 Ziyang (Steve) Yin
Ziyang (Steve) Yin Zhengfeng Wang
Zhengfeng Wang- 1Concordia International School Shanghai, Shanghai, China
- 2Department of Hepatobiliary Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Non-small cell lung cancer (NSCLC) is the predominant form of lung cancer and is characterized by rapid metastasis and high mortality, presenting a challenge for early-stage treatment modalities. The heterogeneity of NSCLC’s tumor microenvironment (TME) significantly influences the efficacy of anti-PD-1 immune checkpoint inhibitors (ICIs) therapy, leading to varied patient responses. This review characterized different strains of oncolytic viruses in NSCLC and the different gene edits in pre-existing oncolytic viruses. This study also aimed to provide strategies to enhance anti-PD-1 therapy in NSCLC by engineering oncolytic viruses (OVs). This study offers insights into the genomic adaptations necessary for OVs targeting NSCLC, identify genetic determinants of anti-PD-1 response variability, and propose genomic edits to bolster therapy effectiveness. The primary goal of this study is to present a theoretically designed OV with a detailed genomic framework capable of enhancing the response to anti-PD-1 therapy, thereby advancing the field of cancer immunotherapy.
1 Introduction
Lung cancer ranks as the second most commonly diagnosed cancer globally, with 2.2 million new cases reported in 2020, accounting for 11.4% of all new cancer diagnoses. It is also the leading cause of cancer-related deaths, with an estimated 1.8 million fatalities per year (Sung et al., 2021). Lung cancer can be split into two types, small-cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). SCLC is the more deadly of the two lung cancer types—with a 5-year survival rate of 7%—while NSCLC is less deadly but the more common of the two (85%) (Wang et al., 2020; ACS, 2022). According to NSCLC can be further grouped into lung adenocarcinomas (40%), squamous cell carcinoma (25%–30%), and large cell carcinoma (10%–15%) (UK, 2022).
Surgery, chemotherapy, radiotherapy, immunotherapy, and targeted therapy are the most common approaches to treat lung cancer. The application of these treatments depends on the stage of cancer the tumor is in upon diagnosis. For stage I and II NSCLC patients, surgical resection is the primary and preferable treatment (Howington et al., 2013). Additionally, neoadjuvant and adjuvant chemotherapy has been approved before and following the resection of stage II or III NSCLCs to increase survival rates (Lackey and Donington, 2013; ACS, 2024a). For patients that have a resectable tumor (typically stages 0, I, II, and IIIa), chemotherapy is usually used neoadjuvant or adjuvant to the operation (Chaft et al., 2021; ACS, 2024c). Typically, cisplatin is used for postoperative chemotherapy, significantly reducing the risk of death in NSCLC (Bradbury et al., 2016). Chemotherapy can also be combined with radiotherapy (also called chemoradiation), prescribed for NSCLC patients in stages IIIa and beyond (ACS, 2024c). Chemotherapy can also synergize with immunotherapy (Usually given to NSCLC patients in stage IIIb and above), with chemotherapy killing tumor cells, reducing immunosuppressive substances released by the tumor, and enhancing the anti-tumor response (ACS, 2024c). PD-L1 expression is also increased after treatment by chemotherapy (Chaft et al., 2021). Although chemotherapy might be efficient at killing cancer cells, the treatment has two major downsides: 1) the chemotherapy resistance in cancerous cells leads to decreased efficacy of the treatment, resulting in an incapability of improving patient outcomes; 2) the cytotoxicity of chemotherapies also indiscriminately kills host cells, resulting in many side effects including nausea and loss of hair (ACS, 2024a). Hence, a more specific alternative has been derived, such as targeted therapy. Angiogenesis inhibitors [inhibit the process of angiogenesis and prevent the formation of blood vessels in tumors, diminishing the cancer’s access to nutrients via the bloodstream, leading to slowed cancer growth and a weaker tumor microenvironment (Daum et al., 2021; ACS, 2024b)], epidermal growth factor receptors (ERBBs) inhibitors [block the signal for cancer cell growth since the EGFR mutations are present in about 31.6% of NSCLC cases (Kumari et al., 2019)], and RAS/MAPK signaling inhibitors [block the signal for cancer cell growth and induce cell death, mutations of KRAS/MAPK singling pathway is the main drivers of NSCLC (Xie et al., 2021; Liu et al., 2022)] are commonly used traditional target therapy drugs for NSCLC. Similar to chemotherapy, however, both primary and acquired drug resistance remains a major problem in targeted therapy approaches (Liu et al., 2022).
2 Immunotherapy in NSCLC
Immunotherapy is a form of targeted therapy that particularly stimulates the host’s immune system in order to fend against foreign cancer cells. Generally, this treatment improves the immune system’s capability of antigen recognition or cytotoxicity, leading to longer survival in patients (Chan and Hughes, 2014). Immune checkpoint inhibitor (ICI)-based therapies have been approved for NSCLC. Immune checkpoints (ICs) are steps in the immune response cycle that regulate self-tolerance. ICs attach to co-stimulators of the T-cell receptors on the surface of cytotoxic T cells, signaling T-cell deactivation and death. Normally, ICs are expressed on the surface of host cells; however, on malignant cells, these ICs are upregulated on the surface, resulting in a weaker immune response (NCI, 2022). ICIs are monoclonal antibodies that attach to either the ligand or the receptor of an IC, resulting in the inhibition of the inhibitory signal that deactivates the T-cell response. One common example of an IC complex is the programmed death ligand-1(PD-L1), which is situated on the cancer cell, and the programmed death receptor-1(PD-1), which is situated on the T cell (Alsaab et al., 2017). When PD-L1 attaches to PD-1, the T cell’s cytolytic activity is inhibited and the T cell is deactivated (Alsaab et al., 2017). To prevent this, a monoclonal antibody that binds to either PD-1 or PD-L1 is introduced, preventing the signaling complex to form and allowing the T cell to release cytotoxic damage upon the target cell (Figure 1).
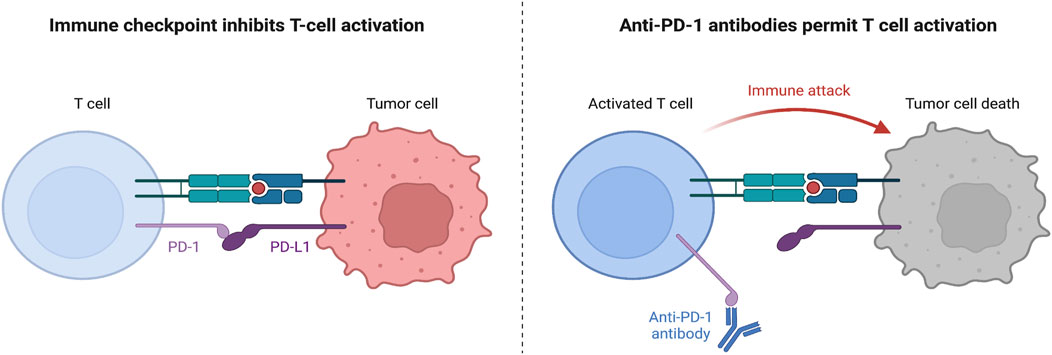
Figure 1. Mechanism of PD-1 and PD-L1 Function. Left: without anti-PD-1 therapy, when CD8+ T cells attach their TCR onto the major histocompatibility complex I (MHC I) expressed on the tumor cell, the tumor cell would express PD-L1 attaching to the PD-1 on the CD8+ T cell surface and inhibiting T cell activation, proliferation, survival, and cytotoxic secretion (Han et al., 2020). Right: however, with anti-PD-1 therapy, a monoclonal antibody can attach to the PD-1 receptor on the T cell surface, preventing signaling with the PD-L1 ligand, and allowing the T cell to secrete granzymes and perforin to kill the tumor cell (Nie et al., 2021).
Beyond ICIs, chimeric antigen receptor (CAR) cell therapy is currently being evaluated in many clinical trials for NSCLC. In CAR-T cells, CARs are receptors formed by merging tumor-specific single chain variable fragments (scFv) with the CD3zeta chain of the T cell receptor (TCR) (Morgenstern and Irwin, 2019). Patient T-cells are extracted and encoded to express these CARs according to the specific genetic coding of their cancer. Upon insertion back into the patient, the new CAR T-cells can initiate cytotoxicity through recognition of tumor-antigens, of which the specificity is defined by the scFv (Xiao et al., 2021). While CAR T cell therapy is a promising treatment, especially for hyper-progressive NSCLC patients, CAR therapy is patient specific and hence cannot be mass produced—resulting in high costs—and could cause self-cytotoxicity due to the scFv fragment recognizing host antigens (Liu et al., 2020). Some alternatives have been developed in hopes of mitigating these consequences. One alternative is CAR natural killer (NK) cell therapy, which has the advantage of possessing broader anti-tumor activity and targeting even MHC downregulated targets, being non-patient specific giving potential to allogeneic CAR NK cell therapy, and less cytokine stimulation decreasing the chances of cytokine release syndrome (Albinger et al., 2021). Another alternative is bi-specific T cell engager (BiTE) therapy, which is made of two antibodies, one targeting tumor-associated antigens and another targeting TCRs, connected through a short constant region in the middle, drawing the tumor cell closer to T cells, increasing antigen recognition and immune response (Goebeler and Bargou, 2020). Recent studies have shown that CAR T-cells can be edited to secrete BiTEs (called CAR.BiTE cells) with the potential of mitigating antigen escape without increasing the toxicity of CAR T cell therapy (Choi et al., 2019).
2.1 Flaw of immunotherapy
However, despite the benefits, immunotherapy has an Achilles heel: tumor cells are able to develop resistance. Less than 30% of NSCLC patients response to current immunotherapies. The three known classifications of immunotherapy resistance in cancers are shown in Table 1 (Sharma et al., 2017; Zhou and Yang, 2023). The progression of resistance is accelerated in NSCLC due to its fast growth rate increasing replicative stress and instances of replicative error—both of which contribute to tumor heterogeneity and genome mutation in immunotherapy resistance (Wang et al., 2012; Zhou and Yang, 2023). Biomarkers that help indicate resistance and immunotherapy efficacy in NSCLC are included in Table 2. Many means of surmounting immunotherapy resistance have been invented, the common ones include: 1) targeting co-inhibitory signals such as the ICIs PD-1, CTLA-4, LAG-3, TIM-3, TIGIT, VISTA, and Siglec-15 to decrease inhibition of immune response (Frisone et al., 2022); 2) enhancing co-stimulatory signals to improve the immune response of cytotoxic cells such as T and NK cells (For patients of NSCLC, the use of CD134 (OX-40), CD137(4-1BB), and IL15 are currently under investigation (Croft, 2009; Frisone et al., 2022); 3) priming the immune system to tumor neoantigens with vaccine therapy to increase antigen recognition and effector functions of T and B cells (Makker et al., 2022); and 4) combining immunotherapy with other therapies such as chemotherapy increases response due to increased neo-antigen release as a result of chemo-induced cell death (Rheinheimer et al., 2020; Frisone et al., 2022). There are many different methods in clinical trial attempting to remedy these resistances in NSCLC, these methods include: targeting co-inhibitory signals such as ICIs and enhancing co-stimulatory signals such as CD134 to increase immune activation, increasing tumor sensitivity through vaccine therapy to increase antigen recognition, and through pre-treatment of other co-therapies such as chemotherapy and radiotherapy (Frisone et al., 2022; Zhou and Yang, 2023). Additionally, nanotechnology has become popular in the search to overcome immunotherapy resistance. Not only can nanoparticles promote T-cell enrichment and activation by using magnetic fields to promote TCR aggregation, they can also improve the bioavailability of insoluble drugs and prolong drug circulation by making the treatments more stable (Shao et al., 2023; Zhang et al., 2023). One specific example are albumin-based nanoparticles that can be encased on proteins to bypass drug-prevention complexes such as drug efflux (Hassanin and Elzoghby, 2020). Zhejiang University had conducted a clinical study to determine the safety and efficacy of platinum-based albumin-bound paclitaxel regimen in the treatment of stage IIB and IIIA NSCLC (Zhao, 2015). However, even with these methods of overcoming resistance, immunotherapeutic treatments fail to treat cancer with consistency and efficacy (Frisone et al., 2022). Hence, in this paper, an additional method of immunotherapy—an oncolytic virus-based cancer vaccine—is proposed.

Table 1. Types and mechanisms of resistance to immunotherapy.
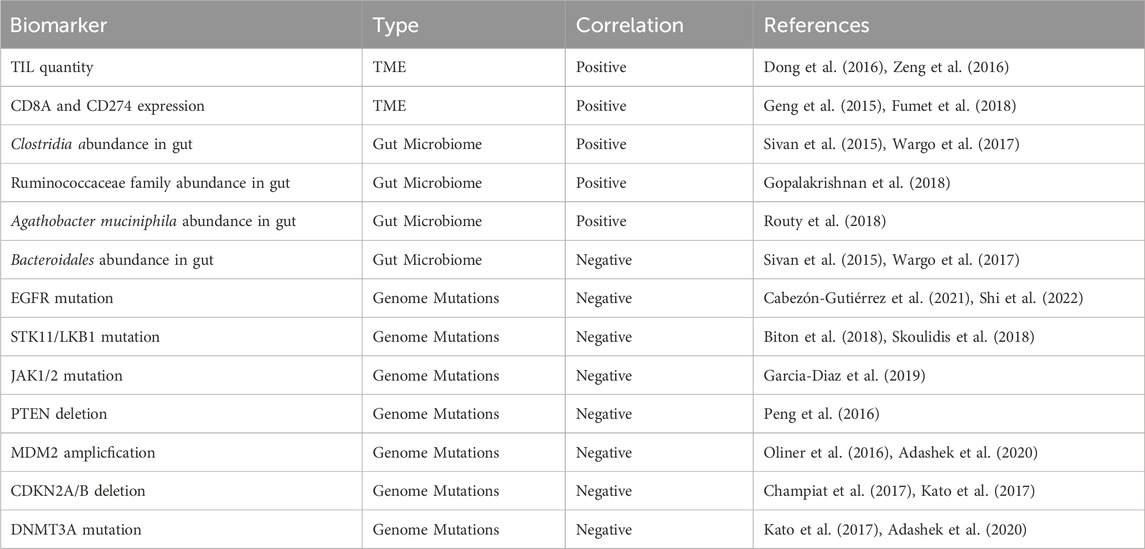
Table 2. Biomarkers for immunotherapy sensitivity.
3 Oncolytic viruses
Oncolytic viruses (OVs) are modified viruses that specifically target tumors, carry and induce the expression of transgenes, and cause direct tumor cell lysis (NCI, 2018). These viruses can be edited to perform certain functions against specific cell types (Kaufman et al., 2015). There are multiple types of oncolytic viruses, each with its unique receptors, genome size, effectiveness, and mechanisms of action (Rahman and McFadden, 2021). Table 3 lists OVs that have shown promise in treating lung cancers. OVs has shown promising results in cancer treatment and have been widely used for cancer treatment, including advanced-stage melanoma and glioblastoma (GBM). Currently, several viruses, including vaccinia virus, coxsackievirus, adenovirus, reovirus, and herpes simplex virus have been tested in NSCLC. The different ongoing clinical trials for oncolytic viruses targeting NSCLC are listed in Table 4.
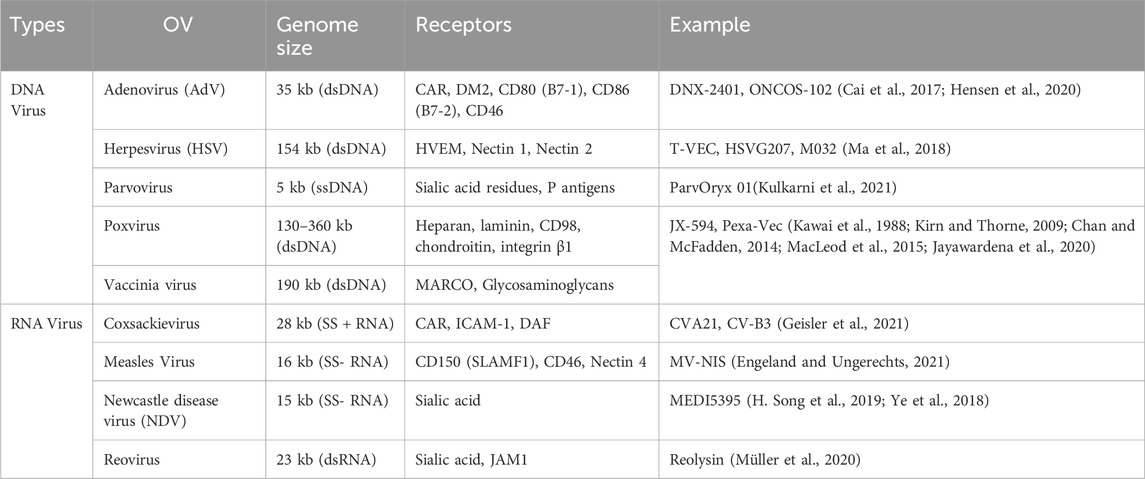
Table 3. Examples of oncolytic viruses.
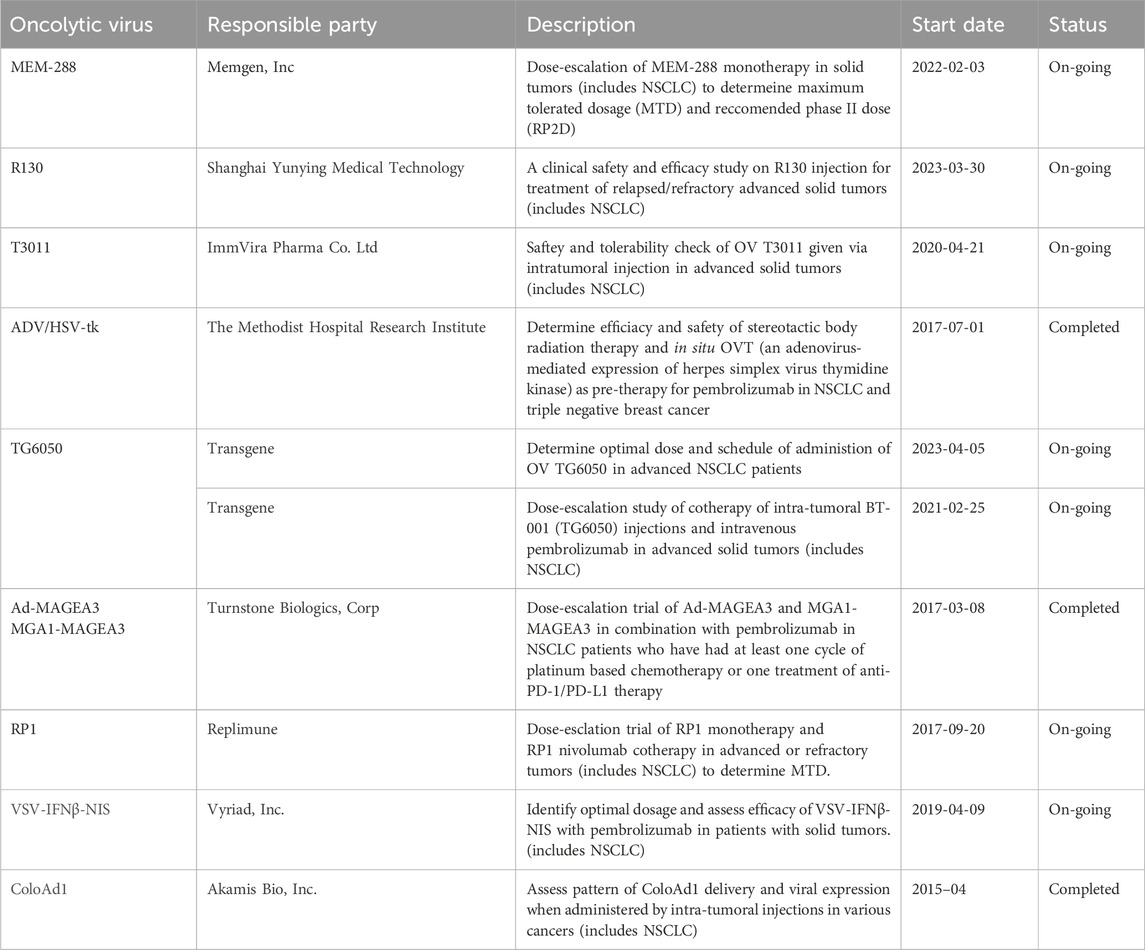
Table 4. Clinical trials involving NSCLC associated OVs.
3.1 Commonly used oncolytic viruses
Vaccinia Virus (VV): VVs are large, enveloped, double-stranded DNA (dsDNA) viruses belonging to the poxvirus family, each containing approximately 190 kbps, encoding around 250 genes. The Lister, Wyeth, and Western Reserve strains of VV are more commonly used in OV research as they can incorporate large amounts of foreign DNA without affecting the virus’s replicative efficacy (Guo et al., 2019; Guse et al., 2011). VVs do not have a specific receptor making them a prominent candidate for multiple types of cancers (Guo et al., 2019; Guse et al., 2011). However, in the interest of lung cancer, VVs preferably target cells with expression of the scavenger receptor MARCO, which, according to the human protein atlas (HPA), is enhanced in lung cancers (Sjöstedt et al., 2020). Additionally, VVs have innate specificity for cancer cells due to their sensitivity toward type-1 IFNs (Guo et al., 2019; Guse et al., 2011). Furthermore, VVs can modulate the TME and evade immunosuppression by secreting virokines (viral proteins that resemble cytokines and chemokines), and viroceptors (viral proteins that act as decoy receptors for cytokines and chemokines), evading immunosuppression within the TME and prolonging viral infection and replication (Truong and Yoo, 2022). VV virion entry is also accelerated by low pH environments due to their low-pH-dependent endosomal pathway, hence making them particularly effective in the TME (Townsley et al., 2006). Additionally, one final feat of VVs is its capabilities to replicate independent of the host cell’s genome due to having its own RNA transcriptase and transcription factors within its viral core (Tolonen et al., 2001). This allows the VV to be even more flexible with its surrounding conditions whilst conducting pathogenesis.
Herpes simplex virus (HSV): HSVs are large, enveloped, dsDNA viruses belonging to the herpesviral family, containing around 152 kbps (Aldrak et al., 2021). The main receptor of HSV is Nectin-1, which according to the HPA, is overexpressed in lung cancers, albeit inconsistently (Farooq et al., 2010; Sjöstedt et al., 2020). Additionally, an alternative receptor for HSV, HVEM, is found to be overexpressed in patients of non-small cell lung cancer with N2 metastasis or later, making HSV relatively specific for lung cancer (Farooq et al., 2010; Ren et al., 2018). Similar to VVs, HSVs have a wide selection of genes that can not only help them navigate the TME and perform immunoevasion but also be manipulated to increase specificity and efficacy (Hong et al., 2022).
Adenovirus Serotype 3 (AdV3): Adenoviruses are a family of viruses capable of holding 25–45 kbps of dsDNA. Many different serotypes of AdVs have been identified, however, the receptors of AdV serotype 3 (AdV3) are more specific toward lung cancer (Zhao et al., 2021). One of the main receptors for AdV3 is desmoglein-2 (DM2), a cadherin that is overexpressed in lung cancers (Wang et al., 2011; Cai et al., 2017; Zhao et al., 2021). Similarly, CD46 is a receptor for AdV3 of which its overexpression is associated with malignant transformation and metastasizing potential (Elvington et al., 2020; Zhao et al., 2021). Co-receptors CD80 (B7-1) and CD86 (B7-2) that are expressed on antigen-presenting cells (APCs) are also receptors of AdV3, allowing for the possibility of AdV3s to infect APCs and provide the tumor-associated antigens directly (if modified to do so) (Zhang and Bergelson, 2005; Zhao et al., 2021). Although AdV3 is very specific for lung cancers, the safety of the virus can be improved by moving the AdV3 fiber knob (a protein that attaches to the virus’s receptor) onto AdV5, replacing the AdV5 fiber knob (5/3 chimerism), taking advantage of the specificity of AdV3 while having the potent but safe lytic activity of AdV5 (Hemminki et al., 2011; Koodie et al., 2019; Zhao et al., 2021).
3.2 Delivery routes and therapeutic effects of OVs
Systemic or local administration of the OVs is another contention of discussion, with local administration limiting the virus to one solid tumor and systemic administration raising many other problems. Currently, intratumoral delivery is the most common route of administration for OVs, being more capable of being controlled, and showing more definite therapeutic effects (Li et al., 2020). Although theoretically, viruses can sustain virion production indefinitely within cancer cells until the cancer is eviscerated, however in practice, multiple doses of the OV are needed to even show tumor regression (Ferguson et al., 2012). Regarding systemic delivery of OVs, the method of administration would be intravenous injection, the limitations of such are as follows: 1) neutralization via immune response; 2) non-specific uptake via non-target tissues; 3) indiscriminate cytotoxic damage (Atasheva and Shayakhmetov, 2021). However, despite these limitations, clinical trials of intravenous delivery of OVs have occasionally shown success, an example of which was the intravenous injection of T-VEC in co-therapy with ipilimumab successfully treating stage IIIb-IV melanoma (Puzanov et al., 2016). Alternatively, there are also additional methods of administration that can be performed according to the type of cancer within the patient. Intraperitoneal injections target organs within the abdominal cavity, intrathecal injections target tumors in the central nervous system, and subcutaneous injections target melanomas and soft tissue sarcomas (Chen et al., 2017; Cohn et al., 2017; Li et al., 2020).
As shown in Figure 2, the OVs first infect the tumor cell through its specific receptor, and subsequently insert its viral genome into the cell which enters the nucleus where the viral genome is replicated, creating virions and the transgene-encoded proteins (Shi et al., 2020). Following this, the OV induces cell lysis of the infected tumor cell and releases tumor antigens, DAMPs, PAMPs, and transgene-encoded proteins. The tumor antigens and DAMPs and PAMPs all result in the increase of immune response to the tumor, with the tumor antigen specifically being uptaken by APCs and presented to immune cells such as the CD8+ cytotoxic T-Cell, increasing tumor recognition and intuitively the anti-tumor immune response (Shi et al., 2020). The transgene-encoded proteins on the other hand can be modified to be combined with co-therapies that can further increase tumor regression (Harrington et al., 2019). To improve CAR therapy performance, the transgenes within the OV can be encoded to express cytokines that increase proliferation and migration and prevent inhibition of the CAR-attached immune cell to increase tumor trafficking and regression (Moon et al., 2018). With recent research, the possibility of having CAR T cells as carriers of small doses of OVs to the tumor site could have promising results, however, more research will need to be conducted before a conclusion can be made (VanSeggelen et al., 2015). Similarly, the transgenes can be encoded with BiTEs, allowing BiTE release upon tumor cell oncolysis, which has been shown to have greater therapeutic effects than BiTE therapy alone (Fajardo et al., 2017). Finally, in combination with ICIs, antibodies targeting against immune checkpoints can be encoded within the transgenes, allowing immune checkpoint inhibition upon the oncolysis of the tumor cell (LaRocca and Warner, 2018). The OV transgenes can also contain other proteins that can help sensitize the tumor environment to ICI therapy, such as increasing immune cell presence in the area or reducing the effects of the TME (LaRocca and Warner, 2018). However, it should be noted that the activation of the immune system could also induce antiviral responses, which could lead to the deactivation of viral replication (Harrington et al., 2019). In summary, with the unique aspects of OVs—tumor specificity, transgene expression, and induction of oncolysis—these special viruses can kill tumor cells, release tumor antigens, increase the effects of combinational therapies, and act as a tumor vaccine for the immune system.
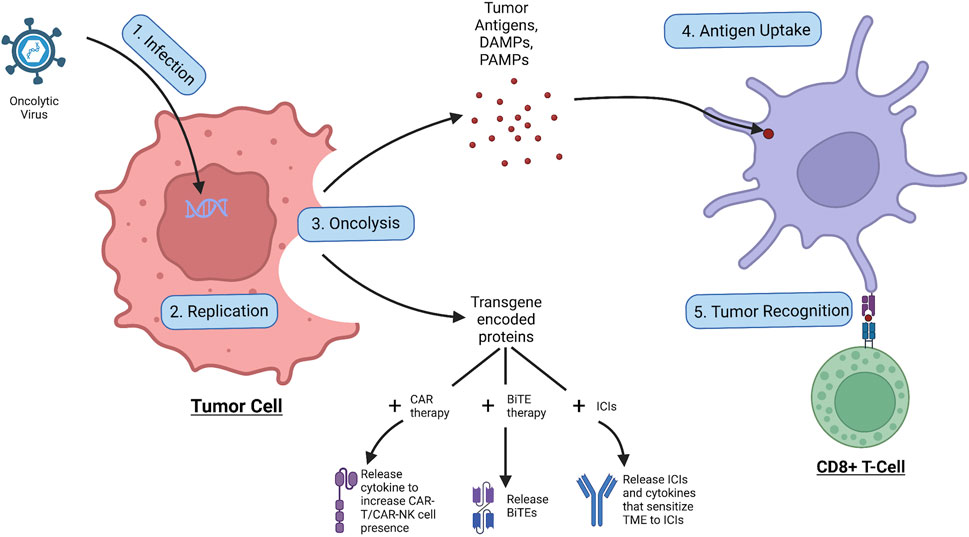
Figure 2. Process of oncolytic virus infection. The OV first infects the tumor cell, allowing for replication of the viral genome, leading to the production of virions and transgene-encoded proteins. Following the OV induces oncolysis, leading to the release of tumor antigens, DAMPs, PAMPs, and transgene-encoded proteins. The tumor antigens are taken up by APCs which use them to activate immune cells such as the CD8+ cytotoxic T cell which go to attack cancer. The released transgene-encoded proteins, depending on how they are encoded, can be cytokines that stimulate CAR-T/CAR-NK cell proliferation and migration, BiTEs that are released upon oncolysis, or ICIs and cytokines that sensitize the TME to the ICI effects.
One example of a clinically approved oncolytic virus is Talimogene Laherparepvec (T-VEC), a modified oncolytic herpes simplex virus that can be used against melanomas and has been recently authorized for glioblastomas (Ando et al., 2008; Ferrucci et al., 2021). T-VEC has four major edits in its genome: 1) Deletion of ICP34.5: Infected cell protein 34.5 (ICP34.5) activates protein phosphatase-1 (PP1) which dephosphorylates elF2alpha, preventing the inhibition of viral replication (Li et al., 2011; Ferrucci et al., 2021). But with the gene deleted, the protein will no longer be produced, meaning the oncolytic HSV will not be able to inhibit elF2alpha phosphorylation, and hence viral replication will come to a halt. However, in tumor cells, due to their highly replicative nature, the phosphorylation of elF2alpha is constantly inhibited, and hence even without ICP34.5, T-VEC is able to replicate in tumor cells (Liu et al., 2003; Kazemi et al., 2004; Guo et al., 2017). This increases the replicative specificity of T-VEC; 2) deletion of ICP47: ICP47 binds to TAP1 and TAP2 residing on the endoplasmic reticulum (ER) membrane, blocking and preventing peptide transport into the ER lumen, resulting in no antigen presentation by MHC I (Goldsmith et al., 1998). Hence, deleting this gene will permit and increase antigen presentation, which can increase antigen recognition and anti-tumor responses (Ferrucci et al., 2021); 3) early Expression of US11: Unique short glycoprotein 11 (US11) blocks the phosphorylation of elF2alpha by binding to PKR (Cassady et al., 1998), The early expression of US11 allows for the promotion of proliferation of virus within tumor cells while not impairing tumor selectivity (Ferrucci et al., 2021); and 4) Insertion of GM-CSF: The granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine that, when secreted, promotes the myeloid cell development and maturation and dendritic cell differentiation and survival (Egea et al., 2010). Hence, when inserted within the transgenes, the GM-CSFs will promote tumor infiltration (Kumar et al., 2022). However, too much GM-CSF has also been shown to exhaust immune cells and promote cancer growth (Kumar et al., 2022).
3.3 OVT co-therapies
Co-therapies are commonly used in conjunction with OVT to increase the efficacy of treatment. One route of co-therapy of OVT is to combine with chemotherapy. Chemotherapy is able to systemically alter the behavior of the tumor and the TME, making it equipped to acts a pre-therapy to OVT (Nguyên et al., 2008). Generally, chemotherapy is able to suppress the immune-surveillance and decrease Treg cell presence (such as MSDC) increasing viral infection rates and decreasing tumor promoting factors respectively. Table 5 lists combinations of chemotherapy and OVT with more specific known synergetic effects summarized from related studies (Nguyên et al., 2008; Heo et al., 2011; Tusell Wennier et al., 2012; Mirbahari et al., 2024). However, the pathways taken in viral replication are very similar to that of those taken by cancer cells. Hence, the synergetic potential of chemotherapy with OVT is limited due to the former inhibiting various functions of the latter in therapy (Nguyên et al., 2008). Besides chemotherapy, OVT can also be in co-therapy with radiotherapy, a combination that has yielded more successful therapeutic results than any of the two alone in many studies (Mansfield et al., 2013; Dai et al., 2014). In Wilkinson et al. (2016) study it was found that not only was vaccinia virus’s viral DNA immune to radiotherapy damage, but there was a slight increase in cytotoxicity when combined. In another study, the increase in cytotoxicity during OV and radiotherapy co-therapy was attributed to the increased release of DAMPs and increased ratio of M1 to M2 macrophages (Chen et al., 2021). Finally, OVT can be in co-therapy with other immunotherapies such as ICIs, angiogenesis inhibitors, CAR therapy, etc (Nguyên et al., 2008). OVT in these instances can act as pre-treatment by either increasing the receptiveness or sensitivity of the environment to the following immunotherapy (Mirbahari et al., 2024). For example, OVs can be deployed first to increase tumor-antigen presence within the TME to increase subsequent CAR T-Cell therapy efficacy due to encouraging antigen uptake and antigen recognition (Shi et al., 2020). This article will focus on the co-therapy of OVs with the ICIs anti-PD-L1/PD-1; this will be expanded more upon in latter sections.
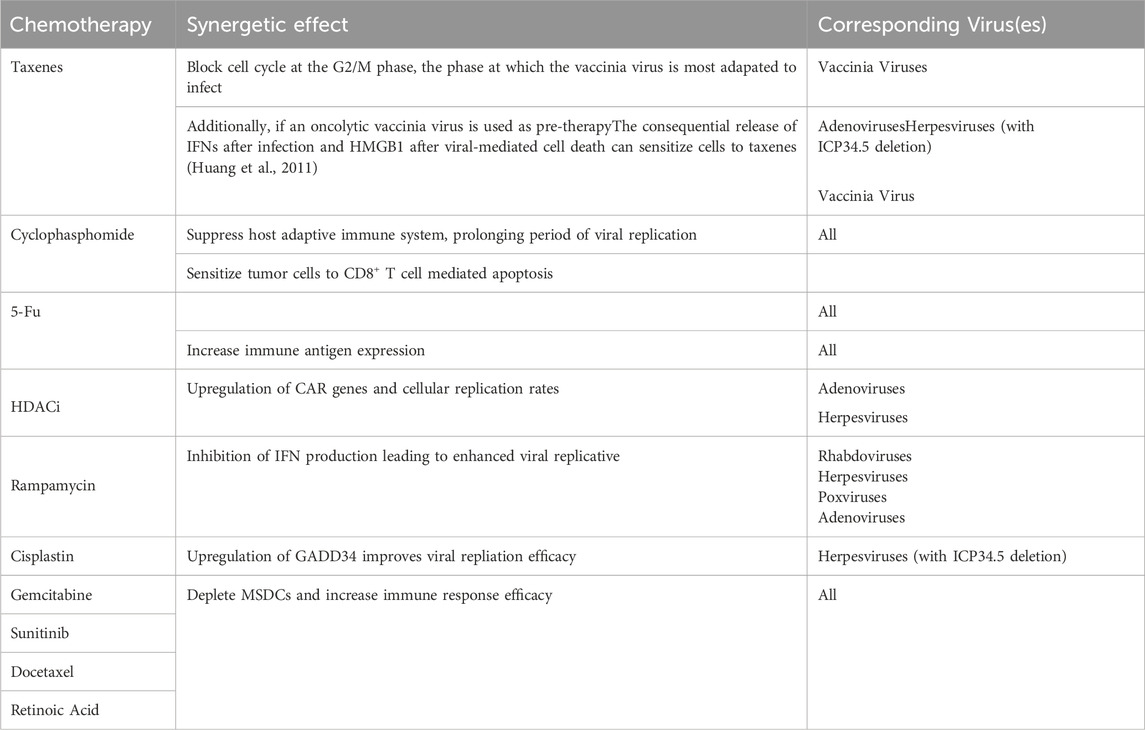
Table 5. Synergies between OVT and chemotherapy.
4 Oncolytic virus engineering
The development of safe, cancer-selective, and efficient OVs against various tumors relies heavily on the genetic engineering of OVs. Both natural and genetically modified viruses have shown promising results in treating various cancers. Understanding the biology and genetics of the virus, the interactions between the virus and the host immunity, and how cancer cells protect themselves from immune cells is critical for the genetic modification of OVs. In general, the aim of engineering OVs is to enable the application of OVs in cancer immunotherapy to broadly activate anti-tumor immune responses, enhance the tumor cell tropism of OVs, and reduce toxicity to normal cells.
4.1 Viral genome edits to increase specificity
While OVs have innate specificity towards cancer cells, without moderation, the OVs can still indiscriminately infect host cells. This is because while some receptors may be overexpressed in cancers, a portion of host cells can still express said receptor and would therefore be susceptible to viral infection of the OV. To mitigate this problem, edits to the viral genome are made, these edits can be generalized into the following groups: a) deactivation of viral replication: In most instances, viruses require the production of specific proteins to disrupt the infected cell’s antiviral pathways to prevent the inhibition of viral replication (Magden et al., 2005; Majdoul and Compton, 2022). At the same time, most, if not all, cancers have a faulty replication pathway that is constantly active resulting in uncontrolled tumor growth (Vassilev and DePamphilis, 2017; Yaacov et al., 2021). Hence, in certain instances, if the virus’s mechanism to induce viral replication is deactivated, the virus will no longer be able to reproduce in regular host cells; However, due to the faulty replicative pathway of cancers, the virus would still be able to reproduce in tumor cells (Everts and van der Poel, 2005). This intuitively increases the specificity of the modified viruses towards cancerous cells via exploiting the cancer cell’s constantly active replicative pathway; b) deactivation of viral immuno-evasion: Viruses have pathways of evading the human immune response, an example of this is the B18R protein of vaccinia viruses which acts as a decoy receptor for interferon-α (IFN-α), preventing the IFN-α signaling cascade from inhibiting viral functions (Kim et al., 2017). At the same time, immune signaling proteins such as interferons (IFNs) and cytokines are downregulated and inhibited in the TME (Fenton et al., 2021). Hence, if the immune evading pathways of the viruses were to be deactivated, the virus may not be able to infect and proliferate in normal host cells, whilst they can function normally in the TME. This increases the specificity of the modified viruses towards cancerous cells via exploiting the TME’s immunosuppressive effects; and c) using tumor-specific promoters: Promoters are sequences of DNA that need to be activated by specific proteins (e.g., growth factors) to initiate transcription of downstream genes (NIH, 2024). Tumor cells can have special proteins that can read promoter sequences unique, or uncommon outside of, tumor cells, creating specificity with promoters (Wang et al., 2018). Hence, if certain genes are placed downstream of unique promoters (such as the human telomerase reverse transcriptase (hTERT) promoter), even if an OV infects a normal host cell, the genetic content downstream of the promoter will not be transcribed within the cell, and hence, will not be expressed. The potential viral genome edits for increasing the specificities of different OVs are listed in Table 6.

Table 6. Choice of viral genome edits.
4.2 Viral genome edits to increase anti-tumor immunity
Tumor cells often exhibit properties such as evasion of immune surveillance and loss of immunological response. Selected genome edits in OVs would have a scientifically explainable positive impact on anti-tumor immune response. Impacts of edits in OVs were judged to be positive or not based on if they met the following criteria: a) increased replication: Increasing the replication of the virus allows the virus to duplicate faster and intuitively infect more cancer cells leading to greater efficacy. Similarly, an increased replication allows for the genes encoded in the viral genome to be expressed more, also increasing the efficacy of treatment. This increased replicative speed can be achieved via the insertion of enhancers or moving certain viral genes for early expression; b) increased antigen recognition: Antigen recognition is a vital step in the process of an immune response, allowing for the engagement of T and B cell effector function (Apavaloaei et al., 2020). One method of increasing recognition of tumor-specific antigens is the deletion of antigen presentation inhibiting pathways in viruses (e.g., the deletion of ICP47 in Herpes Simplex Virus, which prevents the inhibition of antigen transport through TAP1 and TAP2 into ER lumen for attachment to MHC I) (Ren et al., 2018). Another method of increasing recognition of tumor-specific antigens is the insertion of specific cellular markers that would be unique to the tumor upon infection. This would result in the release of these tumor-specific markers upon tumor cell lysis, increasing the number of tumor antigens, and causing T-cells to recognize and kill the OV-infected tumor cells that are also expressing this marker; and c) increased immune cell infiltration: Increasing the presence of immune cells in an area would not only increase the antigen presentation present, but also increase the number of T cells in the area. This would increase the amount of T cell-tumor cell complexes, increasing the efficacy of anti-PD-1 therapy. One method of increasing immune cell concentration would be to release chemokines and cytokines in the interest of increasing the immune cell population near the infected tumor, increasing the migration of APCs and immune cells, subsequently increasing the chances of antigen presentation (Melcher et al., 2021).
4.3 Viral genome edits to increase anti-PD-1 efficacy in NSCLC
As mentioned previously, anti-PD-1 ICI therapy is one form of immunotherapy that has shown great therapeutic results for many cancers; however, ICI therapy has a couple drawbacks that limit its potential effect in patients. Hence, this study aims to design OVs that can enhance and support anti-PD-1 effectiveness for NSCLC patients.
As shown in Figure 3, we analyzed the transcriptomes of NSCLC tumor from 27 patients treated with anti-PD-1 therapy. The 27 patients’ transcriptomes have been grouped into eight responders and 19 non-responders. Gene set enrichment analysis (GSEA) was used to compare the transcriptome data between the responders and non-responders, distinguishing which genes are comparatively altered between the two conditions, providing potential targets for OV design. According to the enrichment plots given by the GSEA, several genes that have shown strong correlation with either the PR or PD groups from the pathways shown in Figures 4A–C have been chosen as targets for editing.
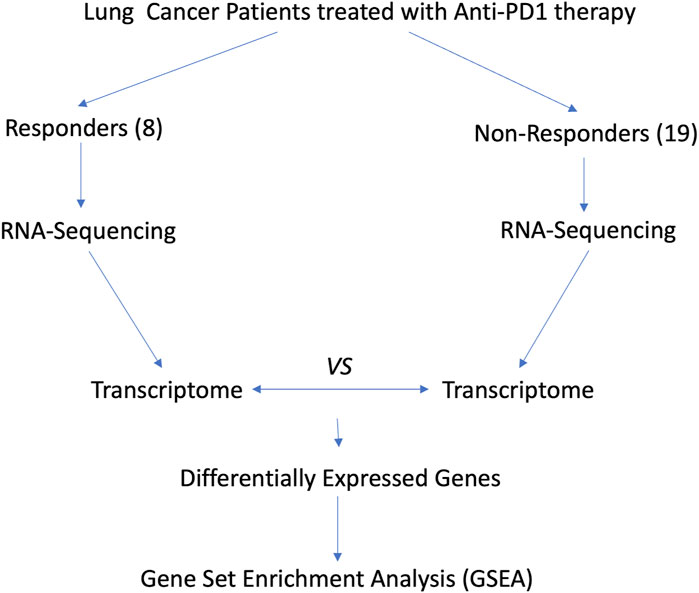
Figure 3. Flow chart for revealing potential targets for designing OVs.
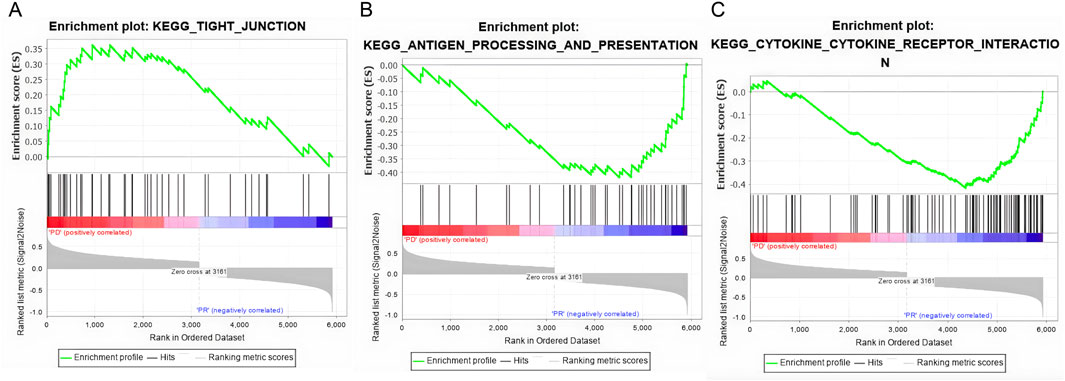
Figure 4. Gene set enrichment plots of three highly correlated pathways. (A) The tight junction pathway shows a positive correlation anti-PD1 non-responsive patients (PD). (B) The antigen process and presenting pathway is positively correlated with responsive patients (PR). (C) The cytokine and cytokine receptor interaction pathway is positively correlated with the PR patients.
4.3.1 Expression of CLDN1
As shown through Figure 5, the KEGG_TIGHT_JUNCTION pathway is positively correlated with non-responsive patients.
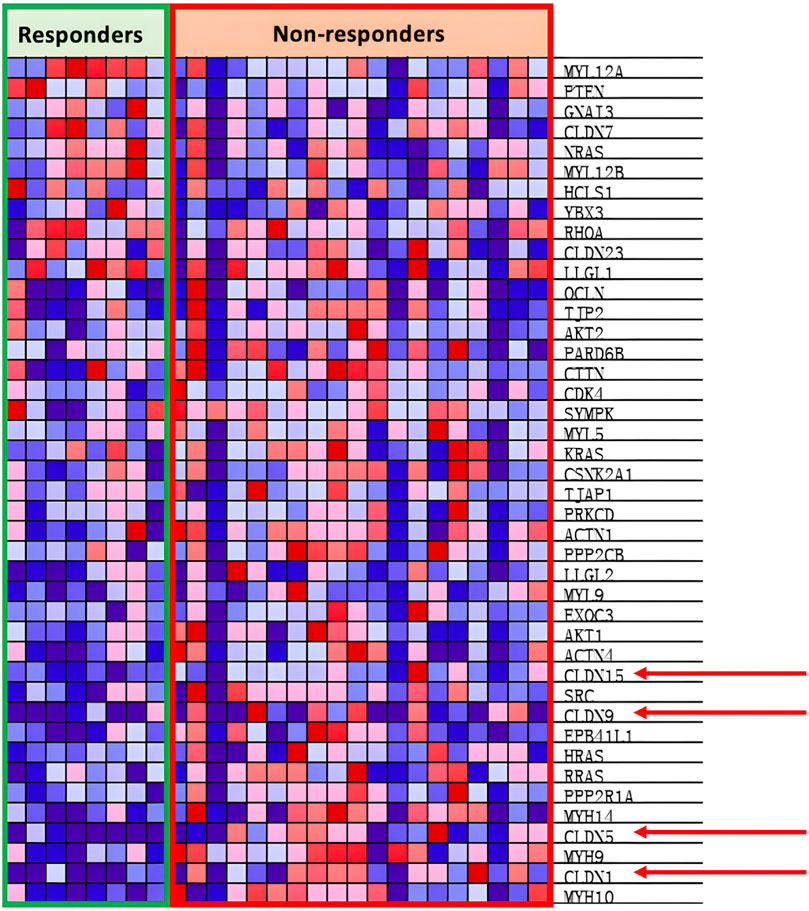
Figure 5. Heat map of KEGG tight junction pathway.
In Figure 5, the heat map shows the overexpression of the CLDN family in non-responsive patients. CLDNs are chosen out of the over-expressed genes as the family are generally tetraspan transmembrane proteins of tight junctions, which allows them to be easily identified thanks to their extracellular domain (Krause et al., 2008). Out of the CLDNs, CLDN1 is chosen as it is widespread within the lung epithelia and endothelia as seen in Figure 5. Furthermore, prior research has shown that CLDN1 promotes drug resistance of NSCLC to cisplatin, showing how CLDN1 is correlated with poor prognosis of NSCLC (Fagerberg et al., 2014). The addition of CLDN1 into the viral genome would result in the release of CLDN1 as cancer antigens upon oncolysis, encouraging cancer recognition as CLDN1 is an easily identifiable surface protein. However, research has also shown that CLDN1 could exert tumor promoter characteristics by increasing the invasion or motility of cancer cells, which could decrease the efficacy of our cancer vaccine (Chao et al., 2009; Sun et al., 2016; Bhat et al., 2020). Experiments from Hutzler et al. (2017) show increased antigen-specific immunity and anti-CLDN6 antibody production when the B16-F10 melanoma cell line was treated with recombinant measles virus-encoded with the CLDN6 gene. In summary, the addition of CLDN1 into the viral genome would increase the immunorecognition of NSCLC cells; however, it may also promote tumor metastasis if the amount is not attenuated.
CLDN1 will act as the main sensitizing antigen in this OV. Hence, the function required of CLDN1 is to be the ligand for HLA1 and HLA2 recognition. However, the full length of the CLDN1 gene spans 633bps. To shorten this gene, only the epitope region will be introduced to the viral genome.
According to the Immune Epitope Database (IEDB), there are two main epitope regions on CLDN1: YPTPRPYPKPAPSSGKD (YPT) and KVFDSLLNL (KVF) (Vita et al., 2018). YPT has affinity for both HLA1 and HLA2 receptors while KVF only has affinity for HLA1 (Goncalves et al., 2021; Marcu et al., 2021). However, as seen on Figures 6A–C, when running the IEDB MHC binding prediction algorithm, KVF showed a much greater affinity for HLA1 than YPT. Additionally, HLA1 is expressed on a wider range of cells than HLA2. Hence, in interest of immune activation, the epitope region KVF was chosen to be inserted into the viral genome. The KVF epitope is 27 bp in length.
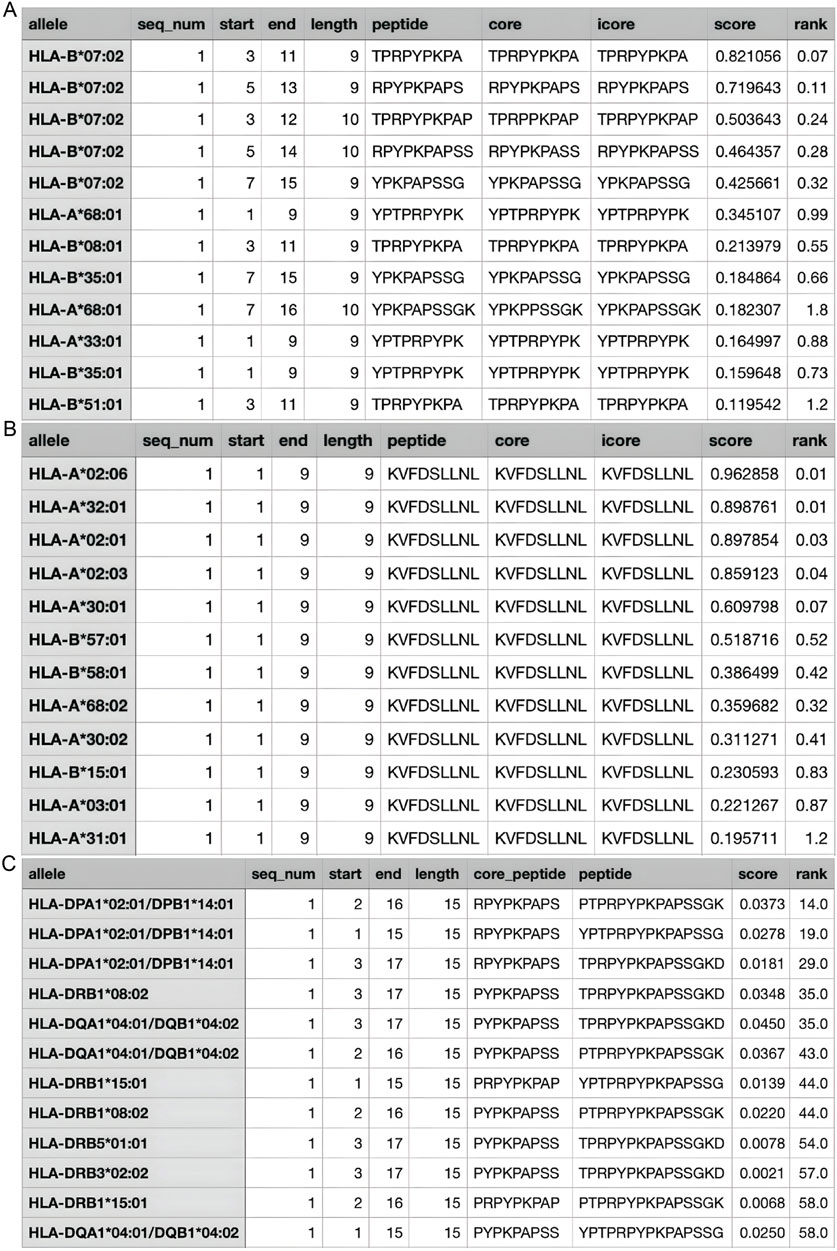
Figure 6. Estimated Values for HLA Affinity. (A) The predicted affinity of CLDN1 epitope YPT to HLA1; (B) The predicted affinity of CLDN1 epitope KVF to HLA1; (C) The predicted affinity of CLDN1 epitope YPT to HLA2. The greater the “score” value, the higher the affinity predicted, and the higher the “rank”—a categorization of the predicted affinity to a relative percentile.
4.3.2 Expression of IFN-gamma
In the antigen presentation pathway, which is enriched in the PR group as shown in Figure 4B, the HLA family of proteins is under expressed within the PD group as seen in Figure 7.
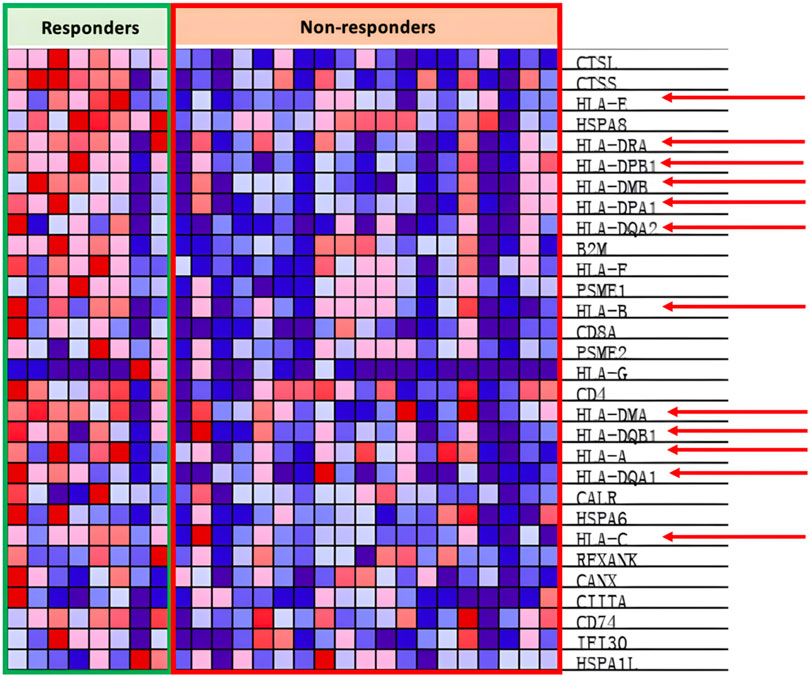
Figure 7. Heat map of KEGG antigen presentation pathway.
HLA proteins, also known as MHCs, are vital in the process of immunorecognition and antigen presentation. Hence, one of the methods of improving response could be to increase MHC presence and stimulation. A prime candidate is interferon-gamma (IFN-gamma), a chemokine that has shown capabilities of restoring HLA to HLA-deficient lung cancer (Traversari et al., 1997). Furthermore, research has already shown that certain oncolytic viruses, such as the vaccinia virus, are capable of inducing IFN-gamma synthesis and secretion within cancer cells and have shown potent tumor regression potential (L. Chen et al., 2021; Li et al., 2022). Samson et al. (2018) conducted an ex-vitro experiment treating high-grade glioma (HGG) cells with reoviruses encoded with IFN-gamma, showing increased PD-L1 expression to be strongly upregulated by IFN-gamma, which benefits future anti-PD-L1 treatment (Li et al., 2022).However, IFN-gamma also activates anti-viral mechanisms with its signaling, resulting in viral degradation in the immune environment, potentially decreasing the anti-tumor efficacy of the OV (Kang et al., 2018; Li et al., 2022). Furthermore, Song et al. (2019) reported that although high doses of IFN-gamma stimulate the classical JAK/STAT pathway, low doses (0.1 ng/mL) of IFN-gamma induce activation of ICAM1-PI3k-Akt-Notch1 signaling in cancer cells, leading to increased cancer cell stemness and CD133 expression (Jorgovanovic et al., 2020). The IFN-gamma induced cancer stemness facilitates NSCLC metastatic growth, and the upregulation of CD133 tumor cells is positively correlated with poor prognosis within NSCLC patients (Song et al., 2019). Hence, although IFN-gamma has the potential to increase tumor regression and anti-PD-L1 treatment efficacy, drawbacks warn about the potential anti-viral and tumor-enhancing side effects of IFN-gamma relating to dosage.
The full IFN-gamma gene is 591 bp long (Pruitt et al., 2009). In this original sequence, there is a signal peptide which is usually cleaved in order for the protein to be secreted and function normally (Wang et al., 2014). In our instance, this cleavage is unnecessary as all intracellular materials will be released upon oncolysis of the cell (LaRocca and Warner, 2018). Hence, to not only decrease the length of the gene sequence but also to ensure the functionality of the protein, the signal peptide will be omitted from the transgene.
Using DTU Health Tech’s signal peptide predicting algorithm SignaIP to predict the signal peptide cleavage site on the IFN-gamma protein, the results in Figure 8 were produced (Teufel et al., 2022). The algorithm predicted that the cleavage site would be between amino acids 23 and 24, meaning that amino acids 1-23 were part of the signal peptide complex. Hence, the remaining 522bps would be included in the viral genome whilst the 69bps that coded for the signaling peptide will be excluded.
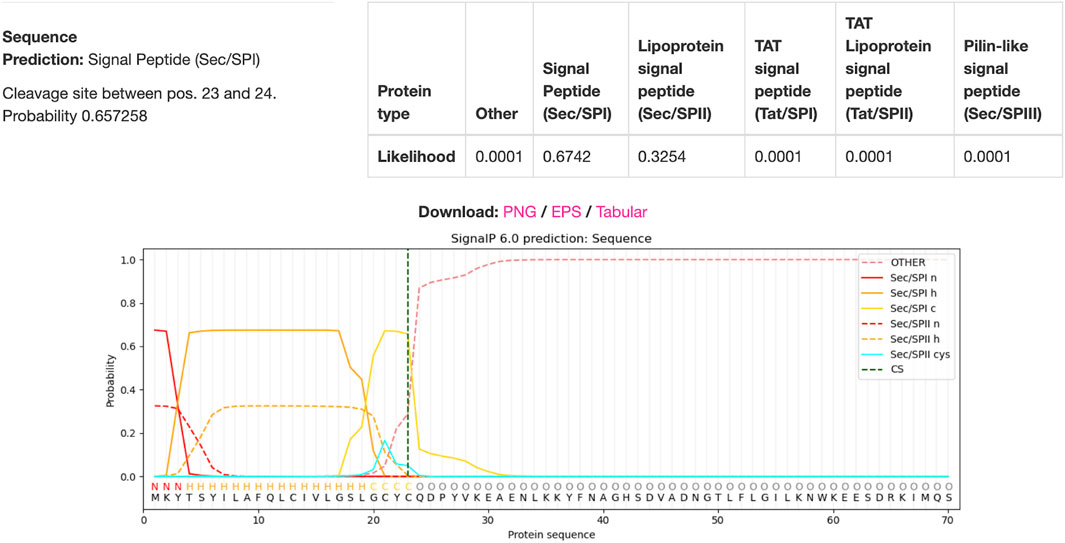
Figure 8. IFN-gamma Signal Peptide Cleavage Site Prediction. DTU Health Tech’s SignalP algorithm predicted that there is a 67.42% chance that there was a signal peptide cleavage site between amino acids 23 and 24.
4.3.3 Expression of IL7
Interleukin seven receptor (IL7R), part of the cytokine cytokine-receptor pathway, is under expressed within the PD groups as seen in Figure 9. IL-7 stimulates anti-tumor responses such as autophagy, migration, proliferation, and angiogenesis (Lin et al., 2017; Zhu et al., 2022). IL-7 is also potent at sensitizing the TME to ICIs and antagonizing the immunosuppressive network, both of which subsequently support the goal to increase response rates to anti-PD-1 treatment (Pellegrini et al., 2009; Ke et al., 2019; Nakao et al., 2020). Nakao et al. (2020) locally injected oncolytic vaccinia viruses with the dual expression of IL-7 and IL-12 into several different types of carcinomas alongside the combinational therapy of ICIs. The results showed that there was an increased presence of tumor-infiltrating lymphocytes (TILs) and increased anti-PD-1 and anti-CTLA4 sensitivity within the injected tumor (Nakao et al., 2020). Similar effects are seen in Kudling et al. (2022) IL-7 expressing oncolytic adenovirus 5, promoting tumor regression, activating CD4+ and CD8+ T cells, and encouraging T cell migration in various cell lines of different cancers. Additionally, Shi et al. (2019) concluded in their experiment that IL-7 promotes the sensitivity of NSCLC cells to cisplatin treatment, opening possibilities of combinational treatment with chemotherapy. Hence, expression of IL-7 in oncolytic viruses shows great potential to induce anti-tumor effects individually and in combination with ICI therapy and chemotherapy.
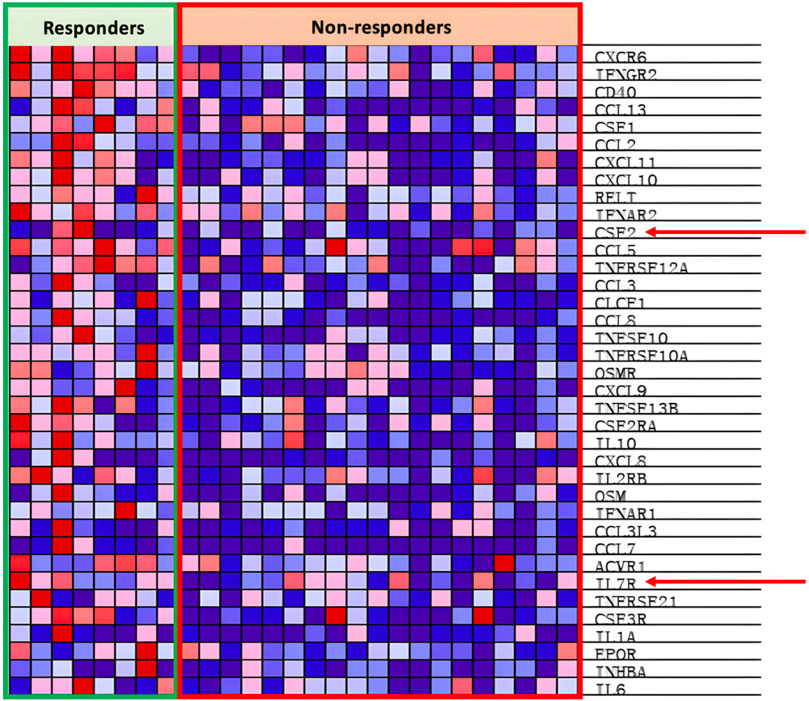
Figure 9. Heat map of cytokine cytokine-receptor pathway.
The IL-7 cytokine is a similar case to IFN-gamma where the full genomic sequence includes a signal peptide which needs to be cleaved for proper function. The original IL-7 gene is 177 amino acids in length (531bps) (Paysan-Lafosse et al., 2022). The signal peptide cleavage site is between amino acids 26 and 27, meaning that amino acids 1-26 code for the signal peptide and amino acids 27-177 code for the functioning IL-7 protein (Ivica et al., 2021). Hence, excluding the 78bps that code for the signal peptide, a nucleotide sequence of 453bps coding for IL-7 will be included in our viral genome.
4.3.4 Expression of GMCSF
Granulocyte-Macrophage Colony-stimulating factor (GM-CSF), encoded by the PR enriched CSF2 gene as seen in Figure 8, is part of the cytokine cytokine-receptor pathway. GM-CSF, as introduced earlier in the article, is vital in the differentiation and proliferation of hematopoietic cells, increasing the presence of neutrophils, effector T-cells, and APCs, leading to increased tumor regression (Ebner et al., 2003; Kumar et al., 2022). As seen by the example of T-VEC, oncolytic viruses can be armed with GM-CSF with the intention of increasing the local anti-tumor immune response. Rangsitratkul et al. (2022) inserted the GM-CSF transgene into oncolytic VSVd51 variants (an attenuated strand of vesicular stomatitis virus) in an experiment to treat bladder cancer. Results from Rangsitratkul et al. (2022) research showed that the GMCSF carrying the OV was able to enhance activation of the innate and adaptive immune system and subsequently improve survival in mice models with bladder cancer (C57Bl/6-MB49). Similarly, Malhotra et al. (2007) compared the effects of oncolytic HSVs with (NV1034) and without (NV1023) the GM-CSF transgene when treating colorectal carcinoma and hepatoma. The NV1034 variant showed significantly better anti-tumor effects compared to NV1023 under normal circumstances; however, in mice depleted of CD4+ and CD8+ T cells, no difference in antitumor effects between the two variants was observed (Malhotra et al., 2007). Hence, the GM-CSF transgene is a prime candidate to be included in an oncolytic virus due to its capabilities of increasing immune cell presence within the TME. However, it should be considered that alongside the increased presence of anti-tumor immune cells, GM-CSF simultaneously increases the presence of myeloid-derived suppressor cells (MDSCs), T-regulator (Treg) cells, M2 macrophages, and other immunosuppressive cells (Kumar et al., 2022).
The GM-CSF protein has one singular domain and is of a compact globular structure. This protein does not have a signaling peptide upon translation and all amino acid sequences are required for the functioning of the protein (Kurzrock and Dranoff, 2003). Correspondingly, the CSF2 gene which codes for the GM-CSF protein is only 432bps in length (Miyatake et al., 1985; The UniProt Consortium, 2023). Hence, the CSF2 gene does not need to be shortened or manipulated and can be introduced into the viral genome due to it being short and not having any unnecessary parts.
4.3.5 Other modifications
Additional to the strategies mentioned above, research was also conducted to find more targets and methods based on recent studies in the field. These targets and methods are seen in Table 7.
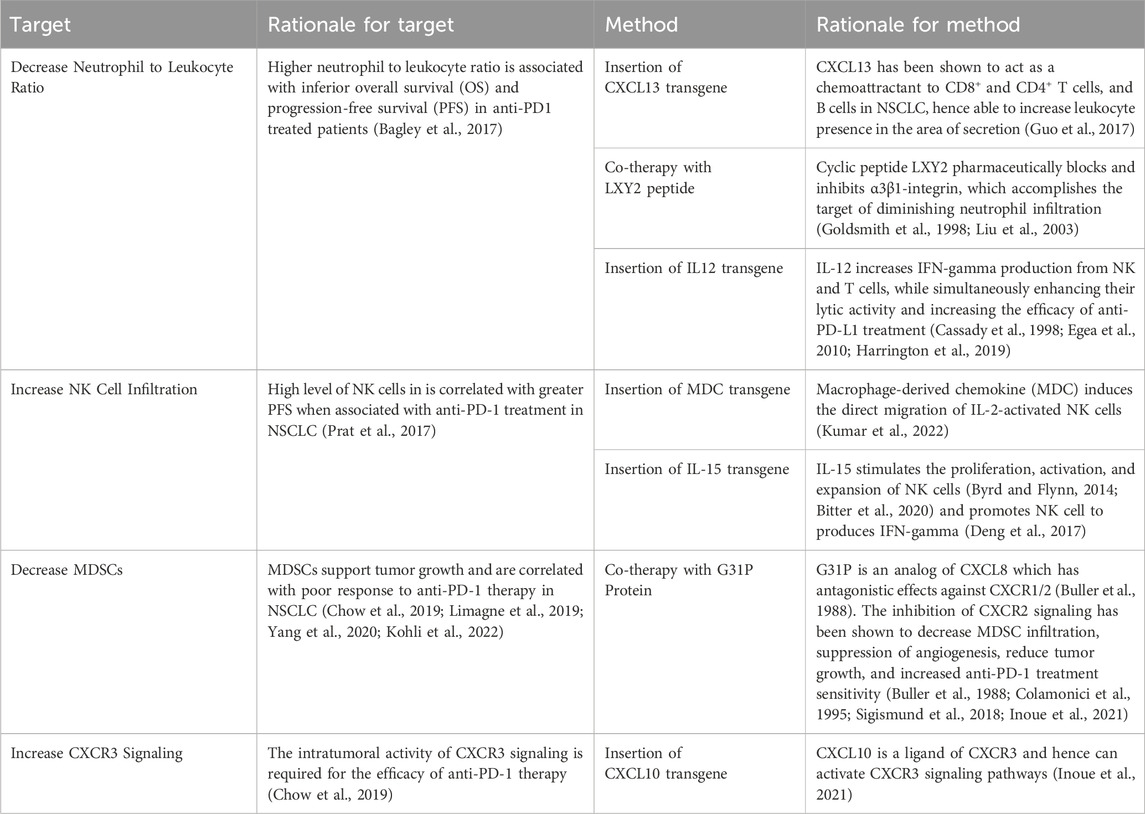
Table 7. Additional potential targets of oncolytic virus editing.
5 Other concerns and future directions
OVs can specifically infect cancer cells and induce the production of transgene proteins, allowing for a controlled and predictable method of regressing tumors. However, despite the various advantages, OVs still have prominent limitations including antiviral immunity, systemic delivery, and dosing strategies. While being able to initiate anti-tumor immunity, the presence of the OV can also evoke anti-viral immunity, which can harm the OV and decrease the efficacy of OV treatment. Furthermore, certain immune signaling pathways overlap in anti-tumor and anti-viral activity, meaning that when considering which transgenes to insert into the OV, anti-tumor efficacy and viral tolerance need to be considered simultaneously. Most modern OVs are administered intralesionally and subcutaneously to ensure the infection of the virus to the local tumor. However, these methods of administration only stimulate local anti-tumor responses and are incapable of activating a systemic reaction. Intravenous (IV) injections of OVs have been considered in order to favor systemic responses to OVs, however, OVs delivered through IV injection face the difficulties of host anti-viral responses and increase the risk of host cell infection. Hence, OVs are still limited when considering the possibility of becoming a systemic treatment. The dosage of OVs per use is also another contention of limitation. While on one side, the dosage cannot be too low or else there will not be enough virions created to sustain infection; however, on the other, if the dosage is too high, the OV may further provoke inflammatory responses or even regain pathogenicity from mutations. Furthermore, certain transgene proteins require dosage control, similar to how IFN-gamma can stimulate anti-tumor responses in high doses but induce pro-tumor effects in low doses. Overall, although OVs are promising, and many of them are being approved by the FDA such as T-VEC, Oncorine, and RIGVIR, there are still many limitations regarding the survival and efficacy of the OV to be worked out and improved upon.
Combinational therapy between oncolytic virotherapy and immune checkpoint inhibitors shows great promise in increasing OS and PFS for cancer patients. The complementary nature of these therapies shows how OVs can sensitize the TME to ICIs, whether by increasing the expression of the immune checkpoints on the tumor or by increasing lymphocyte presence, resulting in the generation of a greater response to said ICI therapy. In this study, a theoretical vaccinia virus OV with capabilities of increasing NSCLC patient response to anti-PD-1 ICI therapy was presented. Further research would be aimed towards producing and testing this OV in vitro on cell lines to observe potential efficacies and defects and test the accuracy of predictions generated in this study.
Author contributions
ZY: Writing–original draft, Writing–review and editing. ZW: Conceptualization, Funding acquisition, Investigation, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the provincial-ministerial co-construction project of the Henan Provincial Medical Science and Technology Research Project (SB201901034).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
ACS (2022). Key statistics for lung cancer. Available at: https://www.cancer.org/cancer/types/lung-cancer/about/key-statistics.html.
ACS (2024a). Chemotherapy for non-small cell lung cancer. American Cancer Society. Available at: https://www.cancer.org/cancer/types/lung-cancer/treating-non-small-cell/chemotherapy.html.
ACS (2024b). Non-small cell lung cancer targeted drug therapy. American Cancer Society. Available at: https://www.cancer.org/cancer/types/lung-cancer/treating-non-small-cell/targeted-therapies.html.
ACS (2024c). Treatment choices for non-small cell lung cancer, by stage. American Cancer Society. Available at: https://www.cancer.org/cancer/types/lung-cancer/treating-non-small-cell/by-stage.html.
Adashek, J. J., Subbiah, I. M., Matos, I., Garralda, E., Menta, A. K., Ganeshan, D. M., et al. (2020). Hyperprogression and immunotherapy: fact, fiction, or alternative fact? Trends Cancer 6 (3), 181–191. doi:10.1016/j.trecan.2020.01.005
Albinger, N., Hartmann, J., and Ullrich, E. (2021). Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 28 (9), 513–527. doi:10.1038/s41434-021-00246-w
Aldrak, N., Alsaab, S., Algethami, A., Bhere, D., Wakimoto, H., Shah, K., et al. (2021). Oncolytic herpes simplex virus-based therapies for cancer. Cells 10 (6), 1541. doi:10.3390/cells10061541
Alsaab, H. O., Sau, S., Alzhrani, R., Tatiparti, K., Bhise, K., Kashaw, S. K., et al. (2017). PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front. Pharmacol. 8, 561. doi:10.3389/fphar.2017.00561
Ando, Y., Kitayama, H., Kawaguchi, Y., and Koyanagi, Y. (2008). Primary target cells of herpes simplex virus type 1 in the hippocampus. Microbes Infect. 10 (14), 1514–1523. doi:10.1016/j.micinf.2008.09.005
Apavaloaei, A., Hardy, M.-P., Thibault, P., and Perreault, C. (2020). The origin and immune recognition of tumor-specific antigens. Cancers 12 (9), 2607. doi:10.3390/cancers12092607
Atasheva, S., and Shayakhmetov, D. M. (2021). Oncolytic viruses for systemic administration: engineering a whole different animal. Mol. Ther. 29 (3), 904–907. doi:10.1016/j.ymthe.2021.02.001
Bagley, S. J., Kothari, S., Aggarwal, C., Bauml, J. M., Alley, E. W., Evans, T. L., et al. (2017). Pretreatment neutrophil-to-lymphocyte ratio as a marker of outcomes in nivolumab-treated patients with advanced non-small-cell lung cancer. Lung Cancer 106, 1–7. doi:10.1016/j.lungcan.2017.01.013
Bhat, A. A., Syed, N., Therachiyil, L., Nisar, S., Hashem, S., Macha, M. A., et al. (2020). Claudin-1, A double-edged sword in cancer. Int. J. Mol. Sci. 21 (2), 569. doi:10.3390/ijms21020569
Biton, J., Mansuet-Lupo, A., Pécuchet, N., Alifano, M., Ouakrim, H., Arrondeau, J., et al. (2018). TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti–PD-1 in lung adenocarcinoma. Clin. Cancer Res. 24 (22), 5710–5723. doi:10.1158/1078-0432.ccr-18-0163
Bitter, E. E., Townsend, M. H., Erickson, R., Allen, C., and O’Neill, K. L. (2020). Thymidine kinase 1 through the ages: a comprehensive review. Cell and Biosci. 10 (1), 138. doi:10.1186/s13578-020-00493-1
Bradbury, P., Sivajohanathan, D., Chan, A., Kulkarni, S., Ung, Y., and Ellis, P. M. (2016). Postoperative adjuvant systemic therapy in completely resected non–small-cell lung cancer: a systematic review. Clin. Lung Cancer 18, 259–273.e8. doi:10.1016/j.cllc.2016.07.002
Buller, R. M., Chakrabarti, S., Cooper, J. A., Twardzik, D. R., and Moss, B. (1988). Deletion of the vaccinia virus growth factor gene reduces virus virulence. J. Virology 62 (3), 866–874. doi:10.1128/jvi.62.3.866-874.1988
Byrd, J. C., and Flynn, J. M. (2014). “102 - chronic lymphocytic leukemia,” in Abeloff's clinical oncology. Editors J. E. Niederhuber, J. O. Armitage, J. H. Doroshow, M. B. Kastan, and J. E. Tepper. Fifth Edition (London, United Kingdom: Churchill Livingstone), 1958–1978.e1957. doi:10.1016/B978-1-4557-2865-7.00102-8
Cabezón-Gutiérrez, L., Custodio-Cabello, S., Palka-Kotlowska, M., Alonso-Viteri, S., and Khosravi-Shahi, P. (2021). Biomarkers of immune checkpoint inhibitors in non–small cell lung cancer: beyond PD-L1. Clin. Lung Cancer 22 (5), 381–389. doi:10.1016/j.cllc.2021.03.006
Cai, F., Zhu, Q., Miao, Y., Shen, S., Su, X., and Shi, Y. (2017). Desmoglein-2 is overexpressed in non-small cell lung cancer tissues and its knockdown suppresses NSCLC growth by regulation of p27 and CDK2. J. Cancer Res. Clin. Oncol. 143 (1), 59–69. doi:10.1007/s00432-016-2250-0
Cassady, K. A., Gross, M., and Roizman, B. (1998). The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virology 72 (11), 8620–8626. doi:10.1128/jvi.72.11.8620-8626.1998
Chaft, J. E., Rimner, A., Weder, W., Azzoli, C. G., Kris, M. G., and Cascone, T. (2021). Evolution of systemic therapy for stages I–III non-metastatic non-small-cell lung cancer. Nat. Rev. Clin. Oncol. 18 (9), 547–557. doi:10.1038/s41571-021-00501-4
Champiat, S., Dercle, L., Ammari, S., Massard, C., Hollebecque, A., Postel-Vinay, S., et al. (2017). Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 23 (8), 1920–1928. doi:10.1158/1078-0432.ccr-16-1741
Chan, B. A., and Hughes, B. G. M. (2014). Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl. Lung Cancer Res. 4 (1), 36–54. doi:10.3978/j.issn.2218-6751.2014.05.01
Chan, W. M., and McFadden, G. (2014). Oncolytic poxviruses. Annu. Rev. Virology 1 (1), 119–141. doi:10.1146/annurev-virology-031413-085442
Chao, Y.-C., Pan, S.-H., Yang, S.-C., Yu, S.-L., Che, T.-F., Lin, C.-W., et al. (2009). Claudin-1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am. J. Respir. Crit. Care Med. 179 (2), 123–133. doi:10.1164/rccm.200803-456OC
Chen, C.-Y., Wang, P.-Y., Hutzen, B., Sprague, L., Swain, H. M., Love, J. K., et al. (2017). Cooperation of oncolytic herpes virotherapy and PD-1 blockade in murine rhabdomyosarcoma models. Sci. Rep. 7 (1), 2396. doi:10.1038/s41598-017-02503-8
Chen, L., Chen, H., Ye, J., Ge, Y., Wang, H., Dai, E., et al. (2021). Intratumoral expression of interleukin 23 variants using oncolytic vaccinia virus elicit potent antitumor effects on multiple tumor models via tumor microenvironment modulation. Theranostics 11 (14), 6668–6681. doi:10.7150/thno.56494
Chen, W.-Y., Chen, Y.-L., Lin, H.-W., Chang, C.-F., Huang, B.-S., Sun, W.-Z., et al. (2021). Stereotactic body radiation combined with oncolytic vaccinia virus induces potent anti-tumor effect by triggering tumor cell necroptosis and DAMPs. Cancer Lett. 523, 149–161. doi:10.1016/j.canlet.2021.09.040
Choi, B. D., Yu, X., Castano, A. P., Bouffard, A. A., Schmidts, A., Larson, R. C., et al. (2019). CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 37 (9), 1049–1058. doi:10.1038/s41587-019-0192-1
Chow, M. T., Ozga, A. J., Servis, R. L., Frederick, D. T., Lo, J. A., Fisher, D. E., et al. (2019). Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity 50 (6), 1498–1512.e5. doi:10.1016/j.immuni.2019.04.010
Cohn, D. E., Sill, M. W., Walker, J. L., O'Malley, D., Nagel, C. I., Rutledge, T. L., et al. (2017). Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin®) in recurrent ovarian, tubal, or peritoneal cancer: an NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 146 (3), 477–483. doi:10.1016/j.ygyno.2017.07.135
Colamonici, O. R., Domanski, P., Sweitzer, S. M., Larner, A., and Buller, R. M. L. (1995). Vaccinia virus B18R gene encodes a type I interferon-binding protein that blocks interferon alpha transmembrane signaling. J. Biol. Chem. 270 (27), 15974–15978. doi:10.1074/jbc.270.27.15974
Croft, M. (2009). The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 9 (4), 271–285. doi:10.1038/nri2526
Dai, M. H., Liu, S. L., Chen, N. G., Zhang, T. P., You, L. Q., Zhang, F., et al. (2014). Oncolytic vaccinia virus in combination with radiation shows synergistic antitumor efficacy in pancreatic cancer. Cancer Lett. 344 (2), 282–290. doi:10.1016/j.canlet.2013.11.007
Daum, S., Hagen, H., Naismith, E., Wolf, D., and Pircher, A. (2021). The role of anti-angiogenesis in the treatment landscape of non-small cell lung cancer - new combinational approaches and strategies of neovessel inhibition. Front. Cell Dev. Biol. 8, 610903. doi:10.3389/fcell.2020.610903
Deng, L., Fan, J., Ding, Y., Zhang, J., Zhou, B., Zhang, Y., et al. (2017). Oncolytic efficacy of thymidine kinase-deleted vaccinia virus strain Guang9. Oncotarget 8 (25), 40533–40543. doi:10.18632/oncotarget.17125
Dong, Z.-Y., Wu, S.-P., Liao, R.-Q., Huang, S.-M., and Wu, Y.-L. (2016). Potential biomarker for checkpoint blockade immunotherapy and treatment strategy. Tumor Biol. 37 (4), 4251–4261. doi:10.1007/s13277-016-4812-9
Ebner, K., Bandion, A., Binder, B. R., de Martin, R., and Schmid, J. A. (2003). GMCSF activates NF-kappaB via direct interaction of the GMCSF receptor with IkappaB kinase beta. Blood 102 (1), 192–199. doi:10.1182/blood-2002-12-3753
Egea, L., Hirata, Y., and Kagnoff, M. F. (2010). GM-CSF: a role in immune and inflammatory reactions in the intestine. Expert Rev. Gastroenterology and Hepatology 4 (6), 723–731. doi:10.1586/egh.10.73
Elvington, M., Liszewski, M. K., and Atkinson, J. P. (2020). CD46 and oncologic interactions: friendly fire against cancer. Antibodies 9 (4), 59. doi:10.3390/antib9040059
Engeland, C. E., and Ungerechts, G. (2021). Measles virus as an oncolytic immunotherapy. Cancers 13 (3), 544. doi:10.3390/cancers13030544
Everts, B., and van der Poel, H. G. (2005). Replication-selective oncolytic viruses in the treatment of cancer. Cancer Gene Ther. 12 (2), 141–161. doi:10.1038/sj.cgt.7700771
Fagerberg, L., Hallström, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteomics 13 (2), 397–406. doi:10.1074/mcp.M113.035600
Fajardo, C. A., Guedan, S., Rojas, L. A., Moreno, R., Arias-Badia, M., de Sostoa, J., et al. (2017). Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res. 77 (8), 2052–2063. doi:10.1158/0008-5472.Can-16-1708
Farooq, A. V., Valyi-Nagy, T., and Shukla, D. (2010). Mediators and mechanisms of herpes simplex virus entry into ocular cells. Curr. Eye Res. 35 (6), 445–450. doi:10.3109/02713681003734841
Fenton, S. E., Saleiro, D., and Platanias, L. C. (2021). Type I and II interferons in the anti-tumor immune response. Cancers 13 (5), 1037. doi:10.3390/cancers13051037
Ferguson, M. S., Lemoine, N. R., and Wang, Y. (2012). Systemic delivery of oncolytic viruses: hopes and hurdles. Adv. Virology 2012, 805629. doi:10.1155/2012/805629
Ferrucci, P. F., Pala, L., Conforti, F., and Cocorocchio, E. (2021). Talimogene Laherparepvec (T-VEC): an intralesional cancer immunotherapy for advanced melanoma. Cancers 13 (6), 1383. doi:10.3390/cancers13061383
Frisone, D., Friedlaender, A., Addeo, A., and Tsantoulis, P. (2022). The landscape of immunotherapy resistance in NSCLC. Front. Oncol. 12, 817548. doi:10.3389/fonc.2022.817548
Fumet, J.-D., Richard, C., Ledys, F., Klopfenstein, Q., Joubert, P., Routy, B., et al. (2018). Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br. J. Cancer 119 (8), 950–960. doi:10.1038/s41416-018-0220-9
Garcia-Diaz, A., Shin, D. S., Moreno, B. H., Saco, J., Escuin-Ordinas, H., Rodriguez, G. A., et al. (2019). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 29 (11), 3766. doi:10.1016/j.celrep.2019.11.113
Geisler, A., Hazini, A., Heimann, L., Kurreck, J., and Fechner, H. (2021). Coxsackievirus B3—its potential as an oncolytic virus. Viruses 13 (5), 718. doi:10.3390/v13050718
Geng, Y., Shao, Y., He, W., Hu, W., Xu, Y., Chen, J., et al. (2015). Prognostic role of tumor-infiltrating lymphocytes in lung cancer: a meta-analysis. Cell. Physiology Biochem. 37 (4), 1560–1571. doi:10.1159/000438523
Goebeler, M.-E., and Bargou, R. C. (2020). T cell-engaging therapies — BiTEs and beyond. Nat. Rev. Clin. Oncol. 17 (7), 418–434. doi:10.1038/s41571-020-0347-5
Goldsmith, K., Chen, W., Johnson, D. C., and Hendricks, R. L. (1998). Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J. Exp. Med. 187 (3), 341–348. doi:10.1084/jem.187.3.341
Goncalves, G., Mullan, K. A., Duscharla, D., Ayala, R., Croft, N. P., Faridi, P., et al. (2021). IFNγ modulates the immunopeptidome of triple negative breast cancer cells by enhancing and diversifying antigen processing and presentation. Front. Immunol. 12, 645770. doi:10.3389/fimmu.2021.645770
Gopalakrishnan, V., Spencer, C. N., Nezi, L., Reuben, A., Andrews, M. C., Karpinets, T. V., et al. (2018). Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359 (6371), 97–103. doi:10.1126/science.aan4236
Guo, L., Chi, Y., Xue, J., Ma, L., Shao, Z., and Wu, J. (2017). Phosphorylated eIF2α predicts disease-free survival in triple-negative breast cancer patients. Sci. Rep. 7 (1), 44674. doi:10.1038/srep44674
Guo, Z. S., Lu, B., Guo, Z., Giehl, E., Feist, M., Dai, E., et al. (2019). Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J. immunotherap. cancer. 7, 6. doi:10.1186/s40425-018-0495-7
Guse, K., Cerullo, V., and Hemminki, A. (2011). Oncolytic vaccinia virus for the treatment of cancer. Expert Opin. Biol. Ther. 11 (5), 595–608. doi:10.1517/14712598.2011.558838
Han, Y., Liu, D., and Li, L. (2020). PD-1/PD-L1 pathway: current researches in cancer. Am. J. Cancer Res. 10 (3), 727–742.
Harrington, K., Freeman, D. J., Kelly, B., Harper, J., and Soria, J.-C. (2019). Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 18 (9), 689–706. doi:10.1038/s41573-019-0029-0
Hassanin, I., and Elzoghby, A. (2020). Albumin-based nanoparticles: a promising strategy to overcome cancer drug resistance. Cancer Drug Resist. 3, 930–946. doi:10.20517/cdr.2020.68
Hemminki, O., Bauerschmitz, G., Hemmi, S., Lavilla-Alonso, S., Diaconu, I., Guse, K., et al. (2011). Oncolytic adenovirus based on serotype 3. Cancer Gene Ther. 18 (4), 288–296. doi:10.1038/cgt.2010.79
Hensen, L. C. M., Hoeben, R. C., and Bots, S. T. F. (2020). Adenovirus receptor expression in cancer and its multifaceted role in oncolytic adenovirus therapy. Int. J. Mol. Sci. 21 (18), 6828. doi:10.3390/ijms21186828
Heo, J., Breitbach, C. J., Moon, A., Kim, C. W., Patt, R., Kim, M. K., et al. (2011). Sequential therapy with JX-594, A targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol. Ther. 19 (6), 1170–1179. doi:10.1038/mt.2011.39
Hong, B., Sahu, U., Mullarkey, M. P., and Kaur, B. (2022). Replication and spread of oncolytic herpes simplex virus in solid tumors. Viruses 14 (1), 118. doi:10.3390/v14010118
Howington, J. A., Blum, M. G., Chang, A. C., Balekian, A. A., and Murthy, S. C. (2013). Treatment of stage I and II non-small cell lung cancer: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines, Am. Coll. Chest Physicians evidence-based Clin. Pract. Guidel. Chest. 143 (5), e278S-e313S. doi:10.1378/chest.12-2359
Huang, B., Sikorski, R., Kirn, D. H., and Thorne, S. H. (2011). Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 18 (2), 164–172. doi:10.1038/gt.2010.121
Hutzler, S., Erbar, S., Jabulowsky, R. A., Hanauer, J. R. H., Schnotz, J. H., Beissert, T., et al. (2017). Antigen-specific oncolytic MV-based tumor vaccines through presentation of selected tumor-associated antigens on infected cells or virus-like particles. Sci. Rep. 7 (1), 16892. doi:10.1038/s41598-017-16928-8
Inoue, T., Byrne, T., Inoue, M., Tait, M. E., Wall, P., Wang, A., et al. (2021). Oncolytic vaccinia virus gene modification and cytokine expression effects on tumor infection, immune response, and killing. Mol. Cancer Ther. 20 (8), 1481–1494. doi:10.1158/1535-7163.Mct-20-0863
Paysan-Lafosse, T., Blum, M., Chuguransky, S., Grego, T., Pinto, B. L., and Salazar, G. A. (2022). InterPro in 2022. Nucleic Acids Res. doi:10.1093/nar/gkac993
Ivica, L., Supriya, K., and Peer, B. (2021). SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 49 (D1), D458–D460. doi:10.1093/nar/gkaa937
Jayawardena, N., Poirier, J. T., Burga, L. N., and Bostina, M. (2020). Virus-receptor interactions and virus neutralization: insights for oncolytic virus development. Oncolytic Virotherapy 9, 1–15. doi:10.2147/ov.s186337
Jorgovanovic, D., Song, M., Wang, L., and Zhang, Y. (2020). Roles of IFN-γ in tumor progression and regression: a review. Biomark. Res. 8 (1), 49. doi:10.1186/s40364-020-00228-x
Kang, S., Brown, H. M., and Hwang, S. (2018). Direct antiviral mechanisms of interferon-gamma. Immune Netw. 18 (5), e33. doi:10.4110/in.2018.18.e33
Kato, S., Goodman, A., Walavalkar, V., Barkauskas, D. A., Sharabi, A., and Kurzrock, R. (2017). Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin. Cancer Res. 23 (15), 4242–4250. doi:10.1158/1078-0432.ccr-16-3133
Kaufman, H. L., Kohlhapp, F. J., and Zloza, A. (2015). Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 14 (9), 642–662. doi:10.1038/nrd4663
Kawai, T., Suzuki, M., and Kageyama, K. (1988). Glycosaminoglycans in lung carcinoma. Hum. Pathol., 19(11), 1288–1292. doi:10.1016/S0046-8177(88)80283-1
Kazemi, S., Papadopoulou, S., Li, S., Su, Q., Wang, S., Yoshimura, A., et al. (2004). Control of alpha subunit of eukaryotic translation initiation factor 2 (eIF2 alpha) phosphorylation by the human papillomavirus type 18 E6 oncoprotein: implications for eIF2 alpha-dependent gene expression and cell death. Mol. Cell. Biol. 24 (8), 3415–3429. doi:10.1128/MCB.24.8.3415-3429.2004
Ke, B., Wei, T., Huang, Y., Gong, Y., Wu, G., Liu, J., et al. (2019). Interleukin-7 resensitizes non-small-cell lung cancer to cisplatin via inhibition of ABCG2. Mediat. Inflamm. 2019, 7241418. doi:10.1155/2019/7241418
Kim, Y. G., Baltabekova, A. Z., Zhiyenbay, E. E., Aksambayeva, A. S., Shagyrova, Z. S., Khannanov, R., et al. (2017). Recombinant Vaccinia virus-coded interferon inhibitor B18R: expression, refolding and a use in a mammalian expression system with a RNA-vector. PLOS ONE 12 (12), e0189308. doi:10.1371/journal.pone.0189308
Kirn, D. H., and Thorne, S. H. (2009). Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 9 (1), 64–71. doi:10.1038/nrc2545
Kohli, K., Pillarisetty, V. G., and Kim, T. S. (2022). Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 29 (1), 10–21. doi:10.1038/s41417-021-00303-x
Koodie, L., Robertson, M. G., Chandrashekar, M., Ruth, G., Dunning, M., Bianco, R. W., et al. (2019). Rodents versus pig model for assessing the performance of serotype chimeric ad5/3 oncolytic Adenoviruses. Cancers 11 (2), 198. doi:10.3390/cancers11020198
Krause, G., Winkler, L., Mueller, S. L., Haseloff, R. F., Piontek, J., and Blasig, I. E. (2008). Structure and function of claudins. Biochimica Biophysica Acta (BBA) - Biomembr., 1778 (3), 631–645. doi:10.1016/j.bbamem.2007.10.018
Kudling, T. V., Clubb, J. H. A., Quixabeira, D. C. A., Santos, J. M., Havunen, R., Kononov, A., et al. (2022). Local delivery of interleukin 7 with an oncolytic adenovirus activates tumor-infiltrating lymphocytes and causes tumor regression. OncoImmunology 11 (1), 2096572. doi:10.1080/2162402X.2022.2096572
Kulkarni, A., Ferreira, T., Bretscher, C., Grewenig, A., El-Andaloussi, N., Bonifati, S., et al. (2021). Oncolytic H-1 parvovirus binds to sialic acid on laminins for cell attachment and entry. Nat. Commun. 12 (1), 3834. doi:10.1038/s41467-021-24034-7
Kumar, A., Taghi Khani, A., Sanchez Ortiz, A., and Swaminathan, S. (2022). GM-CSF: a double-edged sword in cancer immunotherapy. Front. Immunol. 13, 901277. doi:10.3389/fimmu.2022.901277
Kumari, N., Singh, S., Haloi, D., Mishra, S. K., Krishnani, N., Nath, A., et al. (2019). Epidermal growth factor receptor mutation frequency in squamous cell carcinoma and its diagnostic performance in cytological samples: a molecular and immunohistochemical study. World J. Oncol. 10, 142–150. doi:10.14740/wjon1204
Kurzrock, R., and Dranoff, G. (2003). Granulocyte-macrophage colony-stimulating factor. 503–524. doi:10.1016/b978-012689663-3/50025-9
Lackey, A., and Donington, J. (2013). Surgical management of lung cancer. Seminars Interventional Radiology 30 (02), 133–140. doi:10.1055/s-0033-1342954
LaRocca, C. J., and Warner, S. G. (2018). Oncolytic viruses and checkpoint inhibitors: combination therapy in clinical trials. Clin. Transl. Med., 7(1), e35. doi:10.1186/s40169-018-0214-5
Li, L., Liu, S., Han, D., Tang, B., and Ma, J. (2020). Delivery and biosafety of oncolytic virotherapy. Front. Oncol. 10, 475. doi:10.3389/fonc.2020.00475
Li, Q., Tan, F., Wang, Y., Liu, X., Kong, X., Meng, J., et al. (2022). The gamble between oncolytic virus therapy and IFN. Front. Immunol. 13, 971674. doi:10.3389/fimmu.2022.971674
Li, Y., Zhang, C., Chen, X., Yu, J., Wang, Y., Yang, Y., et al. (2011). ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J. Biol. Chem. 286 (28), 24785–24792. doi:10.1074/jbc.M111.232439
Limagne, E., Richard, C., Thibaudin, M., Fumet, J.-D., Truntzer, C., Lagrange, A., et al. (2019). Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. OncoImmunology 8 (4), e1564505. doi:10.1080/2162402X.2018.1564505
Lin, J., Zhu, Z., Xiao, H., Wakefield, M. R., Ding, V. A., Bai, Q., et al. (2017). The role of IL-7 in immunity and cancer. Anticancer Res. 37 (3), 963–967. doi:10.21873/anticanres.11405
Liu, B. L., Robinson, M., Han, Z. Q., Branston, R. H., English, C., Reay, P., et al. (2003). ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 10 (4), 292–303. doi:10.1038/sj.gt.3301885
Liu, J., Kang, R., and Tang, D. (2022). The KRAS-G12C inhibitor: activity and resistance. Cancer Gene Ther. 29 (7), 875–878. doi:10.1038/s41417-021-00383-9
Liu, M., Wang, X., Li, W., Yu, X., Flores-Villanueva, P., Xu-Monette, Z. Y., et al. (2020). Targeting PD-L1 in non-small cell lung cancer using CAR T cells. Oncogenesis 9 (8), 72. doi:10.1038/s41389-020-00257-z
Ma, W., He, H., and Wang, H. (2018). Oncolytic herpes simplex virus and immunotherapy. BMC Immunol. 19 (1), 40. doi:10.1186/s12865-018-0281-9
MacLeod, D. T., Nakatsuji, T., Wang, Z., di Nardo, A., and Gallo, R. L. (2015). Vaccinia virus binds to the scavenger receptor MARCO on the surface of keratinocytes. J. Invest Dermatol 135 (1), 142–150. doi:10.1038/jid.2014.330
Magden, J., Kääriäinen, L., and Ahola, T. (2005). Inhibitors of virus replication: recent developments and prospects. Appl. Microbiol. Biotechnol. 66 (6), 612–621. doi:10.1007/s00253-004-1783-3
Majdoul, S., and Compton, A. A. (2022). Lessons in self-defence: inhibition of virus entry by intrinsic immunity. Nat. Rev. Immunol. 22 (6), 339–352. doi:10.1038/s41577-021-00626-8
Makker, V., Colombo, N., Herráez, A. C., Santin, A. D., Colomba, E., Miller, D. S., et al. (2022). Lenvatinib plus pembrolizumab for advanced endometrial cancer. N. Engl. J. Med., 386(5), 437–448. doi:10.1056/NEJMoa2108330
Malhotra, S., Kim, T., Zager, J., Bennett, J., Ebright, M., D’Angelica, M., et al. (2007). Use of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomas. Surgery 141 (4), 520–529. doi:10.1016/j.surg.2006.10.010
Mansfield, D., Pencavel, T., Kyula, J. N., Zaidi, S., Roulstone, V., Thway, K., et al. (2013). Oncolytic Vaccinia virus and radiotherapy in head and neck cancer. Oral Oncol. 49 (2), 108–118. doi:10.1016/j.oraloncology.2012.07.019
Marcu, A., Bichmann, L., Kuchenbecker, L., Kowalewski, D. J., Freudenmann, L. K., Backert, L., et al. (2021). HLA Ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J. Immunother. Cancer 9 (4), e002071. doi:10.1136/jitc-2020-002071
Melcher, A., Harrington, K., and Vile, R. (2021). Oncolytic virotherapy as immunotherapy. Science 374 (6573), 1325–1326. doi:10.1126/science.abk3436
Mirbahari, S. N., Da Silva, M., Zúñiga, A. I. M., Kooshki Zamani, N., St-Laurent, G., Totonchi, M., et al. (2024). Recent progress in combination therapy of oncolytic vaccinia virus. Front. Immunol. 15, 1272351. doi:10.3389/fimmu.2024.1272351
Miyatake, S., Otsuka, T., Yokota, T., Lee, F., and Arai, K. (1985). Structure of the chromosomal gene for granulocyte-macrophage colony stimulating factor: comparison of the mouse and human genes. EMBO J. 4 (10), 2561–8. doi:10.1002/j.1460-2075.1985.tb03971.x
Moon, E. K., Wang, L.-C. S., Bekdache, K., Lynn, R. C., Lo, A., Thorne, S. H., et al. (2018). Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. OncoImmunology 7 (3), e1395997. doi:10.1080/2162402X.2017.1395997
Morgenstern, D. A., and Irwin, M. S. (2019). Chapter 15 - current and future strategies for treatment of relapsed neuroblastoma. In S. K. Ray (Ed.), Neuroblastoma. 263–281. Academic Press. doi:10.1016/B978-0-12-812005-7.00015-1
Müller, L., Berkeley, R., Barr, T., Ilett, E., and Errington-Mais, F. (2020). Past, present and future of oncolytic reovirus. Cancers 12 (11), 3219. doi:10.3390/cancers12113219
Nakao, S., Arai, Y., Tasaki, M., Yamashita, M., Murakami, R., Kawase, T., et al. (2020). Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci. Transl. Med., 12(526), eaax7992. doi:10.1126/scitranslmed.aax7992
National Cancer Institute at the National Institutes of Health (2018). Available at: https://www.cancer.gov/news-events/cancer-currents-blog/2018/oncolytic-viruses-to-treat-cancer (Accessed August 8, 2024).
National Cancer Institute at the National Institutes of Health (2022). Available at: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/checkpoint-inhibitors (Accessed August 8, 2024).
Nguyên, T. L.-A., Abdelbary, H., Arguello, M., Breitbach, C., Leveille, S., Diallo, J.-S., et al. (2008). Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc. Natl. Acad. Sci. 105 (39), 14981–14986. doi:10.1073/pnas.0803988105
Nie, R.-C., Chen, G.-M., Wang, Y., Zhou, J., Duan, J.-L., Zhou, Z.-W., et al. (2021). Efficacy of anti-PD-1/PD-L1 monotherapy or combinational therapy in patients aged 75 Years or older: a study-level meta-analysis. Front. Oncol. 11, 538174. doi:10.3389/fonc.2021.538174
National Human Genome Research Institute (2024). Available at: https://www.genome.gov/genetics-glossary/Promoter#:∼:text=A%20promoter%2C%20as%20related%20to,molecule%20(such%20as%20mRNA) (Accessed August 8, 2024).
Oliner, J. D., Saiki, A. Y., and Caenepeel, S. (2016). The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb. Perspect. Med. 6 (6), a026336. doi:10.1101/cshperspect.a026336
Pellegrini, M., Calzascia, T., Elford, A. R., Shahinian, A., Lin, A. E., Dissanayake, D., et al. (2009). Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat. Med. 15 (5), 528–536. doi:10.1038/nm.1953
Peng, W., Chen, J. Q., Liu, C., Malu, S., Creasy, C., Tetzlaff, M. T., et al. (2016). Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 6 (2), 202–216. doi:10.1158/2159-8290.cd-15-0283
Prat, A., Navarro, A., Paré, L., Reguart, N., Galván, P., Pascual, T., et al. (2017). Immune-related gene expression profiling after PD-1 blockade in non–small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res. 77 (13), 3540–3550. doi:10.1158/0008-5472.Can-16-3556
Pruitt, K. D., Harrow, J., Harte, R. A., Wallin, C., Diekhans, M., and Maglott, D. R. (2009). The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 19 (7), 1316–23. [Epub 2009 Jun 4. Erratum in: Genome Res. 2009 Aug; 19(8):1506]. doi:10.1101/gr.080531.108
Puzanov, I., Milhem, M. M., Minor, D., Hamid, O., Li, A., Chen, L., et al. (2016). Talimogene Laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 34 (22), 2619–2626. doi:10.1200/jco.2016.67.1529
Rahman, M. M., and McFadden, G. (2021). Oncolytic viruses: newest frontier for cancer immunotherapy. Cancers 13 (21), 5452. doi:10.3390/cancers13215452
Rangsitratkul, C., Lawson, C., Bernier-Godon, F., Niavarani, S.-R., Boudaud, M., Rouleau, S., et al. (2022). Intravesical immunotherapy with a GM-CSF armed oncolytic vesicular stomatitis virus improves outcome in bladder cancer. Mol. Ther. - Oncolytics 24, 507–521. doi:10.1016/j.omto.2022.01.009
Ren, S., Tian, Q., Amar, N., Yu, H., Rivard, C. J., Caldwell, C., et al. (2018). The immune checkpoint, HVEM may contribute to immune escape in non-small cell lung cancer lacking PD-L1 expression. Lung Cancer. 125, 115, 120. doi:10.1016/j.lungcan.2018.09.004
Rheinheimer, S., Heussel, C.-P., Mayer, P., Gaissmaier, L., Bozorgmehr, F., Winter, H., et al. (2020). Oligoprogressive non-small-cell lung cancer under treatment with PD-(L)1 inhibitors. Cancers 12 (4), 1046. doi:10.3390/cancers12041046
Routy, B., Le Chatelier, E., Derosa, L., Duong, C. P. M., Alou, M. T., Daillère, R., et al. (2018). Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 359 (6371), 91–97. doi:10.1126/science.aan3706
Samson, A., Scott, K. J., Taggart, D., West, E. J., Wilson, E., Nuovo, G. J., et al. (2018). Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med., 10(422), eaam7577. doi:10.1126/scitranslmed.aam7577
Shao, J., Jin, Y., and Jin, C. (2023). A new approach to overcoming resistance to immunotherapy: nanotechnology. Front. Oncol. 13, 1210245. doi:10.3389/fonc.2023.1210245
Sharma, P., Hu-Lieskovan, S., Wargo, J. A., and Ribas, A. (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168 (4), 707–723. doi:10.1016/j.cell.2017.01.017
Shi, C., Wang, Y., Xue, J., and Zhou, X. (2022). Immunotherapy for EGFR-mutant advanced non-small-cell lung cancer: current status, possible mechanisms and application prospects. Front. Immunol. 13, 940288. doi:10.3389/fimmu.2022.940288
Shi, L., Xu, Z., Yang, Q., Huang, Y., Gong, Y., Wang, F., et al. (2019). IL-7–Mediated IL-7R-JAK3/STAT5 signalling pathway contributes to chemotherapeutic sensitivity in non–small-cell lung cancer. Cell Prolif. 52(6), e12699. doi:10.1111/cpr.12699
Shi, T., Song, X., Wang, Y., Liu, F., and Wei, J. (2020). Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front. Immunol. 11, 683. doi:10.3389/fimmu.2020.00683
Sigismund, S., Avanzato, D., and Lanzetti, L. (2018). Emerging functions of the EGFR in cancer. Mol. Oncol., 12 (1), 3–20. doi:10.1002/1878-0261.12155
Sivan, A., Corrales, L., Hubert, N., Williams, J. B., Aquino-Michaels, K., Earley, Z. M., et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 350 (6264), 1084–1089. doi:10.1126/science.aac4255
Sjöstedt, E., Zhong, W., Fagerberg, L., Karlsson, M., Mitsiosis, N., Adori, C., et al. (2020). An atlas of the protein-coding genes in the human, pig, and mouse brain. Sci. 367 (6482), 5947. doi:10.1126/science.aay5947
Skoulidis, F., Goldberg, M. E., Greenawalt, D. M., Hellmann, M. D., Awad, M. M., Gainor, J. F., et al. (2018). STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8 (7), 822–835. doi:10.1158/2159-8290.cd-18-0099
Song, H., Zhong, L.-P., He, J., Huang, Y., and Zhao, Y.-X. (2019). Application of Newcastle disease virus in the treatment of colorectal cancer. World J. Clin. Cases 7, 2143–2154. doi:10.12998/wjcc.v7.i16.2143
Song, M., Ping, Y., Zhang, K., Yang, L., Li, F., Zhang, C., et al. (2019). Low-dose IFNγ induces tumor cell stemness in tumor microenvironment of non–small cell lung cancer. Cancer Res. 79 (14), 3737–3748. doi:10.1158/0008-5472.Can-19-0596
Sun, B.-s., Yao, Y.-q., Pei, B.-x., Zhang, Z.-f., and Wang, C.-l. (2016). Claudin-1 correlates with poor prognosis in lung adenocarcinoma. Thorac. Cancer, 7 (5), 556–563. doi:10.1111/1759-7714.12368
Sung, H., Ferlay, J., Siegel, R., and Laversanne, M. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. American Cancer Society. doi:10.3322/caac.21660
Tolonen, N., Doglio, L., Schleich, S., and Locker, J. K. (2001). Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol. Biol. Cell 12 (7), 2031–2046. doi:10.1091/mbc.12.7.2031
Townsley, A. C., Weisberg, A. S., Wagenaar, T. R., and Moss, B. (2006). Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J. Virology 80 (18), 8899–8908. doi:10.1128/jvi.01053-06
Traversari, C., Meazza, R., Coppolecchia, M., Basso, S., Verrecchia, A., van der Bruggen, P., et al. (1997). IFN-γ gene transfer restores HLA-class I expression and MAGE-3 antigen presentation to CTL in HLA-deficient small cell lung cancer. Gene Ther. 4 (10), 1029–1035. doi:10.1038/sj.gt.3300489
Truong, C.-S., and Yoo, S. Y. (2022). Oncolytic vaccinia virus in lung cancer vaccines. Vaccines 10 (2), 240. doi:10.3390/vaccines10020240
Teufel, F., Almagro Armenteros, J. J., Johansen, A. R., Gíslason, M. H., Pihl, S. I., Tsirigos, K. D., et al. (2022). SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025. doi:10.1038/s41587-021-01156-3
Tusell Wennier, S., Liu, J., and McFadden, G. (2012). Bugs and drugs: oncolytic virotherapy in combination with chemotherapy. Curr. Pharm. Biotechnol. 13 (9), 1817–1833. doi:10.2174/138920112800958850
Cancer Research UK (2022). Available at: https://www.cancerresearchuk.org/about-cancer/lung-cancer/stages-types-grades/types (Accessed August 8, 2024).
The UniProt Consortium, UniProt: the Universal Protein Knowledgebase (2023). Nucleic Acids Research, 51 (D1), D523–D531. doi:10.1093/nar/gkac1052
VanSeggelen, H., Tantalo, D. G., Afsahi, A., Hammill, J. A., and Bramson, J. L. (2015). Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol. Ther. Oncolytics 2, 15014. doi:10.1038/mto.2015.14
Vassilev, A., and DePamphilis, M. L. (2017). Links between DNA replication, stem cells and cancer. Genes 8 (2), 45. doi:10.3390/genes8020045
Vita, R., Mahajan, S., Overton, J. A., Dhanda, S. K., Martini, S., Cantrell, J. R, et al. (2018). The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. doi:10.1093/nar/gky1006
Wang, C., Long, Y., Li, W., Dai, W., Xie, S., Liu, Y., et al. (2020). Exploratory study on classification of lung cancer subtypes through a combined K-nearest neighbor classifier in breathomics. Sci. Rep. 10 (1), 5880. doi:10.1038/s41598-020-62803-4
Wang, H., Li, Z.-Y., Liu, Y., Persson, J., Beyer, I., Möller, T., et al. (2011). Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 17 (1), 96–104. doi:10.1038/nm.2270
Wang, J., Mahasittiwat, P., Wong, K. K., Quint, L. E., and Kong, F.-M. (2012). Natural growth and disease progression of non-small cell lung cancer evaluated with 18F-fluorodeoxyglucose PET/CT. Lung Cancer 78 (1), 51–56. doi:10.1016/j.lungcan.2012.06.010
Wang, K., Wang, R.-L., Liu, J.-J., Zhou, J., Li, X., Hu, W.-W., et al. (2018). The prognostic significance of hTERT overexpression in cancers: a systematic review and meta-analysis. Medicine 97 (35), e11794. doi:10.1097/md.0000000000011794
Wang, Z., Zhong, M., Fu, M., Dou, T., and Bian, Z. (2014). Evidence of positive selection at signal peptide region of interferon gamma. Biosci. Biotechnol. Biochem. 78 (4), 588–592. doi:10.1080/09168451.2014.896732
Wargo, J. A., Gopalakrishnan, V., Spencer, C., Karpinets, T., Reuben, A., Andrews, M. C., et al. (2017). Association of the diversity and composition of the gut microbiome with responses and survival (PFS) in metastatic melanoma (MM) patients (pts) on anti-PD-1 therapy. J. Clin. Oncol. 35 (15_Suppl. l), 3008. doi:10.1200/jco.2017.35.15_suppl.3008
Wilkinson, M. J., Smith, H. G., McEntee, G., Kyula-Currie, J., Pencavel, T. D., Mansfield, D. C., et al. (2016). Oncolytic vaccinia virus combined with radiotherapy induces apoptotic cell death in sarcoma cells by down-regulating the inhibitors of apoptosis. Oncotarget 7 (49), 81208–81222. doi:10.18632/oncotarget.12820
Xiao, B.-F., Zhang, J.-T., Zhu, Y.-G., Cui, X.-R., Lu, Z.-M., Yu, B.-T., et al. (2021). Chimeric antigen receptor T-cell therapy in lung cancer: potential and challenges. Front. Immunol. 12, 782775. doi:10.3389/fimmu.2021.782775
Xie, M., Xu, X., and Fan, Y. (2021). KRAS-mutant non-small cell lung cancer: an emerging promisingly treatable subgroup. Front. Oncol. 11, 672612. doi:10.3389/fonc.2021.672612
Yaacov, A., Vardi, O., Blumenfeld, B., Greenberg, A., Massey, D. J., Koren, A., et al. (2021). Cancer mutational processes vary in their association with replication timing and chromatin accessibility. Cancer Res. 81 (24), 6106–6116. doi:10.1158/0008-5472.Can-21-2039
Yang, Z., Guo, J., Weng, L., Tang, W., Jin, S., and Ma, W. (2020). Myeloid-derived suppressor cells—new and exciting players in lung cancer. J. Hematol. and Oncol. 13 (1), 10. doi:10.1186/s13045-020-0843-1
Ye, T., Jiang, K., Wei, L., Barr, M. P., Xu, Q., Zhang, G., et al. (2018). Oncolytic Newcastle disease virus induces autophagy-dependent immunogenic cell death in lung cancer cells. Am. J. Cancer Res. 8 (8), 1514–1527.
Zeng, D.-Q., Yu, Y.-F., Ou, Q.-Y., Li, X.-Y., Zhong, R.-Z., Xie, C.-M., et al. (2016). Prognostic and predictive value of tumor-infiltrating lymphocytes for clinical therapeutic research in patients with non-small cell lung cancer. Oncotarget 7 (12), 13765–13781. doi:10.18632/oncotarget.7282
Zhang, X., Wang, X., Hou, L., Xu, Z., Liu, Y. E., and Wang, X. (2023). Nanoparticles overcome adaptive immune resistance and enhance immunotherapy via targeting tumor microenvironment in lung cancer. Front. Pharmacol. 14, 1130937. doi:10.3389/fphar.2023.1130937
Zhang, Y., and Bergelson, J. M. (2005). Adenovirus receptors. J. Virology 79 (19), 12125–12131. doi:10.1128/jvi.79.19.12125-12131.2005
Zhao, Q. (2015). Neoadjuvant chemotherapy of nanoparticle albumin-bound paclitaxel in lung cancer. NIH.
Zhao, Y., Liu, Z., Li, L., Wu, J., Zhang, H., Zhang, H., et al. (2021). Oncolytic adenovirus: prospects for cancer immunotherapy. Front. Microbiol. 12, 707290. doi:10.3389/fmicb.2021.707290
Zhou, S., and Yang, H. (2023). Immunotherapy resistance in non-small-cell lung cancer: from mechanism to clinical strategies. Front. Immunol. 14, 1129465. doi:10.3389/fimmu.2023.1129465
Keywords: non-small cell lung cancer, immune checkpoint inhibitors, oncolytic viruses, cancer immunotherapy, tumor microenvironment
Citation: Yin ZS and Wang Z (2024) Strategies for engineering oncolytic viruses to enhance cancer immunotherapy. Front. Pharmacol. 15:1450203. doi: 10.3389/fphar.2024.1450203
Received: 17 June 2024; Accepted: 30 July 2024;
Published: 06 September 2024.
Edited by:
Chunbo He, University of Oklahoma Health Sciences Center, United StatesReviewed by:
Gullanki Naga Venkata Charan Tej, University of Oklahoma Health Sciences Center, United StatesJunzhang Zhao, The Sixth Affiliated Hospital of Sun Yat-sen University, China
Copyright © 2024 Yin and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengfeng Wang, ZmNjd2FuZ3pmMUB6enUuZWR1LmNu