 Sanket N. Patel
Sanket N. Patel Kalyani Kulkarni
Kalyani Kulkarni Tahmid Faisal
Tahmid Faisal Tahir Hussain
Tahir Hussain- Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, TX, United States
The consumption of a high-sodium diet (HSD) is injurious and known to elevate blood pressure (BP), especially in obesity. Acute infusion studies depict a functional interdependency between angiotensin-II type 2 receptor (AT2R) and receptor Mas (MasR). Hence, we hypothesize that the subacute blockade of MasR should reverse AT2R-mediated renoprotection in obese Zucker rats (OZRs). Male OZRs were fed an HSD (for 14 days) and treated with the AT2R agonist C21 (100 ng/min) without or with a MasR antagonist A779 (1,000 ng/min). The indices of oxidative stress, proteinuria, kidney injury, and BP were measured before and after, along with the terminal measurements of an array of inflammatory and kidney injury markers. The HSD significantly decreased the estimated glomerular filtration rate and urinary osmolality and increased thirst, diuresis, natriuresis, kaliuresis, plasma creatinine, urinary excretion of H2O2, proteinuria, renal expression and urinary excretion of kidney injury markers (NGAL and KIM-1), and BP indexes. The HSD feeding showed early changes in the renal expression of CRP, ICAM-1, and galectin-1. The C21 treatment prevented these pathological changes. The MasR antagonist A779 attenuated C21-mediated effects on the urinary excretion and renal expression of NGAL and oxidative stress in the absence of inflammation and BP changes. Overall, we conclude that the subacute functional interactions between AT2R and MasR are weak or transient and that the beneficial effects of AT2R activation are independent of the MasR blockade in the kidney of male obese rats fed an HSD.
1 Introduction
The consumption of a high-sodium diet (HSD) is a major health problem not only because it is a known risk factor for salt-sensitive hypertension in obesity and kidney diseases but it can also generally offset the health benefits of an ongoing therapeutic regimen (Smyth et al., 2014). Even the consumption of an HSD for a few days can cause inflammation, oxidative stress, tubular proteinuria, and kidney injury, independent of changes in blood pressure (BP) (Teixeira et al., 2020). Furthermore, HSD consumption is a major modulator of the renin–angiotensin system (RAS). Contrary to numerous reports showing the beneficial effects of the angiotensin-II type 2 receptor (AT2R) and mas receptor (MasR) (Grobe et al., 2006; Grobe et al., 2007; Pinheiro et al., 2009; Shenoy et al., 2010), some reports show their deleterious (Ruster et al., 2011; Burrell et al., 2017; Yu et al., 2019) or lack of effect (Collister and Nahey, 2010; Dilauro et al., 2010; Jehle et al., 2012), and the interactions between AT2R and MasR are increasingly complex than anticipated. For instance, acute infusion studies show that the protective effects of the MasR agonist angiotensin-(1–7) (ang-(1–7)) in normotensive WKY rats and spontaneously hypertensive rats (SHRs) are dependent on AT2R (Walters et al., 2005; Bosnyak et al., 2012). We reported that AT2R and MasR are colocalized and functionally interdependent in the kidney of male obese Zucker rats (OZRs), as evident by the diuretic and natriuretic effects of the AT2R agonist C21 being blocked by the MasR antagonist A779, and those of the MasR agonist ang-(1–7) being blocked by the AT2R antagonist PD123319 (Patel et al., 2017). These cross-blocking effects of the antagonists are not due to the overlap of their affinities with these receptors. One of the mechanisms that play a role in functional interaction is the dimerization of these receptors involving a disulfide bond (Villela et al., 2015; Patel and Hussain, 2018). The functional interdependency between AT2R and MasR has been supported (Tesanovic et al., 2010; Kljajic et al., 2013; Hao P. et al., 2015; Hao P. P. et al., 2015; Liu et al., 2015; Papinska et al., 2015) or refuted (Santos et al., 2003; Silva et al., 2007) by various studies. However, it is unclear whether the AT2R-MasR interactions play a subacute/longer-term role in renal injury, inflammation, and BP regulation.
The acute (single-dose) activation/blockade of a receptor does not necessarily predict longer health outcomes. Therefore, in this study, we attempted to determine the effects of HSD intake in male genetically obese rats. Furthermore, the interactions between AT2R and MasR are studied, i.e., whether 14 days of treatment with the MasR antagonist A779 (1,000 ng/min) reverses the AT2R agonist C21 (100 ng/min)-mediated renoprotection and reduction in oxidative stress, inflammation, and BP in male HSD-fed OZRs. Male rats are generally considered vulnerable to experimental injury (Sullivan et al., 2010). Hence, it is intuitive to believe that the effect size will be pronounced in male cases and allow us to determine the deleterious effects of HSD consumption, protection upon AT2R stimulation, and reversal by the effects of AT2Rs on the blockade by the MasR antagonist. In doing so, compared to other studies that have used higher C21 concentrations, i.e., ≥1 mg/kg, we used a lower dose (0.3 mg/kg) of C21, which has been shown to exert renoprotective and anti-hypertensive effects in vivo (Bosnyak et al., 2010; Kemp et al., 2014). Moreover, compensatory repair processes do occur in vivo; hence, we compared the indexes after HSD feeding with basal values determined before commencing the treatment to infer the actual effect of HSD feeding or drug treatment.
2 Methods
2.1 Experimental protocol
Male obese Zucker rats (HsdHlr:ZUCKER-Leprfa, 6–7 weeks of age) weighing ∼270 g were purchased from Envigo. After arrival, the rats were housed in the University of Houston animal care facility. The animal experimental protocols used in this study were approved by the IACUC at the University of Houston and adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The rats were treated without or with the AT2R agonist C21 (100 ng/min, Alzet 1002, 0.25 μL/h) or MasR antagonist A779 (1,000 ng/min, Alzet 2ML2, 5 μL/h) or together for 14 days using an osmotic pump (DURECT Corporation, Cupertino, CA) surgically implanted in the peritoneal cavity under isoflurane anesthesia. The dose of the AT2R agonist C21 was ∼0.45 mg/kg body weight and that of the MasR antagonist A779 was ∼4.17 mg/kg body weight. C21 was provided by Vicore Pharma (Sweden), and A-779 was purchased from Cayman Chemical, Ann Arbor, MI. The rats were fed a normal chow diet (ND) or HSD, (4% NaCl, Teklad TD.92034, Harlan Laboratories, Madison, WI) for 14 days. The food and water intake was monitored. Blood (∼200 µL) was collected once by pricking the tail (∼1 mm) for the preparation of plasma and baseline measurement of urea nitrogen and creatinine levels. The rats were housed in metabolic cages for 3 days before the implantation of osmotic pumps and during the terminal 3 days for urine collection and measurement of water consumption. The rats were euthanized under isoflurane anesthesia. Urine was also collected directly from the bladder, and blood was collected from a cardiac puncture for separating the plasma. The urine and plasma were stored at −80°C. The kidneys were weighed for the determination of hypertrophy and stored at −80°C for biochemical analysis.
2.2 General parameters
The body weight and food consumption were determined periodically. Two rats were housed per cage. Food was replenished twice a week. The rats were housed in the metabolic cages before the implantation of osmotic pumps (on days −3, −2, and −1) and after the treatment (on days 11, 12, and 13) for urine collection and measurement of water consumption. The water intake and urine output, before and after the treatment period, were presented as the mean ± standard error of the mean (SEM).
2.3 Inflammation array
Inflammation was assessed using sandwich-based semi-quantitative RayBio C-Series Rat Inflammation Array C3 (AAR-INF-3). In brief, kidney tissue was homogenized in a lysis buffer containing a Halt protease–phosphatase inhibitor cocktail without EDTA (78440, Thermo Fisher Scientific). The homogenate was centrifuged at 700 g for 1 h at 4°C. The protein concentration in the clear supernatant homogenate was measured using the Pierce™ BCA Protein Assay Kit (23225, Thermo Fisher Scientific). The membrane was incubated in a blocking buffer for 30 min, followed by incubation with a protein overnight at 4°C under gentle agitation. The sample was aspirated, and the membrane was washed with buffer 1 (5 × 5′) and buffer 2 (5 × 5′), followed by incubation with a biotinylated antibody cocktail overnight at 4°C under gentle agitation. The solution was aspirated, and the membrane was washed with both the buffers again, followed by incubation with an HRP–streptavidin antibody overnight at 4°C. After washing it again, the membrane was incubated with a chemiluminescence detection solution, and an image using the Li-Cor Odyssey Fc Imaging system was acquired. The integrated density values were normalized with two positive controls (POS1 and POS2). Normalized values of inflammatory indices are represented as the mean ± SEM.
2.4 Indices of kidney dysfunction
The urinary excretion of NGAL (ERLCN2) and KIM-1 (ERHAVCR1) and their expression in the kidney were determined using an ELISA kit (Thermo Fisher Scientific). The urinary excretion of NGAL and Kim-1 was presented as ng/day, and their renal expression was presented as ng/mg protein.
Urea nitrogen and creatinine levels in the urine and the plasma were measured in samples collected before the initiation of treatment and after the study period using a QuantiChrom Urea Assay Kit (DIUR-100) and EnzyChrom Creatinine Assay Kit (EIssCT-100), respectively, according to the manufacturer’s instructions (BioAssay Systems). For the urea nitrogen and creatinine measurement, 2.5 or 10 µL plasma (3kD filtrate) and urine (50×) was used, respectively. The estimated glomerular filtration rate (eGFR) was obtained based on plasma urea nitrogen and creatinine concentrations, as previously described (Besseling et al., 2021).
Urinary osmolarity and the excretion of protein, sodium, and potassium were measured in urine collected before the initiation of feeding the HSD and treatment with C21 and/or A779 (on days −3, −2, and −1) and after the study period (on days 11, 12, and 13). The urine samples were diluted 20× for osmolality and 7500× for sodium/potassium measurements. The values of 3 consecutive days were averaged and represented for the study period, i.e., “before” and “after.” Urinary osmolality was measured using an osmometer (model 3250, serial #10010827, Advanced Instruments) and presented as mOsm/kg H2O. The urinary excretion of sodium and potassium was estimated using an atomic absorption spectrometer (AAnalyst 400, PerkinElmer, Inc.) and presented as mmol/day (Ali et al., 2015). The excretion of protein was estimated based on the urinary protein concentration measured by the pyrogallol red-molybdate method and presented as mg/day (Patel et al., 2016).
2.5 Indices of oxidative stress
Urinary hydrogen peroxide levels were measured via the ADHP–peroxidase method (Patel et al., 2016). In brief, 100 µL urine (50×) was mixed with ADHP (50 μL, 20 µM) and anti-rabbit HRP IgG (50 μL, 500×) for 30 min. Fluorescence was read at an excitation wavelength of 540 nm and an emission wavelength of 590 nm using a Varioskan plate reader (Thermo Fisher Scientific) (Patel et al., 2016).
2.6 Tail-cuff plethysmography
BP was measured in a designated quiet area, where the rats were acclimatized for 1 h before experiments began. The BP was measured for 3 days (days −3, −2, and −1) before the surgical implantation of osmotic pumps (day 0) and during terminal 3 days (days 11, 12, and 13) at the same time between 1 p.m. and 4 p.m. by CODA 8 non-invasive tail-cuff plethysmography (Kent Scientific Corporation). The rats were acclimatized to a warm environment for 10–15 min using a warming platform (level 3). Each recording session consisted of 28 inflation and deflation cycles, of which the first three cycles were “acclimation” cycles and not used in the analysis. The minimum volume change was set as 14 µL. The average of at least five accepted runs per rat per day was used for data analysis. The daily CODA measurements acquired before the initiation of HSD feeding and treatment with C21 and/or A779 (on days −3, −2, and −1) and after the study period (on days 11, 12, and 13) were averaged separately and presented as mmHg.
2.7 Statistical analysis
Data are presented as the mean ± SEM. The data were analyzed using GraphPad Prism 6 and subjected to one-way or two-way ANOVA, followed by Fisher’s LSD test, whereas the values “before” and “after” were compared via a non-parametric, two-tailed, Wilcoxon matched paired t-test (p < 0.05; n = 6–7 per group). The values that fell between p > 0.05 and <0.1 were marked in the legend to indicate the lower level of significance in that dataset.
3 Results
3.1 General parameters
The body weight of all rats increased proportionately (Figure 1A), and the food intake remained consistent over time among all study groups (Figure 1B). As expected, the subacute consumption of the HSD significantly increased the water intake (Figure 1C) and diuresis (Figure 1D) (p < 0.05 before vs after and HSD vs OZRs), which was prevented by the AT2R agonist C21 (p < 0.05; HSD + C21 vs HSD). Although the consumption of the HSD did not cause renal hypertrophy per se (Figures 1E, F), C21 decreased the left kidney weight (p < 0.05, HSD + C21 vs HSD), which was reversed by the MasR antagonist A779 (p = ns, HSD + A779 + C21 vs C21). Otherwise, the concurrent blockade of MasR did not affect any of the above indices.
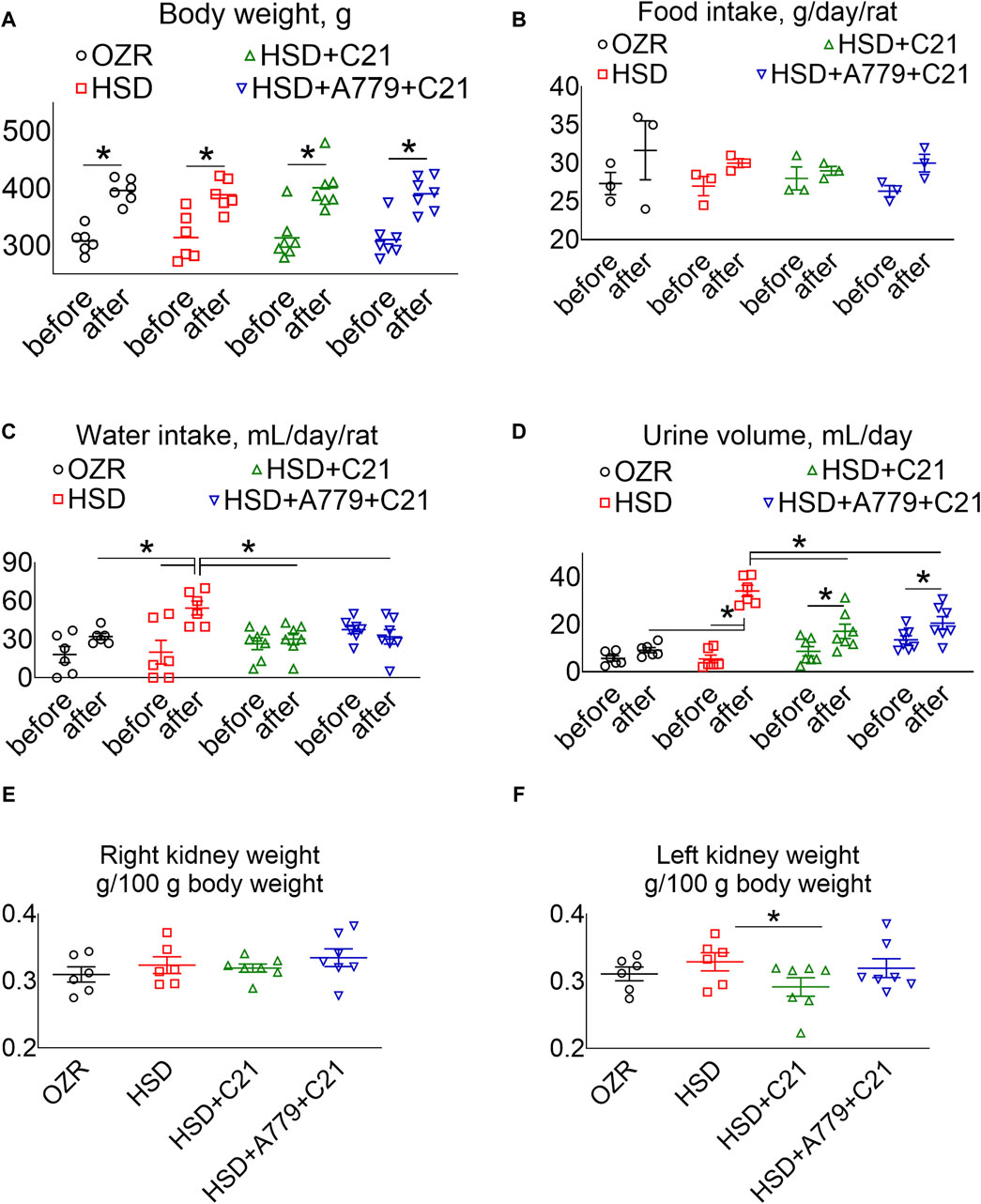
Figure 1. Average body weight (A), food intake (B), and water intake (C) in metabolic cages, and the urine volume (D) of male obese Zucker rats before the initiation of HSD feeding and treatment with the AT2R agonist C21 and/or mas receptor antagonist A779 and after the study period. The body weight normalized weight of the right (E) and left kidneys (F) of the rats are reported. The water intake was determined by housing the rats individually in a metabolic cage before (days −3, −2, and −1) and after the study period (days 11, 12, and 13). The daily food intake, water intake, and urine volume were averaged separately and represented for the study period, i.e., “before” and “after.” Food intake (B): two rats were housed in a cage; hence, each value applies to two rats. The values are represented as the mean ± SEM and analyzed via two-way ANOVA, followed by Fisher’s LSD test, whereas the values “before” and “after” were compared via a non-parametric, two-tailed, Wilcoxon matched paired t-test; *p < 0.05, n = 6–7.
3.2 Indices of oxidative stress and kidney injury
The urinary excretion of H2O2, an indicator of oxidative stress, was increased after HSD intake (Figure 2A) (p < 0.05 before vs after and HSD vs OZRs). However, such an increase in the urinary H2O2 excretion was significantly prevented upon treatment with the AT2R agonist C21 (p < 0.05, HSD + C21 vs HSD, p = ns before vs after). The MasR blockade by A779 modestly, but not significantly, reversed the C21-mediated decrease in oxidative stress (p = 0.08, HSD + A779 + C21 vs HSD + C21, p = ns before vs after and HSD + A779 + C21 vs HSD).
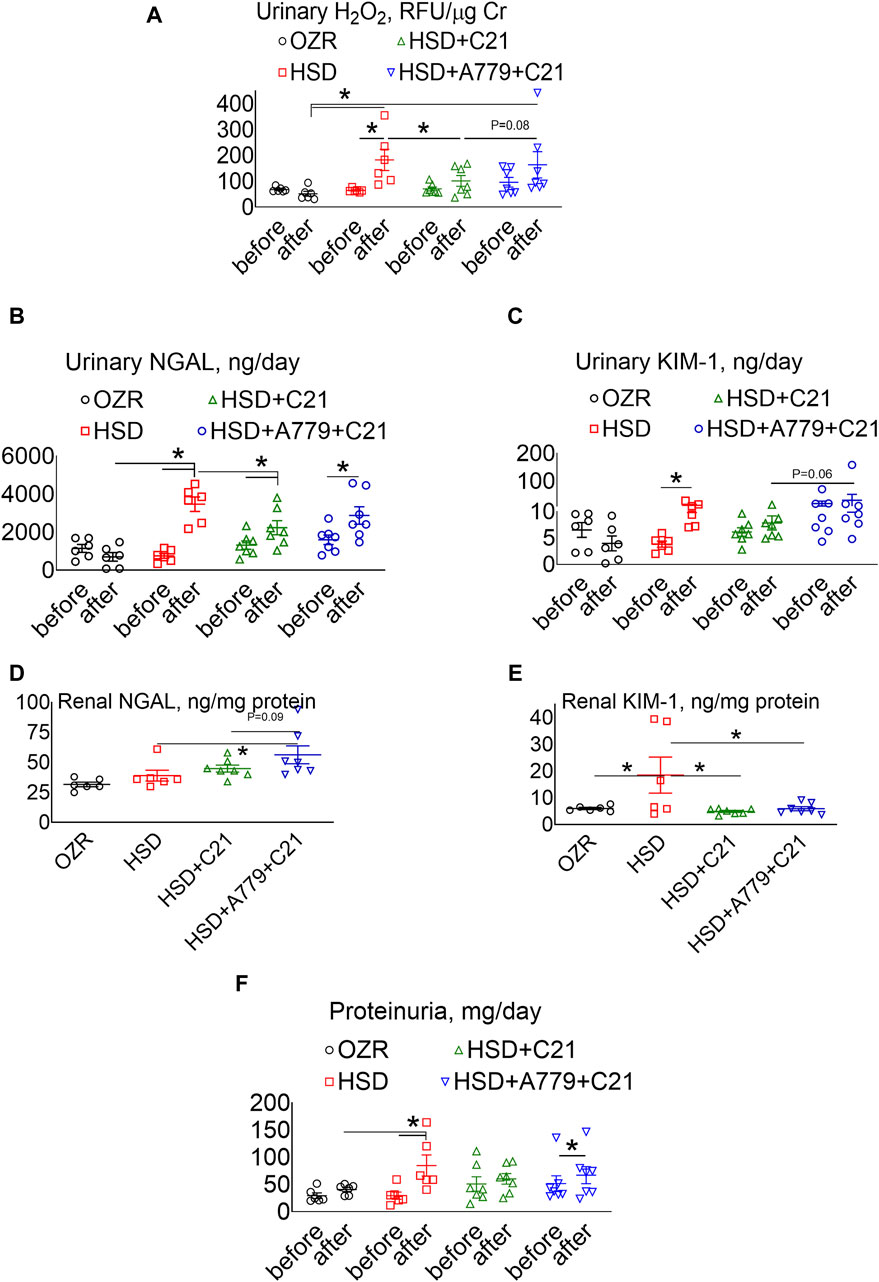
Figure 2. Urinary excretion of hydrogen peroxide (H2O2) (A), NGAL (B), and KIM-1 (C) and the renal expression of NGAL (D) and KIM-1 (E) and proteinuria (F) in male obese Zucker rats. The A–C, F indices were measured before the initiation of HSD feeding and treatment with the AT2R agonist C21 and/or mas receptor antagonist A779 and after the study period. D, E: The indices were measured terminally in the kidney homogenate after the treatment period. Proteinuria was determined in the urine collected before (days −3, −2, and −1) and after the study period (days 11, 12, and 13), averaged, and represented for the study period, i.e., “before” and “after.” The values are represented as the mean ± SEM and analyzed via two-way ANOVA (A–C,F) or one-way ANOVA (D,E), followed by Fisher’s LSD test, whereas the values “before” and “after” were compared via a non-parametric, two-tailed, Wilcoxon matched paired t-test; *p < 0.05, n = 6–7.
The urinary excretion of NGAL (Figure 2B), a marker of renal injury, increased significantly upon HSD consumption (p < 0.5 before vs after and HSD vs OZRs). However, the increase in NGAL excretion was significantly less in OZRs fed the HSD treated with C21 (p < 0.05 before vs after and HSD + C21 vs HSD). The HSD + A779 treatment partially reversed the C21-mediated reduction in NGAL excretion (p < 0.05 before vs after; p = ns, HSD + A779 + C21 vs HSD and HSD + A779 + C21 vs HSD + C21). Similarly, the urinary excretion of KIM-1, another marker of proximal tubular brush border injury, was increased upon HSD intake (Figure 2C) (p < 0.05 before vs after). However, the treatment with C21 prevented such an increase in KIM-1 excretion, which was not impacted during the MasR blockade (p = ns both before vs after).
The renal expression of NGAL (Figure 2D) remained unchanged upon HSD intake in OZRs compared to the kidney of ND-fed OZRs and HSD + C21. However, the MasR blockade by A779 modestly increased the renal NGAL expression (p = 0.6 vs HSD + C21 and p < 0.05 vs HSD). Contrary to NGAL, the renal expression of KIM-1 (Figure 2E) was profoundly increased upon HSD intake (p < 0.05 HSD vs OZRs), which was prevented by treatment with C21 (p < 0.05 vs HSD + C21 vs HSD), but the MasR antagonist A779 did not reverse the C21-mediated reduction in renal KIM-1 expression.
Proteinuria was significantly increased after HSD intake (Figure 2F) (p < 0.05 before vs after and HSD vs OZRs). However, such increases in proteinuria were not evident upon treatment with the AT2R agonist C21 (p = ns before vs after, HSD + C21 vs HSD, and HSD + C21 vs OZRs). The anti-proteinuric effect of C21 was not evident when MasR was blocked with A779 (p < 0.05 before vs after, p = ns before vs after, HSD + C21 vs HSD, and HSD + C21 vs OZRs).
3.3 Indices of kidney dysfunction
As expected, the subacute consumption of the HSD caused a significant decrease in the urinary excretion of creatinine (Figure 3A), which was associated with a significant increase in the plasma creatinine levels (Figure 3B) (both p < 0.05 before vs after and HSD vs OZRs). The C21 treatment partially improved the urinary excretion of creatinine (p < 0.05 HSD + C21 vs HSD), which resulted in a decrease in the plasma creatinine levels (p < 0.05 before vs after and HSD + C21 vs HSD). The MasR blockade by A779 partially prevented the C21-mediated decrease in the urinary excretion of creatinine (p < 0.05 before vs after); however, the plasma creatinine remains unaffected (p < 0.5 HSD + A779 + C21 vs HSD; p = ns before vs after).
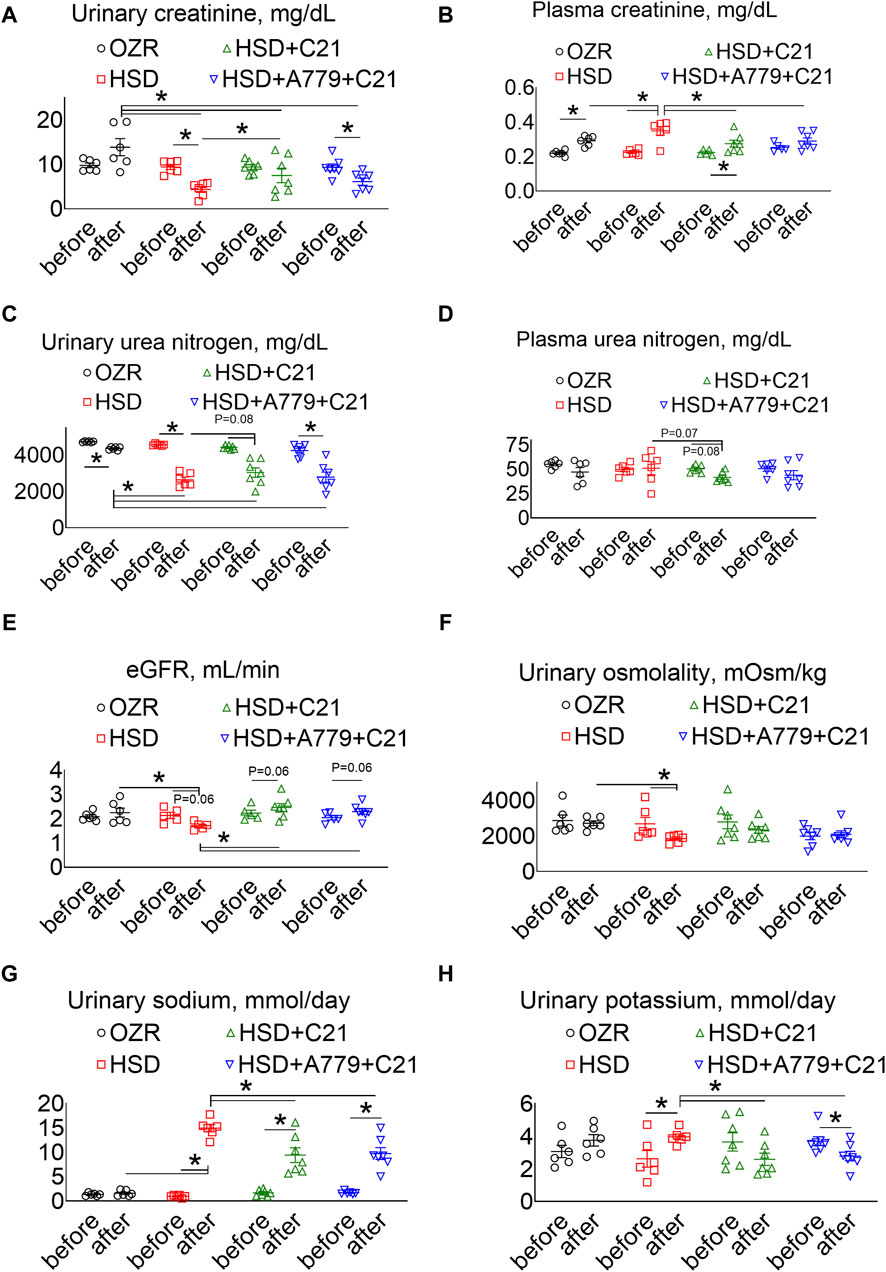
Figure 3. The urinary excretion of creatinine (A), the plasma concentrations of creatinine (B), urinary urea nitrogen (C), plasma urea nitrogen (D), the estimated glomerular filtration rate (eGFR) (E), urinary osmolality (F), urinary sodium (G), and urinary potassium levels (H) were measured in male obese Zucker rats before the initiation of HSD feeding and treatment with the AT2R agonist C21 and/or mas receptor antagonist A779 and after the study period. The plasma values are used to calculate the eGFR, as described previously (Besseling et al., 2021). The urinary osmolality and excretion of sodium and potassium were determined in urine collected before (days −3, −2, and −1) and after the study period (days 11, 12, and 13), averaged, and represented for the study period, i.e., “before” and “after.” The values are represented as the mean ± SEM and analyzed via two-way ANOVA, followed by Fisher’s LSD test, whereas the values “before” and “after” were compared via a non-parametric, two-tailed, Wilcoxon matched paired t-test; *p < 0.05, n = 5–7.
The urinary excretion of urea nitrogen was significantly decreased after HSD consumption (Figure 3C) (p < 0.05 before vs after and HSD vs OZRs), but unexpectedly, it was not associated with an increase in the plasma urea nitrogen levels (Figure 3D). However, C21 treatment partially improved urinary urea nitrogen excretion and decreased plasma urea nitrogen levels (p < 0.05 before vs after and p = 0.07 HSD vs HSD + C21). The MasR blockade by A779 partially prevented the C21-mediated decrease in the urinary excretion of urea nitrogen (p < 0.05 before vs after); however, the plasma urea nitrogen level remains unaffected (p = ns before vs after and HSD + A779 + C21 vs HSD).
The eGFR based on the plasma creatinine and urea nitrogen concentrations showed a similar effect (Besseling et al., 2021). As expected, the eGFR was significantly decreased after HSD consumption (Figure 3E) (p < 0.05 before vs after and HSD vs OZRs), which was modestly, but not significantly, improved with C21 treatment (p = 0.06 before vs after and C21 vs HSD). However, the MasR blockade did not affect the improvement in the eGFR afforded by C21.
Likewise, the HSD intake led to a predicted decrease in osmolality (Figure 3F) (p < 0.05 before vs after and HSD vs OZRs), which was prevented by the AT2R agonist C21 (p = ns before vs after and HSD + C21 vs HSD) but remained unaffected under the MasR blockade (p = ns before vs after, HSD + A779 + C21 vs HSD + C21, and HSD + A779 + C21 vs HSD).
As expected, natriuresis (Figure 3G) was increased upon HSD intake (p < 0.05 before vs after and HSD vs OZRs), which was decreased by C21 (p < 0.05 before vs after and HSD + C21 vs HSD). On the contrary, the HSD intake increased kaliuresis (Figure 3H) (p < 0.05 before vs after), which was significantly prevented by C21 (p < 0.1 HSD + C21 vs HSD; p = ns before vs after). The MasR antagonist A779 did not affect C21-mediated changes in natriuresis and kaliuresis (p < 0.05 before vs after and HSD + A779 + C21 vs HSD; p = ns HSD + A779 + C21 vs HSD + C21).
3.4 Inflammation array
The inflammation array detected ∼30/36 renal proteins involved in inflammation (Figure 4). HSD consumption did not statistically change these inflammatory markers compared to those found in the kidney of ND-fed OZRs. However, the treatment with the AT2R agonist C21 modestly reduced CRP (but not significantly with p = 0.08), ICAM-1 (p < 0.05), and galectin-1 levels (p < 0.05) compared with the HSD, which were not reversed during the MasR blockade by A779. Moreover, A779 further significantly reduced renal gp130, IL-13, IL-3, and IL-6 levels (p < 0.05 HSD + A779 + C21 vs HSD). Specifically, the renal expression of CRP, ICAM-1, and galectin-1 was associated with the renal expression of NGAL (SI Supplementary Table S1 and Supplementary Figure S1) and KIM-1 (Supplementary Table S2 and Supplementary Figure S2), and the renal expression of gp130, IL-13, and IL-6 was associated with changes in the plasma creatinine level and eGFR (Supplementary Table S3 and Supplementary Figure S1).

Figure 4. Sandwich-based semi-quantitative inflammation array in the kidney homogenate of male obese Zucker rats after the subacute feeding of HSD and treatment with the AT2R agonist C21 and/or MasR antagonist A779. The electrochemiluminescent integrated density values are normalized to two positive controls (POS1 and POS2). The values are represented as the mean ± SEM and analyzed via one-way ANOVA, followed by Fisher’s LSD test; *p < 0.1, n = 6.
3.5 Blood pressure
As expected, the systolic BP (SBP) (Figure 5A), diastolic BP (DBP) (Figure 5B), and mean BP (Figure 5C) showed a significant increase after the intake of the HSD (all p < 0.05 HSD vs OZRs). The C21 treatment significantly prevented the increase in these BP indexes, particularly DBP (p = ns before vs after) (compared to SBP, MBP, and PP; p < 0.05 before and after and HSD + C21 vs HSD). Similarly, the MasR antagonist A779 did not affect the C21-mediated prevention of the increase in SBP, DBP, or MBP (p < 0.05 before and after and HSD + A779 + C21 vs HSD). As expected, the heart rate did not change statistically among treatment groups (Figure 5D).
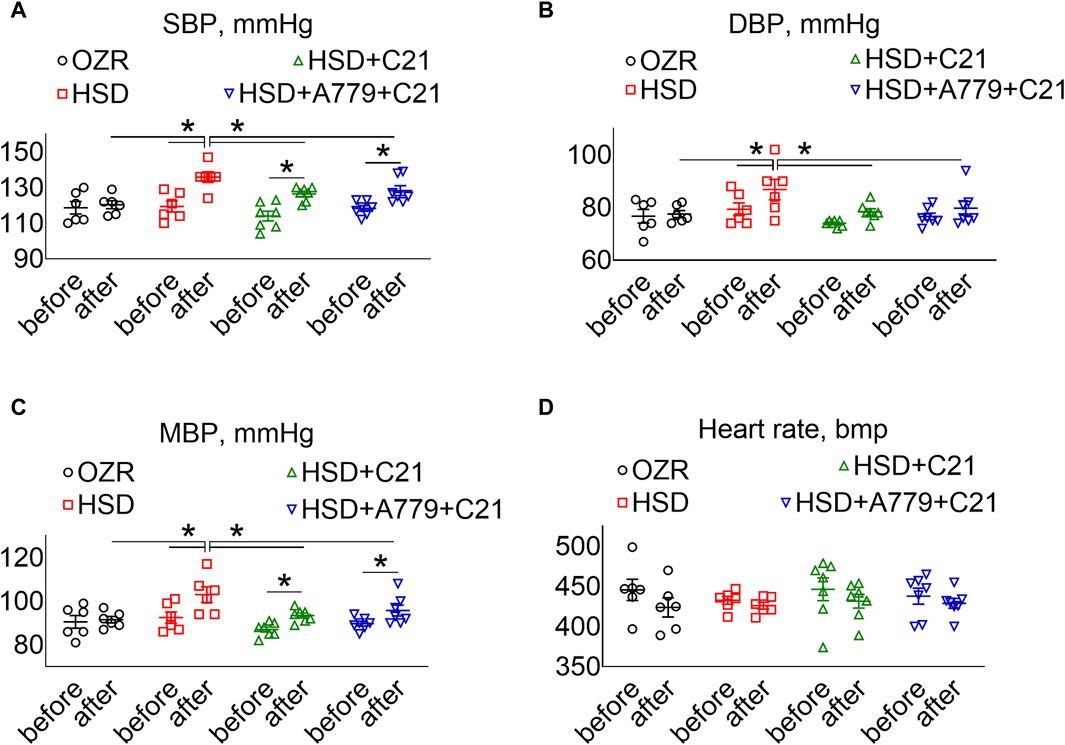
Figure 5. BP indexes: systolic, SBP (A), diastolic, DBP (B), and mean, MBP (C) and heart rate (D) measured via tail-cuff plethysmography before (days −3, −2, and −1) and after the study period (days 11, 12, and 13). The values were averaged and represented for the study period, i.e., “before” and “after.” The values are represented as the mean ± SEM and analyzed via two-way ANOVA, followed by Fisher’s LSD test, whereas the values “before” and “after” were compared via a non-parametric, two-tailed, Wilcoxon matched paired t-test; *p < 0.1, n = 6–7.
As a recap, diuresis–natriuresis was highly correlated with the increase in the urinary excretion of H2O2 and NGAL and a decrease in osmolality and the urinary excretion of urea nitrogen and creatinine, while the decrease in the eGFR was highly correlated with left kidney weight and plasma concentrations of urea nitrogen and creatinine (Supplementary Figure S2).
4 Discussion
The subacute consumption of the HSD by male OZRs increased thirst, diuresis, natriuresis, and kaliuresis; decreased the eGFR and urinary osmolality; and caused oxidative stress, inflammation, proteinuria, kidney injury and dysfunction, and hypertension. Furthermore, the treatment with the AT2R agonist C21 prevented these changes, which aligns with our earlier reports (Patel et al., 2016; Patel et al., 2019) but does not include MasR.
Inflammation array findings do not support the AT2R-MasR interdependency. The effects of AT2R and MasR activation on MAPK proteins involved in inflammation are dependent on the cell type they act upon and are quite opposite, suggesting the anti-inflammatory effects of AT2R and inflammatory effects of MasR, as our findings depict. The AT2R agonist C21 has been reported to exert cardioprotective effects via the phosphorylation of p38 and p42/44 MAPK (Kaschina et al., 2008). However, in rat renal proximal tubule cells, the MasR agonist ang-(1–7) reduced the phosphorylation of p38 and other MAPKs (Su et al., 2006), but in mesangial cells, ang-(1–7) induced p38 phosphorylation and fibrotic effects via MasR, which was independent of angiotensin receptors (Zimpelmann and Burns, 2009). Previously, it was reported that A779 did not affect inflammatory indexes in a rat model of pulmonary arterial hypertension (Ferreira et al., 2009). Another study utilizing a similar dose of A779 (as used here), ang-(1–7) treatment, and masR-null rats showed that MasR activation exerts inflammatory deleterious effects in the kidney (Esteban et al., 2009). Collectively, the findings suggest the inflammatory role of MasR in male OZR kidneys.
The MasR antagonist A779 increased the renal expression and urinary excretion of NGAL, a biomarker of kidney injury (McWilliam et al., 2014; Xiao et al., 2017), which is in contrast to the inflammation array and BP data. NGAL may appear in the urine either due to renal shedding, its reduced uptake by proximal tubules, or its enhanced glomerular clearance. Furthermore, the changes in NGAL levels could also be contributed by its release from immune cells and secretion in urine. In either case, the findings suggest that the blockade of MasR may have caused direct renal injury independent of inflammation and BP changes. The MasR antagonist A779-mediated increase in NGAL levels aligns with the inhibitory effect of the MasR agonist ang-(1–7) on NGAL (Giani et al., 2012). The renal expression of NGAL is positively associated with proteinuria and urinary excretion of H2O2 (an indicator of oxidative stress). The activation of AT2R (Patel et al., 2016) and MasR (Benter et al., 2008; Giani et al., 2011; Patel et al., 2016) has been shown to reduce oxidative stress in the kidney and proteinuria. Overall, it appears that the subacute functional interaction between AT2R and MasR is weak or transient or dependent on the pathology being studied. It is unknown whether AT2R and MasR can physically interact under HSD-fed conditions or upon treatment with their respective ligand. In acute settings, we reported that the MasR antagonist blocked the response of the AT2R agonist and vice versa (Patel et al., 2017). In particular, our study showed that the MasR antagonist A779 [competing with endogenous ang-(1–7)] attenuated the natriuretic/diuretic response to the AT2R agonist C21 in obese rats under acute settings. The AT2R antagonist PD123319 (competing with endogenous angII) attenuated the responses to ang-(1–7). The study showed an interdependence between AT2R and MasR function (Patel et al., 2017). The present study reasonably supports that AT2R-mediated effects were independent of MasR. However, this conclusion should be further verified as we did not include the rat groups with the MasR agonist and antagonist treatment alone. Further studies are required to understand the mechanisms of AT2R and MasR in the regulation of oxidative stress and proteinuria.
Moreover, the renal NGAL expression is negatively correlated with renal CRP, ICAM-1, and galectin-1 expression (Supplementary Figure S2). Hence, counterintuitively, it is likely but unclear whether A779-mediated NGAL is involved in renal recovery (Mori et al., 2005; Schroll et al., 2012) because A779 neither influenced the renal expression or urinary excretion of KIM-1, a proximal tubular scavenger receptor and another biomarker of renal tubular brush border injury (Mori et al., 2021), nor reversed AT2R-mediated anti-inflammation and BP indexes. Notably, renal KIM-1 is positively correlated with CRP, ICAM-1, and galectin-1 (Supplementary Figure S2). Since high salt causes low-grade inflammation, it is likely that major changes occur in the cytokine levels in this subacute setting, and the effects of C21 are not as significant as previously reported in lipopolysaccharide (LPS) and ischemia/perfusion animal models (Patel et al., 2019). It is highly likely that over time, the expression of these inflammatory markers would further increase if the consumption of the HSD continues. Contrarily, it also has been reported that NGAL and KIM-1 findings may not be correlated (Mori et al., 2021). Collectively, the findings suggest the divergent renal effects of AT2Rs and MasRs but not their interdependent mechanisms in renoprotection.
The decrease in HSD-induced urine osmolality could be due to the high water intake, which was reduced in the AT2R agonist C21-treated group, and consistently, the decrease in osmolality was prevented. Another factor that might have prevented a decrease in HSD-induced osmolality could be because AT2R activation inhibits various Na transporters such as Na/K-ATPase in the kidney proximal tubules (Hakam and Hussain, 2006). The effects of the Mas receptor antagonist A779 on diuresis, natriuresis, osmoregulation, eGFR, and BP regulation are acute, biphasic, transient, and often controversial (Hakam and Hussain, 2006). Subacute treatment with A779 did not completely reverse the effects of the AT2R agonist on the BP, diuresis, natriuresis, urinary osmolality, or eGFR, which is in slight contrast with other studies showing the renal effects of A779 in various animal models (Santos and Baracho, 1992; DelliPizzi et al., 1994; Santos et al., 1994; Handa et al., 1996; Santos et al., 1996; Simões-e-Silva et al., 1997; Simões-e-Silva et al., 1998; Widdop et al., 1999; Patel et al., 2017). Moreover, A779 was only able to transiently increase the BP in female SHRs alone (Sullivan et al., 2010). In male SHRs, A779 decreased the BP after 2 weeks of angII infusion (Sullivan et al., 2010), which aligns with our findings.
Likewise, studies utilizing the MasR agonist ang-(1–7) showed AT2R-independent effects in animal models of cardiovascular diseases, showing benefits, injury, or lack of protection (Dilauro et al., 2010). In a model of ischemic injury, the deletion of MasR remained beneficial in the presence of AT2R (Higaki et al., 2018). In an isolated aorta, it was shown that the vasodilator effects of MasR are independent of AT2R (Silva et al., 2007). The activation of MasR alone showed both hypertensive (Han et al., 2012; Li et al., 2015; Ren et al., 2017; Yu et al., 2019) and anti-hypertensive (Shah et al., 2012; Liang et al., 2018) effects, although the MasR agonist or antagonist was delivered to the brain (Han et al., 2012; Li et al., 2015; Ren et al., 2017; Yu et al., 2019). However, the MasR agonist ang-(1–7) itself did show protective effects in DOCA-salt (Grobe et al., 2006) or angII-induced (Grobe et al., 2007) hypertensive rats but were independent of the changes in BP. We showed an interaction of AT2R and MasR in the proximal tubules in the kidney and in vitro. We speculate that such an interaction exists in other cell types as well as these receptors are expressed on the endothelial, epithelial, and vascular smooth muscle cells and share similar signaling and functions such as vasodilatation and natriuresis (Patel and Hussain, 2018). Salt is known to cause damage to the endothelial cells, so the function of these receptors on these cells would likely be impacted, contributing to the blood pressure through the impact on the vascular tone. However, due to the anti-oxidative and anti-inflammatory functions of AT2R, it is likely that endothelial function is protected. However, in the long-term study, this interaction, if any, does not appear to alter the AT2R-mediated renoprotection, but studies to evaluate the physical interaction in various cell types are needed to determine their functional dependence or independence.
Based on the affinity of angiotensin peptides (Bosnyak et al., 2011) and considering the notion that the dose of an antagonist should be ∼10-fold higher than the Ki of the receptor to identify the measurable in vivo response (Macari et al., 1993; Walters et al., 2005), the doses of C21 and A779 we used are within the range of their specificity. Moreover, A779 is a selective MasR antagonist and does not interact with angiotensin receptors; hence, A779 is a better candidate to use to determine interdependency between AT2R and MasR. Likewise, the lower doses of A779 neither affected BP (Walters et al., 2005; Grobe et al., 2007; Collister and Nahey, 2010) nor were consistent in showing the effects of MasR on diuresis–natriuresis (Santos and Baracho, 1992; Santos et al., 1994; Simões-e-Silva et al., 1997; Simões-e-Silva et al., 1998; Widdop et al., 1999). Hence, we can conclude that the lack of reversibility by A779 on BP, diuresis, natriuresis, urinary osmolality, or eGFR is not due to the dose of A779 used in this study.
Earlier, Walters et al. (2005) showed that in AT1R antagonist-treated rats, ang-(1–7) mediated vasodepressor effects through the activation of AT2R and formation of nitric oxide. Later, it was found that ang-(1–7)/MasR is an AT1R antagonist (Mahon et al., 1994; Kostenis et al., 2005), but ang-(1–7) is also a biased AT1R agonist via beta-arrestin (Galandrin et al., 2016; Teixeira et al., 2017). We observed that ang-(1–7) synergizes the nitric oxide response of AT2R agonists in human renal proximal tubule epithelial (human kidney-2) cells, which is dependent on AT1R and AT2R but not MasR (Patel and Hussain, 2020). However, contrary to these findings, previous reports show that A779 did not reverse AT2R agonist (Ocaranza et al., 2014) or AT1R antagonist (Varagic et al., 2013)-mediated anti-hypertensive effects. Further studies are required to dissect the long-term in vivo interactions among AT1R, AT2R, and MasR.
The extensive literature does show the direct causal relationship between HSD intake and the worsening of hypertensive proteinuric kidney injury independent of comorbidities. However, the degree of injury may differ among individuals. Moreover, globally, the consumption of the HSD has increased over time, and the consumption of an HSD over time can offset the health benefits of ongoing therapeutic regimens. Therefore, considering the therapeutic implications of an ∼10-fold lower dose of AT2R agonist C21 (compared to the typically used dose of C21) in renoprotection in male obese rats, the current work holds high significance. It is important to note that despite numerous reports describing the beneficial role of the MasR agonist ang-(1–7), we show that the MasR blockade did not alter the BP indexes and renal inflammation, and at large, the subacute effects of the AT2R agonist C21 are independent of MasR. The AT2R-MasR functional interdependence appears transient, and MasR may neither be a potent regulator of BP nor has a sustained effect on BP, which is a dynamic phenomenon regulated by multiple organ systems. In addition, the findings show that MasR may play a direct role in the regulation of NGAL and oxidative stress independent of inflammation and BP regulation. Although the present study suggests that AT2R-mediated beneficial effects are independent of the functional interaction with MasR in the subacute setting, other components of the RAS, particularly a reduction in angII/AT1R and increase in ang-(1–7) in the kidney, may play a role in the net effects of AT2R activation, as reported earlier (Samuel et al., 2012; Patel et al., 2017). Although we did not measure RAS components, particularly angII and AT1R expression in the kidney of the C21 + A779 group, it is likely that the C21 effects on angII/AT1R reduction will continue even in the presence of A779, which does not alter the C21 effects in the present study.
5 New and noteworthy
The subacute consumption of the HSD caused renal injury, as evident by KIM-1 and NGAL levels. KIM-1 has shown a positive correlation with CRP, ICAM-1, and galectin-1, while NGAL has shown a negative correlation with CRP, ICAM-1, and galectin-1. The AT2R agonist C21 prevented the subacute deleterious effects of HSD consumption in male OZRs, which are not reversed by the MasR antagonist A779. However, A779 increased proteinuria, oxidative stress, and NGAL, independent of inflammation, BP changes, and AT2R.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by the Institutional Animal Care and Use Committee, University of Houston. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SP: conceptualization, data curation, formal analysis, investigation, methodology, project administration, software, supervision, validation, visualization, writing–original draft, and writing–review and editing. KK: conceptualization, formal analysis, investigation, methodology, and writing–review and editing. TF: formal analysis, investigation, methodology, and writing–review and editing. TH: conceptualization, formal analysis, funding acquisition, investigation, resources, supervision, validation, visualization, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. TH was supported by the National Institutes of Health R01 grants DK061578 and DK117495.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1409313/full#supplementary-material
References
Ali, Q., Patel, S., and Hussain, T. (2015). Angiotensin AT2 receptor agonist prevents salt-sensitive hypertension in obese Zucker rats. Am. J. Physiol. Ren. Physiol. 308 (12), F1379–F1385. doi:10.1152/ajprenal.00002.2015
Benter, I. F., Yousif, M. H. M., Dhaunsi, G. S., Kaur, J., Chappell, M. C., and Diz, D. I. (2008). Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am. J. Nephrol. 28 (1), 25–33. doi:10.1159/000108758
Besseling, P. J., Pieters, T. T., Nguyen, I. T. N., de Bree, P. M., Willekes, N., Dijk, A. H., et al. (2021). A plasma creatinine- and urea-based equation to estimate glomerular filtration rate in rats. Am. J. Physiol. Ren. Physiol. 320 (3), F518–F524. doi:10.1152/ajprenal.00656.2020
Bosnyak, S., Jones, E. S., Christopoulos, A., Aguilar, M. I., Thomas, W. G., and Widdop, R. E. (2011). Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin. Sci. (Lond) 121 (7), 297–303. doi:10.1042/CS20110036
Bosnyak, S., Welungoda, I. K., Hallberg, A., Alterman, M., Widdop, R. E., and Jones, E. S. (2010). Stimulation of angiotensin AT2 receptors by the non-peptide agonist, Compound 21, evokes vasodepressor effects in conscious spontaneously hypertensive rats. Br. J. Pharmacol. 159 (3), 709–716. doi:10.1111/j.1476-5381.2009.00575.x
Bosnyak, S., Widdop, R. E., Denton, K. M., and Jones, E. S. (2012). Differential mechanisms of ang (1-7)-mediated vasodepressor effect in adult and aged candesartan-treated rats. Int. J. Hypertens. 2012, 192567. doi:10.1155/2012/192567
Burrell, L. M., Gayed, D., Griggs, K., Patel, S. K., and Velkoska, E. (2017). Adverse cardiac effects of exogenous angiotensin 1-7 in rats with subtotal nephrectomy are prevented by ACE inhibition. PLoS One 12 (2), e0171975. doi:10.1371/journal.pone.0171975
Collister, J. P., and Nahey, D. B. (2010). Simultaneous administration of Ang(1-7) or A-779 does not affect the chronic hypertensive effects of angiotensin II in normal rats. J. Renin Angiotensin-Aldosterone Syst. 11 (2), 99–102. doi:10.1177/1470320309359928
DelliPizzi, A., Hilchey, S. D., and Bell-Quilley, C. P. (1994). Natriuretic action of angiotensin(1-7). Br. J. Pharmacol. 111, 1–3. doi:10.1111/j.1476-5381.1994.tb14014.x
Dilauro, M., Zimpelmann, J., Robertson, S. J., Genest, D., and Burns, K. D. (2010). Effect of ACE2 and angiotensin-(1-7) in a mouse model of early chronic kidney disease. Am. J. Physiol. Ren. Physiol. 298 (6), F1523–F1532. doi:10.1152/ajprenal.00426.2009
Esteban, V., Heringer-Walther, S., Sterner-Kock, A., de Bruin, R., van den Engel, S., Wang, Y., et al. (2009). Angiotensin-(1-7) and the g protein-coupled receptor MAS are key players in renal inflammation. PLoS One 4 (4), e5406. doi:10.1371/journal.pone.0005406
Ferreira, A. J., Shenoy, V., Yamazato, Y., Sriramula, S., Francis, J., Yuan, L., et al. (2009). Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 179 (11), 1048–1054. doi:10.1164/rccm.200811-1678OC
Galandrin, S., Denis, C., Boularan, C., Marie, J., M'Kadmi, C., Pilette, C., et al. (2016). Cardioprotective angiotensin-(1-7) peptide acts as a natural-biased ligand at the angiotensin II type 1 receptor. Hypertension 68 (6), 1365–1374. doi:10.1161/HYPERTENSIONAHA.116.08118
Giani, J. F., Burghi, V., Veiras, L. C., Tomat, A., Muñoz, M. C., Cao, G., et al. (2012). Angiotensin-(1-7) attenuates diabetic nephropathy in Zucker diabetic fatty rats. Am. J. Physiol. Ren. Physiol. 302 (12), F1606–F1615. doi:10.1152/ajprenal.00063.2012
Giani, J. F., Muñoz, M. C., Pons, R. A., Cao, G., Toblli, J. E., Turyn, D., et al. (2011). Angiotensin-(1-7) reduces proteinuria and diminishes structural damage in renal tissue of stroke-prone spontaneously hypertensive rats. Am. J. Physiol. Ren. Physiol. 300 (1), F272–F282. doi:10.1152/ajprenal.00278.2010
Grobe, J. L., Mecca, A. P., Lingis, M., Shenoy, V., Bolton, T. A., Machado, J. M., et al. (2007). Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1-7). Am. J. Physiol. Heart Circ. Physiol. 292 (2), H736–H742. doi:10.1152/ajpheart.00937.2006
Grobe, J. L., Mecca, A. P., Mao, H., and Katovich, M. J. (2006). Chronic angiotensin-(1-7) prevents cardiac fibrosis in DOCA-salt model of hypertension. Am. J. Physiol. Heart Circ. Physiol. 290 (6), H2417–H2423. doi:10.1152/ajpheart.01170.2005
Hakam, A. C., and Hussain, T. (2006). Angiotensin II AT2 receptors inhibit proximal tubular Na+-K+-ATPase activity via a NO/cGMP-dependent pathway. Am. J. Physiol. Ren. Physiol. 290 (6), F1430–F1436. doi:10.1152/ajprenal.00218.2005
Han, Y., Sun, H. J., Li, P., Gao, Q., Zhou, Y. b., Zhang, F., et al. (2012). Angiotensin-(1-7) in paraventricular nucleus modulates sympathetic activity and cardiac sympathetic afferent reflex in renovascular hypertensive rats. PLoS One 7 (11), e48966. doi:10.1371/journal.pone.0048966
Handa, R. K., Ferrario, C. M., and Strandhoy, J. W. (1996). Renal actions of angiotensin-(1-7) - in vivo and in vitro studies. Am. J. Physiology Ren. Fluid Electrolyte Physiology 39, F141–F147. doi:10.1152/ajprenal.1996.270.1.F141
Hao, P., Yang, J., Liu, Y., Zhang, M., Zhang, K., Gao, F., et al. (2015a). Combination of angiotensin-(1-7) with perindopril is better than single therapy in ameliorating diabetic cardiomyopathy. Sci. Rep. 5, 8794. doi:10.1038/srep08794
Hao, P. P., Yang, J. M., Zhang, M. X., Zhang, K., Chen, Y. G., Zhang, C., et al. (2015b). Angiotensin-(1-7) treatment mitigates right ventricular fibrosis as a distinctive feature of diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 308 (9), H1007–H1019. doi:10.1152/ajpheart.00563.2014
Higaki, A., Mogi, M., Iwanami, J., Min, L. J., Bai, H. Y., Shan, B. S., et al. (2018). Beneficial effect of mas receptor deficiency on vascular cognitive impairment in the presence of angiotensin II type 2 receptor. J. Am. Heart Assoc. 7 (3), e008121. doi:10.1161/JAHA.117.008121
Jehle, A. B., Xu, Y., Dimaria, J. M., French, B. A., Epstein, F. H., Berr, S. S., et al. (2012). A nonpeptide angiotensin II type 2 receptor agonist does not attenuate postmyocardial infarction left ventricular remodeling in mice. J. Cardiovasc Pharmacol. 59 (4), 363–368. doi:10.1097/FJC.0b013e3182444110
Kaschina, E., Grzesiak, A., Li, J., Foryst-Ludwig, A., Timm, M., Rompe, F., et al. (2008). Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 118 (24), 2523–2532. doi:10.1161/CIRCULATIONAHA.108.784868
Kemp, B. A., Howell, N. L., Gildea, J. J., Keller, S. R., Padia, S. H., and Carey, R. M. (2014). AT₂ receptor activation induces natriuresis and lowers blood pressure. Circ. Res. 115 (3), 388–399. doi:10.1161/CIRCRESAHA.115.304110
Kljajic, S. T., Widdop, R. E., Vinh, A., Welungoda, I., Bosnyak, S., Jones, E. S., et al. (2013). Direct AT₂ receptor stimulation is athero-protective and stabilizes plaque in apolipoprotein E-deficient mice. Int. J. Cardiol. 169 (4), 281–287. doi:10.1016/j.ijcard.2013.09.015
Kostenis, E., Milligan, G., Christopoulos, A., Sanchez-Ferrer, C. F., Heringer-Walther, S., Sexton, P. M., et al. (2005). G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 111 (14), 1806–1813. doi:10.1161/01.CIR.0000160867.23556.7D
Li, P., Zhang, F., Sun, H. J., Zhang, F., and Han, Y. (2015). Angiotensin-(1-7) enhances the effects of angiotensin II on the cardiac sympathetic afferent reflex and sympathetic activity in rostral ventrolateral medulla in renovascular hypertensive rats. J. Am. Soc. Hypertens. 9 (11), 865–877. doi:10.1016/j.jash.2015.08.005
Liang, B., Zhao, Y. N., Wang, X., Yu, X. J., Li, Y., Yang, H. Y., et al. (2018). Angiotensin-(1-7) attenuates hypertension and cardiac hypertrophy via modulation of nitric oxide and neurotransmitter levels in the paraventricular nucleus in salt-sensitive hypertensive rats. RSC Adv. 8 (16), 8779–8786. doi:10.1039/c7ra09136b
Liu, Y., Li, B., Wang, X., Li, G., Shang, R., Yang, J., et al. (2015). Angiotensin-(1-7) suppresses hepatocellular carcinoma growth and angiogenesis via complex interactions of angiotensin II type 1 receptor, angiotensin II type 2 receptor and mas receptor. Mol. Med. 21 (1), 626–636. doi:10.2119/molmed.2015.00022
Macari, D., Bottari, S., Whitebread, S., De Gasparo, M., and Levens, N. (1993). Renal actions of the selective angiotensin AT 2 receptor ligands CGP 42112B and PD 123319 in the sodium-depleted rat. Eur. J. Pharmacol. 249, 85–93. doi:10.1016/0014-2999(93)90665-5
Mahon, J. M., Carr, R. D., Nicol, A. K., and Henderson, I. W. (1994). Angiotensin(1-7) is an antagonist at the type 1 angiotensin II receptor. J. Hypertens. 12, 1377–1382. doi:10.1097/00004872-199412000-00010
McWilliam, S. J., Antoine, D. J., Sabbisetti, V., Pearce, R. E., Jorgensen, A. L., Lin, Y., et al. (2014). Reference intervals for urinary renal injury biomarkers KIM-1 and NGAL in healthy children. Biomarkers Med. 8 (10), 1189–1197. doi:10.2217/bmm.14.36
Mori, K., Lee, H. T., Rapoport, D., Drexler, I. R., Foster, K., Yang, J., et al. (2005). Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J. Clin. Investigation 115 (3), 610–621. doi:10.1172/JCI23056
Mori, Y., Ajay, A. K., Chang, J. H., Mou, S., Zhao, H., Kishi, S., et al. (2021). KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 33 (5), 1042–1061 e7. doi:10.1016/j.cmet.2021.04.004
Ocaranza, M. P., Moya, J., Barrientos, V., Alzamora, R., Hevia, D., Morales, C., et al. (2014). Angiotensin-(1-9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis. J. Hypertens. 32 (4), 771–783. doi:10.1097/HJH.0000000000000094
Papinska, A. M., Mordwinkin, N. M., Meeks, C. J., Jadhav, S. S., and Rodgers, K. E. (2015). Angiotensin-(1-7) administration benefits cardiac, renal and progenitor cell function in db/db mice. Br. J. Pharmacol. 172 (18), 4443–4453. doi:10.1111/bph.13225
Patel, S., Dhande, I., Gray, E. A., Ali, Q., and Hussain, T. (2019). Prevention of lipopolysaccharide-induced CD11b(+) immune cell infiltration in the kidney: role of AT(2) receptors. Biosci. Rep. 39 (5). doi:10.1042/BSR20190429
Patel, S., and Hussain, T. (2018). Dimerization of AT(2) and mas receptors in control of blood pressure. Curr. Hypertens. Rep. 20 (5), 41. doi:10.1007/s11906-018-0845-3
Patel, S., and Hussain, T. (2020). Synergism between Angiotensin receptors ligands: role of Angiotensin-(1-7) in modulating AT(2) R agonist response on nitric oxide in kidney cells. Pharmacol. Res. Perspect. 8 (6), e00667. doi:10.1002/prp2.667
Patel, S. N., Ali, Q., and Hussain, T. (2016). Angiotensin II type 2-receptor agonist C21 reduces proteinuria and oxidative stress in kidney of high-salt-fed obese zucker rats. Hypertension 67 (5), 906–915. doi:10.1161/HYPERTENSIONAHA.115.06881
Patel, S. N., Ali, Q., Samuel, P., Steckelings, U. M., and Hussain, T. (2017). Angiotensin II type 2 receptor and receptor mas are colocalized and functionally interdependent in obese zucker rat kidney. Hypertension 70 (4), 831–838. doi:10.1161/HYPERTENSIONAHA.117.09679
Pinheiro, S. V. B., Ferreira, A. J., Kitten, G. T., da Silveira, K. D., da Silva, D. A., Santos, S. H. S., et al. (2009). Genetic deletion of the angiotensin-(1-7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int. 75 (11), 1184–1193. doi:10.1038/ki.2009.61
Ren, X., Zhang, F., Zhao, M., Zhao, Z., Sun, S., Fraidenburg, D. R., et al. (2017). Angiotensin-(1-7) in paraventricular nucleus contributes to the enhanced cardiac sympathetic afferent reflex and sympathetic activity in chronic heart failure rats. Cell Physiol. Biochem. 42 (6), 2523–2539. doi:10.1159/000480214
Ruster, C., Franke, S., Wenzel, U., Schmidthaupt, R., Fraune, C., Krebs, C., et al. (2011). Podocytes of AT2 receptor knockout mice are protected from angiotensin II-mediated RAGE induction. Am. J. Nephrol. 34 (4), 309–317. doi:10.1159/000329321
Samuel, P., Ali, Q., Sabuhi, R., Wu, Y., and Hussain, T. (2012). High Na intake increases renal angiotensin II levels and reduces expression of the ACE2-AT(2)R-MasR axis in obese Zucker rats. Am. J. Physiol. Ren. Physiol. 303 (3), F412–F419. doi:10.1152/ajprenal.00097.2012
Santos, R. A. S., and Baracho, N. C. V. (1992). Angiotensin-(1-7) is a potent antidiuretic peptide in rats. Braz. J. Med. Biol. Res. 25, 651–654.
Santos, R. A. S., Campagnole-Santos, M. J., Baracho, N. C., Fontes, M. A., Silva, L. C., Neves, L. A., et al. (1994). Characterization of a new angiotensin antagonist selective for angiotensin-(1-7): evidence that the actions of angiotensin-(1-7) are mediated by specific angiotensin receptors. Brain Res. Bull. 35 (4), 293–298. doi:10.1016/0361-9230(94)90104-x
Santos, R. A. S., Simões e Silva, A. C., Magaldi, A. J., Khosla, M. C., Cesar, K. R., Passaglio, K. T., et al. (1996). Evidence for a physiological role of angiotensin-(1-7) in the control of hydroelectrolyte balance. Hypertension 27 (4), 875–884. doi:10.1161/01.hyp.27.4.875
Santos, R. A. S., Simoes e Silva, A. C., Maric, C., Silva, D. M. R., Machado, R. P., de Buhr, I., et al. (2003). Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. PNAS 100 (14), 8258–8263. doi:10.1073/pnas.1432869100
Schroll, A., Eller, K., Feistritzer, C., Nairz, M., Sonnweber, T., Moser, P. A., et al. (2012). Lipocalin-2 ameliorates granulocyte functionality. Eur. J. Immunol. 42 (12), 3346–3357. doi:10.1002/eji.201142351
Shah, A., Oh, Y. B., Lee, S. H., Lim, J. M., and Kim, S. H. (2012). Angiotensin-(1-7) attenuates hypertension in exercise-trained renal hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 302 (11), H2372–H2380. doi:10.1152/ajpheart.00846.2011
Shenoy, V., Ferreira, A. J., Qi, Y., Fraga-Silva, R. A., Díez-Freire, C., Dooies, A., et al. (2010). The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 182 (8), 1065–1072. doi:10.1164/rccm.200912-1840OC
Silva, D. M., Vianna, H. R., Cortes, S. F., Campagnole-Santos, M. J., Santos, R. A. S., and Lemos, V. S. (2007). Evidence for a new angiotensin-(1-7) receptor subtype in the aorta of Sprague-Dawley rats. Peptides 28 (3), 702–707. doi:10.1016/j.peptides.2006.10.007
Simões-e-Silva, A. C., Baracho, N. C. V., Passaglio, K. T., and Santos, R. A. S. (1997). Renal actions of angiotensin-(1-7). Braz. J. Med. Biol. Res. 30, 503–513. doi:10.1590/s0100-879x1997000400012
Simões-e-Silva, A. C., Bello, A. P., Baracho, N. C., Khosla, M. C., and Santos, R. A. (1998). Diuresis and natriuresis produced by long term administration of a selective Angiotensin-(1–7) antagonist in normotensive and hypertensive rats. Regul. Pept. 74, 177–184. doi:10.1016/s0167-0115(98)00038-x
Smyth, A., O'Donnell, M. J., Yusuf, S., Clase, C. M., Teo, K. K., Canavan, M., et al. (2014). Sodium intake and renal outcomes: a systematic review. Am. J. Hypertens. 27 (10), 1277–1284. doi:10.1093/ajh/hpt294
Su, Z., Zimpelmann, J., and Burns, K. D. (2006). Angiotensin-(1-7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int. 69 (12), 2212–2218. doi:10.1038/sj.ki.5001509
Sullivan, J. C., Bhatia, K., Yamamoto, T., and Elmarakby, A. A. (2010). Angiotensin (1-7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension 56 (4), 658–666. doi:10.1161/HYPERTENSIONAHA.110.153668
Teixeira, D. E., Peruchetti, D. B., Souza, M. C., das Graças Henriques, M. G., Pinheiro, A. A. S., and Caruso-Neves, C. (2020). A high salt diet induces tubular damage associated with a pro-inflammatory and pro-fibrotic response in a hypertension-independent manner. Biochim. Biophys. Acta Mol. Basis Dis. 1866 (11), 165907. doi:10.1016/j.bbadis.2020.165907
Teixeira, L. B., Parreiras-E-Silva, L. T., Bruder-Nascimento, T., Duarte, D. A., Simões, S. C., Costa, R. M., et al. (2017). Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 7 (1), 11903. doi:10.1038/s41598-017-12074-3
Tesanovic, S., Vinh, A., Gaspari, T. A., Casley, D., and Widdop, R. E. (2010). Vasoprotective and atheroprotective effects of angiotensin (1-7) in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 30 (8), 1606–1613. doi:10.1161/ATVBAHA.110.204453
Varagic, J., Ahmad, S., VonCannon, J. L., Moniwa, N., Brosnihan, K. B., Wysocki, J., et al. (2013). Predominance of AT(1) blockade over mas-mediated angiotensin-(1-7) mechanisms in the regulation of blood pressure and renin-angiotensin system in mRen2.Lewis rats. Am. J. Hypertens. 26 (5), 583–590. doi:10.1093/ajh/hps090
Villela, D., Leonhardt, J., Patel, N., Joseph, J., Kirsch, S., Hallberg, A., et al. (2015). Angiotensin type 2 receptor (AT2R) and receptor Mas: a complex liaison. Clin. Sci. (Lond) 128 (4), 227–234. doi:10.1042/CS20130515
Walters, P. E., Gaspari, T. A., and Widdop, R. E. (2005). Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 45 (5), 960–966. doi:10.1161/01.HYP.0000160325.59323.b8
Widdop, R. E., Sampey, D. B., and Jarrott, B. (1999). Cardiovascular effects of angiotensin-(1-7) in conscious spontaneously hypertensive rats. Hypertension 34 (4 Pt 2), 964–968. doi:10.1161/01.hyp.34.4.964
Xiao, X., Yeoh, B. S., and Vijay-Kumar, M. (2017). Lipocalin 2: an emerging player in iron Homeostasis and inflammation. Annu. Rev. Nutr. 37, 103–130. doi:10.1146/annurev-nutr-071816-064559
Yu, X. J., Miao, Y. W., Li, H. B., Su, Q., Liu, K. L., Fu, L. Y., et al. (2019). Blockade of endogenous angiotensin-(1-7) in hypothalamic paraventricular nucleus attenuates high salt-induced sympathoexcitation and hypertension. Neurosci. Bull. 35 (1), 47–56. doi:10.1007/s12264-018-0297-4
Keywords: angiotensin-II type 2 receptor, receptor Mas, high-sodium diet, C21, kidney, interdependency
Citation: Patel SN, Kulkarni K, Faisal T and Hussain T (2024) Angiotensin-II type 2 receptor-mediated renoprotection is independent of receptor Mas in obese Zucker rats fed high-sodium diet. Front. Pharmacol. 15:1409313. doi: 10.3389/fphar.2024.1409313
Received: 29 March 2024; Accepted: 27 June 2024;
Published: 29 July 2024.
Edited by:
Utpal Sen, University of Louisville, United StatesReviewed by:
Belisario Enrique Fernandez, Héctor Alejandro Barceló Foundation, ArgentinaPatricio Araos, Universidad Autónoma de Chile, Chile
Copyright © 2024 Patel, Kulkarni, Faisal and Hussain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tahir Hussain, dGh1c3NhaW5AY2VudHJhbC51aC5lZHU=