 Chun-Yue Liu1,2
Chun-Yue Liu1,2 Si-Xuan Liu
Si-Xuan Liu Chang Li
Chang Li Yue-Hu Pei
Yue-Hu Pei- 1Department of Medicinal Chemistry and Natural Medicine Chemistry, College of Pharmacy, Harbin Medical University, Harbin, China
- 2Department of Chemistry, College of Pharmacy, Harbin Medical University, Daqing, China
Human fungal pathogens could cause a broad plethora of infections in both the immunocompetent and immunocompromised host. Fungal infections have become important causes of morbidity and mortality in recent years, the current arsenal of anti-fungal therapies was restricted. Ibrexafungerp was a novel, highly bioavailable glucan synthase inhibitor formulated for both intravenous and oral administration being developed by Scynexis; it was also the first novel anti-fungal drug class approved in more than 20 years. Ibrexafungerp was one semi-synthetic derivative of enfumafungin, a natural product isolated from fungi. This review reported the discovery of enfumafungin and ibrexafungerp, their anti-fungal mechanism, summed up 63 fernane-type triterpenoids from natural products, including 49 from plants, 9 from fungi and 5 from lichen. In addition, the review summarized the progress of enzymes responsible for the biosynthesis of type II fernane triterpenoid (enfumafungin skeleton) and type I fernane triterpenoid (polytolypin skeleton). The good example kept our confidence up for searching for new leading compounds and discovering drugs from fungi.
Introduction
Human fungal pathogens could cause a broad plethora of infections in both the immunocompetent and immunocompromised host. The incidence of fungal infections has increased significantly over the last several decades (Yu et al., 2021; Li Z. et al., 2023). Consequently, fungal infections have become important causes of morbidity and mortality (Gamal et al., 2021). The most common clinical pathogenic fungi were Candida albicans, Aspergillus fumigatus and Cryptococcus neoformans (Liu et al., 2023). Among them, C. albicans was an opportunistic pathogen, which could cause superficial infection and lead to mortal systemic infections, especially in immunocompromised patients (Gamal et al., 2021). It was reported that up to 70%–75% of women experienced an episode of Vulvovaginal candidiasis (VVC) and it has been estimated that approximately 10%–15% of asymptomatic women were colonized with a Candida species (Anderson et al., 2004; Sobel, 2007).
The current arsenal of anti-fungal therapies was restricted, with only three major drug classes available as first-line therapies (Armstrong-James, 2022). The first ones were the polyenes. They had very broad spectrum anti-fungal activity, but their clinical use was often limited due to their nephrotoxicity caused by non-specific binding to mammalian sterols (Armstrong-James et al., 2020); The second ones were azole derivatives. They were better tolerated than the polyenes and had the advantage of being orally bio-available; However, inhibition of host P450 enzymes resulted in a plethora of drug-drug interactions (DDIs) that present challenges for patients taking multiple medications (Nwankwo et al., 2022); The third ones were the echnocandins, which were the most recent class of anti-fungals, compromised the integrity of the fungal cell wall, eventually leading to cell lysis under osmotic stress, through the inhibition of β-1,3-glucan synthase (GS), which produced β-1,3-glucan, a critical component of the cell wall. Due to the specificity of their target for fungal cells, the echinocandins have demonstrated improved tolerability and fewer DDIs compared to the polyenes and azoles. The echinocandins were now recommended as first line therapy for invasive candidiasis, as a result of these advantages and because of their broad spectrum of activity for Candida species. Restriction to parenteral administration due to poor oral bioavailability constituted a significant limitation for echinocandin therapy (Perlin, 2011). Besides, significant resistance of Candida glabrata and Candida auris to echinocandins has emerged (Armstrong-James, 2022). Therefore, developing new drugs and strategies to combat fungal infections was crucial.
Fortunately, ibrexafungerp (formerly named SCY-078) was a novel, highly bio-available glucan synthase inhibitor formulated for both intravenous and oral administration currently being developed by Scynexis (Jersey City, NJ, United States). It has been approved on June 2021 in the United States for the treatment of vulvovaginal candidiasis (VVC), and it was the first novel anti-fungal drug class approved in the past 20 years (Azie et al., 2020; Waterer, 2021; Angulo et al., 2022; Sucher et al., 2022). Ibrexafungerp exerted concentration-dependent fungicidal activity against Candida species and retained good in vitro activity against most echinocandin-resistant strains and fluconazole-resistant strains (Nunnally et al., 2019; Davis et al., 2020; Gamal et al., 2021; Mesquida et al., 2021; Sobel et al., 2021; Wiederhold et al., 2021). In addition, Ibrexafungerp has been shown to be safe and effective in the treatment of vulvovaginal candidiasis caused by C. albicans in phase II and phase III clinical trials (Vazquez et al., 2021; Daraskevicius et al., 2022; Schwebke et al., 2022; Sobel et al., 2022; Goje et al., 2023). It was approved for vulvovaginal candidiasis in adult and postmenarchal pediatric females and was given as two 150-mg tablets orally, administered 12 h apart (Sucher et al., 2022). Clinical trials were ongoing for recurrent and complicated vulvovaginal candidiasis as well as invasive candidiasis and pulmonary aspergillosis (Colombo and Vazquez, 2021; Phillips et al., 2022; Sucher et al., 2022).
Structurally, ibrexafungerp was one semi-synthetic derivative originated from fernane-type triterpenoid glycoside enfumafungin (Figure 1), which was firstly isolated from an endophytic fungus in 2000 (Peláez et al., 2000; Apgar et al., 2021; Lee, 2021). Thus, herein we summarized the discovery procedure of ibrexafungerp from enfumafungin, the modified derivatives of enfumafungin, the mechanism of them, as well as the fernane type triterpenoids isolated from natural products up to now, and the biosynthesis clues of this kind of triterpenoids.
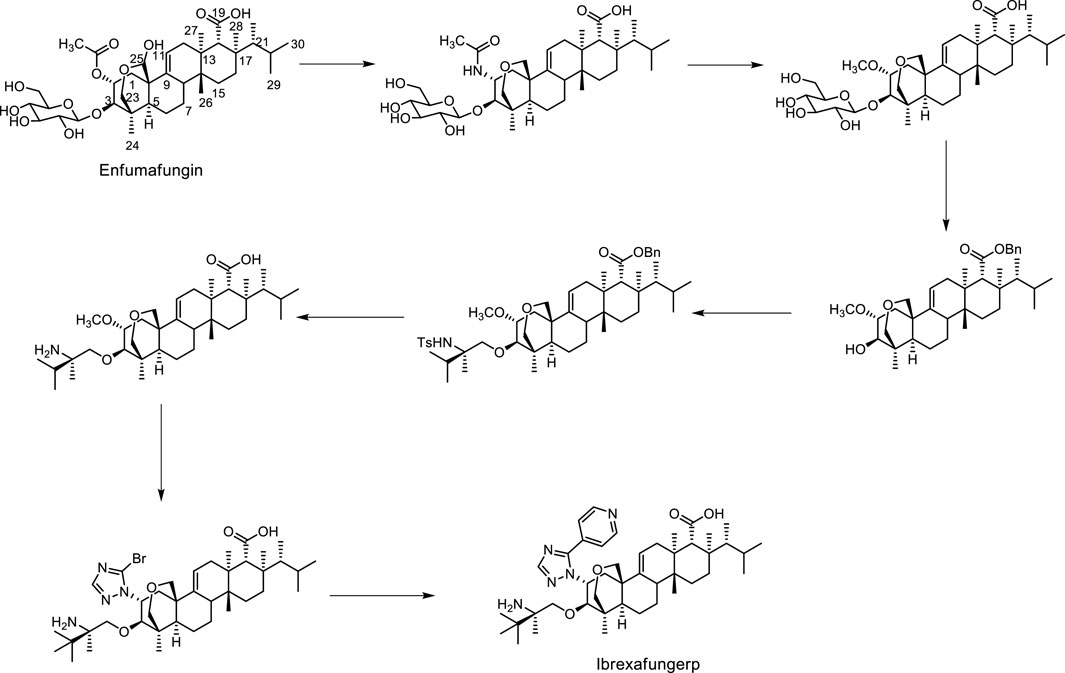
Figure 1. The synthetic procedure from enfumafungin to ibrexafungerp.
The discovery procedure, from enfumafungin to ibrexafungerp
Enfumafungin was a fernane-type triterpene glycoside that was originally isolated from the fermentation of Hormonema sp. by Merck in 2000 (Peláez et al., 2000; Schwartz et al., 2000; Hector and Bierer, 2011). Then it was found out that the natural product inhibited the synthesis of 1,3-β-D-glucan, a necessary component of the fungal cell wall, by inhibiting the enzyme glucan synthase (Onishi et al., 2000). Unfortunately, it showed weak activity in a murine model of disseminated candidiasis due to limited stability in vivo. Therefore, since its isolation, Merck undertook the challenge of improving oral efficacy and pharmacokinetic properties of enfumafungin through semi-synthetic modification of the natural product. Firstly, enfumafungin was elaborated by a series of chemical transformations such as reduction of the hemiacetal, protection of the carboxylic acid functionality at C-19 as a benzyl ester and deglycosylation to produce several key intermediates, shown in Figure 1, from which most semi-synthetic derivatives reported in the patent were prepared. Then, In 2010, the patent WO2010019204 described 318 examples of 25-deoxyenfumafungins, with a substituted triazole group or 1,2,3-triazole, pyrazole, imidazole at C-2, and an amino-containing alkoxy group at C-3 (Merck and Co, 2010). The triazole ring of the exemplified compounds often contained an aryl or heteroaryl substituent. Biological activity for many of the derivatives against C. albicans were reported, with nine of the exemplified compounds having reported activities <1 ng/mL. One of these compounds was MK-3118 (the formerly name of ibrexafungerp), an orally active enfumafungin derivative, which was example 173 in the application (Liberator et al., 2010; Merck and Co, 2010). Thus, MK-3118 has been evaluated using various in vitro and in vivo assays. In a glucan synthase assay, MK-3118 resulted in an half maximal inhibitory concentration (IC50) of 0.6 ng/mL for partially purified microsomal enzyme derived from C. albicans, an improvement of three orders of magnitude compared with the 25-deoxyenfumafungin parent compound (Balkovej et al., 2009; Hector and Bierer, 2011). Using glucan synthase prepared from A. fumigatus, an IC50 of 0.0017 μg/mL was determined for MK-3118, compared with 0.5 ng/mL for caspofungin (Motyl et al., 2010; Hector and Bierer, 2011). This compound showed minimum inhibitory concentration (MIC) values of ≤1 μg/mL and ≤0.015 μg/mL against 160 strains of 7 Candida spp. and 40 Aspergillus spp., respectively. Moreover, MK-3118 has been evaluated in standard rodent models of candidiasis and aspergillosis. Following administration to mice, the half-life of MK-3118 was determined to be 4.4 h, with 34% oral bioavailability. Overall, the preclinical results demonstrate a comparable level of activity for MK-3118 against Candida spp. compared with the echinocandin caspofungin, while mouse efficacy results for aspergillosis suggested a somewhat inferior response compared with caspofungin (Hector and Bierer, 2011; Jiménez-Ortigosa et al., 2013; Pfaller et al., 2013a; Pfaller et al., 2013b).
Since 2015, several clinical trials have been conducted to determine the optimal dosing regimen and to assess the efficacy and safety of ibrexafungerp in the treatment of acute VVC, including phase II clinical trial SCY-078-203 (Nyirjesy et al., 2021; Sucher et al., 2022), and phase III clinical trial VANISH 303 and VANISH 306 (Sobel et al., 2022; Sucher et al., 2022). As expected, ibrexafungerp was approved by Food and Drug Administration (FDA) for the treatment of acute VVC in 2021 (Figure 2). It was currently the only oral non-azole agent approved for the treatment of VVC, it was also the first drug in a new class of anti-fungal agents known as fernane-type triterpenoids. In addition, ibrexafungerp was being studied for the treatment of invasive candidiasis and aspergillosis in ongoing clinical trials, and was approved for recurrent VVC (RVCC) in December 2022 (Sucher et al., 2022; Phillips et al., 2023).
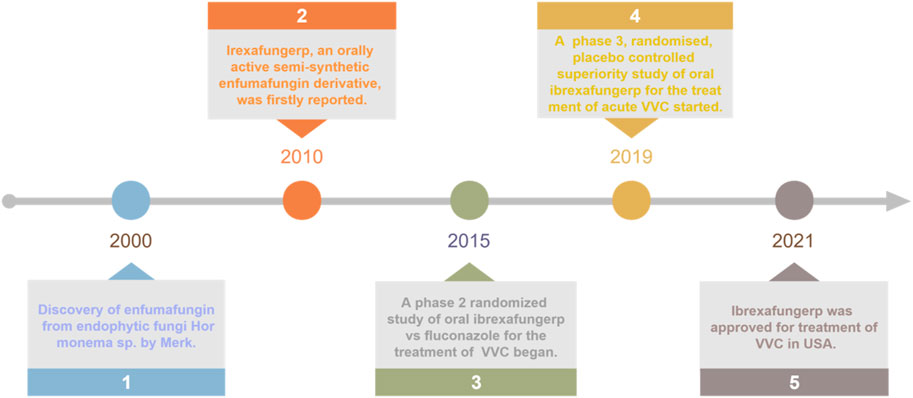
Figure 2. Key milestones in the development of oral ibrexafungerp from enfumafungin.
The mechanism of ibrexafungerp and enfumafungin
In 2000, Onishi group reported the identification of four triterpenoids as anti-fungal agents. One of the four compounds, enfumafungin, showed significant bioactivity, which could be comparable to that of the control L-733560 (Onishi et al., 2000). Moreover, they also found that enfumafungin specifically inhibited glucan synthesis in whole cells and in (1,3)-β-D-glucan synthase assays, altered the morphologies of yeasts and molds. To further explore the effects of enfumafungin on glucan synthase, enfumafungin was tested against Saccharomyces cerevisiae strains with point mutations in FKS1 (Zhao et al., 2023), the gene encoding the vegetatively expressed large subunit of glucan synthase. The result indicated that enfumafungin produced a unique response in S. cerevisiae strains with point mutations in FKS1, which support the conclusion that enfumafungin was specific inhibitors of glucan synthase. Thus, enfumafungin represented a new group of (1,3)-β-D-glucan synthase inhibitors (Douglas et al., 1997; Onishi et al., 2000). Since ibrexafungerp was reported in 2010, it has been evaluated using various in vitro and in vivo assays, its mechanism was also discussed (Hector and Bierer, 2011; Jiménez-Ortigosa et al., 2013; Pfaller et al., 2017). It was the first triterpenoid of (1, 3)-β-D-glucan inhibitor with good oral bioavailability (Hector and Bierer, 2011).
Fernane-type triterpenoids from natural products
Fernane-type triterpenoids were a rare type triterpenoids, with unique 6/6/6/6/5 skeleton. They have been isolated from plants, fungi and bacteria. It was unusual migrated hopane triterpenoids. A number of fernane-type triterpenoids were isolated and identified since 1980s. Several of them showed anti-fungal bio-activity.
In the course of search for the biologically active constituents of Euphorbia supina Rafin, a toxic annual weed which was used as a folk medicine for the treatment of gastroenteric diseases and for healing suppurated swelling, Tanaka et al. found that the plant contained a number of triterpenoids bearing a novel and biogenetically interesting fernane skeleton, migrated fernane skeleton and seco-fernane skeleton. Their structures were established as fern-8-en-3β-ol (1), 3β-methoxy-fern-9 (11)-ene (2), ferna-7,9 (11)-dien-3β-ol (3), 3β-hydroxyfern-8-en-7,11-dione (4), 3β/7a-dihydroxyfern-8-en-1l-one (5), 3β/11β-dihydroxyfern-8-en-7-one (6), and spirosupinanonediol (7) (Tanaka and Shunyo, 1988; Tanaka and Shunyo, 1989) as well as neospirosupinanonediol (8), supinenolone D (9) and neospirosupinanetrione (10), supinenolone E (11) (Tanaka and Shunyo, 1991a; Tanaka and Shunyo, 1991b) (Figure 3).
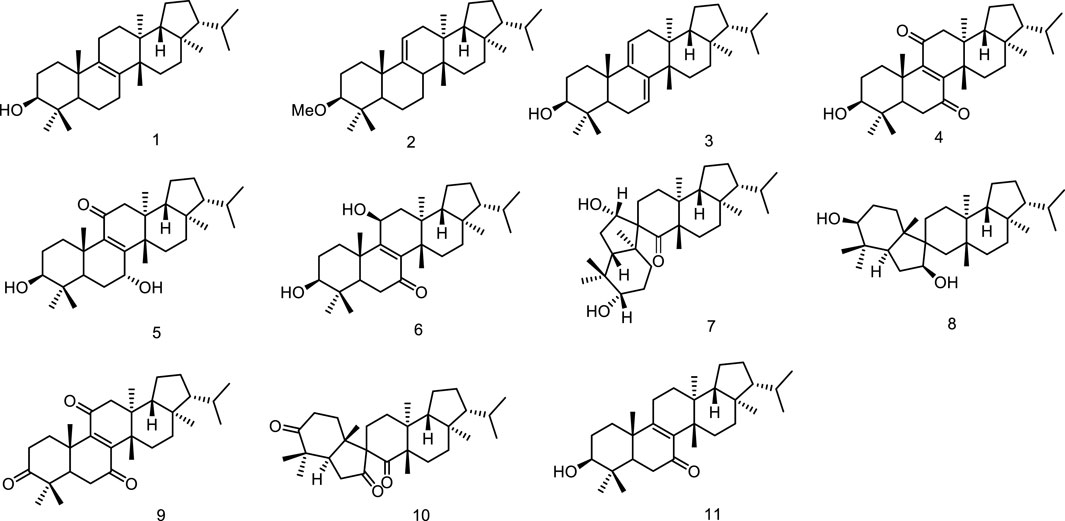
Figure 3. Fernane type tri-terpenoids (1–11) from natural products.
Phytochemical studies on Sericostoma pauciflorum provided a new fernane-type triterpenoid sericostinyl acetate (12) (Ayatollahi et al., 1991). A new fernane type triterpene has been isolated from the fern Adiantum venustum. Its structure has been elucidated to be fern-9 (1l)-en-25-oic acid (13) by Banerjee et al. (1991). A new seco-triterpene-dienoic acid was isolated from Euphorbia Chamaesyce, its structure was established as 3,4-seco-8βH-ferna-4 (23),9 (11)-dien-3-oic acid (14) (Tanaka et al., 1996). Ageta et al. isolated a series of fernane triterpenoids from Adiantum cuneatum, including 7β,25-epoxyfern-8-ene (15), 25-norfern-7-en-10β-yl formate (16), fern-9 (11)-ene (17), ferna-7,9 (11)-diene (18), fern-7-ene (19) (Shiojima et al., 1997a), fern-7-en-25-ol (20), fern-9 (11)-en-25-ol (21), 7β,25-epoxyfern-9 (11)-en-8α-ol (22), 7α,8α-epoxyfernan-25-ol (23) (Shiojima et al., 1997b). Five new fernane type triterpenoids with α hydroxy group at C-3, ferna-7, 9 (11)-diene-3α, 16α-diol (24), 3α 16α-dihydroxyferna-7, 9 (11)-dien-12-one (25), ferna-7, 9 (11)-diene-3α, 16α, 19α-triol (26), 3α, 16α-dihydroxyfern-8-en-11-one (27) and 3α, 16α-dihydroxyfern-8-ene-7, 11-dione (28), were isolated from the leaves of Lonicera gracilipes var. glandulosa MAXIM (Kikuchi et al., 1999). A new fernane-type triterpene, named integnfolin (29), was isolated and characterized from the aerial parts of Teucrium integnfolium, the structure of the new compound was deduced to be 3β-hydroxy-fern-9 (11)-en-23-oic acid through its spectral properties and X-ray crystallographic analysis (Chen et al., 2000) (Figure 4).
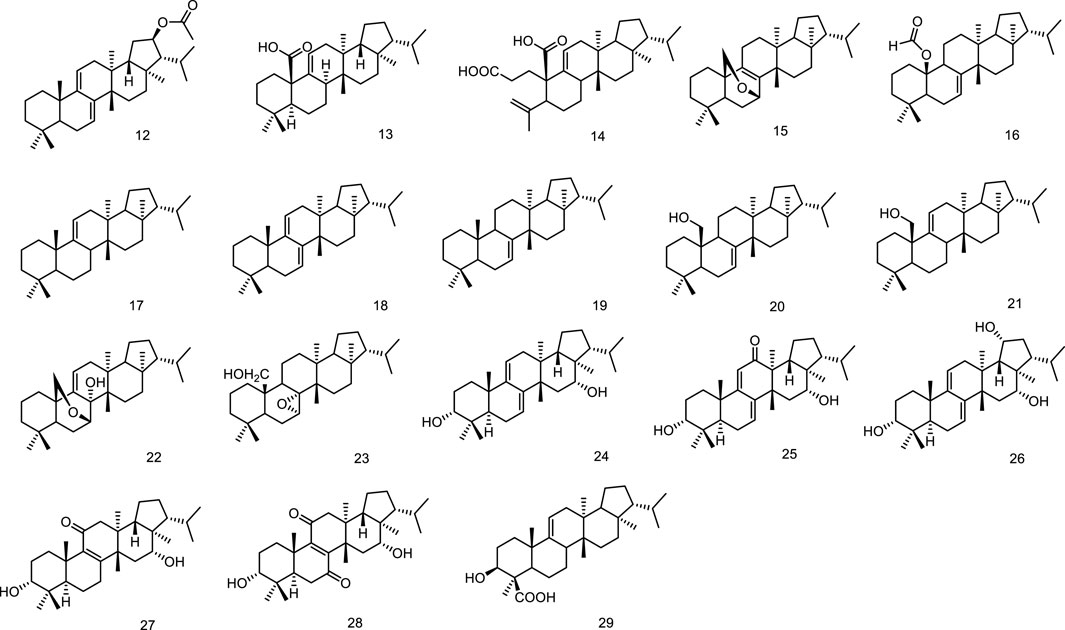
Figure 4. Fernane type tri-terpenoids (12–29) from natural products.
Two new fernane-type triterpenes, namely betufernanediol A (30) and betufernanediol B (isomers) (31) have been isolated from the stem bark of Betula pendula (Mukhtar et al., 2003). Two new fernane triterpenoids, 7α-hydroxyfern-8-en-11-one (32) and 11β-hydroxyfern-8-en-7-one (33), were isolated from the methyl alcohol extract of the leaves of Angiopteris palmiformis (Chen et al., 2010). The aerial parts of Spergula fallax afforded one new fernane class triterpenoid, 3-O-[β-D-glucopyranoside-(1→4)-O-α-L-(2-O-acetyl)-arabinopyranosyl]-2α,3β,19β,20β-tetrahydroxyfern-7en-6-oxo-29-oic acid (34) (Hamed et al., 2014). Belamcandaoids C-N (35–46), twelve new 3,4-seco-triterpenoids belonging to fernane type, were isolated from the seeds of Belamcanda chinensis (Song et al., 2021). Extensive phytochemical investigation on the whole herbs of Euphorbia hypericifolia led to the isolation of lots of structurally diverse triterpenoids, including two fernanes isomotiol (47) and teuviscin A (48) (Hu et al., 2021). One fernane-type triterpenoid, fern-9 (11)-ene-2α,3β-diol (49) was isolated from the stem bark of Ancistrocarpus densispinosus Oliv (Figure 5).
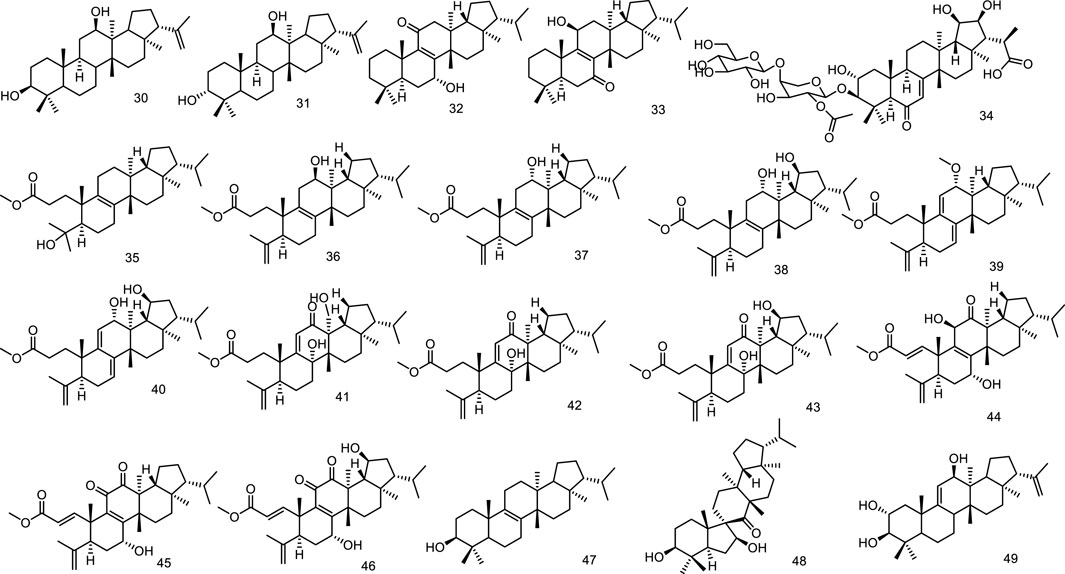
Figure 5. Fernane type tri-terpenoids (30–49) from natural products.
A thin-layer chromatography-biography screening guided phytochemical investigation for anti-fungal constituents from a lichen, Lobaria kurokawae Yoshim., led to the isolation of a pentacyclic triterpenoid, retigeric acid B (50). It exhibited anti-fungal activity alone against both azole-sensitive and -resistant C. albicans isolates. Furthermore, when it was combined with azoles, strong synergy was observed against azole-resistant strains, with synergistic or indifferent effects observed against azole-sensitive strains. 50 was an acid with anti-fungal activity that possibly has activity either in facilitating the uptake of azoles or in enhancing the membrane damage associated with the action of the azoles (Sun et al., 2009). Three novel fernane-type triterpenoids lobarialides A-C (51–53), together with two known ones retigeric acids A and B (54 and 50), were isolated by anti-fungal bioassay-guided from fractionation of the lichen Lobaria kurokawae. The isolated compounds were evaluated against the sensitive strain of the pathogenic fungus Candida albicans (CA2) according to the NCCLS method. 50 showed the best bioactivity with MIC values at 4.0 μg/mL. These results indicated that the anti-fungal activity of these compounds was closely related to the number of COO groups in the respective compounds. As the number of the COO groups increases, the activity is magnified. Although compound 50 exhibited weaker anti-fungal activity than these glycosides, it is the first anti-fungal fernane-type triterpenoid aglycone reported until 2009 (Wang et al., 2009).
Fernane-type triterpenoids are relatively uncommon as fungal metabolites, while WF11605 (55) from the strain F11605, displayed anti-inflammatory effects (Shigematsu et al., 1992; Tsujii et al., 1992). Polytolypin (56), exhibiting anti-fungal and antibiotic activity, has been isolated from cultures of Polytofypa bystricis (JS189), a fungal colonist of porcupine dung. Polytolypin was found to be active against C. albicans (ATCC 14053), producing an 11-mm zone of inhibition in a standard disk assay at 80 pg/disk (Gamble et al., 1995). Enfumafungin (57) from Hormonema sp. (Schwartz et al., 2000) and fuscoatroside (58) from Aspergillus flavussclerotia (Joshi et al., 2002) displayed significant anti-fungal activities. Then four new triterpenoid glycosides, kolokosides A-D (59–62), were isolated from cultures of a Hawaiian wood-decay fungus (Xylaria sp.). 59 exhibited activity against Gram-positive bacteria (Deyrup et al., 2007). Peniciside (63), a new fernane triterpenoid glycoside, was isolated from the EtOAc extract of the solid-state fermented rice culture of the fungus Penicillium sp. 169. 63 was the first example of a fernane triterpenoid glycoside with two hydroxyls at C-19 and C-20 (Yuan et al., 2012) (Figure 6).
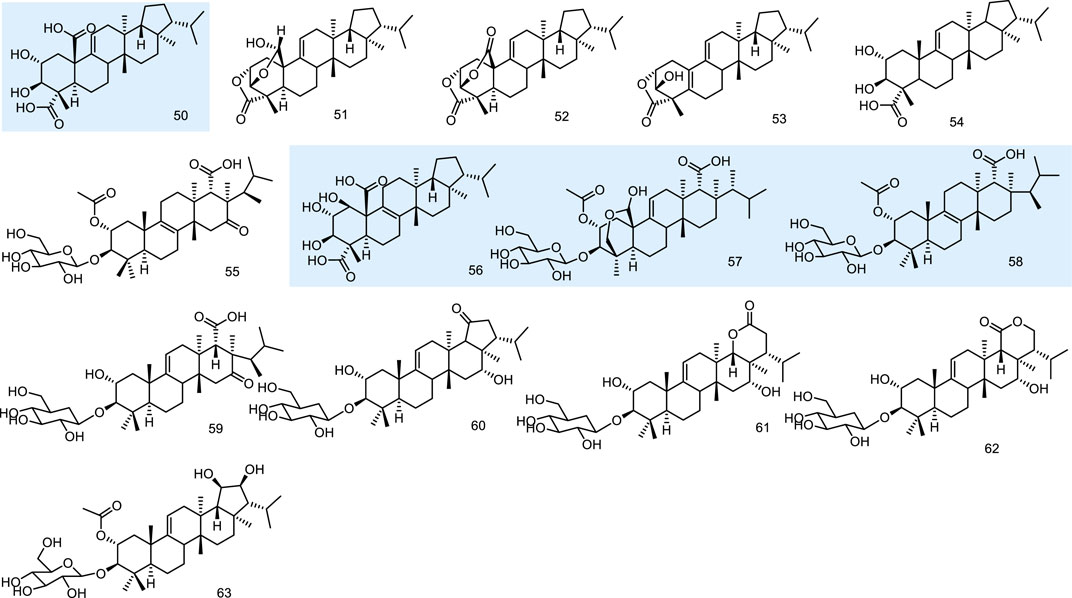
Figure 6. Fernane type tri-terpenoids (50–63) from natural products.
Biosynthesis of enfumafungin
The biosynthesis of enfumafungin was still unclear until Kuhnert et al. reported on the preliminary identification of the enfumafungin biosynthetic gene cluster (BGC) based on genome sequencing, phylogenetic reconstruction, gene disruption, and cDNA sequencing studies. It was interesting to find out that enfumafungin synthase (efuA) consisted of a terpene cyclase domain (TC) fused to a glycosyltransferase (GT) domain and thus represented a novel multifunctional enzyme (Figure 7). Moreover, the TC domain bore a phylogenetic relationship to bacterial squalene-hopene cyclases (SHC) and included a typical DXDD motif within the active center suggesting that efuA evolved from SHCs. Phylogenetic reconstruction of the GT domain indicated that this portion of the fusion gene originated from fungal sterol GTs (Kuhnert et al., 2018).
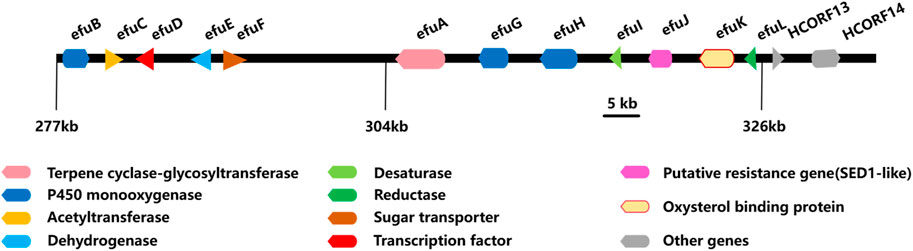
Figure 7. Biosynthesis gene cluster of enfumafungin.
Then, in 2021, Li et al. reported the biosynthesis of group II fernane type triterpenoid, polytolypin. The authors have identified the biosynthetic gene cluster of polytolypin from its producer Polytolypa hystricis UAMH7299 and characterized its biosynthetic pathway. A new fernane-type triterpene cyclase was responsible for the biosynthesis of motiol, which was subsequently oxidized by three cytochrome P450s to give polytolypin. Moreover, the three P450 enzymes were employed in combination with two other new fungal fernane-type cyclases for isomotiol and fernenol biosynthesis to generate six polytolypin analogues (Li et al., 2023) (Figure 8).
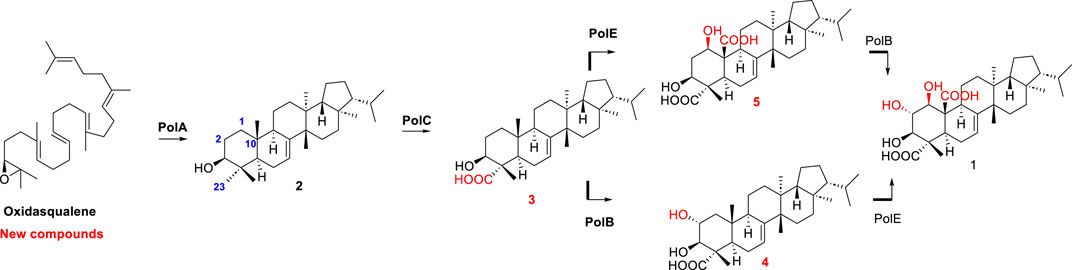
Figure 8. Biosynthesis pathway of polytolypin.
Discussion
Inventing and developing a new medicine is a long, complex, costly and highly risky process that has few peers in the commercial world. Research and development (R&D) for most of the medicines available today has required 12–24 years for a single new medicine, from starting a project to the launch of a drug product. In addition, many expensive, long-term research projects completely fail to produce a marketable medicine (Lombardino and Lowe, 2004). Fortunately, it took only 21 years from discovery of enfumafungin to use of ibrexafungerp as an anti-fungal drug approved by FDA.
Literature reviews demonstrated that natural products account for around 50% of approved new drugs in past few years (Newman and Giddings, 2013; Newman and Cragg, 2020). In recent years, since the discovery of penicillin, the first β-lactam antibiotic, fungi provided modern medicine with important antibiotics for curing life threatening infectious diseases, fungal secondary metabolites have revolutionized medicine yielding blockbuster drugs and drug leads of enormous therapeutic (Elander, 2003; Aly and Debbab, 2011). Ibrexafungerp, whose leading compound was enfumafungin, which was firstly isolated from fungus Hormonema sp., also represented one example of typical agents originated from fungi. In addition, it was still challenge for drug discovery from microorganism in recent years, especially using the classical bioactivity-guided isolation method to search for the leading compounds from natural products. With the development of genome sequencing, recent genome mining and biosynthetic engineering could enable faster and more efficient production of more leading compounds (Méndez et al., 2015; Hindra et al., 2017; Atanasov et al., 2021). Besides, co-cultivation using helper strains could also promote access to discovery of leading compounds (Abdel- Razek et al., 2018; Moussa et al., 2019).
Triterpenoids have been clinically used widely, such as fusidic acid, ginsenoside Rg3 et al. Among them, fusidic acid, which was used for treatment of skin infections caused by Staphylococcus aureus, was a group of fungi-derived 29-nor protostane triterpenoid antibiotics, targeting elongation factor G to inhibit bacterial protein synthesis (Cao et al., 2020; Chavez et al., 2021). Similarly, enfumafungin, represented the first fernane type-triterpenoid with anti-fungal activity, blocking the synthesis of the fungal cell wall polymer β-(1,3)-D-glucan, whose derivative ibrexafungerp received FDA for the treatment of Vulvovaginal candidiasis (VVC) in June 2021, and was subsequently approved for recurrent VVC (RVCC) in December 2022. Fluconazole is the only oral medication FDA approved for VVC before. None of these treatments are FDA approved for RVVC. Ibrexafungerp, a triterpenoid fungicidal agent, was FDA approved in 2021, becoming the first oral non-azole agent for VVC (Phillips et al., 2023). Ibrexafungerp provides an alternative oral option for treatment of acute, severe VVC. It is the only FDA approved anti-fungal for RVVC. Currently, the population could benefit from this drug are those with azole allergy, non-albicans or azole resistant albicans species, or other azole contraindications such as drug interactions (like statins or tricyclics) (Phillips et al., 2022; Phillips et al., 2023).
In conclusion, from enfumafungin, a metabolite from fungi, to ibrexafungerp, one “first-in-class” anti-fungal agent. It took 21 years from discovery of enfumafungin to use of ibrexafungerp as an marketable anti-fungal medicine approved by FDA, during this period, a number of derivatives and similar type natural products were synthesized or isolated, their bioactivity-structure relationship were discussed. The good example kept our confidence up for searching for new leading compounds and discovering drugs from fungi. Moreover, close attention has been paid to the biosynthetic pathway of fernane-type triterpenoids, it will become possible that ibrexafungerp be produced by bio-synthetic way soon.
Author contributions
C-YL: Conceptualization, Investigation, Writing–original draft. LZ: Conceptualization, Investigation, Writing–original draft. S-XL: Writing–original draft. Y-FL: Writing–original draft. CL: Supervision, Writing–review and editing, Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization. Y-HP: Project administration, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdel- Razek, A. S., Hamed, A., Frese, M., Sewald, N., and Shaaban, M. (2018). Penicisteroid C: new polyoxygenated steroid produced by co-culturing of Streptomyces piomogenuswith Aspergillus Niger. Steroids 138, 21–25. doi:10.1016/j.steroids.2018.06.005
Aly, A. H., Debbab, A., and Proksch, P. (2011). Fifty years of drug discovery from fungi. Fungal Divers. 50, 3–19. doi:10.1007/s13225-011-0116-y
Anderson, M. R., Klink, K., and Cohrssen, A. (2004). Evaluation of vaginal complaints. JAMA 291 (11), 1368–1379. doi:10.1001/jama.291.11.1368
Angulo, D. A., Alexander, B., Rautemaa-Richardson, R., Alastruey-Izquierdo, A., Hoenigl, M., Ibrahim, A. S., et al. (2022). Ibrexafungerp, a novel triterpenoid antifungal in development for the treatment of mold infections. J. Fungi 8 (11), 1121. doi:10.3390/jof8111121
Apgar, J. M., Wilkening, R. R., Parker, D. L. J., Meng, D., Wildonger, K. J., Sperbeck, D., et al. (2021). Ibrexafungerp: an orally active β-1,3-glucan synthesis inhibitor. BIOORG Med. Chem. Lett. 32, 127661. doi:10.1016/j.bmcl.2020.127661
Armstrong-James, D. (2022). Antifungal chemotherapies and immunotherapies for the future. Parasite Immunol. 45 (2), e12960. doi:10.1111/pim.12960
Armstrong-James, D., Koh, M., Ostermann, M., and Cockwell, P. (2020). Optimal management of acute kidney injury in critically ill patients with invasive fungal infections being treated with liposomal amphotericin B. BMJ Case Rep. 13 (5), e233072. doi:10.1136/bcr-2019-233072
Atanasov, A. G., Zotchev, S. B., Dirsch, V. M., and Supuran, C. T. (2021). Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug Discov. 20, 200–216. doi:10.1038/s41573-020-00114-z
Ayatollahi, A. M., Ahmed, Z., Malik, A., Afza, N., and Badar, Y. (1991). A fernane-type triterpene from Sericostoma pauciflorum. J. Nat. Prod. 54 (2), 570–572. doi:10.1021/np50074a033
Azie, N., David, A., Barbara, D., and Jack, D. S. (2020). Oral ibrexafungerp: an investigational agent for the treatment of vulvovaginal candidiasis. Expert Opin. Inv Drug 29 (9), 893–900. doi:10.1080/13543784.2020.1791820
Balkovej, J., Bouffard, J. A., and Dropkinski, J. F. (2009). “Derivatives of the β-1,3-glucan synthase (GS) inhibitor enfumafungin with potent antifungal activity - initial studies,” in Proceedings of the 49th, San Francisco, CA.
Banerjee, J., Datta, G., Dutta, C. P., Eguchi, T., Fujimoto, Y., and Kakinuma, K. (1991). Fern-9(11)-en-25-oic acid, a triterpene from Adiantum venustum. Phytochemistry 30 (10), 3478–3480. doi:10.1016/0031-9422(91)83238-g
Cao, Z. Q., Lv, J. M., Liu, Q., Qin, S. Y., Chen, G. D., Dai, P., et al. (2020). Biosynthetic study of cephalosporin P1 reveals a multifunctional P450 enzyme and a site-selective acetyltransferase. ACS Chem. Biol. 15 (1), 44–51. doi:10.1021/acschembio.9b00863
Chavez, M. G., Garcia, A., Lee, H. Y., Lau, G. W., Parker, E. N., Komnick, K. E., et al. (2021). Synthesis of fusidic acid derivatives yields a potent antibiotic with an improved resistance profile. ACS Infect. Dis. 7 (2), 493–505. doi:10.1021/acsinfecdis.0c00869
Chen, C. R., Liao, Y. W., Wu, H. T., Shih, W. L., Tzeng, C. Y., Yang, S. Z., et al. (2010). Triterpenoids from Angiopteris palmiformis. Chem. Pharm. Bull. 58 (3), 408–411. doi:10.1248/cpb.58.408
Chen, X. L., Wang, T. E., Jiang, B., Lin, Z. W., Lu, Y., Zheng, Q. T., et al. (2000). A new fernane-type triterpenoid from Teucrium integrifolium. Nat. Prod. Lett. 14 (6), 459–462. doi:10.1080/10575630008043785
Colombo, R. E., and Vazquez, J. A. (2021). An evaluation of ibrexafungerp for the treatment of invasive candidiasis: the evidence to date. Expert Opin. Pharmacother. 22 (7), 797–807. doi:10.1080/14656566.2021.1890026
Daraskevicius, J., Petraitis, V., Davainis, L., and Zucenka, A. (2022). The feasibility of ibrexafungerp for the treatment of fungal infections in patients with hematological malignancies. J. Fungi 8 (5), 440. doi:10.3390/jof8050440
Davis, M. R., Donnelley, M. A., and Thompson, G. R. (2020). Ibrexafungerp: a novel oral glucan synthase inhibitor. Med. Mycol. 58 (5), 579–592. doi:10.1093/mmy/myz083
Deyrup, S. T., Gloer, J. B., Donnell, K. O., and Wicklow, D. T. (2007). Kolokosides A-D: triterpenoid glycosides from a Hawaiian isolate of xylaria sp. J. Nat. Prod. 70, 378–382. doi:10.1021/np060546k
Douglas, C. M., D’Ippolito, J. A., Shei, G. J., Meinz, M., Onishi, J., Marrinan, J. A., et al. (1997). Identification of the FKS1 gene of Candida albicans as the essential target of 1,3-beta-D-glucan synthase inhibitors. Antimicrob. Agents Chemother. 41 (11), 2471–2479. doi:10.1128/AAC.41.11.2471
Elander, R. P. (2003). Industrial production of beta-lactam antibiotics. Appl. Microbiol. Biotechnol. 61, 385–392. doi:10.1007/s00253-003-1274-y
Gamal, A., Chu, S., McCormick, T. S., Borroto-Esoda, K., Angulo, D., and Ghannoum, M. A. (2021). Ibrexafungerp, a novel oral triterpenoid antifungal in development: overview of antifungal activity against Candida glabrata. Front. Cell Infect. Microbiol. 11, 642358. doi:10.3389/fcimb.2021.642358
Gamble, W. R., Gloer, J. B., Scott, J. A., and Malloch, D. (1995). Polytolypin, a new antifungal triterpenoid from the coprophilous fungus Polytolypa Hystricis. J. Nat. Prod. 58 (12), 1983–1986. doi:10.1021/np50126a034
Goje, O., Sobel, R., Nyirjesy, P., Goldstein, S. R., Spitzer, M., Faught, B., et al. (2023). Oral ibrexafungerp for vulvovaginal candidiasis treatment: an analysis of VANISH 303 and VANISH 306. J. Women’s Health. 32 (2), 178–186. doi:10.1089/jwh.2022.0132
Hamed, A. I., Masullo, M., Pecio, L., Gallotta, D., Mahalel, U. A., Pawelec, S., et al. (2014). Unusual fernane and gammacerane glycosides from the aerial parts of Spergula fallax. J. Nat. Prod. 77 (3), 657–662. doi:10.1021/np4008415
Hector, R. F., and Bierer, D. E. (2011). New β-glucan inhibitors as antifungal drugs. Expert Opin. Ther. Pat. 21 (10), 1597–1610. doi:10.1517/13543776.2011.603899
Hindra, D. Y., Teng, Q. H., Dong, L. B., Crnovcic, I., Huang, T. T., Ge, H. M., et al. (2017). Genome mining of Streptomyces mobaraensis DSM40847 as a bleomycin producer providing a biotechnology platform to engineer designer bleomycin analogues. Org. Lett. 19, 1386–1389. doi:10.1021/acs.orglett.7b00283
Hu, R., Sang, J., Li, W., Tian, Y., Zou, M. F., Tang, G. H., et al. (2021). Structurally diverse triterpenoids with cytotoxicity from Euphorbia hypericifolia. Fitoterapia 151, 104888. doi:10.1016/j.fitote.2021.104888
Jiménez-Ortigosa, C., Padmaja, P., Mary, R. M., and David, S. P. (2013). Enfumafungin derivative MK-3118 shows increased in vitro potency against clinical echinocandin-resistant Candida species and Aspergillus species isolates. Antimicrob Agents Chemother 58 (2), 1248–1251. doi:10.1128/AAC.02145-13
Joshi, B. K., Gloer, G. B., and Wicklow, D. T. (2002). Bioactive natural products from a sclerotium-colonizing isolate of Humicola fuscoatra. J. Nat. Prod. 65, 1734–1737. doi:10.1021/np020295p
Kikuchi, M., Kawarada, N., and Yaoita, Y. (1999). Studies on the constituents of Lonicera species. XIII. New fernane type triterpenoids from the leaves of Lonicera gracilipes var. glandulosa MAXIM. Chem. Pharm. Bull. 47 (5), 663–666. doi:10.1248/cpb.47.663
Kuhnert, E., Li, Y., Lan, N., Yue, Q., Chen, L., Cox, R. J., et al. (2018). Enfumafungin synthase represents a novel lineage of fungal triterpene cyclases. Environ. Microbiol. 20 (9), 3325–3342. doi:10.1111/1462-2920.14333
Lee, A. (2021). Ibrexafungerp: first approval. Drugs 81 (12), 1445–1450. doi:10.1007/s40265-021-01571-5
Li, X. Y., Lv, J. M., Cao, Z. Q., Wang, G. Q., Lin, F. L., Chen, G. D., et al. (2023). Biosynthetic characterization of the antifungal fernane-type triterpenoid polytolypin for generation of new analogues via combinatorial biosynthesis. Org. Biomol. Chem. 21 (4), 851–857. doi:10.1039/d2ob02158g
Li, Z., Huang, Y., Tu, J., Yang, W., Liu, N., Wang, W., et al. (2023). Discovery of BRD4–HDAC dual Inhibitors with improved fungal selectivity and potent synergistic antifungal activity against fluconazole-resistant candida albicans. J. Med. Chem. 66 (8), 5950–5964. doi:10.1021/acs.jmedchem.3c00165
Liberator, P., Giacobbe, R., and Racine, F. (2010). “Semi-synthetic analogs of enfumafungin: novel inhibitors of B-1,2-glucan synthase (GS) with potent in vitro antifungal activity,” in Proceedings of the 50th interscience cong, Boston, MA, Antimicrob Agents Chemother. 50th ICAAC.
Liu, C., Chen, C. Q., Mao, S. S., Han, Z. B., Zhu, Z. B., and Li, Y. (2023). Use of probiotic lactobacilli in the treatment of vaginal infections: in vitro and in vivo investigations. Front. Cell Infect. Microbiol. 13, 1187831. doi:10.3389/fcimb.2023.1153894
Lombardino, J. G., and Lowe, J. A. 3rd. (2004). The role of the medicinal chemist in drug discovery-then and now. Nat. Rev. Drug Discov. 3 (10), 853–862. doi:10.1038/nrd1523
Méndez, C., González- Sabín, J., Morís, F., and Salas, J. A. (2015). Expanding the chemical diversity of the antitumoral compound mithramycin by combinatorial biosynthesis and biocatalysis: the quest for mithralogs with improved therapeutic window. Planta Med. 81, 1326–1338. doi:10.1055/s-0035-1557876
Mesquida, A., Vicente, T., Reigadas, E., Palomo, M., Sánchez-Carrillo, C., Muñoz, P., et al. (2021). In vitro activity of ibrexafungerp and comparators against candida albicans genotypes from vaginal samples and blood cultures. Clin. Microbiol. Infect. 27 (6), 915.e5–e8. doi:10.1016/j.cmi.2021.02.006
Motyl, M., Tan, C., and Liberator, P. (2010). “MK-3118, an orally active enfumafungin with potent in vitro anti-fungal activity,” in Proceedings of the 50th interscience cong, Boston, MA, Antimicrob Agents Chemother. 50th ICAAC.
Moussa, M., Ebrahim, W., Bonus, M., Gohlke, H., Mandi, A., Kurtan, T., et al. (2019). Co-culture of the fungus Fusarium tricinctum with Streptomyces lividans induces production of cryptic naphthoquinone dimers. RSC Adv. 9 (3), 1491–1500. doi:10.1039/c8ra09067j
Mukhtar, H. M., Ansari, S. H., Ali, M., and Naved, T. (2003). New oleanene and fernane-type triterpenes from the stem bark of Betula Pendula Roth. Pharmazie 58, 671–673.
Newman, D. J., and Cragg, G. M. (2020). Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 83, 770–803. doi:10.1021/acs.jnatprod.9b01285
Newman, D. J., and Giddings, L. A. (2013). Natural products as leads to antitumor drugs. Phytochem. Rev. 13 (1), 123–137. doi:10.1007/s11101-013-9292-6
Nunnally, N. S., Etienne, K. A., Angulo, D., Lockhart, S. R., and Berkow, E. L. (2019). In vitro activity of ibrexafungerp, a novel glucan synthase inhibitor against Candida glabrata isolates with FKS mutations. Antimicrob. Agents Ch. 63 (11), e01692–19. doi:10.1128/AAC.01692-19
Nwankwo, L., Gilmartin, D., Matharu, S., Nuh, A., Donovan, J., Armstrong-James, D., et al. (2022). Experience of Isavuconazole as a salvage therapy in chronic pulmonary fungal disease. J. Fungi (Basel) 8 (4), 362. doi:10.3390/jof8040362
Nyirjesy, P., Schwebke, J. R., Angulo, D. A., Harriott, I. A., Azie, N. E., and Sobel, J. D. (2021). Phase 2 randomized study of oral ibrexafungerp versus fluconazole in vulvovaginal candidiasis. Clin. Infect. Dis. 74 (12), 2129–2135. doi:10.1093/cid/ciab841
Onishi, J., Meinz, M., Thompson, J., Curotto, J., Dreikorn, S., Rosenbach, M., et al. (2000). Discovery of novel antifungal (1,3)-beta-D-glucan synthase inhibitors. Antimicrob. Agents Chemother. 44 (2), 368–377. doi:10.1128/aac.44.2.368-377.2000
Peláez, F., Cabello, A., Platas, G., Díez, M. T., González, V. A., Basilio, A., et al. (2000). The discovery of enfumafungin, a novel antifungal compound produced by an endophytic Hormonema species biological activity and taxonomy of the producing organisms. Syst. Appl. Microbiol. 23 (3), 333–343. doi:10.1016/s0723-2020(00)80062-4
Perlin, D. S. (2011). Current perspectives on echinocandin class drugs. Future Microbiol. 6 (4), 441–457. doi:10.2217/fmb.11.19
Pfaller, M., Shawn, A. M., Paul, R. R., Katyna, B., and Mariana, C. (2017). Differential activity of the oral glucan synthase inhibitor SCY-078 against wild-type and echinocandin-resistant strains of Candida species. Antimicrob. Agents Chemother. 61 (8), e001611–17. doi:10.1128/AAC.00161-17
Pfaller, M. A., Messer, S. A., Motyl, M. R., Jones, R. N., and Castanheira, M. (2013a). Activity of MK-3118, a new oral glucan synthase inhibitor, tested against Candida spp. by two international methods (CLSI and EUCAST). J. Antimicrob. Chemother. 68 (4), 858–863. doi:10.1093/jac/dks466
Pfaller, M. A., Messer, S. A., Motyl, M. R., Jones, R. N., and Castanheira, M. (2013b). In vitro activity of a new oral glucan synthase inhibitor (MK-3118) tested against Aspergillus spp. by CLSI and EUCAST broth microdilution methods. Antimicrob. Agents Chemother. 57 (2), 1065–1068. doi:10.1128/AAC.01588-12
Phillips, N. A., Bachmann, G., Haefner, H., Martens, M., and Stockdale, C. (2022). Topical treatment of recurrent vulvovaginal candidiasis: an expert consensus. Women’s Health Rep. 3 (1), 38–42. doi:10.1089/whr.2021.0065
Phillips, N. A., Rocktashel, M., and Merjanian, L. (2023). Ibrexafungerp for the treatment of vulvovaginal candidiasis: design, development and place in therapy. Drug Des. Devel Ther. 7 (17), 363–367. doi:10.2147/DDDT.S339349
Schwartz, R. E., Smith, S. K., Onishi, J. C., Meinz, M., Kurtz, M., Giacobbe, R. A., et al. (2000). Isolation and structural determination of enfumafungin, a triterpene glycoside antifungal agent that is a specific inhibitor of glucan synthesis. JACS 122 (20), 4882–4886. doi:10.1021/ja9944296
Schwebke, J. R., Sobel, R., Gersten, J. K., Sussman, S. A., Lederman, S. N., Jacobs, M. A., et al. (2022). Ibrexafungerp versus placebo for vulvovaginal candidiasis treatment: a phase 3, randomized, controlled superiority trial (VANISH 303). Clin. Infect. Dis. 74 (11), 1979–1985. doi:10.1093/cid/ciab750
Shigematsu, N., Tsujii, E., Kayakiri, N., Takase, S., Tanaka, H., and Tada, T. (1992). WF1 1605, an antagonist of leukotriene B4 produced by a fungus II. Structure determination. J. Antibiot. 45 (5), 704–708. doi:10.7164/antibiotics.45.704
Shiojima, K., Arai, Y., Nakane, T., and Ageta, H. (1997a). Fern constituents: Adiantum cuneatum. I. Three new triterpenoids, glaucanol B acetate, 7.BETA.,25-Epoxyfern-8-ene and 25-Norfern-7-en-10.BETA.-yl formate. Chem. Pharm. Bull. 45 (4), 636–638. doi:10.1248/cpb.45.636
Shiojima, K., Arai, Y., Nakane, T., Ageta, H., and Cai, S. Q. (1997b). Fern constituents: Adiantum cuneatum. III. Four new triterpenoids,4,23-bisnor-3,4-secofilic-5(24)-en-3-al, 4,23-Bisnor-3,3-dimethoxy-3,4-secofilic-5(24)-ene, 7.BETA.,25-Epoxyfern-9(11)-en-8.ALPHA.-ol and 7.ALPHA.,8.ALPHA.-Epoxyfernan-25-ol. Chem. Pharm. Bull. 45 (10), 1608–1610. doi:10.1248/cpb.45.1608
Sobel, J. D. (2007). Vulvovaginal candidosis. Lancet. 369 (9577), 1961–1971. doi:10.1016/S0140-6736(07)60917-9
Sobel, J. D., Borroto-Esoda, K., Azie, N., and Angulo, D. (2021). In vitro pH activity of ibrexafungerp against fluconazole-susceptible and -resistant Candida isolates from women with vulvovaginal candidiasis. Antimicrob. Agents Chemother. 65 (8), e0056221. doi:10.1128/AAC.00562-21
Sobel, R., Nyirjesy, P., Ghannoum, M. A., Delchev, D. A., Azie, N. E., Angulo, D., et al. (2022). Efficacy and safety of oral ibrexafungerp for the treatment of acute vulvovaginal candidiasis: a global phase 3, randomised, placebo-controlled superiority study (VANISH 306). BJOG 129 (3), 412–420. doi:10.1111/1471-0528.16972
Song, Y. Y., Jiang, J. B., Yan, Y. M., and Cheng, Y. X. (2021). Isolation and identification of belamcandaoids A-N from Belamcanda chinensis seeds and their inhibition on extracellular matrix in TGF-β1 induced kidney proximal tubular cells. Bioorg Chem. 114, 105067. doi:10.1016/j.bioorg.2021.105067
Sucher, A. J., Annie, T., Charlene, T., Netra, M., Allwyn, N., and Elias, B. C. (2022). Ibrexafungerp: a new triterpenoid antifungal. Am. J. Health Syst. Pharm. 79 (24), 2208–2221. doi:10.1093/ajhp/zxac256
Sun, L. M., Sun, S. J., Cheng, A. X., Wu, X. Z., Zhang, Y., and Lou, H. X. (2009). In vitro activities of retigeric acid B alone and in combination with azole antifungal agents against Candida albicans. Antimicrob. Agents Chemother. 53 (4), 1586–1591. doi:10.1128/AAC.00940-08
Tanaka, R., Ida, T., Kita, S., Kamisako, W., and Matsunaga, S. (1996). A 3,4-seco-8βH-fernadienoic acid and other constituents from Euphorbia Chamaesyce. Phytochemistry 41 (4), 1163–1168. doi:10.1016/0031-9422(95)00721-0
Tanaka, R., and Shunyo, M. (1988). Triterpene constituents from Euphorbia supina. Phytochemistry 27 (11), 3579–3584. doi:10.1016/0031-9422(88)80772-6
Tanaka, R., and Shunyo, M. (1989). Supinenolones A, B and C, fernane type triterpenoids from Euphorbia supina. Phytochemistry 28 (11), 3149–3154. doi:10.1016/0031-9422(89)80296-1
Tanaka, R., and Shunyo, M. (1991a). Fernane and unusually migrated fernane triterpene-triones from Euphorbia Supina. Phytochemistry 30 (1), 293–296. doi:10.1016/0031-9422(91)84140-n
Tanaka, R., and Shunyo, M. (1991b). Fernane and multiflorrane triterpene ketols from Euphorbia Supina. Phytochemistry 30 (12), 4093–4097.
Tsujii, E., Tsurumi, Y., Miyata, S., Fujie, K., Kawakami, A., Okamoto, M., et al. (1992). WF11605, an antagonist of leukotriene B4 produced by a fungus. I. Producing strain, fermentation, isolation and biological activity. J. Antibiot. 45 (5), 698–703. doi:10.7164/antibiotics.45.698
Vazquez, J. A., Oliver, C., Philipp, K., Riina, R. R., Rohit, B., Marshall, L. G., et al. (2021). 123. Oral ibrexafungerp outcomes by fungal disease in patients from an interim analysis of a phase 3 open-label study (FURI). Open Forum Infect. Dis. 4 (8), S73–S74. doi:10.1093/ofid/ofab466.123
Wang, X. N., Zhang, H. J., Ren, D. M., Ji, M., Yu, W. T., and Lou, H. X. (2009). Lobarialides A-C, antifungal triterpenoids from the lichen Lobaria kurokawae. Chem and Biodivers 6 (5), 746–753. doi:10.1002/cbdv.200800054
Waterer, G. (2021). Advances in anti-fungal therapies. Mycopathologia 186 (5), 665–672. doi:10.1007/s11046-021-00560-2
Wiederhold, N. P., Najvar, L. K., Olivo, M., Morris, K. N., Patterson, H. P., Catano, G., et al. (2021). Ibrexafungerp demonstrates in vitro activity against fluconazole-resistant Candida auris and in vivo efficacy with delayed initiation of therapy in an experimental model of invasive candidiasis. Antimicrob. Agents Chemother. 65 (6), e02694–20. doi:10.1128/AAC.02694-20
Yu, Y., Wolf, A. K., Thusek, S., Heinekamp, T., Bromley, M., Krappmann, S., et al. (2021). Direct visualization of fungal burden in filamentous fungus-infected silkworms. J. Fungi 7 (2), 136. doi:10.3390/jof7020136
Yuan, X. H., Xu, G. B., Wu, W. L., Yang, T., and Li, G. Y. (2012). Peniciside, a new triterpenoid glycoside, from the fungus Penicillium sp. 169. Arch Pharm Res. 35 (2), 311–314. doi:10.1007/s12272-012-0210-z
Keywords: triterpenoid, enfumafungin, ibrexafungerp, anti-fungal, fernane type
Citation: Liu C-Y, Zhang L, Liu S-X, Lu Y-F, Li C and Pei Y-H (2024) A review of the fernane-type triterpenoids as anti-fungal drugs. Front. Pharmacol. 15:1447450. doi: 10.3389/fphar.2024.1447450
Received: 11 June 2024; Accepted: 08 August 2024;
Published: 21 August 2024.
Edited by:
Yuxiang Dong, University of Nebraska Medical Center, United StatesReviewed by:
Wangbin Wu, University of Nebraska Medical Center, United StatesHui Lu, Tongji University, China
Copyright © 2024 Liu, Zhang, Liu, Lu, Li and Pei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chang Li, bGljaGFuZzY2MUAxMjYuY29t; Yue-Hu Pei, cGVpeXVlaEB2aXAuMTYzLmNvbQ==