 Caifeng Zhang
Caifeng Zhang Keke Su1,2
Keke Su1,2- 1Department of Gastroenterology, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, China
- 2First College for Clinical Medicine, Xinxiang Medical University, Xinxiang, Henan, China
Niemann-Pick disease type C1 (NP-C1) is a rare and devastating recessive inherited lysosomal lipid and cholesterol storage disorder caused by mutations in the NPC1 or NPC2 gene. These two proteins bind to cholesterol and cooperate in endosomal cholesterol transport. Characteristic clinical manifestations of NP-C1 include hepatosplenomegaly, progressive neurodegeneration, and ataxia. While the rarity of NP-C1 presents a significant obstacle to progress, researchers have developed numerous potential therapeutic approaches over the past two decades to address this condition. Various methods have been proposed and continuously improved to slow the progression of NP-C1, although they are currently at an animal or clinical experimental stage. This overview of NP-C1 therapy will delve into different theoretical treatment strategies, such as small molecule therapies, cell-based approaches, and gene therapy, highlighting the complex therapeutic challenges associated with this disorder.
Introduction
Niemann-Pick type C1 disease (NP-C1) was first described in the early twentieth century and further elucidated by Alan Crocker in the 1960s (Crocker, 1961). Peter Pentchev, two decades later, conducted a series of investigations on a spontaneous mouse model, shedding light on the molecular processes underlying this enigmatic disorder (Pentchev et al., 1986). NP-C1 is now recognized as a prototypical lysosomal storage disorder, characterized by the abnormal accumulation of cholesterol and other lipids in late endosomes and lysosomes (LE/LY) (Brauer et al., 2019). While lipid buildup is observed in various tissues such as the liver, spleen, lungs, and bone marrow, the most critical disease manifestations stem from progressive neurodegeneration (Vanier, 2010; Patterson and Walkley, 2017).
The NP-C1 is a rare condition with a worldwide estimated incidence ranging from 1/1,00,000 to 1/1,20,000 live births (Mengel et al., 2013). However, the actual incidence is likely higher due to frequent misdiagnoses or cases that go undetected (Stampfer et al., 2013). NP-C1 presents with varying ages of onset, categorized into infancy, childhood, juvenile, adolescence, and adulthood. Most patients experience symptoms in early childhood and typically pass away within 5–20 years after the onset of the disease (Hung et al., 2014). The clinical diagnosis of NP-C1 currently relies on laboratory analyses, including filipin staining of skin fibroblasts and biomarker assessments. However, a definitive diagnosis is confirmed through next-generation sequencing (NGS) analysis (Encarnacao et al., 2023). Patients with NP-C1 typically present with liver and spleen enlargement in the early stages, followed by progressive neurodegeneration and other symptoms as the disease advances (Nadjar et al., 2018). Unfortunately, there are limited well-established pharmacological treatments available for NP-C1. Over the past two decades, researchers have explored diverse approaches for managing NP-C1 through animal or clinical experiments (Patterson and Platt, 2004). This review aims to provide a comprehensive overview of NP-C disease, delving into various treatment strategies from multiple angles, including traditional small molecules, cell-based therapies, and gene therapy. The discussion highlights the intricate therapeutic challenges associated with combating this debilitating condition.
NPC protein function
Cholesterol serves multiple crucial functions in the body, acting as a key component of cell membranes and plasma lipoproteins while also serving as a precursor for bile acids, hormones, and vitamin D3. As such, the synthesis and transport of cholesterol are essential for maintaining cellular integrity (Lamri et al., 2018). In NP-C1, there is a disruption in the intracellular transport and balance of free cholesterol. The accumulation of excess free cholesterol in late endosomes and lysosomes can lead to significant cellular and tissue damage in the nervous system and other organs (Lopez et al., 2014). NP-C1 in humans is primarily caused by mutations in two genes, NPC1 and NPC2, with approximately 95% of cases linked to NPC1 mutations and 5% to NPC2 mutations (Vanier, 2010).
NPC1 is a large transmembrane protein located on the boundary membrane of late endosomes and lysosomes, consisting of 13 transmembrane domains encoding 1,278 amino acids crucial for intracellular cholesterol transport. On the other hand, NPC2 is a soluble lumen protein responsible for transporting cholesterol from lysosomal vesicles to the N-terminal domain of NPC1. Both proteins are essential for cholesterol export from lysosomes (Naureckiene et al., 2000; Infante et al., 2008a; Scott and Ioannou, 2004). While the precise roles of NPC1 and NPC2 in lysosomal cholesterol transport are not fully understood, it is believed that they are involved in pathways regulating cholesterol transport within lysosomes or promoting movement of lysosomal lipid substrates (Lloyd-Evans and Platt, 2010; Infante et al., 2008b).
A prevailing hypothesis suggests that when a lipid cargo reaches late endosomes/lysosomes, it is broken down into its constituent molecules. NPC2 facilitates the transfer of cholesterol or other lipids to organelle boundary membranes, while NPC1 detects an increase in cholesterol at the cell membrane and initiates transport of the cargo to its designated destinations (Wang et al., 2010; Kwon et al., 2009). NPC1, working in conjunction with NPC2, facilitates the removal of low-density lipoprotein (LDL)-derived cholesterol from the endosomal compartment through a yet-to-be-defined mechanism, although significant progress has been achieved (Somers et al., 2003). The generally accepted model posits that NPC2 binds free cholesterol post-hydrolysis of LDL cholesterol esterase and transfers it to NPC1, which then mediates the extraction of cholesterol from the lysosome (Zampieri et al., 2014) (Figure 1A). Consequently, the absence of either NPC1 or NPC2 protein could lead to the manifestation of this genetic disease, as there are no substitutes for these two proteins in NP-C1.
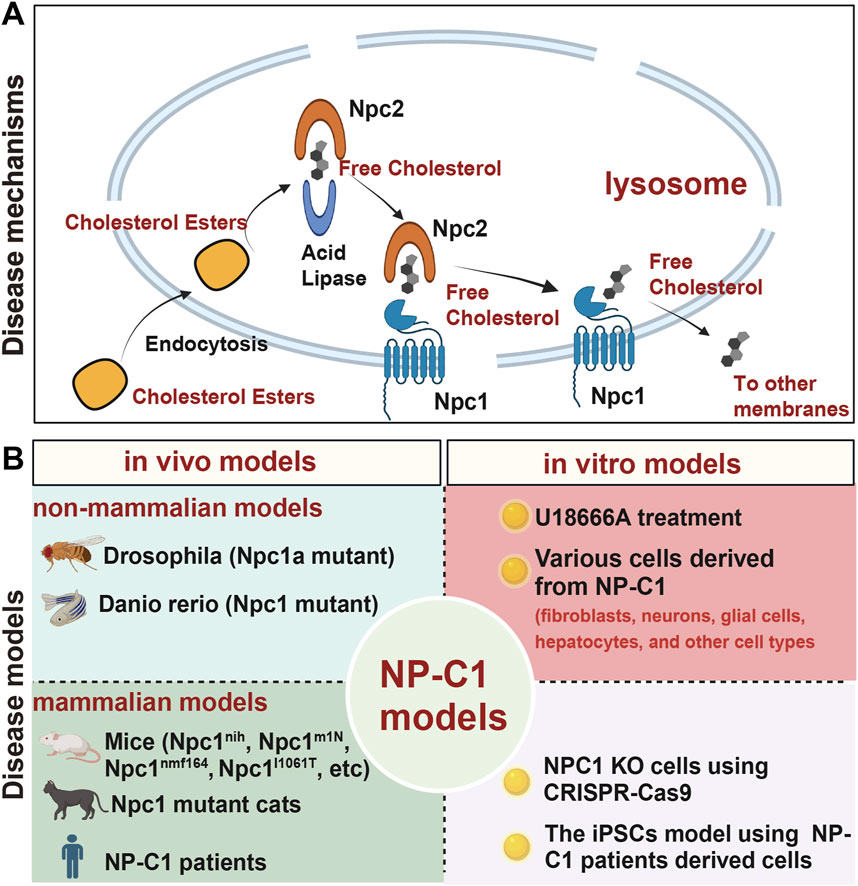
Figure 1. A brief overview of the mechanisms and models of NP-C1. (A) The process by which NPC1 and NPC2 proteins transport cholesterol from lysosomes to other organelle membranes. (B) Current in vivo and in vitro models used for researching NP-C1.
Currently used NP-C1 model systems
The clinical spectrum of NP-C1 varies from a severe neonatal disorder to a chronic neurodegenerative disease that can onset in adulthood. The most severe cases present in early childhood and result in death during childhood or adolescence. The onset of NP-C1 symptoms can vary among individuals, with some experiencing disease manifestation in adolescence or adulthood due to different genetic information influencing disease progression. Given ethical considerations, the timing of symptom onset, and the complexities involved, the development of animal and cell models is essential for studying NP-C1 and advancing our understanding of this disorder (Figure 1B). The evolving NP-C1 models should offer several advantages, including ease of gene manipulation, straightforward modification and screening processes, and are particularly well-suited for establishing platforms for the discovery and development of high-throughput drugs.
In vivo models
Numerous animal models have been employed in the study of NP-C1, owing to the relatively conserved nature of the genes and functions of Npc1 across non-mammalian and mammalian populations. A disease model has been developed by mutating Drosophila Npc1a. Null mutants exhibit early lethality, movement impairments, neuronal cholesterol deposits, accumulation of multilamellar bodies, and age-dependent neurodegeneration that mimics the human neurodegenerative condition (Phillips et al., 2008). In zebrafish (Danio rerio), Npc1 morphants and mutants show high lethality, reduced steroidogenesis, abnormal cell movements, and a severe neurometabolic phenotype (Quelle-Regaldie et al., 2023; Schwend et al., 2011). In addition, the nematode Caenorhabditis elegans and the yeast Saccharomyces cerevisiae have been developed and utilized to gain insights into NPC cellular pathways (Boland et al., 2017; Frain et al., 2024). Currently, investigations into non-mammalian NP-C1 predominantly concentrate on Drosophila and D. rerio. Nevertheless, research in this area has slowed in recent years, largely due to the rapid advancement of mammalian and in vitro models.
Studies on NP-C1 in mammals predominantly focus on mice and cats, which are frequently used in laboratory research and clinical drug development studies. Different mutations in the Npc1 gene in mice can lead to varying phenotypes. These mice are currently the most common NPC1 rodent model for assessing potential small molecules for this debilitating disease. The initial mouse strains used for studying NP-C1, namely C57BLKS/Jspm and BALB/cNpc1nih, originated as spontaneous mutations. Through crossbreeding, researchers confirmed them as allelic and independently positioned them on mouse chromosome 18, in a region syntenic to the human NPC1 locus (Loftus et al., 1997; Pentchev et al., 1984). These two commonly used mice models display typical NP-C1 neurological symptoms such as hepatomegaly, splenomegaly, decreased weight gain, increased lung mass, disturbed motor coordination, tremor, ataxia and loss of Purkinje cells (Loftus et al., 1997). The pathology of NP-C1 in these mouse models initiates at 4–6 weeks of age, closely resembling the onset of NP-C1 in infants and young children. They serve as valuable research tools for studying the severe infantile onset forms of NP-C1.
Maue et al. established a new mouse model (Npc1nmf164) with a point mutation in the Npc1 gene, specifically an A to G change at cDNA base pair 3,163, resulting in an aspartate to glycine substitution at position 1005 (D1005G). The lifespan analyses show that these mice begin developing the disease at 4 weeks and typically survive until around 16 weeks, indicating a lifespan extension of approximately 5 weeks compared to Npc1nih mice. The histological analyses reveal abnormal cholesterol accumulation, glial activation and Purkinje cell loss at a slower rate than in the Npc1nih mouse model (Maue et al., 2012). Praggastis and colleagues developed an Npc1I1061T knock-in mouse model that shows a less severe and delayed form of NP-C1. This model is characterized by reduced weight loss, improved motor coordination, decreased Purkinje cell death, reduced lipid storage, and delayed premature death compared to the Npc1nih mice (Praggastis et al., 2015). Meanwhile, the lifespan analyses show that these mice begin developing the disease at 8 weeks and pass away within 17–18 weeks. Taken together, these two mice models offer many advantages as a model for the late-onset, more slowly progressing forms of NP-C1 that comprise the large majority of human cases. Furthermore, current research is exploring the effects of cholesterol transport abnormalities on organ development and homeostasis through using the Cre-loxP system to selectively target Npc1 gene in specific tissues (Elrick et al., 2010). NP-C1 in humans is a genetic disorder commonly associated with widespread organ dysfunction. Therefore, mice with systemic Npc1 mutations are considered more appropriate candidates for NP-C1 drug development. However, different mutations in Npc1 mutant mice can result in varying phenotypes, reflecting the complexity observed in clinical cases. These diverse mutations in the NPC1 gene result in the production of unique proteins, triggering a cascade of biochemical responses in the body and giving rise to a spectrum of disease presentations.
Additionally, a feline model of NP-C1 has been meticulously characterized, exhibiting phenotypic, morphological, and biochemical similarities to human NPC1. The disease manifestation in NP-C1 felines mirrors the juvenile form of human NP-C1, replicating numerous clinical features such as hepatomegaly, pulmonary complications, and central nervous system (CNS) involvement, notably ataxia, among other symptoms (Rakib et al., 2023). Research on Npc2 gene mutations causing NP-C1 is relatively limited compared to studies on the Npc1 gene. However, in mouse and cat models, mutations in the Npc2 gene have been shown to result in symptoms similar to those observed in clinical NP-C1, including weight loss, decreased motor coordination, cerebellar Purkinje neuron death, lipid storage, and premature death (Rakib et al., 2023; Lee and Hong, 2023; Pallottini and Pfrieger, 2020).
In vitro models
In addition to the NP-C1 animal models mentioned above, researchers have developed cell models for NP-C1, emphasizing the direct role of lysosomal lipid accumulation in cellular signal transduction and phenotype analysis. Researchers isolated fibroblasts, neurons, glial cells, hepatocytes, and other cell types from Npc1 mutant mice with diverse genotypes to investigate cellular states throughout the progression of NP-C1 (Peake and Vance, 2012; Malara et al., 2024; Kulinski and Vance, 2007). Findings from these studies using cells from mutant mice suggest that the absence of the Npc1 disrupts cholesterol metabolism, impacting intracellular processes such as autophagy, endoplasmic reticulum (ER), vesicle sorting, and multiple signaling pathways (Hoglinger et al., 2019; Schwerd et al., 2017; Sarkar et al., 2013).
In humans, obtaining a comprehensive understanding of the majority of NP-C1 variants using patient-derived fibroblasts remains challenging due to their infrequent occurrence in isolation. Recent advances in induced pluripotent stem cells (iPSCs) technology have made it possible to create cell-based disease models using human cells derived from patient iPSCs. Utilizing a fibroblast-induced iPSCs model from NP-C1 patients can offer valuable insights into the pathological mechanisms of NP-C1 (Trilck et al., 2013). The iPSCs derived from NP-C1 fibroblasts express various stem cell markers, differentiate into cells of all three germ layers, and induce teratoma formation in immunodeficient mice. Additionally, the iPSCs from NP-C1 patients exhibit cholesterol accumulation in the cytoplasm, a characteristic not seen in cells from healthy individuals. These observations demonstrate that iPSCs derived from patient cells retain pluripotency while displaying disease-specific features, highlighting their potential as a valuable tool for studying and modeling diseases (Trilck et al., 2013; Volkner et al., 2022). While the iPSCs model established using fibroblasts provides valuable insights into NP-C1, it may be limited in studying organ-specific pathologies such as neuronal loss and hepatocyte damage. Therefore, it is crucial to differentiate the iPSCs into specific cell types like neurons, glial cells and hepatocyte-like cells using specialized methods during in vitro modeling. These approaches are essential for gaining a deeper understanding of the mechanisms underlying brain and liver dysfunction in NP-C1 (Prabhu et al., 2021; Volkner et al., 2021; Peter et al., 2017).
The use of CRISPR/Cas9 technology to knockout specific genes within the genome of mammals has indeed become routine in recent years (Du et al., 2017). Research has indeed shown that knocking out the Npc1 gene using technology can lead to cholesterol accumulation characteristics in various cells. Deletion of the Npc1 has been associated with enhanced cell connectivity in 293T cells and may promote inflammation response and apoptosis in N2a cells (Du et al., 2017; Jia et al., 2023; Yang et al., 2023). Furthermore, researchers employed the saturation prime editing (SPE) platform to edit the Npc1 gene, revealing that 410 out of the 706 assayed missense mutations present significant risks for the disease (Erwood et al., 2022). Additionally, a haploid cell model established using CRISPR/Cas9 technology serves as a valuable platform for studying the pathogenesis of NP-C1 (Erwood et al., 2019). These findings highlight the importance of Npc1 in cellular function and the significant potential of gene editing technology in developing NPC1 models. With the advancement of CRISPR/Cas9 technology, achieving single-base mutations has become feasible (Chen and Liu, 2023). By integrating CRISPR/Cas9 with iPSCs technology (Xu et al., 2019), a wide range of NP-C1 cell models will be generated. This potent combination enables accurate genetic alterations, rendering it an invaluable asset for exploring gene functionality and disease mechanisms across diverse organisms.
Potential therapeutic approaches for NP-C1
Currently, there are limited effective therapies for NP-C1. Treatment mainly focuses on symptom management and slowing down the progression of the disease. Some treatment methods include drug therapy, nutritional support and physical therapy, aiming to improve the quality of life and delay the progression of the disease. In recent years, advancements in NP-C1 animal and cell models have provided a more profound insight into the pathogenesis of NP-C1, facilitating high-throughput drug screening. The introduction of innovative technologies like gene editing and stem cell therapy has expanded the scope of potential treatment options for the disease. Nevertheless, additional research and clinical trials are imperative to pinpoint more effective treatment strategies.
Small molecule therapeutics
Indeed, as our understanding of the molecular mechanisms of NP-C1 has improved, researchers have been able to explore a variety of small molecules for treating the condition. By conducting studies and clinical trials, researchers have made significant progress in developing treatments for NP-C1. These efforts are directed towards offering improved management and potential therapeutic options for individuals affected by NP-C1 (Table 1).
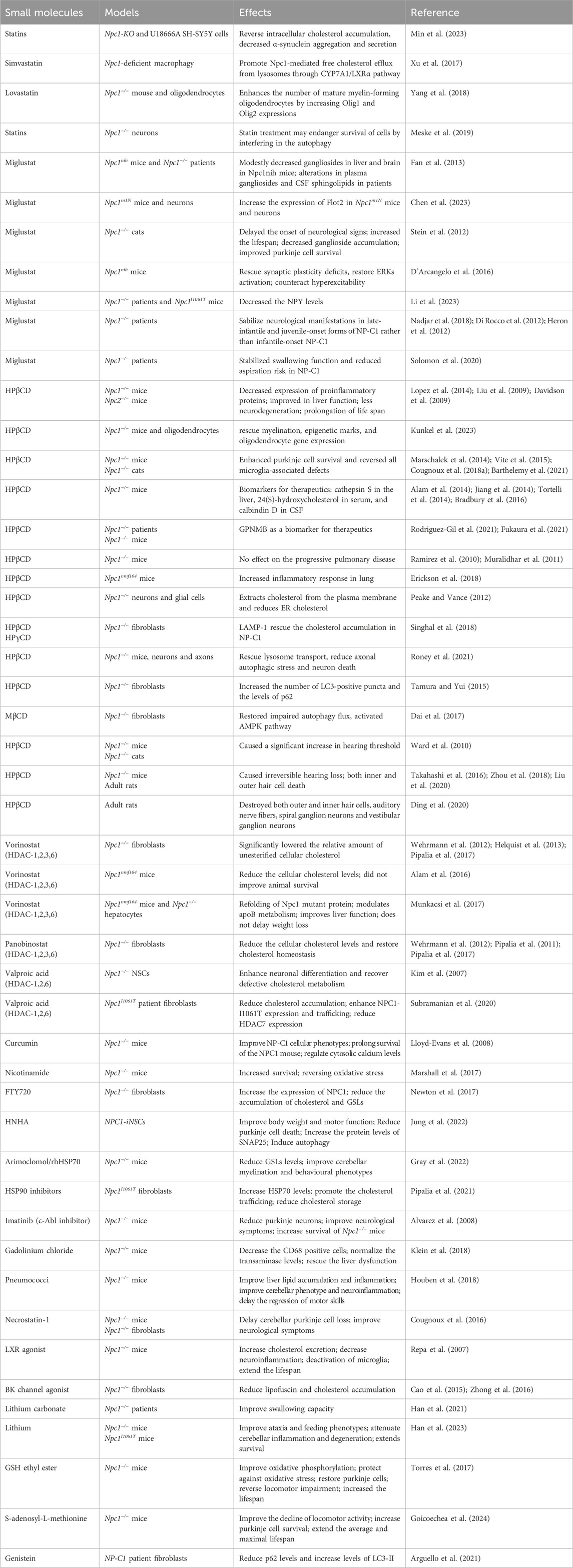
Table 1. Small molecule therapeutics in NP-C1 treatment.
Statins
Statins, 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, are a vital class of drugs used to lower cholesterol levels. Researchers are investigating the potential therapeutic benefits of statins for NP-C1, as they believe that these drugs may have a positive impact on the accumulation and impaired trafficking of cholesterol in Npc1-deficient cells. Studies have demonstrated that statins can reverse intracellular cholesterol accumulation and α-synuclein aggregation induced by Npc1-KO or U18666A treatment (Min et al., 2023). In macrophages, simvastatin promotes Npc1-mediated free cholesterol efflux from lysosomes through CYP7A1/LXRα pathway (Xu et al., 2017). While lovastatin has been shown to restore the number of mature myelin-forming oligodendrocytes (Yang et al., 2018), Meske et al. reported that statin treatment may pose a risk to the survival of Npc1 mutant neurons (Meske et al., 2019). These findings suggest that statin drugs may have the potential to improve symptoms in NP-C1 due to their cholesterol-lowering properties, but they do not seem to effectively alter the neurological progression.
Miglustat
In addition to the cholesterol accumulation, Npc1-deficient cells also accumulate gangliosides and other glycosphingolipids (GSLs), which are key players in the pathogenesis of NPC disease (Zervas et al., 2001). Miglustat, a reversible inhibitor of GSLs synthesis recognized for its ability to reduce GSLs buildup in type 1 Gaucher’s disease, has demonstrated positive effects through reducing GSLs levels on the progression of NP-C1 (Pineda et al., 2018). In addition to GSLs, there was a notable accumulation of gangliosides in the liver and brain. Following miglustat treatment in NP-C1 animal models and NP-C1 patients, the levels of gangliosides in plasma and cerebrospinal fluid (CSF) decreased significantly (Fan et al., 2013). In Npc1−/− mice, gangliosides sequestration and the loss of lipid rafts lead to cell dysfunction and symptoms of NP-C1. Miglustat increased the expression of Flot2, a marker for lipid rafts, which was found to be diminished in neurons of Npc1−/− mice (Chen et al., 2023).
Accumulation of gangliosides and GSLs in neurons is closely linked to cell metabolism, maintaining homeostasis, and cell death. Research in the NPC1 feline disease model indicates that miglustat delayed the onset of neurological symptoms, extended the lifespan of treated cats, and correlated with reduced ganglioside buildup in the cerebellum and enhanced Purkinje cell survival. Analyzing microglia from treated cats decreased production of reactive oxygen species (ROS) (Stein et al., 2012). Furthermore, the administration of miglustat in Npc1−/− mice demonstrated the ability to rescue deficits in synaptic plasticity, restore ERK activation, and alleviate hyperexcitability (D’Arcangelo et al., 2016). Collectively, these results indicate that the prolonged survival of Purkinje cells, reduction in ganglioside accumulation, and restored synaptic plasticity are key factors contributing to the neurological enhancements observed in NPC-1 individuals treated with miglustat.
Intense inflammation is a significant factor that triggers multi-organ damage in NP-C1. Miglustat has been shown to suppress astrocyte pathogenic activities in CNS inflammation by inhibiting the production of pro-inflammatory cytokines and restoring lactate generation in astrocytes (Chao et al., 2019). While there are currently few specific data confirming the anti-inflammatory role of miglustat in NPC1, several studies have confirmed its combined use with anti-inflammatory drugs in this condition. Pro-neuropeptide Y (NPY) levels were significantly elevated in individuals with NPC1 compared to healthy controls. Miglustat demonstrated efficacy in mitigating neuroinflammation and decreasing excitotoxicity by modulating NPY levels (Li et al., 2023).
For NP-C1 with varying onset periods, research indicates that miglustat can stabilize neurological manifestations in pediatric patients with late-infantile and juvenile-onset forms of NP-C1, rather than specifically targeting neurologic manifestations in infantile-onset NP-C1 (Di Rocco et al., 2012; Heron et al., 2012). Adolescent/adult-onset NP-C1 often initially presents with non-specific isolated neuro-psychiatric manifestations (motor, cognitive, or psychotic). Patients with milder neurological disabilities tend to respond more positively to miglustat therapy (Nadjar et al., 2018). Furthermore, the use of miglustat is linked to stabilized swallowing function and reduced aspiration risk in NP-C1, underscoring the potential for quantifying swallowing dysfunction as a clinically relevant functional outcome measure in future therapeutic trials for NP-C1 (Solomon et al., 2020).
2-hydroxypropyl--cyclodextrin (HPβCD)
Miglustat has shown limited effectiveness in reducing substrates and does not affect the accumulation of cholesterol in the body. Furthermore, its ability to slow the progression of neurological symptoms is also constrained (Pineda et al., 2018). Cyclodextrins (CDs), a family of cyclic oligosaccharides, have the ability to form complexes with cholesterol, effectively replacing dysfunctional cholesterol transport proteins. In NP-C1, 2-hydroxypropyl-β-cyclodextrin (HPβCD) is being considered as an alternative treatment option. A range of studies have demonstrated the effectiveness of HPβCD in treating NP-C1. A single dose of HPβCD has been shown to suppress sterol synthesis, down-regulate SREBP2 and its target genes, and reduce the expression of macrophage-associated inflammatory genes in the liver and brain (Liu et al., 2009; Liu et al., 2010). Additionally, HPβCD was able to rescue myelination, epigenetic marks, and oligodendrocyte gene expression, emphasizing the critical role of HPβCD in oligodendrocyte lineage maturation and epigenetic regulation in NP-C1 (Kunkel et al., 2023). Prolonged administration of HPβCD has shown improvements in liver function tests, reduced neurodegeneration, amelioration of cholesterol or GSLs storage, and a significant extension of lifespan (Lopez et al., 2014; Liu et al., 2009; Liu et al., 2010; Davidson et al., 2009). Importantly, treatment with HPβCD resulted in a reduction in Purkinje cell loss, reversed microglia-mediated neuroinflammation, and induced a concerted action of neurons and glial cells to restore lipid homeostasis in the CNS (Marschalek et al., 2014; Vite et al., 2015; Cougnoux et al., 2018a; Barthelemy et al., 2021). Several biomarkers in serum and body fluids have been developed for predicting the prognosis of NP-C1 with HPβCD treatment. These include cathepsin S in the liver, 24(S)-hydroxycholesterol in serum, and Calbindin D in CSF (Alam et al., 2014; Jiang et al., 2014; Tortelli et al., 2014; Bradbury et al., 2016). Transcriptome sequencing analysis of NPC1 patients receiving cyclodextrin therapy identified GPNMB as a key factor in evaluating treatment efficacy (Rodriguez-Gil et al., 2021; Fukaura et al., 2021). While HPβCD treatment shows significant protective effects in the brain and liver in NP-C1, its influence on lung dysfunction is minimal (Ramirez et al., 2010; Muralidhar et al., 2011; Erickson et al., 2018). This implies that lung cells may have the capability to resist the effects of CDs on cholesterol trafficking.
A mutation in NPC1 leads to the sequestration of unesterified cholesterol in the late endosomal/lysosomal compartment. HPβCD binds to unesterified cholesterol and facilitates its delivery from the lysosome to the ER, resulting in a significant increase in ACAT-mediated cholesterol esterification and a decrease in unesterified cholesterol levels (Liu et al., 2009; Aqul et al., 2011). The accumulation of cholesteryl esters in the cytosol is anticipated to be considerably less toxic than the accumulation of free cholesterol in NP-C1 patients (Abi-Mosleh et al., 2009). LAMP1, situated in the membranes of lysosomes, was upregulated in response to HPβCD treatment. This upregulation facilitated cholesterol trafficking at the late endosome/lysosome compartments, effectively rescuing the cholesterol accumulation defect observed in fibroblast cells derived from NPC1 patients (Singhal et al., 2018). On the other hand, HPβCD treatment significantly increased autophagic flux and restored lysosome transport, leading to a reduction in axonal autophagic stress and neuronal death in NP-C1 (Roney et al., 2021; Tamura and Yui, 2015) Methyl-β-cyclodextrin (MβCD), a potent analog of HPβCD, was also found to restore impaired macroautophagy/autophagy flux in NP-C1 by activating the AMPK pathway (Dai et al., 2017). These findings demonstrate the translational promise of HPβCD in enhancing impaired autophagic flux and reinstating axonal homeostasis in the initial phases of NP-C1.
HDAC inhibitors (HDACi)
Histone deacetylase inhibitors (HDACi) are emerging as promising therapeutics for a diverse array of diseases, encompassing cancer and neurodegenerative conditions. Recently, a genome-wide, conditional synthetic lethality screen was conducted using the yeast model of NP-C1 (Munkacsi et al., 2011). Additionally, a high-content screen targeting the reduction of lysosomal cholesterol storage was performed with Npc11I1061T patient fibroblasts (Pugach et al., 2018). Both studies suggest that HDACi emerges as a promising candidate therapy for NP-C1.
Two HDAC inhibitors, vorinostat and panobinostat, have been reported to be effective in treating various cancers and are showing promise in the management of cholesterol metabolism disorders. Both vorinostat and panobinostat have demonstrated the ability to reduce levels of unesterified cellular cholesterol and restore cholesterol homeostasis in Npc1−/− fibroblasts (Wehrmann et al., 2012; Helquist et al., 2013; Pipalia et al., 2011). Vorinostat additionally improves liver function and modulates apoB metabolism, although it does not delay weight loss or enhance animal survival in Npc1nmf164 mice (Alam et al., 2016; Munkacsi et al., 2017). Mechanistically, vorinostat and panobinostat enhance the expression and trafficking of the NPC1 mutant protein, revealing unanticipated epigenomic plasticity in spatial covariance relationships that restore NPC1 functionality (Pipalia et al., 2017; Wang et al., 2019). Valproic acid (VPA), a histone deacetylase inhibitor, has the potential to promote neuronal differentiation and restore impaired cholesterol metabolism in neural stem cells (NSCs) derived from Npc1-deficient mice (Kim et al., 2007). In Npc11I1061T models, VPA enhances NPC1-I1061T protein expression and trafficking, resulting in the restoration of cholesterol homeostasis by reducing HDAC7 expression (Subramanian et al., 2020). These studies suggest that FDA-approved HDAC inhibitors can improve the development of NP-C1 to some extent in preclinical research.
In addition, other HDAC inhibitors have been reported to improve NPC1. Treatment with curcumin, a natural compound, has been shown to normalize cellular phenotypes associated with NP-C1 and prolong the survival of NP-C1 mice by regulating cytosolic calcium levels (Lloyd-Evans et al., 2008). Nicotinamide has been shown to prolong NP-C1 mouse survival and prevent oxidative stress (Marshall et al., 2017). FTY720 (fingolimod) and its active phosphorylated form (FTY720-P) act as HDAC inhibitors, increasing the expression of NPC1 and significantly reducing the accumulation of cholesterol and GSLs in human NPC1 mutant fibroblasts (Newton et al., 2017). A new synthetic HDAC inhibitor, N-hydroxy-7-(2-naphthylthio) heptanomide (HNHA), has been found to ameliorate NPC1 phenotypes such as body weight, motor function, and Purkinje cell death through autophagy induction (Jung et al., 2022). Furthermore, most HDAC inhibitors enhance the NPC1 mutant protein and promote the exit of the NPC1 protein from the ER, facilitating its delivery to late endosomes/lysosomes to stabilize cholesterol metabolism in NPC1. This process involves the refolding of the NPC1 mutant protein through changes in protein chaperones. Treatment with arimoclomol, a well-characterized heat shock protein (HSP) amplifier, or recombinant human heat shock protein 70 (rhHSP70) has been shown to reduce GSLs levels in the CNS, leading to improved cerebellar myelination and behavioral phenotypes in Npc1−/− mice (Gray et al., 2022). On the other hand, inhibiting HSP90 with several HSP90 inhibitors has been found to increase the expression of HSP70, promote the clearance of cholesterol from late endosomes/lysosomes, and reduce cholesterol storage in NPC1I1061T skin fibroblasts (Pipalia et al., 2021). These studies suggest that heat shock protein-based therapies hold promise and should be clinically evaluated for treating NP-C1.
Other types of drugs
At present, drug development for NP-C1 predominantly centers on diminishing cholesterol buildup. Nevertheless, NP-C1 manifests several other pathological characteristics, including neuroinflammation occurs, heightened oxidative stress, disturbed ion balance, among others. Each of these pathological processes presents an opportunity for targeted drug development or combination therapy. In recent years, there has been significant advancement in the development of NPC1 drugs incorporating anti-inflammatory strategies. In a previous study, it was demonstrated that the proapoptotic tyrosine kinase c-Abl signaling is activated in Npc1−/− neurons (Contreras et al., 2016). Treatment with the c-Abl-specific inhibitor imatinib resulted in the preservation of Purkinje neurons, a reduction in general cell apoptosis in the cerebellum, improvement of neurological symptoms, and increased survival of Npc1−/− mice (Alvarez et al., 2008). Furthermore, treatment with gadolinium chloride (GdCl3) or heat-inactivated pneumococci was found to effectively reduce liver lipid accumulation and inflammation, leading to the rescue of some parameters of liver dysfunction in NP-C1 mice (Klein et al., 2018; Houben et al., 2018). In NP-C1, the necroptosis-related genes RIP3 and RIP1 are upregulated. Inhibition of necroptosis has been shown to significantly delay cerebellar Purkinje cell loss, slow the progression of neurological symptoms, and prolong survival in Npc1−/− mice (Cougnoux et al., 2016). Moreover, incorporating drugs such as ibuprofen, aspirin, metformin, and others that possess anti-inflammatory properties in combination therapy with currently known effective drugs may provide additional benefits in managing NP-C1.
Defective Ca2+ release has been associated with a number of lysosomal storage diseases (LSDs), including NP-C1. Lysosomes express big-conductance Ca2+-activated potassium (BK) channels that regulate lysosomal Ca2+ release (Cao et al., 2015). Activation of BK by NS1619 reduces lipofuscin and cholesterol accumulation in NPC1 cells in a Ca2+-dependent manner (Zhong et al., 2016). Lithium decreases STING/SREBP2 activation by reducing intracellular Ca2+ levels. Treatment with lithium has been shown to improve NP-C1 phenotypes, extend survival in Npc1 mouse models, and enhance swallowing capacity in NP-C1 patients (Han et al., 2021; Han et al., 2023). The buildup of cholesterol in mitochondria is recognized to hinder the entry of glutathione (GSH) into mitochondria, leading to the depletion of mitochondrial GSH. However, the supplementation of GSH ethyl ester and S-Adenosyl-L-methionine has been shown to restore the mitochondrial GSH levels in the liver and brain, consequently increasing the median survival and maximum lifespan of Npc1−/− mice (Torres et al., 2017; Goicoechea et al., 2024). Furthermore, treatment with Genistein enhanced lysosomal protein expression and autophagic flux, resulting in reduced p62 levels and increased levels of LC3-II in NP-C1 patient fibroblasts (Arguello et al., 2021). These findings indicate that strategies involving anti-inflammatory actions, oxidative stress mitigation, and autophagy activation could be considered as potential treatments for NP-C1 or as complementary therapeutic approaches.
Clinical trials
As previously noted, preclinical investigations into the efficacy of conventional small molecules for managing NP-C1 have predominantly centered on the mentioned medications. In recent years, significant strides have been made in clinical research concerning NP-C1. A review of clinical trial registrations indicates that investigations into the efficacy of HPβCD, Vorinostat, Lithium Carbonate, and other pharmaceuticals for NP-C1 are currently in progress (Table 2).
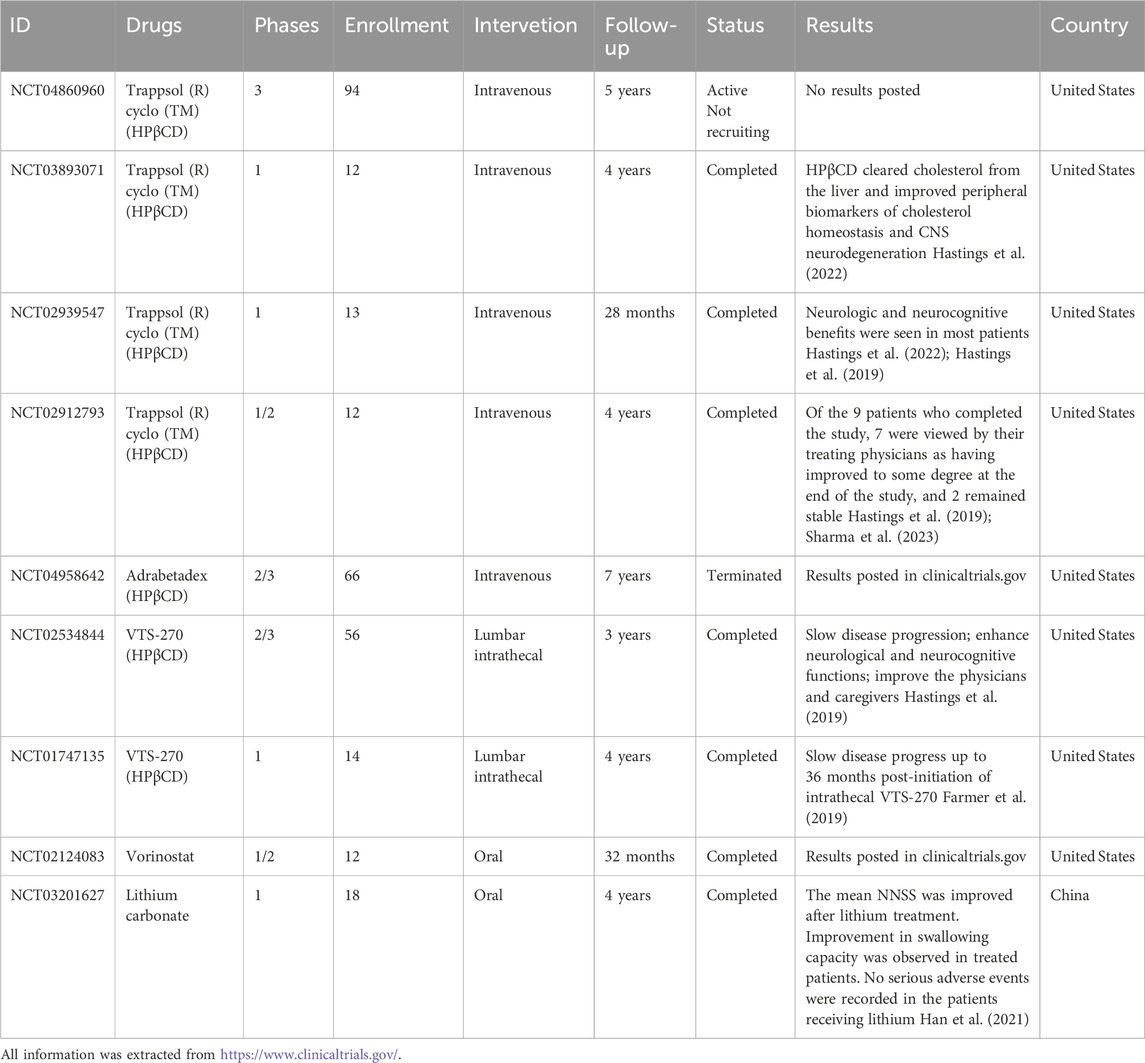
Table 2. Clinical trials for NP-C1.
HPβCD has been administered to NP-C1 patients with approved Investigational New Drugs (INDs) globally since 2009. In 2015, a clinical study on the intrathecal administration of HPβCD in a 12-year-old subject with mildly symptomatic NPC demonstrated that it was generally safe and well tolerated (Maarup et al., 2015). Subsequently, open-label phase I/IIa studies of VTS-270, a formulation of HPβCD, were conducted in individuals with NP-C1 across various age groups. The research demonstrated an acceptable safety profile for VTS-270 and offered evidence of restoring neuronal cholesterol homeostasis and decelerating the progression of neurological disease (Ory et al., 2017; Farmer et al., 2019). Several case reports studying the impact of intravenous administration of HPβCD in NP-C1 children and young adults demonstrate both the safety and potential benefits of HPβCD, including an improvement in liver function and cholesterol metabolism (Hastings et al., 2022; Hastings et al., 2019; Reynolds et al., 2021). However, HPβCD is excreted rapidly from the body and has poor penetration across the human blood-brain barrier. The long-term injection of HPβCD can pose challenges due to difficulties in metabolism, potentially resulting in negative effects such as increased hearing threshold (Ward et al., 2010), inner and outer hair cells (Takahashi et al., 2016; Zhou et al., 2018; Liu et al., 2020) and significant damage to the auditory and vestibular systems (Ding et al., 2020).
Furthermore, Vorinostat and Lithium Carbonate are being utilized in clinical research for treating NP-C1. Vorinostat, as an HDACi, faces challenges in crossing the blood-brain barrier, requiring higher dosages for clinical efficacy, which may pose significant risks to patients. In comparison, Lithium Carbonate shows inferior treatment outcomes compared to HPβCD (Han et al., 2021). Therefore, to establish these drugs as effective treatments for NP-C1, further researchs are essential to overcome these obstacles and optimize combined therapeutic strategies in NP-C1 treatment.
Combination therapy
Given the broad impact of NP-C1 on various organs and biological processes, a comprehensive treatment strategy targeting cholesterol accumulation with anti-inflammatory agents, antioxidants, and the promotion of autophagy holds great promise for addressing NP-C1. Studies have shown that non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin and ibuprofen can significantly prolong the lifespan of NPC1 mice and delay the onset of neuro-inflammation (Smith et al., 2009; Williams et al., 2014). Co-treatment with HPβCD and metformin has been found to reduce the inflammatory response in the liver, brain, and spleen of Npc1−/− mice, though it did not lead to an extension of survival time or an increase in body weight (Du et al., 2021). Furthermore, the beneficial effects of RIPK1 inhibition on Npc1−/− mice may be attributed to its role in neuroinflammation and cytokine production (Cougnoux et al., 2018b). While the combination of antioxidant drugs with HPβCD may reduce cholesterol accumulation, it does not improve NP-C1 lung pathology or alleviate cochlear damage and associated hearing loss caused by HPβCD treatment alone (Manohar et al., 2022; Erickson and Borbon, 2019). These studies show targeting inflammation in the brain represents a promising clinical intervention strategy (Table 3).
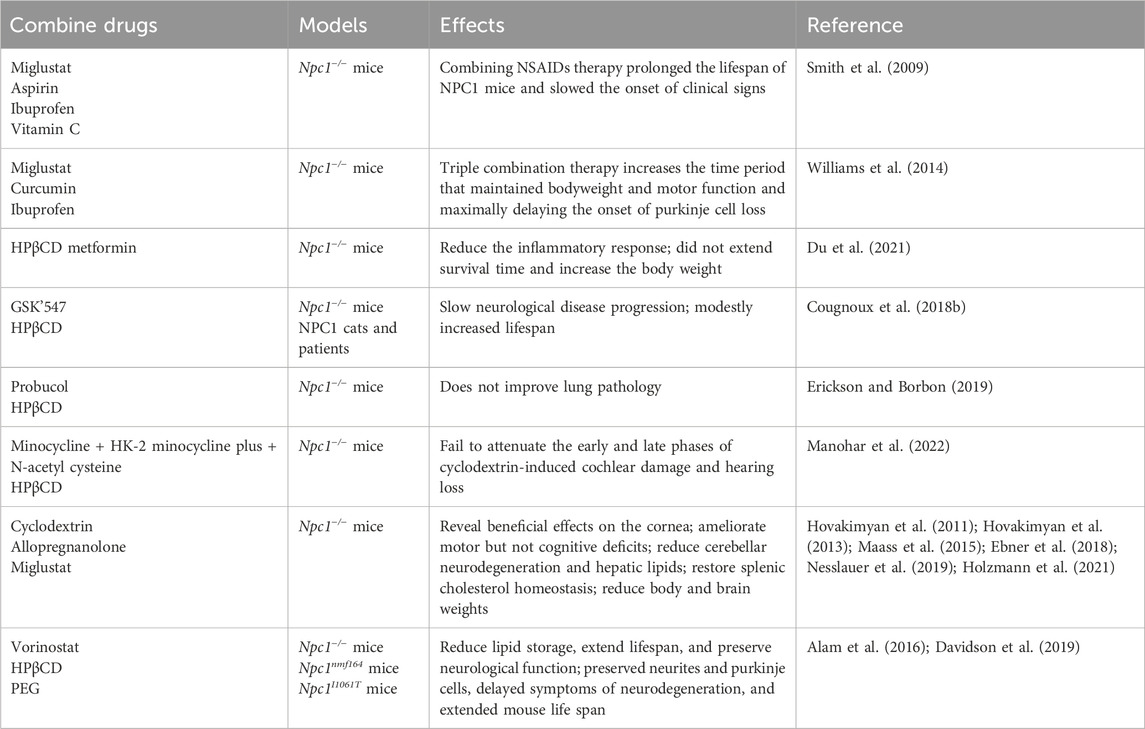
Table 3. Combination therapy in NP-C1 treatment.
Numerous studies have employed a combination therapy involving Cyclodextrin, Allopregnanolone, and Miglustat, unveiling positive effects across multiple domains. These include enhancements in corneal health, amelioration of motor deficits (although not cognitive impairments), reduction in cerebellar neurodegeneration and hepatic lipid accumulation, restoration of splenic cholesterol balance, and reductions in body and brain weights (Hovakimyan et al., 2011; Hovakimyan et al., 2013; Maass et al., 2015; Ebner et al., 2018; Nesslauer et al., 2019; Holzmann et al., 2021). Furthermore, the combination of HDAC inhibitors with HPβCD has shown promising therapeutic effects (Alam et al., 2016; Davidson et al., 2019) (Table 3).
Gene therapy
In recent years, advancements in gene editing technology and drug delivery have propelled gene therapy for NP-C1 as a promising treatment strategy aimed at correcting the defective NPC1 gene within the patient’s body. In studies using Npc1−/− mice, researchers developed AAV9 vectors to transport the NPC1 gene under the transcriptional control of neuronal-specific (CamKII) or ubiquitous (EF1a) promoters. The results showed that treatment with AAV9-EF1a-NPC1 led to improved survival, growth, and reduced hepatic cholesterol accumulation compared to AAV9-CamKII-NPC1 (Chandler et al., 2017). This suggests that systemic AAV gene delivery may be a preferred option for NPC1 therapy.
Systemic AAV9-mediated gene therapy can significantly extend lifespan, enhance Purkinje cell survival, restore locomotor activity and coordination, prevent or alleviate neurodegeneration, reduce biochemical abnormalities, and normalize various motor function indicators through different injection approaches (Xie et al., 2017; Hughes et al., 2018; Kurokawa et al., 2021). Moreover, the meticulous selection and enhancement of AAV, as demonstrated by AAV-PHP.B shell proteins, enable the effective transfer from the periphery to the CNS. This progression results in a notably superior alleviation of disease symptoms when contrasted with a similar AAV9 vector in Npc1m1N/m1N mice (Davidson et al., 2021).
Indeed, the development of non-viral gene delivery methods is crucial in advancing gene therapy for NP-C1. A study revealed that the DNA of NPC1 is encapsulated within Trojan horse lipoproteins (THLs) that selectively target organs using monoclonal antibodies. THLs treatment reduced tissue inclusion bodies in the brain and peripheral organs but did not extend the lifespan in Npc1−/− mice (Jiang et al., 2020). Messenger RNA holds great potential as a disease-modifying treatment for NP-C1. Engineered NPC1 mRNA with optimized codons and N1-methylpseudouridine base modification has been demonstrated to correct the cholesterol transport defect in NP-C1 patient cells, confirming the promising potential of engineered mRNA in the treatment of inherited disease (Furtado et al., 2022). Compared to miglustat and CDs treatments for NP-C1, currently, there have been no reports of adverse effects resulting from the over-expression of human NPC1 through gene therapy.
Cell-based therapy
Cell therapy is an innovative treatment approach that involves introducing healthy cells or repairing damaged cells to address a wide range of diseases, including neurodegenerative disorders. A study assessing the therapeutic impact of transplanted murine NSCs on Npc1−/− mice revealed that the implanted cells survived in the cerebellum and prolonged the lifespan of the mice. However, there was no significant improvement in body weight or ataxic symptoms, suggesting that the therapeutic effect of NSCs transplantation on NP-C1 is only partially effective (Ahmad et al., 2007). However, transplantation of human amniotic epithelial stem cells has been shown to extend the lifespan, reduce rapid weight loss, and decrease cholesterol deposition in NP-C1 mice, demonstrating promising therapeutic effects (Hong et al., 2012).
Mesenchymal stem cells (MSCs) have the ability to secrete various growth factors and contribute to tissue repair in diseases. Recent studies have shown that the transplantation of bone marrow-derived MSCs promotes the formation of neuronal networks with functional synaptic transmission, restores SphK activity, and reduces pathology in Purkinje neurons through the secretion of VEGF (Bae et al., 2005; Bae et al., 2007; Lee et al., 2014; Lee et al., 2010a). Transplantation of adipose tissue-derived MSCs has been shown to rescue Purkinje neurons, restore motor coordination, and alleviate inflammatory responses in NP-C1 mice (Bae et al., 2010). Human umbilical cord blood-derived MSCs have been demonstrated to protect against neuronal cell death and improve motor deficits by modulating neuroinflammatory conditions (Lee et al., 2010b; Seo et al., 2011). Additionally, these stem cells have the ability to suppress cholesterol synthesis and improve impaired autophagic flux in NP-C1 through the secretion of 14,15-epoxyeicosatrienoic acid (Kang et al., 2018). Recent research has suggested that conditioned medium from human menstrual blood-derived MSCs can protect against cell inflammation and apoptosis of Npc1 mutant neurons in vitro (Yang et al., 2023). Additionally, extracellular vesicles, which are important components of MSCs, have been demonstrated to reduce inflammation, decrease microglial and astrocyte proliferation, and modify the pathophysiological processes of NP-C1 (Van Hoecke et al., 2021). These findings offer new perspectives on the potential therapeutic use of MSCs and their extracellular vesicles in the management of NP-C1.
Conclusion and outlook
In this review, we offer a brief overview of the current research on disease models used for NP-C1. We also conduct a comprehensive analysis of both preclinical and clinical data related to NP-C1 treatment, including traditional chemical drug therapy, gene therapy, and cell-based therapy. By systematically presenting the range of treatment options for NP-C1, this review not only consolidates current knowledge but also highlights potential directions for future research and therapeutic interventions.
NP-C1 is a rare genetic disorder primarily caused by gene mutations, characterized by the degeneration of brain nerves and damage to various organs such as the liver and spleen. Treatment challenges arise due to the necessity for therapies to effectively penetrate the blood-brain barrier to address CNS damage. Currently, the primary treatments for NP-C1 are oral miglustat and intracranial injection of HPβCD. However, both of these methods can lead to some side effects. Hence, there is an urgent requirement to develop treatment strategies utilizing innovative small molecule therapeutics. The development of gene editing technology has enabled researchers to repair mutated genes in patients within the body through precise gene delivery. This direct gene repair method holds promise as a leading approach for treating various genetic disorders. However, the heterogeneity of the Npc1 gene among patients, coupled with the influence of diverse genetic backgrounds, leads to varying degrees of disease characteristics (Guatibonza Moreno et al., 2023). Therefore, analyzing the genetic backgrounds of individual patients is essential for the development of tailored treatments in the future (Las Heras et al., 2023). In NP-C1 patients, mutations in the Npc1 gene often present as single nucleotide variations that result in missense mutations. Advancements in next-generation genome editors, such as base and prime editors, have demonstrated the potential to correct these mutations in NP-C1, paving the way for personalized treatment approaches. Adeno-associated viruses have shown both safety and efficacy in the body, indicating that using AAV viruses as vectors for gene therapy could be a crucial treatment option for NP-C1. Furthermore, preclinical studies on cell-based therapy have demonstrated that mesenchymal stem cell transplantation can effectively preserve Purkinje cells in NP-C1.
While current treatments for NP-C1 primarily focus on drug therapies and do not provide a definitive cure, the emergence of diverse treatment options offers possibilities for the comprehensive management of the condition in the future. A multifaceted approach that combines gene therapy for repairing mutated genes at the DNA level, traditional drug adjuncts to enhance cholesterol excretion and cellular phenotypes, and mesenchymal stem cell transplantation to regulate tissues and organs through vital cytokine secretion may be crucial in effectively managing this complex disorder (Figure 2). Using a combination of treatment strategies could be crucial in addressing the intricate nature of NP-C1.
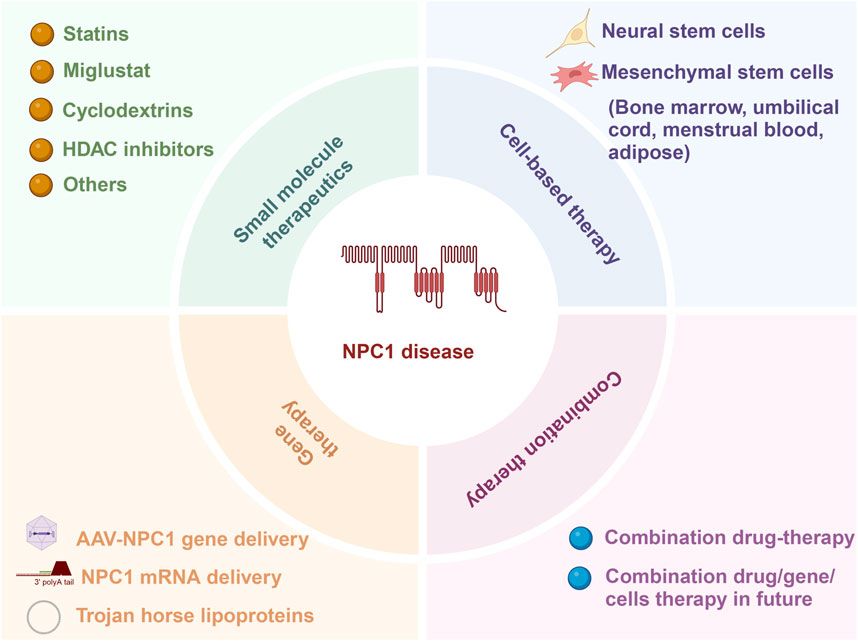
Figure 2. Current approaches for addressing NP-C1 encompass small molecule therapy, gene therapy, cell-based therapy, and combination therapy.
Author contributions
CZ: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing. KS: Writing–original draft. XJ: Writing–original draft. YT: Writing–original draft. KL: Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the research project on medical science and technology in Henan Province (LHGJ20210531).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
NP-C1, Niemann-Pick disease type C1; LE/LY, late endosomes and lysosomes; LDL, low-density lipoprotein; CNS, central nervous system; ER, endoplasmic reticulum; iPSCs, induced pluripotent stem cells; GSLs, glycosphingolipids; CSF, cerebrospinal fluid; ROS, reactive oxygen species; NPY, Pro-neuropeptide Y; HPβCD, 2-hydroxypropyl-β-cyclodextrin; CDs, Cyclodextrins; MβCD, Methyl-β-cyclodextrin; INDs, investigational new drugs; HDACi, Histone deacetylase inhibitors; VPA, valproic acid; NSCs, neural stem cells; HSP, heat shock protein; LSDs, lysosomal storage diseases; GSH, glutathione; NSAIDs, non-steroidal anti-inflammatory drugs; MSCs, mesenchymal stem cells.
References
Abi-Mosleh, L., Infante, R. E., Radhakrishnan, A., Goldstein, J. L., and Brown, M. S. (2009). Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. U. S. A. 106, 19316–19321. doi:10.1073/pnas.0910916106
Ahmad, I., Hunter, R. E., Flax, J. D., Snyder, E. Y., and Erickson, R. P. (2007). Neural stem cell implantation extends life in Niemann-Pick C1 mice. J. Appl. Genet. 48, 269–272. doi:10.1007/BF03195222
Alam, M. S., Getz, M., and Haldar, K. (2016). Chronic administration of an HDAC inhibitor treats both neurological and systemic Niemann-Pick type C disease in a mouse model. Sci. Transl. Med. 8, 326ra23. doi:10.1126/scitranslmed.aad9407
Alam, M. S., Getz, M., Yi, S., Kurkewich, J., Safeukui, I., and Haldar, K. (2014). Plasma signature of neurological disease in the monogenetic disorder Niemann-Pick Type C. J. Biol. Chem. 289, 8051–8066. doi:10.1074/jbc.M113.526392
Alvarez, A. R., Klein, A., Castro, J., Cancino, G. I., Amigo, J., Mosqueira, M., et al. (2008). Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 22, 3617–3627. doi:10.1096/fj.07-102715
Aqul, A., Liu, B., Ramirez, C. M., Pieper, A. A., Estill, S. J., Burns, D. K., et al. (2011). Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 31, 9404–9413. doi:10.1523/JNEUROSCI.1317-11.2011
Arguello, G., Balboa, E., Tapia, P. J., Castro, J., Yanez, M. J., Mattar, P., et al. (2021). Genistein activates transcription factor EB and corrects niemann-pick C phenotype. Int. J. Mol. Sci. 22, 4220. doi:10.3390/ijms22084220
Bae, J. S., Carter, J. E., and Jin, H. K. (2010). Adipose tissue-derived stem cells rescue Purkinje neurons and alleviate inflammatory responses in Niemann-Pick disease type C mice. Cell. Tissue Res. 340, 357–369. doi:10.1007/s00441-010-0942-3
Bae, J. S., Furuya, S., Shinoda, Y., Endo, S., Schuchman, E. H., Hirabayashi, Y., et al. (2005). Neurodegeneration augments the ability of bone marrow-derived mesenchymal stem cells to fuse with Purkinje neurons in Niemann-Pick type C mice. Hum. Gene Ther. 16, 1006–1011. doi:10.1089/hum.2005.16.1006
Bae, J. S., Han, H. S., Youn, D. H., Carter, J. E., Modo, M., Schuchman, E. H., et al. (2007). Bone marrow-derived mesenchymal stem cells promote neuronal networks with functional synaptic transmission after transplantation into mice with neurodegeneration. Stem Cells 25, 1307–1316. doi:10.1634/stemcells.2006-0561
Barthelemy, A., Demais, V., Stancu, I. C., Vasile, E., Houben, T., Reber, M., et al. (2021). Glial contribution to cyclodextrin-mediated reversal of cholesterol accumulation in murine NPC1-deficient neurons in vivo. Neurobiol. Dis. 158, 105469. doi:10.1016/j.nbd.2021.105469
Boland, S., Schmidt, U., Zagoriy, V., Sampaio, J. L., Fritsche, R. F., Czerwonka, R., et al. (2017). Phosphorylated glycosphingolipids essential for cholesterol mobilization in Caenorhabditis elegans. Nat. Chem. Biol. 13, 647–654. doi:10.1038/nchembio.2347
Bradbury, A., Bagel, J., Sampson, M., Farhat, N., Ding, W., Swain, G., et al. (2016). Cerebrospinal fluid Calbindin D concentration as a biomarker of cerebellar disease progression in niemann-pick type C1 disease. J. Pharmacol. Exp. Ther. 358, 254–261. doi:10.1124/jpet.116.232975
Brauer, A. U., Kuhla, A., Holzmann, C., Wree, A., and Witt, M. (2019). Current challenges in understanding the cellular and molecular mechanisms in niemann-pick disease type C1. Int. J. Mol. Sci. 20, 4392. doi:10.3390/ijms20184392
Cao, Q., Zhong, X. Z., Zou, Y., Zhang, Z., Toro, L., and Dong, X. P. (2015). BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev. Cell. 33, 427–441. doi:10.1016/j.devcel.2015.04.010
Chandler, R. J., Williams, I. M., Gibson, A. L., Davidson, C. D., Incao, A. A., Hubbard, B. T., et al. (2017). Systemic AAV9 gene therapy improves the lifespan of mice with Niemann-Pick disease, type C1. Hum. Mol. Genet. 26, 52–64. doi:10.1093/hmg/ddw367
Chao, C. C., Gutierrez-Vazquez, C., Rothhammer, V., Mayo, L., Wheeler, M. A., Tjon, E. C., et al. (2019). Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell. 179, 1483–1498. doi:10.1016/j.cell.2019.11.016
Chen, P. J., and Liu, D. R. (2023). Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 24, 161–177. doi:10.1038/s41576-022-00541-1
Chen, T. I., Hsu, P. C., Lee, N. C., Liu, Y. H., Wang, H. C., Lu, Y. H., et al. (2023). Loss of Flot2 expression in deep cerebellar nuclei neurons of mice with Niemann-Pick disease type C. Heliyon 9, e18082. doi:10.1016/j.heliyon.2023.e18082
Contreras, P. S., Gonzalez-Zuniga, M., Gonzalez-Hodar, L., Yanez, M. J., Dulcey, A., Marugan, J., et al. (2016). Neuronal gene repression in Niemann-Pick type C models is mediated by the c-Abl/HDAC2 signaling pathway. Biochim. Biophys. Acta 1859, 269–279. doi:10.1016/j.bbagrm.2015.11.006
Cougnoux, A., Clifford, S., Salman, A., Ng, S. L., Bertin, J., and Porter, F. D. (2018b). Necroptosis inhibition as a therapy for Niemann-Pick disease, type C1: inhibition of RIP kinases and combination therapy with 2-hydroxypropyl-β-cyclodextrin. Mol. Genet. Metab. 125, 345–350. doi:10.1016/j.ymgme.2018.10.009
Cougnoux, A., Cluzeau, C., Mitra, S., Li, R., Williams, I., Burkert, K., et al. (2016). Necroptosis in Niemann-Pick disease, type C1: a potential therapeutic target. Cell. Death Dis. 7, e2147. doi:10.1038/cddis.2016.16
Cougnoux, A., Drummond, R. A., Collar, A. L., Iben, J. R., Salman, A., Westgarth, H., et al. (2018a). Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum. Mol. Genet. 27, 2076–2089. doi:10.1093/hmg/ddy112
Crocker, A. C. (1961). The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J. Neurochem. 7, 69–80. doi:10.1111/j.1471-4159.1961.tb13499.x
Dai, S., Dulcey, A. E., Hu, X., Wassif, C. A., Porter, F. D., Austin, C. P., et al. (2017). Methyl-β-cyclodextrin restores impaired autophagy flux in Niemann-Pick C1-deficient cells through activation of AMPK. Autophagy 13, 1435–1451. doi:10.1080/15548627.2017.1329081
D’Arcangelo, G., Grossi, D., Racaniello, M., Cardinale, A., Zaratti, A., Rufini, S., et al. (2016). Miglustat reverts the impairment of synaptic plasticity in a mouse model of NPC disease. Neural Plast. 2016, 3830424. doi:10.1155/2016/3830424
Davidson, C. D., Ali, N. F., Micsenyi, M. C., Stephney, G., Renault, S., Dobrenis, K., et al. (2009). Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 4, e6951. doi:10.1371/journal.pone.0006951
Davidson, C. D., Gibson, A. L., Gu, T., Baxter, L. L., Deverman, B. E., Beadle, K., et al. (2021). Improved systemic AAV gene therapy with a neurotrophic capsid in Niemann-Pick disease type C1 mice. Life Sci. Alliance 4, e202101040. doi:10.26508/lsa.202101040
Davidson, J., Molitor, E., Moores, S., Gale, S. E., Subramanian, K., Jiang, X., et al. (2019). 2-Hydroxypropyl-β-cyclodextrin is the active component in a triple combination formulation for treatment of Niemann-Pick C1 disease. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1864, 1545–1561. doi:10.1016/j.bbalip.2019.04.011
Ding, D., Manohar, S., Jiang, H., and Salvi, R. (2020). Hydroxypropyl-β-cyclodextrin causes massive damage to the developing auditory and vestibular system. Hear Res. 396, 108073. doi:10.1016/j.heares.2020.108073
Di Rocco, M., Dardis, A., Madeo, A., Barone, R., and Fiumara, A. (2012). Early miglustat therapy in infantile Niemann-Pick disease type C. Pediatr. Neurol. 47, 40–43. doi:10.1016/j.pediatrneurol.2012.04.005
Du, J., Liu, X., Zhang, Y., Han, X., Ma, C., Liu, Y., et al. (2021). The effects of combined therapy with metformin and hydroxypropyl-β-cyclodextrin in a mouse model of niemann-pick disease type C1. Front. Pharmacol. 12, 825425. doi:10.3389/fphar.2021.825425
Du, X., Lukmantara, I., and Yang, H. (2017). CRISPR/Cas9-Mediated generation of niemann-pick C1 knockout cell line. Methods Mol. Biol. 1583, 73–83. doi:10.1007/978-1-4939-6875-6_7
Ebner, L., Glaser, A., Brauer, A., Witt, M., Wree, A., Rolfs, A., et al. (2018). Evaluation of two liver treatment strategies in a mouse model of niemann-pick-disease type C1. Int. J. Mol. Sci. 19, 972. doi:10.3390/ijms19040972
Elrick, M. J., Pacheco, C. D., Yu, T., Dadgar, N., Shakkottai, V. G., Ware, C., et al. (2010). Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 19, 837–847. doi:10.1093/hmg/ddp552
Encarnacao, M., Ribeiro, I., David, H., Coutinho, M. F., Quelhas, D., and Alves, S. (2023). Challenges in the definitive diagnosis of niemann-pick type C-leaky variants and alternative transcripts. Genes (Basel) 14, 1990. doi:10.3390/genes14111990
Erickson, R. P., and Borbon, I. A. (2019). Co-treatment with probucol does not improve lung pathology in hydroxypropyl-β-cyclodextrin-treated Npc1-/- mice. J. Appl. Genet. 60, 175–178. doi:10.1007/s13353-019-00487-x
Erickson, R. P., Deutsch, G., and Patil, R. (2018). A pilot study of direct delivery of hydroxypropyl-beta-cyclodextrin to the lung by the nasal route in a mouse model of Niemann-Pick C1 disease: motor performance is unaltered and lung disease is worsened. J. Appl. Genet. 59, 187–191. doi:10.1007/s13353-018-0431-z
Erwood, S., Bily, T. M. I., Lequyer, J., Yan, J., Gulati, N., Brewer, R. A., et al. (2022). Saturation variant interpretation using CRISPR prime editing. Nat. Biotechnol. 40, 885–895. doi:10.1038/s41587-021-01201-1
Erwood, S., Brewer, R. A., Bily, T. M. I., Maino, E., Zhou, L., Cohn, R. D., et al. (2019). Modeling Niemann-Pick disease type C in a human haploid cell line allows for patient variant characterization and clinical interpretation. Genome Res. 29, 2010–2019. doi:10.1101/gr.250720.119
Fan, M., Sidhu, R., Fujiwara, H., Tortelli, B., Zhang, J., Davidson, C., et al. (2013). Identification of Niemann-Pick C1 disease biomarkers through sphingolipid profiling. J. Lipid Res. 54, 2800–2814. doi:10.1194/jlr.M040618
Farmer, C. A., Thurm, A., Farhat, N., Bianconi, S., Keener, L. A., and Porter, F. D. (2019). Long-term neuropsychological outcomes from an open-label phase I/IIa trial of 2-Hydroxypropyl-β-Cyclodextrins (VTS-270) in niemann-pick disease, type C1. CNS Drugs 33, 677–683. doi:10.1007/s40263-019-00642-2
Frain, K. M., Dedic, E., Nel, L., Bohush, A., Olesen, E., Thaysen, K., et al. (2024). Conformational changes in the Niemann-Pick type C1 protein NCR1 drive sterol translocation. Proc. Natl. Acad. Sci. U. S. A. 121, e2315575121. doi:10.1073/pnas.2315575121
Fukaura, M., Ishitsuka, Y., Shirakawa, S., Ushihama, N., Yamada, Y., Kondo, Y., et al. (2021). Intracerebroventricular treatment with 2-Hydroxypropyl-β-Cyclodextrin decreased cerebellar and hepatic glycoprotein nonmetastatic melanoma protein B (GPNMB) expression in niemann-pick disease type C model mice. Int. J. Mol. Sci. 22, 452. doi:10.3390/ijms22010452
Furtado, D., Cortez-Jugo, C., Hung, Y. H., Bush, A. I., and Caruso, F. (2022). mRNA treatment rescues niemann-pick disease type C1 in patient fibroblasts. Mol. Pharm. 19, 3987–3999. doi:10.1021/acs.molpharmaceut.2c00463
Goicoechea, L., Torres, S., Fabrega, L., Barrios, M., Nunez, S., Casas, J., et al. (2024). S-Adenosyl-l-methionine restores brain mitochondrial membrane fluidity and GSH content improving Niemann-Pick type C disease. Redox Biol. 72, 103150. doi:10.1016/j.redox.2024.103150
Gray, J., Fernandez-Suarez, M. E., Falah, M., Smith, D., Smith, C., Kaya, E., et al. (2022). Heat shock protein amplification improves cerebellar myelination in the Npc1(nih) mouse model. EBioMedicine 86, 104374. doi:10.1016/j.ebiom.2022.104374
Guatibonza Moreno, P., Pardo, L. M., Pereira, C., Schroeder, S., Vagiri, D., Almeida, L. S., et al. (2023). At a glance: the largest Niemann-Pick type C1 cohort with 602 patients diagnosed over 15 years. Eur. J. Hum. Genet. 31, 1108–1116. doi:10.1038/s41431-023-01408-7
Han, S., Wang, Q., Song, Y., Pang, M., Ren, C., Wang, J., et al. (2023). Lithium ameliorates Niemann-Pick C1 disease phenotypes by impeding STING/SREBP2 activation. iScience 26, 106613. doi:10.1016/j.isci.2023.106613
Han, S., Zhang, H., Yi, M., Liu, X., Maegawa, G. H. B., Zou, Y., et al. (2021). Potential disease-modifying effects of lithium carbonate in niemann-pick disease, type C1. Front. Pharmacol. 12, 667361. doi:10.3389/fphar.2021.667361
Hastings, C., Liu, B., Hurst, B., Cox, G. F., and Hrynkow, S. (2022). Intravenous 2-hydroxypropyl-β-cyclodextrin (Trappsol® Cyclo™) demonstrates biological activity and impacts cholesterol metabolism in the central nervous system and peripheral tissues in adult subjects with Niemann-Pick Disease Type C1: results of a phase 1 trial. Mol. Genet. Metab. 137, 309–319. doi:10.1016/j.ymgme.2022.10.004
Hastings, C., Vieira, C., Liu, B., Bascon, C., Gao, C., Wang, R. Y., et al. (2019). Expanded access with intravenous hydroxypropyl-β-cyclodextrin to treat children and young adults with Niemann-Pick disease type C1: a case report analysis. Orphanet J. Rare Dis. 14, 228. doi:10.1186/s13023-019-1207-1
Helquist, P., Maxfield, F. R., Wiech, N. L., and Wiest, O. (2013). Treatment of Niemann--pick type C disease by histone deacetylase inhibitors. Neurotherapeutics 10, 688–697. doi:10.1007/s13311-013-0217-2
Heron, B., Valayannopoulos, V., Baruteau, J., Chabrol, B., Ogier, H., Latour, P., et al. (2012). Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C. Orphanet J. Rare Dis. 7, 36. doi:10.1186/1750-1172-7-36
Hoglinger, D., Burgoyne, T., Sanchez-Heras, E., Hartwig, P., Colaco, A., Newton, J., et al. (2019). NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun. 10, 4276. doi:10.1038/s41467-019-12152-2
Holzmann, C., Witt, M., Rolfs, A., Antipova, V., and Wree, A. (2021). Gender-specific effects of two treatment strategies in a mouse model of niemann-pick disease type C1. Int. J. Mol. Sci. 22, 2539. doi:10.3390/ijms22052539
Hong, S. B., Seo, M. S., Park, S. B., Seo, Y. J., Kim, J. S., and Kang, K. S. (2012). Therapeutic effects of human amniotic epithelial stem cells in Niemann-Pick type C1 mice. Cytotherapy 14, 630–638. doi:10.3109/14653249.2012.663485
Houben, T., Magro Dos Reis, I., Oligschlaeger, Y., Steinbusch, H., Gijbels, M. J. J., Hendrikx, T., et al. (2018). Pneumococcal immunization reduces neurological and hepatic symptoms in a mouse model for niemann-pick type C1 disease. Front. Immunol. 9, 3089. doi:10.3389/fimmu.2018.03089
Hovakimyan, M., Maass, F., Petersen, J., Holzmann, C., Witt, M., Lukas, J., et al. (2013). Combined therapy with cyclodextrin/allopregnanolone and miglustat improves motor but not cognitive functions in Niemann-Pick Type C1 mice. Neuroscience 252, 201–211. doi:10.1016/j.neuroscience.2013.08.001
Hovakimyan, M., Petersen, J., Maass, F., Reichard, M., Witt, M., Lukas, J., et al. (2011). Corneal alterations during combined therapy with cyclodextrin/allopregnanolone and miglustat in a knock-out mouse model of NPC1 disease. PLoS One 6, e28418. doi:10.1371/journal.pone.0028418
Hughes, M. P., Smith, D. A., Morris, L., Fletcher, C., Colaco, A., Huebecker, M., et al. (2018). AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann-Pick type C1 disease. Hum. Mol. Genet. 27, 3079–3098. doi:10.1093/hmg/ddy212
Hung, Y. H., Faux, N. G., Killilea, D. W., Yanjanin, N., Firnkes, S., Volitakis, I., et al. (2014). Altered transition metal homeostasis in Niemann-Pick disease, type C1. Metallomics 6, 542–553. doi:10.1039/c3mt00308f
Infante, R. E., Radhakrishnan, A., Abi-Mosleh, L., Kinch, L. N., Wang, M. L., Grishin, N. V., et al. (2008a). Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 283, 1064–1075. doi:10.1074/jbc.M707944200
Infante, R. E., Wang, M. L., Radhakrishnan, A., Kwon, H. J., Brown, M. S., and Goldstein, J. L. (2008b). NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. U. S. A. 105, 15287–15292. doi:10.1073/pnas.0807328105
Jia, Z., Yang, M., Zhao, Y., Li, X., Yang, C., Qiao, L., et al. (2023). CRISPR-Cas9-Mediated NPC1 gene deletion enhances HEK 293 T cell adhesion by regulating E-cadherin. Mol. Biotechnol. 65, 252–262. doi:10.1007/s12033-022-00503-2
Jiang, D., Lee, H., and Pardridge, W. M. (2020). Plasmid DNA gene therapy of the Niemann-Pick C1 mouse with transferrin receptor-targeted Trojan horse liposomes. Sci. Rep. 10, 13334. doi:10.1038/s41598-020-70290-w
Jiang, H., Sidhu, R., Fujiwara, H., De Meulder, M., de Vries, R., Gong, Y., et al. (2014). Development and validation of sensitive LC-MS/MS assays for quantification of HP-β-CD in human plasma and CSF. J. Lipid Res. 55, 1537–1548. doi:10.1194/jlr.D050278
Jung, Y., Lee, S. E., Kang, I., Cho, S. M., Kang, K. S., and Kwon, H. J. (2022). Upregulation of SNAP25 by HDAC inhibition ameliorates Niemann-Pick Type C disease phenotypes via autophagy induction. Clin. Transl. Med. 12, e776. doi:10.1002/ctm2.776
Kang, I., Lee, B. C., Lee, J. Y., Kim, J. J., Sung, E. A., Lee, S. E., et al. (2018). Stem cell-secreted 14,15- epoxyeicosatrienoic acid rescues cholesterol homeostasis and autophagic flux in Niemann-Pick-type C disease. Exp. Mol. Med. 50, 1–14. doi:10.1038/s12276-018-0176-0
Kim, S. J., Lee, B. H., Lee, Y. S., and Kang, K. S. (2007). Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 360, 593–599. doi:10.1016/j.bbrc.2007.06.116
Klein, A. D., Oyarzun, J. E., Cortez, C., and Zanlungo, S. (2018). Gadolinium chloride rescues Niemann(-)Pick type C liver damage. Int. J. Mol. Sci. 19, 3599. doi:10.3390/ijms19113599
Kulinski, A., and Vance, J. E. (2007). Lipid homeostasis and lipoprotein secretion in Niemann-Pick C1-deficient hepatocytes. J. Biol. Chem. 282, 1627–1637. doi:10.1074/jbc.M610001200
Kunkel, T. J., Townsend, A., Sullivan, K. A., Merlet, J., Schuchman, E. H., Jacobson, D. A., et al. (2023). The cholesterol transporter NPC1 is essential for epigenetic regulation and maturation of oligodendrocyte lineage cells. Nat. Commun. 14, 3964. doi:10.1038/s41467-023-39733-6
Kurokawa, Y., Osaka, H., Kouga, T., Jimbo, E., Muramatsu, K., Nakamura, S., et al. (2021). Gene therapy in a mouse model of niemann-pick disease type C1. Hum. Gene Ther. 32, 589–598. doi:10.1089/hum.2020.175
Kwon, H. J., Abi-Mosleh, L., Wang, M. L., Deisenhofer, J., Goldstein, J. L., Brown, M. S., et al. (2009). Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 137, 1213–1224. doi:10.1016/j.cell.2009.03.049
Lamri, A., Pigeyre, M., Garver, W. S., and Meyre, D. (2018). The extending spectrum of NPC1-related human disorders: from niemann-pick C1 disease to obesity. Endocr. Rev. 39, 192–220. doi:10.1210/er.2017-00176
Las Heras, M., Szenfeld, B., Ballout, R. A., Buratti, E., Zanlungo, S., Dardis, A., et al. (2023). Understanding the phenotypic variability in Niemann-Pick disease type C (NPC): a need for precision medicine. NPJ Genom Med. 8, 21. doi:10.1038/s41525-023-00365-w
Lee, D., and Hong, J. H. (2023). Niemann-Pick disease type C (NPDC) by mutation of NPC1 and NPC2: aberrant lysosomal cholesterol trafficking and oxidative stress. Antioxidants (Basel) 12, 2021. doi:10.3390/antiox12122021
Lee, H., Bae, J. S., and Jin, H. K. (2010b). Human umbilical cord blood-derived mesenchymal stem cells improve neurological abnormalities of Niemann-Pick type C mouse by modulation of neuroinflammatory condition. J. Vet. Med. Sci. 72, 709–717. doi:10.1292/jvms.09-0495
Lee, H., Lee, J. K., Min, W. K., Bae, J. H., He, X., Schuchman, E. H., et al. (2010a). Bone marrow-derived mesenchymal stem cells prevent the loss of Niemann-Pick type C mouse Purkinje neurons by correcting sphingolipid metabolism and increasing sphingosine-1-phosphate. Stem Cells 28, 821–831. doi:10.1002/stem.401
Lee, H., Lee, J. K., Park, M. H., Hong, Y. R., Marti, H. H., Kim, H., et al. (2014). Pathological roles of the VEGF/SphK pathway in Niemann-Pick type C neurons. Nat. Commun. 5, 5514. doi:10.1038/ncomms6514
Li, W., Pergande, M. R., Crutchfield, C. A., Searle, B. C., Backlund, P. S., Picache, J. A., et al. (2023). A differential proteomics study of cerebrospinal fluid from individuals with Niemann-Pick disease, Type C1. Proteomics 23, e2200378. doi:10.1002/pmic.202200378
Liu, B., Ramirez, C. M., Miller, A. M., Repa, J. J., Turley, S. D., and Dietschy, J. M. (2010). Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 51, 933–944. doi:10.1194/jlr.M000257
Liu, B., Turley, S. D., Burns, D. K., Miller, A. M., Repa, J. J., and Dietschy, J. M. (2009). Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl. Acad. Sci. U. S. A. 106, 2377–2382. doi:10.1073/pnas.0810895106
Liu, X., Ding, D., Chen, G. D., Li, L., Jiang, H., and Salvi, R. (2020). 2-Hydroxypropyl-β-cyclodextrin ototoxicity in adult rats: rapid onset and massive destruction of both inner and outer hair cells above a critical dose. Neurotox. Res. 38, 808–823. doi:10.1007/s12640-020-00252-7
Lloyd-Evans, E., Morgan, A. J., He, X., Smith, D. A., Elliot-Smith, E., Sillence, D. J., et al. (2008). Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255. doi:10.1038/nm.1876
Lloyd-Evans, E., and Platt, F. M. (2010). Lipids on trial: the search for the offending metabolite in Niemann-Pick type C disease. Traffic 11, 419–428. doi:10.1111/j.1600-0854.2010.01032.x
Loftus, S. K., Morris, J. A., Carstea, E. D., Gu, J. Z., Cummings, C., Brown, A., et al. (1997). Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 277, 232–235. doi:10.1126/science.277.5323.232
Lopez, A. M., Terpack, S. J., Posey, K. S., Liu, B., Ramirez, C. M., and Turley, S. D. (2014). Systemic administration of 2-hydroxypropyl-β-cyclodextrin to symptomatic Npc1-deficient mice slows cholesterol sequestration in the major organs and improves liver function. Clin. Exp. Pharmacol. Physiol. 41, 780–787. doi:10.1111/1440-1681.12285
Maarup, T. J., Chen, A. H., Porter, F. D., Farhat, N. Y., Ory, D. S., Sidhu, R., et al. (2015). Intrathecal 2-hydroxypropyl-beta-cyclodextrin in a single patient with Niemann-Pick C1. Mol. Genet. Metab. 116, 75–79. doi:10.1016/j.ymgme.2015.07.001
Maass, F., Petersen, J., Hovakimyan, M., Schmitt, O., Witt, M., Hawlitschka, A., et al. (2015). Reduced cerebellar neurodegeneration after combined therapy with cyclodextrin/allopregnanolone and miglustat in NPC1: a mouse model of Niemann-Pick type C1 disease. J. Neurosci. Res. 93, 433–442. doi:10.1002/jnr.23509
Malara, M., Prestel, M., and Tahirovic, S. (2024). Endo-lysosomal dysfunction and neuronal-glial crosstalk in Niemann-Pick type C disease. Philos. Trans. R. Soc. Lond B Biol. Sci. 379, 20220388. doi:10.1098/rstb.2022.0388
Manohar, S., Ding, D., Jiang, H., Li, L., Chen, G. D., Kador, P., et al. (2022). Combined antioxidants and anti-inflammatory therapies fail to attenuate the early and late phases of cyclodextrin-induced cochlear damage and hearing loss. Hear Res. 414, 108409. doi:10.1016/j.heares.2021.108409
Marschalek, N., Albert, F., Meske, V., and Ohm, T. G. (2014). The natural history of cerebellar degeneration of Niemann-Pick C mice monitored in vitro. Neuropathol. Appl. Neurobiol. 40, 933–945. doi:10.1111/nan.12154
Marshall, C. A., Borbon, I. A., and Erickson, R. P. (2017). Relative efficacy of nicotinamide treatment of a mouse model of infantile Niemann-Pick C1 disease. J. Appl. Genet. 58, 99–102. doi:10.1007/s13353-016-0367-0
Maue, R. A., Burgess, R. W., Wang, B., Wooley, C. M., Seburn, K. L., Vanier, M. T., et al. (2012). A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 21, 730–750. doi:10.1093/hmg/ddr505
Mengel, E., Klunemann, H. H., Lourenco, C. M., Hendriksz, C. J., Sedel, F., Walterfang, M., et al. (2013). Niemann-Pick disease type C symptomatology: an expert-based clinical description. Orphanet J. Rare Dis. 8, 166. doi:10.1186/1750-1172-8-166
Meske, V., Albert, F., Gerstenberg, S., Kallwellis, K., and Ohm, T. G. (2019). NPC1-deficient neurons are selectively vulnerable for statin treatment. Neuropharmacology 151, 159–170. doi:10.1016/j.neuropharm.2019.04.012
Min, J. O., Ho, H. A., Lee, W., Jung, B. C., Park, S. J., Kim, S., et al. (2023). Statins suppress cell-to-cell propagation of α-synuclein by lowering cholesterol. Cell. Death Dis. 14, 474. doi:10.1038/s41419-023-05977-9
Munkacsi, A. B., Chen, F. W., Brinkman, M. A., Higaki, K., Gutierrez, G. D., Chaudhari, J., et al. (2011). An “exacerbate-reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J. Biol. Chem. 286, 23842–23851. doi:10.1074/jbc.M111.227645
Munkacsi, A. B., Hammond, N., Schneider, R. T., Senanayake, D. S., Higaki, K., Lagutin, K., et al. (2017). Normalization of hepatic homeostasis in the npc1(nmf164) mouse model of niemann-pick type C disease treated with the histone deacetylase inhibitor vorinostat. J. Biol. Chem. 292, 4395–4410. doi:10.1074/jbc.M116.770578
Muralidhar, A., Borbon, I. A., Esharif, D. M., Ke, W., Manacheril, R., Daines, M., et al. (2011). Pulmonary function and pathology in hydroxypropyl-beta-cyclodextin-treated and untreated Npc1(-)/(-) mice. Mol. Genet. Metab. 103, 142–147. doi:10.1016/j.ymgme.2011.03.001
Nadjar, Y., Hutter-Moncada, A. L., Latour, P., Ayrignac, X., Kaphan, E., Tranchant, C., et al. (2018). Adult Niemann-Pick disease type C in France: clinical phenotypes and long-term miglustat treatment effect. Orphanet J. Rare Dis. 13, 175. doi:10.1186/s13023-018-0913-4
Naureckiene, S., Sleat, D. E., Lackland, H., Fensom, A., Vanier, M. T., Wattiaux, R., et al. (2000). Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290, 2298–2301. doi:10.1126/science.290.5500.2298
Nesslauer, A. M., Glaser, A., Graler, M., Engelmann, R., Muller-Hilke, B., Frank, M., et al. (2019). A therapy with miglustat, 2-hydroxypropyl-ß-cyclodextrin and allopregnanolone restores splenic cholesterol homeostasis in Niemann-pick disease type C1. Lipids Health Dis. 18, 146. doi:10.1186/s12944-019-1088-2
Newton, J., Hait, N. C., Maceyka, M., Colaco, A., Maczis, M., Wassif, C. A., et al. (2017). FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. 31, 1719–1730. doi:10.1096/fj.201601041R
Ory, D. S., Ottinger, E. A., Farhat, N. Y., King, K. A., Jiang, X., Weissfeld, L., et al. (2017). Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1-2 trial. Lancet 390, 1758–1768. doi:10.1016/S0140-6736(17)31465-4
Pallottini, V., and Pfrieger, F. W. (2020). Understanding and treating niemann-pick type C disease: models matter. Int. J. Mol. Sci. 21, 8979. doi:10.3390/ijms21238979
Patterson, M. C., and Platt, F. (2004). Therapy of Niemann-Pick disease, type C. Biochim. Biophys. Acta 1685, 77–82. doi:10.1016/j.bbalip.2004.08.013
Patterson, M. C., and Walkley, S. U. (2017). Niemann-Pick disease, type C and roscoe brady. Mol. Genet. Metab. 120, 34–37. doi:10.1016/j.ymgme.2016.11.008
Peake, K. B., and Vance, J. E. (2012). Normalization of cholesterol homeostasis by 2-hydroxypropyl-β-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 287, 9290–9298. doi:10.1074/jbc.M111.326405
Pentchev, P. G., Boothe, A. D., Kruth, H. S., Weintroub, H., Stivers, J., and Brady, R. O. (1984). A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J. Biol. Chem. 259, 5784–5791. doi:10.1016/s0021-9258(18)91082-3
Pentchev, P. G., Comly, M. E., Kruth, H. S., Patel, S., Proestel, M., and Weintroub, H. (1986). The cholesterol storage disorder of the mutant BALB/c mouse. A primary genetic lesion closely linked to defective esterification of exogenously derived cholesterol and its relationship to human type C Niemann-Pick disease. J. Biol. Chem. 261, 2772–2777. doi:10.1016/s0021-9258(17)35852-0
Peter, F., Rost, S., Rolfs, A., and Frech, M. J. (2017). Activation of PKC triggers rescue of NPC1 patient specific iPSC derived glial cells from gliosis. Orphanet J. Rare Dis. 12, 145. doi:10.1186/s13023-017-0697-y
Phillips, S. E., Woodruff, E. A., Liang, P., Patten, M., and Broadie, K. (2008). Neuronal loss of Drosophila NPC1a causes cholesterol aggregation and age-progressive neurodegeneration. J. Neurosci. 28, 6569–6582. doi:10.1523/JNEUROSCI.5529-07.2008
Pineda, M., Walterfang, M., and Patterson, M. C. (2018). Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J. Rare Dis. 13, 140. doi:10.1186/s13023-018-0844-0
Pipalia, N. H., Cosner, C. C., Huang, A., Chatterjee, A., Bourbon, P., Farley, N., et al. (2011). Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 108, 5620–5625. doi:10.1073/pnas.1014890108
Pipalia, N. H., Saad, S. Z., Subramanian, K., Cross, A., Al-Motawa, A., Garg, K., et al. (2021). HSP90 inhibitors reduce cholesterol storage in Niemann-Pick type C1 mutant fibroblasts. J. Lipid Res. 62, 100114. doi:10.1016/j.jlr.2021.100114
Pipalia, N. H., Subramanian, K., Mao, S., Ralph, H., Hutt, D. M., Scott, S. M., et al. (2017). Histone deacetylase inhibitors correct the cholesterol storage defect in most Niemann-Pick C1 mutant cells. J. Lipid Res. 58, 695–708. doi:10.1194/jlr.M072140
Prabhu, A. V., Kang, I., De Pace, R., Wassif, C. A., Fujiwara, H., Kell, P., et al. (2021). A human iPSC-derived inducible neuronal model of Niemann-Pick disease, type C1. BMC Biol. 19, 218. doi:10.1186/s12915-021-01133-x
Praggastis, M., Tortelli, B., Zhang, J., Fujiwara, H., Sidhu, R., Chacko, A., et al. (2015). A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 35, 8091–8106. doi:10.1523/JNEUROSCI.4173-14.2015
Pugach, E. K., Feltes, M., Kaufman, R. J., Ory, D. S., and Bang, A. G. (2018). High-content screen for modifiers of Niemann-Pick type C disease in patient cells. Hum. Mol. Genet. 27, 2101–2112. doi:10.1093/hmg/ddy117
Quelle-Regaldie, A., Gandoy-Fieiras, N., Rodriguez-Villamayor, P., Maceiras, S., Losada, A. P., Folgueira, M., et al. (2023). Severe neurometabolic phenotype in npc1-/- zebrafish with a C-terminal mutation. Front. Mol. Neurosci. 16, 1078634. doi:10.3389/fnmol.2023.1078634
Rakib, T. M., Islam, M. S., Uddin, M. M., Rahman, M. M., Yabuki, A., Yamagami, T., et al. (2023). Novel mutation in the feline NPC2 gene in cats with niemann-pick disease. Anim. (Basel) 13, 1744. doi:10.3390/ani13111744
Ramirez, C. M., Liu, B., Taylor, A. M., Repa, J. J., Burns, D. K., Weinberg, A. G., et al. (2010). Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr. Res. 68, 309–315. doi:10.1203/PDR.0b013e3181ee4dd2
Repa, J. J., Li, H., Frank-Cannon, T. C., Valasek, M. A., Turley, S. D., Tansey, M. G., et al. (2007). Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J. Neurosci. 27, 14470–14480. doi:10.1523/JNEUROSCI.4823-07.2007
Reynolds, M., Linneman, L. A., Luna, S., Warner, B. B., Turmelle, Y. P., Kulkarni, S. S., et al. (2021). A phase 1/2 open label nonrandomized clinical trial of intravenous 2-hydroxypropyl-β-cyclodextrin for acute liver disease in infants with Niemann-Pick C1. Mol. Genet. Metab. Rep. 28, 100772. doi:10.1016/j.ymgmr.2021.100772
Rodriguez-Gil, J. L., Baxter, L. L., Watkins-Chow, D. E., Johnson, N. L., Davidson, C. D., Carlson, S. R., et al. (2021). Transcriptome of HPβCD-treated Niemann-Pick disease type C1 cells highlights GPNMB as a biomarker for therapeutics. Hum. Mol. Genet. 30, 2456–2468. doi:10.1093/hmg/ddab194
Roney, J. C., Li, S., Farfel-Becker, T., Huang, N., Sun, T., Xie, Y., et al. (2021). Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C. Dev. Cell. 56, 1452–1468.e8. doi:10.1016/j.devcel.2021.03.032
Sarkar, S., Carroll, B., Buganim, Y., Maetzel, D., Ng, A. H., Cassady, J. P., et al. (2013). Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell. Rep. 5, 1302–1315. doi:10.1016/j.celrep.2013.10.042
Schwend, T., Loucks, E. J., Snyder, D., and Ahlgren, S. C. (2011). Requirement of Npc1 and availability of cholesterol for early embryonic cell movements in zebrafish. J. Lipid Res. 52, 1328–1344. doi:10.1194/jlr.M012377
Schwerd, T., Pandey, S., Yang, H. T., Bagola, K., Jameson, E., Jung, J., et al. (2017). Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann-Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn's disease. Gut 66, 1060–1073. doi:10.1136/gutjnl-2015-310382
Scott, C., and Ioannou, Y. A. (2004). The NPC1 protein: structure implies function. Biochim. Biophys. Acta 1685, 8–13. doi:10.1016/j.bbalip.2004.08.006
Seo, Y., Yang, S. R., Jee, M. K., Joo, E. K., Roh, K. H., Seo, M. S., et al. (2011). Human umbilical cord blood-derived mesenchymal stem cells protect against neuronal cell death and ameliorate motor deficits in Niemann Pick type C1 mice. Cell. Transpl. 20, 1033–1047. doi:10.3727/096368910X545086
Sharma, R., Hastings, C., Staretz-Chacham, O., Raiman, J., Paucar, M., Spiegel, R., et al. (2023). Long-term administration of intravenous Trappsol® Cyclo™ (HP-β-CD) results in clinical benefits and stabilization or slowing of disease progression in patients with Niemann-Pick disease type C1: results of an international 48-week Phase I/II trial. Mol. Genet. Metab. Rep. 36, 100988. doi:10.1016/j.ymgmr.2023.100988
Singhal, A., Szente, L., Hildreth, J. E. K., and Song, B. (2018). Hydroxypropyl-beta and -gamma cyclodextrins rescue cholesterol accumulation in Niemann-Pick C1 mutant cell via lysosome-associated membrane protein 1. Cell. Death Dis. 9, 1019. doi:10.1038/s41419-018-1056-1
Smith, D., Wallom, K. L., Williams, I. M., Jeyakumar, M., and Platt, F. M. (2009). Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol. Dis. 36, 242–251. doi:10.1016/j.nbd.2009.07.010
Solomon, B. I., Smith, A. C., Sinaii, N., Farhat, N., King, M. C., Machielse, L., et al. (2020). Association of miglustat with swallowing outcomes in niemann-pick disease, type C1. JAMA Neurol. 77, 1564–1568. doi:10.1001/jamaneurol.2020.3241
Somers, K. L., Royals, M. A., Carstea, E. D., Rafi, M. A., Wenger, D. A., and Thrall, M. A. (2003). Mutation analysis of feline Niemann-Pick C1 disease. Mol. Genet. Metab. 79, 99–103. doi:10.1016/s1096-7192(03)00074-x
Stampfer, M., Theiss, S., Amraoui, Y., Jiang, X., Keller, S., Ory, D. S., et al. (2013). Niemann-Pick disease type C clinical database: cognitive and coordination deficits are early disease indicators. Orphanet J. Rare Dis. 8, 35. doi:10.1186/1750-1172-8-35
Stein, V. M., Crooks, A., Ding, W., Prociuk, M., O'Donnell, P., Bryan, C., et al. (2012). Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C. J. Neuropathol. Exp. Neurol. 71, 434–448. doi:10.1097/NEN.0b013e31825414a6
Subramanian, K., Hutt, D. M., Scott, S. M., Gupta, V., Mao, S., and Balch, W. E. (2020). Correction of Niemann-Pick type C1 trafficking and activity with the histone deacetylase inhibitor valproic acid. J. Biol. Chem. 295, 8017–8035. doi:10.1074/jbc.RA119.010524
Takahashi, S., Homma, K., Zhou, Y., Nishimura, S., Duan, C., Chen, J., et al. (2016). Susceptibility of outer hair cells to cholesterol chelator 2-hydroxypropyl-β-cyclodextrine is prestin-dependent. Sci. Rep. 6, 21973. doi:10.1038/srep21973
Tamura, A., and Yui, N. (2015). β-Cyclodextrin-threaded biocleavable polyrotaxanes ameliorate impaired autophagic flux in Niemann-Pick type C disease. J. Biol. Chem. 290, 9442–9454. doi:10.1074/jbc.M115.636803
Torres, S., Matias, N., Baulies, A., Nunez, S., Alarcon-Vila, C., Martinez, L., et al. (2017). Mitochondrial GSH replenishment as a potential therapeutic approach for Niemann Pick type C disease. Redox Biol. 11, 60–72. doi:10.1016/j.redox.2016.11.010
Tortelli, B., Fujiwara, H., Bagel, J. H., Zhang, J., Sidhu, R., Jiang, X., et al. (2014). Cholesterol homeostatic responses provide biomarkers for monitoring treatment for the neurodegenerative disease Niemann-Pick C1 (NPC1). Hum. Mol. Genet. 23, 6022–6033. doi:10.1093/hmg/ddu331
Trilck, M., Hubner, R., Seibler, P., Klein, C., Rolfs, A., and Frech, M. J. (2013). Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J. Rare Dis. 8, 144. doi:10.1186/1750-1172-8-144
Van Hoecke, L., Van Cauwenberghe, C., Borger, V., Bruggeman, A., Castelein, J., Van Imschoot, G., et al. (2021). Anti-inflammatory mesenchymal stromal cell-derived extracellular vesicles improve pathology in niemann-pick type C disease. Biomedicines 9, 1864. doi:10.3390/biomedicines9121864
Vanier, M. T. (2010). Niemann-Pick disease type C. Orphanet J. Rare Dis. 5, 16. doi:10.1186/1750-1172-5-16
Vite, C. H., Bagel, J. H., Swain, G. P., Prociuk, M., Sikora, T. U., Stein, V. M., et al. (2015). Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl. Med. 7, 276ra26. doi:10.1126/scitranslmed.3010101
Volkner, C., Liedtke, M., Untucht, R., Hermann, A., and Frech, M. J. (2021). Patient-specific iPSC-derived neural differentiated and hepatocyte-like cells, carrying the compound heterozygous mutation p.V1023Sfs*15/p.G992R, present the “variant” biochemical phenotype of niemann-pick type C1 disease. Int. J. Mol. Sci. 22, 12184. doi:10.3390/ijms222212184
Volkner, C., Pantoom, S., Liedtke, M., Lukas, J., Hermann, A., and Frech, M. J. (2022). Assessment of FDA-approved drugs as a therapeutic approach for niemann-pick disease type C1 using patient-specific iPSC-based model systems. Cells 11, 319. doi:10.3390/cells11030319
Wang, C., Scott, S. M., Subramanian, K., Loguercio, S., Zhao, P., Hutt, D. M., et al. (2019). Quantitating the epigenetic transformation contributing to cholesterol homeostasis using Gaussian process. Nat. Commun. 10, 5052. doi:10.1038/s41467-019-12969-x
Wang, M. L., Motamed, M., Infante, R. E., Abi-Mosleh, L., Kwon, H. J., Brown, M. S., et al. (2010). Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell. Metab. 12, 166–173. doi:10.1016/j.cmet.2010.05.016
Ward, S., O'Donnell, P., Fernandez, S., and Vite, C. H. (2010). 2-hydroxypropyl-beta-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr. Res. 68, 52–56. doi:10.1203/PDR.0b013e3181df4623
Wehrmann, Z. T., Hulett, T. W., Huegel, K. L., Vaughan, K. T., Wiest, O., Helquist, P., et al. (2012). Quantitative comparison of the efficacy of various compounds in lowering intracellular cholesterol levels in Niemann-Pick type C fibroblasts. PLoS One 7, e48561. doi:10.1371/journal.pone.0048561
Williams, I. M., Wallom, K. L., Smith, D. A., Al Eisa, N., Smith, C., and Platt, F. M. (2014). Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol. Dis. 67, 9–17. doi:10.1016/j.nbd.2014.03.001
Xie, C., Gong, X. M., Luo, J., Li, B. L., and Song, B. L. (2017). AAV9-NPC1 significantly ameliorates Purkinje cell death and behavioral abnormalities in mouse NPC disease. J. Lipid Res. 58, 512–518. doi:10.1194/jlr.M071274
Xu, H., Wang, B., Ono, M., Kagita, A., Fujii, K., Sasakawa, N., et al. (2019). Targeted disruption of HLA genes via CRISPR-cas9 generates iPSCs with enhanced immune compatibility. Cell. Stem Cell. 24, 566–578. doi:10.1016/j.stem.2019.02.005
Xu, X., Zhang, A., Halquist, M. S., Yuan, X., Henderson, S. C., Dewey, W. L., et al. (2017). Simvastatin promotes NPC1-mediated free cholesterol efflux from lysosomes through CYP7A1/LXRα signalling pathway in oxLDL-loaded macrophages. J. Cell. Mol. Med. 21, 364–374. doi:10.1111/jcmm.12970
Yang, F., Feng, X., Rolfs, A., and Luo, J. (2018). Lovastatin promotes myelin formation in NPC1 mutant oligodendrocytes. J. Neurol. Sci. 386, 56–63. doi:10.1016/j.jns.2018.01.015
Yang, M., Zhao, Y., Li, X., Li, H., Cheng, F., Liu, Y., et al. (2023). Conditioned medium of human menstrual blood-derived endometrial stem cells protects against cell inflammation and apoptosis of Npc1(KO) N2a cells. Metab. Brain Dis. 38, 2301–2313. doi:10.1007/s11011-023-01243-1
Zampieri, S., Bianchi, E., Cantile, C., Saleri, R., Bembi, B., and Dardis, A. (2014). Characterization of a spontaneous novel mutation in the NPC2 gene in a cat affected by Niemann Pick type C disease. PLoS One 9, e112503. doi:10.1371/journal.pone.0112503
Zervas, M., Somers, K. L., Thrall, M. A., and Walkley, S. U. (2001). Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 11, 1283–1287. doi:10.1016/s0960-9822(01)00396-7
Zhong, X. Z., Sun, X., Cao, Q., Dong, G., Schiffmann, R., and Dong, X. P. (2016). BK channel agonist represents a potential therapeutic approach for lysosomal storage diseases. Sci. Rep. 6, 33684. doi:10.1038/srep33684
Keywords: cell-based therapy, combination therapy, small molecule therapy, disease models, gene therapy, Niemann-Pick disease type C1
Citation: Zhang C, Su K, Jiang X, Tian Y and Li K (2024) Advances in research on potential therapeutic approaches for Niemann-Pick C1 disease. Front. Pharmacol. 15:1465872. doi: 10.3389/fphar.2024.1465872
Received: 17 July 2024; Accepted: 20 August 2024;
Published: 28 August 2024.
Edited by:
Robert Powers, University of Nebraska-Lincoln, United StatesReviewed by:
Xuntian Jiang, Washington University in St. Louis, United StatesSreekanth Vedagopuram, Brigham and Women’s Hospital and Harvard Medical School, United States
Copyright © 2024 Zhang, Su, Jiang, Tian and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caifeng Zhang, emhhbmdjYWlmZW5nNjY2QDE2My5jb20=