21249436
Wang, B., Nie, J., Wu, L., Hu, Y., Wen, Z., Dong, L., et al. (2018a). AMPKα2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ. Res. 122 (5), 712–729. doi:10.1161/CIRCRESAHA.117.312317
Wang, C., Zhu, L., Yuan, W., Sun, L., Xia, Z., Zhang, Z., et al. (2020a). Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J. Cell Mol. Med. 24 (12), 6670–6679. doi:10.1111/jcmm.15318
Wang, D., Li, J., Luo, G., Zhou, J., Wang, N., Wang, S., et al. (2023a). Nox4 as a novel therapeutic target for diabetic vascular complications. Redox Biol. 64, 102781. doi:10.1016/j.redox.2023.102781
Wang, L., Bi, X., and Han, J. (2020b). Silencing of peroxisome proliferator-activated receptor-alpha alleviates myocardial injury in diabetic cardiomyopathy by downregulating 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 expression. IUBMB Life 72 (9), 1997–2009. doi:10.1002/iub.2337
Wang, N., Wang, W., Wang, X., Mang, G., Chen, J., Yan, X., et al. (2022b). Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ. Res. 131 (11), 893–908. doi:10.1161/CIRCRESAHA.122.320488
Wang, X., Elksnis, A., Wikström, P., Walum, E., Welsh, N., and Carlsson, P. O. (2018b). The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS One 13 (9), e0204271. doi:10.1371/journal.pone.0204271
Wang, X., Zhang, G., Dasgupta, S., Niewold, E. L., Li, C., Li, Q., et al. (2022a). ATF4 protects the heart from failure by antagonizing oxidative stress. Circ. Res. 131 (1), 91–105. doi:10.1161/CIRCRESAHA.122.321050
Wang, Y., Liu, X., Zhang, C., and Wang, Z. (2018c). High salt diet induces metabolic alterations in multiple biological processes of Dahl salt-sensitive rats. J. Nutr. Biochem. 56, 133–141. doi:10.1016/j.jnutbio.2018.01.007
Wang, Y., Liu, X. Y., Wang, Y., Zhao, W. X., Li, F. D., Guo, P. R., et al. (2023b). NOX2 inhibition stabilizes vulnerable plaques by enhancing macrophage efferocytosis via MertK/PI3K/AKT pathway. Redox Biol. 64, 102763. doi:10.1016/j.redox.2023.102763
Wu, X., and Williams, K. J. (2012). NOX4 pathway as a source of selective insulin resistance and responsiveness. Arterioscler. Thromb. Vasc. Biol. 32 (5), 1236–1245. doi:10.1161/ATVBAHA.111.244525
Xu, S., Chamseddine, A. H., Carrell, S., and Miller, F. J. (2014). Nox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biol. 2, 642–650. doi:10.1016/j.redox.2014.04.004
Yamamoto, T., and Sano, M. (2022). Deranged myocardial fatty acid metabolism in heart failure. Int. J. Mol. Sci. 23 (2), 996. doi:10.3390/ijms23020996
Yang, J., Brown, M. E., Zhang, H., Martinez, M., Zhao, Z., Bhutani, S., et al. (2017). High-throughput screening identifies microRNAs that target Nox2 and improve function after acute myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 312 (5), H1002-H1012–h1012. doi:10.1152/ajpheart.00685.2016
Yu, F., Zhang, Q., Liu, H., Liu, J., Yang, S., Luo, X., et al. (2022). Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov. 8 (1), 40. doi:10.1038/s41421-022-00390-6
Zhang, D., Li, Y., Wang, W., Lang, X., Zhang, Y., Zhao, Q., et al. (2022). NOX1 promotes myocardial fibrosis and cardiac dysfunction via activating the TLR2/NF-κB pathway in diabetic cardiomyopathy. Front. Pharmacol. 13, 928762. doi:10.3389/fphar.2022.928762
Zhang, M., Brewer, A. C., Schröder, K., Santos, C. X., Grieve, D. J., Wang, M., et al. (2010). NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 107 (42), 18121–18126. doi:10.1073/pnas.1009700107
Zhang, M., Mongue-Din, H., Martin, D., Catibog, N., Smyrnias, I., Zhang, X., et al. (2018). Both cardiomyocyte and endothelial cell Nox4 mediate protection against hemodynamic overload-induced remodelling. Cardiovasc Res. 114 (3), 401–408. doi:10.1093/cvr/cvx204
Zhang, N., Zhang, Y., Xu, J., Wang, P., Wu, B., Lu, S., et al. (2023). α-myosin heavy chain lactylation maintains sarcomeric structure and function and alleviates the development of heart failure. Cell Res. 33 (9), 679–698. doi:10.1038/s41422-023-00844-w
Zhang, S. X., Khalyfa, A., Wang, Y., Carreras, A., Hakim, F., Neel, B. A., et al. (2014). Sleep fragmentation promotes NADPH oxidase 2-mediated adipose tissue inflammation leading to insulin resistance in mice. Int. J. Obes. (Lond) 38 (4), 619–624. doi:10.1038/ijo.2013.139
Zhang, X., Dong, X., Jie, H., Li, S., Li, H., Su, Y., et al. (2024). Downregulation of the (pro)renin receptor alleviates ferroptosis-associated cardiac pathological changes via the NCOA 4-mediated ferritinophagy pathway in diabetic cardiomyopathy. Int. Immunopharmacol. 138, 112605. doi:10.1016/j.intimp.2024.112605
Zhang, Y., Murugesan, P., Huang, K., and Cai, H. (2020). NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat. Rev. Cardiol. 17 (3), 170–194. doi:10.1038/s41569-019-0260-8
Zhao, G. J., Zhao, C. L., Ouyang, S., Deng, K. Q., Zhu, L., Montezano, A. C., et al. (2020). Ca(2+)-Dependent NOX5 (NADPH oxidase 5) exaggerates cardiac hypertrophy through reactive oxygen species production. Hypertension 76 (3), 827–838. doi:10.1161/HYPERTENSIONAHA.120.15558
Zhao, Q. D., Viswanadhapalli, S., Williams, P., Shi, Q., Tan, C., Yi, X., et al. (2015). NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 131 (7), 643–655. doi:10.1161/CIRCULATIONAHA.114.011079
Ziaeian, B., and Fonarow, G. C. (2016). Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 13 (6), 368–378. doi:10.1038/nrcardio.2016.25
Keywords: NADPH oxidases, oxidative stress, energy metabolism, cardiovascular diseases, ferroptosis
Citation: Jie H, Zhang J, Wu S, Yu L, Li S, Dong B and Yan F (2025) Interplay between energy metabolism and NADPH oxidase-mediated pathophysiology in cardiovascular diseases. Front. Pharmacol. 15:1503824. doi: 10.3389/fphar.2024.1503824
Received: 29 September 2024; Accepted: 27 December 2024;
Published: 10 January 2025.
Edited by:
Xiangxiang Wei, Fudan University, ChinaReviewed by:
Dorota Katarzyna Dymkowska, Polish Academy of Sciences, PolandThomas P. Flagg, Uniformed Services University of the Health Sciences, United States
Copyright © 2025 Jie, Zhang, Wu, Yu, Li, Dong and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Yan, eWFuZmVuZ0BzZHUuZWR1LmNu; Bo Dong, Ym9kb25nQHNkdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship