 Zhimin Mao
Zhimin Mao Qun Ji
Qun Ji Xuhui Zeng
Xuhui Zeng- Institute of Reproductive Medicine, Medical School, Nantong University, Nantong, Jiangsu, China
Acrolein (ACR) is a ubiquitous environmental pollutant and byproduct of lipid peroxidation that has been implicated in male infertility. However, the molecular mechanisms underlying ACR-induced toxicity in Sertoli cells remain unclear. Given its role in inducing oxidative stress, we examined whether ferroptosis, an iron-dependent form of regulated cell death, could mediate ACR toxicity in Sertoli cells. We also tested if hydrogen sulfide (H2S), which has antioxidant and ACR detoxifying properties, could protect Sertoli cells from ACR-induced ferroptosis. ACR exposure decreased Sertoli cell viability, increased protein carbonylation and p38 MAPK phosphorylation, indicating oxidative injury. ACR also depleted glutathione (GSH), downregulated the cystine importer SLC7A11, increased intracellular ferrous iron (Fe2+) and lipid peroxidation, suggesting activation of ferroptosis. Consistently, the ferroptosis inhibitor deferoxamine (DFO) markedly attenuates ACR-induced cell death. Further studies revealed that ACR-induced ferroptotic changes were prevented by exogenous H2S and exaggerated by inhibition of endogenous H2S production. Furthermore, H2S also suppressed GPX4 inhibitor RSL3-induced intracellular ACR accumulation and ferroptosis. In summary, our study demonstrates that ACR induces ferroptotic cell death in Sertoli cells, which can be prevented by H2S through multiple mechanisms. Targeting the H2S pathway may represent a therapeutic strategy to mitigate ACR-induced Sertoli cell injury and preserve male fertility.
1 Introduction
Infertility affects more than 15% of couples of reproductive age worldwide, posing a serious global health problem. Male factor infertility, often due to declines in sperm quantity and quality, accounts for up to 50% of infertile couples (Agarwal et al., 2021). Environmental toxicants and unhealthy lifestyles are major contributors to impaired sperm production and function (Ilacqua et al., 2018; Rodprasert et al., 2023). Therefore, understanding the effects and mechanisms of these factors on testicular function, particularly spermatogenesis, is imperative to develop preventive strategies against male infertility.
Sertoli cells are the somatic cells of the testes that support spermatogenesis by providing structural and nutritional support to developing germ cells. As a component of the blood-testis barrier (BTB), Sertoli cells also protect germ cells from toxins and other insults (Ghafouri-Fard et al., 2021; Thumfart and Mansuy, 2023). Because of this reason, Sertoli cells themselves are vulnerable to damage from toxicants like cadmium, arsenic, bisphenol A and reactive aldehydes, resulting in impaired spermatogenesis and infertility (Liu et al., 2011; Zhang et al., 2017; Ramos-Treviño et al., 2018; Mao et al., 2023a). Elucidating the mechanisms of Sertoli cell injury and identifying protective agents are thus important areas of research.
Acrolein (ACR) is a ubiquitous α,β-unsaturated aldehyde found in the environment, diet, and endogenously produced through lipid peroxidation and other metabolic processes (Stevens and Maier, 2008). ACR exposure is associated with smoking, alcohol consumption, pollution, diabetes, and cardiovascular diseases—all risk factors for male infertility (Mahalingam et al., 2017; Sansone et al., 2018; Del Giudice et al., 2020; Huang et al., 2024). Importantly, ACR levels are exceptionally high in the male reproductive system as sperm membranes are rich in polyunsaturated fatty acids (PUFAs), which produce high levels of endogenous ACR during lipid peroxidation (Gautier and Aurich, 2022). Consistently, high ACR levels have been detected in mature sperm (Moazamian et al., 2015). The presence of ACR in the male reproductive system could cause toxic effects and lead to infertility. The cytotoxicity of ACR has been shown to be stems mainly from its reactivity with proteins, lipids, and DNA. ACR readily forms adducts with these biomolecules, disrupting their structure and function. Additionally, ACR induces oxidative stress by depleting glutathione (GSH) and inhibiting antioxidant enzymes (Moghe et al., 2015). We and others have shown ACR causes oxidative damage in testicular Sertoli cells, which can lead to the impairment of spermatogenesis (Liu et al., 2011; Mao et al., 2023a). Despite the established link between ACR and impaired male reproductive function, the precise molecular mechanisms underlying its detrimental effects on Sertoli cells and spermatogenesis remain elusive. Further research is warranted to unravel these mechanisms and identify potential therapeutic interventions to mitigate ACR-induced reproductive toxicity.
As of the downstream cellular event, ferroptosis has been drawn great attention. Ferroptosis is an iron-dependent form of programmed cell death caused by a redox imbalance between the production of oxidants and antioxidants. Ferroptosis is characterized by depletion of GSH, inactivation of the enzyme glutathione peroxidase 4 (GPX4), and unchecked lipid peroxidation. This results in the accumulation of toxic lipid peroxidation products such as ACR and 4-hydroxynonenal (4-HNE) (Wang et al., 2023). As a reactive species, ACR is implicated not only in the induction of oxidative stress, but also as a driving factor for oxidative stress and an effector molecule for ferroptosis. It is therefore conceivable that ACR could be critically involved in Sertoli cell injury, potentially through induction of oxidative stress and ferroptosis.
Hydrogen sulfide (H2S) is an endogenous gasotransmitter with antioxidant and cytoprotective effects in many tissues. It is generated enzymatically in cells by cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST) (Kimura, 2010). All three H2S-producing enzymes are expressed in the testes, implying a physiological role in the male reproductive system (Sugiura et al., 2005; Li et al., 2015). Emerging evidence indicates H2S counters oxidative stress in various cell models through multiple mechanisms (Wu et al., 2015; Mao et al., 2019; Hu et al., 2020). The multi-faceted effects of H2S on oxidative stress make it an intriguing potential therapy for ACR-induced toxicity. In this study, we speculated that the toxicity of ACR on Sertoli cells could be related to ferroptosis, which could be prevented by H2S. The aim of this study was to test this hypothesis.
Here, we present our results that ACR caused Sertoli cell injury by inducing ferroptosis, and H2S protected against Sertoli cell ferroptosis induced by ACR and other stimuli. H2S could be developed to prevent and treat ferroptotic Sertoli cell injury.
2 Materials and methods
2.1 Materials
Acrolein (ACR, #ZD816) was obtained from Xianding Biotechnology Co., Ltd. (Shanghai, China). Sodium hydrosulfide hydrate (NaHS, #161527) and deferoxamine (DFO, #D9533) were purchased from Sigma-Aldrich (St Louis, MO, United States). β-cyano-L-Alanine (BCA, #10010947) was bought from Cayman Chemical (Ann Arbor, MI, United States). RSL3 (#HY-100218A) was from MedChemExpress (MCE, Shanghai, China). Anti-rabbit antibody against SLC7A11 (#26864-1-AP) was from Proteintech (Chicago, IL, United States). Anti-rabbit antibody against phospho-p38MAPK (Thr180/Tyr182) (#8203S) and anti-mouse antibody against α-tubulin (#3873T) were from Cell Signaling Technology (Danvers, MA, United States). Anti-mouse β-actin (#GB12001) antibody was from Servicebio Technology (Wuhan, China). Anti-rabbit IgG (#5366), and anti-mouse IgG (#5470) were from Cell Signaling Technology (Danvers, MA, United States).
2.2 Cell culture
Mouse Sertoli TM4 cells (#CRL-1715) were obtained from the American Type Culture Collection (ATCC, Rockville, MD, United States). Cell culture was performed as we previously reported (Mao et al., 2023a). For routine maintenance, the cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F-12 (#11320-033, Invitrogen) supplemented with 10% fetal bovine serum (FBS, #F8687, Sigma-Aldrich). For experiments, the cells were cultured in DMEM/F-12 containing 1% FBS.
2.3 Assessment of cell viability
To evaluate cell viability, a double staining protocol with Calcein-AM and propidium iodide (PI) was utilized (#C542, Dojindo). Briefly, cells in 96-well plates were incubated with a mixture of Calcein-AM and PI for 10–20 min. Viable cells were stained green with Calcein-AM due to intracellular esterase activity, while dead cells with compromised plasma membranes were stained red with PI. Stained cells were visualized and imaged under a fluorescence microscope.
Alternatively, cell viability was quantified using the WST reagent (#CK04, Dojindo). Cells cultured in 96-well plates were incubated with WST for approximately 30 min, and the absorbance at 450 nm was measured using a spectrometer.
2.4 Western blot analysis
Western blot was performed as described previously (Mao et al., 2023a). Briefly, equal amounts of protein samples were separated by SDS-PAGE gels and transferred to PVDF membranes (#IPVH00010, Merck Millipore). The membranes were blocked with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) and incubated with primary antibodies followed by secondary antibodies. Protein bands were detected using Amersham Imager 600 (GE Healthcare). To confirm equal protein loading, the membranes were reprobed with anti-α-tubulin or β-actin antibody. Densitometry analysis was performed using ImageJ software and normalized to the respective control.
2.5 Assessment of protein carbonylation with oxyblot
Protein carbonylation was examined using the OxyBlot Protein Oxidation Detection Kit (#S7150, EMD Millipore) as described previously (Mao et al., 2023a). Briefly, 5–10 μg of protein samples were denatured with 12% SDS and derivatized with 2,4-dinitrophenylhydrazine (DNPH). The DNP-derivatized protein samples were processed for standard Western blotting. To confirm equal protein loading, the blots were stained with EZBlue Gel Staining Reagent (#G1041-500ML, Sigma-Aldrich).
2.6 Quantification of GSH
Intracellular GSH levels were measured using a GSSG/GSH quantification kit from Dojindo (#G263). Briefly, the treated cells were lysed with 80 µL 10 mM HCl, followed by addition of 20 µL of 5% 5-sulfosalicylic acid. After collecting the supernatant by centrifuging the lysate at 4°C, GSH content in the supernatants was quantified by measuring absorbance at 405 nm using a microplate reader, according to the manufacturer’s protocol.
2.7 Detection of intracellular ferrous iron (Fe2+)
Intracellular Fe2+ was detected using the FerroOrange fluorescent probe from Dojindo (#F374). After treatments, cells were washed and incubated with 1 μM FerroOrange solution for 30 min at 37°C. Fluorescence microscopy images were acquired and analyzed using ImageJ software. The mean fluorescence intensity of each treatment group was normalized to that of the control.
2.8 Assessment of lipid peroxidation
Lipid peroxidation was assessed utilizing the fluorescent probe C11 BODIPY 581/591 (#GC40165, GlpBio). Briefly, after treatments, cells were incubated with 2.5 μM C11 BODIPY 581/591 dye in fresh culture medium for 30 min at 37°C. Subsequently, the cells were washed, imaged under a fluorescence microscope, and fluorescence intensity was quantified using ImageJ software. The mean fluorescence intensity of each treatment group was normalized to that of the control group.
2.9 Measurement of intracellular ACR
Relative intracellular ACR levels were examined utilizing AcroleinRED from funakoshi (#FDV-0022). After treatments, culture medium was removed and replaced with medium containing 20 μM AcroleinRED. Following 30–60 min incubation, cells were washed, imaged under a fluorescence microscope, and fluorescence intensity was quantified using ImageJ software. The mean fluorescence intensity of each treatment group was normalized to that of the control.
2.10 Statistical analysis
The data are expressed as mean ± standard error (S.E.). Statistical comparisons between two groups were performed using Student’s t-test. For comparisons of multiple groups, one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test was utilized. Statistical analyses were conducted using either Microsoft Excel (Microsoft Corporation, Redmond, WA, United States) or Sigmaplot software (Systat Software Inc., San Jose, CA, United States). In all cases, a P-value less than 0.05 was considered statistically significant.
3 Results
3.1 ACR induces hallmarks of ferroptosis in Sertoli cells
As we have reported that ACR induces cell death in cultured Sertoli cells (Mao et al., 2023a). Here, we repeated some of the experiments and found that ACR caused concentration-dependent cell death (Supplementary Figures S1A, B). This effect of ACR was associated with an early elevation in protein carbonylation and activation of the oxidative stress-sensitive P38, suggesting induction of oxidative cell death (Supplementary Figure S1C).
To investigate whether ferroptosis underlies ACR cytotoxicity, we analyzed several ferroptosis markers. ACR exposure caused a concentration-dependent reduction in intracellular GSH, which was associated with a downregulation of the cystine transporter SLC7A11 (Figures 1A–C). Further Investigation demonstrated that ACR exposure led to an increase in intracellular Fe2+ levels. This was evidenced by the heightened fluorescence intensity observed within the cells following the addition of FerroOrange, a fluorescent probe specifically designed to detect Fe2+ (Figures 1D, E). Additionally, ACR also caused lipid peroxidation (Figures 1F, G) as detected by using a fluorescent dye C11 BODIPY 581/591. This dye emits red fluorescence when bound to non-oxidized lipids. However, when lipids become oxidized, the dye shifts its fluorescence to the green range. ACR exposure decreased the red fluorescence, while it increased green fluorescence, indicating an induction of lipid oxidation. In further support of an involvement of ferroptosis in ACR-induced cell death, the ferroptosis inhibitor deferoxamine (DFO, an iron chelator that prevents iron-dependent lipid peroxidation) significantly prevented ACR-induced cell death, as revealed by assessment of cell viability using Calcein-AM/PI staining and WST assay (Figures 1H, I). Collectively, these data show that ACR triggered ferroptotic cell death in Sertoli cells.
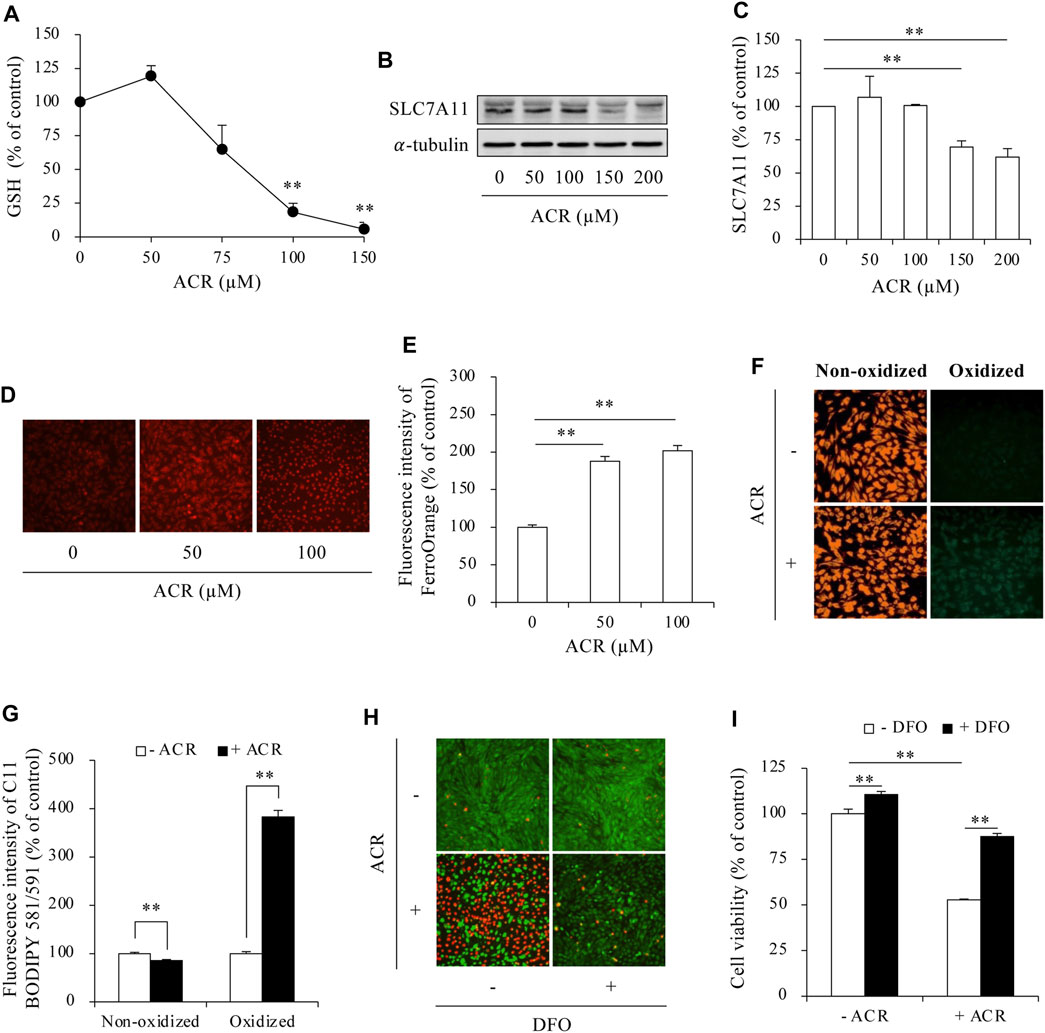
Figure 1. ACR induced ferroptosis in Sertoli cells. (A) Effect of ACR on intracellular GSH level. TM4 cells were cultured with the indicated concentrations of ACR for 3 h. The cellular lysates were analyzed for GSH levels with GSSG/GSH quantification kit. Data are mean ± S.E. (n = 3; **P < 0.01 vs. control). (B,C) Effect of ACR on the protein level of SLC7A11. TM4 cells were incubated with the indicated concentrations of ACR for 3 h, and the cellular lysates were used for protein expression of SLC7A11 (B). Densitometric quantitation of these bands was shown in (C). Data in (C) are mean ± S.E. (n = 3; **P < 0.01). (D,E) Effect of ACR on intracellular Fe2+ level. TM4 cells were stimulated with the indicated concentrations of ACR for 3 h. FerroOrange staining was conducted for Fe2+ detection. (F,G) Induction of lipid peroxidation by ACR. TM4 cells were cultured with 100 µM ACR for 3 h, followed by staining with C11 BODIPY 581/591 for detection of lipid oxidation. Densitometric quantitation of relative fluorescence intensity in (D,F) was performed on 20 cells randomly selected from each group, and the data were shown in (E,G), respectively. (n = 20; **P < 0.01). (H,I) Effect of DFO on ACR-induced Sertoli cell death. TM4 cells were pretreated with or without 400 µM DFO for 30 min before exposing to 100 µM ACR for an additional 6 h. The cell viability was determined by Calcein-AM/PI staining (H) and WST assay (I). Data in (I) are mean ± S.E. (n = 5; **P < 0.01).
3.2 H2S inhibits ACR-induced ferroptosis in Sertoli cells
We have reported that H2S protects against ACR-induced oxidative injury in Sertoli cells (Mao et al., 2023a). Here, we tested whether the effect of H2S could be through suppressing ferroptosis. First, we confirmed that H2S donor NaHS prevented ACR-induced oxidative cell death, as evidenced by the obviously reduced number of PI-positive red cells, as well as the increased formazan formation (Figures 2A, B). This protective effects of H2S donor were associated with a reduction in the level of protein carbonylation and P38 phosphorylation (Figure 2C).
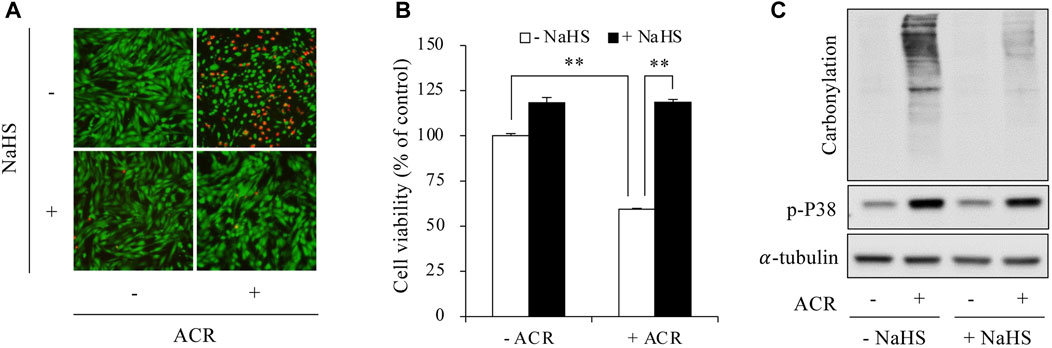
Figure 2. H2S prevented ACR-induced oxidative injury in Sertoli cells. (A,B) Amelioration of Sertoli cell viability by sodium hydrosulfide (NaHS). TM4 cells were pretreated with or without 1 mM NaHS for 15 min before exposing to 100 µM ACR for an additional 6 h. The cell viability was determined by Calcein-AM/PI staining (A) and WST assay (B). Data in (B) are mean ± S.E. (n = 5; **P < 0.01). (C) Inhibition of protein carbonylation and P38 activation by NaHS. TM4 cells were exposed to 50 µM ACR with or without 1 mM NaHS for 6 h. Cellular lysates were used to analysis of protein carbonylation and P38 phosphorylation.
Then, we determined the possible involvement of ferroptosis. As shown in Figure 3, supplementing cells with exogenous H2S elevated intracellular GSH levels, upregulated SLC7A11 expression, and blocked Fe2+ and lipid peroxidation accumulation. Consistently, inhibiting endogenous H2S synthesis with BCA exerted the opposite effects. It exacerbated ACR toxicity (Figures 4A, B) and ACR-induced cellular oxidation (Figure 4C) as we have previously documented (Mao et al., 2023a). It also significantly potentiated ACR-induced changes in ferroptosis markers, including markedly reduced GSH/SLC7A11 level (Figures 4D–F) and elevated lipid peroxidation (Figures 4G, H). Together, these findings indicate H2S protects Sertoli cells from ACR by inhibiting ferroptosis.
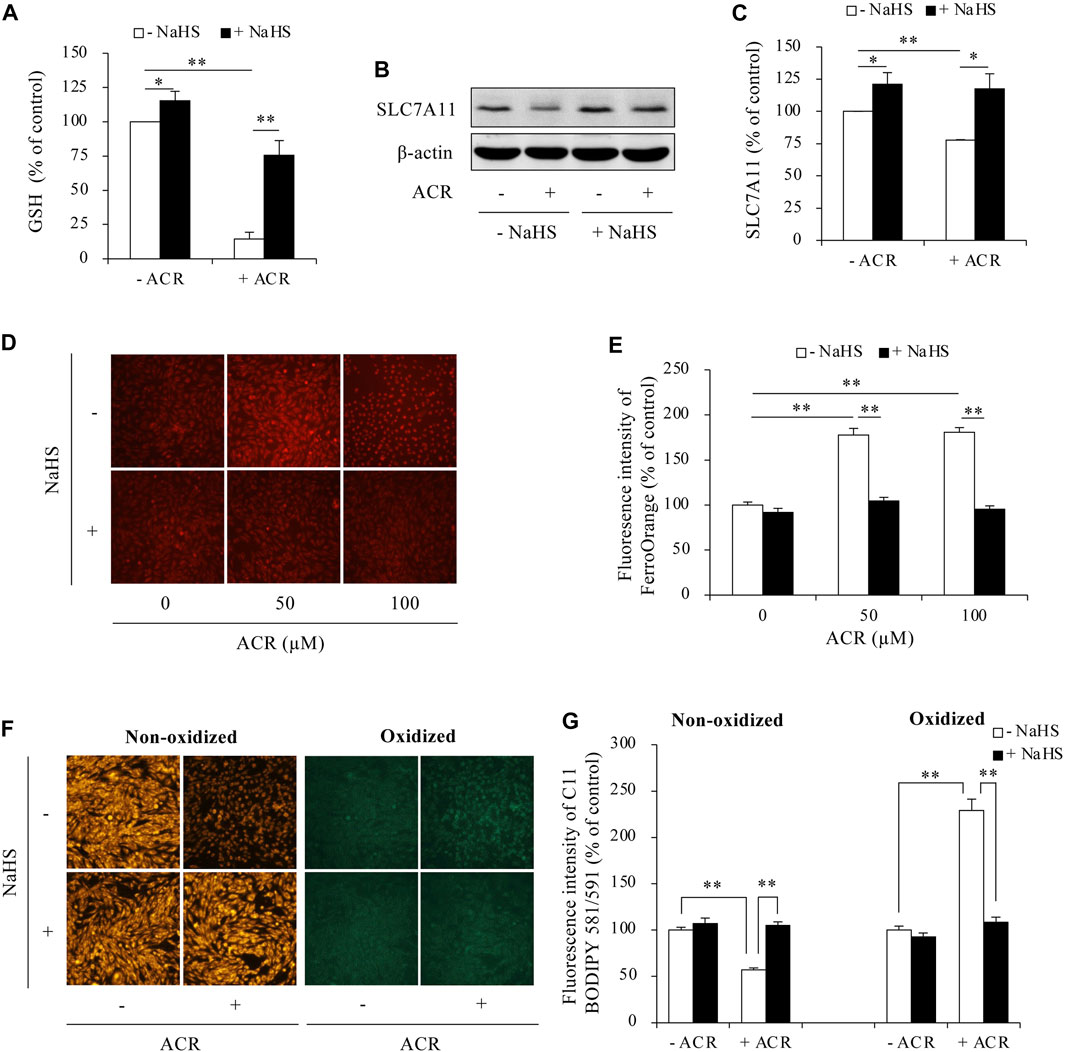
Figure 3. H2S inhibited ACR-induced ferroptosis in Sertoli cells. (A) Reversion of GSH level by NaHS. TM4 cells were exposed to 100 µM ACR with or without 1 mM NaHS for 3 h. The cellular lysates were analyzed for GSH levels with GSSG/GSH quantification kit. Data are mean ± S.E. (n = 4; *P < 0.05, **P < 0.01). (B,C) Upregulation of SLC7A11 by NaHS. TM4 cells were exposed to 100 µM ACR with or without 1 mM NaHS for 3 h. Cellular lysates were used to analysis of protein expression of SLC7A11 (B). Densitometric quantitation of these bands was shown in (C). Data in (C) are mean ± S.E. (n = 3; *P < 0.05, **P < 0.01). (D,E) Clearance of Fe2+ accumulation by NaHS. TM4 cells were stimulated with the indicated concentrations of ACR with or without NaHS for 3 h. FerroOrange staining was performed for Fe2+ detection. (F,G) Prevention of lipid peroxidation by NaHS. TM4 cells were exposed to 100 µM ACR with or without NaHS for 3 h. After that, C11 BODIPY 581/591 staining were used for indication of lipid oxidation. Densitometric quantitation of relative fluorescence intensity in (D,F) was performed on 20 cells randomly selected from each group, and the data were shown in (E,G), respectively (n = 20; **P < 0.01).
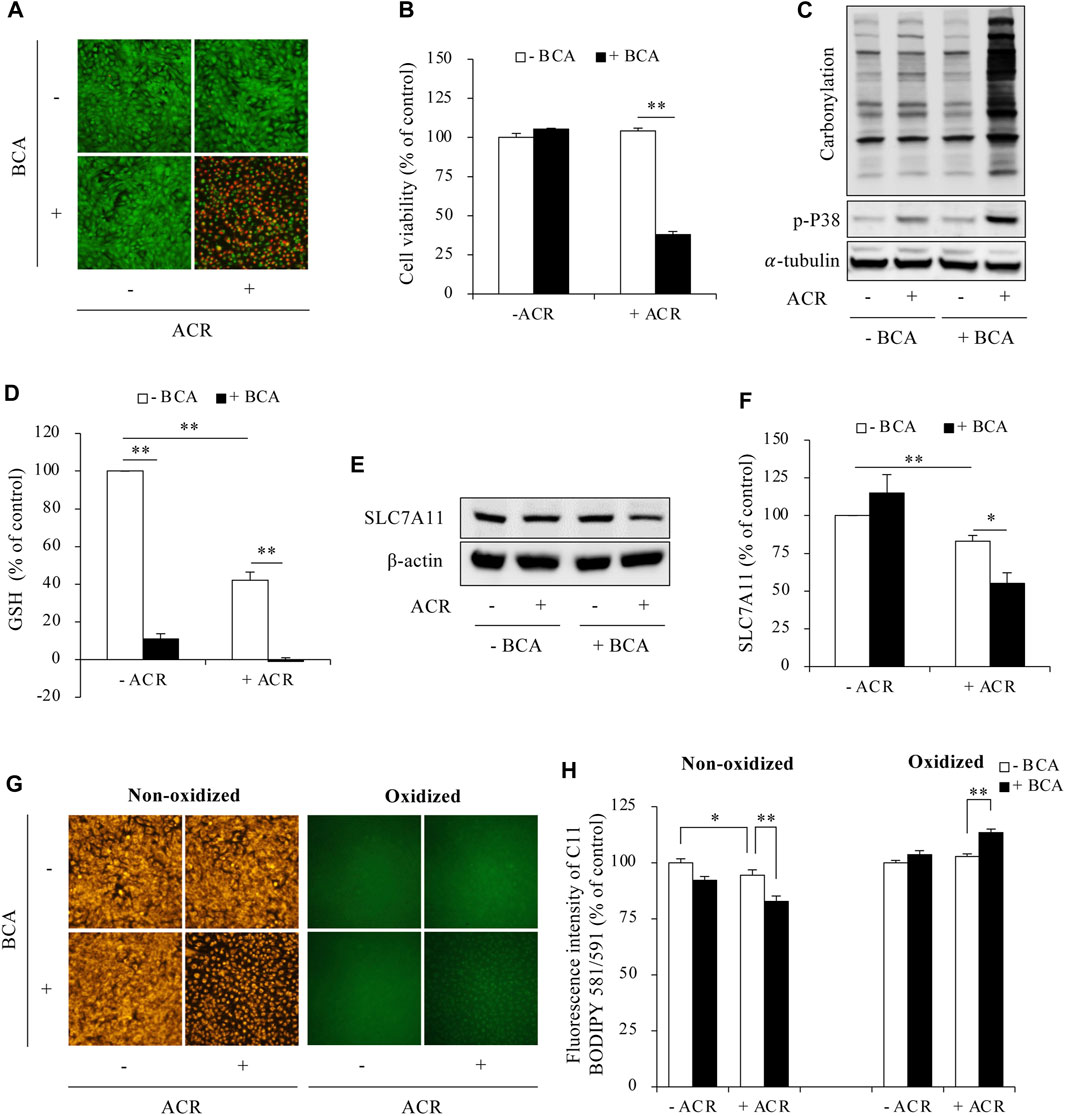
Figure 4. Inhibition of H2S promoted ferroptosis in ACR-treated Sertoli cells. (A,B) Acceleration of Sertoli cell death by BCA. TM4 cells were pretreated with or without 2 mM BCA for 12 h before exposing to 50 µM ACR for an additional 5 h. The cell viability was determined by Calcein-AM/PI staining (A) and WST assay (B). Data in B are mean ± S.E. (n = 5; **P < 0.01). (C) Aggravation of protein carbonylation and P38 activation by BCA. TM4 cells were exposed to 50 µM ACR with or without 2 mM BCA for 2 h. Cellular lysates were subjected to analysis of protein carbonylation and P38 phosphorylation. (D) Acceleration of GSH consumption by BCA. TM4 cells were pretreated with or without 2 mM BCA for 6 h before exposing to 50 µM ACR for an additional 3 h. The cellular lysates were analyzed for GSH levels with GSSG/GSH quantification kit. Data are mean ± S.E. (n = 4; **P < 0.01). (E,F) Aggravation of downregulated protein expression of SLC7A11 by BCA. TM4 cells were incubated with 75 µM ACR for 3 h, and the cellular lysates were used for protein expression of SLC7A11 (E). Densitometric quantitation of these bands was shown in (F). Data in (F) are mean ± S.E. (n = 3; *P < 0.05, **P < 0.01). (G,H) Promotion of lipid peroxidation by BCA. TM4 cells were exposed to 50 µM ACR with or without 2 mM BCA pretreatment for 3 h. After that, C11 BODIPY 581/591 staining were used for analyzing lipid oxidation. Densitometric quantitation of relative fluorescence intensity in (G) was calculated on 20 cells randomly selected from each group, and the data were shown in (H) (n = 20; *P < 0.05, **P < 0.01).
3.3 H2S inhibits RSL3-induced ferroptosis in Sertoli cells
Given the suppressive effect of H2S on ACR-induced ferroptosis, we examined whether this extends to other ferroptosis triggers using the GPX4 inhibitor RSL3. Figure 5 shows that exogenous H2S donor NaHS also prevented RSL3-induced cell death (Figures 5A, B). On the contrary, inhibition of endogenous H2S with BCA exacerbated cell death (Figures 5C, D). Consistent with the protective actions of H2S donor on cell death, NaHS also significantly suppressed RSL3-induced changes in ferroptotic markers. It significantly reduced RSL3-triggered lipid peroxidation (Figures 5E, F). Intriguingly, as one of the lipid peroxidation products, RSL3 inhibition dramatically elevated the endogenous level of ACR. However, in the presence of H2S donor, this accumulation of ACR could be completely abolished (Figures 5G, H). These observations suggest that besides ACR, H2S could also inhibit ferroptosis induced by other stimuli in Sertoli cells.
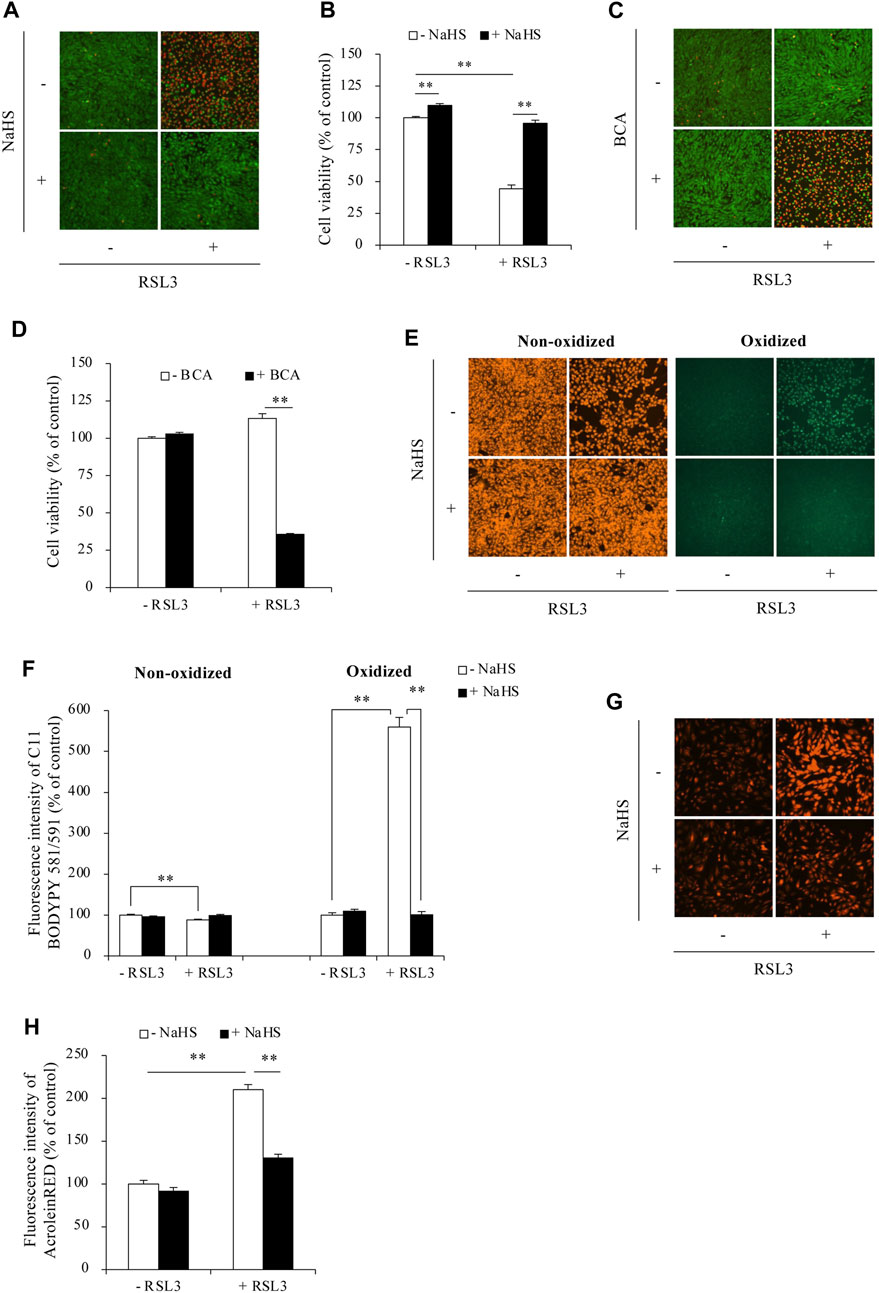
Figure 5. H2S protected against RSL3-induced ferroptosis in Sertoli cells. (A–D) Effect of H2S on RSL3-induced Sertoli cell death. TM4 cells were incubated with 5 µM RSL3 with or without 1 mM NaHS for 24 h (A) or in the presence or absence of 2 mM BCA for 16 h (B). The cell viability was determined by Calcein-AM/PI staining (A,C) and WST assay (B,D). Data in (B,D) are mean ± S.E. (n = 4; **P < 0.01). (E,F) Effect of H2S on RSL3-induced lipid peroxidation. TM4 cells were exposed to 5 µM RSL3 with or without 1 mM NaHS for 16 h. After that, C11 BODIPY 581/591 staining were subjected to analysis of lipid oxidation. (G,H) Effect of H2S on RSL3-induced ACR accumulation. TM4 cells were incubated with 10 µM RSL3 with or without 1 mM NaHS for 6 h. AcroleinRED staining was performed for ACR detection. Densitometric quantitation of relative influorescence intensity in (E,G) was performed on 20 cells randomly selected from each group, and the data were shown in (F,H), respectively (n = 20; **P < 0.01).
4 Discussion
In this study, we demonstrated that H2S prevented ACR-induced ferroptosis in Sertoli cells via multiple mechanisms. Given that the ACR is a key toxic chemical underlying reproductive injury under various situations, our finding could have significant implications.
ACR is a ubiquitous toxicant implicated in reproductive damage from various pathological factors. Cigarette smoke, dietary fat peroxidation, high-temperature cooking, automotive exhaust, industrial pollution, and water disinfection byproducts could all result in an increased level of ACR in vivo. In addition, ACR also arises endogenously through lipid peroxidation, oxidative stress, inflammation, and polyamine metabolism. Conditions like diabetes, atherosclerosis, neurodegenerative diseases, and trauma are associated with increased ACR (Stevens and Maier, 2008; Moghe et al., 2015). The elevated ACR could readily cross the BTB and exert toxic effects in testes. The highly reactive ACR has many toxic effects, such as formation of adducts with proteins, lipids and DNA, depletion of GSH, suppression of other antioxidants, and activation of cell death signaling (Stevens and Maier, 2008; Moghe et al., 2015). These molecular events interfere the process of sperm production and impairs sperm function. Moreover, damaged cells themselves can generate additional ACR, thus propagating a vicious cycle of oxidative damage that compromises male fertility (Aitken et al., 2012; Moazamian et al., 2015). Given all the factors leading to the elevated ACR have been documented to be closely related to male infertility, our findings about the effect and mechanisms of ACR toxicity and the protective strategy in Sertoli cells should have close clinical relevance.
Our results showed that ACR induced oxidative stress and triggered ferroptosis in Sertoli cells. It depleted GSH, induced protein carbonylation, activated redox signaling. These findings align with previous studies from ours and others showing that ACR induces oxidative stress via elevating reactive oxygen species (ROS) production and suppressing antioxidant enzymes levels (Taghiabadi et al., 2012; Kang, 2013; Mao et al., 2023a). One of the key mechanisms underlying the toxicity of ACR is its ability to deplete intracellular GSH levels via formation of conjugates with GSH (Shah et al., 2014; Xiong et al., 2021). In this study, we found that ACR could also reduce the protein level of SLC7A11, the cystine/glutamate antiporter responsible for cystine uptake. The effect of ACR could be an additional mechanism contributing to the reduced intracellular GSH level.
Our study further characterized that one form of the cell death initiated by ACR in Sertoli cells is ferroptosis. Besides its reported actions on ferroptosis-related molecular events, such as GSH depletion and oxidative stress induction, ACR also increased intracellular iron accumulation and promoted lipid peroxidation in Sertoli cells. ACR exposure markedly increased redox-active Fe2+ levels. The mechanisms by which ACR disrupted iron homeostasis are currently unclear. Potentially, it could be through 1) upregulating iron import proteins, 2) downregulating iron storage and export proteins, and/or 3) releasing iron from biomolecules by adduct formation. We have recently reported that cyclophosphamide (CYP)-induced cystitis was associated with decreased expression of the cellular iron export protein ferroportin 1 (FPN1) in bladder tissues (Mao et al., 2023b). Given that ACR is a metabolite of CYP and mediates CYP-induced cystitis, it is conceivable that ACR may exert the similar effect to affect cellular iron export, thus resulting in iron accumulation. Further analysis of the effects of ACR on the regulators of iron import/export and iron release will provide important insights into mechanisms of ACR-mediated ferroptosis and tissue injury.
Iron acts as a crucial catalyst in the process of lipid peroxidation. Consequently, the observed increase in intracellular Fe2+ levels induced by ACR further exacerbated lipid peroxidation, leading to the accumulation of lipid peroxidation products, including ACR itself. This creates a detrimental cycle where ACR-initiated oxidative damage fuels further ACR generation, thereby perpetuating cellular injury and ultimately culminating in cell death. Breaking this vicious cycle is therefore essential for mitigating the cytotoxic effects of ACR.
Our results revealed that H2S could be an ideal candidate for counteracting ACR-induced ferroptosis. It worked through multiple mechanisms. First, H2S enhanced cellular oxidative defenses. In contrary to the effects of ACR, H2S upregulated SLC7A11, increased intracellular GSH level and suppressed the activation of redox signaling pathway. In addition, H2S can also upregulate GPX4 expression (Zhang et al., 2024), a key enzyme that inhibits ferroptosis by converting lipid hydroperoxides into non-toxic lipid alcohols. Second, H2S restricted iron-mediated-catalytic ROS generation. H2S limited the availability of catalytic iron, essential for the Fenton reaction, thereby reducing ROS production and subsequent lipid peroxidation. Third, H2S directly eliminated lipid peroxidation products. Previous studies, including our own, have documented that H2S can directly scavenge ROS and lipid-derived aldehydes such as ACR and 4-hydroxynonenal (4-HNE) (Schreier et al., 2009; Mao et al., 2022; Mao et al., 2023a). In the current study, we also demonstrated that H2S dramatically suppressed ACR accumulation resulting from GPX4 inhibition. Collectively, our findings indicate that H2S offered multiple protective mechanisms against ferroptotic cell death induced by ACR.
Apart from ACR, H2S also inhibited GPX4 inhibitor RSL3-induced ferroptosis in Sertoli cells. Because ferroptosis induced by various insults shares similar molecular mechanisms, it is conceivable that H2S could act as a potent ferroptosis inhibitor irrespective of the specific insult. Indeed, there are several reports showing that H2S suppressed ferroptosis in several different cell types induced by different insults (Wang et al., 2020; Wang et al., 2022; Zhang et al., 2024). For examples, H2S inhibits specifically RSL3-mediated cell death in muscle and lung cells (Wang et al., 2020; Wang et al., 2022). Thus, our results add to the emerging evidence that H2S is a powerful inhibitor of ferroptosis across multiple cell types and different ferroptotic triggers. Our study provided the first evidence showing a critical involvement of H2S in protecting cells from ACR-induced ferroptosis in Sertoli cells. It may contribute to the defense against reproductive injuries. Notably, ferroptosis has been implicated in many diseases, including kidney dysfunction, neurodegeneration, and ischemia-reperfusion injury (Jiang et al., 2021). Thus, the findings in this study could also be used to mitigate ferroptosis and oxidative damage in these contexts.
It is worth mentioning that we have reported that H2S is a potent scavenger of ACR (Mao et al., 2022). The question naturally occurs as to whether the direct reaction between ACR and NaHS could influence the interpretation of the changes in the downstream molecular events. If the reaction between ACR and H2S caused a complete elimination of ACR, the downstream events should also be significantly affected. However, even so, it should not invalidate the overall conclusions because 1) NaHS alone significantly increases GSH and SLC7A11, suggesting that H2S can directly modulate ferroptosis pathways; 2) inhibiting endogenous H2S generation with BCA also significantly potentiated ACR-induced ferroptosis; 3) H2S also blunted RSL3-induced cell death, lipid peroxidation, and ACR accumulation (Figure 5), indicating that the protective effect of H2S extends beyond simply scavenging ACR. Collectively, this evidence supports the role of H2S in protecting against ferroptosis induced by ACR and other stimuli.
In this study, we chose to investigate Sertoli cells, as they are the main components of the BTB and provide the first line of defense against various insults (Kaur et al., 2014). Sertoli cells are known to be susceptible to both oxidative stress and inflammatory damage (Wong and Cheng, 2011; Dutta et al., 2021). Since Sertoli cells play a critical role in supporting germ cell growth and development through supplying nutrients, growth factors, and other biomolecules (Petersen and Söder, 2006), their dysfunction could greatly impact germ cell activity. Recently, ferroptosis has been shown to mediate Sertoli cell death induced by diverse factors. For example, ferroptosis drives oxygen-glucose deprivation/reoxygenation (OGD/R)-induced Sertoli cell death (Li et al., 2018). It also has a key role in mono-2-ethylhexyl ester (MEHP)-induced Sertoli cell injury (Wu et al., 2022). In addition, Hepatitis B virus decreases human Sertoli cell viability by promoting ferroptosis through TRIM37-mediated ubiquitination and loss of GPX4 activity (Pan et al., 2023). In this context, our study reveals a novel role for H2S as a master regulator of lipid peroxidation and ferroptosis in Sertoli cells. The finding may enhance our understanding of self-defensive mechanisms in Sertoli cells. As the results of the current study are based on in vitro cell models, further research is necessary to determine whether these findings can be replicated in vivo. This will be the primary focus of our future investigations.
It is worth mentioning that, as a ferroptosis inhibitor, H2S has several advantages. First, its inhibitory effect on ferroptosis is not insult-specific. It suppressed ferroptosis triggered by multiple inducers. Second, H2S blocks ferroptosis through diverse mechanisms. Third, as a gaseous transmitter, H2S readily permeates cell membranes (Riahi and Rowley, 2014), making it uniquely equipped to access lipid peroxides that form at membrane lesions.
In conclusion, our findings establish ferroptosis as a potentially important mechanism driving ACR-induced toxicity in Sertoli cells. Moreover, we have identified H2S as a potent protector against ACR-mediated ferroptotic cell death. Our study indicates that targeting the H2S pathway holds promise as a therapeutic strategy to alleviate oxidative damage and ferroptosis triggered by ACR and other insults as well.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
ZM: Writing–review and editing, Writing–original draft, Validation, Software, Methodology, Investigation, Funding acquisition, Data curation, Conceptualization. QJ: Writing–original draft, Methodology, Investigation, Data curation. PC: Writing–original draft, Methodology, Investigation, Data curation. KZ: Writing–original draft, Methodology, Investigation. XZ: Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China to ZM (grant number 82201754) and the Natural Science Foundation of Jiangsu Province to ZM (grant number BK20210845).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1440147/full#supplementary-material
References
Agarwal, A., Baskaran, S., Parekh, N., Cho, C.-L., Henkel, R., Vij, S., et al. (2021). Male infertility. Lancet 397 (10271), 319–333. doi:10.1016/S0140-6736(20)32667-2
Aitken, R. J., Whiting, S., De Iuliis, G. N., McClymont, S., Mitchell, L. A., and Baker, M. A. (2012). Electrophilic aldehydes generated by sperm metabolism activate mitochondrial reactive oxygen species generation and apoptosis by targeting succinate dehydrogenase. J. Biol. Chem. 287 (39), 33048–33060. doi:10.1074/jbc.M112.366690
Del Giudice, F., Kasman, A. M., Ferro, M., Sciarra, A., De Berardinis, E., Belladelli, F., et al. (2020). Clinical correlation among male infertility and overall male health: a systematic review of the literature. Investigative Clin. Urology 61 (4), 355–371. doi:10.4111/icu.2020.61.4.355
Dutta, S., Sengupta, P., Slama, P., and Roychoudhury, S. (2021). Oxidative stress, testicular inflammatory pathways, and male reproduction. Int. J. Mol. Sci. 22 (18), 10043. doi:10.3390/ijms221810043
Gautier, C., and Aurich, C. (2022). “Fine feathers make fine birds” – the mammalian sperm plasma membrane lipid composition and effects on assisted reproduction. Animal Reproduction Sci. 246, 106884. doi:10.1016/j.anireprosci.2021.106884
Ghafouri-Fard, S., Shoorei, H., Mohaqiq, M., Haidar Abbas Raza, S., and Taheri, M. (2021). The role of different compounds on the integrity of blood-testis barrier: a concise review based on in vitro and in vivo studies. Gene 780, 145531. doi:10.1016/j.gene.2021.145531
Hu, L., Guo, J., Zhou, L., Zhu, S., Wang, C., Liu, J., et al. (2020). Hydrogen sulfide protects retinal pigment epithelial cells from oxidative stress-induced apoptosis and affects autophagy. Oxidative Med. Cell. Longev. 2020, 8868564–8868615. doi:10.1155/2020/8868564
Huang, R., Chen, J., Guo, B., Jiang, C., and Sun, W. (2024). Diabetes-induced male infertility: potential mechanisms and treatment options. Mol. Med. 30 (1), 11. doi:10.1186/s10020-023-00771-x
Ilacqua, A., Izzo, G., Emerenziani, G. P., Baldari, C., and Aversa, A. (2018). Lifestyle and fertility: the influence of stress and quality of life on male fertility. Reproductive Biol. Endocrinol. 16 (1), 115. doi:10.1186/s12958-018-0436-9
Jiang, X., Stockwell, B. R., and Conrad, M. (2021). Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22 (4), 266–282. doi:10.1038/s41580-020-00324-8
Kang, J. H. (2013). Modification and inactivation of Cu,Zn-superoxide dismutase by the lipid peroxidation product, acrolein. BMB Rep. 46 (11), 555–560. doi:10.5483/bmbrep.2013.46.11.138
Kaur, G., Thompson, L. A., and Dufour, J. M. (2014). Sertoli cells – immunological sentinels of spermatogenesis. Seminars Cell & Dev. Biol. 30, 36–44. doi:10.1016/j.semcdb.2014.02.011
Kimura, H. (2010). Hydrogen sulfide: its production, release and functions. Amino Acids 41 (1), 113–121. doi:10.1007/s00726-010-0510-x
Li, G., Xie, Z. Z., Chua, J. M., Wong, P. C., and Bian, J. (2015). Hydrogen sulfide protects testicular germ cells against heat-induced injury. Nitric Oxide 46, 165–171. doi:10.1016/j.niox.2014.10.005
Li, L., Hao, Y., Zhao, Y., Wang, H., Zhao, X., Jiang, Y., et al. (2018). Ferroptosis is associated with oxygen-glucose deprivation/reoxygenation-induced Sertoli cell death. Int. J. Mol. Med. 41, 3051–3062. doi:10.3892/ijmm.2018.3469
Liu, F., Li, X.-L., Lin, T., He, D.-W., Wei, G.-H., Liu, J.-H., et al. (2011). The cyclophosphamide metabolite, acrolein, induces cytoskeletal changes and oxidative stress in Sertoli cells. Mol. Biol. Rep. 39 (1), 493–500. doi:10.1007/s11033-011-0763-9
Mahalingam, S., Chiang, C., and Flaws, J. (2017). Environmental contaminants affecting fertility and somatic health. Seminars Reproductive Med. 35 (03), 241–249. doi:10.1055/s-0037-1603569
Mao, Z., Huang, Y., Li, B., Tomoya, K., Shinmori, H., Zeng, X., et al. (2022). Hydrogen sulfide as a potent scavenger of toxicant acrolein. Ecotoxicol. Environ. Saf. 229, 113111. doi:10.1016/j.ecoenv.2021.113111
Mao, Z., Huang, Y., Zhang, Z., Yang, X., Zhang, X., Huang, Y., et al. (2019). Pharmacological levels of hydrogen sulfide inhibit oxidative cell injury through regulating the redox state of thioredoxin. Free Radic. Biol. Med. 134, 190–199. doi:10.1016/j.freeradbiomed.2019.01.009
Mao, Z., Li, H., Zhao, X.-L., and Zeng, X.-H. (2023a). Hydrogen sulfide protects Sertoli cells against toxicant Acrolein-induced cell injury. Food Chem. Toxicol. 176, 113784. doi:10.1016/j.fct.2023.113784
Mao, Z., Zhong, K., Liu, X., and Zeng, X. (2023b). Ferroptosis contributes to cyclophosphamide-induced hemorrhagic cystitis. Chemico-Biological Interact. 384, 110701. doi:10.1016/j.cbi.2023.110701
Moazamian, R., Polhemus, A., Connaughton, H., Fraser, B., Whiting, S., Gharagozloo, P., et al. (2015). Oxidative stress and human spermatozoa: diagnostic and functional significance of aldehydes generated as a result of lipid peroxidation. MHR Basic Sci. reproductive Med. 21 (6), 502–515. doi:10.1093/molehr/gav014
Moghe, A., Ghare, S., Lamoreau, B., Mohammad, M., Barve, S., McClain, C., et al. (2015). Molecular mechanisms of acrolein toxicity: relevance to human disease. Toxicol. Sci. 143 (2), 242–255. doi:10.1093/toxsci/kfu233
Pan, C., Kong, X., Wu, Z., and Fei, Q. (2023). The role of hepatitis B virus surface protein in inducing Sertoli cell ferroptosis. Andrology 12 (3), 643–654. doi:10.1111/andr.13520
Petersen, C., and Söder, O. (2006). The sertoli cell – a hormonal target and ‘super’ nurse for germ cells that determines testicular size. Hormone Res. Paediatr. 66 (4), 153–161. doi:10.1159/000094142
Ramos-Treviño, J., Bassol-Mayagoitia, S., Hernández-Ibarra, J. A., Ruiz-Flores, P., and Nava-Hernández, M. P. (2018). Toxic effect of cadmium, lead, and arsenic on the sertoli cell: mechanisms of damage involved. DNA Cell Biol. 37 (7), 600–608. doi:10.1089/dna.2017.4081
Riahi, S., and Rowley, C. N. (2014). Why can hydrogen sulfide permeate cell membranes? J. Am. Chem. Soc. 136 (43), 15111–15113. doi:10.1021/ja508063s
Rodprasert, W., Toppari, J., and Virtanen, H. E. (2023). Environmental toxicants and male fertility. Best Pract. Res. Clin. Obstetrics Gynaecol. 86, 102298. doi:10.1016/j.bpobgyn.2022.102298
Sansone, A., Di Dato, C., de Angelis, C., Menafra, D., Pozza, C., Pivonello, R., et al. (2018). Smoke, alcohol and drug addiction and male fertility. Reproductive Biol. Endocrinol. 16 (1), 3. doi:10.1186/s12958-018-0320-7
Schreier, S. M., Muellner, M. K., Steinkellner, H., Hermann, M., Esterbauer, H., Exner, M., et al. (2009). Hydrogen sulfide scavenges the cytotoxic lipid oxidation product 4-HNE. Neurotox. Res. 17 (3), 249–256. doi:10.1007/s12640-009-9099-9
Shah, H., Speen, A. M., Saunders, C., Brooke, E. A. S., Nallasamy, P., Zhu, H., et al. (2014). Protection of HepG2 cells against acrolein toxicity by 2-cyano-3,12-dioxooleana-1,9-dien-28-imidazolide via glutathione-mediated mechanism. Exp. Biol. Med. 240 (10), 1340–1351. doi:10.1177/1535370214563900
Stevens, J. F., and Maier, C. S. (2008). Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 52 (1), 7–25. doi:10.1002/mnfr.200700412
Sugiura, Y., Kashiba, M., Maruyama, K., Hoshikawa, K., Sasaki, R., Saito, K., et al. (2005). Cadmium exposure alters metabolomics of sulfur-containing amino acids in rat testes. Antioxidants Redox Signal. 7, 781–787. doi:10.1089/ars.2005.7.781
Taghiabadi, E., Imenshahidi, M., Abnous, K., Mosafa, F., Sankian, M., Memar, B., et al. (2012). Protective effect of silymarin against acrolein-induced cardiotoxicity in mice. Evidence-Based Complementary Altern. Med. 2012, 352091–352114. doi:10.1155/2012/352091
Thumfart, K. M., and Mansuy, I. M. (2023). What are Sertoli cells? Historical, methodological, and functional aspects. Andrology 11 (5), 849–859. doi:10.1111/andr.13386
Wang, B., Wang, Y., Zhang, J., Hu, C., Jiang, J., Li, Y., et al. (2023). ROS-induced lipid peroxidation modulates cell death outcome: mechanisms behind apoptosis, autophagy, and ferroptosis. Archives Toxicol. 97 (6), 1439–1451. doi:10.1007/s00204-023-03476-6
Wang, Y., Liao, S., Pan, Z., Jiang, S., Fan, J., Yu, S., et al. (2022). Hydrogen sulfide alleviates particulate matter-induced emphysema and airway inflammation by suppressing ferroptosis. Free Radic. Biol. Med. 186, 1–16. doi:10.1016/j.freeradbiomed.2022.04.014
Wang, Y., Yu, R., Wu, L., and Yang, G. (2020). Hydrogen sulfide guards myoblasts from ferroptosis by inhibiting ALOX12 acetylation. Cell. Signal. 78, 109870. doi:10.1016/j.cellsig.2020.109870
Wong, E. W. P., and Cheng, C. Y. (2011). Impacts of environmental toxicants on male reproductive dysfunction. Trends Pharmacol. Sci. 32 (5), 290–299. doi:10.1016/j.tips.2011.01.001
Wu, D., Hu, Q., Liu, X., Pan, L., Xiong, Q., and Zhu, Y. Z. (2015). Hydrogen sulfide protects against apoptosis under oxidative stress through SIRT1 pathway in H9c2 cardiomyocytes. Nitric Oxide 46, 204–212. doi:10.1016/j.niox.2014.11.006
Wu, Y., Wang, J., Zhao, T., Chen, J., Kang, L., Wei, Y., et al. (2022). Di-(2-ethylhexyl) phthalate exposure leads to ferroptosis via the HIF-1α/HO-1 signaling pathway in mouse testes. J. Hazard. Mater. 426, 127807. doi:10.1016/j.jhazmat.2021.127807
Xiong, R., Wu, Q., Bryant, M., Rosenfeldt, H., Healy, S., and Cao, X. (2021). In vitro dosimetry analyses for acrolein exposure in normal human lung epithelial cells and human lung cancer cells. Environ. Toxicol. Pharmacol. 83, 103576. doi:10.1016/j.etap.2020.103576
Zhang, H., Pan, J., Huang, S., Chen, X., Chang, A. C. Y., Wang, C., et al. (2024). Hydrogen sulfide protects cardiomyocytes from doxorubicin-induced ferroptosis through the SLC7A11/GSH/GPx4 pathway by Keap1 S-sulfhydration and Nrf2 activation. Redox Biol. 70, 103066. doi:10.1016/j.redox.2024.103066
Keywords: acrolein, male infertility, Sertoli cell, ferroptosis, hydrogen sulfide, oxidative stress
Citation: Mao Z, Ji Q, Chen P, Zhong K and Zeng X (2024) Hydrogen sulfide protects against toxicant acrolein-induced ferroptotic cell death in Sertoli cells. Front. Pharmacol. 15:1440147. doi: 10.3389/fphar.2024.1440147
Received: 29 May 2024; Accepted: 17 July 2024;
Published: 01 August 2024.
Edited by:
Luchen Shan, Jinan University, ChinaReviewed by:
Andres A. Caro, Hendrix College, United StatesMilton Prabu, Annamalai University, India
Fei Sun, Tongji Medical College, China
Copyright © 2024 Mao, Ji, Chen, Zhong and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhimin Mao, bWFvemhpbWluQG50dS5lZHUuY24=