 Natalie Jayne Kurganovs
Natalie Jayne Kurganovs Nikolai Engedal
Nikolai Engedal- Autophagy in Cancer Lab, Institute for Cancer Research, Department of Tumor Biology, Oslo University Hospital, Oslo, Norway
Around 1 in 7 men will be diagnosed with prostate cancer during their lifetime. Many strides have been made in the understanding and treatment of this malignancy over the years, however, despite this; treatment resistance and disease progression remain major clinical concerns. Recent evidence indicate that autophagy can affect cancer formation, progression, and therapeutic resistance. Autophagy is an evolutionarily conserved process that can remove unnecessary or dysfunctional components of the cell as a response to metabolic or environmental stress. Due to the emerging importance of autophagy in cancer, targeting autophagy should be considered as a potential option in disease management. In this review, along with exploring the advances made on understanding the role of autophagy in prostate carcinogenesis and therapeutics, we will critically consider the conflicting evidence observed in the literature and suggest how to obtain stronger experimental evidence, as the application of current findings in clinical practice is presently not viable.
1 Prostate cancer and current aspects of prostate cancer therapy
The prostate is the most cancer-prone internal organ in men and is the highest cause of cancer-associated mortalities in Western countries (Siegel et al., 2023). The survival rate of patients diagnosed with prostate cancer (commonly abbreviated as PCa) is dependent on the clinical stage. Whilst early stage and confined disease may be managed by surgery, more advanced stages of prostate cancer require additional therapy, and frequently becomes very challenging to treat (Moul, 2004). The non-surgical standard of care for advanced prostate cancer is androgen deprivation therapy (ADT) (Parker et al., 2020), due to the pivotal role the androgen receptor (AR) plays in prostate cancer progression (Ferro et al., 2021), and radiation therapy (Parker et al., 2020), which causes cell death through either direct or indirect nuclear DNA damage via the generation of free radicals (Zou et al., 2017; Chaiswing et al., 2018) (Figure 1). While both ADT and radiation are administered with curative intent for men with localised prostate cancer, treatment resistance remains a serious clinical concern. Around 70% of patients treated with ADT show indications of disease progression within 2 years, despite the therapy resulting in exceptionally low levels of circulating testosterone (Crawford et al., 1989; Eisenberger et al., 1998; Mottet et al., 2011; James et al., 2015). 25%–50% of high-risk prostate cancer patients treated with radiation therapy develop biochemical recurrence within 5 years following treatment, with up to 30% succumbing to their disease within 10 years (Zietman et al., 2005; Agarwal et al., 2008; Boorjian et al., 2011). Following the development of treatment resistance, or metastatic lesions, chemotherapies and targeted therapies are also utilized in prostate cancer patients. Chemotherapies are systemic medications that inhibit cell proliferation and/or promote cancer cell death. First identified to have an effect on prostate cancer in the 1990s, chemotherapies are currently advised for use in both hormone-sensitive and -insensitive metastatic prostate cancer (Tannock et al., 1996; Tannock et al., 2004; de Bono et al., 2010; Eisenberger et al., 2017). Alas, resistance to chemotherapy occurs frequently and only half of the patients respond to docetaxel, which is the current mainstay of chemotherapy for prostate cancer patients. As a result, docetaxel only improves median survival by 2 months (Petrylak et al., 2004; Tannock et al., 2004). Thus, more efficient therapies are needed. Newer chemotherapeutics are utilised as second line therapies, following the initial failure of a first line such as docetaxel. These include mitoxantrone and cabazitaxel, which can extend median overall survival just over a year (de Bono et al., 2010). More recently, targeted therapies such as Poly-ADP ribose polymerase inhibitors (PARPi) have become a promising therapeutic option for metastatic prostate cancer (Pezaro, 2020; Risdon et al., 2021; Taylor et al., 2023) due to the induction of synthetic lethality in cells with homologous recombination repair deficiency (HRD) (de Bono et al., 2020; de Bono et al., 2021; Smith et al., 2022). HRD is found in approximately 30% of metastatic prostate cancer patients (Mateo et al., 2015; Robinson et al., 2015). PARPi can also be employed in combination with ADT (Hussain et al., 2018). However, in spite of very encouraging clinical effects, the development of treatment resistance is a major challenge also with PARPi therapies (Rose et al., 2020).
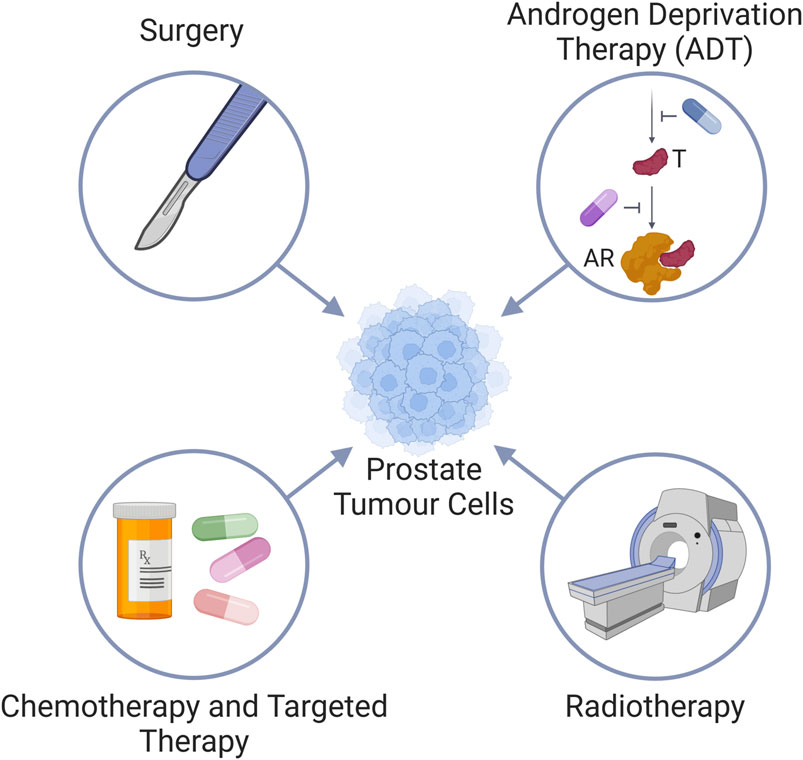
Figure 1. Standard treatment modalities for prostate cancer. Prostate cancer is treated via (i) surgery, (ii) Androgen deprivation therapy (ADT) by either inhibiting the androgen synthesis pathway (blue pill) to reduce testosterone (T) levels, or use of androgen receptor (AR) antagonists (purple pill); (iii) radiotherapy, and (iv) chemotherapy and targeted therapies.
The mechanisms controlling the development and persistence of treatment resistance in prostate cancer remain poorly characterised, and filling this knowledge gap is imperative to improve prostate cancer therapeutic options. There have been many indications in the literature that autophagy may play a key role in carcinogenesis (cancer formation and progression) and treatment resistance. Below, we will discuss the available data concerning the role of autophagy in prostate carcinogenesis and prostate cancer therapeutics.
2 Autophagy
Autophagy (meaning “self-eating”) is a vital homeostatic process required for essential functioning of the cell and organism, and can affect various cellular activities. It is an evolutionarily conserved process that was first discovered in the 1950s and 60s through the emergence of electron microscopy (Novikoff and Essner, 1962; De Duve and Wattiaux, 1966). In this mechanism, redundant or dysfunctional cellular components are recycled in a lysosomal-dependent manner (Xie and Klionsky, 2007; Ohsumi, 2014; Trelford and Di Guglielmo, 2021). Autophagy can be classed as either bulk autophagy (largely non-selective degradation and recycling of bulk cytoplasm) or selective autophagy (targeted recycling of organelles, protein aggregates, or other cellular components).
There are three main types of autophagy; chaperone-mediated autophagy, microautophagy, and macroautophagy. Chaperone-mediated autophagy refers to a type of selective autophagy that identifies individual proteins for unfolding and direct import to the lysosome via a protein complex involving LAMP2A (Tekirdag and Cuervo, 2018). Microautophagy is the least characterised out of the three subtypes and involves lysosomal or endosomal uptake of cytoplasmic contents through direct invagination of the lysosomal or endosomal (in which case the process is termed endosomal microautophagy) limiting membrane (Sahu et al., 2011; Li et al., 2012; Schuck, 2020). The final and main type of autophagy is known as macroautophagy.
Macroautophagy (from now on referred to simply as “autophagy” unless otherwise specified) is distinct from the other two types of autophagy in that the site of sequestration occurs away from the limiting membrane of the lysosome and, instead, involves the formation of double/multi-membrane cytoplasmic vacuoles which transport cargo to the lysosome. These sequestering vacuoles, termed autophagosomes form de novo through expansion instead of budding from a pre-existing organelle containing cargo (Yang and Klionsky, 2009). The initiation step frequently occurs in association with an endoplasmic reticulum subdomain called the omegasome (although there importantly also are other sites and other membrane sources for autophagy initiation), which is enriched for the lipid phosphatidylinositol 3-phosphate (PI(3)P) (Axe et al., 2008). The machinery for autophagosome formation (Figure 2), is highly conserved and contains two major initiation complexes: the ULK1 (unc-51 like autophagy initiating activating kinase 1) complex and the class III PI-3-kinase complex 1 (PI3K3-C1) (Mizushima et al., 2011; Hurley and Schulman, 2014; Suzuki et al., 2017). The membrane then begins to expand and creates the primary double/multi-membrane sequestering structure referred to as the “phagophore” (Seglen, 1987; He and Klionsky, 2009). The expansion of the phagophore is driven by the PI(3)P binding WIPI-1-4 proteins and the ATG (autophagy-related)8 family and ATG12 conjugation systems (altogether involving the following ATG proteins in mammals: ATG3, ATG4A-D, ATG5, ATG7, ATG10, ATG12, ATG16L1, and the mammalian ATG8 (mATG8) homologues MAP1LC3A, MAP1LC3B, MAP1LC3C, GABARAP, GABARAPL1, and GABARAPL2).
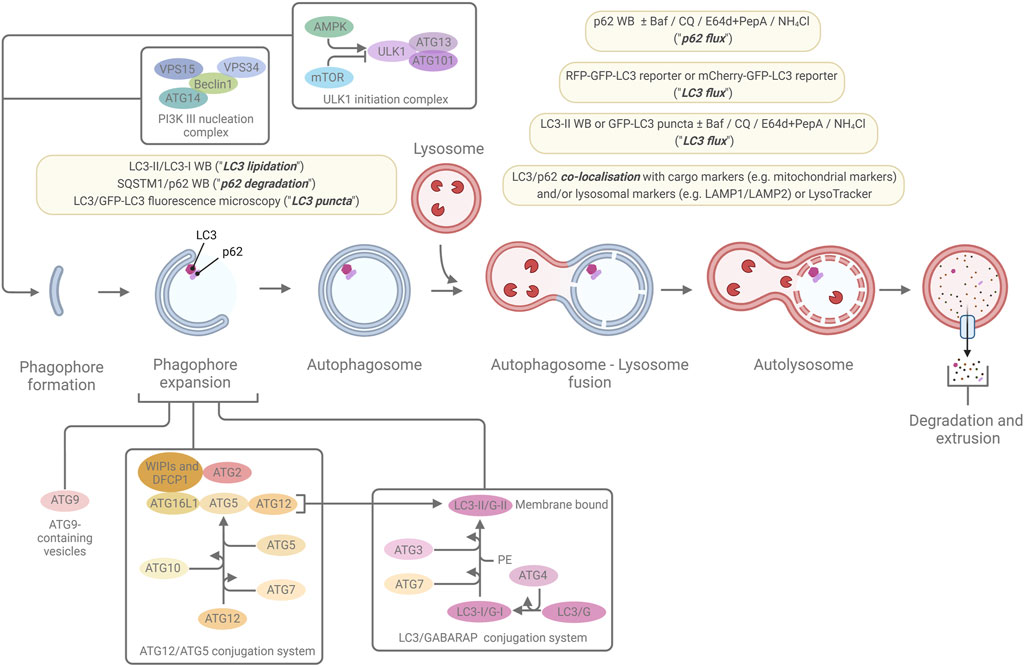
Figure 2. The macroautophagic pathway and the most commonly used marker-based autophagy assays. A simplified overview of the macroautophagic pathway and some of the major proteins, protein complexes, and conjugation systems that are involved in autophagosome formation, are shown. The light-yellow boxes highlight the most common ways to monitor macroautophagy through marker-based assays that indicate levels of LC3 lipidation, p62 degradation, LC3 puncta, LC3 flux, p62 flux, and LC3 or p62 co-localisation with cargo marker or lysosomal markers. For further details, see the main text and references within.
MAP1LC3B (microtubule-associated protein 1 light-chain 3B; commonly referred to as LC3B, or simply LC3) and the other mATG8 are cleaved by ATG4 proteases, exposing a C-terminal glycine residue. The cleaved forms (termed mATG8-I, for instance LC3-I or GABARAP-I) can then be conjugated to phosphatidylethanolamine (PE) (Kabeya et al., 2004; Sou et al., 2006) (termed “lipidation”) by sequential ubiquitylation-like reactions catalysed by ATG7 (E1-like enzyme), ATG3 (E2-like enzyme), and the ATG5/ATG12-ATG16L1 complex to generate a membrane-bound form of mATG8-I, referred to as mATG8-II (for instance LC3-II or GABARAP-II) (Kabeya et al., 2000; Tanida et al., 2001; Tanida et al., 2002; Tanida et al., 2004). Initially, both the LC3- and GABARAP subfamilies of the mATG8 proteins were thought to be essential for autophagosome formation (Weidberg et al., 2010), however, recent studies by us and others indicate that the GABARAPs play a dominant role, and that the LC3s are frequently not required for the autophagic pathway to go to completion (Szalai et al., 2015; Nguyen et al., 2016; Vaites et al., 2018; Luhr et al., 2019; Sønstevold et al., 2021). An important other function of the mAGT8s is to recruit various types of autophagy receptors to the inner phagophore membrane via interaction with specific motifs/regions in the receptors, the best described being the so-called LC3/ATG8-interacting region/motif (LIR/AIM). This interaction can mediate selective autophagy when the autophagy receptors in turn are bound to specific cargo. Selective autophagy receptors include: i) soluble receptors that bind to ubiquitinated cargo, e.g., SQSTM1 (commonly referred to as p62), NBR1, NDP52, TAX1BP1, and OPTN, ii) soluble, ubiquitin-independent receptors, e.g., NCOA4 and NUFIP1, and iii) membrane-bound receptors, e.g., the mitochondrially-residing receptors NIX, BNIP3, and FUNDC1, and the ER-residing receptors FAM134B, RTN3L, CCPG1, SEC62, TEX264 and ATL3 (Lamark and Johansen, 2021).
During late-stage expansion of the phagophore, the membrane bends to generate a spherical structure and, upon completion, the phagophore fully surrounds its cargo and forms a closed structure called the “autophagosome.” The outer membrane of the autophagosome may fuse with a lysosomal membrane, the product of which is referred to as an “autolysosome” (Yang and Klionsky, 2009). The inner autophagosome membrane and the autophagic cargo are subsequently degraded due to exposure to the acidic lumen and hydrolases of the lysosome. Component parts are transported back to the cytoplasm via lysosomal permeases and can be utilized by the cell or the organism as building blocks or to generate energy (Yorimitsu and Klionsky, 2005).
As the names imply, the ATG proteins were identified through their roles in autophagy. Importantly, however, it has become increasingly evident that most, if not all, ATG proteins also have non-autophagic functions (Velikkakath et al., 2012; Elgendy et al., 2014; Rohatgi et al., 2015; Kaizuka and Mizushima, 2016; Nunes et al., 2016; Yang et al., 2016; Cadwell and Debnath, 2018; Galluzzi and Green, 2019; Hu et al., 2020; Fang et al., 2021; Mailler et al., 2021; Zhang et al., 2022a; Hamaoui and Subtil, 2022; Tedesco et al., 2024; Tran et al., 2024). This includes the by far most used autophagic marker LC3B (Baisamy et al., 2009; Hanson et al., 2010; Chung et al., 2012; Ramkumar et al., 2017; Lindner et al., 2020; Nieto-Torres et al., 2021; Yoon et al., 2024), which originally was identified as a microtubule-associated protein in the 1980s (Vallee and Davis, 1983) before its revelation as a mammalian homologue of yeast ATG8 in 2000 (Kabeya et al., 2000). Also, autophagy receptors such as p62 have non-autophagic functions (Moscat et al., 2007; Moscat et al., 2016). Together, this has important implications for the interpretation of results that are based on methods which employ ATGs like LC3 or other markers to explore the function of autophagy, as described further below.
3 Methods to monitor autophagy
In order to critically review the role of autophagy in prostate cancer formation and therapy, it is essential to be aware of the principles of various methods that are used to monitor autophagy, and the difference between them. To that end, we here provide a brief overview of various autophagy assays, with key references and a particular emphasis on methods that have been employed in publications that specifically have explored the role of autophagy in prostate carcinogenesis and prostate cancer therapeutics.
In the first era of autophagy research (1950s–1980s), autophagy was predominantly assessed by morphometric analyses of electron microscopy images. To date, this remains one of the gold standard methods. However, since autophagy is a dynamic process, determination of autophagic activity requires quantitative analyses of cargo flux through the pathway. In the late 1970s and 80s, Per O. Seglen identified the first autophagy inhibitors and pioneered the development of functional assays to quantify bulk autophagic flux (Seglen et al., 1979; Seglen and Gordon, 1982; Kopitz et al., 1990; Seglen et al., 2009; Seglen et al., 2015), some of which have been revived, optimized, and extensively validated by us, e.g., the LDH sequestration assay (Luhr et al., 2018a) and the long-lived protein degradation assay (Luhr et al., 2018b) (Figure 3). Later, genetically engineered cargo reporters, such as those that utilize the fluorescent coral protein mKeima, have been developed to monitor both bulk and selective autophagy (Katayama et al., 2011; An and Harper, 2018; Engedal et al., 2022) (Figure 3).
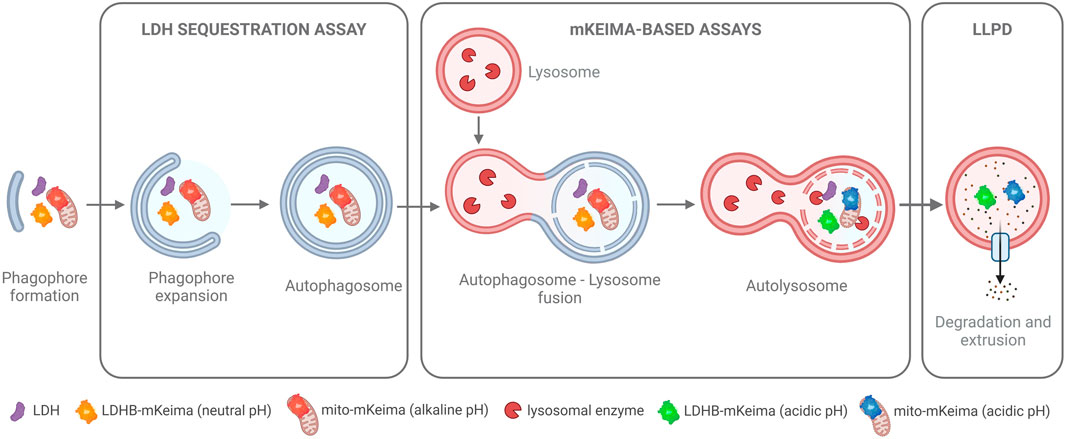
Figure 3. Functional Autophagy Assays. An overview of cargo-based methods to measure autophagy at different steps of the pathway. The LDH sequestration assay measures the sequestration of the ubiquitously expressed, soluble cytosolic protein lactate dehydrogenase (LDH) into autophagosomes. The mKeima-based assays measure the autophagic flux of either cytosol (with the LDHB-mKeima probe) or of specific cargo (e.g., of mitochondria as illustrated here with the mito-mKeima probe). Upon flux of the cargo probes to the acidic environment of the autolysosome, mKeima, which is resistant to lysosomal enzymes, shifts its fluorescence excitation maximum, thus allowing detection and ratiometric quantification of the autophagic flux of the cargo probes. The long-lived protein degradation (LLPD) assay is an endpoint assay that utilizes radiolabeling of cellular proteins to track the extrusion of amino acid products from autolysosomes into the cytoplasm, which occurs as a result of the autophagic degradation of long-lived proteins in the autolysosomes. For further details, see the main text and references within.
Although cargo-based autophagy methods thus have been re-introduced to autophagy research, the last two decades have been overwhelmingly dominated by the use of autophagic markers to monitor autophagy (Figure 2)—primarily LC3 and secondarily p62. Cellular LC3-II levels can be assessed by Western blotting, since LC3-II travels quicker through SDS-PAGE gel than its cytosolic counterpart (LC3-I). Accumulation of LC3-II on phagophores and autophagosomes can also be detected by fluorescence microscopy in the form of “LC3-positive puncta.” Whilst most phagophore-interacting proteins dissociate during autophagosome formation, LC3s and GABARAPs stay attached to the inner autophagosome membrane throughout the pathway until they are degraded in the autolysosomes. Similarly, p62, which is an autophagy receptor that on the one hand interacts with LC3s and GABARAPs and on the other hand can interact with specific cargo, stays associated with the inner autophagosome membrane until degraded in the autolysosomes. Therefore, by assessing LC3-II or p62 levels or LC3- or p62-positive puncta in the absence or presence of lysosomal inhibitors such as Bafilomycin A1 (Baf A1; blocks the lysosomal proton pump and thereby neutralises lysosomal pH and therefore autolysosomal function), Chloroquine (CQ; weak base that temporarily neutralises lysosomal pH and blocks autophagosome-lysosome fusion (Lu et al., 2017; Mauthe et al., 2018)), or E64d + Pepstatin A (PepA) (lysosomal protease inhibitors), the autophagic flux of LC3 and p62 can be inferred. Such measurements are frequently used as proxies for autophagic cargo flux. However, there are a number of important limitations and potential confounding factors in the use of autophagic markers like LC3 and p62 in assessing autophagic activity (Klionsky et al., 2021) including potential influences from transcriptional and translational regulation of LC3 and p62 (Engedal et al., 2013; Klionsky et al., 2021), conjugation of LC3 to other membrane compartments than phagophores/autophagosomes (Malhotra, 2013; Pimentel-Muiños and Boada-Romero, 2014; Durgan et al., 2021), conjugation of LC3 to proteins (termed “protein ATG8ylation”) (Carosi et al., 2021), puncta formation due to LC3/p62 aggregation in the absence of phagophores (Szeto et al., 2006; Kuma et al., 2007; Runwal et al., 2019), a high degree of uncertainty in the correlation between LC3/p62 marker flux versus actual cargo flux, and that LC3 and p62 have many other cellular functions than autophagy (Moscat et al., 2007; Baisamy et al., 2009; Hanson et al., 2010; Chung et al., 2012; Moscat et al., 2016; Ramkumar et al., 2017; Lindner et al., 2020; Kumar et al., 2021; Nieto-Torres et al., 2021; Yoon et al., 2024) and are not always necessary for autophagy (Sala et al., 2014; Szalai et al., 2015; Xu et al., 2015; Nguyen et al., 2016; Vaites et al., 2018; Luhr et al., 2019; Sønstevold et al., 2021). Therefore, the use of autophagic markers alone is insufficient for drawing solid conclusions about autophagic activity (Klionsky et al., 2021). Indeed, upon direct comparison of cargo-based versus LC3-based assays, we have identified conditions where LC3 flux measurements show a complete lack of positive correlation with actual autophagic cargo flux in mammalian cells (Szalai et al., 2015; Sønstevold et al., 2021).
Alterations in autophagy may also be assessed by dyes that stain autophagic vacuoles, such as the acidotropic dyes monodansylcadaverine (MDC), its derivative Monodansylpentane (also referred to as “AUTOdot”), and acridine orange, or with the amphiphilic dye “CYTO-ID”. In general, these dyes display a substantial lack of specificity towards autophagic vacuoles and are not recommended to be used on their own to monitor changes in autophagy (Klionsky et al., 2021).
Various autophagy modulators (inhibitors and activators) are used as tools in the autophagy field. Those mentioned in this review are listed in Table 1. For further reading, methods to monitor autophagy are excellently presented and referenced in the fourth Edition of the autophagy community’s “Guidelines for the use and interpretation of assays for monitoring autophagy” (Klionsky et al., 2021).

Table 1. Overview of autophagy-modulating drugs mentioned in this review.
4 The role of autophagy in cancer—a brief overview
Traditionally, autophagy has been viewed as a pathway that enables the survival of cells, in particular under starvation or energy deprivation conditions. This is ascribed to its function in nutrient recycling and metabolic adaptation (Lum et al., 2005; Onodera and Ohsumi, 2005; Lecker et al., 2006; Poillet-Perez et al., 2018). It has been suggested that autophagy plays a tumour suppressive role in non-transformed cells via removing damaged organelles, reactive oxygen species, and misfolded proteins, which otherwise could promote cellular transformation (Gozuacik and Kimchi, 2004; Mizushima, 2007; Ávalos et al., 2014; Das et al., 2020). This is in line with early work from the group of Per O. Seglen, who almost 40 years ago demonstrated that autophagy is downregulated during liver carcinogenesis in rats (Schwarze and Seglen, 1985; Kisen et al., 1993), marking the first documented association between autophagy and cancer. A putative genetic link was identified ∼15 years later by Beth Levine’s lab who showed that monoallelic loss of Becn1 (homologue of yeast ATG6) can contribute to several types of malignancies in mice (Liang et al., 1999; Qu et al., 2003). Another ∼12 years later, the group of Noboru Mizushima observed development of benign liver tumours in mice with either mosaic deletion of Atg5 or liver-specific deletion of Atg7 (Takamura et al., 2011). The tumour cells in these mice display swelling of the mitochondria, accumulation of p62, oxidative stress and genomic damage responses (Takamura et al., 2011), indicating the global effect a defect in autophagy can have on cellular transformation. In humans, loss of BECN1 is limited to a few cancer types, like those of the breast and ovary, and co-occurs with the loss of BRCA1 (Laddha et al., 2014). Furthermore, at a general level, copy-number alterations or mutations in ATG genes are very rare across human cancers (Lebovitz et al., 2015). Therefore, cancer-related changes in autophagic activity levels predominantly have other underlying causes, which are yet to be defined. Although the above-mentioned and other studies (Galluzzi et al., 2015) support the notion that autophagy suppresses malignant transformation, conclusive encompassing evidence is still lacking, in part due to the increasing realisation of the important non-autophagic functions of the ATGs.
With the progression of cancer, it has been proposed that autophagy proficiency might be restored and elevated in the cancer cells, and/or in other cells in the tumour microenvironment and host tissues, to fulfill the metabolic needs of the tumour cells to survive and grow (Galluzzi et al., 2015; Sousa et al., 2016; Katheder et al., 2017). Additionally, autophagy may be utilized by the cancer cells to survive therapeutic insults (Moosavi and Djavaheri-Mergny, 2019; Chang and Zou, 2020; Mele et al., 2020; Vempati and Malla, 2020; Ahmadi-Dehlaghi et al., 2023). Conversely, it has been reported that autophagy can play tumour suppressive roles also in established tumours. For instance, autophagy may enhance anti-tumour immune responses (Zhong et al., 2016), and limit cancer metastasis (Marsh et al., 2021). Moreover, autophagy may contribute to cell death under various conditions, including in response to some types of anti-cancer therapies (Linder and Kögel, 2019). In sum, the function of autophagy in cancer appears to be highly complex and may have different roles in different cancer types, cancer stages and conditions.
5 Autophagy in prostate carcinogenesis
It is unclear whether autophagy levels are altered during prostate tumorigenesis and how that may affect cancer development and progression. However, analyses of autophagic markers and links between expression levels of ATGs and prostate carcinogenesis have been explored in various settings, including patient tissue, mouse models, and cell lines. Moreover, studies have used marker-based assays to investigate how major genetic drivers of prostate carcinogenesis, e.g., androgens and TP53, affect autophagy. These studies are discussed below (in Section 5.1 and 5.2, respectively).
5.1 Links between expression levels of ATGs and prostate carcinogenesis
An early study indicated loss of heterozygosity of the BRCA1 loci and other loci on chromosome 17q in human prostate cancer (Gao et al., 1995), suggesting monoallelic loss of BECN1, which is located at 17q23.31 (Liang et al., 1999) (Table 2). However, a more detailed study on a larger set of patients (from the TCGA cohort) revealed that loss of BECN1 is a very rare event in prostate cancer (Laddha et al., 2014). Moreover, Becn1+/−mice do not develop prostate tumours, unlike the spontaneous generation of liver and lung carcinomas observed in these mice (Liang et al., 1999; Qu et al., 2003). On the other hand, in a genetically engineered mouse model of prostate cancer with inducible prostate-specific deficiency in the Pten tumour suppressor gene (which is frequently lost in human prostate cancers), combined prostate-specific deficiency in Atg7 led to a delay in prostate tumour development and growth of both primary and castrate-resistant tumours (Santanam et al., 2016). These results may indicate a tumour promoting role of autophagy in prostate carcinogenesis. However, before concluding this, it is imperative to examine the effects of depletion of several other ATGs, as well as their effect in different models of prostate cancer. It is important to note that ATG7 has several non-autophagic functions (DeSelm et al., 2011; Lee et al., 2012; Chen et al., 2023; Deng et al., 2023), and in relation to this, an alternative explanation for the delayed development and growth of the Pten deficiency-induced mouse prostate tumours mentioned above (Santanam et al., 2016), could for instance, be related to the autophagy-independent effect that ATG7 has upon binding to the tumour suppressor TP53, which results in enhanced apoptotic cell death in the absence of Atg7 (Lee et al., 2012). It should also be noted that prostate cancer has not been associated with alterations in ATG7 expression or function.

Table 2. Overview of published studies on the role of autophagy in prostate carcinogenesis.
In terms of cell line studies, 3 human prostate cancer cell lines are predominantly utilized in the prostate cancer field, namely, the hormone-responsive LNCaP cells (originating from a lymph node metastasis, but retaining many of the phenotypes of primary disease) and the hormone-insensitive cell lines PC3 and DU145 (originating from bone and brain metastases, respectively). LNCaP cells have less aggressive traits than PC3 and DU145 in terms of poorer in vitro proliferation, migration and invasion abilities, as well as lower tumorigenicity and metastatic potential in mouse models (Sobel and Sadar, 2005; Shi et al., 2022). PC3 and DU145 cells are AR-negative (Sobel and Sadar, 2005) and therefore not prototypic CRPC cells, since clinically developed CRPC cells predominantly retain AR expression and activity. CRPC versions of LNCaP cells have been developed from LNCaP mouse xenografts, where resistant xenograft tumour cells that appeared after castration have been termed C4 (Wu et al., 1994). A second round of xenografting, where C4 cells were xenografted in castrated mice produced castration-resistant cells with metastatic potential, termed C4-2. Xenografting of C4-2 cells in castrated mice produced tumour cells that had metastasised to bone and are termed C4-2B (Thalmann et al., 1994). The LNCaP-C4 model resembles the clinical progression from androgen-dependence to castration-resistance, where the CRPC C4 cell lines retain AR expression and activity (Dehm and Tindall, 2006; Decker et al., 2012). More recently, the VCaP cell line was generated from a vertebral metastasis of a patient with CRPC (Korenchuk et al., 2001). VCaP is androgen-responsive and expresses high amounts of AR (Korenchuk et al., 2001; van Bokhoven et al., 2003).
Interestingly, due to alternative ATG5 mRNA splicing, DU145 cells do not express any functional ATG5 protein, and therefore display autophagy defects as assessed by LC3/p62 Western blotting, LC3 immunostaining, and LDH sequestration (Ouyang et al., 2013; Luhr et al., 2018a; Wible et al., 2019; Peng et al., 2021). It has been proposed that the defective autophagy is a key contributing factor to the highly migratory and metastatic phenotype of DU145 cells, since stable knockdown of ATG5 in LNCaP cells, which reduced LC3-II levels and elevated p62 protein levels, led to increased LNCaP cell migration, invasion and wound healing in vitro, and a marked appearance of LNCaP lung metastases in an in vivo NOD/SCID mouse model (Shi et al., 2022). This indicates that loss of autophagy in prostate cancer cells promotes the metastatic process and thus prostate cancer progression. However, since ATG5 has several highly cancer-relevant non-autophagic functions (Yousefi et al., 2006; Maskey et al., 2013; Guo et al., 2017; Li et al., 2021), it will be essential to evaluate whether the increase in migration, invasion and metastasis that was observed upon ATG5 knockdown also occurs upon knockdown of other central ATGs. Moreover, the proposal above is apparently in conflict with the results from another study, which reported that overexpression of wild type HA-tagged ATG5 in DU145 cells, which restored LC3 lipidation, LC3-II flux (as assessed by LC3 WB ± CQ) and LC3 puncta formation, led to enhanced in vitro cell proliferation and migration as assessed by wound healing (Peng et al., 2021). It remains to be determined how overexpression of HA-ATG5 affects the non-autophagic functions of ATG5, as well as the effect the overexpression will have on other assays for migration and invasion in vitro, and on metastasis in in vivo models. It also remains to be determined whether different degrees of ATG5 overexpression would have different cellular effects. Of relevance, two reports have indicated that ATG5 protein is frequently highly overexpressed in prostate tumours compared to normal prostate tissue (Kim et al., 2011; Kharaziha et al., 2015). The overexpression did not correlate with pathologic parameters such as Gleason grade, and no mutations in ATG5 were found (Kim et al., 2011). Of note, however, loss of ATG5 was observed in 18% of the castration-resistant patients in one of the studies (Kharaziha et al., 2015). Although a rather limited number of patient samples were included in the latter report (only 28 samples were quantified), overall, the results warrant additional studies on the deregulation of ATG5 protein expression in prostate cancer. At the mRNA level, TCGA data reveal that the average expression of full-length ATG5 mRNA is increased in prostate tumours compared to normal prostatic tissue (Wible et al., 2019). However, also at the mRNA level, a small subset of patients show reduced or complete loss of ATG5 (Wible et al., 2019).
As opposed to that observed for ATG5, high tumour protein expression of ULK1 has been found to significantly associate with biochemical recurrence following radical prostatectomy of prostate cancer patients (Liu et al., 2015). Similarly, another study found that high concomitant expression of ULK1 and the mitochondrion-associated protein LRPPR correlated with serum prostate-specific antigen (PSA) levels, Gleason score, PSA levels following ADT, and the amount of metastatic lesions (Zhang et al., 2017). However, it is unclear whether these associations are related to alterations in autophagy levels or not, since changes in the expression of other ATG proteins (ATG5, BECLIN1, LC3B, or ATG9) did not show any significant correlations with any clinicopathological parameters (Liu et al., 2015; Zhang et al., 2017). Moreover, ULK1 exerts a number of non-autophagic functions (Joo et al., 2016; Joshi et al., 2016; Wang and Kundu, 2017; Saleiro et al., 2018; Wu et al., 2020; Rajak et al., 2022; Liang et al., 2023), which may be altered upon its increased expression, and overexpression of ULK1 may, somewhat counterintuitively, actually result in reduced autophagy levels (Chan et al., 2007). Correlations between ULK1 tumour expression levels and clinical parameters are therefore very difficult to interpret.
A potential role of decreased autophagy in promoting prostate cancer progression has been suggested through correlations between low prostate tumour expression of the core ATG genes FIP200, ATG16L1, or GABARAPL1 and poor prognosis in clinical prostate cancer cohorts (Li et al., 2014; Huang et al., 2015; Su et al., 2017). It is important to note also here that it remains to be determined whether, and to what extent, the decreased expressions observed translate into alterations in functional autophagy. Of note, however, we have previously found that knockdown of FIP200 strongly reduces functional autophagy in LNCaP cells, as assessed by the LDH sequestration and long-lived protein degradation assays (Seglen et al., 2015; Luhr et al., 2018a). Furthermore, FIP200 silencing with an siRNA oligo was reported to promote cell cycle progression and reduce cell death in LNCaP, suggesting a tumour suppressive function of autophagy in these cells, in concordance with the observed increased biochemical recurrence in prostate cancer patients who had undetectable FIP200 protein expression in their tumours (Li et al., 2014). It is however very important to note that FIP200 has various non-autophagic functions that are highly relevant to both cell proliferation and cell death (Gan and Guan, 2008; Chen et al., 2016).
5.2 Links between genetic drivers of prostate carcinogenesis and autophagy
Androgens, which play a crucial role in prostate carcinogenesis, have been shown to increase LC3-II levels and GFP-LC3 puncta, as well as mCherry-GFP-LC3 flux in LNCaP and VCaP cells (Shi et al., 2013). Moreover, androgen-stimulated cell proliferation was sensitive to the autophagy-inhibiting drugs Baf A1, 3-Methyladenine (3MA) and CQ, or siRNA-mediated knockdown of ATG7 (Shi et al., 2013). 3MA is a pan-PI3K inhibitor that blocks autophagy through PI3K class III inhibition (Seglen and Gordon, 1982; Blommaart et al., 1997). In a follow-up study (Blessing et al., 2017), androgens were found to increase the expression of ATG4B, ATG4D, ULK1 and ULK2, as well as the transcription factor TFEB, which regulates genes involved in autophagy and lysosomal biogenesis. In clinical prostate cancer cohorts, a combined gene expression score of these 5 genes was increased in metastatic disease. Moreover, prostate adenocarcinoma patients from the TCGA dataset displaying a tumour mRNA expression level greater than 2-fold above the mean of either ATG4B, ATG4D, ULK1, or ULK2, had poor prognosis. Finally, knockdown of TFEB or any of the 4 ATG genes diminished androgen-mediated proliferation of prostate cancer cell lines, and overexpression of any of the 5 genes increased cell proliferation independently of androgens (Blessing et al., 2017). These findings suggest that androgens promote prostate cancer growth and progression via an AR-mediated increase in autophagy. However, it remains to be determined whether alterations in the expression of any of the 5 genes identified lead to changes in functional autophagic activity. For instance, there is a high degree of redundancy among ATGA, ATG4B, ATG4C and ATG4D (Agrotis et al., 2019), and overall autophagic flux appears to be unchanged in Atg4D knockout mice (Tamargo-Gómez et al., 2021). Furthermore, ULK2 is not always necessary for autophagy (due to redundancy with ULK1), and, as mentioned above, overexpression of ULK1 can lead to inhibition of autophagic activity (Chan et al., 2007). Adding to the complexity, other studies performed in prostate cancer cell lines reported that AR efficiently blocks mTOR-inhibitor-induced generation of autophagosomes (Jiang et al., 2012), and in another study, androgens were found to decrease serum starvation-induced LC3-II flux and mRFP-GFP-LC3 flux via upregulation of the endoplasmic reticulum chaperone glucose-regulated protein 78/BiP, with the effect of AR being linked to counteraction of starvation-induced cell death (Bennett et al., 2010). This suggests an autophagy-inhibitory role of AR, which might promote prostate cancer progression by preventing excessive, cell death-promoting autophagy during nutrient limitation in the tumour microenvironment (Eisenberg-Lerner et al., 2009; Bennett et al., 2010).
Loss of TP53 or acquisition of TP53 mutations has previously been implicated in driving metastatic castration-resistant prostate cancer (Gundem et al., 2015; Hong et al., 2015). Further research has indicated an association between TP53 loss-of-function mutations and increased levels of LC3B, ULK1 and BECLIN1 in castration-resistant prostate tumour specimens, and androgen-independent tumour organoids were found to be highly vulnerable to the autophagy inhibitors CQ or ULK101 (an ULK inhibitor) in combination with the anti-androgen enzalutamide (Zhang et al., 2022b). It remains to be determined whether and how the changes in LC3B, ULK1 and BECLIN1 expression translates into alterations in autophagic activity or capacity. Moreover, as a general cautionary note, none of the existing autophagy-inhibitory drugs are specific, and thus the observed reduction in organoid growth could alternatively arise from non-autophagy related effects. Additional tests involving genetic interference with different ATGs would help clarify the role of autophagy in this setting.
In summary, there are several indications that alterations in autophagy may strongly affect the development and progression of prostate cancer, but more research is needed to elucidate its specific effects, as well as to whether, and in which contexts, autophagy plays a tumour suppressor or pro-oncogenic role during prostate carcinogenesis.
6 Autophagy in prostate cancer therapeutics
Several studies have explored the role of autophagy in prostate cancer therapy, particularly in the context of the two main non-surgical treatments for advanced prostate cancer; ADT and radiation, but also with regards to chemo- and targeted therapeutics. For an overview, see Table 3 and the remainder of this chapter.
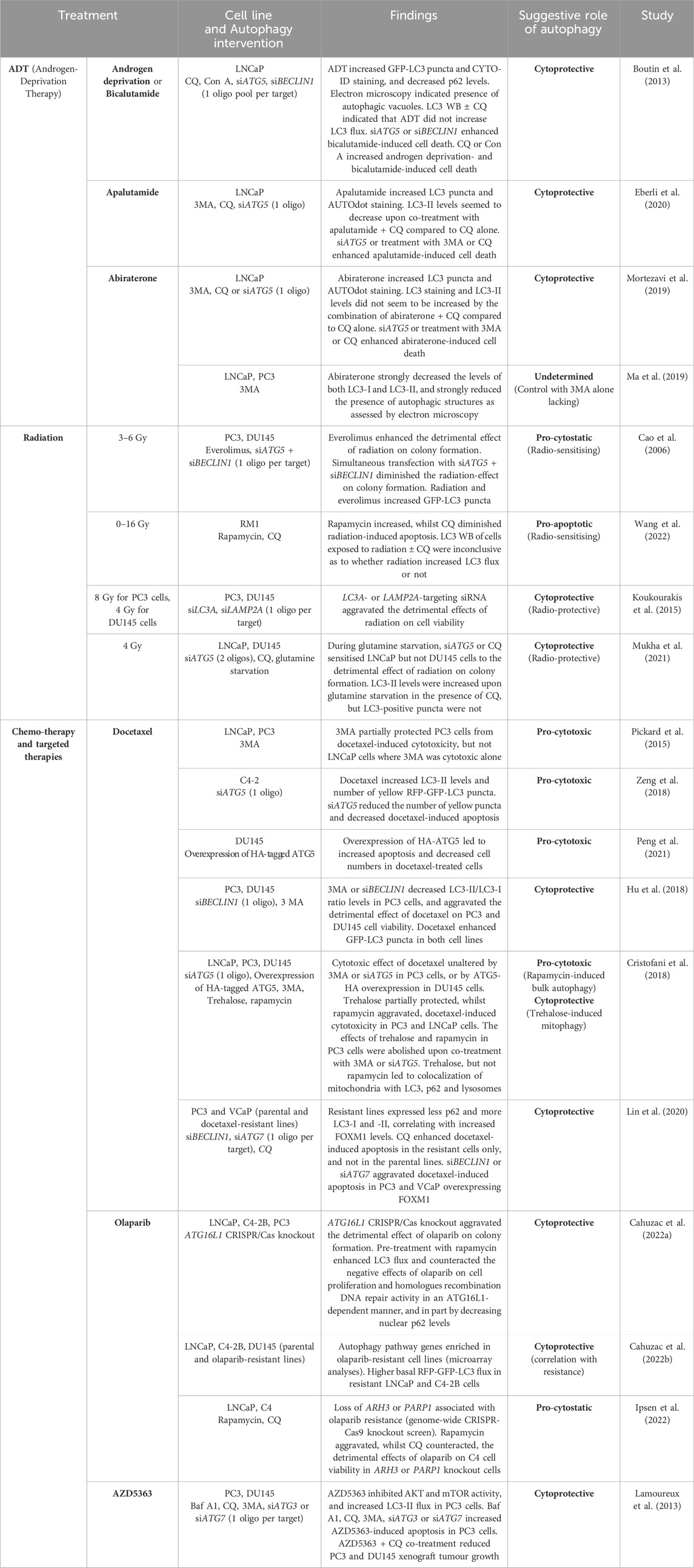
Table 3. Overview of published studies on the role of autophagy in prostate cancer therapeutics.
6.1 Androgen deprivation therapy (ADT)
In the androgen-responsive LNCaP prostate cancer cell line, it was found that androgen deprivation or treatment with the anti-androgen bicalutamide (an AR antagonist) led to accumulation of GFP-LC3 puncta and the presence of double- and multi-membrane structures, as assessed by electron microscopy (Boutin et al., 2013). Moreover, LC3-II/LC3-I ratio levels were increased by androgen deprivation or bicalutamide treatment in the presence of the lysosomal protease inhibitors E64d and PepA. In further support of autophagy promotion, androgen deprivation or bicalutamide treatment decreased p62 levels in an E64d/PepA-sensitive manner, and enhanced CYTO-ID fluorescent staining. In contrast, bicalutamide failed to increase LC3-II levels or the LC3-II/LC3-I ratio in the presence of CQ (Boutin et al., 2013), indicating that bicalutamide does not increase LC3 flux. It should be noted that in these experiments, the treatment with E64d/PepA or CQ lasted for 48 h which can lead to many indirect and non-specific effects in the cells and thereby affect the results (Klionsky et al., 2021). Moreover, no functional autophagy assays were included, and it is uncertain whether the cyto-ID assay specifically and reliably reflects autophagic activity. Therefore, additional experiments are required to draw more firm conclusions on the effect of androgen deprivation or bicalutamide on autophagic activity in LNCaP cells. A protective role of autophagy in bicalutamide-treated LNCaP cells was suggested by the observation that siRNA-mediated knockdown of ATG5 or BECLIN1, which decreased the number of GFP-LC3 puncta and the LC3-II/LC3-I ratio, led to enhanced bicalutamide-induced cell death (Boutin et al., 2013). Moreover, CQ or Concanamycin A (Con A; acts like Baf A1 to block the lysosomal proton pump and thereby neutralise lysosomal pH) increased cell death in both androgen-deprived and bicalutamide-treated cells (Boutin et al., 2013). Thus, autophagy may serve to prevent or limit ADT-induced cell death in LNCaP cells. As a cautionary note, it should be mentioned that CQ and Con A may promote cell death independently of autophagy inhibition. Moreover, siRNAs may promote cell death via off-target effects, and therefore results from using only one siRNA (or only one pool of siRNAs) per target are uncertain without being accompanied by rescue experiments.
Similarly, it has been reported that treatment with 3MA or CQ, or transfection with an ATG5-targeting siRNA led to enhanced cell death in LNCaP cells exposed to the AR-antagonistic anti-androgen apalutamide (Eberli et al., 2020) or the androgen biosynthesis inhibitor abiraterone (Mortezavi et al., 2019). While the conclusion about the potential protective role of autophagy in these conditions are associated with the same uncertainties as mentioned above, the effects of apalutamide or abiraterone on autophagic activity in LNCaP cells also remain unclear thus far, since functional autophagy assays have not been used, and inconsistent and seemingly contradictory results have been obtained with non-functional assays. Thus, while LC3 puncta and AUTOdot staining was increased upon treatment with apalutamide or abiraterone in LNCaP cells (Mortezavi et al., 2019; Eberli et al., 2020), LC3-II levels seemed to be decreased upon co-treatment of apalutamide or abiraterone with CQ compared to that observed with CQ alone (Mortezavi et al., 2019; Eberli et al., 2020), and immunofluorescent LC3 staining did not seem to be increased by the combination of abiraterone + CQ compared to CQ alone (Mortezavi et al., 2019). Monitoring autophagy with LC3 or other markers is associated with variation and many potentially confounding effects (Klionsky et al., 2021). Moreover, the AUTOdot dye (Monodansylpentane), which stains lipid droplets (Chen et al., 2017), lacks specificity to autophagy. Further illustrating the apparently conflicting results that can be obtained when using non-functional autophagy assays, another study reported that treatment of LNCaP or PC3 cells with abiraterone strongly decreased the levels of both LC3-I and LC3-II, and nearly abolished the presence of autophagic structures as assessed by electron microscopy (Ma et al., 2019). Whilst these findings contradict those described above on the effect of abiraterone in LNCaP cells (Mortezavi et al., 2019), and are opposite of the ADT-effects observed with androgen deprivation or bicalutamide in LNCaP cells (Boutin et al., 2013), it remains to be determined whether the changes observed with non-functional assays are a result of increased or decreased autophagic cargo flux. In summary, the effect of ADT on autophagy in prostate cancer cells remains to be fully characterised, and whilst there are indications that autophagy may protect against ADT-induced cell death, such studies should be followed up by knockdown (or knockout) of many different ATGs and with more than one siRNA (or guide RNA) per target, to control for the effect of abolishing non-autophagic functions of ATGs and for off-target effects.
6.2 Radiation therapy
The role of autophagy in radiation treatment of prostate cancer cells has also been investigated. One of the main actions of therapeutic ionizing radiation is to damage genomic DNA, leading to defective cancer cell proliferation and potentially cell death. In regards to DNA damage, autophagy is able to either directly or indirectly regulate genomic stability through modulation of reactive oxygen species (Karantza-Wadsworth et al., 2007; Mathew et al., 2007; Hewitt and Korolchuk, 2017), through directly targeting double stranded break repair-associated proteins (Hewitt et al., 2016; Wang et al., 2016; Sharma et al., 2018) or through the selective degradation of nuclear components (Park et al., 2009; Dou et al., 2015). However, other mechanisms related to autophagy may lead to increased sensitivity to radiation (Roy et al., 2022). In line with the latter, the rapamycin derivative everolimus, which is expected to activate autophagy via direct mTOR inhibition, enhanced the effect of radiation in reducing the ability of PC3 and DU145 prostate cancer cells to form colonies from single cells (Cao et al., 2006). Furthermore, simultaneous knockdown of ATG5 and BECLIN1 rendered radiation less efficient in reducing colony formation (Cao et al., 2006). Although everolimus and radiation increased the number of GFP-LC3 puncta, no LC3 flux measurements or functional autophagy assays were included, and thus it remains to be assessed whether autophagic activity is increased by the treatments in these cells. Furthermore, it was not tested whether the knockdown of ATG5 and BECLIN1 was successful in reducing autophagic activity. In relation to findings in the DU145 cell line, it is important to keep in mind that these cells are defective in ATG5-dependent autophagy, as they do not express any functional ATG5 protein due to alternative ATG5 mRNA splicing (Ouyang et al., 2013; Wible et al., 2019; Peng et al., 2021). Hence, although the radiosensitising and radioprotective effects of everolimus and ATG5+BECLIN1 knockdown, respectively, were more pronounced in PC3 cells than in DU145 cells (Cao et al., 2006), at least part of the effects is likely to be unrelated to changes in autophagy. In support of the notion that autophagy may contribute to the radiosensitivity of prostate cancer cells, rapamycin was found to elevate radiation-induced apoptosis in RM1 murine prostate cancer cells, whilst CQ rendered the cells less sensitive to radiation (Wang et al., 2022). These effects were associated with downregulated expression of the deacetylase SIRT1 upon radiation, which was aggravated by rapamycin and diminished by CQ (Wang et al., 2022). LC3 Western blotting of RM1 cells treated with radiation ± CQ were inconclusive in determining whether radiation increased LC3 flux or not (Wang et al., 2022). In contrast to the two studies mentioned above, another report proposed that autophagy contributes to radioresistance in prostate cancer cells, since siRNA-mediated knockdown of LC3A or LAMP2A aggravated the detrimental effects of radiation on cell viability in PC3 and DU145 cells (Koukourakis et al., 2015). It is difficult to ascertain whether radiation altered autophagic activity or not in this study, since changes in autophagy were assessed by LC3A/LAMP2A localization and steady-state measurements of LC3A-II, LAMP2A and p62 levels, without the use of any flux measurements or functional autophagy assays. It is also unclear whether LC3A or LAMP2A knockdown inhibited autophagy, as the effects of the LC3A- and LAMP2A-targeting siRNAs on autophagic activity were not tested. Furthermore, the ability of the two siRNAs to also radiosensitise DU145 cells, which lack functional ATG5, suggest that their effects may be unrelated to alterations in autophagy. However, another study found that during glutamine starvation, two different ATG5-targeting siRNAs sensitised LNCaP but not DU145 cells to the effects of radiation on colony formation (Mukha et al., 2021). The authors proposed that glutamine starvation activates radioprotective autophagy. However, LC3-based assays were inconclusive in determining whether glutamine starvation induced LC3 flux in LNCaP cells, since LC3-II levels were increased upon glutamine starvation in the presence of CQ, but LC3-positive puncta were not. Functional autophagy assays were not used. Moreover, the effect of radiation on autophagy and the result of knocking down other ATGs than ATG5 on radiosensitivity were not assessed. The influence of ATG5 knockdown on radiosensitivity in the presence of glutamine was also not tested. Taken together, the published reports on the role of autophagy in prostate cancer radiotherapy are incomplete and partly contradictory. More research is required to clarify i) how radiation affects autophagic activity and ii) whether autophagy promotes or counteracts the detrimental effects of radiation in prostate cancer cells.
6.3 Chemotherapy and targeted therapeutics
6.3.1 Chemotherapy: Docetaxel
Docetaxel is a chemotherapeutic drug in the class of taxanes, which inhibits microtubule depolymerization and thereby blocks cancer cell division and promotes cell death. Docetaxel is the mainstay first-line treatment of metastatic, castration-resistant prostate cancer. However, limited therapeutic efficacy and development of treatment resistance seriously restrict and shorten the clinical benefits. Several studies have examined how autophagy may influence the effects of docetaxel on prostate cancer cells. One study observed that 3MA partially protected PC3 cells from docetaxel-induced cytotoxicity, but not LNCaP cells, where 3MA was substantially cytotoxic by itself (Pickard et al., 2015). This suggested a pro-cytotoxic role of autophagy in docetaxel-treated cells. In line, another study found that docetaxel-induced cytotoxicity, caspase activity and apoptosis in C4-2 cells was dampened upon transfection with an ATG5-targeting siRNA (Zeng et al., 2018). Docetaxel increased LC3-II levels and the number of yellow puncta in C4-2 cells expressing RFP-GFP-LC3. Whilst siATG5 strongly reduced the number of yellow puncta, the effect of docetaxel on LC3 flux was not assessed. A pro-cytotoxic role of autophagy in docetaxel-treated cells was likewise indicated in third study, which observed that docetaxel-induced apoptosis and decrease in cell numbers were strongly increased upon overexpression of HA-tagged ATG5 in DU145 cells (Peng et al., 2021). The authors verified that HA-ATG5 overexpression restored LC3 lipidation and flux in the DU145 cells. In contrast, a fourth study reported that treatment of PC3 cells with 3MA or a BECLIN1-targeting siRNA, which decreased LC3-II/LC3-I ratio levels, aggravated the detrimental effect of docetaxel on cell viability (Hu et al., 2018). However, very similar results were obtained in the ATG5-deficient DU145 cell line, which suggests that the observed effects of 3MA and the BECLIN1-targeting siRNA on docetaxel-induced cytotoxicity may have been unrelated to changes in autophagy. Of note, docetaxel was reported to strongly enhance GFP-LC3 puncta in both cell lines, indicating that this was an ATG5-independent phenomenon. In PC3 cells, docetaxel slightly increased LC3-II levels in the presence of CQ, indicating a modest induction of LC3 flux (Hu et al., 2018).
Inconsistent with the four papers described above, a fifth study observed that the detrimental effect of docetaxel on cell viability in PC3 cells was unaltered by treatment with 3MA or transfection with an ATG5-targeting siRNA (Cristofani et al., 2018). Moreover, docetaxel-induced cytotoxicity was unchanged upon overexpression of HA-tagged ATG5 in DU145 cells. Docetaxel increased the mRNA and protein expression levels of both LC3 and p62, but no flux measurements were performed. In either case, the putative ability of docetaxel to activate or inhibit autophagy was apparently not sufficient to alter the cytotoxic effects of docetaxel. However, the disaccharide trehalose, which has been proposed to induce autophagy independently of mTOR inhibition (Sarkar et al., 2007), was found to partially protect PC3 and LNCaP cells, but not DU145, from docetaxel. In contrast, the mTOR inhibitor rapamycin aggravated the detrimental effects of docetaxel in PC3 and LNCaP cells, but not in DU145. The effects of trehalose and rapamycin in PC3 cells were abolished upon co-treatment with 3MA or transfection with an ATG5-targeting siRNA. Treatment with trehalose, but not rapamycin, led to colocalization of mitochondria (MitoTracker) with LC3, p62 and lysosomes (LysoTracker). This led the authors to propose that trehalose induced its cytoprotective effect via autophagic degradation of mitochondria (mitophagy), whereas rapamycin promoted cell death via bulk autophagy. It should be noted that the authors only used one ATG5-targeting siRNA oligo, and that they did not test whether the siATG5 reduced trehalose-induced mitochondrial colocalization with LC3, p62 or lysosomes. Moreover, the authors did not examine whether treatment with trehalose led to completion of the mitophagic process, i.e., flux of mitochondria to acidic autolysosomes and their degradation - as opposed to a putative trehalose-mediated block in the fusion of autophagosomes with lysosomes. Of relevance, the autophagy-inducing effect of trehalose has been questioned by data that indicates lack of autophagy induction and instead a trehalose-induced impairment of lysosomal membrane integrity, leading to inhibition of autophagosome–lysosome fusion (Yoon et al., 2017). Finally, a sixth study examined autophagy in partially docetaxel-resistant PC3 and VCaP prostate cancer cells (Lin et al., 2020). Compared to the parental cell lines, the more resistant cells displayed lower expression of p62, and higher expression of LC3-I and -II, along with indications of higher LC3 flux as assessed by Western blotting of CQ-treated cells. Additionally, the resistant lines expressed higher levels of the transcription factor Forkhead box protein M1 (FOXM1), which contributed to resistance as well as to the changes in p62 and LC3 levels. Treatment with CQ led to an increase in docetaxel-induced apoptosis in the resistant, but not the parental cell lines. Moreover, in PC3 and VCaP cells with enforced FOXM1 overexpression, a BECLIN1- or an ATG7-targeting siRNA aggravated docetaxel-induced apoptosis. Together, this study suggests autophagy as a cytoprotective mechanism that contributes to docetaxel resistance. However, since the results from the six studies mentioned here vary considerably and partly contradict one another, it is impossible to draw a consensus conclusion on the role of autophagy in the regulation of docetaxel cytotoxicity and treatment resistance in prostate cancer cells. In order to obtain more decisive evidence, the use of functional autophagy assays, more than one siRNA per target (and/or rescue experiments) and targeting of several different ATGs along with verification of successful interference with functional autophagy, is needed. Very little is known about the role of autophagy during treatment with chemotherapies other than docetaxel in prostate cancer.
6.3.2 Targeted therapy
Clinically approved targeted therapies for prostate cancer comprise various PARP inhibitors (olaparib, niraparib, rucaparib, and talazoparib) and the PSMA-targeted radioligand therapy lutetium (177Lu) vipivotide tetraxetan (Lu-PSMA). There is very limited knowledge about how such therapies affect autophagy in prostate cancer cells and how autophagy may influence their therapeutic efficacy and/or the development of treatment resistance. The exception is for olaparib, where a recent study demonstrated that ATG16L1 CRISPR/Cas knockout sensitised LNCaP, C4-2B and PC3 cells to the detrimental effect of olaparib on colony formation from single cells (Cahuzac et al., 2022a). Furthermore, pre-treatment with rapamycin, which enhanced LC3 flux, counteracted the negative effects of olaparib on cell proliferation and homologues recombination (HR) DNA repair activity in an ATG16L1-dependent manner. This effect of rapamycin was lost in the ATG16L1 KO cells and rescued upon re-introduction of HA-tagged ATG16L1, demonstrating specific relation to ATG16L1 and not to any off-target effect introduced by the knockout. Mechanistically, the effects of rapamycin and ATG16L1 KO were linked to variations in nuclear p62 levels, which displayed an inverse correlation with nuclear levels of the FLNA protein that promotes the recruitment of BRCA1 and Rad51 to DNA break sites. Together, this indicates that autophagy plays a protective role by increasing HR-mediated DNA repair in olaparib-treated cells, which may contribute to olaparib resistance. As a cautionary note, however, the direct link to autophagy was not tested with other autophagy inducers than rapamycin or with knockdown/knockout of other ATG genes than ATG16L1. Moreover, the effect of olaparib on autophagy was not assessed, and since no functional autophagy assays were employed, it remains to be determined whether a fully functional autophagy pathway would be required for the observed effects. In another study from the same group, microarray analyses revealed that autophagy pathway genes were enriched in olaparib-resistant versions of LNCaP, C4-2B and DU145 cells (Cahuzac et al., 2022b). Moreover, resistant LNCaP and C4-2B cells displayed higher basal LC3 flux as assessed upon transient transfection with an RFP-GFP-LC3 reporter. Contrarily, a different study that performed a genome-wide CRISPR-Cas9 knockout screen, identified that loss of ARH3 or PARP1 was associated with olaparib resistance, as well as with reduced basal LC3 flux, in LNCaP and C4 cells (Ipsen et al., 2022). Moreover, in ARH3 or PARP1 knockout cells, rapamycin exacerbated the detrimental effects of olaparib on C4 cell viability, whilst CQ did the opposite. Those results suggested a pro-cytostatic instead of cytoprotective role of autophagy in olaparib-treated prostate cancer cells, albeit validations with more specific modulation of autophagy were not performed. In summary, additional studies, which should include functional autophagy assays and additional ways to activate and inhibit autophagy (by targeting several different ATGs), are required to delineate and further clarify the role of autophagy in the regulation of prostate cancer cells’ sensitivity to olaparib. The potential influence of autophagy on the effects of other PARP inhibitors and Lu-PSMA also remains to be explored.
Among emerging drug-based targeted therapies that are not yet approved for clinical use in prostate cancer patients, the AKT inhibitor AZD5363 (capivasertib) is one of the most promising. AZD5363 was recently FDA-approved for use in combination with fulvestrant in a subset of breast cancer patients with one or more alterations in PIK3CA/AKT1/PTEN (Shirley, 2024). Such alterations, and especially mutations in PTEN, which renders the PI3K-AKT pathway constitutively active, are frequently found in prostate cancer (Choudhury, 2022). Two phase III clinical trials are ongoing for the use of AZD5363 in combination with either docetaxel + prednisolone or abiraterone in prostate cancer patients (Shirley, 2024). AKT inhibitors are also being tested in combination with PARP inhibitors in clinical trials (Choudhury, 2022). It has been suggested that autophagy mediates cytoprotective effects to counteract AZD5363-induced cell death in prostate cancer cells (Lamoureux et al., 2013). In that study, AZD5363 inhibited AKT and downstream mTOR activity in PC3 cells, and increased LC3 flux as assessed by Western blotting in the presence of Baf A1 or CQ. The detrimental effect of AZD5363 on cell viability was exacerbated along with increased caspase activity in PC3 cells co-treated with AZD5363 and Baf A1, CQ or 3MA. Moreover, transfection with an ATG3- or an ATG7-targeting siRNA increased AZD5363-induced apoptosis and caspase activation. Lastly, the authors demonstrated that the growth of PC3 xenograft tumours in mice was strongly reduced upon CQ and AZD5363 co-treatment (Lamoureux et al., 2013). These results suggested that targeting autophagy may enhance the efficacy of AZD5363 in prostate cancer treatment. As a cautionary note, however, it was not assessed whether the knockdown of ATG3 or ATG7 inhibited autophagy (neither with marker-based nor with functional autophagy assays), and since only one siRNA oligo was used per target, it cannot be excluded that potential off-target effects could have influenced the results. Moreover, since very similar effects were observed with the combination of AZD5363 and CQ in DU145 xenografts (which are defective in ATG5-dependent autophagy) as in PC3 xenografts (Lamoureux et al., 2013), the growth-inhibitory effects of CQ under those conditions are, at least in part, likely to be unrelated to inhibition of autophagy.
7 Discussion—limitations of current studies, main challenges and solutions
As evident from the overview and critical examination of current studies above, the role that autophagy may play in prostate carcinogenesis and prostate therapeutics remains highly uncertain. The conclusions drawn in the different studies range from autophagy impeding to promoting prostate cancer formation and progression, as well as from contributing to counteracting the effects of various therapeutic treatments. The same applies to the autophagy in cancer field in general (i.e., across all cancer types). The reason for the apparent paradoxical/dichotomous function of autophagy in cancer is often ascribed to its role being highly context- and cancer type-specific. Whereas this indeed very likely explains some of the apparent contradictory findings, we believe that above all, the main reason lies in the highly underestimated and under-communicated limitations in the methodological approaches that are used in the vast majority of current studies. Two aspects are of particular importance:
(i) First of all, the field is seriously impeded by being extremely reliant on the use of autophagic markers to assess alterations in autophagic activity, with LC3 being the by far most used marker. The problem with this is, as explained in the paragraph on autophagy methods above, that autophagy markers are insufficient and unreliable when used alone (Klionsky et al., 2021). Moreover, in most studies, there is uncertainty as to whether, or to which degree, the various pharmacological or genetic interferences/modulations of autophagy that have been employed have actually had their intended or anticipated effects on autophagic activity. In sum, we believe that this has resulted in many apparent contradictory findings, and, overall, in a critical lack of knowledge on the function of autophagy in cancer and on how autophagic activity changes during carcinogenesis as well as during anti-cancer therapy and development of treatment resistance.
(ii) Secondly, progress in the field is critically impeded by a general underestimation of what is required to firmly demonstrate causal relations between autophagy modulation and cellular/phenotypic effects. Many studies solely used pharmacological means to infer causality, which is associated with a very high degree of uncertainty due to the plethora of non-specific effects that all autophagy-modulating drugs exert on cells. Therefore, in this review, the majority of studies that have been selected from the literature are the ones that use at least one type of genetic interference with autophagy to infer causal relations. However, the extent to which this is necessary in order to firmly prove causality should not be underestimated. The reason for this is firstly that, as opposed to what was assumed upon their initial identification in the 1990s, most, if not all, ATG proteins have non-autophagic cellular functions (Yousefi et al., 2006; Gan and Guan, 2008; Baisamy et al., 2009; Hanson et al., 2010; DeSelm et al., 2011; Chung et al., 2012; Lee et al., 2012; Velikkakath et al., 2012; Maskey et al., 2013; Elgendy et al., 2014; Rohatgi et al., 2015; Chen et al., 2016; Joo et al., 2016; Joshi et al., 2016; Kaizuka and Mizushima, 2016; Nunes et al., 2016; Yang et al., 2016; Guo et al., 2017; Ramkumar et al., 2017; Wang and Kundu, 2017; Cadwell and Debnath, 2018; Saleiro et al., 2018; Galluzzi and Green, 2019; Hu et al., 2020; Lindner et al., 2020; Wu et al., 2020; Fang et al., 2021; Li et al., 2021; Mailler et al., 2021; Nieto-Torres et al., 2021; Sun et al., 2021; Zhang et al., 2022a; Hamaoui and Subtil, 2022; Rajak et al., 2022; Chen et al., 2023; Deng et al., 2023; Liang et al., 2023; Wang et al., 2023; Tedesco et al., 2024; Tran et al., 2024; Yoon et al., 2024) that are likely to influence the cellular/phenotypic effects that are observed upon their knockdown, knockout or overexpression, and many of these functions affect pathways that are highly relevant to cancer. Therefore, in order to infer causality, the same cellular/phenotypic effect must be observed upon interference with a number of different ATGs. Alas, this is very rarely done. Moreover, as mentioned in point (i) above, it is very important to use appropriate autophagy assays to verify that the knockdown or knockout (or overexpression) in question has actually had the intended effect on reducing (or increasing) autophagic activity. It cannot be taken for granted, since there are many different ATGs on which cells may show differential reliance, and on which different cell types may show different dependencies on in relation to cellular autophagic capacity. For instance, even very efficient knockdown of ATG5, whose gene product is one of the most central and critical in macroautophagy, has been shown to be insufficient or inefficient in reducing LC3-II levels in several types of mammalian cells (Hosokawa et al., 2006; Shin et al., 2013; Domagala et al., 2018), whereas ATG5 knockout leads to complete disappearance of LC3-II (Hosokawa et al., 2006; Domagala et al., 2018; Szalai et al., 2015), and lipidated GABARAPs (Szalai et al., 2015). Thus, the fraction of cellular ATG5 protein required to maintain its role in autophagy seems to be very small. The same may not be the case for non-autophagic functions of ATG5. Hence, the possibility exists that ATG5 knockdown may abolish several of the non-autophagic effects of ATG5, while at the same time failing to affect autophagic activity. In the studies discussed in the current review, all, except one study, have maximally interfered with two different ATGs to infer causality. Whilst some studies have not assessed how that affected autophagy (and instead just assumed that autophagy was impaired), others have used autophagic markers, but none have used functional assays to assess the effect of ATG interference on autophagic activity. This is a very important point, since, as mentioned above, autophagic markers are unreliable when used alone. Another important point, which applies to the whole autophagy field, is that few if any studies justify their choice of which ATG(s) they interfere with to infer causality between autophagy and cellular effects/phenotypes. The studies on autophagy in prostate cancer that have been discussed in the current review range from interfering with ULK1, ULK2, ATG3, ATG4B, ATG4D, ATG5, BECLIN1, ATG7, LC3A, ATG16L1, FIP200, TFEB, LAMP2A, or p62. While it is good that many different ATGs or autophagy regulators have been targeted in these studies, the problem is that the various studies have chosen just one or two targets in a seemingly random way to infer causality. Hence, it becomes very difficult to compare the results, since different ATGs and autophagy regulators have different non-autophagic functions, which are also likely to be affected upon knockdown/knockout. Additionally, the importance of using more than one different siRNA oligo or guide RNA and/or rescue experiments to rule out off-target effects should not be underestimated, as most studies discussed in this review have not done so. Finally, attention should be given to the fact that the stability of several of the ATGs depend on one another. For instance, as has been demonstrated in prostate cancer cell lines, knockout of ATG7 leads to a severe depletion of ATG5 and ATG12 protein levels (which is at least in part due to ATG7 being required for ATG5-ATG12 conjugation) and partial depletion of ATG16L1 protein expression, whilst knockout of ATG5 leads to a severe depletion of ATG12 and ATG16L1 proteins (Wible et al., 2019). Knockout or knockdown of ATG5 and ATG7 are therefore likely to result in many of the same influences on non-autophagic cellular processes that are associated with changes in the expression levels of ATG5, ATG12 and ATG16L1. Hence, demonstration of similar phenotypic effects upon genetic interference with ATG5 and ATG7 is not very strong evidence that those effects are specifically caused by autophagy inhibition. When deciding upon which ATGs to target in order to infer causal relationships with autophagy, it is advisable to choose ATGs who display minimal co-dependencies.Another limitation in the field of autophagy in cancer relates to the fact that most studies so far have focussed on the role of cancer cell-intrinsic autophagy in carcinogenesis and cancer therapy. It will be important for future studies to also explore the role of autophagy in the crosstalk between different cells of the tumour microenvironment, e.g., in relation to cancer-associated fibroblasts and various cells of the immune compartment. At present, the existing studies on this topic in prostate cancer are too sparse for inclusion into this review.
Lastly, whilst studies in the field have predominantly aimed to understand the role of classical macroautophagy in cancer, an emerging theme is the roles played by non-canonical, alternative autophagy pathways, for instance secretory autophagy (Gonzalez et al., 2020). Further explorations of such pathways, but also of selective types of autophagy and the relatively understudied CMA and microautophagy pathways, will lead to a more complete understanding of the role of autophagy in prostate cancer.
A number of clinical trials involving drugs that may alter autophagic activity are ongoing on prostate cancer cohorts (www.clinicaltrials.gov). These include drugs that may either inhibit (such as hydroxychloroquine, HCQ) or activate autophagy (such as rapamycin and derivatives). A main challenge is that the pre-clinical research data on which the clinical trials are grounded are suffering from the aforementioned limitations in understanding the causalities between autophagy modulation and biological effects. Therefore, it is at present not feasible to predict whether and in which contexts one should aim to inhibit or activate autophagy in prostate cancer therapy. Alas, the results from completed clinical trials involving autophagy-modulating drugs in prostate cancer patients (such as HCQ, pantoprazole, everolimus, and rapamycin) have not led to clarification of this point, since they have not been successful (NCT00786682) (Lemos et al., 2024; Mehnert et al., 2019). Another challenge with the current clinical trials is that none of the existing autophagy-modulating drugs are specific and could exert anti-cancer effects in an autophagy-independent manner—as has been reported for HCQ in melanoma and breast cancer cells (Maycotte et al., 2012; Maes et al., 2014). Therefore, in addition to the requirement for a better basic understanding of the role of autophagy in prostate cancer biology and therapy, drugs that are more specific at inhibiting or activating autophagy are needed for future testing in the clinical setting. Finally, a remaining challenge is to identify reliable methods to determine autophagy levels in patient tissue (Humbert et al., 2020) to verify that the autophagy-modulating drugs have had their intended effects.
8 Conclusion
Overall, current studies indicate that the macroautophagic pathway may play an important role in regulating prostate carcinogenesis as well as the response and development of resistance to prostate cancer therapeutics. However, and largely due to insufficient methodological approaches, it is still highly uncertain whether autophagy predominantly plays a tumour -suppressor or -promoting role, as well as whether autophagy plays a therapy-promoting or -resistant role. To overcome this, a shift in the field to focus on utilising functional autophagy assays, rather than the current general bias on almost exclusively using marker-based assays, is required. Moreover, the approaches for elucidating causality between autophagy modulation and biological effects through pharmacological and genetic interference with various ATGs and other central autophagy-regulating genes, must be substantially improved. Finally, future focus on the role of autophagy in the crosstalk between cells of the tumour microenvironment, as well as the role of selective autophagy, CMA, microautophagy, and various non-canonical autophagy pathways will lead to a more comprehensive understanding on the role of autophagy in prostate cancer.
Author contributions
NJK: Conceptualization, Visualization, Writing—original draft, Writing–review and editing. NE: Conceptualization, Funding acquisition, Project administration, Supervision, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the South-Eastern Norway Regional Health Authority (AUTOprost; Project number 2021088).
Acknowledgments
The authors acknowledge BioRender.com, which was used to create Figures 1–3.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agarwal, P. K., Sadetsky, N., Konety, B. R., Resnick, M. I., and Carroll, P. R.Cancer of the Prostate Strategic Urological Research Endeavor CaPSURE (2008). Treatment failure after primary and salvage therapy for prostate cancer: likelihood, patterns of care, and outcomes. Cancer 112 (2), 307–314. doi:10.1002/cncr.23161
Agrotis, A., Pengo, N., Burden, J. J., and Ketteler, R. (2019). Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 15 (6), 976–997. doi:10.1080/15548627.2019.1569925
Ahmadi-Dehlaghi, F., Mohammadi, P., Valipour, E., Pournaghi, P., Kiani, S., and Mansouri, K. (2023). Autophagy: a challengeable paradox in cancer treatment. Cancer Med. 12 (10), 11542–11569. doi:10.1002/cam4.5577
An, H., and Harper, J. W. (2018). Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 20 (2), 135–143. doi:10.1038/s41556-017-0007-x
Ávalos, Y., Canales, J., Bravo-Sagua, R., Criollo, A., Lavandero, S., and Quest, A. F. (2014). Tumor suppression and promotion by autophagy. Biomed. Res. Int. 2014, 603980. doi:10.1155/2014/603980
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182 (4), 685–701. doi:10.1083/jcb.200803137
Baisamy, L., Cavin, S., Jurisch, N., and Diviani, D. (2009). The ubiquitin-like protein LC3 regulates the Rho-GEF activity of AKAP-Lbc. J. Biol. Chem. 284 (41), 28232–28242. doi:10.1074/jbc.M109.054668
Bennett, H. L., Fleming, J. T., O'Prey, J., Ryan, K. M., and Leung, H. Y. (2010). Androgens modulate autophagy and cell death via regulation of the endoplasmic reticulum chaperone glucose-regulated protein 78/BiP in prostate cancer cells. Cell Death Dis. 1 (9), e72. doi:10.1038/cddis.2010.50
Blessing, A. M., Rajapakshe, K., Reddy Bollu, L., Shi, Y., White, M. A., Pham, A. H., et al. (2017). Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy 13 (3), 506–521. doi:10.1080/15548627.2016.1268300
Blommaart, E. F., Krause, U., Schellens, J. P., Vreeling-Sindelárová, H., and Meijer, A. J. (1997). The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 243 (1-2), 240–246. doi:10.1111/j.1432-1033.1997.0240a.x
Boorjian, S. A., Karnes, R. J., Viterbo, R., Rangel, L. J., Bergstralh, E. J., Horwitz, E. M., et al. (2011). Long-term survival after radical prostatectomy versus external-beam radiotherapy for patients with high-risk prostate cancer. Cancer 117 (13), 2883–2891. doi:10.1002/cncr.25900
Boutin, B., Tajeddine, N., Vandersmissen, P., Zanou, N., Van Schoor, M., Mondin, L., et al. (2013). Androgen deprivation and androgen receptor competition by bicalutamide induce autophagy of hormone-resistant prostate cancer cells and confer resistance to apoptosis. Prostate 73 (10), 1090–1102. doi:10.1002/pros.22658
Cadwell, K., and Debnath, J. (2018). Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol. 217 (3), 813–822. doi:10.1083/jcb.201706157
Cahuzac, M., Langlois, P., Péant, B., Fleury, H., Mes-Masson, A. M., and Saad, F. (2022a). Pre-activation of autophagy impacts response to olaparib in prostate cancer cells. Commun. Biol. 5 (1), 251. doi:10.1038/s42003-022-03210-5
Cahuzac, M., Péant, B., Mes-Masson, A. M., and Saad, F. (2022b). Development of olaparib-resistance prostate cancer cell lines to identify mechanisms associated with acquired resistance. Cancers (Basel) 14 (16), 3877. doi:10.3390/cancers14163877
Cao, C., Subhawong, T., Albert, J. M., Kim, K. W., Geng, L., Sekhar, K. R., et al. (2006). Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 66 (20), 10040–10047. doi:10.1158/0008-5472.Can-06-0802
Carosi, J. M., Nguyen, T. N., Lazarou, M., Kumar, S., and Sargeant, T. J. (2021). ATG8ylation of proteins: a way to cope with cell stress? J. Cell Biol. 220 (11), e202108120. doi:10.1083/jcb.202108120
Chaiswing, L., Weiss, H. L., Jayswal, R. D., Clair, D. K. S., and Kyprianou, N. (2018). Profiles of radioresistance mechanisms in prostate cancer. Crit. Rev. Oncog. 23 (1-2), 39–67. doi:10.1615/CritRevOncog.2018025946
Chan, E. Y., Kir, S., and Tooze, S. A. (2007). siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 282 (35), 25464–25474. doi:10.1074/jbc.M703663200
Chang, H., and Zou, Z. (2020). Targeting autophagy to overcome drug resistance: further developments. J. Hematol. Oncol. 13 (1), 159. doi:10.1186/s13045-020-01000-2
Chen, B. H., Yang, H. J., Chou, H. Y., Chen, G. C., and Yang, W. Y. (2017). Staining of lipid droplets with Monodansylpentane. Methods Mol. Biol. 1560, 231–236. doi:10.1007/978-1-4939-6788-9_17
Chen, J., Liang, Y., Hu, S., Jiang, J., Zeng, M., and Luo, M. (2023). Role of ATG7-dependent non-autophagic pathway in angiogenesis. Front. Pharmacol. 14, 1266311. doi:10.3389/fphar.2023.1266311
Chen, S., Wang, C., Yeo, S., Liang, C. C., Okamoto, T., Sun, S., et al. (2016). Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev. 30 (7), 856–869. doi:10.1101/gad.276428.115
Choudhury, A. D. (2022). PTEN-PI3K pathway alterations in advanced prostate cancer and clinical implications. Prostate 82 (Suppl. 1), S60–s72. doi:10.1002/pros.24372
Chung, Y. H., Yoon, S. Y., Choi, B., Sohn, D. H., Yoon, K. H., Kim, W. J., et al. (2012). Microtubule-associated protein light chain 3 regulates Cdc42-dependent actin ring formation in osteoclast. Int. J. Biochem. Cell Biol. 44 (6), 989–997. doi:10.1016/j.biocel.2012.03.007
Crawford, E. D., Eisenberger, M. A., McLeod, D. G., Spaulding, J. T., Benson, R., Dorr, F. A., et al. (1989). A controlled trial of leuprolide with and without flutamide in prostatic carcinoma. N. Engl. J. Med. 321 (7), 419–424. doi:10.1056/nejm198908173210702
Cristofani, R., Montagnani Marelli, M., Cicardi, M. E., Fontana, F., Marzagalli, M., Limonta, P., et al. (2018). Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis. 9 (9), 889. doi:10.1038/s41419-018-0866-5
Das, C. K., Banerjee, I., and Mandal, M. (2020). Pro-survival autophagy: an emerging candidate of tumor progression through maintaining hallmarks of cancer. Semin. Cancer Biol. 66, 59–74. doi:10.1016/j.semcancer.2019.08.020
de Bono, J., Mateo, J., Fizazi, K., Saad, F., Shore, N., Sandhu, S., et al. (2020). Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 382 (22), 2091–2102. doi:10.1056/NEJMoa1911440
de Bono, J. S., Mehra, N., Scagliotti, G. V., Castro, E., Dorff, T., Stirling, A., et al. (2021). Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol. 22 (9), 1250–1264. doi:10.1016/s1470-2045(21)00376-4
de Bono, J. S., Oudard, S., Ozguroglu, M., Hansen, S., Machiels, J. P., Kocak, I., et al. (2010). Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 376 (9747), 1147–1154. doi:10.1016/s0140-6736(10)61389-x
Decker, K. F., Zheng, D., He, Y., Bowman, T., Edwards, J. R., and Jia, L. (2012). Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 40 (21), 10765–10779. doi:10.1093/nar/gks888
De Duve, C., and Wattiaux, R. (1966). Functions of lysosomes. Annu. Rev. Physiol. 28, 435–492. doi:10.1146/annurev.ph.28.030166.002251
Dehm, S. M., and Tindall, D. J. (2006). Ligand-independent androgen receptor activity is activation function-2-independent and resistant to antiandrogens in androgen refractory prostate cancer cells. J. Biol. Chem. 281 (38), 27882–27893. doi:10.1074/jbc.M605002200
Deng, C., Li, C., Dong, X., Yu, Y., Guo, W., Guan, Y., et al. (2023). Atg7 senses ATP levels and regulates AKT(1)-PDCD4 phosphorylation-ubiquitination axis to promote survival during metabolic stress. Commun. Biol. 6 (1), 1252. doi:10.1038/s42003-023-05656-7
DeSelm, C. J., Miller, B. C., Zou, W., Beatty, W. L., van Meel, E., Takahata, Y., et al. (2011). Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell 21 (5), 966–974. doi:10.1016/j.devcel.2011.08.016
Domagala, A., Stachura, J., Gabrysiak, M., Muchowicz, A., Zagozdzon, R., Golab, J., et al. (2018). Inhibition of autophagy sensitizes cancer cells to Photofrin-based photodynamic therapy. BMC Cancer 18 (1), 210. doi:10.1186/s12885-018-4126-y
Dou, Z., Xu, C., Donahue, G., Shimi, T., Pan, J. A., Zhu, J., et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527 (7576), 105–109. doi:10.1038/nature15548
Durgan, J., Lystad, A. H., Sloan, K., Carlsson, S. R., Wilson, M. I., Marcassa, E., et al. (2021). Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol. Cell 81 (9), 2031–2040.e8. doi:10.1016/j.molcel.2021.03.020
Eberli, D., Kranzbühler, B., Mortezavi, A., Sulser, T., and Salemi, S. (2020). Apalutamide in combination with autophagy inhibitors improves treatment effects in prostate cancer cells. Urol. Oncol. 38 (8), 683.e619–683. doi:10.1016/j.urolonc.2020.04.030
Eisenberger, M., Hardy-Bessard, A. C., Kim, C. S., Géczi, L., Ford, D., Mourey, L., et al. (2017). Phase III study comparing a reduced dose of cabazitaxel (20 mg/m(2)) and the currently approved dose (25 mg/m(2)) in postdocetaxel patients with metastatic castration-resistant prostate cancer-PROSELICA. J. Clin. Oncol. 35 (28), 3198–3206. doi:10.1200/jco.2016.72.1076
Eisenberger, M. A., Blumenstein, B. A., Crawford, E. D., Miller, G., McLeod, D. G., Loehrer, P. J., et al. (1998). Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N. Engl. J. Med. 339 (15), 1036–1042. doi:10.1056/nejm199810083391504
Eisenberg-Lerner, A., Bialik, S., Simon, H. U., and Kimchi, A. (2009). Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 16 (7), 966–975. doi:10.1038/cdd.2009.33
Elgendy, M., Ciro, M., Abdel-Aziz, A. K., Belmonte, G., Dal Zuffo, R., Mercurio, C., et al. (2014). Beclin 1 restrains tumorigenesis through Mcl-1 destabilization in an autophagy-independent reciprocal manner. Nat. Commun. 5, 5637. doi:10.1038/ncomms6637
Engedal, N., Sønstevold, T., Beese, C. J., Selladurai, S., Melcher, T., Simensen, J. E., et al. (2022). Measuring autophagic cargo flux with keima-based probes. Methods Mol. Biol. 2445, 99–115. doi:10.1007/978-1-0716-2071-7_7
Engedal, N., Torgersen, M. L., Guldvik, I. J., Barfeld, S. J., Bakula, D., Sætre, F., et al. (2013). Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 9 (10), 1475–1490. doi:10.4161/auto.25900
Fang, D., Xie, H., Hu, T., Shan, H., and Li, M. (2021). Binding features and functions of ATG3. Front. Cell Dev. Biol. 9, 685625. doi:10.3389/fcell.2021.685625
Ferro, M., Lucarelli, G., Crocetto, F., Dolce, P., Verde, A., La Civita, E., et al. (2021). First-line systemic therapy for metastatic castration-sensitive prostate cancer: an updated systematic review with novel findings. Crit. Rev. Oncol. Hematol. 157, 103198. doi:10.1016/j.critrevonc.2020.103198
Galluzzi, L., and Green, D. R. (2019). Autophagy-independent functions of the autophagy machinery. Cell 177 (7), 1682–1699. doi:10.1016/j.cell.2019.05.026
Galluzzi, L., Pietrocola, F., Bravo-San Pedro, J. M., Amaravadi, R. K., Baehrecke, E. H., Cecconi, F., et al. (2015). Autophagy in malignant transformation and cancer progression. Embo J. 34 (7), 856–880. doi:10.15252/embj.201490784
Gan, B., and Guan, J. L. (2008). FIP200, a key signaling node to coordinately regulate various cellular processes. Cell Signal 20 (5), 787–794. doi:10.1016/j.cellsig.2007.10.021
Gao, X., Zacharek, A., Salkowski, A., Grignon, D. J., Sakr, W., Porter, A. T., et al. (1995). Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer. Cancer Res. 55 (5), 1002–1005.
Gonzalez, C. D., Resnik, R., and Vaccaro, M. I. (2020). Secretory autophagy and its relevance in metabolic and degenerative disease. Front. Endocrinol. (Lausanne) 11, 266. doi:10.3389/fendo.2020.00266
Gozuacik, D., and Kimchi, A. (2004). Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23 (16), 2891–2906. doi:10.1038/sj.onc.1207521
Gundem, G., Van Loo, P., Kremeyer, B., Alexandrov, L. B., Tubio, J. M. C., Papaemmanuil, E., et al. (2015). The evolutionary history of lethal metastatic prostate cancer. Nature 520 (7547), 353–357. doi:10.1038/nature14347
Guo, H., Chitiprolu, M., Roncevic, L., Javalet, C., Hemming, F. J., Trung, M. T., et al. (2017). Atg5 disassociates the V(1)V(0)-ATPase to promote exosome production and tumor metastasis independent of canonical macroautophagy. Dev. Cell 43 (6), 716–730. doi:10.1016/j.devcel.2017.11.018
Hamaoui, D., and Subtil, A. (2022). ATG16L1 functions in cell homeostasis beyond autophagy. Febs J. 289 (7), 1779–1800. doi:10.1111/febs.15833
Hanson, H. H., Kang, S., Fernández-Monreal, M., Oung, T., Yildirim, M., Lee, R., et al. (2010). LC3-dependent intracellular membrane tubules induced by gamma-protocadherins A3 and B2: a role for intraluminal interactions. J. Biol. Chem. 285 (27), 20982–20992. doi:10.1074/jbc.M109.092031
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi:10.1146/annurev-genet-102808-114910
Hewitt, G., Carroll, B., Sarallah, R., Correia-Melo, C., Ogrodnik, M., Nelson, G., et al. (2016). SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 12 (10), 1917–1930. doi:10.1080/15548627.2016.1210368
Hewitt, G., and Korolchuk, V. I. (2017). Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol. 27 (5), 340–351. doi:10.1016/j.tcb.2016.11.011
Hong, M. K., Macintyre, G., Wedge, D. C., Van Loo, P., Patel, K., Lunke, S., et al. (2015). Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun. 6, 6605. doi:10.1038/ncomms7605
Hosokawa, N., Hara, Y., and Mizushima, N. (2006). Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett. 580 (11), 2623–2629. doi:10.1016/j.febslet.2006.04.008
Hu, F., Li, G., Huang, C., Hou, Z., Yang, X., Luo, X., et al. (2020). The autophagy-independent role of BECN1 in colorectal cancer metastasis through regulating STAT3 signaling pathway activation. Cell Death Dis. 11 (5), 304. doi:10.1038/s41419-020-2467-3
Hu, F., Zhao, Y., Yu, Y., Fang, J. M., Cui, R., Liu, Z. Q., et al. (2018). Docetaxel-mediated autophagy promotes chemoresistance in castration-resistant prostate cancer cells by inhibiting STAT3. Cancer Lett. 416, 24–30. doi:10.1016/j.canlet.2017.12.013
Huang, C. Y., Huang, S. P., Lin, V. C., Yu, C. C., Chang, T. Y., Lu, T. L., et al. (2015). Genetic variants of the autophagy pathway as prognostic indicators for prostate cancer. Sci. Rep. 5, 14045. doi:10.1038/srep14045
Humbert, M., Morán, M., de la Cruz-Ojeda, P., Muntané, J., Wiedmer, T., Apostolova, N., et al. (2020). Assessing autophagy in archived tissue or how to capture autophagic flux from a tissue snapshot. Biol. (Basel) 9 (3), 59. doi:10.3390/biology9030059
Hurley, J. H., and Schulman, B. A. (2014). Atomistic autophagy: the structures of cellular self-digestion. Cell 157 (2), 300–311. doi:10.1016/j.cell.2014.01.070
Hussain, M., Daignault-Newton, S., Twardowski, P. W., Albany, C., Stein, M. N., Kunju, L. P., et al. (2018). Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J. Clin. Oncol. 36 (10), 991–999. doi:10.1200/jco.2017.75.7310
Ipsen, M. B., Sørensen, E. M. G., Thomsen, E. A., Weiss, S., Haldrup, J., Dalby, A., et al. (2022). A genome-wide CRISPR-Cas9 knockout screen identifies novel PARP inhibitor resistance genes in prostate cancer. Oncogene 41 (37), 4271–4281. doi:10.1038/s41388-022-02427-2
James, N. D., Spears, M. R., Clarke, N. W., Dearnaley, D. P., De Bono, J. S., Gale, J., et al. (2015). Survival with newly diagnosed metastatic prostate cancer in the "docetaxel era": data from 917 patients in the control arm of the STAMPEDE trial (MRC PR08, CRUK/06/019). Eur. Urol. 67 (6), 1028–1038. doi:10.1016/j.eururo.2014.09.032
Jiang, Q., Yeh, S., Wang, X., Xu, D., Zhang, Q., Wen, X., et al. (2012). Targeting androgen receptor leads to suppression of prostate cancer via induction of autophagy. J. Urol. 188 (4), 1361–1368. doi:10.1016/j.juro.2012.06.004
Joo, J. H., Wang, B., Frankel, E., Ge, L., Xu, L., Iyengar, R., et al. (2016). The noncanonical role of ULK/ATG1 in ER-to-golgi trafficking is essential for cellular homeostasis. Mol. Cell 62 (4), 491–506. doi:10.1016/j.molcel.2016.04.020
Joshi, A., Iyengar, R., Joo, J. H., Li-Harms, X. J., Wright, C., Marino, R., et al. (2016). Nuclear ULK1 promotes cell death in response to oxidative stress through PARP1. Cell Death Differ. 23 (2), 216–230. doi:10.1038/cdd.2015.88
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 19 (21), 5720–5728. doi:10.1093/emboj/19.21.5720
Kabeya, Y., Mizushima, N., Yamamoto, A., Oshitani-Okamoto, S., Ohsumi, Y., and Yoshimori, T. (2004). LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117 (Pt 13), 2805–2812. doi:10.1242/jcs.01131
Kaizuka, T., and Mizushima, N. (2016). Atg13 is essential for autophagy and cardiac development in mice. Mol. Cell Biol. 36 (4), 585–595. doi:10.1128/mcb.01005-15
Karantza-Wadsworth, V., Patel, S., Kravchuk, O., Chen, G., Mathew, R., Jin, S., et al. (2007). Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 21 (13), 1621–1635. doi:10.1101/gad.1565707
Katayama, H., Kogure, T., Mizushima, N., Yoshimori, T., and Miyawaki, A. (2011). A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 18 (8), 1042–1052. doi:10.1016/j.chembiol.2011.05.013
Katheder, N. S., Khezri, R., O'Farrell, F., Schultz, S. W., Jain, A., Rahman, M. M., et al. (2017). Microenvironmental autophagy promotes tumour growth. Nature 541 (7637), 417–420. doi:10.1038/nature20815
Kharaziha, P., Chioureas, D., Baltatzis, G., Fonseca, P., Rodriguez, P., Gogvadze, V., et al. (2015). Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget 6 (35), 37066–37082. doi:10.18632/oncotarget.5797
Kim, M. S., Song, S. Y., Lee, J. Y., Yoo, N. J., and Lee, S. H. (2011). Expressional and mutational analyses of ATG5 gene in prostate cancers. Apmis 119 (11), 802–807. doi:10.1111/j.1600-0463.2011.02812.x
Kisen, G. O., Tessitore, L., Costelli, P., Gordon, P. B., Schwarze, P. E., Baccino, F. M., et al. (1993). Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis 14 (12), 2501–2505. doi:10.1093/carcin/14.12.2501
Klionsky, D. J., Abdel-Aziz, A. K., Abdelfatah, S., Abdellatif, M., Abdoli, A., Abel, S., et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17 (1), 1–382. doi:10.1080/15548627.2020.1797280
Kopitz, J., Kisen, G. O., Gordon, P. B., Bohley, P., and Seglen, P. O. (1990). Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol. 111 (3), 941–953. doi:10.1083/jcb.111.3.941
Korenchuk, S., Lehr, J. E., Mclean, L., Lee, Y. G., Whitney, S., Vessella, R., et al. (2001). VCaP, a cell-based model system of human prostate cancer. Vivo 15 (2), 163–168.
Koukourakis, M. I., Kalamida, D., Mitrakas, A., Pouliliou, S., Kalamida, S., Sivridis, E., et al. (2015). Intensified autophagy compromises the efficacy of radiotherapy against prostate cancer. Biochem. Biophys. Res. Commun. 461 (2), 268–274. doi:10.1016/j.bbrc.2015.04.014
Kuma, A., Matsui, M., and Mizushima, N. (2007). LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 3 (4), 323–328. doi:10.4161/auto.4012
Kumar, S., Jia, J., and Deretic, V. (2021). Atg8ylation as a general membrane stress and remodeling response. Cell Stress 5 (9), 128–142. doi:10.15698/cst2021.09.255
Laddha, S. V., Ganesan, S., Chan, C. S., and White, E. (2014). Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol. Cancer Res. 12 (4), 485–490. doi:10.1158/1541-7786.Mcr-13-0614
Lamark, T., and Johansen, T. (2021). Mechanisms of selective autophagy. Annu. Rev. Cell Dev. Biol. 37, 143–169. doi:10.1146/annurev-cellbio-120219-035530
Lamoureux, F., Thomas, C., Crafter, C., Kumano, M., Zhang, F., Davies, B. R., et al. (2013). Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin. Cancer Res. 19 (4), 833–844. doi:10.1158/1078-0432.Ccr-12-3114
Lebovitz, C. B., Robertson, A. G., Goya, R., Jones, S. J., Morin, R. D., Marra, M. A., et al. (2015). Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11 (9), 1668–1687. doi:10.1080/15548627.2015.1067362
Lecker, S. H., Goldberg, A. L., and Mitch, W. E. (2006). Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 17 (7), 1807–1819. doi:10.1681/asn.2006010083
Lee, I. H., Kawai, Y., Fergusson, M. M., Rovira, I. I., Bishop, A. J., Motoyama, N., et al. (2012). Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 336 (6078), 225–228. doi:10.1126/science.1218395
Lemos, G., Fernandes, C., Silva, F. H., and Calmasini, F. B. (2024). The role of autophagy in prostate cancer and prostatic diseases: a new therapeutic strategy. Prostate Cancer Prostatic Dis. 27, 230–238. doi:10.1038/s41391-024-00793-4
Li, S., Zhang, L., Zhang, G., Shangguan, G., Hou, X., Duan, W., et al. (2021). A nonautophagic role of ATG5 in regulating cell growth by targeting c-Myc for proteasome-mediated degradation. iScience 24 (11), 103296. doi:10.1016/j.isci.2021.103296
Li, W. W., Li, J., and Bao, J. K. (2012). Microautophagy: lesser-known self-eating. Cell Mol. Life Sci. 69 (7), 1125–1136. doi:10.1007/s00018-011-0865-5
Li, X., Wan, X., Chen, H., Yang, S., Liu, Y., Mo, W., et al. (2014). Identification of miR-133b and RB1CC1 as independent predictors for biochemical recurrence and potential therapeutic targets for prostate cancer. Clin. Cancer Res. 20 (9), 2312–2325. doi:10.1158/1078-0432.Ccr-13-1588
Liang, P., Zhang, J., Wu, Y., Zheng, S., Xu, Z., Yang, S., et al. (2023). An ULK1/2-PXN mechanotransduction pathway suppresses breast cancer cell migration. EMBO Rep. 24 (11), e56850. doi:10.15252/embr.202356850
Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., et al. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402 (6762), 672–676. doi:10.1038/45257
Lin, J. Z., Wang, W. W., Hu, T. T., Zhu, G. Y., Li, L. N., Zhang, C. Y., et al. (2020). FOXM1 contributes to docetaxel resistance in castration-resistant prostate cancer by inducing AMPK/mTOR-mediated autophagy. Cancer Lett. 469, 481–489. doi:10.1016/j.canlet.2019.11.014
Linder, B., and Kögel, D. (2019). Autophagy in cancer cell death. Biol. (Basel) 8 (4), 82. doi:10.3390/biology8040082
Lindner, P., Christensen, S. B., Nissen, P., Møller, J. V., and Engedal, N. (2020). Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal 18 (1), 12. doi:10.1186/s12964-019-0499-z
Liu, B., Miyake, H., Nishikawa, M., Tei, H., and Fujisawa, M. (2015). Expression profile of autophagy-related markers in localized prostate cancer: correlation with biochemical recurrence after radical prostatectomy. Urology 85 (6), 1424–1430. doi:10.1016/j.urology.2015.03.006
Lu, S., Sung, T., Lin, N., Abraham, R. T., and Jessen, B. A. (2017). Lysosomal adaptation: how cells respond to lysosomotropic compounds. PLoS One 12 (3), e0173771. doi:10.1371/journal.pone.0173771
Luhr, M., Sætre, F., and Engedal, N. (2018b). The long-lived protein degradation assay: an efficient method for quantitative determination of the autophagic flux of endogenous proteins in adherent cell lines. Bio Protoc. 8 (9), e2836. doi:10.21769/BioProtoc.2836
Luhr, M., Szalai, P., and Engedal, N. (2018a). The lactate dehydrogenase sequestration assay - a simple and reliable method to determine bulk autophagic sequestration activity in mammalian cells. J. Vis. Exp. 137, 57971. doi:10.3791/57971
Luhr, M., Torgersen, M. L., Szalai, P., Hashim, A., Brech, A., Staerk, J., et al. (2019). The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J. Biol. Chem. 294 (20), 8197–8217. doi:10.1074/jbc.RA118.002829
Lum, J. J., Bauer, D. E., Kong, M., Harris, M. H., Li, C., Lindsten, T., et al. (2005). Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120 (2), 237–248. doi:10.1016/j.cell.2004.11.046
Ma, X., Zou, L., Li, X., Chen, Z., Lin, Z., and Wu, X. (2019). Inhibition of autophagy improves the efficacy of abiraterone for the treatment of prostate cancer. Cancer Biother Radiopharm. 34 (3), 181–188. doi:10.1089/cbr.2018.2559
Maes, H., Kuchnio, A., Peric, A., Moens, S., Nys, K., De Bock, K., et al. (2014). Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26 (2), 190–206. doi:10.1016/j.ccr.2014.06.025
Mailler, E., Guardia, C. M., Bai, X., Jarnik, M., Williamson, C. D., Li, Y., et al. (2021). The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat. Commun. 12 (1), 6750. doi:10.1038/s41467-021-26999-x
Malhotra, V. (2013). Unconventional protein secretion: an evolving mechanism. Embo J. 32 (12), 1660–1664. doi:10.1038/emboj.2013.104
Marsh, T., Tolani, B., and Debnath, J. (2021). The pleiotropic functions of autophagy in metastasis. J. Cell Sci. 134 (2), jcs247056. doi:10.1242/jcs.247056
Maskey, D., Yousefi, S., Schmid, I., Zlobec, I., Perren, A., Friis, R., et al. (2013). ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat. Commun. 4, 2130. doi:10.1038/ncomms3130
Mateo, J., Carreira, S., Sandhu, S., Miranda, S., Mossop, H., Perez-Lopez, R., et al. (2015). DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373 (18), 1697–1708. doi:10.1056/NEJMoa1506859
Mathew, R., Kongara, S., Beaudoin, B., Karp, C. M., Bray, K., Degenhardt, K., et al. (2007). Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 21 (11), 1367–1381. doi:10.1101/gad.1545107
Mauthe, M., Orhon, I., Rocchi, C., Zhou, X., Luhr, M., Hijlkema, K. J., et al. (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14 (8), 1435–1455. doi:10.1080/15548627.2018.1474314
Maycotte, P., Aryal, S., Cummings, C. T., Thorburn, J., Morgan, M. J., and Thorburn, A. (2012). Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 8 (2), 200–212. doi:10.4161/auto.8.2.18554
Mehnert, J. M., Kaveney, A. D., Malhotra, J., Spencer, K., Portal, D., Goodin, S., et al. (2019). A phase I trial of MK-2206 and hydroxychloroquine in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 84 (4), 899–907. doi:10.1007/s00280-019-03919-x
Mele, L., Del Vecchio, V., Liccardo, D., Prisco, C., Schwerdtfeger, M., Robinson, N., et al. (2020). The role of autophagy in resistance to targeted therapies. Cancer Treat. Rev. 88, 102043. doi:10.1016/j.ctrv.2020.102043
Mizushima, N. (2007). Autophagy: process and function. Genes Dev. 21 (22), 2861–2873. doi:10.1101/gad.1599207
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. doi:10.1146/annurev-cellbio-092910-154005
Moosavi, M. A., and Djavaheri-Mergny, M. (2019). Autophagy: new insights into mechanisms of action and resistance of treatment in acute promyelocytic leukemia. Int. J. Mol. Sci. 20 (14), 3559. doi:10.3390/ijms20143559
Mortezavi, A., Salemi, S., Kranzbühler, B., Gross, O., Sulser, T., Simon, H. U., et al. (2019). Inhibition of autophagy significantly increases the antitumor effect of Abiraterone in prostate cancer. World J. Urol. 37 (2), 351–358. doi:10.1007/s00345-018-2385-5
Moscat, J., Diaz-Meco, M. T., and Wooten, M. W. (2007). Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci. 32 (2), 95–100. doi:10.1016/j.tibs.2006.12.002
Moscat, J., Karin, M., and Diaz-Meco, M. T. (2016). p62 in cancer: signaling adaptor beyond autophagy. Cell 167 (3), 606–609. doi:10.1016/j.cell.2016.09.030
Mottet, N., Bellmunt, J., Bolla, M., Joniau, S., Mason, M., Matveev, V., et al. (2011). EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur. Urol. 59 (4), 572–583. doi:10.1016/j.eururo.2011.01.025
Moul, J. W. (2004). The evolving definition of advanced prostate cancer. Rev. Urol. 6 (Suppl. 8), S10–S17.
Mukha, A., Kahya, U., Linge, A., Chen, O., Löck, S., Lukiyanchuk, V., et al. (2021). GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 11 (16), 7844–7868. doi:10.7150/thno.58655
Nguyen, T. N., Padman, B. S., Usher, J., Oorschot, V., Ramm, G., and Lazarou, M. (2016). Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215 (6), 857–874. doi:10.1083/jcb.201607039
Nieto-Torres, J. L., Leidal, A. M., Debnath, J., and Hansen, M. (2021). Beyond autophagy: the expanding roles of ATG8 proteins. Trends Biochem. Sci. 46 (8), 673–686. doi:10.1016/j.tibs.2021.01.004
Novikoff, A. B., and Essner, E. (1962). Cytolysomes and mitochondrial degeneration. J. Cell Biol. 15 (1), 140–146. doi:10.1083/jcb.15.1.140
Nunes, J., Zhang, H., Angelopoulos, N., Chhetri, J., Osipo, C., Grothey, A., et al. (2016). ATG9A loss confers resistance to trastuzumab via c-Cbl mediated Her2 degradation. Oncotarget 7 (19), 27599–27612. doi:10.18632/oncotarget.8504
Ohsumi, Y. (2014). Historical landmarks of autophagy research. Cell Res. 24 (1), 9–23. doi:10.1038/cr.2013.169
Onodera, J., and Ohsumi, Y. (2005). Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J. Biol. Chem. 280 (36), 31582–31586. doi:10.1074/jbc.M506736200
Ouyang, D. Y., Xu, L. H., He, X. H., Zhang, Y. T., Zeng, L. H., Cai, J. Y., et al. (2013). Autophagy is differentially induced in prostate cancer LNCaP, DU145 and PC-3 cells via distinct splicing profiles of ATG5. Autophagy 9 (1), 20–32. doi:10.4161/auto.22397
Park, Y. E., Hayashi, Y. K., Bonne, G., Arimura, T., Noguchi, S., Nonaka, I., et al. (2009). Autophagic degradation of nuclear components in mammalian cells. Autophagy 5 (6), 795–804. doi:10.4161/auto.8901
Parker, C., Castro, E., Fizazi, K., Heidenreich, A., Ost, P., Procopio, G., et al. (2020). Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 31 (9), 1119–1134. doi:10.1016/j.annonc.2020.06.011
Peng, K., Sun, A., Zhu, J., Gao, J., Li, Y., Shao, G., et al. (2021). Restoration of the ATG5-dependent autophagy sensitizes DU145 prostate cancer cells to chemotherapeutic drugs. Oncol. Lett. 22 (3), 638. doi:10.3892/ol.2021.12899
Petrylak, D. P., Tangen, C. M., Hussain, M. H., Lara, P. N., Jones, J. A., Taplin, M. E., et al. (2004). Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 351 (15), 1513–1520. doi:10.1056/NEJMoa041318
Pezaro, C. (2020). PARP inhibitor combinations in prostate cancer. Ther. Adv. Med. Oncol. 12, 1758835919897537. doi:10.1177/1758835919897537
Pickard, R. D., Spencer, B. H., McFarland, A. J., Bernaitis, N., Davey, A. K., Perkins, A. V., et al. (2015). Paradoxical effects of the autophagy inhibitor 3-methyladenine on docetaxel-induced toxicity in PC-3 and LNCaP prostate cancer cells. Naunyn Schmiedeb. Arch. Pharmacol. 388 (7), 793–799. doi:10.1007/s00210-015-1104-7
Pimentel-Muiños, F. X., and Boada-Romero, E. (2014). Selective autophagy against membranous compartments: canonical and unconventional purposes and mechanisms. Autophagy 10 (3), 397–407. doi:10.4161/auto.27244
Poillet-Perez, L., Xie, X., Zhan, L., Yang, Y., Sharp, D. W., Hu, Z. S., et al. (2018). Autophagy maintains tumour growth through circulating arginine. Nature 563 (7732), 569–573. doi:10.1038/s41586-018-0697-7
Qu, X., Yu, J., Bhagat, G., Furuya, N., Hibshoosh, H., Troxel, A., et al. (2003). Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 112 (12), 1809–1820. doi:10.1172/jci20039
Rajak, S., Raza, S., and Sinha, R. A. (2022). ULK1 signaling in the liver: autophagy dependent and independent actions. Front. Cell Dev. Biol. 10, 836021. doi:10.3389/fcell.2022.836021
Ramkumar, A., Murthy, D., Raja, D. A., Singh, A., Krishnan, A., Khanna, S., et al. (2017). Classical autophagy proteins LC3B and ATG4B facilitate melanosome movement on cytoskeletal tracks. Autophagy 13 (8), 1331–1347. doi:10.1080/15548627.2017.1327509
Risdon, E. N., Chau, C. H., Price, D. K., Sartor, O., and Figg, W. D. (2021). PARP inhibitors and prostate cancer: to infinity and beyond BRCA. Oncologist 26 (1), e115–e129. doi:10.1634/theoncologist.2020-0697
Robinson, D., Van Allen, E. M., Wu, Y. M., Schultz, N., Lonigro, R. J., Mosquera, J. M., et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 161 (5), 1215–1228. doi:10.1016/j.cell.2015.05.001
Rohatgi, R. A., Janusis, J., Leonard, D., Bellvé, K. D., Fogarty, K. E., Baehrecke, E. H., et al. (2015). Beclin 1 regulates growth factor receptor signaling in breast cancer. Oncogene 34 (42), 5352–5362. doi:10.1038/onc.2014.454
Rose, M., Burgess, J. T., O'Byrne, K., Richard, D. J., and Bolderson, E. (2020). PARP inhibitors: clinical relevance, mechanisms of action and tumor resistance. Front. Cell Dev. Biol. 8, 564601. doi:10.3389/fcell.2020.564601
Roy, A., Bera, S., Saso, L., and Dwarakanath, B. S. (2022). Role of autophagy in tumor response to radiation: implications for improving radiotherapy. Front. Oncol. 12, 957373. doi:10.3389/fonc.2022.957373
Runwal, G., Stamatakou, E., Siddiqi, F. H., Puri, C., Zhu, Y., and Rubinsztein, D. C. (2019). LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 9 (1), 10147. doi:10.1038/s41598-019-46657-z
Sahu, R., Kaushik, S., Clement, C. C., Cannizzo, E. S., Scharf, B., Follenzi, A., et al. (2011). Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20 (1), 131–139. doi:10.1016/j.devcel.2010.12.003
Sala, D., Ivanova, S., Plana, N., Ribas, V., Duran, J., Bach, D., et al. (2014). Autophagy-regulating TP53INP2 mediates muscle wasting and is repressed in diabetes. J. Clin. Invest. 124 (5), 1914–1927. doi:10.1172/jci72327
Saleiro, D., Blyth, G. T., Kosciuczuk, E. M., Ozark, P. A., Majchrzak-Kita, B., Arslan, A. D., et al. (2018). IFN-γ-inducible antiviral responses require ULK1-mediated activation of MLK3 and ERK5. Sci. Signal 11 (557), eaap9921. doi:10.1126/scisignal.aap9921
Santanam, U., Banach-Petrosky, W., Abate-Shen, C., Shen, M. M., White, E., and DiPaola, R. S. (2016). Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 30 (4), 399–407. doi:10.1101/gad.274134.115
Sarkar, S., Davies, J. E., Huang, Z., Tunnacliffe, A., and Rubinsztein, D. C. (2007). Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 282 (8), 5641–5652. doi:10.1074/jbc.M609532200
Schuck, S. (2020). Microautophagy - distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 133 (17), jcs246322. doi:10.1242/jcs.246322
Schwarze, P. E., and Seglen, P. O. (1985). Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes isolated from carcinogen-treated rats. Exp. Cell Res. 157 (1), 15–28. doi:10.1016/0014-4827(85)90148-x
Seglen, P. O. (1987). “Regulation of autophagic protein degradation in isolated liver cells,” in Lysosomes: their role in protein breakdown. Editors H. Glaumann, and F. J. Ballard (Academic Press), 371–414.
Seglen, P. O., and Gordon, P. B. (1982). 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 79 (6), 1889–1892. doi:10.1073/pnas.79.6.1889
Seglen, P. O., Grinde, B., and Solheim, A. E. (1979). Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur. J. Biochem. 95 (2), 215–225. doi:10.1111/j.1432-1033.1979.tb12956.x
Seglen, P. O., Luhr, M., Mills, I. G., Sætre, F., Szalai, P., and Engedal, N. (2015). Macroautophagic cargo sequestration assays. Methods 75, 25–36. doi:10.1016/j.ymeth.2014.12.021
Seglen, P. O., Øverbye, A., and Saetre, F. (2009). Sequestration assays for mammalian autophagy. Methods Enzymol. 452, 63–83. doi:10.1016/s0076-6879(08)03605-7
Sharma, A., Alswillah, T., Singh, K., Chatterjee, P., Willard, B., Venere, M., et al. (2018). USP14 regulates DNA damage repair by targeting RNF168-dependent ubiquitination. Autophagy 14 (11), 1976–1990. doi:10.1080/15548627.2018.1496877
Shi, Y., Han, J. J., Tennakoon, J. B., Mehta, F. F., Merchant, F. A., Burns, A. R., et al. (2013). Androgens promote prostate cancer cell growth through induction of autophagy. Mol. Endocrinol. 27 (2), 280–295. doi:10.1210/me.2012-1260
Shi, Y. X., Sun, Z. W., Jia, D. L., and Wang, H. B. (2022). Autophagy deficiency promotes lung metastasis of prostate cancer via stabilization of TWIST1. Clin. Transl. Oncol. 24 (7), 1403–1412. doi:10.1007/s12094-022-02786-y
Shin, J. H., Min, S. H., Kim, S. J., Kim, Y. I., Park, J., Lee, H. K., et al. (2013). TAK1 regulates autophagic cell death by suppressing the phosphorylation of p70 S6 kinase 1. Sci. Rep. 3, 1561. doi:10.1038/srep01561
Shirley, M. (2024). Capivasertib: first approval. Drugs 84 (3), 337–346. doi:10.1007/s40265-024-01998-6
Siegel, R. L., Miller, K. D., Wagle, N. S., and Jemal, A. (2023). Cancer statistics, 2023. CA Cancer J. Clin. 73 (1), 17–48. doi:10.3322/caac.21763
Smith, M. R., Scher, H. I., Sandhu, S., Efstathiou, E., Lara, P. N., Yu, E. Y., et al. (2022). Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol. 23 (3), 362–373. doi:10.1016/s1470-2045(21)00757-9
Sobel, R. E., and Sadar, M. D. (2005). Cell lines used in prostate cancer research: a compendium of old and new lines--part 1. J. Urol. 173 (2), 342–359. doi:10.1097/01.ju.0000141580.30910.57
Sønstevold, T., Engedal, N., and Torgersen, M. L. (2021). Perturbation of cellular redox homeostasis dictates divergent effects of polybutyl cyanoacrylate (PBCA) nanoparticles on autophagy. Cells 10 (12), 3432. doi:10.3390/cells10123432
Sou, Y. S., Tanida, I., Komatsu, M., Ueno, T., and Kominami, E. (2006). Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J. Biol. Chem. 281 (6), 3017–3024. doi:10.1074/jbc.M505888200
Sousa, C. M., Biancur, D. E., Wang, X., Halbrook, C. J., Sherman, M. H., Zhang, L., et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536 (7617), 479–483. doi:10.1038/nature19084
Su, W., Li, S., Chen, X., Yin, L., Ma, P., Ma, Y., et al. (2017). GABARAPL1 suppresses metastasis by counteracting PI3K/Akt pathway in prostate cancer. Oncotarget 8 (3), 4449–4459. doi:10.18632/oncotarget.13879
Sun, S. Y., Hu, X. T., Yu, X. F., Zhang, Y. Y., Liu, X. H., Liu, Y. H., et al. (2021). Nuclear translocation of ATG5 induces DNA mismatch repair deficiency (MMR-D)/microsatellite instability (MSI) via interacting with Mis18α in colorectal cancer. Br. J. Pharmacol. 178 (11), 2351–2369. doi:10.1111/bph.15422
Suzuki, H., Osawa, T., Fujioka, Y., and Noda, N. N. (2017). Structural biology of the core autophagy machinery. Curr. Opin. Struct. Biol. 43, 10–17. doi:10.1016/j.sbi.2016.09.010
Szalai, P., Hagen, L. K., Sætre, F., Luhr, M., Sponheim, M., Øverbye, A., et al. (2015). Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp. Cell Res. 333 (1), 21–38. doi:10.1016/j.yexcr.2015.02.003
Szeto, J., Kaniuk, N. A., Canadien, V., Nisman, R., Mizushima, N., Yoshimori, T., et al. (2006). ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2 (3), 189–199. doi:10.4161/auto.2731
Takamura, A., Komatsu, M., Hara, T., Sakamoto, A., Kishi, C., Waguri, S., et al. (2011). Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25 (8), 795–800. doi:10.1101/gad.2016211
Tamargo-Gómez, I., Martínez-García, G. G., Suárez, M. F., Rey, V., Fueyo, A., Codina-Martínez, H., et al. (2021). ATG4D is the main ATG8 delipidating enzyme in mammalian cells and protects against cerebellar neurodegeneration. Cell Death Differ. 28 (9), 2651–2672. doi:10.1038/s41418-021-00776-1
Tanida, I., Tanida-Miyake, E., Komatsu, M., Ueno, T., and Kominami, E. (2002). Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J. Biol. Chem. 277 (16), 13739–13744. doi:10.1074/jbc.M200385200
Tanida, I., Tanida-Miyake, E., Ueno, T., and Kominami, E. (2001). The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem. 276 (3), 1701–1706. doi:10.1074/jbc.C000752200
Tanida, I., Ueno, T., and Kominami, E. (2004). LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36 (12), 2503–2518. doi:10.1016/j.biocel.2004.05.009
Tannock, I. F., de Wit, R., Berry, W. R., Horti, J., Pluzanska, A., Chi, K. N., et al. (2004). Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 351 (15), 1502–1512. doi:10.1056/NEJMoa040720
Tannock, I. F., Osoba, D., Stockler, M. R., Ernst, D. S., Neville, A. J., Moore, M. J., et al. (1996). Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J. Clin. Oncol. 14 (6), 1756–1764. doi:10.1200/jco.1996.14.6.1756
Taylor, A. K., Kosoff, D., Emamekhoo, H., Lang, J. M., and Kyriakopoulos, C. E. (2023). PARP inhibitors in metastatic prostate cancer. Front. Oncol. 13, 1159557. doi:10.3389/fonc.2023.1159557
Tedesco, G., Santarosa, M., and Maestro, R. (2024). Beyond self-eating: emerging autophagy-independent functions for the autophagy molecules in cancer (Review). Int. J. Oncol. 64 (6), 57. doi:10.3892/ijo.2024.5645
Tekirdag, K., and Cuervo, A. M. (2018). Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J. Biol. Chem. 293 (15), 5414–5424. doi:10.1074/jbc.R117.818237
Thalmann, G. N., Anezinis, P. E., Chang, S. M., Zhau, H. E., Kim, E. E., Hopwood, V. L., et al. (1994). Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 54 (10), 2577–2581.
Tran, S., Juliani, J., Harris, T. J., Evangelista, M., Ratcliffe, J., Ellis, S. L., et al. (2024). BECLIN1 is essential for intestinal homeostasis involving autophagy-independent mechanisms through its function in endocytic trafficking. Commun. Biol. 7 (1), 209. doi:10.1038/s42003-024-05890-7
Trelford, C. B., and Di Guglielmo, G. M. (2021). Molecular mechanisms of mammalian autophagy. Biochem. J. 478 (18), 3395–3421. doi:10.1042/bcj20210314
Vaites, L. P., Paulo, J. A., Huttlin, E. L., and Harper, J. W. (2018). Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/Mon1 regulator C18orf8/RMC1 in macroautophagic and selective autophagic flux. Mol. Cell Biol. 38 (1), 003922–e417. doi:10.1128/mcb.00392-17
Vallee, R. B., and Davis, S. E. (1983). Low molecular weight microtubule-associated proteins are light chains of microtubule-associated protein 1 (MAP 1). Proc. Natl. Acad. Sci. U. S. A. 80 (5), 1342–1346. doi:10.1073/pnas.80.5.1342
van Bokhoven, A., Varella-Garcia, M., Korch, C., Johannes, W. U., Smith, E. E., Miller, H. L., et al. (2003). Molecular characterization of human prostate carcinoma cell lines. Prostate 57 (3), 205–225. doi:10.1002/pros.10290
Velikkakath, A. K., Nishimura, T., Oita, E., Ishihara, N., and Mizushima, N. (2012). Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 23 (5), 896–909. doi:10.1091/mbc.E11-09-0785
Vempati, R. K., and Malla, R. R. (2020). Autophagy-induced drug resistance in liver cancer. Crit. Rev. Oncog. 25 (1), 21–30. doi:10.1615/CritRevOncog.2020034969
Wang, B., and Kundu, M. (2017). Canonical and noncanonical functions of ULK/Atg1. Curr. Opin. Cell Biol. 45, 47–54. doi:10.1016/j.ceb.2017.02.011
Wang, F., Peters, R., Jia, J., Mudd, M., Salemi, M., Allers, L., et al. (2023). ATG5 provides host protection acting as a switch in the atg8ylation cascade between autophagy and secretion. Dev. Cell 58 (10), 866–884.e8. doi:10.1016/j.devcel.2023.03.014
Wang, K. X., Yan, C., Yang, X., Zhu, P. Y., Cui, W. W., Ye, C., et al. (2022). Enhanced autophagy promotes radiosensitivity by mediating Sirt1 downregulation in RM-1 prostate cancer cells. Biochem. Biophys. Res. Commun. 609, 84–92. doi:10.1016/j.bbrc.2022.03.142
Wang, Y., Zhang, N., Zhang, L., Li, R., Fu, W., Ma, K., et al. (2016). Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol. Cell 63 (1), 34–48. doi:10.1016/j.molcel.2016.05.027
Weidberg, H., Shvets, E., Shpilka, T., Shimron, F., Shinder, V., and Elazar, Z. (2010). LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. Embo J. 29 (11), 1792–1802. doi:10.1038/emboj.2010.74
Wible, D. J., Chao, H. P., Tang, D. G., and Bratton, S. B. (2019). ATG5 cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12-ATG5-ATG16L1 complex assembly and autophagy. Cell Discov. 5, 42. doi:10.1038/s41421-019-0110-1
Wu, H. C., Hsieh, J. T., Gleave, M. E., Brown, N. M., Pathak, S., and Chung, L. W. (1994). Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int. J. Cancer 57 (3), 406–412. doi:10.1002/ijc.2910570319
Wu, W., Wang, X., Berleth, N., Deitersen, J., Wallot-Hieke, N., Böhler, P., et al. (2020). The autophagy-initiating kinase ULK1 controls RIPK1-mediated cell death. Cell Rep. 31 (3), 107547. doi:10.1016/j.celrep.2020.107547
Xie, Z., and Klionsky, D. J. (2007). Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9 (10), 1102–1109. doi:10.1038/ncb1007-1102
Xu, T., Nicolson, S., Denton, D., and Kumar, S. (2015). Distinct requirements of Autophagy-related genes in programmed cell death. Cell Death Differ. 22 (11), 1792–1802. doi:10.1038/cdd.2015.28
Yang, S. W., Ping, Y. F., Jiang, Y. X., Luo, X., Zhang, X., Bian, X. W., et al. (2016). ATG4A promotes tumor metastasis by inducing the epithelial-mesenchymal transition and stem-like properties in gastric cells. Oncotarget 7 (26), 39279–39292. doi:10.18632/oncotarget.9827
Yang, Z., and Klionsky, D. J. (2009). An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 335, 1–32. doi:10.1007/978-3-642-00302-8_1
Yoon, J., Hwang, Y., Yun, H., Chung, J. M., Kim, S., Kim, G., et al. (2024). LC3B drives transcription-associated homologous recombination via direct interaction with R-loops. Nucleic Acids Res., gkae156. doi:10.1093/nar/gkae156
Yoon, Y. S., Cho, E. D., Jung Ahn, W., Won Lee, K., Lee, S. J., and Lee, H. J. (2017). Is trehalose an autophagic inducer? Unraveling the roles of non-reducing disaccharides on autophagic flux and alpha-synuclein aggregation. Cell Death Dis. 8 (10), e3091. doi:10.1038/cddis.2017.501
Yorimitsu, T., and Klionsky, D. J. (2005). Autophagy: molecular machinery for self-eating. Cell Death Differ. 12 (Suppl. 2), 1542–1552. doi:10.1038/sj.cdd.4401765
Yousefi, S., Perozzo, R., Schmid, I., Ziemiecki, A., Schaffner, T., Scapozza, L., et al. (2006). Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 8 (10), 1124–1132. doi:10.1038/ncb1482
Zeng, J., Liu, W., Fan, Y. Z., He, D. L., and Li, L. (2018). PrLZ increases prostate cancer docetaxel resistance by inhibiting LKB1/AMPK-mediated autophagy. Theranostics 8 (1), 109–123. doi:10.7150/thno.20356
Zhang, H. Y., Ma, Y. D., Zhang, Y., Cui, J., and Wang, Z. M. (2017). Elevated levels of autophagy-related marker ULK1 and mitochondrion-associated autophagy inhibitor LRPPRC are associated with biochemical progression and overall survival after androgen deprivation therapy in patients with metastatic prostate cancer. J. Clin. Pathol. 70 (5), 383–389. doi:10.1136/jclinpath-2016-203926
Zhang, Y., Guo, R., Wang, S. S., Jiang, X. Y., Cui, H. Y., Guo, Y., et al. (2022a). Autophagy-related proteins in genome stability: autophagy-dependent and independent actions. Int. J. Biol. Sci. 18 (14), 5329–5344. doi:10.7150/ijbs.76134
Zhang, Y., Song, X. L., Yu, B., Foong, L. C., Shu, Y., Mai, C. W., et al. (2022b). TP53 loss-of-function causes vulnerability to autophagy inhibition in aggressive prostate cancer. Int. J. Urol. 29 (9), 1085–1094. doi:10.1111/iju.15021
Zhong, Z., Sanchez-Lopez, E., and Karin, M. (2016). Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 166 (2), 288–298. doi:10.1016/j.cell.2016.05.051
Zietman, A. L., DeSilvio, M. L., Slater, J. D., Rossi, C. J., Miller, D. W., Adams, J. A., et al. (2005). Comparison of conventional-dose vs high-dose conformal radiation therapy in clinically localized adenocarcinoma of the prostate: a randomized controlled trial. Jama 294 (10), 1233–1239. doi:10.1001/jama.294.10.1233
Keywords: autophagy, prostate, cancer, carcinogenesis, therapy, androgen deprivation, radiation, chemotherapy
Citation: Kurganovs NJ and Engedal N (2024) To eat or not to eat: a critical review on the role of autophagy in prostate carcinogenesis and prostate cancer therapeutics. Front. Pharmacol. 15:1419806. doi: 10.3389/fphar.2024.1419806
Received: 18 April 2024; Accepted: 20 May 2024;
Published: 07 June 2024.
Edited by:
Valerie R. Wiersma, University Medical Center Groningen, NetherlandsReviewed by:
Vildan Bozok Cetintas, Ege University, TürkiyeEvangelos Koustas, National and Kapodistrian University of Athens, Greece
Yunus Akkoc, Research Center for Translational Medicine—Koç University, Türkiye
Copyright © 2024 Kurganovs and Engedal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalie Jayne Kurganovs, bmF0YWxpZS5qYXluZS5rdXJnYW5vdnNAcnItcmVzZWFyY2gubm8=; Nikolai Engedal, bmlrb2xhaS5lbmdlZGFsQHJyLXJlc2VhcmNoLm5v