 Mingli Sun
Mingli Sun Xin Zhang1†
Xin Zhang1† Xiaopeng Zhao
Xiaopeng Zhao- 1College of Exercise and Health, Shenyang Sport University, Shenyang, Liaoning, China
- 2College of Basic Medical Science, China Medical University, Shenyang, Liaoning, China
- 3Innovation Institute, China Medical University, Shenyang, Liaoning, China
As a chemotherapy agent, doxorubicin is used to combat cancer. However, cardiotoxicity has limited its use. The existing strategies fail to eliminate doxorubicin-induced cardiotoxicity, and an in-depth exploration of its pathogenesis is in urgent need to address the issue. Endoplasmic reticulum stress (ERS) occurs when Endoplasmic Reticulum (ER) dysfunction results in the accumulation of unfolded or misfolded proteins. Adaptive ERS helps regulate protein synthesis to maintain cellular homeostasis, while prolonged ERS stimulation may induce cell apoptosis, leading to dysfunction and damage to tissue and organs. Numerous studies on doxorubicin-induced cardiotoxicity strongly link excessive activation of the ERS to mechanisms including oxidative stress, calcium imbalance, autophagy, ubiquitination, and apoptosis. The researchers also found several clinical drugs, chemical compounds, phytochemicals, and miRNAs inhibited doxorubicin-induced cardiotoxicity by targeting ERS. The present review aims to outline the interactions between ERS and other mechanisms in doxorubicin-induced cardiotoxicity and summarize ERS’s role in this type of cardiotoxicity. Additionally, the review enumerates several clinical drugs, phytochemicals, chemical compounds, and miRNAs targeting ERS for considering therapeutic regimens that address doxorubicin-induced cardiotoxicity.
1 Introduction
Doxorubicin (DOX), an anthracycline anticancer drug obtained from the Streptomyces Peutius (Damiani et al., 2016), has been widely applied to the treatment of various types of cancer, such as breast cancer, ovarian cancer, and multiple myeloma (Duggan and Keating, 2011). However, cumulative doxorubicin dose exceeding 400–700 mg/m2 in adults and 300 mg/m2 in children may lead to cardiac damage (Li and Hill, 2014), manifested mainly by arrhythmia, cardiomyopathy, left ventricular dysfunction, and even congestive heart failure, by which its use has been limited. Clinically, echocardiogram, cardiac troponin T, and other cardiac injury-related indicators are detected to diagnose doxorubicin-induced cardiotoxicity (Mitry and Edwards, 2016). Currently, combination therapy of doxorubicin with other anticancer drugs is put into use to alleviate this side effect, and another antioxidant drug, Dexrazoxane, is also beneficial for its prevention (Ludke et al., 2009). Nevertheless, such strategies fail to eliminate doxorubicin-induced cardiotoxicity, and an in-depth exploration of its pathogenesis is in urgent need to address the issue. Some molecular pathological mechanisms, such as inflammation, oxidative stress, programmed cell death, metabolic disturbance, and endoplasmic reticulum stress, etc., are involved in doxorubicin-induced cardiotoxicity (Renu et al., 2018), among which ERS is gradually brought to the forefront. The principal pathological mechanism of doxorubicin-induced cardiotoxicity is illustrated in Figure 1.
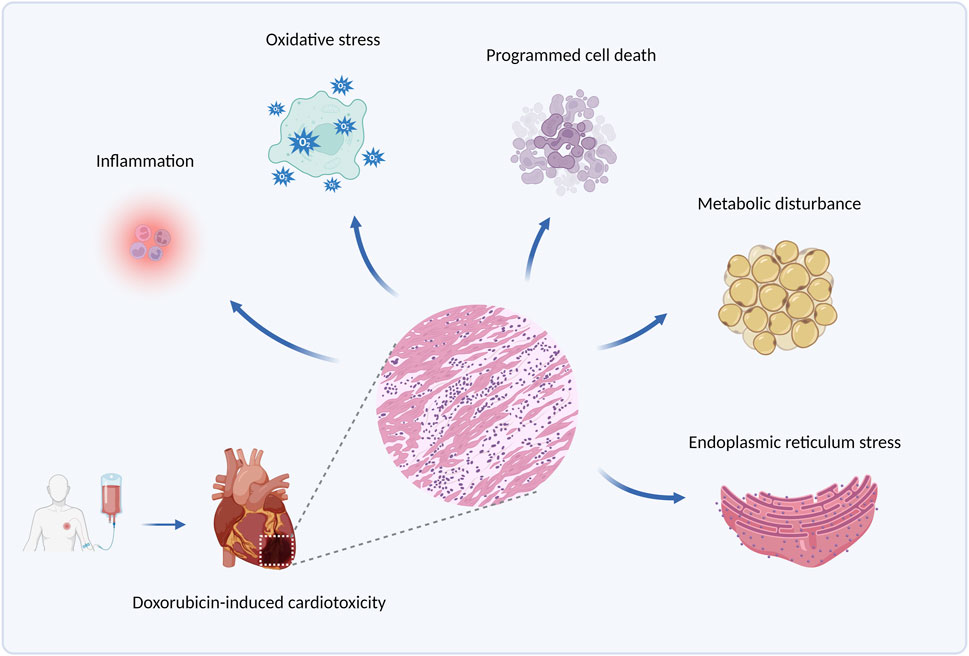
Figure 1. The principal pathological mechanisms underlying doxorubicin-induced cardiotoxicity. Produced using BioRender.com (www.biorender.com). Pathological mechanisms underlying doxorubicin-induced cardiotoxicity involve inflammation, oxidative stress, programmed cell death (such as apoptosis, autophagy, and ferroptosis), metabolic disturbance, endoplasmic reticulum stress, etc.
Endoplasmic reticulum stress (ERS) is a state of cells under various stimulations that cause unfolded or misfolded proteins to accumulate in the Endoplasmic Reticulum (ER). In the short term, adaptive ERS helps regulate protein synthesis to maintain cellular homeostasis (Yoshida, 2007), while prolonged ERS stimulation may induce cell apoptosis, leading to dysfunction and damage to tissue and organs (Ayaub et al., 2016). ERS is associated with the onset and progression of multiple diseases, including diabetes (Oakes and Papa, 2015), bone disease (Wen et al., 2023), intervertebral disc degeneration (Huang et al., 2022b), and cardiovascular disease (Zhu and Zhou, 2021a). Recent studies have also identified ERS as one of the contributors to doxorubicin-induced cardiotoxicity (Renu et al., 2018; Yarmohammadi et al., 2024), and a review in 2021 summarized the research progress on the role of ERS in doxorubicin-induced cardiotoxicity, providing us with a promising target for its treatment (Yarmohammadi et al., 2021). Subsequently, a growing number of studies investigated the potential role of ERS in doxorubicin-induced cardiotoxicity, so it is necessary for an updated summary of this topic.
In this review, the activation of ERS and its participation in doxorubicin-induced cardiotoxicity via various mechanisms including autophagy, ubiquitination, oxidative stress, calcium homeostasis disorder, and apoptosis will be elaborated in sequence. Moreover, potential drugs, compounds, and miRNAs targeting ERS to address doxorubicin-induced cardiotoxicity will be enumerated, to provide direct evidence for its feasibility.
2 ERS and the unfolded protein responses
ER is an essential intracellular protein-processing organelle, whose homeostasis is crucial for normal cellular function (Ellgaard and Helenius, 2003). Under certain conditions, unfolded or misfolded proteins accumulate in ER and induce ERS (Yap et al., 2021). Once ERS happens, the cell activates a process called unfolded protein responses (UPR) to relieve ERS and assist ER back to the state of homeostasis (Malhotra and Kaufman, 2007). UPR consists of three critical ERS receptors: PKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) (Meyerovich et al., 2016). In mammalian cells, ERS activation is regulated by 78-kDa glucose-regulated protein (GRP78), a molecular chaperone in ER (Kimata and Kohno, 2011). Under physiological state, the aforementioned three ERS sensors bind to GRP78 and lose their functions temporarily (Ren et al., 2021). However, the accumulation of unfolded or misfolded proteins promotes the dissociation of GRP78 from ERS sensors and its interaction with unfolded proteins, resulting in the occurrence of UPR (Yap et al., 2021). It has also been suggested that unfolded or misfolded proteins can directly activate IRE1 (Gardner et al., 2013), whereas the activation of ATF6 requires the protein disulfide bond isomerase A5 (PDIA5) (Higa et al., 2014).
PERK is a type I transmembrane protein on ER with a protein kinase structural domain, which is structurally similar to eukaryotic translation-initiation factor 2α (elF2α) kinase (Harding et al., 1999). When dissociation happens between GRP78 and PERK, PERK undergoes homodimerization and trans-phosphorylation, which in turn causes elF2α phosphorylation and inactivation. Inactivation of elF2α inhibits most mRNA translations and protein productions, in which way ERS could be mitigated (Bertolotti et al., 2000; Raven et al., 2008). However, phosphorylated elF2α also upregulates the translation of activating transcription factor 4 (ATF4) (Harding et al., 2003), which turns out to induce apoptosis by increasing the expression of C/EBP homologous protein (CHOP) (Harding et al., 2003; Hu et al., 2018). More than that, ATF4 also enhances elF2α dephosphorylation by increasing GADD34 expression, thereby restoring the impaired translation process (Hu et al., 2018). ATF6 is another transcriptional factor (Glembotski, 2014) with a distinct activation pattern from PERK and IRE1. After the dissociation of ATF6 and GRP78, Golgi localization signalling is activated, allowing ATF6 to enter and obtain its active fragment through the degradation by resident proteases s1 and s2 (Shen et al., 2002). The fragment, ATF6 (p50), enters the nucleus and regulates ERS-related gene transcription (Endres and Reinhardt, 2013). IRE1, another transmembrane protein on ER with the protein kinase and RNase potential (Lee et al., 2008), activates after dissociation with GRP78 and cleaves the mRNA of X-box binding protein 1 (XBP1), resulting in the formation of sheared XBP1 (XBP1s) (Yoshida et al., 2001). Meanwhile, the RNA endonuclease structural domain of IRE1 tears up other mRNAs to destroy them, thus mitigating ERS, a process we call regulated IRE1-dependent decay (RIDD) (Almanza et al., 2022). In addition, IRE1 is capable of regulating cell apoptosis, by not only the activation of CHOPs through the IRE1-XBP1 pathway (Peña and Harris, 2011) but also the awakening of JUK and caspase-12 (Yoneda et al., 2001; Chakrabarti et al., 2011).
An adaptive UPR positively works in maintaining normal cellular function (Hetz, 2012). However, prolonged or excessive ERS turns adaptive UPR into a “maladaptive UPR” that drives apoptosis (Hetz et al., 2015). Excessive UPR correlates with various diseases, including diabetes, metabolic disorders, and inflammation (Wang and Kaufman, 2012a). In doxorubicin-induced cardiotoxicity, UPR acts as a double-edged sword. On one hand, maladaptive UPR participates in various pathological processes such as apoptosis, and aggravating doxorubicin-induced cardiac injury (Yarmohammadi et al., 2021). On the other hand, activating adaptive UPR plays a protective role (Maiuolo et al., 2021; Fa et al., 2022). Figure 2 summarizes the principal process of UPR response.
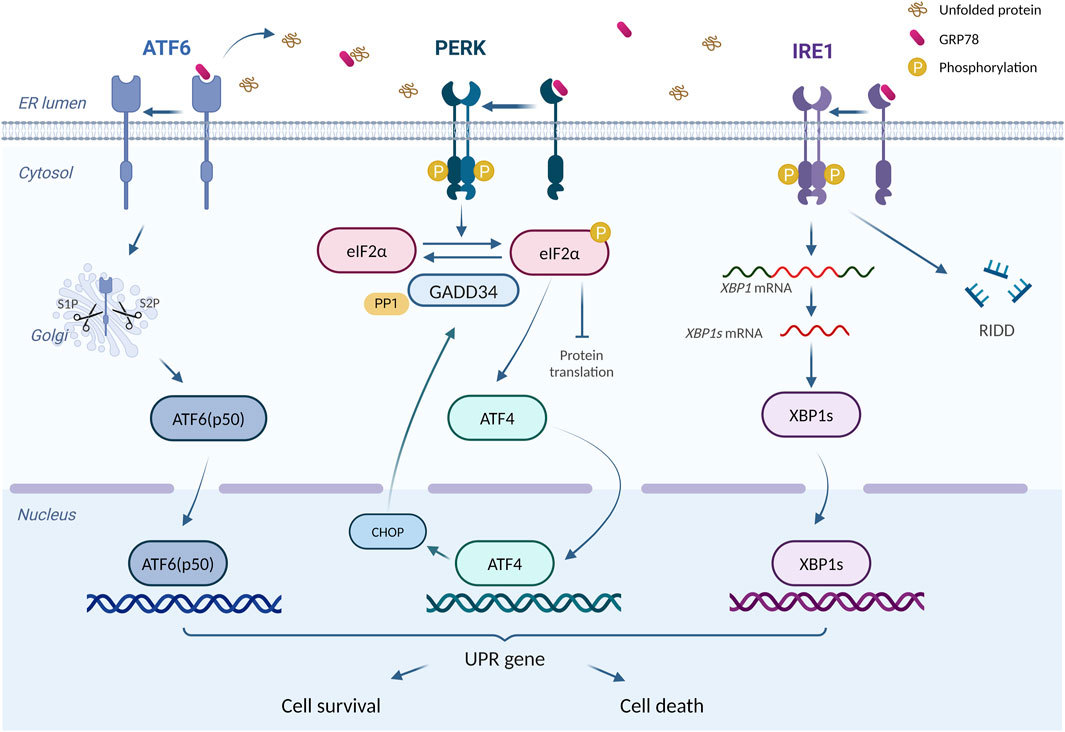
Figure 2. The principal process of UPR response. “UPR Signalling (ATF6, PERK, IRE1)” is produced using BioRender.com (www.biorender.com). Retrieved from https://app.biorender.com/biorender-templates. Dissociates from GRP78, ATF6, IRE1, and PERK trigger the UPR response. Adaptive UPR response restores ER function and cellular homeostasis, whereas excessive ERS leads to cell death.
3 The role of ERS in doxorubicin-induced cardiotoxicity
Numerous factors and intricate mechanisms are involved in the pathogenesis of doxorubicin-induced cardiotoxicity, among which ERS plays a vital role. In this section, the intricate mechanisms of ERS involved in doxorubicin-induced cardiotoxicity will be illustrated. Figure 3 summarizes the principal role of ERS in doxorubicin-induced cardiotoxicity.
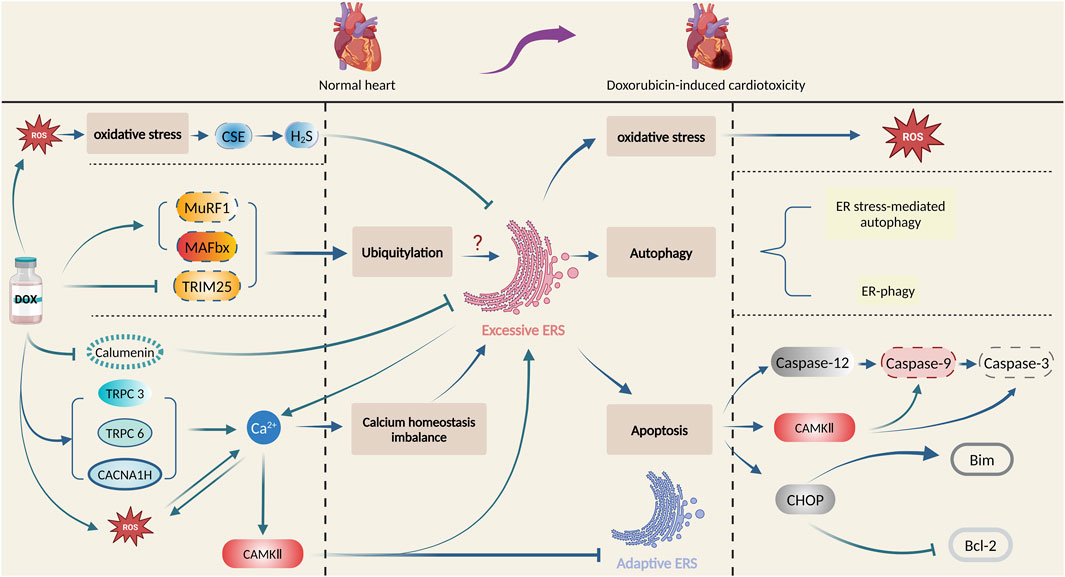
Figure 3. Principal role of ERS in doxorubicin-induced cardiotoxicity. Produced using BioRender.com (www.biorender.com). Doxorubicin promotes oxidative stress, ubiquitination, and calcium homeostasis imbalance, and then activates ERS, which leads to cardiac damage and pathological changes by multiple mechanisms such as oxidative stress, autophagy, and apoptosis.
3.1 ERS and oxidative stress
Increased levels of oxidants in cells may disturb the balance of oxidants and antioxidants, causing an interruption in redox signalling conduction and control as well as molecular damage (Sies, 2015). Oxidative stress is associated with various diseases, including atherosclerosis, chronic obstructive pulmonary disease (COPD), Alzheimer’s disease, and cancer (Forman and Zhang, 2021), also found to be essential in myocardial damage caused by doxorubicin administration (Rawat et al., 2021). Oxidative stress induces ERS (Cubillos-Ruiz et al., 2017), while ERS also promotes reactive oxygen species (ROS) generation, leading to oxidative stress (Zeeshan et al., 2016), indicating a mutual promotion of the two processes. Moreover, they are also inextricably linked to doxorubicin-induced cardiotoxicity. Recently, scientists discovered that doxorubicin-treated cardiomyocytes experienced damage and elevated the quantities of both ROS and ERS-associated proteins, indicating that both oxidative stress and ERS are implicated in doxorubicin-induced cardiotoxicity (Wang et al., 2018e; Kim et al., 2022b). They also discovered that activation of protein arginine methyltransferase 1 (PRMT1) or inhibition of the transient receptor potential ankyrin 1 (TRPA1) channel reduced ROS production and ERS and decreased their harmful effects on doxorubicin-treated cardiomyocytes (Wang et al., 2018e; Kim et al., 2022b). These findings suggest that activating PRMT1 or inhibiting TRPA1 could be potential treatments for doxorubicin-induced cardiotoxicity by targeting oxidative stress and ERS.
Cystathionine-c-lyase (CSE) is one of the key enzymes for H2S synthesis (Filipovic, 2015). Wang et al. (2012b) revealed that doxorubicin treatment led to impaired cardiac function and diminished CSE, increased ROS accumulation, and expression of ERS-associated proteins. However, administration of ROS scavenger attenuated intracellular ROS accumulation, and cardiotoxicity and corrected the activity of CSE. Moreover, H2S markedly suppressed doxorubicin-induced ERS and cardiotoxicity. This suggests that oxidative stress may lead to the inhibition of the CSE-H2S pathway, and induce ERS in doxorubicin-induced cardiotoxicity (Wang et al., 2012b).
Serum and glucocorticoid-regulated protein kinase 1 (SGK1) is a serine-threonine protein kinase from the AGC protein kinase subfamily (Sang et al., 2020). Scientists found increased SGK1 expression attenuated doxorubicin-induced cardiotoxicity in rats and simultaneously inhibited the production of ROS and ERS-associated proteins such as GRP78, while overexpressed GRP78 reverses this inhibitive effect on ROS and cardiotoxicity, suggesting that ERS exacerbates the effects of doxorubicin on cardiomyocyte through enhanced oxidative stress (Wang and Han, 2022b).
In summary, the interaction between oxidative stress and ERS amplified the cardiac injury effect caused by doxorubicin, implying a new treatment for doxorubicin-induced cardiotoxicity through an intervention on oxidative stress and ERS.
3.2 ERS and calcium homeostasis imbalance
Calcium ions, key molecules in regulating cardiac electrical and mechanical activities, may sometimes develop homeostasis imbalance in cardiomyocytes. Continuous imbalance in intracellular calcium entry and removal leads to cellular deterioration, resulting in calcium homeostasis imbalance and cell death (Berridge et al., 2003; Vassalle and Lin, 2004). Some calcium-dependent signalling pathways, such as calmodulin kinase, are crucial in the regulation of calcium homeostasis imbalance (Takemoto-Kimura et al., 2017). Imbalances in calcium homeostasis contribute to diverse pathological conditions, including ischemia-reperfusion injury, neurological disorders, pancreatitis, and doxorubicin-induced cardiotoxicity (Calvo-Rodriguez et al., 2020; Li et al., 2021; Petersen et al., 2021; Rawat et al., 2021). Excessive calcium efflux from ER leads to high cytoplasmic calcium ion levels and low levels in ER, followed by ERS induction (Ali and Petrovsky, 2019). Following doxorubicin exposure, intracellular Ca2+ concentration of cardiomyocytes is increased, while pretreatment with sarcoplasmic reticulum (SR) Ca2+ channel blockers, the Ca2+ increase is inhibited, meanwhile, doxorubicin-mediated activation of caspase-3 is significantly inhibited (Kim et al., 2006). In addition, doxorubicin exposure induces calcium-calmodulin (CaM)-dependent protein kinase II (CaMKII) -dependent SR Ca leakage, which partially contributes to cellular calcium homeostasis imbalance (Sag et al., 2011). This indicates that ERS and calcium homeostatic imbalance may be involved in doxorubicin-induced cardiotoxicity.
Altered calcium ion concentration induced by doxorubicin has a complex effect on the role of ERS in cardiomyocytes. Studies have shown that calcium channel proteins may affect ERS and doxorubicin-induced cardiotoxicity via calcium ions. Hu et al. (2019) discovered a significant increase in the expression of Cav3.2(CACNA1H) T-type calcium channel in doxorubicin-induced model rats, along with increased ERS proteins, calcium ion concentration, and developed cardiac dysfunction, while inhibition of CACNA1H channel by ABT-639 resulted in reduced ERS protein level and calcium ion concentration, and recovery of cardiac function (Hu et al., 2019). Transient Receptor Potential (TRP) protein is capable of assembling its subunits C1, C3, C5, and C6 into a calcium channel that regulates intracellular calcium ion concentration (Ohba et al., 2007). In another study, the data show that doxorubicin can disrupt calcium homeostasis and increase ERS levels in cardiomyocytes. Moreover, Salvianolic acid B is responsible for alleviating calcium overload, ERS, and doxorubicin-induced cardiotoxicity by inhibiting TRPC3 and TRPC6 (Chen et al., 2017). These suggest that calcium channel proteins CACNA1H, TRPC3, and TRPC6 may affect ERS via calcium ions in cardiomyocytes following doxorubicin exposure and are expected to become a potential target for doxorubicin-induced cardiotoxicity.
Upon doxorubicin stimulation, calcium-binding protein calumenin is also involved in regulating ERS of cardiomyocytes. Recent research has indicated that there is decreased calumenin level in cardiomyocytes following doxorubicin exposure, and several interventions such as miR378*, astragalus injection, and creatine phosphate disodium salt could inhibit doxorubicin-induced ERS and cardiotoxicity by targeting calumenin (Ma et al., 2016; Wang et al., 2018a; Wang et al., 2018c). These studies indicate that calumenin-regulated calcium homeostasis could alleviate doxorubicin-induced ERS and cardiotoxicity.
CaMKII also regulates ERS in doxorubicin-induced cardiotoxicity. Researchers found Ca2+ dependent CaMKII activation is involved in doxorubicin-induced cardiotoxicity by promoting apoptosis, while ERS sensor GRP78 overexpression partly protects cardiomyocytes from doxorubicin-induced cell death by regulating Ca2+ dependent pathways (Tscheschner et al., 2019). Scientists also found that CaMKII activation is involved in doxorubicin-induced cardiotoxicity by triggering endoplasmic reticulum stress and apoptosis by regulating the IRE1α/XBP1s pathway (Kong et al., 2022). These have provided evidence that CaMKII might hinder defensive ERS in doxorubicin-induced cardiotoxicity.
There is the ER-mitochondria interconnection in cardiomyocytes following doxorubicin exposure, mediating the interaction of ROS and calcium homeostasis disturbance upon ERS. ERS can be triggered by the dysfunction of mitochondria-associated membranes (MAMs), where Ca2+ transduction between the ER and mitochondria takes place (Kim and Choi, 2021). Mitochondria is an important source of ROS during ERS (Cao and Kaufman, 2014). During ERS, there is a vicious cycle between Ca2+ release from the ER and mitochondrial ROS production that disrupts cellular homeostasis and induces apoptosis (Csordás and Hajnóczky, 2009). Upon the stimulation by doxorubicin, the intracellular Ca2+ concentration of cardiomyocytes is increased, while the Ca2+ increase is inhibited by pretreatment with sarcoplasmic reticulum (SR) Ca2+ channel blockers and antioxidants. In addition, doxorubicin also induces ROS generation of cardiomyocytes in a time-dependent manner, while ROS production is blocked by the pretreatment of the SR Ca2+ channel blockers and the antioxidants. These results demonstrate that doxorubicin could induce ROS generation of cardiomyocytes, thus resulting in an increase in intracellular Ca2+ and that the increased intracellular Ca2+ further induces ROS production of cardiomyocytes (Kim et al., 2006).
In conclusion, following doxorubicin-induced ERS, the calcium homeostasis imbalance is produced, and altered calcium ion concentration has also a complex effect on the role of ERS, with the participation of multiple calcium-dependent signalling pathways. Regulation of calcium homeostasis imbalance and ERS could be an effective solution for doxorubicin-induced cardiotoxicity. Figure 4 summarizes the interaction between ERS and calcium homeostasis imbalance in doxorubicin-induced cardiotoxicity.
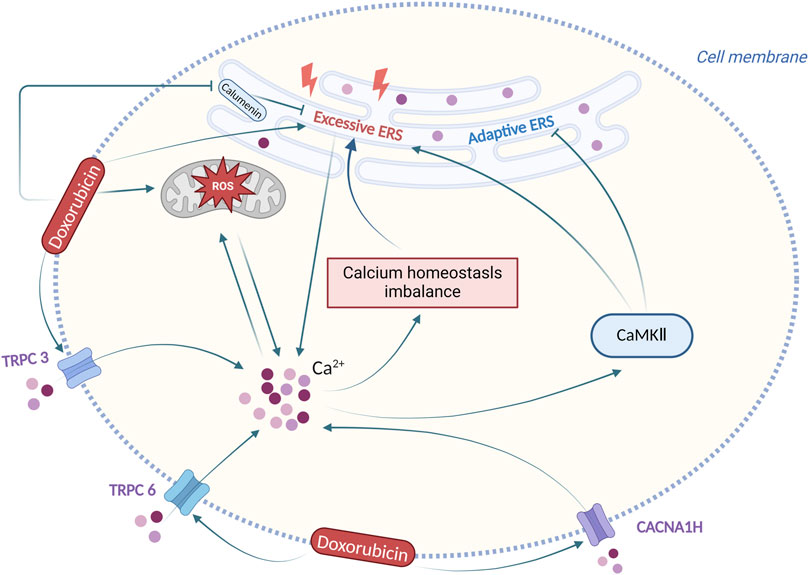
Figure 4. Interaction between ERS and calcium homeostasis imbalance in doxorubicin-induced cardiotoxicity. Produced using BioRender.com (www.biorender.com). Following doxorubicin stimulation, the ERS and the calcium homeostasis imbalance are produced, and altered calcium ion concentration has also a complex effect on ERS.
3.3 ERS and autophagy
Autophagy, a process of degradation of damaged cellular components and macromolecules as well as the recycling of catabolic products, is crucial for maintaining intracellular homeostasis (Parzych and Klionsky, 2014). Studies have shown that doxorubicin leads to cell death by affecting the expression of autophagy-related genes to initiate excessive autophagy while restoring normal autophagy protects the heart from doxorubicin-induced cardiotoxicity (Zhu et al., 2009; Bartlett et al., 2016; Li et al., 2016; He et al., 2021). However, the underlying mechanism of autophagy in the development of doxorubicin-induced cardiotoxicity remains unclear. Autophagy is closely related to ERS response. On one hand, autophagy facilitates the restoration of ER homeostasis by eliminating accumulated unfolded and misfolded proteins from ER (Senft and Ronai, 2015). On the other hand, ERS can trigger two types of autophagy: ERS-mediated autophagy and ER-phagy (Song et al., 2018). The UPR signalling pathway is a key promoter in ERS-mediated autophagy. Receptor-mediated selective ER-phagy is initiated by multiple ER surface proteins including FAM134B, RTN3L, TEX264, and ATL3 through the LC3/GABARAP interaction region (LIR/GIM) (Liang et al., 2020). It is worth noting that ERS-associated autophagy is strongly correlated with diseases like non-alcoholic fatty liver disease (NAFLD), vascular calcification, and disc degeneration (Lebeaupin et al., 2018; Huang et al., 2022b; Rao et al., 2022).
In recent years, researchers have also been focusing on the role of ERS-mediated autophagy in doxorubicin-induced cardiotoxicity. One study investigating the impact of interleukin-12p35 on doxorubicin-induced cardiotoxicity reveals that interleukin-12p35 knockdown escalates cardiac injury in mice treated with doxorubicin, along with the occurrence of ERS and autophagy pathway activation. Conversely, mice with doxorubicin-induced cardiotoxicity were treated with interleukin-12 or interleukin-35 experienced diminished cardiac injury along with inhibited autophagy pathways and ERS, suggesting that ERS and autophagy are implicated in the pathogenesis of doxorubicin-induced cardiotoxicity, and interleukin-12p35 might be a contributor to the suppression of ERS and autophagy pathways (Ye et al., 2018). Apart from this, another study found that both FAM134B-mediated ER-phagy and non-FAM134B-mediated autophagy are involved in doxorubicin-induced cardiotoxicity. In an ex vivo doxorubicin-induced cardiotoxicity model, Qu et al. found that the overexpression of gasdermin D (GSDMD), a pyroptosis-inducing agent, promoted FAM134B-mediated ER-phagy, leading to the death of cardiomyocytes and worsening of myocardial damage. In contrast, the opposite effect was observed upon inhibition of GSDMD, indicating that FAM134B-mediated ER-phagy is involved in the pathogenesis of doxorubicin-induced cardiotoxicity, and inhibiting GSDMD can reverse the effect (Qu et al., 2022). In addition, scientists found cell-cycle progression gene 1 (CCPG1) -mediated ER-phagy could protect cardiomyocytes from ER stress induced by doxorubicin (Nakagama et al., 2023). Nevertheless, Xu et al. examined the protective effect of Rg1 against doxorubicin-induced cardiac injury in mice, concluding that the protective impact of Rg1 is accomplished by ERS-mediated autophagy (Xu et al., 2018). To summarize, ERS-induced autophagy encourages the pathogenesis of doxorubicin-induced cardiotoxicity, while GSDMD, interleukin-12p35, and Rg1 may affect such a process.
3.4 ERS and ubiquitination
Ubiquitin, a small protein molecule comprised of 76 amino acids, undergoes ubiquitination through the action of E1, E2, and E3 ubiquitin ligases, thereby taking part in the regulation of several cellular processes, such as gene transcription, cell cycle progression, and apoptosis (Shaid et al., 2013). Ubiquitination plays a significant role in tumors, neurodegenerative diseases, myasthenia gravis, diabetes, and also doxorubicin-induced cardiotoxicity (Popovic et al., 2014; Wang et al., 2022a). One study indicated that UPR could potentially regulate the ubiquitin-proteasome pathway (Schiattarella et al., 2021). However, other studies detected an elevated level of ERS proteins with impaired ubiquitination (Gerakis et al., 2019; Liu et al., 2020). These findings point to a reciprocal relationship between ubiquitination and ERS. As above, ERS is a significant risk factor in doxorubicin-induced cardiotoxicity (Renu et al., 2018). Ubiquitination may also be involved in doxorubicin-induced cardiotoxicity. In one study, mice suffering doxorubicin-induced cardiotoxicity exhibited elevated levels of E3 ligases, MuRF1 and MAFbx, followed by increased proteasomal degradation markers, and a significant rise in ERS levels, suggesting that doxorubicin could increase ERS by promoting ubiquitination, thereby contributing to cardiotoxicity (Sishi et al., 2013). Another study discovered that inhibiting ubiquitin ligase tripartite motif 25 (TRIM25) in doxorubicin-induced cardiomyocytes led to a decrease of XBP1 nuclear translocation, which increased ERS and exacerbated cardiac injury. Additionally, doxorubicin-treated mice with TRIM25 overexpression developed cardioprotective effects and improved ERS while weakened cardioprotective effects after being injected with the ER stress inducer. It was demonstrated that TRIM25 could inhibit doxorubicin-induced ERS in cardiomyocytes by affecting the activity of the p85α-XBP1 axis through ubiquitination of p85α (Shen et al., 2022), which has proved that ubiquitination has something to do with ERS that contributes to cardiotoxicity caused by doxorubicin.
In summary, doxorubicin-induced cardiotoxicity does have some association with ubiquitination-promoted ERS, and regulating the ubiquitin ligases and their downstream pathways serves as a latent way of intervening doxorubicin-induced cardiotoxicity.
3.5 ERS and apoptosis
Apoptosis is a genetically regulated process of programmed cell death crucial in internal environment homeostasis maintaining and proper functioning of organisms. It is categorized into three pathways: the endogenous pathway (mitochondrial pathway), the exogenous pathway (death receptor pathway), and the ERS-induced apoptosis pathway (Hu et al., 2018). Among them, the ERS-induced apoptosis pathway contributes to various diseases, including liver injury (Zhang et al., 2022b), pulmonary fibrosis (Burman et al., 2018), and neurodegenerative disorders (Ghemrawi and Khair, 2020). In doxorubicin-induced cardiotoxicity, ERS also plays a role in cardiac injury by initiating apoptosis (Yarmohammadi et al., 2021).
3.5.1 ERS induces apoptosis through the CHOP pathway
CHOP belongs to the family of CCAAT/enhancer-binding proteins (C/EBPs), regulating cell proliferation, differentiation, and energy metabolism (Ubeda et al., 1996), whereas it can also be regulated by ERS to induce apoptosis through the PERK, ATF6, and IRE1 pathways (Hu et al., 2018). The PERK/eIF2α/ATF4 signalling pathway predominates in the induction of CHOP (Yarmohammadi et al., 2021). CHOP enhances apoptosis mainly through two pathways. One is to upregulate pro-apoptotic proteins such as Bim and downregulate the anti-apoptotic protein Bcl-2. The other is to promote hyperoxidation of the intracellular environment via ERS oxidase 1α(ERO1α) (Tabas and Ron, 2011). During the onset of doxorubicin-induced cardiotoxicity, ERS increases the expression of CHOP through the PERK/eIF2α/ATF4 pathway, thus regulating the levels of apoptotic protein to promote apoptosis.
In one study, researchers found that doxorubicin-induced the expression of CHOP along with an enhancement of apoptosis in isolated cardiomyocytes, indicating ERS-induced apoptosis was involved in doxorubicin-induced cardiotoxicity (Bagchi et al., 2021). Another study found that in cardiomyocytes treated by doxorubicin, overexpression of stress-inducible protein insulin-like growth factor II receptor α (IGF-IIRα) could result in their apoptosis followed by enhanced ERS and induced CHOP. Nevertheless, ERS-mediated apoptosis was attenuated with increasing doses of CHOP-targeted siRNA (Pandey et al., 2022). This suggests that ERS induces apoptosis by increasing CHOP in doxorubicin-induced cardiotoxicity, and IGF-IIRα could initiate this process.
The mechanism of the increase in CHOP expression in doxorubicin-induced cardiomyocytes was further analysed. Zhu et al. (2021b) found that doxorubicin-treated mice developed impaired cardiac function with increased expression of p-PERK, p-eIF2α, ATF4, and CHOP, and the knockdown of stim1 could further aggravate this condition, leading to cardiac cell apoptosis. Kim et al. (2022b) discovered that there was a rise in ATF4 and CHOP expression along with impaired cardiac function in doxorubicin-treated mice, whereas PRMT1 overexpression led to the modification of ATF4, decreased CHOP expression, and a relief in the heart. Another study focused on the role of general control nonderepressible 2 (GCN2), a serine/threonine kinase responsible for the phosphorylation of eIF2α, and found that GCN2 knockdown in doxorubicin-treated mouse cardiomyocytes resulted in an inhibition of ATF4 and CHOP through eIF2α phosphorylation suppression, and restored cardiac function (Wang et al., 2018b).
All of the above has demonstrated that ERS promotes apoptosis by facilitating CHOP expression through the PERK/eIF2α/ATF4 pathway in doxorubicin-induced cardiotoxicity, and modulating PERK/eIF2α/ATF4 pathway may become a new intervention modality for doxorubicin-induced cardiotoxicity.
3.5.2 ERS induces apoptosis through the Caspase-12 pathway
Belonging to an inflammatory caspase, caspase-12 is a protein encoded by the human CASP12 gene that has shown a correlation with ERS-mediated apoptosis (Sahoo et al., 2023). The underlying mechanisms indicated that ERS could activate caspase-12 through an interaction between pro-caspase-12 and IRE1α via the bridging molecule TRAF2. In this way, pro-caspase-12 dissociates from TRAF2 and becomes caspase-12. Another mechanism of pro-caspase-12 activation is through the activation of m-calpain by Ca2+ released from ER during ERS. M-calpain is transferred from the cytoplasm into ER to cleave the structural domain of the caspase-recruitment domain (CARD) precursor in caspase-12 and activate it (García de la Cadena and Massieu, 2016). Upon activation, caspase-12 is translocated from ER to the cytoplasm to cleave the caspase-9 precursor, activating the effector caspase-3 and inducing apoptosis (Szegezdi et al., 2003).
In one study, doxorubicin-induced increased expression of ERS markers and chemoattractant receptor-homologous molecule expressed on T helper type 2 cells (CRTH2) in mouse cardiomyocytes, while CRTH2 knockdown could improve the survival rate of doxorubicin treated mice, decrease caspase-12 and caspase-3 levels and reduce apoptosis. In addition, blockage of m-calpain prevented the CRTH2-mediated effect, suggesting that ERS possibly promotes apoptosis through the m-calpain/caspase-12 signalling pathway and CRTH2 could regulate this process (Zuo et al., 2018). In addition, IGF-IIRα might contribute to ERS induction and promote cardiomyocyte apoptosis upon the stimulation of doxorubicin by caspase-12 (Pandey et al., 2022). Other studies found that an increase of caspase-12 as well as some ERS-related proteins in doxorubicin-treated cardiomyocytes, whereas Resolvin D1, interleukin-10 and Shengmai injections ameliorated the doxorubicin-induced apoptosis, along with a significant reduction in both ERS and caspase-12 levels (Chen et al., 2015; Wang et al., 2021; Malik et al., 2022). These suggest ERS induces apoptosis through caspase-12 in doxorubicin-induced cardiotoxicity, and the underlying mechanism of its activation is probably related to Ca2+-mediated m-calpain activation. In addition to the above mechanisms, doxorubicin-induced ERS can also be involved in caspase-dependent apoptosis by activating Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Tscheschner et al., 2019).
In summary, ERS is involved in doxorubicin-induced cardiotoxicity by promoting apoptosis through CHOP, caspase-12, and CaMKII. Targeting ERS-induced apoptosis may serve as a new intervention to treat doxorubicin-induced cardiotoxicity. Figure 5 summarizes the principal mechanisms of ERS-induced apoptosis in doxorubicin-induced cardiotoxicity.
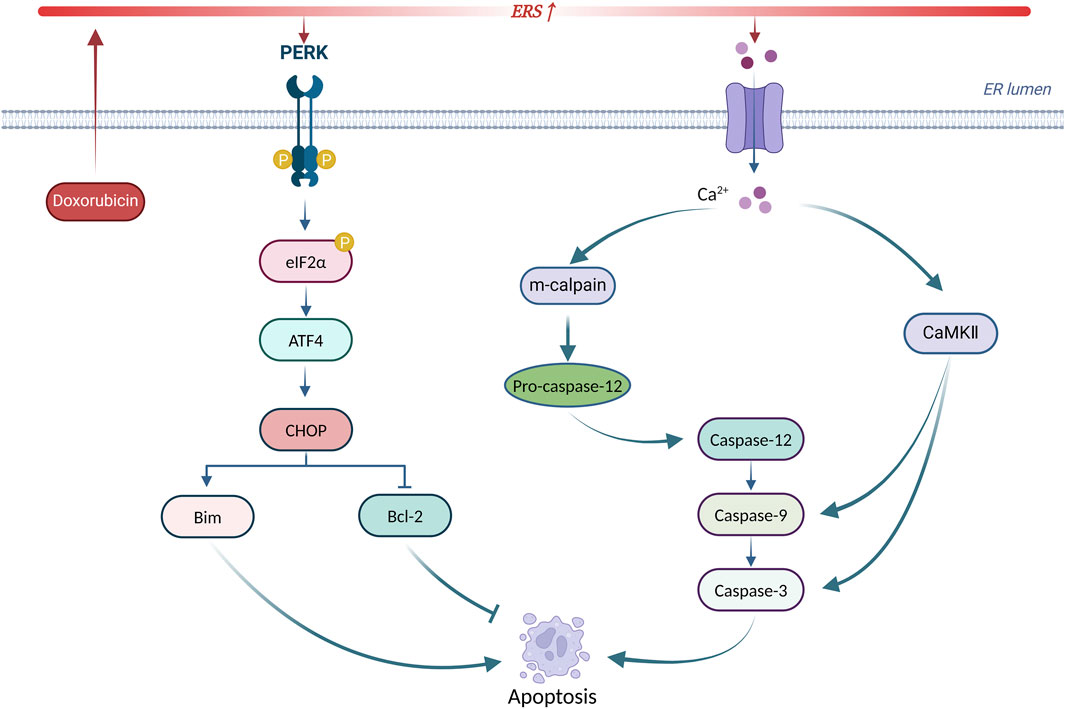
Figure 5. Principal mechanisms of ERS-induced apoptosis in doxorubicin-induced cardiotoxicity. Produced using BioRender.com (www.biorender.com). Doxorubicin activates ERS, leading to UPR receptor activation and calcium efflux. On the one hand, the PERK/eIF2α/ATF4 pathway increases CHOP expression to initiate apoptosis. On the other hand, calcium efflux activates the caspase-dependent apoptotic pathway via m-calpain leading to apoptosis. In addition, calcium efflux activates CaMKII to trigger apoptosis.
4 Targeting ERS for doxorubicin-induced cardiotoxicity
4.1 Targeting ERS by clinical drugs
The above has outlined the contribution of ERS to doxorubicin-induced cardiotoxicity, suggesting a putative target of ERS for certain interventions. Researchers have found a variety of clinical drugs that could affect doxorubicin-induced cardiotoxicity by modulating ERS. For example, Sacubitril/valsartan, Folic acid, Doxycycline, Dapagliflozin, Empagliflozin, and Diacerein could reduce ERS and apoptosis, thus inhibiting doxorubicin-induced cardiotoxicity (Lai et al., 2010; Chang et al., 2021; Ye et al., 2021; Kim et al., 2022a; El-Gohary et al., 2024; Malik et al., 2024). Astragalus injection might inhibit doxorubicin-induced excessive ERS by raising calumenin levels and inhibiting phosphorylation of connexin 43 (Cx43) (Ma et al., 2016; He et al., 2018). Moreover, creatine phosphate disodium salt could also attenuate ERS-mediated apoptosis and cardiotoxicity by regulating calumenin (Miao et al., 2016; Wang et al., 2018c). These studies have shed light on novel ways of using those conventional drugs to inhibit doxorubicin-induced cardiotoxicity.
4.2 Targeting ERS by chemical compounds
Several newly discovered chemical compounds targeting ERS have been examined the activity against doxorubicin-induced cardiotoxicity recently. For instance, 1910, Resolvin D1 and Sodium hydrosulfide (NaHS) may inhibit doxorubicin-induced cardiotoxicity by modulating CHOP-mediated apoptosis (Lu et al., 2011; Wang et al., 2012b; Wang et al., 2021). 4-phenylbutyrate (4-PBA), Pifithrin-α and Resolvin D1 could also regulate ERS-induced apoptosis through caspase-12, thus attenuating doxorubicin-induced cardiotoxicity (Chua et al., 2006; Fu et al., 2016; Wang et al., 2021).1910, Resolvin D1, and NaHS may also attenuate doxorubicin-induced apoptosis by regulating GRP78 (Lu et al., 2011; Wang et al., 2012b; Wang et al., 2021).
4.3 Targeting ERS by phytochemicals
In addition, many phytochemicals have been extracted and utilized to alleviate doxorubicin-induced cardiotoxicity. For example, total flavonoids of Selaginella tamariscina (P. Beauv.) Spring, Ononin, Salvianolic acid B, Resveratrol, Ophiopogonin D and Alginate Oligosaccharide may alleviate doxorubicin-induced cardiotoxicity by modulating CHOP (Meng et al., 2014; Lou et al., 2015; Chen et al., 2016; Guo et al., 2016; Chen et al., 2017; Zhang et al., 2022a; Gao et al., 2022). Similarly, Caffeic acid phenethyl ester may alleviate doxorubicin-induced cardiotoxicity by inhibiting ERS-associated degradation (Zhang et al., 2022c). Apart from the above effects on apoptosis, another compound, Ginsenoside Rg1 may improve doxorubicin-induced cardiac damage by inhibiting ERS and autophagy (Xu et al., 2018). Additionally, Bergamot Polyphenolic Fraction, Olea europea L. Extract, and Cynara cardunculus may reduce doxorubicin-induced cardiotoxicity by modulating ATF6 (Maiuolo et al., 2021). Biotin-conjugated ADTM analog (BAA) may attenuate doxorubicin-induced apoptosis by regulating GRP78 (Zhang et al., 2019).
4.4 Targeting ERS by microRNAs
Apart from the above-mentioned, non-coding RNAs especially microRNAs also show a potential to manage ERS and reduce doxorubicin-induced cardiotoxicity. miR-378 plays a crucial role in various cancers (Zeng et al., 2017), and it also plays a key role in doxorubicin-induced cardiotoxicity by regulating ERS-induced apoptosis. The underlying mechanisms indicate that miR-378 regulates cyclophilin A (PPIA), a regulator of protein folding (Nigro et al., 2013), to restrain the PERK-CHOP pathway and inhibit doxorubicin-induced cardiotoxicity (Wang et al., 2018d). Another study demonstrated that miR-378 could increase the level of calumenin to mitigate ERS induced by calcium homeostasis imbalance, thus alleviating doxorubicin-induced cardiotoxicity. Moreover, Huang et al. (2022a) discovered that miR-181a-5p, another microRNA whose content could be improved by intravenous injection of mesenchymal stromal cells, reduced ERS-induced apoptosis in doxorubicin treated cardiomyocytes via inhibiting GRP78. In addition, scientists found inhibition of miR-194-5p could alleviate doxorubicin-induced cardiotoxicity via P21-activated kinase 2 (PAK2) and XBP1s, and miR-194-5p/PAK2/XBP1s axis might be the potential targets for doxorubicin-induced cardiotoxicity (Fa et al., 2022).
In summary, various medications have been proven accessible to relieve doxorubicin-induced cardiotoxicity by altering ERS, and modulation of ERS may serve as a new target for the treatment of doxorubicin-induced cardiotoxicity. With consistently advanced and updated science and technology, more pieces of the puzzle of ERS in doxorubicin-induced cardiotoxicity will be found, which will provide a brighter future in the treatment of doxorubicin-induced cardiotoxicity by targeting ERS. Table 1 summarizes principal compounds targeting ERS to improve doxorubicin-induced cardiotoxicity, and Figure 6 summarizes the interventions targeting ERS in doxorubicin-induced cardiotoxicity.
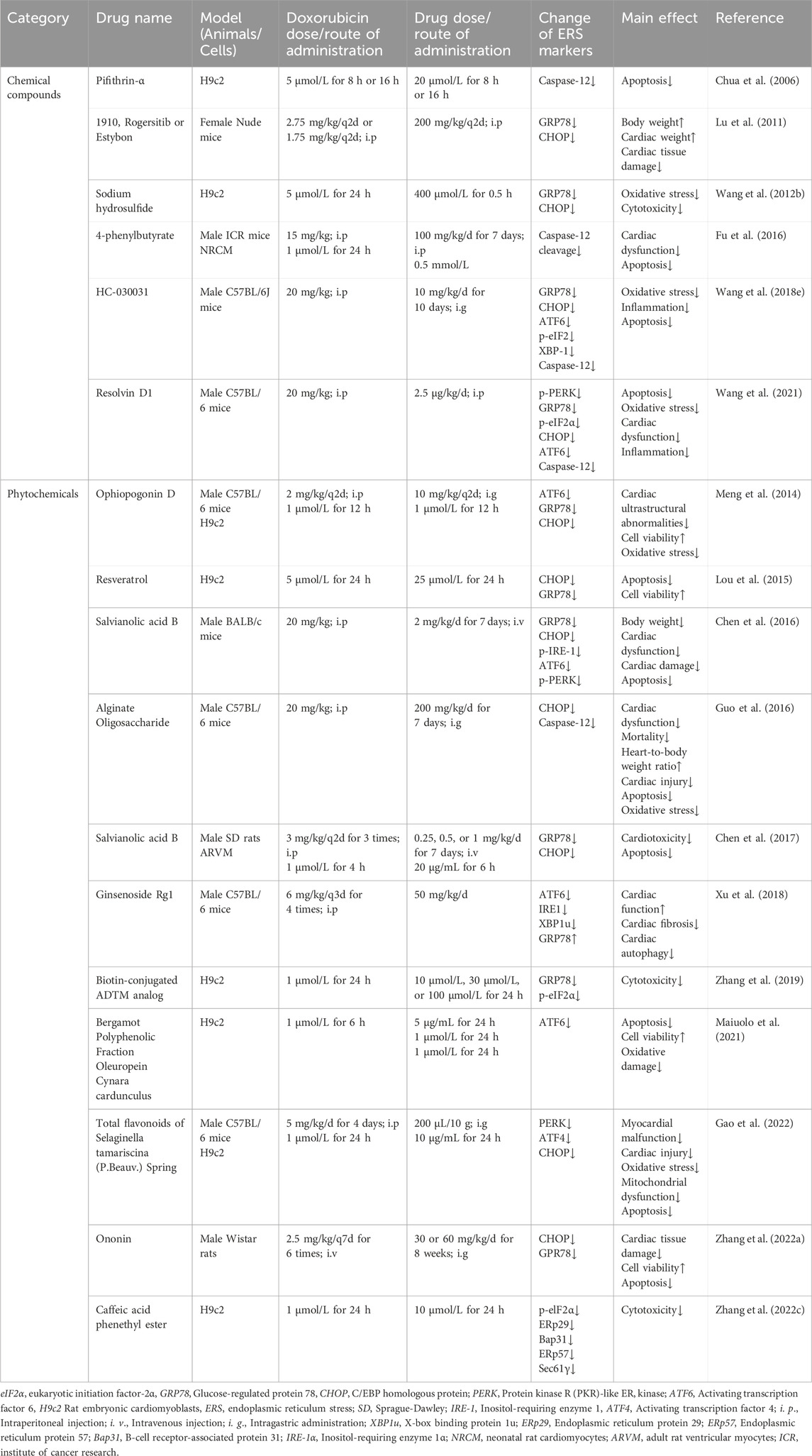
Table 1. Principal compounds targeting ERS to improve doxorubicin-induced cardiotoxicity.
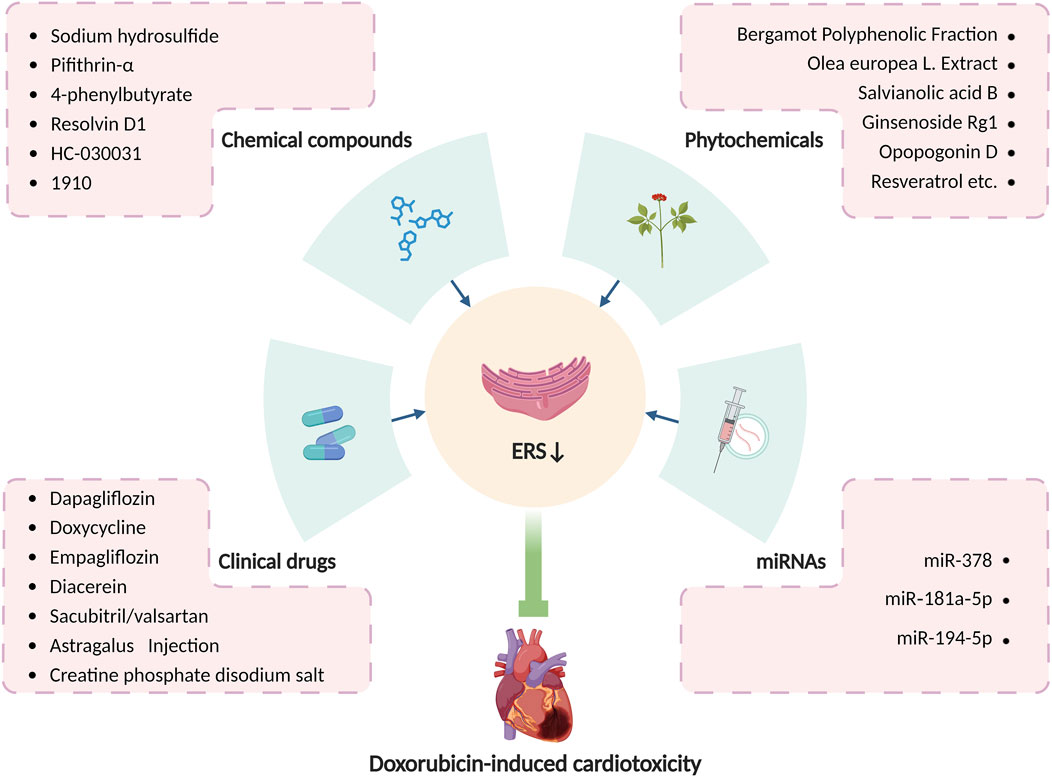
Figure 6. Interventions targeting ERS in doxorubicin-induced cardiotoxicity. Produced using BioRender.com (www.biorender.com). Many clinical drugs, chemical compounds, phytochemicals, and some microRNAs can ameliorate doxorubicin-induced cardiotoxicity by targeting ERS.
5 Future and prospect
Current studies suggest that ERS is a crucial factor in the development of doxorubicin-induced cardiotoxicity. Various studies have shown that doxorubicin causes cardiac injury through numerous mechanisms related to ERS, including autophagy, ubiquitination, oxidative stress, and apoptosis, demonstrating a possibility that ERS inhibition may come to an effect in the intervention of doxorubicin-induced cardiotoxicity. As evidence, newly discovered drugs and methods targeting ERS have been proven to reduce doxorubicin-induced cardiotoxicity. However, the ERS-UPR system, which serves as a mechanism for the cell to restore homeostasis, is ubiquitous in cells. Therefore, excessive inhibition of ERS may exacerbate the disease. So, it is of great value to investigate how to regulate it appropriately. In addition, it remains unclear whether ERS inhibitors adversely affect other parts of the human body, which may bring multiple side effects. Therefore, it is important to evaluate the side effects of ERS inhibitors. The present studies mainly focus on the role of ERS in doxorubicin-treated animal cardiac tissues or cardiomyocytes, not the accurate role of ERS in cardiac tissues of patients treated by doxorubicin, resulting in an urgent need for further clinical trials to confirm whether the ERS inhibitor can be used for treatment effectively. Moreover, experimental evidence indicates that the UPR pathway suppresses doxorubicin-induced cardiotoxicity in the early stages, but a conversion from beneficial UPR to the excessive ERS response will lead to cardiac impairment, with a still perplexing mechanism, and how the three UPR pathways activate and interact with each other, need further investigation.
This review has presented an overview of the interaction of ERS with various mechanisms, including autophagy, ubiquitination, oxidative stress, calcium homeostasis imbalance, and apoptosis in the development of doxorubicin-induced cardiotoxicity. Furthermore, different types of potential interventions targeting ERS for doxorubicin-induced cardiotoxicity, including clinical drugs, chemical compounds, phytochemicals as well as miRNA, are also embraced. We are confident that as soon as all speculations turn into certainties, modulation of ERS will show its capabilities in the treatment of doxorubicin-induced cardiotoxicity.
Author contributions
MS: Writing–review and editing, Writing–original draft, Supervision, Conceptualization. XZ: Writing–original draft, Writing–review and editing, Visualization. BT: Writing–original draft. QZ: Writing–original draft. XPZ: Writing–original draft. DD: Writing–original draft.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ali, E. S., and Petrovsky, N. (2019). Calcium signaling as a therapeutic target for liver steatosis. Trends Endocrinol. Metab. 30 (4), 270–281. doi:10.1016/j.tem.2019.02.005
Almanza, A., Mnich, K., Blomme, A., Robinson, C. M., Rodriguez-Blanco, G., Kierszniowska, S., et al. (2022). Regulated IRE1α-dependent decay (RIDD)-mediated reprograming of lipid metabolism in cancer. Nat. Commun. 13 (1), 2493. doi:10.1038/s41467-022-30159-0
Ayaub, E. A., Kolb, P. S., Mohammed-Ali, Z., Tat, V., Murphy, J., Bellaye, P. S., et al. (2016). GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J. Pathol. 239 (4), 411–425. doi:10.1002/path.4738
Bagchi, A. K., Malik, A., Akolkar, G., Jassal, D. S., and Singal, P. K. (2021). Endoplasmic reticulum stress promotes iNOS/NO and influences inflammation in the development of doxorubicin-induced cardiomyopathy. Antioxidants (Basel) 10 (12), 1897. doi:10.3390/antiox10121897
Bartlett, J. J., Trivedi, P. C., Yeung, P., Kienesberger, P. C., and Pulinilkunnil, T. (2016). Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 473 (21), 3769–3789. doi:10.1042/bcj20160385
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4 (7), 517–529. doi:10.1038/nrm1155
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2 (6), 326–332. doi:10.1038/35014014
Burman, A., Tanjore, H., and Blackwell, T. S. (2018). Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 68-69, 355–365. doi:10.1016/j.matbio.2018.03.015
Calvo-Rodriguez, M., Hou, S. S., Snyder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer's disease. Nat. Commun. 11 (1), 2146. doi:10.1038/s41467-020-16074-2
Cao, S. S., and Kaufman, R. J. (2014). Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal 21 (3), 396–413. doi:10.1089/ars.2014.5851
Chakrabarti, A., Chen, A. W., and Varner, J. D. (2011). A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 108 (12), 2777–2793. doi:10.1002/bit.23282
Chang, W. T., Lin, Y. W., Ho, C. H., Chen, Z. C., Liu, P. Y., and Shih, J. Y. (2021). Dapagliflozin suppresses ER stress and protects doxorubicin-induced cardiotoxicity in breast cancer patients. Arch. Toxicol. 95 (2), 659–671. doi:10.1007/s00204-020-02951-8
Chen, R., Sun, G., Yang, L., Wang, J., and Sun, X. (2016). Salvianolic acid B protects against doxorubicin induced cardiac dysfunction via inhibition of ER stress mediated cardiomyocyte apoptosis. Toxicol. Res. (Camb) 5 (5), 1335–1345. doi:10.1039/c6tx00111d
Chen, R. C., Sun, G. B., Ye, J. X., Wang, J., Zhang, M. D., and Sun, X. B. (2017). Salvianolic acid B attenuates doxorubicin-induced ER stress by inhibiting TRPC3 and TRPC6 mediated Ca(2+) overload in rat cardiomyocytes. Toxicol. Lett. 276, 21–30. doi:10.1016/j.toxlet.2017.04.010
Chen, Y., Tang, Y., Xiang, Y., Xie, Y. Q., Huang, X. H., and Zhang, Y. C. (2015). Shengmai injection improved doxorubicin-induced cardiomyopathy by alleviating myocardial endoplasmic reticulum stress and caspase-12 dependent apoptosis. Biomed. Res. Int. 2015, 952671. doi:10.1155/2015/952671
Chua, C. C., Liu, X., Gao, J., Hamdy, R. C., and Chua, B. H. (2006). Multiple actions of pifithrin-alpha on doxorubicin-induced apoptosis in rat myoblastic H9c2 cells. Am. J. Physiol. Heart Circ. Physiol. 290 (6), H2606–H2613. doi:10.1152/ajpheart.01138.2005
Csordás, G., and Hajnóczky, G. (2009). SR/ER-mitochondrial local communication: calcium and ROS. Biochim. Biophys. Acta 1787 (11), 1352–1362. doi:10.1016/j.bbabio.2009.06.004
Cubillos-Ruiz, J. R., Bettigole, S. E., and Glimcher, L. H. (2017). Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 168 (4), 692–706. doi:10.1016/j.cell.2016.12.004
Damiani, R. M., Moura, D. J., Viau, C. M., Caceres, R. A., Henriques, J. A. P., and Saffi, J. (2016). Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch. Toxicol. 90 (9), 2063–2076. doi:10.1007/s00204-016-1759-y
Duggan, S. T., and Keating, G. M. (2011). Pegylated liposomal doxorubicin: a review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related Kaposi's sarcoma. Drugs 71 (18), 2531–2558. doi:10.2165/11207510-000000000-00000
El-Gohary, R. M., Okasha, A. H., Abd El-Azeem, A. H., Abdel Ghafar, M. T., Ibrahim, S., Hegab, I. I., et al. (2024). Uncovering the cardioprotective potential of Diacerein in doxorubicin cardiotoxicity: mitigating ferritinophagy-mediated ferroptosis via upregulating NRF2/SLC7A11/GPX4 Axis. Antioxidants (Basel) 13 (4), 493. doi:10.3390/antiox13040493
Ellgaard, L., and Helenius, A. (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4 (3), 181–191. doi:10.1038/nrm1052
Endres, K., and Reinhardt, S. (2013). ER-stress in Alzheimer's disease: turning the scale? Am. J. Neurodegener. Dis. 2 (4), 247–265.
Fa, H., Xiao, D., Chang, W., Ding, L., Yang, L., Wang, Y., et al. (2022). MicroRNA-194-5p attenuates doxorubicin-induced cardiomyocyte apoptosis and endoplasmic reticulum stress by targeting P21-activated kinase 2. Front. Cardiovasc Med. 9, 815916. doi:10.3389/fcvm.2022.815916
Filipovic, M. R. (2015). Persulfidation (S-sulfhydration) and H2S. Handb. Exp. Pharmacol. 230, 29–59. doi:10.1007/978-3-319-18144-8_2
Forman, H. J., and Zhang, H. (2021). Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20 (9), 689–709. doi:10.1038/s41573-021-00233-1
Fu, H. Y., Sanada, S., Matsuzaki, T., Liao, Y., Okuda, K., Yamato, M., et al. (2016). Chemical endoplasmic reticulum chaperone alleviates doxorubicin-induced cardiac dysfunction. Circ. Res. 118 (5), 798–809. doi:10.1161/circresaha.115.307604
Gao, L., Yuan, P., Wei, Y., Fu, Y., Hou, Y., Li, P., et al. (2022). Total flavonoids of Selaginella tamariscina (P.Beauv.) Spring ameliorates doxorubicin-induced cardiotoxicity by modulating mitochondrial dysfunction and endoplasmic reticulum stress via activating MFN2/PERK. Phytomedicine 100, 154065. doi:10.1016/j.phymed.2022.154065
García de la Cadena, S., and Massieu, L. (2016). Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Focus caspase-12. Apoptosis 21 (7), 763–777. doi:10.1007/s10495-016-1247-0
Gardner, B. M., Pincus, D., Gotthardt, K., Gallagher, C. M., and Walter, P. (2013). Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5 (3), a013169. doi:10.1101/cshperspect.a013169
Gerakis, Y., Quintero, M., Li, H., and Hetz, C. (2019). The UFMylation system in proteostasis and beyond. Trends Cell Biol. 29 (12), 974–986. doi:10.1016/j.tcb.2019.09.005
Ghemrawi, R., and Khair, M. (2020). Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int. J. Mol. Sci. 21 (17), 6127. doi:10.3390/ijms21176127
Glembotski, C. C. (2014). Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J. Mol. Cell Cardiol. 71, 11–15. doi:10.1016/j.yjmcc.2013.09.018
Guo, J. J., Ma, L. L., Shi, H. T., Zhu, J. B., Wu, J., Ding, Z. W., et al. (2016). Alginate Oligosaccharide prevents acute doxorubicin cardiotoxicity by suppressing oxidative stress and endoplasmic reticulum-mediated apoptosis. Mar. Drugs 14 (12), 231. doi:10.3390/md14120231
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397 (6716), 271–274. doi:10.1038/16729
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11 (3), 619–633. doi:10.1016/s1097-2765(03)00105-9
He, H., Wang, L., Qiao, Y., Yang, B., Yin, D., and He, M. (2021). Epigallocatechin-3-gallate pretreatment alleviates doxorubicin-induced ferroptosis and cardiotoxicity by upregulating AMPKα2 and activating adaptive autophagy. Redox Biol. 48, 102185. doi:10.1016/j.redox.2021.102185
He, Z. H., Shao, L. Q., Xuan, L. Y., Wang, C. G., Wei, C. X., Wang, Y., et al. (2018). Effect of Astragalus injection on cardiomyocyte apoptosis, endoplasmic reticulum stress and expression of connexin in cardiomyopathy rats induced by adriamycin. Zhongguo Ying Yong Sheng Li Xue Za Zhi 34 (2), 159–163. doi:10.12047/j.cjap.5576.2018.039
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13 (2), 89–102. doi:10.1038/nrm3270
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17 (7), 829–838. doi:10.1038/ncb3184
Higa, A., Taouji, S., Lhomond, S., Jensen, D., Fernandez-Zapico, M. E., Simpson, J. C., et al. (2014). Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell Biol. 34 (10), 1839–1849. doi:10.1128/mcb.01484-13
Hu, H., Tian, M., Ding, C., and Yu, S. (2018). The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 9, 3083. doi:10.3389/fimmu.2018.03083
Hu, J., Wu, Q., Wang, Z., Hong, J., Chen, R., Li, B., et al. (2019). Inhibition of CACNA1H attenuates doxorubicin-induced acute cardiotoxicity by affecting endoplasmic reticulum stress. Biomed. Pharmacother. 120, 109475. doi:10.1016/j.biopha.2019.109475
Huang, A., Liu, Y., Qi, X., Chen, S., Huang, H., Zhang, J., et al. (2022a). Intravenously transplanted mesenchymal stromal cells: a new endocrine reservoir for cardioprotection. Stem Cell Res. Ther. 13 (1), 253. doi:10.1186/s13287-022-02922-z
Huang, J., Zhou, Q., Ren, Q., Luo, L., Ji, G., and Zheng, T. (2022b). Endoplasmic reticulum stress associates with the development of intervertebral disc degeneration. Front. Endocrinol. (Lausanne) 13, 1094394. doi:10.3389/fendo.2022.1094394
Kim, B. S., Park, I. H., Lee, A. H., Kim, H. J., Lim, Y. H., and Shin, J. H. (2022a). Sacubitril/valsartan reduces endoplasmic reticulum stress in a rat model of doxorubicin-induced cardiotoxicity. Arch. Toxicol. 96 (4), 1065–1074. doi:10.1007/s00204-022-03241-1
Kim, C. W., and Choi, K. C. (2021). Effects of anticancer drugs on the cardiac mitochondrial toxicity and their underlying mechanisms for novel cardiac protective strategies. Life Sci. 277, 119607. doi:10.1016/j.lfs.2021.119607
Kim, S. W., Ahn, B. Y., Tran, T. T. V., Pyun, J. H., Kang, J. S., and Leem, Y. E. (2022b). PRMT1 suppresses doxorubicin-induced cardiotoxicity by inhibiting endoplasmic reticulum stress. Cell Signal 98, 110412. doi:10.1016/j.cellsig.2022.110412
Kim, S. Y., Kim, S. J., Kim, B. J., Rah, S. Y., Chung, S. M., Im, M. J., et al. (2006). Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp. Mol. Med. 38 (5), 535–545. doi:10.1038/emm.2006.63
Kimata, Y., and Kohno, K. (2011). Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol. 23 (2), 135–142. doi:10.1016/j.ceb.2010.10.008
Kong, L., Zhang, Y., Ning, J., Xu, C., Wang, Z., Yang, J., et al. (2022). CaMKII orchestrates endoplasmic reticulum stress and apoptosis in doxorubicin-induced cardiotoxicity by regulating the IRE1α/XBP1s pathway. J. Cell Mol. Med. 26 (20), 5303–5314. doi:10.1111/jcmm.17560
Lai, H. C., Yeh, Y. C., Ting, C. T., Lee, W. L., Lee, H. W., Wang, L. C., et al. (2010). Doxycycline suppresses doxorubicin-induced oxidative stress and cellular apoptosis in mouse hearts. Eur. J. Pharmacol. 644 (1-3), 176–187. doi:10.1016/j.ejphar.2010.07.010
Lebeaupin, C., Vallée, D., Hazari, Y., Hetz, C., Chevet, E., and Bailly-Maitre, B. (2018). Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 69 (4), 927–947. doi:10.1016/j.jhep.2018.06.008
Lee, K. P., Dey, M., Neculai, D., Cao, C., Dever, T. E., and Sicheri, F. (2008). Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132 (1), 89–100. doi:10.1016/j.cell.2007.10.057
Li, D. L., and Hill, J. A. (2014). Cardiomyocyte autophagy and cancer chemotherapy. J. Mol. Cell Cardiol. 71, 54–61. doi:10.1016/j.yjmcc.2013.11.007
Li, D. L., Wang, Z. V., Ding, G., Tan, W., Luo, X., Criollo, A., et al. (2016). Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133 (17), 1668–1687. doi:10.1161/circulationaha.115.017443
Li, S., Chen, J., Liu, M., Chen, Y., Wu, Y., Li, Q., et al. (2021). Protective effect of HINT2 on mitochondrial function via repressing MCU complex activation attenuates cardiac microvascular ischemia-reperfusion injury. Basic Res. Cardiol. 116 (1), 65. doi:10.1007/s00395-021-00905-4
Liang, J. R., Lingeman, E., Luong, T., Ahmed, S., Muhar, M., Nguyen, T., et al. (2020). A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 180 (6), 1160–1177.e20. doi:10.1016/j.cell.2020.02.017
Liu, Y., Tao, S., Liao, L., Li, Y., Li, H., Li, Z., et al. (2020). TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 11 (1), 348. doi:10.1038/s41467-019-14190-2
Lou, Y., Wang, Z., Xu, Y., Zhou, P., Cao, J., Li, Y., et al. (2015). Resveratrol prevents doxorubicin-induced cardiotoxicity in H9c2 cells through the inhibition of endoplasmic reticulum stress and the activation of the Sirt1 pathway. Int. J. Mol. Med. 36 (3), 873–880. doi:10.3892/ijmm.2015.2291
Lu, M., Merali, S., Gordon, R., Jiang, J., Li, Y., Mandeli, J., et al. (2011). Prevention of Doxorubicin cardiopathic changes by a benzyl styryl sulfone in mice. Genes Cancer 2 (10), 985–992. doi:10.1177/1947601911436199
Ludke, A. R., Al-Shudiefat, A. A., Dhingra, S., Jassal, D. S., and Singal, P. K. (2009). A concise description of cardioprotective strategies in doxorubicin-induced cardiotoxicity. Can. J. Physiol. Pharmacol. 87 (10), 756–763. doi:10.1139/y09-059
Ma, K. Y., Wan, Q., Wang, Y. L., Tao, X. X., Huang, X., Chai, H., et al. (2016). The effect of astragalus injection on the expression of endoplasmic reticulum stress chaperonin GRP78 and GRP94 mRNA in adriamycin-injured cardiomyocytes with calumenin silencing by shRNA. Zhongguo Ying Yong Sheng Li Xue Za Zhi 32 (2), 154–157. doi:10.13459/j.cnki.cjap.2016.02.015
Maiuolo, J., Bava, I., Carresi, C., Gliozzi, M., Musolino, V., Scarano, F., et al. (2021). The effects of Bergamot polyphenolic fraction, Cynara cardunculus, and Olea europea L. Extract on doxorubicin-induced cardiotoxicity. Nutrients 13 (7), 2158. doi:10.3390/nu13072158
Malhotra, J. D., and Kaufman, R. J. (2007). The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18 (6), 716–731. doi:10.1016/j.semcdb.2007.09.003
Malik, A., Bagchi, A. K., Jassal, D. S., and Singal, P. K. (2022). Interleukin-10 mitigates doxorubicin-induced endoplasmic reticulum stress as well as cardiomyopathy. Biomedicines 10 (4), 890. doi:10.3390/biomedicines10040890
Malik, A., Bagchi, A. K., Jassal, D. S., and Singal, P. K. (2024). Doxorubicin-induced cardiomyopathy is mitigated by empagliflozin via the modulation of endoplasmic reticulum stress pathways. Mol. Med. Rep. 29 (5), 74. doi:10.3892/mmr.2024.13198
Meng, C., Yuan, C. H., Zhang, C. C., Wen, M. D., Gao, Y. H., Ding, X. Y., et al. (2014). Ophiopogonin D protects cardiomyocytes against doxorubicin-induced injury through suppressing endoplasmic reticulum stress. Yao Xue Xue Bao 49 (8), 1117–1123.
Meyerovich, K., Ortis, F., Allagnat, F., and Cardozo, A. K. (2016). Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. J. Mol. Endocrinol. 57 (1), R1–r17. doi:10.1530/jme-15-0306
Miao, Y. U., Zhi-Hui, H. E., Li-Ying, X., Xiao-Tong, S., Jie, L., Jing-Yi, F., et al. (2016). Effect of creatine phosphate sodium on miRNA378, miRNA378(*) and calumenin mRNA in adriamycin-injured cardiomyocytes. Zhongguo Ying Yong Sheng Li Xue Za Zhi 32 (6), 514–518. doi:10.13459/j.cnki.cjap.2016.06.007
Mitry, M. A., and Edwards, J. G. (2016). Doxorubicin induced heart failure: phenotype and molecular mechanisms. Int. J. Cardiol. Heart Vasc. 10, 17–24. doi:10.1016/j.ijcha.2015.11.004
Nakagama, S., Maejima, Y., Fan, Q., Shiheido-Watanabe, Y., Tamura, N., Ihara, K., et al. (2023). Endoplasmic reticulum selective autophagy alleviates anthracycline-induced cardiotoxicity. JACC CardioOncol 5 (5), 656–670. doi:10.1016/j.jaccao.2023.05.009
Nigro, P., Pompilio, G., and Capogrossi, M. C. (2013). Cyclophilin A: a key player for human disease. Cell Death Dis. 4 (10), e888. doi:10.1038/cddis.2013.410
Oakes, S. A., and Papa, F. R. (2015). The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 10, 173–194. doi:10.1146/annurev-pathol-012513-104649
Ohba, T., Watanabe, H., Murakami, M., Takahashi, Y., Iino, K., Kuromitsu, S., et al. (2007). Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell Cardiol. 42 (3), 498–507. doi:10.1016/j.yjmcc.2006.10.020
Pandey, S., Kuo, C. H., Chen, W. S., Yeh, Y. L., Kuo, W. W., Chen, R. J., et al. (2022). Perturbed ER homeostasis by IGF-IIRα promotes cardiac damage under stresses. Mol. Cell Biochem. 477 (1), 143–152. doi:10.1007/s11010-021-04261-8
Parzych, K. R., and Klionsky, D. J. (2014). An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal 20 (3), 460–473. doi:10.1089/ars.2013.5371
Peña, J., and Harris, E. (2011). Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 286 (16), 14226–14236. doi:10.1074/jbc.M111.222703
Petersen, O. H., Gerasimenko, J. V., Gerasimenko, O. V., Gryshchenko, O., and Peng, S. (2021). The roles of calcium and ATP in the physiology and pathology of the exocrine pancreas. Physiol. Rev. 101 (4), 1691–1744. doi:10.1152/physrev.00003.2021
Popovic, D., Vucic, D., and Dikic, I. (2014). Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20 (11), 1242–1253. doi:10.1038/nm.3739
Qu, Y., Gao, R., Wei, X., Sun, X., Yang, K., Shi, H., et al. (2022). Gasdermin D mediates endoplasmic reticulum stress via FAM134B to regulate cardiomyocyte autophagy and apoptosis in doxorubicin-induced cardiotoxicity. Cell Death Dis. 13 (10), 901. doi:10.1038/s41419-022-05333-3
Rao, Z., Zheng, Y., Xu, L., Wang, Z., Zhou, Y., Chen, M., et al. (2022). Endoplasmic reticulum stress and pathogenesis of vascular calcification. Front. Cardiovasc Med. 9, 918056. doi:10.3389/fcvm.2022.918056
Raven, J. F., Baltzis, D., Wang, S., Mounir, Z., Papadakis, A. I., Gao, H. Q., et al. (2008). PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J. Biol. Chem. 283 (6), 3097–3108. doi:10.1074/jbc.M709677200
Rawat, P. S., Jaiswal, A., Khurana, A., Bhatti, J. S., and Navik, U. (2021). Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 139, 111708. doi:10.1016/j.biopha.2021.111708
Ren, J., Bi, Y., Sowers, J. R., Hetz, C., and Zhang, Y. (2021). Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 18 (7), 499–521. doi:10.1038/s41569-021-00511-w
Renu, K., V, G. A., P, B. T., and Arunachalam, S. (2018). Molecular mechanism of doxorubicin-induced cardiomyopathy - an update. Eur. J. Pharmacol. 818, 241–253. doi:10.1016/j.ejphar.2017.10.043
Sag, C. M., Köhler, A. C., Anderson, M. E., Backs, J., and Maier, L. S. (2011). CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell Cardiol. 51 (5), 749–759. doi:10.1016/j.yjmcc.2011.07.016
Sahoo, G., Samal, D., Khandayataray, P., and Murthy, M. K. (2023). A review on caspases: key regulators of biological activities and apoptosis. Mol. Neurobiol. 60 (10), 5805–5837. doi:10.1007/s12035-023-03433-5
Sang, Y., Kong, P., Zhang, S., Zhang, L., Cao, Y., Duan, X., et al. (2020). SGK1 in human cancer: emerging roles and mechanisms. Front. Oncol. 10, 608722. doi:10.3389/fonc.2020.608722
Schiattarella, G. G., Altamirano, F., Kim, S. Y., Tong, D., Ferdous, A., Piristine, H., et al. (2021). Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat. Commun. 12 (1), 1684. doi:10.1038/s41467-021-21931-9
Senft, D., and Ronai, Z. A. (2015). UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40 (3), 141–148. doi:10.1016/j.tibs.2015.01.002
Shaid, S., Brandts, C. H., Serve, H., and Dikic, I. (2013). Ubiquitination and selective autophagy. Cell Death Differ. 20 (1), 21–30. doi:10.1038/cdd.2012.72
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3 (1), 99–111. doi:10.1016/s1534-5807(02)00203-4
Shen, Y., Zhang, H., Ni, Y., Wang, X., Chen, Y., Chen, J., et al. (2022). Tripartite motif 25 ameliorates doxorubicin-induced cardiotoxicity by degrading p85α. Cell Death Dis. 13 (7), 643. doi:10.1038/s41419-022-05100-4
Sies, H. (2015). Oxidative stress: a concept in redox biology and medicine. Redox Biol. 4, 180–183. doi:10.1016/j.redox.2015.01.002
Sishi, B. J., Loos, B., van Rooyen, J., and Engelbrecht, A. M. (2013). Doxorubicin induces protein ubiquitination and inhibits proteasome activity during cardiotoxicity. Toxicology 309, 23–29. doi:10.1016/j.tox.2013.04.016
Song, S., Tan, J., Miao, Y., and Zhang, Q. (2018). Crosstalk of ER stress-mediated autophagy and ER-phagy: involvement of UPR and the core autophagy machinery. J. Cell Physiol. 233 (5), 3867–3874. doi:10.1002/jcp.26137
Szegezdi, E., Fitzgerald, U., and Samali, A. (2003). Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann. N. Y. Acad. Sci. 1010, 186–194. doi:10.1196/annals.1299.032
Tabas, I., and Ron, D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13 (3), 184–190. doi:10.1038/ncb0311-184
Takemoto-Kimura, S., Suzuki, K., Horigane, S. I., Kamijo, S., Inoue, M., Sakamoto, M., et al. (2017). Calmodulin kinases: essential regulators in health and disease. J. Neurochem. 141 (6), 808–818. doi:10.1111/jnc.14020
Tscheschner, H., Meinhardt, E., Schlegel, P., Jungmann, A., Lehmann, L. H., Müller, O. J., et al. (2019). CaMKII activation participates in doxorubicin cardiotoxicity and is attenuated by moderate GRP78 overexpression. PLoS One 14 (4), e0215992. doi:10.1371/journal.pone.0215992
Ubeda, M., Wang, X. Z., Zinszner, H., Wu, I., Habener, J. F., and Ron, D. (1996). Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol. Cell Biol. 16 (4), 1479–1489. doi:10.1128/mcb.16.4.1479
Vassalle, M., and Lin, C. I. (2004). Calcium overload and cardiac function. J. Biomed. Sci. 11 (5), 542–565. doi:10.1007/bf02256119
Wang, A. J., Tang, Y., Zhang, J., Wang, B. J., Xiao, M., Lu, G., et al. (2022a). Cardiac SIRT1 ameliorates doxorubicin-induced cardiotoxicity by targeting sestrin 2. Redox Biol. 52, 102310. doi:10.1016/j.redox.2022.102310
Wang, F., and Han, L. (2022b). Upregulation of serum and glucocorticoid-regulated kinase 1 (SGK1) ameliorates doxorubicin-induced cardiotoxic injury, apoptosis, inflammation and oxidative stress by suppressing glucose regulated protein 78 (GRP78)-mediated endoplasmic reticulum stress. Bioengineered 13 (1), 844–855. doi:10.1080/21655979.2021.2013109
Wang, M., Zhang, J., Zhao, M., Liu, J., Ye, J., Xu, Y., et al. (2021). Resolvin D1 attenuates doxorubicin-induced cardiotoxicity by inhibiting inflammation, oxidative and endoplasmic reticulum stress. Front. Pharmacol. 12, 749899. doi:10.3389/fphar.2021.749899
Wang, S., and Kaufman, R. J. (2012a). The impact of the unfolded protein response on human disease. J. Cell Biol. 197 (7), 857–867. doi:10.1083/jcb.201110131
Wang, X. Y., Yang, C. T., Zheng, D. D., Mo, L. Q., Lan, A. P., Yang, Z. L., et al. (2012b). Hydrogen sulfide protects H9c2 cells against doxorubicin-induced cardiotoxicity through inhibition of endoplasmic reticulum stress. Mol. Cell Biochem. 363 (1-2), 419–426. doi:10.1007/s11010-011-1194-6
Wang, Y., Cui, X., Wang, Y., Fu, Y., Guo, X., Long, J., et al. (2018a). Protective effect of miR378* on doxorubicin-induced cardiomyocyte injury via calumenin. J. Cell Physiol. 233 (10), 6344–6351. doi:10.1002/jcp.26615
Wang, Y., Lei, T., Yuan, J., Wu, Y., Shen, X., Gao, J., et al. (2018b). GCN2 deficiency ameliorates doxorubicin-induced cardiotoxicity by decreasing cardiomyocyte apoptosis and myocardial oxidative stress. Redox Biol. 17, 25–34. doi:10.1016/j.redox.2018.04.009
Wang, Y., Sun, Y., Guo, X., Fu, Y., Long, J., Wei, C. X., et al. (2018c). Creatine phosphate disodium salt protects against Dox-induced cardiotoxicity by increasing calumenin. Med. Mol. Morphol. 51 (2), 96–101. doi:10.1007/s00795-017-0176-5
Wang, Y., Zhang, Q., Wei, C., Zhao, L., Guo, X., Cui, X., et al. (2018d). MiR-378 modulates energy imbalance and apoptosis of mitochondria induced by doxorubicin. Am. J. Transl. Res. 10 (11), 3600–3609.
Wang, Z., Wang, M., Liu, J., Ye, J., Jiang, H., Xu, Y., et al. (2018e). Inhibition of TRPA1 attenuates doxorubicin-induced acute cardiotoxicity by suppressing oxidative stress, the inflammatory response, and endoplasmic reticulum stress. Oxid. Med. Cell Longev. 2018, 5179468. doi:10.1155/2018/5179468
Wen, Z. Q., Lin, J., Xie, W. Q., Shan, Y. H., Zhen, G. H., and Li, Y. S. (2023). Insights into the underlying pathogenesis and therapeutic potential of endoplasmic reticulum stress in degenerative musculoskeletal diseases. Mil. Med. Res. 10 (1), 54. doi:10.1186/s40779-023-00485-5
Xu, Z. M., Li, C. B., Liu, Q. L., Li, P., and Yang, H. (2018). Ginsenoside Rg1 prevents doxorubicin-induced cardiotoxicity through the inhibition of autophagy and endoplasmic reticulum stress in mice. Int. J. Mol. Sci. 19 (11), 3658. doi:10.3390/ijms19113658
Yap, K. N., Yamada, K., Zikeli, S., Kiaris, H., and Hood, W. R. (2021). Evaluating endoplasmic reticulum stress and unfolded protein response through the lens of ecology and evolution. Biol. Rev. Camb Philos. Soc. 96 (2), 541–556. doi:10.1111/brv.12667
Yarmohammadi, F., Rezaee, R., Haye, A. W., and Karimi, G. (2021). Endoplasmic reticulum stress in doxorubicin-induced cardiotoxicity may be therapeutically targeted by natural and chemical compounds: a review. Pharmacol. Res. 164, 105383. doi:10.1016/j.phrs.2020.105383
Yarmohammadi, F., Wallace Hayes, A., and Karimi, G. (2024). Molecular mechanisms involved in doxorubicin-induced cardiotoxicity: a bibliometrics analysis by VOSviewer. Naunyn Schmiedeb. Arch. Pharmacol. 397 (4), 1971–1984. doi:10.1007/s00210-023-02773-2
Ye, J., Huang, Y., Que, B., Chang, C., Liu, W., Hu, H., et al. (2018). Interleukin-12p35 knock out aggravates doxorubicin-induced cardiac injury and dysfunction by aggravating the inflammatory response, oxidative stress, apoptosis and autophagy in mice. EBioMedicine 35, 29–39. doi:10.1016/j.ebiom.2018.06.009
Ye, S., Zhou, X., Chen, P., and Lin, J. F. (2021). Folic acid attenuates remodeling and dysfunction in the aging heart through the ER stress pathway. Life Sci. 264, 118718. doi:10.1016/j.lfs.2020.118718
Yoneda, T., Imaizumi, K., Oono, K., Yui, D., Gomi, F., Katayama, T., et al. (2001). Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276 (17), 13935–13940. doi:10.1074/jbc.M010677200
Yoshida, H. (2007). ER stress and diseases. Febs J. 274 (3), 630–658. doi:10.1111/j.1742-4658.2007.05639.x
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107 (7), 881–891. doi:10.1016/s0092-8674(01)00611-0
Zeeshan, H. M., Lee, G. H., Kim, H. R., and Chae, H. J. (2016). Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 17 (3), 327. doi:10.3390/ijms17030327
Zeng, M., Zhu, L., Li, L., and Kang, C. (2017). miR-378 suppresses the proliferation, migration and invasion of colon cancer cells by inhibiting SDAD1. Cell Mol. Biol. Lett. 22, 12. doi:10.1186/s11658-017-0041-5
Zhang, H., Weng, J., Sun, S., Zhou, J., Yang, Q., Huang, X., et al. (2022a). Ononin alleviates endoplasmic reticulum stress in doxorubicin-induced cardiotoxicity by activating SIRT3. Toxicol. Appl. Pharmacol. 452, 116179. doi:10.1016/j.taap.2022.116179
Zhang, J., Guo, J., Yang, N., Huang, Y., Hu, T., and Rao, C. (2022b). Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 13 (12), 1051. doi:10.1038/s41419-022-05444-x
Zhang, Y., Deng, H., Zhou, H., Lu, Y., Shan, L., Lee, S. M., et al. (2019). A novel agent attenuates cardiotoxicity and improves antitumor activity of doxorubicin in breast cancer cells. J. Cell Biochem. 120 (4), 5913–5922. doi:10.1002/jcb.27880
Zhang, Y., Kong, D., Han, H., Cao, Y., Zhu, H., and Cui, G. (2022c). Caffeic acid phenethyl ester protects against doxorubicin-induced cardiotoxicity and increases chemotherapeutic efficacy by regulating the unfolded protein response. Food Chem. Toxicol. 159, 112770. doi:10.1016/j.fct.2021.112770
Zhu, H., and Zhou, H. (2021a). Novel insight into the role of endoplasmic reticulum stress in the pathogenesis of myocardial ischemia-reperfusion injury. Oxid. Med. Cell Longev. 2021, 5529810. doi:10.1155/2021/5529810
Zhu, J., Zhang, X., Xie, H., Wang, Y., Zhang, X., and Lin, Z. (2021b). Cardiomyocyte Stim1 deficiency exacerbates doxorubicin cardiotoxicity by magnification of endoplasmic reticulum stress. J. Inflamm. Res. 14, 3945–3958. doi:10.2147/jir.S304520
Zhu, W., Soonpaa, M. H., Chen, H., Shen, W., Payne, R. M., Liechty, E. A., et al. (2009). Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119 (1), 99–106. doi:10.1161/circulationaha.108.799700
Keywords: endoplasmic reticulum stress, doxorubicin-induced cardiotoxicity, apoptosis, autophagy, targeted therapy
Citation: Sun M, Zhang X, Tan B, Zhang Q, Zhao X and Dong D (2024) Potential role of endoplasmic reticulum stress in doxorubicin-induced cardiotoxicity-an update. Front. Pharmacol. 15:1415108. doi: 10.3389/fphar.2024.1415108
Received: 09 April 2024; Accepted: 01 August 2024;
Published: 12 August 2024.
Edited by:
Antonio Lax, University of Murcia, SpainReviewed by:
Maja Ðanic, University of Novi Sad, SerbiaClara Quiroga, Pontificia Universidad Católica de Chile, Chile
Copyright © 2024 Sun, Zhang, Tan, Zhang, Zhao and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingli Sun, c3VuMTg5NDAxMDMxMjZAMTYzLmNvbQ==
†These authors have contributed equally to this work