
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 05 August 2024
Sec. Translational Pharmacology
Volume 15 - 2024 | https://doi.org/10.3389/fphar.2024.1409210
This article is part of the Research TopicMolecular Targets in Oncological and Hematological Disease Management: Innovations in Precision MedicineView all 10 articles
 Monika Chomczyk1†
Monika Chomczyk1† Luca Gazzola2†
Luca Gazzola2† Shubhankar Dash1
Shubhankar Dash1 Patryk Firmanty1
Patryk Firmanty1 Binsah S. George3
Binsah S. George3 Vakul Mohanty4Hussein A. Abbas5
Vakul Mohanty4Hussein A. Abbas5 Natalia Baran1,5*
Natalia Baran1,5*Acute myeloid leukemia (AML), an aggressive malignancy of hematopoietic stem cells, is characterized by the blockade of cell differentiation, uncontrolled proliferation, and cell expansion that impairs healthy hematopoiesis and results in pancytopenia and susceptibility to infections. Several genetic and chromosomal aberrations play a role in AML and influence patient outcomes. TP53 is a key tumor suppressor gene involved in a variety of cell features, such as cell-cycle regulation, genome stability, proliferation, differentiation, stem-cell homeostasis, apoptosis, metabolism, senescence, and the repair of DNA damage in response to cellular stress. In AML, TP53 alterations occur in 5%–12% of de novo AML cases. These mutations form an important molecular subgroup, and patients with these mutations have the worst prognosis and shortest overall survival among patients with AML, even when treated with aggressive chemotherapy and allogeneic stem cell transplant. The frequency of TP53-mutations increases in relapsed and recurrent AML and is associated with chemoresistance. Progress in AML genetics and biology has brought the novel therapies, however, the clinical benefit of these agents for patients whose disease is driven by TP53 mutations remains largely unexplored. This review focuses on the molecular characteristics of TP53-mutated disease; the impact of TP53 on selected hallmarks of leukemia, particularly metabolic rewiring and immune evasion, the clinical importance of TP53 mutations; and the current progress in the development of preclinical and clinical therapeutic strategies to treat TP53-mutated disease.
Acute myeloid leukemia (AML) is a malignant, clonal, hematological disease (Caruso et al., 2022) that develops from transformed hematopoietic stem cells (HSCs) and is characterized by cells’ uncontrolled proliferation, expansion, and an unlimited capacity for self-renewal (George et al., 2021). AML occurs mainly in adults over 40 years old, with the peak incidence in patients above 70 years of age (Daver et al., 2023a). Most patients with AML harbor mutations that cause cells’ malignant proliferation and enhance their ability to evade cell death (Chen et al., 2022). AML shows different metabolic and physiologic hallmarks, depending on the types of harbored mutations (Kennedy and Lowe, 2022). The most commonly mutated genes related to AML initiation and progression include FMS-related receptor tyrosine kinase 3 (FLT3), DNA methyltransferase three alpha (DNMT3A), nucleophosmin 1 (NPM1), and tet methylcytosine dioxygenase 2 (TET2) (Villatoro et al., 2020). AML mutations, together with cytogenetic abnormalities, have critical implications for clinical outcomes (Olivier et al., 2010). It is estimated that 50% of de novo AML cases show cytogenetic abnormalities, and the number and frequency of mutations increase in patients developing therapy-related AML, who have been treated previously with cytotoxic therapies such as alkylating agents, topoisomerase II inhibitors, and radiotherapy (Hong et al., 2016). Therapy-related AML is also frequently characterized by complex karyotypes and mutations in the tumor protein p53 (TP53) gene (Olivier et al., 2010; Hong et al., 2016). The incidence of TP53 mutations in AML can vary. While the frequency of TP53 mutations is estimated to account for 10% of de novo AML cases, it rises strikingly in therapy-related AML or relapsing/refractory (R/R) AML cases, reaching up to 30% and 25%, respectively, in these groups (Daver et al., 2023a). An even higher frequency of mutant TP53 is associated with the complex karyotype subtype of AML, in which the frequency of TP53 mutations reaches up to 70%, mainly due to selective pressure caused by acquired resistance to DNA damage following chemotherapy and radiotherapy.
Identifying potential therapeutic vulnerabilities and therapeutic targets in AML is challenging due to AML’s genetic heterogeneity (Zhou et al., 2020). The exploration of novel genomic targets has led to the development of only a few potent targeted therapies, such as IDH, FLT3, or KMT2A inhibitors for genomically defined AML subsets (Appelbaum et al., 2024). However, due to the genotypic and phenotypic diversity of mutant TP53, finding targeted therapies against TP53 remains an unresolved challenge.
TP53 is a 20-kbp gene located on chromosome 17p13.1 (Olivier et al., 2010). So far, 15 isoforms of p53 have been identified (Haaland et al., 2021). Despite some differences, each p53 isoform consists of five common domains: an N-terminal, a proline-rich domain, a DNA-binding domain (DBD), a regulatory domain, and a C-terminal (George et al., 2021). Activation of p53 occurs in response to diverse cellular stress factors such as hypoxia, DNA damage, oncogene expression, or replicative stress (Daver et al., 2022; Wang H. et al., 2023). Through its DBD domain, p53 regulates the transcription of genes involved in cell-cycle regulation, genome stability, proliferation, stem-cell homeostasis and differentiation, and cell-death regulation (Daver et al., 2022; Wang H. et al., 2023). P53 also has antiangiogenic properties; it represses metastases, controls tumor-promoting inflammation, facilitates the immune response, promotes replicative senescence, enhances the effects of growth suppressors, and regulates energetics and metabolism (Daver et al., 2022; Wang H. et al., 2023). The p53 inhibitors MDM2 and MDM4, which control p53 s ubiquitination and ubiquitin–proteasome system activity, tightly regulate p53 levels. (Wang H. et al., 2023). In AML, p53 is mainly silenced by the upregulation of MDM2, MDM4/MDMX, ARF, and E6 (Abramowitz et al., 2017; Latif et al., 2021; Tashakori et al., 2022). The precisely controlled level of p53 might be also disturbed due to somatic mutations in TP53 or to imbalances in the gene products interacting with p53, leading to its inactivation (Olivier et al., 2010). Most somatic mutations occur mainly as point missense mutations, frameshift insertions or deletions, splice sites, and nonsense mutations; these mutations are observed in leukemia and in many other types of cancer (Olivier et al., 2010). Studies have shown that almost 90% of TP53 mutations detected in patients with therapy-related myeloid neoplasms have variant allele frequencies (VAFs) greater than 10%, and that these VAFs frequently occurred with the loss of 17p across the TP53 locus (loss of heterozygosity) or as copy-neutral loss of heterozygosity (Donehower et al., 2019; Shah et al., 2023). Compared to wild-type TP53, TP53 mutation with a VAF greater than10% was associated with inferior outcomes and worse survival (Donehower et al., 2019; Shah et al., 2023).
TP53 mutations are a strong indicator of prognosis, and studies have shown that, in AML, multi-hit mutated TP53 is associated with genomic instability, thrombocytopenia, and a higher blast count, independent of the VAF (Deng et al., 2020). Further studies have shown that, unlike AML cells that carry multi-hit mutated TP53, those that carry monoallelic TP53 mutations frequently harbor co-mutations in genes like tet methylcytosine dioxygenase 2 (TET2), splicing factor 3b subunit 1 (SF3B1), ASXL transcriptional regulator 1 (ASXL1), RUNX family transcription factor 1 (RUNX1), isocitrate dehydrogenase (NADP(+)) 2 (IDH2), serine and arginine rich splicing factor 2 (SRSF2) (Shah et al., 2023), Mitogen-activated protein kinase kinase kinase 7 (TAK1), BCL6 corepressor (BCOR), and Cbl proto-oncogene (CBL) (Daver et al., 2023a). Finally, TP53 monoallelic mutations with co-mutated RUNX1, KRAS proto-oncogene, GTPase (KRAS), or CBL are correlated with poor prognosis more frequently than monoallelic TP53 mutations (Daver et al., 2023a). Although performing a TP53 status analysis is not yet considered standard procedure, Cox multivariate hazard models have shown that heavy alterations of TP53 allele status independently predict a poor prognosis (Daver et al., 2023a). The 2022 findings of European LeukemiaNet support the consideration of a TP53 mutation as a distinct AML entity with a “very-adverse” risk profile like that listed for European LeukemiaNet in the 2022 International Consensus Classification (Tashakori et al., 2022; Fleming et al., 2023).
In AML, TP53 mutations are mainly found in 6 DBD hotspots: R175H, G245S, R248Q/W, R249S, R273H/S, and R282W (Kennedy and Lowe, 2022). These mutations lead to reduced activity, complete loss of function (LOF), or, less frequently, to a switch or gain of function (GOF), suggesting that there may be some tissue-specific requirements for the loss of wild-type (wt) or gain of mutant p53 functions (Boettcher et al., 2019; Daver et al., 2022; Kennedy and Lowe, 2022).
Beyond the most common TP53 mutations, the Y220C mutation frequently appears in various solid tumors and leukemias (Barnoud et al., 2021). Research suggests that this mutation is the ninth-most-frequent p53 cancer mutation (Barnoud et al., 2021; Hassin and Oren, 2023). It creates a cavity on the p53’s surface, making it highly unstable. Interestingly, the Y220C mutation has been linked to 3 cases of familial cancer, and it appears to grant the p53 new cancer-promoting abilities. These abilities include stimulating the growth of blood vessels (angiogenesis) and making cancer cells resistant to the chemotherapy drug doxorubicin (Loke et al., 2022).
In general, TP53 mutations substantially impact tumor development. In fact, over half of all human cancers have some form of TP53 mutation. The mutations can be particularly severe in Li-Fraumeni syndrome, in which a mutated TP53 gene dramatically increases the risk of cancers like osteosarcoma, leukemia, breast cancer, brain tumors, and adrenal tumors (Vaddavalli and Schumacher, 2022). Since p53 plays a role in many cellular processes, mutations in this gene can disrupt a wide range of functions and ultimately make cells more likely to acquire the characteristics needed to become cancerous, as illustrated in Figure 1.
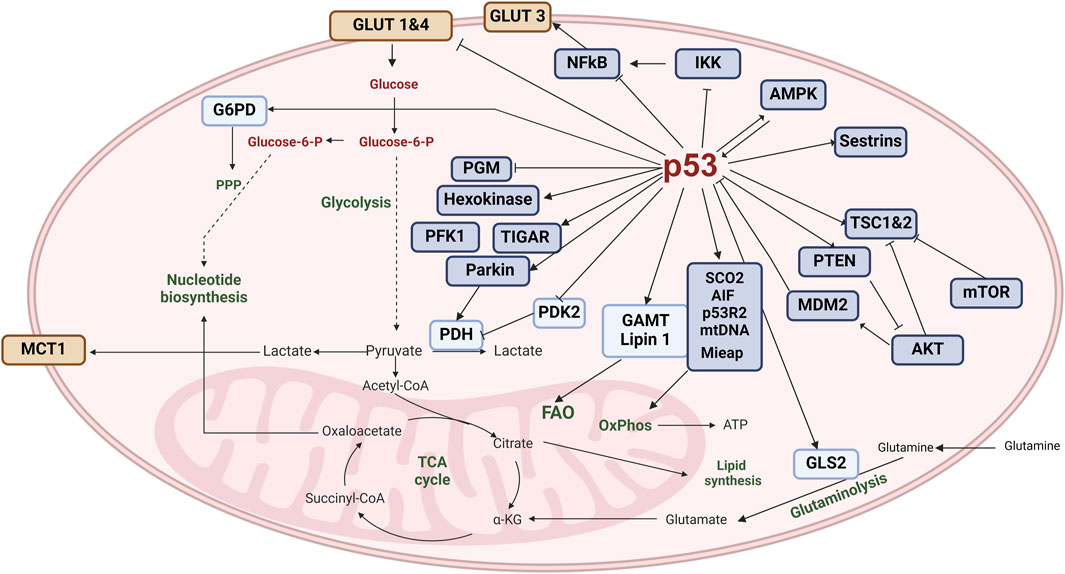
Figure 1. The role of p53 in the hallmarks of acute myeloid leukemia. The wild-type and mutant forms of p53 play distinct roles in controlling the diverse hallmarks of cancer. Wild-type p53 (wtp53) maintains effective anti-tumor immunity, mainly through its role in cell death (apoptosis or autophagy), senescence, or regulation of reactive oxygen species (ROS). It is also involved in regulating the immune response. In contrast, mutant p53 exerts negative effects on the immune environment by activating inflammatory pathways; this process can be enhanced by an excessive production of ROS. In consequence, mutant p53 contributes to cancer-associated chronic inflammation and protects leukemia cells from immune-mediated destruction and phagocytosis. It causes genome instability and the accumulation of new mutations that further perturbate the DNA repair machinery and enable uncontrolled growth and arrest of the cell cycle. Mutant p53 also sustains proliferative signaling, leading to replicative immortality, resistance to cell death, and finally metabolic rewiring to meet new energetic and biosynthetic demands. (Image created with BioRender.com).
Mutations in TP53 affect homeostasis, not only via altering protein activity, but also by altering the isoforms ratio, which may diminish a patient’s response to chemotherapy (Haaland et al., 2021). Among the most common TP53 mutations, mutations in the hotspots R175 and R248 are frequently detected in diverse solid tumors but occur less frequently in AML (Barnoud et al., 2021). Interestingly, AML patients with heterozygous TP53 mutations have shown similar responses to therapy as those harboring wt TP53 (Barnoud et al., 2021; Kennedy and Lowe, 2022; Daver et al., 2023a). However, the sequential acquisition of the mutation of one allele, followed by mutation of the other allele or loss of the entire 17p chromosome, leads to a complete LOF of wt TP53 and has been identified as the key progression mechanism involving TP53 mutation. (Hong et al., 2016; Boettcher et al., 2019; Yang et al., 2022; Shah et al., 2023). Furthermore, wt TP53 LOF is seen in patients with Li-Fraumeni syndrome; affected individuals develop AML at a frequency comparable to that of healthy individuals with other TP53 mutations, but they manifest more aggressive disease (Prokocimer et al., 2017; Boettcher et al., 2019; Daver et al., 2022; Shah et al., 2023). TP53 LOF is often correlated with nonautonomous effects on the tumor immune microenvironment; it subverts the wt p53 effect and allows the evasion of attack from the immune system (Prokocimer et al., 2017; Loizou et al., 2019; Hassin and Oren, 2023; Rajagopalan et al., 2023). Many point mutations in TP53 have been studied by overexpressing the missense allele in TP53 null tumor cells; specifically, an increase in growth independence, tumor progression, metastasis, and drug resistance (Singh et al., 2019). These changes have been associated with missense TP53 variants, indicating that they have novel functions that promote tumor growth and contribute to tumorigenesis. (Loizou et al., 2019; Singh et al., 2019; Rajagopalan et al., 2023). While the GOF of mutated p53 in AML harboring TP53 mut/mut is still debated, an alternative mechanism called separation of function seems to contribute to AML pathogenesis (Kennedy and Lowe, 2022). Table 1 summarizes the two main types of mutations that can affect the p53 protein, a critical tumor suppressor in the human body. Understanding these mutations leads to a fuller understanding of how cancers develop and progress.

Table 1. Differences between the loss-of-function and gain-of-function mutations affecting the p53 protein.
p53 activity is tightly regulated by posttranslational modifications, with phosphorylation being a crucial event (Ashcroft et al., 1999). Understanding p53 phosphorylation at multiple serine, threonine, and tyrosine residues by specific kinases [ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR), and checkpoint kinase 2 (CHK2)] in response to diverse cellular stresses (e.g., DNA damage, oxidative stress) and examining the multifaceted consequences of p53 phosphorylation, including enhanced protein stability, augmented transcriptional activity, and modulation of subcellular localization, may lead to the development of new therapeutic strategies (Sakaguchi et al., 1998; Ashcroft et al., 1999). These phosphorylation events orchestrate the activation of various target genes involved in cell-cycle arrest, DNA repair, and apoptosis, ultimately determining cellular fate following stress induction (Loughery and Meek, 2013). A significant percentage of AML cases have mutations in the TP53 gene, and these mutations can disrupt the normal phosphorylation and regulation of p53, leading to its dysfunction (Ni et al., 2024). Mutant p53 proteins may have altered phosphorylation patterns or impaired interactions with kinases and other regulatory proteins, affecting their ability to properly respond to cellular stresses (Hales et al., 2014).
Several signaling pathways, such as the PI3K/AKT/mTOR pathway, MAPK pathway, and JAK/STAT pathway, can modulate p53 phosphorylation and activity in leukemias (Leu et al., 2020). Dysregulation of these pathways in AML can influence p53 phosphorylation and its downstream functions, potentially promoting leukemic cell survival or drug resistance (Motlagh et al., 2022).
p53 is a master regulator of cancer-relevant pathways, governing genomic stability (DNA repair), cell fate (cell cycle arrest, senescence, apoptosis), and cellular processes (metabolism, autophagy, ferroptosis) (Figure 1) (Singh et al., 2019).
The TP53 gene plays a crucial role in regulating cellular energy and metabolism (Leu et al., 2020). As a transcription factor, TP53 controls the expression of various genes involved in metabolic pathways (Motlagh et al., 2022; Luo et al., 2023). It influences the balance between glycolysis and oxidative phosphorylation (Motlagh et al., 2022; Luo et al., 2023). TP53 also impacts glycolysis through reduction or downregulation of key glycolytic enzymes, or transporters for glucose, pyruvate or other essential for glycolysis nutrients, and through suppression of the AKT/mTOR and NF-κB signaling pathways. (Motlagh et al., 2022; Luo et al., 2023; McClure et al., 2023; Roche et al., 2023). Additionally, TP53 controls glucose-regulating cellular energetics and metabolism by suppressing the glucose transporters (GLUT1 and GLUT4) that bring glucose into the cell. TP53 can also induce TP53-induced glycolysis and apoptosis regulator, which diverts glucose away from glycolysis and towards the pentose-phosphate pathway, involved in nucleotide synthesis, lipids synthesis, and amino acids synthesis to meet the energy demands of the cell (Motlagh et al., 2022; Luo et al., 2023; McClure et al., 2023; Roche et al., 2023). TP53 can induce the expression of genes involved in antioxidant defense, protecting cells from oxidative stress (Sakaguchi et al., 1998; Ashcroft et al., 1999; Loughery and Meek, 2013; Hales et al., 2014; Ni et al., 2024). This shift towards oxidative phosphorylation increases ATP production and provides more efficient energy for cellular processes (Luo et al., 2023; McClure et al., 2023; Roche et al., 2023). TP53 also inhibits the expression of genes involved in fatty-acid synthesis, promoting lipid breakdown and utilization (Luo et al., 2023; McClure et al., 2023; Roche et al., 2023). Furthermore, TP53 can modulate the activity of enzymes involved in energy metabolism, such as AMP-activated protein kinase (AMPK) (Motlagh et al., 2022; Luo et al., 2023; McClure et al., 2023; Roche et al., 2023). In addition, it can influence mitochondrial function and biogenesis, affecting cellular energy production (Motlagh et al., 2022; Luo et al., 2023; McClure et al., 2023; Roche et al., 2023; Sanford et al., 2023; Li et al., 2024). Overall, TP53 plays a multifaceted role in cellular energy and metabolism, maintaining the balance between energy production and utilization and the protection against cellular stress. Dysregulation of TP53 can lead to metabolic reprogramming, which is often observed in cancer and other diseases (Leu et al., 2020; Motlagh et al., 2022; Luo et al., 2023; Roche et al., 2023) and can also lead to decreased mitochondrial activity, further compromising energy production and forcing cells to increasingly rely on glycolysis, which is inefficient (Figure 2) (Motlagh et al., 2022; Luo et al., 2023; Roche et al., 2023).
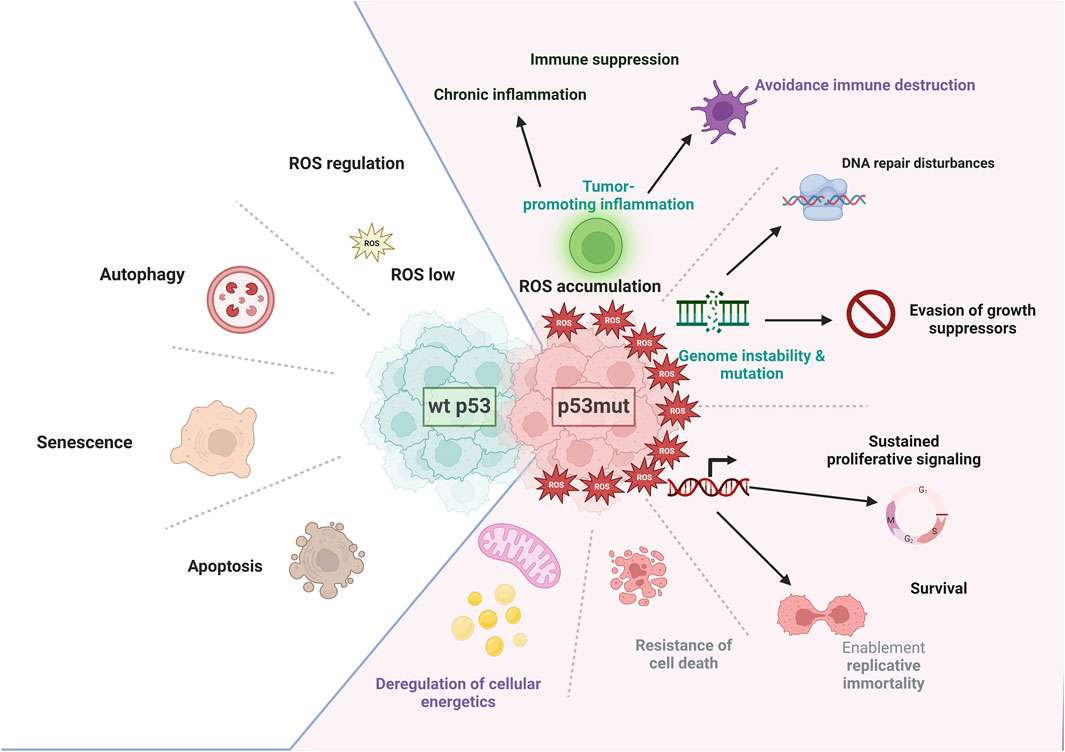
Figure 2. p53 and metabolism. A crucial protein in cellular regulation, p53 exerts control over several metabolic pathways involved in energy production and cellular homeostasis. It also plays a substantial role in regulating mitochondrial oxidative phosphorylation, glycolysis, glutaminolysis, fatty-acid oxidation, and nucleotide synthesis. To maintain mitochondrial integrity and promote oxidative phosphorylation, p53 transcriptionally induces the expression of SCO2, AIF1, and p53R2, while also physically interacting with mitochondrial DNA polymerase γ and MIAP (mitochondrial import associated protein). Additionally, p53 reduces glucose uptake by directly repressing the transcription of GLUT1 and GLUT4 and indirectly repressing the expression of GLUT3. Furthermore, p53 negatively regulates phosphoglycerate mutase, activates hexokinase and phosphofructokinase one at the protein level, and transcriptionally induces TP53-induced glycolysis and apoptosis regulator (TIGAR) and Parkin, which inhibit glycolysis. Parkin, in turn, positively regulates pyruvate dehydrogenase and the expression of monocarboxylate transporter 1. In contrast, p53 negatively regulates the expression of pyruvate dehydrogenase kinase isoform 2, which inhibits PDH activity. It also induces the expression of glutaminase 2 (GLS2), which catalyzes the hydrolysis of glutamine to glutamate. Glutamate is converted to α-ketoglutarate, which promotes the tricarboxylic acid (TCA) cycle and oxidative phosphorylation. p53 also interacts with glucose-6-phosphate dehydrogenase (G6PD) to negatively regulate its activity, leading to the downregulation of the pentose phosphate pathway, nucleotide synthesis, and nicotinamide adenine dinucleotide phosphate (NADPH) production. p53 induces the expression of GAMT and Lipin1, promoting fatty-acid oxidation and the production of acetyl-CoA. (Image created with BioRender.com).
TP53 mutations can lead to the downregulation of major histocompatibility complex (MHC) molecules, which present antigens to immune cells (Alos et al., 2020; Motlagh et al., 2022; Hassin and Oren, 2023). By reducing MHC expression, cancer cells can evade recognition by cytotoxic T cells, which rely on MHC-antigen complexes to identify and eliminate abnormal cells (Alos et al., 2020; Wang et al., 2024). Compared to patients with TP53-WT, patients who have AML with TP53 mutations have shown higher expression levels of IFNG, FOXP3 in blast cells of primary BM samples, immune checkpoints, markers of immune senescence, and phosphatidylinositol 3-kinase-Akt and NF-κB signaling intermediates (Vadakekolathu et al., 2020). TP53 mutations can also impair the activation of T cells, which are crucial for mounting an effective immune response against cancer cells (Desai et al., 2023; Wang et al., 2024). TP53 regulates the expression of costimulatory molecules and cytokines involved in T-cell activation (Desai et al., 2023; Wang et al., 2024). Mutations in TP53 can disrupt this regulation, leading to insufficient T-cell activation and compromised antitumor immune responses (Alos et al., 2020; Desai et al., 2023). TP53 mutations can also negatively impact the effector functions of immune cells such as cytotoxic T cells and natural killer (NK) cells (Alos et al., 2020). These mutations can result in the downregulation of cytotoxic molecules such as perforin and granzyme B, which are responsible for killing cancer cells (Desai et al., 2023). As a result, cancer cells can evade immune-mediated cell death.
Programmed cell death protein one and its ligand, programmed cell death ligand 1 (PD-L1), play a role in immune tolerance and the suppression of antitumor immune responses, and TP53 has been shown to regulate the expression of these immune checkpoint molecules (Alos et al., 2020). Dysregulation of TP53 can lead to abnormal expression of PD-L1, which can inhibit T-cell function and promote immune evasion (Alos et al., 2020). TP53 mutations induce immunosuppressive factors such as transforming growth factor beta and interleukin-10 (Daver et al., 2021). These factors can inhibit the activation and function of immune cells, creating an immunosuppressive microenvironment that favors tumor growth and immune evasion (Alos et al., 2020; Daver et al., 2021). TP53 mutations also can disrupt the normal processes of tumor immune surveillance, which is the mechanism by which the immune system detects and eliminates cancer cells (Alos et al., 2020; Daver et al., 2021).
TP53 can activate immune cells such as macrophages, dendritic cells, and NK cells (Motlagh et al., 2022). It regulates the expression of cytokines such as interferons and interleukins, chemokines, and costimulatory molecules involved in immune-cell activation, immune signaling, immune cell function, and coordination (Motlagh et al., 2022). Proper T-cell activation is crucial for an effective immune response (Hassin and Oren, 2023; Wang et al., 2024). TP53 controls the recruitment of immune cells to the site of infection or inflammation by regulating the expression of chemokines (Wang et al., 2024). This helps in mobilizing immune cells and directing them to the specific locations where they are needed (Wang et al., 2024). In addition, TP53 influences the differentiation of immune cells such as macrophages and dendritic cells, which are responsible for phagocytosis, antigen presentation, and immune regulation, and ensures an effective immune response (Singh et al., 2019; Motlagh et al., 2022; Wang et al., 2024).
Tp53 participates in tumor immune surveillance and is involved in the resolution of inflammation by promoting the clearance of inflammatory cells and the restoration of tissue homeostasis. This helps in preventing chronic inflammation, which can have detrimental effects on the immune system (Alos et al., 2020). Tumor-promoting inflammation, also known as chronic inflammation, is caused by an overproduction of proinflammatory cytokines such as tumor necrosis factor-α, interleukin-1, and interleukin-6 (Shi and Jiang, 2021; Morganti et al., 2022; Miller et al., 2023; Muto et al., 2023; Qin et al., 2023). These cytokines can promote tumor growth by stimulating cell proliferation, angiogenesis, and tissue remodeling (Shi and Jiang, 2021; Morganti et al., 2022; Muto et al., 2023; Qin et al., 2023). Immune cells, including macrophages, neutrophils, and lymphocytes, release additional proinflammatory molecules, creating a positive feedback loop that sustains inflammation and promotes tumor growth (Herbrich et al., 2021; Muto et al., 2023; Qin et al., 2023). Inflammatory cells release reactive oxygen species and reactive nitrogen species, which can cause DNA damage and genomic instability (Shi and Jiang, 2021; Muto et al., 2023; Qin et al., 2023). Inflammatory mediators can suppress the function of immune cells such as T cells and NK cells (Vadakekolathu et al., 2020; Shi and Jiang, 2021; Muto et al., 2023; Qin et al., 2023). The main events impacted by p53 mutations or p53 loss are summarized in Figure 3.
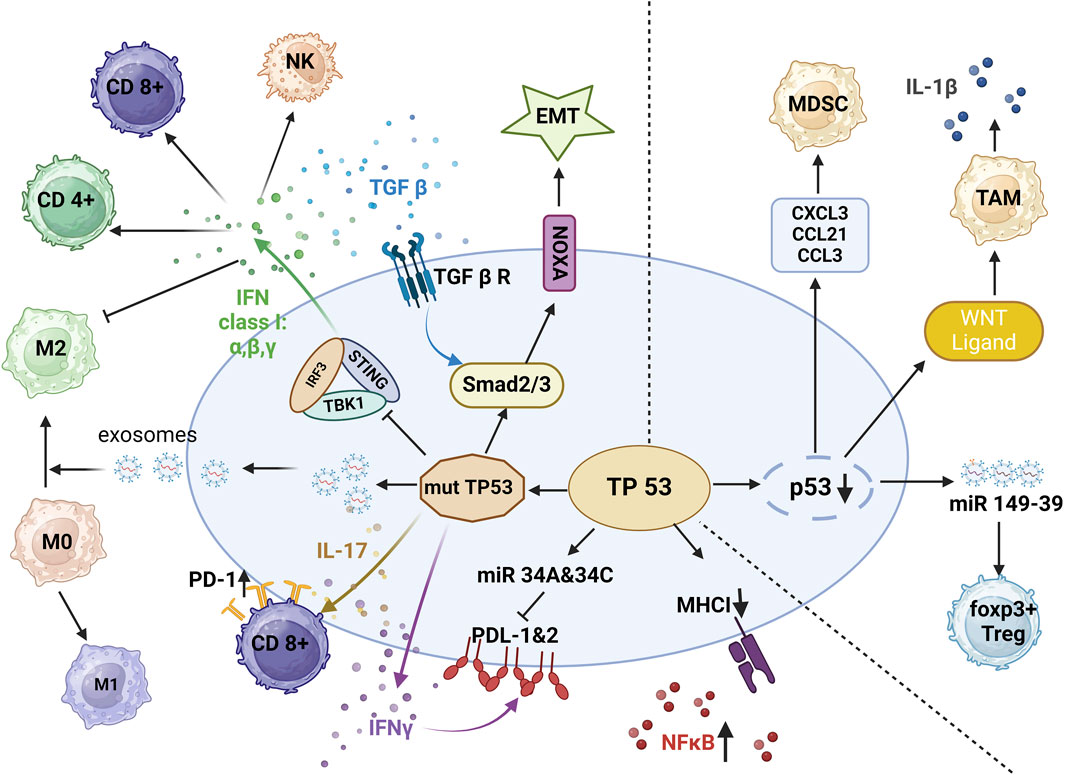
Figure 3. p53 and immune suppression. In the setting of mutant p53, decreased release of IFN-I reduces infiltration by CD4+ T cells, CD8+ T cells, and natural killer (NK) cells, and interferon gamma (IFNγ) increases the expression of programmed cell death ligand 1 (PD-L1) and programmed cell death ligand 2 (PD-L2). By activation of interleukin 17 (IL-17) signaling, mutant p53 leads to CD8+T-cell exhaustion. In concert with transforming growth factor beta (TGF-β), mutant p53 promotes epithelial-to-mesenchymal transition EMT. With the release of exosomes, mutant p53 also promotes the polarization of M1 macrophages to M2 macrophages, protecting tumor cells from phagocytosis. In the setting of p53 deletion, tumor cells release WNT ligands, chemokine ligand 1 (CXCL1), chemokine ligand 3 (CCL3) and chemokine ligand 21 (CCL21), and miR-149–39 to enhance the differentiation of tumor-associated macrophages (TAMs) and regulatory T cells (Tregs) (Image created with BioRender.com).
One of the hallmarks of cancer is uncontrolled cell proliferation; p53 controls cell division and prevents excessive proliferation (Mantovani et al., 2019). When DNA damage is detected, p53 halts the cell cycle to allow for repair, or it triggers apoptosis if the damage is too severe. Mutations in p53 render cells incapable of this checkpoint control, leading to uncontrolled proliferation (Mantovani et al., 2019; Capaci et al., 2020). In regard to the cancer hallmarks of stem-cell homeostasis and differentiation, p53 maintains a delicate balance between stem cell self-renewal and differentiation into mature cells (Shi and Jiang, 2021; Qin et al., 2023). It ensures the proper development of stem cells and prevents uncontrolled stem-cell expansion that could lead to tumors (Perez Montero et al., 2024; Scott et al., 2024).
Nutlin-3a, an MDM2 inhibitor and a selective activator of the p53 pathway, has been shown to exhibit dose-dependent antiproliferative and cytotoxic activity in OCI-AML-3 and MOLM-13 cells with wt p53 but to produce no response in HL-60 and NB4 cells expressing mutant p53 (Haaland et al., 2014; Borthakur et al., 2015; Trino et al., 2016; Fontana et al., 2021). The lack of response to Nutlin-3a indicated that the p53 pathway can be activated by Nutlin-3a only in cells with wt p53 (Kojima et al., 2005; Cheng et al., 2023; Zhou et al., 2023; Liu et al., 2024; Tomiyasu et al., 2024; Varineau and Calo, 2024).
In response to severe DNA damage or cellular stress, p53 triggers apoptosis, a process of controlled cell suicide. This eliminates potentially dangerous cells and prevents the spread of mutations (Cheng et al., 2023; Zhou et al., 2023; Liu et al., 2024; Tomiyasu et al., 2024; Varineau and Calo, 2024). Mutant p53 can malfunction in this pathway, allowing damaged cells to survive (Cheng et al., 2023; Zhou et al., 2023; Liu et al., 2024; Tomiyasu et al., 2024; Varineau and Calo, 2024). Furthermore, mutations that specifically keep the proliferation-promoting features or survival-preserving functions of wild-type p53, such as adaptation to metabolic stress, while disrupting the canonical tumor suppressive activities (such as apoptosis and senescence), can result in phenotypes that resemble those associated with gain-of-function mutations (Pfister and Prives, 2017). Controlling genome stability by p53 is another hallmark of AML and plays a central role in maintaining genomic integrity. It helps repair DNA damage through various mechanisms and activates checkpoints to halt cell division if DNA repair is incomplete (Vaddavalli and Schumacher, 2022). Dysfunction of p53 leads to an accumulation of mutations, increasing the risk of cancer (Wiederschain et al., 2005; Meyer et al., 2009), and alterations such as microsatellite instability or p53 mutations were enriched substantially in patients with therapy-related AML (Wiederschain et al., 2005; Meyer et al., 2009). Normal cells have a limited lifespan and eventually stop dividing after reaching a certain number of cell divisions (replicative senescence), but p53 can induce this senescence state in damaged or stressed cells, preventing them from becoming cancerous (Wiederschain et al., 2005; Meyer et al., 2009). Mutations in TP53 can bypass this safeguard, allowing abnormal cell proliferation and leading to uncontrolled cell growth (Gutu et al., 2023). Research suggests a potential link between p53 function and lifespan regulation, indicating that heightened p53 activity is associated with a shorter lifespan as shown in murine models. Conversely, experiments in flies suggest that reduced p53 activity appears to extend lifespan (Wiederschain et al., 2005). TP53 is well known as a “gatekeeper” of the cell cycle; it is capable of blocking cells in the G1/S phase if activated by hypoxia, heat shock, or other extrinsic or intrinsic stress signals (Olivier et al., 2010; Wang H. et al., 2023). In healthy cells, in case of DNA damage, ATM or ATR kinases activate checkpoint kinases (CHK1 and CHK2), which activate p53 and p21 and initiate G1 arrest and senescence phenotype (Wang H. et al., 2023). p53 activates cyclin-dependent kinase inhibitor 1A and p14 alternate open reading frame, which are involved in senescence and growth arrest, and targets several genes involved in the apoptosis and necrosis pathway (Barnoud et al., 2021). Mutations in TP53 have shown partial or complete alteration of the target gene set, depending on the position and the type of mutation (Olivier et al., 2010; Barnoud et al., 2021; Kennedy and Lowe, 2022).
As previously mentioned, choosing and managing therapy for patients with AML who harbor TP53 mutations or have complex karyotypes remains challenging (Abramowitz et al., 2017; Latif et al., 2021; Hassin and Oren, 2023). Moreover, the current therapeutic management of AML considers several risk factors that are included in prognostic models predicting therapy responses and outcomes, and these models classify patients with TP53 mutations as poor responders (Haase et al., 2019). Here we will discuss the outcomes of patients with TP53 mutations after treatment with selected therapy regimens (Olivier et al., 2010).
Intense chemotherapy is based on the combination of antimetabolic and antiproliferative agents like cytarabine and anthracyclines, and it is a backbone of AML therapy (DiNardo et al., 2019; Kadia et al., 2021). Intense chemotherapy based on high-dose cytarabine (AraC)/daunorubicin, known as the 7 + 3 regime, is still considered the gold standard of care (Kantarjian et al., 2021; Daver et al., 2022). Therapy modification by dose reduction of AraC or daunorubicine and the further introduction of fludarabine or cladribine has been shown to improve the response to therapy, patient outcomes, and therapy safety profile (Kadia et al., 2021). Intense chemotherapy is mainly followed by consolidation and/or maintenance therapy, and in some cases is followed by HSC transplantation (Guerra et al., 2019). A study has shown that drug-induced pancytopenia, which frequently occurs in patients treated with intense chemotherapy, became managable with protocol administration of granulocyte colony-stimulation factor (Kadia et al., 2021). Also, a new liposomal formulation of AraC/daunorubicin, known as CPX-351, has been shown to reduce the risk of cardiotoxicity in patients treated with that combination (Daver et al., 2020). However, none of these regimens specifically address the challenges of drug resistance linked to TP53 (DiNardo et al., 2019; Daver et al., 2022). Mutations in the TP53 gene render Acute Myeloid Leukemia (AML) resistant to traditional chemotherapies. Consequently, effective treatment strategies for TP53-mutated AML either bypass the need for wild-type p53 function altogether or aim to restore its normal activity (Daver et al., 2020). Most patients with TP53-mutated AML have a median overall survival of only a few months, despite receiving active anticancer treatment.
Counterintuitively, although chemotherapy and radiation (cytotoxic stress) aim to damage cancer cells, they do not directly cause TP53 mutations (Yan et al., 2020). Individuals with TP53 mutations in their blood stem cells (hematopoietic clones) face a significantly increased risk of developing Acute Myeloid Leukemia (AML). The median time to AML diagnosis after detecting a TP53 mutation is approximately 4.9 years (Desai et al., 2019; Young et al., 2019; Saygin et al., 2023). While TP53 mutations are rare in blood stem cells, these mutated cells have a survival advantage. This allows them to outcompete healthy cells under pressure from chemotherapy or stem cell transplant (Desai et al., 2019; Young et al., 2019; Saygin et al., 2023).
In the last decade, many hypomethylating agents (HMAs) have been developed for patients who cannot undergo intense chemotherapy, especially those over the age of 60 with high-risk features (Haase et al., 2019; Daver et al., 2023a; Wang H. et al., 2023). The most relevant approved drugs belonging to this category are azacytidine (AZA) and decitabine (DEC), which have gained considerable interest in the last decade due to the possibility of combining them with other types of therapies. (Daver et al., 2020; George et al., 2021; Kennedy and Lowe, 2022; Daver et al., 2023a; Wang H. et al., 2023). For instance, a 10-day decitabine regimen in patients with AML led to an excellent 100% ORR in patients with mutated TP53 disease compared to 41% in those with wt TP53 (p < 0.001), however was not sufficient for mutational clearance.
Allogenic stem cell transplantation (allo-SCT) is used in secondary AML following other therapies such as intense chemotherapy and HMA treatments. Allo-SCT can have a curative effect and can lower the probability of disease relapse in patients with a poor prognosis, but it requires early minimal residual disease (MRD) monitoring, and the incidence of complications is still high (Loke et al., 2022; Dekker et al., 2023; Gutman et al., 2023; Tettero et al., 2023). Genetic aberrations involving TP53, FLT3, NPM1, RUNX1, and ASXL1, together with factors such as age, sex, and cytogenetic characteristics, are the main risk factors affecting the outcome of patients undergoing allo-SCT (Yan et al., 2020; Loke et al., 2022; Malagola et al., 2023; Chattopadhyay et al., 2024; Park et al., 2024). TP53 positive MRD status in patients with AML, for example, has been associated with a significantly worse survival (median overall survival, 6.4 months vs 21.7 months, p = 0.042) both in patients with TP53-mutated AML and myelodysplastic syndrome receiving HMA as frontline therapy (n = 24) prior allo-SCT (Olivier et al., 2010; Malagola et al., 2023; Song et al., 2023; Chattopadhyay et al., 2024; Park et al., 2024; Sahasrabudhe and Mims, 2024). Thus, the early detection of these mutations using complex MRD monitoring and testing for the loss of chimerism (LoC after allo-SCT is a serious concern as it can be an early indicator of relapse) after stem cell transplant are the key strategies for addressing the otherwise-poor survival of patients relapsing after allo-SCT (Sahasrabudhe and Mims, 2024).
Bcl-2 family genes are composed of antiapoptotic and proapoptotic genes such as Bcl-2 and BAX/BAK, respectively (DiNardo et al., 2019; Guerra et al., 2019). Bcl-2 inhibitor–based therapy relies on altering the equilibrium of Bcl-2 components by depleting the antiapoptotic members and allowing the p53-mediated activation of BAX and BAK following the permeabilization of the outer mitochondrial membrane and caspase cascade (DiNardo et al., 2019; Guerra et al., 2019; Carter et al., 2023a). Venetoclax (VEN) has proven to be as effective as monotherapy, especially in AML harboring IDH1/2 and SRSF2/ZRSF2 mutations (Daver et al., 2020; George et al., 2021; Thijssen et al., 2021). Further synergistic activity was observed with AZA in the VIALE-A trial and with low-dose cytarabine in the VIALE-C trial (Thijssen et al., 2021; Carter et al., 2023a; Malagola et al., 2023; Song et al., 2023; Pratz et al., 2024; Sahasrabudhe and Mims, 2024). However, the best outcome was obtained when VEN was combined with MCL-1 inhibitor in therapies (Thijssen et al., 2021; Casado and Cutillas, 2023). A cohort analysis of patients harboring TP53 mutations demonstrated a lack of improvement among patients on the VEN + AZA regimen compared with historical controls (Hong et al., 2016; DiNardo et al., 2019). However, given the limited number of patients, further studies in this specific patient cohort are warranted. Although therapy with VEN + AZA has a good safety profile in elderly patients, the short response duration in patients with TP53 mutation and rapidly acquired resistance to VEN warrants further investigations on the mechanisms of resistance in these patients as well as further work on novel combinatorial strategies (DiNardo et al., 2019; Guerra et al., 2019; Kadia et al., 2021; Zhang et al., 2022; Gutman et al., 2023). However, due to the rewiring of other BH3-mimetic family members upon Bcl-2 pharmacological inhibition, other therapeutic strategies with molecules targeting Bcl-2, Bcl-XL, and Bcl-W were evaluated. Navitoclax (ABT-263) is an antagonist of the antiapoptotic members of the Bcl-2 family—Bcl-2, Bcl-XL, and Bcl-W—and prevents their binding to the apoptotic effectors proteins Bax and Bak, thereby triggering apoptotic processes in cells overexpressing these proteins (Gress et al., 2024). However, the application of navitoclax in patients is challenging because its induction of severe thrombocytopenia limits its utility. Besides navitoclax, several other molecules have been tested in phase I and Ib clinical trials, including the Mcl-1 inhibitors AZD5991, MIK665, AMG176, and AMG397, alone or in combination with VEN (Luedtke et al., 2017; Tron et al., 2018; Carter et al., 2022; Fang et al., 2022; Liu et al., 2022; Carter et al., 2023a; Wang Y. et al., 2023; Torka et al., 2023). Furthermore, several dual inhibitors targeting Bcl-2/-XL have been developed, including LP-118, AZD0466, and dual Bcl-2/-XL PROTAC degraders (Khan et al., 2022; Gress et al., 2024). Finally, VEN has been tested with several FLT3 inhibitors such as quizartinib or gliteritinib in patients with FLT3-mutated AML as well as in those with wt FLT. VEN improved survival in a cohort of patients with mutated FLT3, successfully impairing leukemia progression (Yilmaz et al., 2022). While the list of potential combinations of BCL2 and FLT3 co-inhibition in acute myeloid leukemia tested preclinically expands rapidly, none of these so far has shown improved efficacy in patients with TP53 mutations (Perl, 2019; Brinton et al., 2020; Carter et al., 2021; Zhu et al., 2021; Carter et al., 2023b; Popescu et al., 2023).
In recent years, increased knowledge regarding the features about AML—obtained through genome-wide association studies, single-cell RNA sequencing, and proteomic approaches—has enabled the consideration of several novel therapeutic approaches for the treatment of patients with AML. These approaches could pave the way for new generations of HMA and BCL-2 inhibitors; such inhibitors are currently being evaluated as monotherapies or in combination with approved therapies (Daver et al., 2020).
The main inhibitors of p53, MDM2 and MDM4, are often upregulated in many malignancies harboring wt p53 (Wang H. et al., 2023). While several molecules such as HDM201, MK-8242, BI-907828, RG7388, and RG7112 have been developed to interfere with p53 activity, most remain in the preclinical testing phase (Wang H. et al., 2023). Idasanutlin (RG7388), an MDM2 antagonist with a pyrrolidine structure, has demonstrated better efficacy, selectivity, and availability for the treatment of AML compared with the other drugs of the nutlin family (Trino et al., 2016; Fontana et al., 2021), and in a phase III trial with cytarabine, it improved survival and recovery in patients with R/R AML (Italiano et al., 2021; Konopleva et al., 2022; Daver et al., 2023b; Smalley et al., 2024). Mutant p53 tends to aggregate with p73, p63, or other p53 molecules, inactivating or reducing their activity (Kennedy and Lowe, 2022). Interference with these interactions would restore the activity of p73 and residual wtp53 (Ramos et al., 2020; Klein et al., 2021; Cai et al., 2022). ReACp53, for example, is a small peptide that interferes with the aggregation of mutant p53 with p73 and p63, and it has been shown to restore the wt conformation and nuclear localization of p53, promoting apoptosis in vitro and tumor suppression in vivo (Spaety et al., 2019; Zhang et al., 2019; Ramos et al., 2020; Klein et al., 2021; Rozenberg et al., 2021; Cai et al., 2022; Wang H. et al., 2023; Hassin and Oren, 2023).
Another strategy to address the severe outcomes of mutant TP53 and the consequent LOF is to reverse the altered mutant conformation to one resembling that of wt TP53 (Prokocimer et al., 2017; Hassin and Oren, 2023). Eprenetapopt (APR-246), also known as PRIMA1-MET, is a small molecule that causes selective apoptosis in cancer cells with TP53 mutations (Hassin and Oren, 2023). Eprenetapopt binds covalently to cysteine residues in the DBD and forces conformational changes in the wt conformation, leading to the depletion of antioxidants and d-nucleotides and the induction of ROS(Reactive Oxygen Species)-linked cell death through ferroptosis (Sallman, 2020; Birsen et al., 2022; Daver et al., 2022). Eprenetapopt was evaluated with AZA in phase III clinical trial, but it showed no significant benefits in patients with TP53 mutations (Daver et al., 2022; Hassin and Oren, 2023). In contrast, in combination with AZA and VEN, this combination therapy demonstrated an acceptable safety profile and promising signs of effectiveness. These findings support further investigation of this approach as a first-line treatment for TP53-mutated AML (Garcia-Manero et al., 2023; Wang H. et al., 2023). Other trial have evaluated APR-246 as post-transplant maintenance therapy and focused on patients with TP53-mutated acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS) who had undergone allogeneic hematopoietic stem cell transplantation (allo-SCT) leading to encouraging RFS and OS outcomes in this high-risk population (Spaety et al., 2019). APR-246 was also evaluated with DEC, VEN, and low-dose cytarabine in patients with AML who were over 60 years old and ineligible for intense chemotherapy. The drug combination produced encouraging results and had an acceptable safety profile (Mishra et al., 2022).
In addition, APR-548, an orally available TP53 reactivator, has undergone clinical evaluation in patients with solid tumors and hematological malignancies, indicating that p53 mutants differ in functionality and form from typical AML cases and subsequently display inconsistent responses to therapy with APR-548 at the cellular level (George et al., 2021).
Finally, ZMC1, ZMC2, and ZMC3, which belong to a new class of zinc metallochaperones, sequester zinc ions crucial for DNA recognition from mutated p53, promoting wt-like behavior, p53-dependent apoptosis in vitro, and tumor regression in vivo (Wang H. et al., 2023; Hassin and Oren, 2023). These zinc metallochaperones have been shown to reactivate mutant p53 using an on/off switch, and they have shown specificity for mutant p53 (Hassin and Oren, 2023).
Trisenox (ATO) (As2O3) is a small molecule that binds to allosteric sites on a wide subset of p53 mutants and induces p53 proteasome–mediated degradation via structural stabilization (Gummlich, 2021; Song et al., 2023). It has been observed that a few p53 mutants treated with ATO demonstrate restored wt p53 activity (Daver et al., 2022; Hassin and Oren, 2023). Trisenox is mainly used in certain subtypes of AML, including acute promyeloid leukemia, in which it has shown dose-dependent dual effects, including differentiation at low concentrations and apoptosis at high concentrations, on Leukemia Stem Cells (LSC) (Yilmaz et al., 2021). Clinical trials involving treatments with trisenox + HMA + all-trans retinoic acid are ongoing (Yilmaz et al., 2021).
Development of adoptive T cell therapy for relapsed/refractory acute myeloid leukemia (R/R AML) has shown limited progress to date. Chimeric antigen receptors (CARs) implemented in vitro in T cells have proven to be effective for R/R B-cell lymphoid malignancies (CD19+) and multiple myeloma (Caruso et al., 2022; Gurney and O’Dwyer, 2021; Mueller et al., 2024). Several proteins commonly overexpressed in AML, such as CD38, CD123, TIM3, CD7, CD19, and NKG2D, have been considered as targets for evaluation in AML treatments, with the aim of treatment being to eradicate the LSC-like population (Gurney and O’Dwyer, 2021; Valeri et al., 2022; Cao et al., 2022; Cui et al., 2021; El et al., 2021; Jetani et al., 2021; Mai et al., 2023). However, it has been difficult to find suitable tumor-associated antigens for CAR T-cell administration in patients with AML (Cui et al., 2021; El et al., 2021; Yilmaz et al., 2021; Sanford et al., 2023); thus, clinical responses to CAR T-cell therapy are seen in only one-fourth of treated subjects (Atilla and Benabdellah, 2023). Recent studies have tried to overcome TP53 deficiency–linked resistance to CAR T cells by targeting the lipids metabolism aiming at blocking cholesterol metabolism or activity of carnitine o-octanoyltransferase and at improving CAR T-cell anti-leukemic properties. While the specific results of targeting these pathways are not provided here, further investigation might lead to more effective and personalized treatment options in the future. (Mueller et al., 2023; Roche et al., 2023; Sanford et al., 2023; Albinger et al., 2024). Although CAR T-cell therapy alone might not be sufficient to achieve a complete eradication of residual disease, some studies suggest that combined CAR T-cell therapy and pharmacological blockade with demethylating agents or venetoclax (VEN) might be a promising strategy. This approach could lead to more effective and better-tolerated cellular therapies for patients with TP53-mutated myeloid neoplasms (Leick et al., 2022; Mandeville et al., 2023; Mueller et al., 2023; Sanford et al., 2023).
NK cells are the first line of defense against tumor cells (Mandeville et al., 2023; Albinger et al., 2024). They induce cell death in two ways: by releasing tumor necrosis factor-α and IFN-γ, which activate the extrinsic apoptosis pathway, and by triggering cell death using the tumor necrosis factor–related apoptosis-inducing ligand or Fas ligand. Compared to the use of CAR T cells, the use of CAR NK cells has significantly fewer side effects (Gurney and O’Dwyer, 2021; Albinger et al., 2024; Albinger et al., 2022). Until now, CAR NK therapy has shown good efficacy in clinical trials against circulating AML cells, but it displays a low penetrative ability in bone marrow niches; thus, it is considered as a maintenance or consolidation therapy before or following an allo-SCT rather than as induction therapy (Gurney and O’Dwyer, 2021). After intensive therapy and allogenic hematopoietic cell transplantation, the outcomes of CAR NK cell therapy in AML patients with TP53 mutations remains poor, those patients with lower TP35 VAFs at diagnosis might still benefit from transplantation. combined with CAR NK therapy (Zhao et al., 2023). One possible mechanism of overcoming resistance to CAR NK therapy would be to select AML clones that resist or even suppress NK cell activity and mobilize them from Bone marrow, making them more susceptible to therapy (Gurney and O’Dwyer, 2021; Valeri et al., 2022; Albinger et al., 2024; Li et al., 2023). Persistent hypoxia and bone marrow remodeling with poor vascularization might also be obstacles to the efficacy of CAR NK therapy (Caruso et al., 2022). These and other factors contributing to therapy resistance need further investigation to improve the chances of disease eradication.
Since the approval of gemtuzumab ozogamicin (GO, a monoclonal antibody targeting CD33, conjugated with calicheamicin) (Laszlo et al., 2019; Bouvier et al., 2021; Casado and Cutillas, 2023), immunotherapy for AML has advanced substantially. New monoclonal antibodies, used along with other types of therapies in the induction phase or as a part of consolidation therapy after intense chemotherapy, have been added to the available treatment options (Hassin and Oren, 2023). For example, sabatolimab (MBC453) is a humanized, high-affinity, IgG4, anti-TIM3 antibody that uses an autocrine signaling loop via galactin-9 and promotes LSC renewal; it is currently being combined with HMA in an ongoing clinical trial (Brunner et al., 2024; Zeidan et al., 2024). Its observed side effects have been minimal, and the parameters of recovery and patients’ OS have been encouraging, especially in patients with AML harboring mutations such as RUNX1 and ASXL1 (Kantarjian et al., 2021; Daver et al., 2022). Another antibody, magrolimab, is a humanized antibody against CD47, a surface receptor expressed by myeloid malignancies that helps tumor cells evade phagocytosis (Bouvier et al., 2021; Zhao et al., 2023). Magrolimab has been evaluated in AML patients not eligible for intense chemotherapy who were in the early stages of treatments with AZA + VEN, and it demonstrated good tolerability, but it also caused frequent side effects such as anemia and fatigue (George et al., 2021; Zeidan et al., 2022; Daver et al., 2023c).
Bispecific and trispecific antibodies are a promising new area of immunotherapy for AML (Arvindam et al., 2021; Bouvier et al., 2021; Boyiadzis et al., 2023). These engineered molecules, which offer a targeted approach of attacking AML cells by harnessing the immune system, include flotetuzumab (targets CD33 on AML cells and CD16 on NK cells), AMG 330 (targets CD33 on AML cells and CD3 on T cells), and JNJ-63709178 (targets CD33; bispecific antibody) (Krupka et al., 2016; Jitschin et al., 2018; Laszlo et al., 2019; Vadakekolathu et al., 2020; Uy et al., 2021; Barwe et al., 2022; Boyiadzis et al., 2023; Marcinek et al., 2023; Rimando et al., 2023). Study investigating the CD123 × CD3 bispecific, dual-affinity, retargeting antibody flotetuzumab (CP-MGD006-01; NCT02152956) demonstrated complete remission in almost 50% of patients with TP53-mutated, R/R AML, and these patients had significantly higher tumor inflammation signature, FOXP3, CD8, inflammatory chemokine, and PD1 gene expression scores at baseline compared with nonresponders (Vadakekolathu et al., 2020). Examples of relevant clinical trials are given in Table 2.
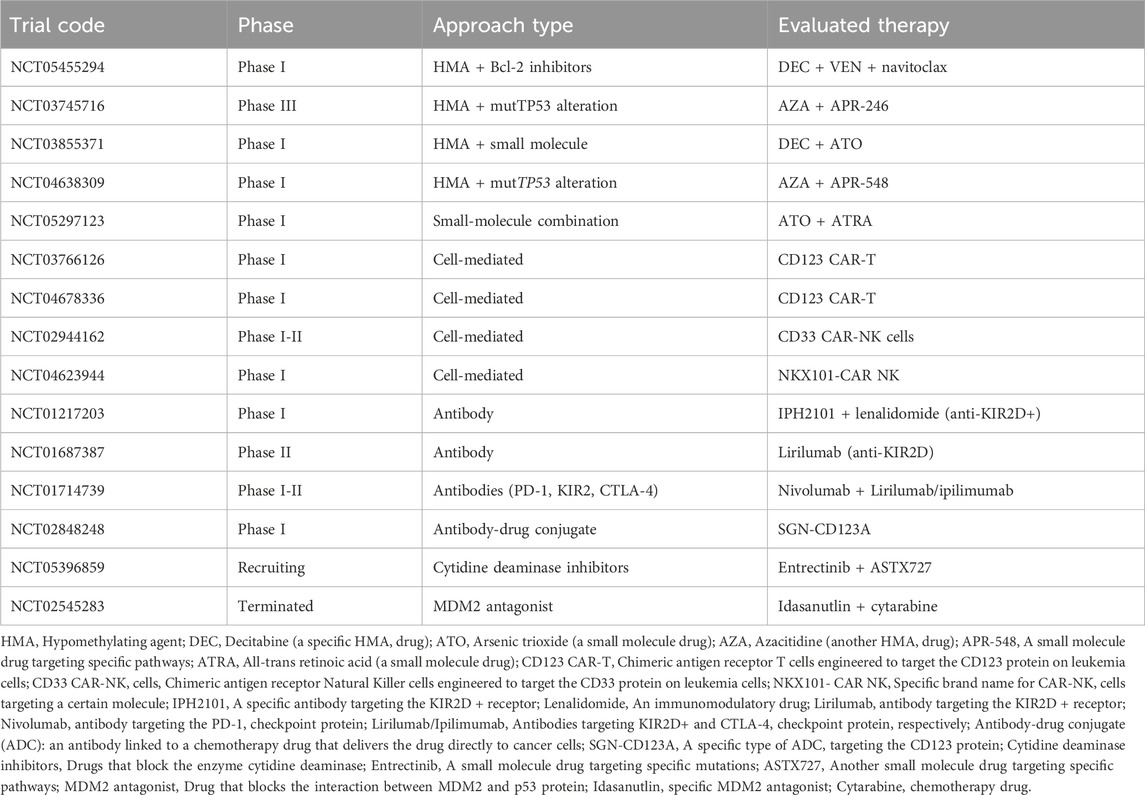
Table 2. Examples of clinical trials in acute myeloid leukemia with the potential to target TP53.
For over four decades, high-dose cytotoxic chemotherapy remained the mainstay treatment for AML. However, recent scientific breakthroughs have revolutionized our understanding of this leukemia’s molecular basis. This newfound knowledge has not only shed light on the underlying causes of AML but also led to the development of several targeted therapies. These novel agents offer greater efficacy and reduced toxicity compared to conventional chemotherapy. With novel compounds selectively targeting TP53, therapeutic approaches targeting the TP53 pathway are progressing in early clinical testing and could soon be considered the standard of care for individuals with AML who are 60 and older and ineligible for intense therapy or who have been diagnosed with advanced disease. Moreover, many targeted approaches and combinations are currently being tested in clinical trials with the aims of reducing the rate of disease recurrence and minimizing drug toxicities, making the prospects of new AML therapies promising. However, further investigation of the factors contributing to therapy resistance is warranted, and efforts to understand the metabolic and immune mechanisms contributing to therapy failure with the help of single-cell, high-throughput technology and spatial analysis are ongoing.
Acute myeloid leukemia (AML), an aggressive malignancy of hematopoietic stem cells, is associated with poor outcomes, especially in elderly patients, due to several genetic and chromosomal aberrations. Tumor protein p53 (TP53) is a key tumor-suppressor gene involved in a variety of cellular processes, including the regulation of apoptosis, metabolism, and the rewiring of the immune environment. Although TP53 mutations are relatively rare in patients with de novo AML, these mutations has been identified as an important molecular subgroup, and patients with these mutations have the worst prognosis and shortest overall survival among patients with AML, even when treated with aggressive chemotherapy and allogeneic stem cell transplant for relapsed or therapy-related AML. Progress in AML genetics and biology has brought the novel therapies, however, the clinical benefit of these agents for patients whose disease is driven by TP53 mutations remains largely unexplored. This review focuses on examining the role of TP53 mutations on such hallmarks of leukemia like metabolic rewiring and immune evasion, the clinical significance of these changes, and the current progress in the therapeutic targeting of mutated p53 and its downstream effects.
MC: Writing–review and editing, Writing–original draft, Visualization, Methodology, Conceptualization. LG: Writing–original draft. SD: Writing–review and editing, Visualization, Methodology, Conceptualization. PF: Writing–review and editing, Visualization, Methodology, Conceptualization. BG: Writing–review and editing. VM: Writing–review and editing, Visualization, Methodology, Conceptualization. HA: Writing–review and editing. NB: Writing–review and editing, Writing–original draft, Visualization, Validation, Supervision, Project administration, Methodology, Funding acquisition, Conceptualization.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Science Centre, Poland (2021/43/B/NZ5/03345) (NB), and in part by the NIH/NCI Cancer Center Support Grant P30CA016672, as well as by NIH/NCI R01CA231364 (NB); HA was supported by the Physician Scientist Program and Cancer Prevention and Research Institute of Texas Funding.
All figures were created using BioRender software. We thank Laura L. Russell, scientific editor, Research Medical Library, The University of Texas MD Anderson Cancer Center for editing this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abramowitz, J., Neuman, T., Perlman, R., and Ben-Yehuda, D. (2017). Gene and protein analysis reveals that p53 pathway is functionally inactivated in cytogenetically normal Acute Myeloid Leukemia and Acute Promyelocytic Leukemia. BMC Med. Genomics 10 (1), 18. doi:10.1186/s12920-017-0249-2
Albinger, N., Muller, S., Kostyra, J., Kuska, J., Mertlitz, S., Penack, O., et al. (2024). Manufacturing of primary CAR-NK cells in an automated system for the treatment of acute myeloid leukemia. Bone Marrow Transpl. 59 (4), 489–495. doi:10.1038/s41409-023-02180-4
Albinger, N., Pfeifer, R., Nitsche, M., Mertlitz, S., Campe, J., Stein, K., et al. (2022). Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 12 (4), 61. doi:10.1038/s41408-022-00660-2
Alos, L., Fuster, C., Castillo, P., Jares, P., Garcia-Herrera, A., Marginet, M., et al. (2020). TP53 mutation and tumoral PD-L1 expression are associated with depth of invasion in desmoplastic melanomas. Ann. Transl. Med. 8 (19), 1218. doi:10.21037/atm-20-1846
Appelbaum, J. S., Wei, A. H., Mandrekar, S. J., Tiong, I. S., Chua, C. C., Teh, T. C., et al. (2024). Clinical evaluation of complete remission (CR) with partial hematologic recovery (CRh) in acute myeloid leukemia: a report of 7235 patients from seven cohorts. Leukemia 38 (2), 389–392. doi:10.1038/s41375-024-02143-8
Arvindam, U. S., van Hauten, P. M. M., Schirm, D., Schaap, N., Hobo, W., Blazar, B. R., et al. (2021). A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia 35 (6), 1586–1596. doi:10.1038/s41375-020-01065-5
Ashcroft, M., Kubbutat, M. H., and Vousden, K. H. (1999). Regulation of p53 function and stability by phosphorylation. Mol. Cell Biol. 19 (3), 1751–1758. PMID: 10022862; PMCID: PMC83968. doi:10.1128/MCB.19.3.1751
Atilla, E., and Benabdellah, K. (2023). The black hole: CAR T cell therapy in AML. Cancers (Basel) 15 (10), 2713. doi:10.3390/cancers15102713
Barnoud, T., Indeglia, A., and Murphy, M. E. (2021). Shifting the paradigms for tumor suppression: lessons from the p53 field. Oncogene 40 (25), 4281–4290. doi:10.1038/s41388-021-01852-z
Barwe, S. P., Kisielewski, A., Bonvini, E., Muth, J., Davidson-Moncada, J., Kolb, E. A., et al. (2022). Efficacy of flotetuzumab in combination with cytarabine in patient-derived xenograft models of pediatric acute myeloid leukemia. J. Clin. Med. 11 (5), 1333. doi:10.3390/jcm11051333
Birsen, R., Larrue, C., Decroocq, J., Johnson, N., Guiraud, N., Gotanegre, M., et al. (2022). APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 107 (2), 403–416. doi:10.3324/haematol.2020.259531
Boettcher, S., Miller, P. G., Sharma, R., McConkey, M., Leventhal, M., Krivtsov, A. V., et al. (2019). A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365 (6453), 599–604. doi:10.1126/science.aax3649
Borthakur, G., Duvvuri, S., Ruvolo, V., Tripathi, D. N., Piya, S., Burks, J., et al. (2015). MDM2 inhibitor, nutlin 3a, induces p53 dependent autophagy in acute leukemia by AMP kinase activation. PLoS One 10 (10), e0139254. doi:10.1371/journal.pone.0139254
Bouvier, A., Hamel, J. F., Delaunay, J., Delabesse, E., Dumas, P. Y., Ledoux, M. P., et al. (2021). Molecular classification and prognosis in younger adults with acute myeloid leukemia and intermediate-risk cytogenetics treated or not by gemtuzumab ozogamycin: final results of the GOELAMS/FILO acute myeloid leukemia 2006-intermediate-risk trial. Eur. J. Haematol. 107 (1), 111–121. doi:10.1111/ejh.13626
Boyiadzis, M., Desai, P., Daskalakis, N., Donnellan, W., Ferrante, L., Goldberg, J. D., et al. (2023). First-in-human study of JNJ-63709178, a CD123/CD3 targeting antibody, in relapsed/refractory acute myeloid leukemia. Clin. Transl. Sci. 16 (3), 429–435. doi:10.1111/cts.13467
Brinton, L. T., Zhang, P., Williams, K., Canfield, D., Orwick, S., Sher, S., et al. (2020). Synergistic effect of BCL2 and FLT3 co-inhibition in acute myeloid leukemia. J. Hematol. Oncol. 13 (1), 139. doi:10.1186/s13045-020-00973-4
Brunner, A. M., Esteve, J., Porkka, K., Knapper, S., Traer, E., Scholl, S., et al. (2024). Phase Ib study of sabatolimab (MBG453), a novel immunotherapy targeting TIM-3 antibody, in combination with decitabine or azacitidine in high- or very high-risk myelodysplastic syndromes. Am. J. Hematol. 99 (2), E32–E36. doi:10.1002/ajh.27161
Cai, B. H., Hsu, Y. C., Yeh, F. Y., Lin, Y. R., Lu, R. Y., Yu, S. J., et al. (2022). P63 and P73 activation in cancers with p53 mutation. Biomedicines 10 (7), 1490. doi:10.3390/biomedicines10071490
Cao, X., Dai, H., Cui, Q., Li, Z., Shen, W., Pan, J., et al. (2022). CD7-directed CAR T-cell therapy: a potential immunotherapy strategy for relapsed/refractory acute myeloid leukemia. Exp. Hematol. Oncol. 11 (1), 67. doi:10.1186/s40164-022-00318-6
Capaci, V., Mantovani, F., and Sal, G. D. (2020). A mutant p53/Hif1α/miR-30d axis reprograms the secretory pathway promoting the release of a prometastatic secretome. Cell Stress 4 (11), 261–264. doi:10.15698/cst2020.11.235
Carter, B. Z., Mak, P. Y., Tao, W., Ayoub, E., Ostermann, L. B., Huang, X., et al. (2023a). Combined inhibition of BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance of TP53-mutant acute myeloid leukemia to individual BH3 mimetics. Blood Cancer J. 13 (1), 57. doi:10.1038/s41408-023-00830-w
Carter, B. Z., Mak, P. Y., Tao, W., Ostermann, L. B., Mak, D. H., Ke, B., et al. (2023b). Inhibition of menin, BCL-2, and FLT3 combined with a hypomethylating agent cures NPM1/FLT3-ITD/-TKD mutant acute myeloid leukemia in a patient-derived xenograft model. Haematologica 108 (9), 2513–2519. doi:10.3324/haematol.2022.281927
Carter, B. Z., Mak, P. Y., Tao, W., Warmoes, M., Lorenzi, P. L., Mak, D., et al. (2022). Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and resensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica 107 (1), 58–76. doi:10.3324/haematol.2020.260331
Carter, B. Z., Tao, W., Mak, P. Y., Ostermann, L. B., Mak, D., McGeehan, G., et al. (2021). Menin inhibition decreases Bcl-2 and synergizes with venetoclax in NPM1/FLT3-mutated AML. Blood 138 (17), 1637–1641. doi:10.1182/blood.2021011917
Caruso, S., De Angelis, B., Del Bufalo, F., Ciccone, R., Donsante, S., Volpe, G., et al. (2022). Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J. Hematol. Oncol. 15 (1), 163. doi:10.1186/s13045-022-01376-3
Casado, P., and Cutillas, P. R. (2023). Proteomic characterization of acute myeloid leukemia for precision medicine. Mol. Cell Proteomics 22 (4), 100517. doi:10.1016/j.mcpro.2023.100517
Chattopadhyay, S., Lionel, S., Selvarajan, S., Devasia, A. J., Korula, A., Kulkarni, U., et al. (2024). Relapse and transformation to myelodysplastic syndrome and acute myeloid leukemia following immunosuppressive therapy for aplastic anemia is more common as compared to allogeneic stem cell transplantation with a negative impact on survival. Ann. Hematol. 103 (3), 749–758. doi:10.1007/s00277-024-05621-2
Chen, X., Zhang, T., Su, W., Dou, Z., Zhao, D., Jin, X., et al. (2022). Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death Dis. 13 (11), 974. doi:10.1038/s41419-022-05408-1
Cheng, J., Yan, Z., Jiang, K., Liu, C., Xu, D., Lyu, X., et al. (2023). Discovery of JN122, a spiroindoline-containing molecule that inhibits MDM2/p53 protein-protein interaction and exerts robust in vivo antitumor efficacy. J. Med. Chem. 66 (24), 16991–17025. doi:10.1021/acs.jmedchem.3c01815
Cui, Q., Qian, C., Xu, N., Kang, L., Dai, H., Cui, W., et al. (2021). CD38-directed CAR-T cell therapy: a novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J. Hematol. Oncol. 14 (1), 82. doi:10.1186/s13045-021-01092-4
Daver, N., Alotaibi, A. S., Bucklein, V., and Subklewe, M. (2021). T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia 35 (7), 1843–1863. doi:10.1038/s41375-021-01253-x
Daver, N., Wei, A. H., Pollyea, D. A., Fathi, A. T., Vyas, P., and DiNardo, C. D. (2020). New directions for emerging therapies in acute myeloid leukemia: the next chapter. Blood Cancer J. 10 (10), 107. doi:10.1038/s41408-020-00376-1
Daver, N. G., Dail, M., Garcia, J. S., Jonas, B. A., Yee, K. W. L., Kelly, K. R., et al. (2023b). Venetoclax and idasanutlin in relapsed/refractory AML: a nonrandomized, open-label phase 1b trial. Blood 141 (11), 1265–1276. doi:10.1182/blood.2022016362
Daver, N. G., Iqbal, S., Renard, C., Chan, R. J., Hasegawa, K., Hu, H., et al. (2023a). Treatment outcomes for newly diagnosed, treatment-naive TP53-mutated acute myeloid leukemia: a systematic review and meta-analysis. J. Hematol. Oncol. 16 (1), 19. doi:10.1186/s13045-023-01417-5
Daver, N. G., Maiti, A., Kadia, T. M., Vyas, P., Majeti, R., Wei, A. H., et al. (2022). TP53-Mutated myelodysplastic syndrome and acute myeloid leukemia: biology, current therapy, and future directions. Cancer Discov. 12 (11), 2516–2529. doi:10.1158/2159-8290.CD-22-0332
Daver, N. G., Vyas, P., Kambhampati, S., Al Malki, M. M., Larson, R. A., Asch, A. S., et al. (2023c). Tolerability and efficacy of the anticluster of differentiation 47 antibody magrolimab combined with azacitidine in patients with previously untreated AML: phase Ib results. J. Clin. Oncol. 41 (31), 4893–4904. doi:10.1200/JCO.22.02604
Dekker, S. E., Rea, D., Cayuela, J. M., Arnhardt, I., Leonard, J., and Heuser, M. (2023). Using measurable residual disease to optimize management of AML, ALL, and chronic myeloid leukemia. Am. Soc. Clin. Oncol. Educ. Book 43, e390010. doi:10.1200/EDBK_390010
Deng, J., Wu, X., Ling, Y., Liu, X., Zheng, X., Ye, W., et al. (2020). The prognostic impact of variant allele frequency (VAF) in TP53 mutant patients with MDS: a systematic review and meta-analysis. Eur. J. Haematol. 105 (5), 524–539. doi:10.1111/ejh.13483
Desai, P., Hassane, D., and Roboz, G. J. (2019). Clonal hematopoiesis and risk of acute myeloid leukemia. Best. Pract. Res. Clin. Haematol. 32 (2), 177–185. doi:10.1016/j.beha.2019.05.007
Desai, P. N., Wang, B., Fonseca, A., Borges, P., Jelloul, F. Z., Reville, P. K., et al. (2023). Single-cell profiling of CD8+ T cells in acute myeloid leukemia reveals a continuous spectrum of differentiation and clonal hyperexpansion. Cancer Immunol. Res. 11, 1011–1028. doi:10.1158/2326-6066.CIR-22-0961
DiNardo, C. D., Pratz, K., Pullarkat, V., Jonas, B. A., Arellano, M., Becker, P. S., et al. (2019). Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133 (1), 7–17. doi:10.1182/blood-2018-08-868752
Donehower, L. A., Soussi, T., Korkut, A., Liu, Y., Schultz, A., Cardenas, M., et al. (2019). Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 28 (11), 1370–1384. doi:10.1016/j.celrep.2019.07.001
El, K. N., Hughes, A., Yu, W., Myburgh, R., Matschulla, T., Taromi, S., et al. (2021). Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat. Commun. 12 (1), 6436. doi:10.1038/s41467-021-26683-0
Fang, D. D., Zhu, H., Tang, Q., Wang, G., Min, P., Wang, Q., et al. (2022). FLT3 inhibition by olverembatinib (HQP1351) downregulates MCL-1 and synergizes with BCL-2 inhibitor lisaftoclax (APG-2575) in preclinical models of FLT3-ITD mutant acute myeloid leukemia. Transl. Oncol. 15 (1), 101244. doi:10.1016/j.tranon.2021.101244
Fleming, S., Tsai, X. C., Morris, R., Hou, H. A., and Wei, A. H. (2023). TP53 status and impact on AML prognosis within the ELN 2022 risk classification. Blood 142 (23), 2029–2033. doi:10.1182/blood.2023020855
Fontana, M. C., Nanni, J., Ghelli Luserna di Rora, A., Petracci, E., Padella, A., Ghetti, M., et al. (2021). Pharmacological inhibition of WIP1 sensitizes acute myeloid leukemia cells to the MDM2 inhibitor nutlin-3a. Biomedicines 9 (4), 388. doi:10.3390/biomedicines9040388
Garcia-Manero, G., Goldberg, A. D., Winer, E. S., Altman, J. K., Fathi, A. T., Odenike, O., et al. (2023). Eprenetapopt combined with venetoclax and azacitidine in TP53-mutated acute myeloid leukaemia: a phase 1, dose-finding and expansion study. Lancet Haematol. 10 (4), e272–e283. doi:10.1016/S2352-3026(22)00403-3
George, B., Kantarjian, H., Baran, N., Krocker, J. D., and Rios, A. (2021). TP53 in acute myeloid leukemia: molecular aspects and patterns of mutation. Int. J. Mol. Sci. 22 (19), 10782. doi:10.3390/ijms221910782
Gress, V., Roussy, M., Boulianne, L., Bilodeau, M., Cardin, S., El-Hachem, N., et al. (2024). CBFA2T3::GLIS2 pediatric acute megakaryoblastic leukemia is sensitive to BCL-XL inhibition by navitoclax and DT2216. Blood Adv. 8 (1), 112–129. doi:10.1182/bloodadvances.2022008899
Guerra, V. A., DiNardo, C., and Konopleva, M. (2019). Venetoclax-based therapies for acute myeloid leukemia. Best. Pract. Res. Clin. Haematol. 32 (2), 145–153. doi:10.1016/j.beha.2019.05.008
Gummlich, L. (2021). ATO stabilizes structural p53 mutants. Nat. Rev. Cancer 21 (3), 141. doi:10.1038/s41568-021-00337-1
Gurney, M., and O’Dwyer, M. (2021). Realizing innate potential: CAR-NK cell therapies for acute myeloid leukemia. Cancers (Basel) 13 (7), 1568. doi:10.3390/cancers13071568
Gutman, J. A., Winters, A., Kent, A., Amaya, M., McMahon, C., Smith, C., et al. (2023). Higher-dose venetoclax with measurable residual disease-guided azacitidine discontinuation in newly diagnosed acute myeloid leukemia. Haematologica 108 (10), 2616–2625. doi:10.3324/haematol.2023.282681
Gutu, N., Binish, N., Keilholz, U., Herzel, H., and Granada, A. E. (2023). p53 and p21 dynamics encode single-cell DNA damage levels, fine-tuning proliferation and shaping population heterogeneity. Commun. Biol. 6 (1), 1196. doi:10.1038/s42003-023-05585-5
Haaland, I., Hjelle, S. M., Reikvam, H., Sulen, A., Ryningen, A., McCormack, E., et al. (2021). p53 protein isoform profiles in AML: correlation with distinct differentiation stages and response to epigenetic differentiation therapy. Cells 10 (4), 833. doi:10.3390/cells10040833
Haaland, I., Opsahl, J. A., Berven, F. S., Reikvam, H., Fredly, H. K., Haugse, R., et al. (2014). Molecular mechanisms of nutlin-3 involve acetylation of p53, histones and heat shock proteins in acute myeloid leukemia. Mol. Cancer 13, 116. doi:10.1186/1476-4598-13-116
Haase, D., Stevenson, K. E., Neuberg, D., Maciejewski, J. P., Nazha, A., Sekeres, M. A., et al. (2019). TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 33 (7), 1747–1758. doi:10.1038/s41375-018-0351-2
Hales, E. C., Taub, J. W., and Matherly, L. H. (2014). New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal 26 (1), 149–161. Epub 2013 Oct 16. PMID: 24140475. doi:10.1016/j.cellsig.2013.09.021
Hassin, O., and Oren, M. (2023). Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug Discov. 22 (2), 127–144. doi:10.1038/s41573-022-00571-8
Herbrich, S., Baran, N., Cai, T., Weng, C., Aitken, M. J. L., Post, S. M., et al. (2021). Overexpression of CD200 is a stem cell-specific mechanism of immune evasion in AML. J. Immunother. Cancer 9 (7), e002968. doi:10.1136/jitc-2021-002968
Hong, M., Hao, S., Patel, K. P., Kantarjian, H. M., Garcia-Manero, G., Yin, C. C., et al. (2016). Whole-arm translocation of der(5;17)(p10;q10) with concurrent TP53 mutations in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS): A unique molecular-cytogenetic subgroup. Cancer Genet. 209 (5), 205–214. doi:10.1016/j.cancergen.2016.04.001
Italiano, A., Miller, W. H., Blay, J. Y., Gietema, J. A., Bang, Y. J., Mileshkin, L. R., et al. (2021). Phase I study of daily and weekly regimens of the orally administered MDM2 antagonist idasanutlin in patients with advanced tumors. Invest. New Drugs 39 (6), 1587–1597. doi:10.1007/s10637-021-01141-2
Jetani, H., Navarro-Bailon, A., Maucher, M., Frenz, S., Verbruggen, C., Yeguas, A., et al. (2021). Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. Blood 138 (19), 1830–1842. doi:10.1182/blood.2020009192
Jitschin, R., Saul, D., Braun, M., Tohumeken, S., Volkl, S., Kischel, R., et al. (2018). CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J. Immunother. Cancer 6 (1), 116. doi:10.1186/s40425-018-0432-9
Kadia, T. M., Reville, P. K., Borthakur, G., Yilmaz, M., Kornblau, S., Alvarado, Y., et al. (2021). Venetoclax plus intensive chemotherapy with cladribine, idarubicin, and cytarabine in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: a cohort from a single-centre, single-arm, phase 2 trial. Lancet Haematol. 8 (8), e552–e561. doi:10.1016/S2352-3026(21)00192-7
Kantarjian, H. M., Kadia, T. M., DiNardo, C. D., Welch, M. A., and Ravandi, F. (2021). Acute myeloid leukemia: treatment and research outlook for 2021 and the MD Anderson approach. Cancer 127 (8), 1186–1207. doi:10.1002/cncr.33477
Kennedy, M. C., and Lowe, S. W. (2022). Mutant p53: it's not all one and the same. Cell Death Differ. 29 (5), 983–987. doi:10.1038/s41418-022-00989-y
Khan, S., Wiegand, J., Zhang, P., Hu, W., Thummuri, D., Budamagunta, V., et al. (2022). BCL-X(L) PROTAC degrader DT2216 synergizes with sotorasib in preclinical models of KRAS(G12C)-mutated cancers. J. Hematol. Oncol. 15 (1), 23. doi:10.1186/s13045-022-01241-3
Klein, A. M., Biderman, L., Tong, D., Alaghebandan, B., Plumber, S. A., Mueller, H. S., et al. (2021). MDM2, MDMX, and p73 regulate cell-cycle progression in the absence of wild-type p53. Proc. Natl. Acad. Sci. U. S. A. 118 (44), e2102420118. doi:10.1073/pnas.2102420118
Kojima, K., Konopleva, M., Samudio, I. J., Shikami, M., Cabreira-Hansen, M., McQueen, T., et al. (2005). MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood 106 (9), 3150–3159. doi:10.1182/blood-2005-02-0553
Konopleva, M. Y., Rollig, C., Cavenagh, J., Deeren, D., Girshova, L., Krauter, J., et al. (2022). Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv. 6 (14), 4147–4156. doi:10.1182/bloodadvances.2021006303
Krupka, C., Kufer, P., Kischel, R., Zugmaier, G., Lichtenegger, F. S., Kohnke, T., et al. (2016). Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia 30 (2), 484–491. doi:10.1038/leu.2015.214
Laszlo, G. S., Beddoe, M. E., Godwin, C. D., Bates, O. M., Gudgeon, C. J., Harrington, K. H., et al. (2019). Relationship between CD33 expression, splicing polymorphism, and in vitro cytotoxicity of gemtuzumab ozogamicin and the CD33/CD3 BiTE® AMG 330. Haematologica 104 (2), e59–e62. doi:10.3324/haematol.2018.202069
Latif, A. L., Newcombe, A., Li, S., Gilroy, K., Robertson, N. A., Lei, X., et al. (2021). BRD4-mediated repression of p53 is a target for combination therapy in AML. Nat. Commun. 12 (1), 241. doi:10.1038/s41467-020-20378-8
Leick, M. B., Silva, H., Scarfo, I., Larson, R., Choi, B. D., Bouffard, A. A., et al. (2022). Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell 40 (5), 494–508 e5. doi:10.1016/j.ccell.2022.04.001
Leu, J. I., Murphy, M. E., and George, D. L. (2020). Functional interplay among thiol-based redox signaling, metabolism, and ferroptosis unveiled by a genetic variant of TP53. Proc. Natl. Acad. Sci. U. S. A. 117 (43), 26804–26811. doi:10.1073/pnas.2009943117
Li, F., Zhang, F., Wang, T., Xie, Z., Luo, H., Dong, W., et al. (2024). A self-amplifying loop of TP53INP1 and P53 drives oxidative stress-induced apoptosis of bone marrow mesenchymal stem cells. Apoptosis 29, 882–897. doi:10.1007/s10495-023-01934-1
Li, L., Mohanty, V., Dou, J., Huang, Y., Banerjee, P. P., Miao, Q., et al. (2023). Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci. Adv. 9 (30), eadd6997. doi:10.1126/sciadv.add6997
Liu, S., Qiao, X., Wu, S., Gai, Y., Su, Y., Edwards, H., et al. (2022). c-Myc plays a critical role in the antileukemic activity of the Mcl-1-selective inhibitor AZD5991 in acute myeloid leukemia. Apoptosis 27 (11-12), 913–928. doi:10.1007/s10495-022-01756-7
Liu, Z., Yang, Y., Sun, X., Ma, R., Zhang, W., Wang, W., et al. (2024). Discovery of novel antitumor small-molecule agent with dual action of CDK2/p-RB and MDM2/p53. Molecules 29 (3), 725. doi:10.3390/molecules29030725
Loizou, E., Banito, A., Livshits, G., Ho, Y. J., Koche, R. P., Sanchez-Rivera, F. J., et al. (2019). A gain-of-function p53-mutant oncogene promotes cell fate plasticity and myeloid leukemia through the pluripotency factor FOXH1. Cancer Discov. 9 (7), 962–979. doi:10.1158/2159-8290.CD-18-1391
Loke, J., Labopin, M., Craddock, C., Cornelissen, J. J., Labussiere-Wallet, H., Wagner-Drouet, E. M., et al. (2022). Additional cytogenetic features determine outcome in patients allografted for TP53 mutant acute myeloid leukemia. Cancer 128 (15), 2922–2931. doi:10.1002/cncr.34268
Loughery, J. E. P., and Meek, D. W. (2013). Switching on p53: an essential role for protein phosphorylation? Biodiscovery. doi:10.7750/biodiscovery.2013.8.1
Luedtke, D. A., Niu, X., Pan, Y., Zhao, J., Liu, S., Edwards, H., et al. (2017). Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct. Target Ther. 2, 17012. doi:10.1038/sigtrans.2017.12
Luo, K., Qian, Z., Jiang, Y., Lv, D., Zhu, K., Shao, J., et al. (2023). Characterization of the metabolic alteration-modulated tumor microenvironment mediated by TP53 mutation and hypoxia. Comput. Biol. Med. 163, 107078. doi:10.1016/j.compbiomed.2023.107078
Mai, S., Hodges, A., Chen, H. M., Zhang, J., Wang, Y. L., Liu, Y., et al. (2023). LILRB3 modulates acute myeloid leukemia progression and acts as an effective target for CAR T-cell therapy. Cancer Res. 83 (24), 4047–4062. doi:10.1158/0008-5472.CAN-22-2483
Malagola, M., Polverelli, N., Beghin, A., Bolda, F., Comini, M., Farina, M., et al. (2023). Bone marrow CD34+ molecular chimerism as an early predictor of relapse after allogeneic stem cell transplantation in patients with acute myeloid leukemia. Front. Oncol. 13, 1133418. doi:10.3389/fonc.2023.1133418
Mandeville, T. K., Mavis, C., Gu, J., Bowman, K., Olejniczak, S., Dey, P., et al. (2023). Mitochondrial reprogramming by bcl-2 inhibitor venetoclax enhances αCD19 CAR-T cell fitness and anti-tumor efficacy. Blood 142, 6845. doi:10.1182/blood-2023-191051
Mantovani, F., Collavin, L., and Del Sal, G. (2019). Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26 (2), 199–212. doi:10.1038/s41418-018-0246-9
Marcinek, A., Brauchle, B., Rohrbacher, L., Hanel, G., Philipp, N., Markl, F., et al. (2023). CD33 BiTE® molecule-mediated immune synapse formation and subsequent T-cell activation is determined by the expression profile of activating and inhibitory checkpoint molecules on AML cells. Cancer Immunol. Immunother. 72 (7), 2499–2512. doi:10.1007/s00262-023-03439-x
McClure, M. B., Kogure, Y., Ansari-Pour, N., Saito, Y., Chao, H. H., Shepherd, J., et al. (2023). Landscape of genetic alterations underlying hallmark signature changes in cancer reveals TP53 aneuploidy-driven metabolic reprogramming. Cancer Res. Commun. 3 (2), 281–296. doi:10.1158/2767-9764.CRC-22-0073
Meyer, M., Rubsamen, D., Slany, R., Illmer, T., Stabla, K., Roth, P., et al. (2009). Oncogenic RAS enables DNA damage- and p53-dependent differentiation of acute myeloid leukemia cells in response to chemotherapy. PLoS One 4 (11), e7768. doi:10.1371/journal.pone.0007768
Miller, K. N., Li, B., Pierce-Hoffman, H. R., Lei, X., Havas, A. P., Patel, S., et al. (2023). A mitochondria-regulated p53-CCF circuit integrates genome integrity with inflammation. bioRxiv, 2023.11.20.567963. Nov 21:2023.11.20.567963. doi:10.1101/2023.11.20.567963
Mishra, A., Tamari, R., DeZern, A. E., Byrne, M. T., Gooptu, M., Chen, Y. B., et al. (2022). Eprenetapopt plus azacitidine after allogeneic hematopoietic stem-cell transplantation for TP53-mutant acute myeloid leukemia and myelodysplastic syndromes. J. Clin. Oncol. 40 (34), 3985–3993. doi:10.1200/JCO.22.00181
Morganti, C., Ito, K., Yanase, C., Verma, A., Teruya-Feldstein, J., and Ito, K. (2022). NPM1 ablation induces HSC aging and inflammation to develop myelodysplastic syndrome exacerbated by p53 loss. EMBO Rep. 23 (5), e54262. doi:10.15252/embr.202154262
Motlagh, A. V., Mahdevar, M., Mirzaei, S., Entezari, M., Hashemi, M., Hushmandi, K., et al. (2022). Introduction of mutant TP53 related genes in metabolic pathways and evaluation their correlation with immune cells, drug resistance and sensitivity. Life Sci. 303, 120650. doi:10.1016/j.lfs.2022.120650
Mueller, J., Schimmer, R., Schneiter, F., Fulin, J., Lysenko, V., Myburgh, R., et al. (2023). Deficiency in human aml confers resistance to car T-cells that can Be overcome by synergistical pharmacological interventions targeting the cholesterol or wnt pathway. Ann. Hematol. 102 (Suppl. 1), S63–S. doi:10.1038/s44321-024-00024-2
Mueller, J., Schimmer, R. R., Koch, C., Schneiter, F., Fullin, J., Lysenko, V., et al. (2024). Targeting the mevalonate or Wnt pathways to overcome CAR T-cell resistance in TP53-mutant AML cells. EMBO Mol. Med. 16 (3), 445–474. doi:10.1038/s44321-024-00024-2
Muto, T., Walker, C. S., Agarwal, P., Vick, E., Sampson, A., Choi, K., et al. (2023). Inactivation of p53 provides a competitive advantage to del(5q) myelodysplastic syndrome hematopoietic stem cells during inflammation. Haematologica 108 (10), 2715–2729. doi:10.3324/haematol.2022.282349
Ni, X., Lu, C. P., Xu, G. Q., and Ma, J. J. (2024). Transcriptional regulation and post-translational modifications in the glycolytic pathway for targeted cancer therapy. Acta Pharmacol. Sin. Epub ahead of print. PMID: 38622288. doi:10.1038/s41401-024-01264-1
Olivier, M., Hollstein, M., and Hainaut, P. (2010). TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2 (1), a001008. doi:10.1101/cshperspect.a001008
Park, S., Bang, S. Y., Kwag, D., Lee, J. H., Kim, T. Y., Lee, J., et al. (2024). Reduced toxicity (FluBu3) versus myeloablative (BuCy) conditioning in acute myeloid leukemia patients who received first allogeneic hematopoietic stem cell transplantation in measurable residual disease-negative CR1. Bone Marrow Transpl. 59 (6), 813–823. doi:10.1038/s41409-024-02255-w
Perez Montero, S., Paul, P. K., di Gregorio, A., Bowling, S., Shepherd, S., Fernandes, N. J., et al. (2024). Mutation of p53 increases the competitive ability of pluripotent stem cells. Development 151 (2), dev202503. doi:10.1242/dev.202503
Perl, A. E. (2019). Improving response to FLT3 inhibitors-BCL2 the rescue? Clin. Cancer Res. 25 (22), 6567–6569. doi:10.1158/1078-0432.CCR-19-2339
Pfister, N. T., and Prives, C. (2017). Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harb. Perspect. Med. 7 (2), a026054. doi:10.1101/cshperspect.a026054
Popescu, B., Stahlhut, C., Tarver, T. C., Wishner, S., Lee, B. J., Peretz, C. A. C., et al. (2023). Allosteric SHP2 inhibition increases apoptotic dependency on BCL2 and synergizes with venetoclax in FLT3-and KIT-mutant AML. Cell Rep. Med. 4 (11), 101290. doi:10.1016/j.xcrm.2023.101290
Pratz, K. W., Jonas, B. A., Pullarkat, V., Thirman, M. J., Garcia, J. S., Dohner, H., et al. (2024). Long-term follow-up of VIALE-A: venetoclax and azacitidine in chemotherapy-ineligible untreated acute myeloid leukemia. Am. J. Hematol. 99 (4), 615–624. doi:10.1002/ajh.27246
Prokocimer, M., Molchadsky, A., and Rotter, V. (2017). Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood 130 (6), 699–712. doi:10.1182/blood-2017-02-763086
Qin, Z., Liu, H., Sheng, Q., Dan, J., Wu, X., Li, H., et al. (2023). Mutant p53 leads to low-grade IFN-I-induced inflammation and impairs cGAS-STING signalling in mice. Eur. J. Immunol. 53 (9), e2250211. doi:10.1002/eji.202250211
Rajagopalan, A., Feng, Y., Gayatri, M. B., Ranheim, E. A., Klungness, T., Matson, D. R., et al. (2023). A gain-of-function p53 mutant synergizes with oncogenic NRAS to promote acute myeloid leukemia in mice. J. Clin. Invest. 133 (24), e173116. doi:10.1172/JCI173116
Ramos, H., Raimundo, L., and Saraiva, L. (2020). p73: from the p53 shadow to a major pharmacological target in anticancer therapy. Pharmacol. Res. 162, 105245. doi:10.1016/j.phrs.2020.105245
Rimando, J. C., Chendamarai, E., Rettig, M. P., Jayasinghe, R., Christopher, M. J., Ritchey, J. K., et al. (2023). Flotetuzumab and other T-cell immunotherapies upregulate MHC class II expression on acute myeloid leukemia cells. Blood 141 (14), 1718–1723. doi:10.1182/blood.2022017795
Roche, M. E., Ko, Y. H., Domingo-Vidal, M., Lin, Z., Whitaker-Menezes, D., Birbe, R. C., et al. (2023). TP53 Induced Glycolysis and Apoptosis Regulator and Monocarboxylate Transporter 4 drive metabolic reprogramming with c-MYC and NFkB activation in breast cancer. Int. J. Cancer 153 (9), 1671–1683. doi:10.1002/ijc.34660
Rozenberg, J. M., Zvereva, S., Dalina, A., Blatov, I., Zubarev, I., Luppov, D., et al. (2021). The p53 family member p73 in the regulation of cell stress response. Biol. Direct 16 (1), 23. doi:10.1186/s13062-021-00307-5
Sahasrabudhe, K. D., and Mims, A. S. (2024). MRD in AML: who, what, when, where, and how? Blood 143 (4), 296–298. doi:10.1182/blood.2023022226
Sakaguchi, K., Herrera, J. E., Saito, S., Miki, T., Bustin, M., Vassilev, A., et al. (1998). DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 12 (18), 2831–2841. PMID: 9744860; PMCID: PMC317174. doi:10.1101/gad.12.18.2831
Sallman, D. A. (2020). To target the untargetable: elucidation of synergy of APR-246 and azacitidine in TP53 mutant myelodysplastic syndromes and acute myeloid leukemia. Haematologica 105 (6), 1470–1472. doi:10.3324/haematol.2020.249060
Sanford, J. D., Franklin, D., Grois, G. A., Jin, A., and Zhang, Y. (2023). Carnitine o-octanoyltransferase is a p53 target that promotes oxidative metabolism and cell survival following nutrient starvation. J. Biol. Chem. 299 (7), 104908. doi:10.1016/j.jbc.2023.104908
Saygin, C., Zhang, P., Stauber, J., Aldoss, I., Sperling, A. S., Weeks, L. D., et al. (2023). Acute lymphoblastic leukemia with myeloid mutations is a high-risk disease associated with clonal hematopoiesis. Blood Cancer Discov. 5 (3), 164–179. doi:10.1158/2643-3230.BCD-23-0106
Scott, M. T., Liu, W., Mitchell, R., Clarke, C. J., Kinstrie, R., Warren, F., et al. (2024). Activating p53 abolishes self-renewal of quiescent leukaemic stem cells in residual CML disease. Nat. Commun. 15 (1), 651. doi:10.1038/s41467-024-44771-9
Shah, M. V., Tran, E. N. H., Shah, S., Chhetri, R., Baranwal, A., Ladon, D., et al. (2023). TP53 mutation variant allele frequency of ≥10% is associated with poor prognosis in therapy-related myeloid neoplasms. Blood Cancer J. 13 (1), 51. doi:10.1038/s41408-023-00821-x
Shi, D., and Jiang, P. (2021). A different facet of p53 function: regulation of immunity and inflammation during tumor development. Front. Cell Dev. Biol. 9, 762651. doi:10.3389/fcell.2021.762651
Singh, S., Kumar, M., Kumar, S., Sen, S., Upadhyay, P., Bhattacharjee, S., et al. (2019). The cancer-associated, gain-of-function TP53 variant P152Lp53 activates multiple signaling pathways implicated in tumorigenesis. J. Biol. Chem. 294 (38), 14081–14095. doi:10.1074/jbc.RA118.007265
Smalley, J. P., Cowley, S. M., and Hodgkinson, J. T. (2024). MDM2 antagonist idasanutlin reduces HDAC1/2 abundance and corepressor Partners but not HDAC3. ACS Med. Chem. Lett. 15 (1), 93–98. doi:10.1021/acsmedchemlett.3c00449
Song, H., Wu, J., Tang, Y., Dai, Y., Xiang, X., Li, Y., et al. (2023). Diverse rescue potencies of p53 mutations to ATO are predetermined by intrinsic mutational properties. Sci. Transl. Med. 15 (690), eabn9155. doi:10.1126/scitranslmed.abn9155
Spaety, M. E., Gries, A., Badie, A., Venkatasamy, A., Romain, B., Orvain, C., et al. (2019). HDAC4 levels control sensibility toward cisplatin in gastric cancer via the p53-p73/BIK pathway. Cancers (Basel) 11 (11), 1747. doi:10.3390/cancers11111747
Tashakori, M., Kadia, T., Loghavi, S., Daver, N., Kanagal-Shamanna, R., Pierce, S., et al. (2022). TP53 copy number and protein expression inform mutation status across risk categories in acute myeloid leukemia. Blood 140 (1), 58–72. doi:10.1182/blood.2021013983
Tettero, J. M., Ngai, L. L., Bachas, C., Breems, D. A., Fischer, T., Gjertsen, B. T., et al. (2023). Measurable residual disease-guided therapy in intermediate-risk acute myeloid leukemia patients is a valuable strategy in reducing allogeneic transplantation without negatively affecting survival. Haematologica 108 (10), 2794–2798. doi:10.3324/haematol.2022.282639
Thijssen, R., Diepstraten, S. T., Moujalled, D., Chew, E., Flensburg, C., Shi, M. X., et al. (2021). Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood 137 (20), 2721–2735. doi:10.1182/blood.2020010167
Tomiyasu, H., Habara, M., Hanaki, S., Sato, Y., Miki, Y., and Shimada, M. (2024). FOXO1 promotes cancer cell growth through MDM2-mediated p53 degradation. J. Biol. Chem. 300 (4), 107209. doi:10.1016/j.jbc.2024.107209
Torka, P., Russell, T., Mavis, C., Gu, J., Ghione, P., Barth, M., et al. (2023). AMG176, an MCL-1 inhibitor, is active in pre-clinical models of aggressive B-cell lymphomas. Leuk. Lymphoma 64 (6), 1175–1185. doi:10.1080/10428194.2023.2200876
Trino, S., Iacobucci, I., Erriquez, D., Laurenzana, I., De Luca, L., Ferrari, A., et al. (2016). Targeting the p53-MDM2 interaction by the small-molecule MDM2 antagonist Nutlin-3a: a new challenged target therapy in adult Philadelphia positive acute lymphoblastic leukemia patients. Oncotarget 7 (11), 12951–12961. doi:10.18632/oncotarget.7339
Tron, A. E., Belmonte, M. A., Adam, A., Aquila, B. M., Boise, L. H., Chiarparin, E., et al. (2018). Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 9 (1), 5341. doi:10.1038/s41467-018-07551-w
Uy, G. L., Aldoss, I., Foster, M. C., Sayre, P. H., Wieduwilt, M. J., Advani, A. S., et al. (2021). Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 137 (6), 751–762. doi:10.1182/blood.2020007732
Vadakekolathu, J., Lai, C., Reeder, S., Church, S. E., Hood, T., Lourdusamy, A., et al. (2020). TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 4 (20), 5011–5024. doi:10.1182/bloodadvances.2020002512
Vaddavalli, P. L., and Schumacher, B. (2022). The p53 network: cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 38 (6), 598–612. doi:10.1016/j.tig.2022.02.010
Valeri, A., Garcia-Ortiz, A., Castellano, E., Cordoba, L., Maroto-Martin, E., Encinas, J., et al. (2022). Overcoming tumor resistance mechanisms in CAR-NK cell therapy. Front. Immunol. 13, 953849. doi:10.3389/fimmu.2022.953849
Varineau, J. E., and Calo, E. (2024). A common cellular response to broad splicing perturbations is characterized by metabolic transcript downregulation driven by the Mdm2-p53 axis. Dis. Model Mech. 17 (2), dmm050356. doi:10.1242/dmm.050356
Villatoro, A., Konieczny, J., Cuminetti, V., and Arranz, L. (2020). Leukemia stem cell release from the stem cell niche to treat acute myeloid leukemia. Front. Cell Dev. Biol. 8, 607. doi:10.3389/fcell.2020.00607
Wang, B., Reville, P. K., Yassouf, M. Y., Jelloul, F. Z., Ly, C., Desai, P. N., et al. (2024). Comprehensive characterization of IFNγ signaling in acute myeloid leukemia reveals prognostic and therapeutic strategies. Nat. Commun. 15 (1), 1821. doi:10.1038/s41467-024-45916-6
Wang, H., Guo, M., Wei, H., and Chen, Y. (2023a). Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduct. Target Ther. 8 (1), 92. doi:10.1038/s41392-023-01347-1
Wang, Y., Wang, D., Wang, Y., Yang, H., Wang, G., and Wu, S. (2023b). Synergistic activity and mechanism of cytarabine and MCL-1 inhibitor AZD5991 against acute myeloid leukemia. Neoplasma 70 (2), 287–293. doi:10.4149/neo_2023_221217N1185
Wiederschain, D., Kawai, H., Shilatifard, A., and Yuan, Z. M. (2005). Multiple mixed lineage leukemia (MLL) fusion proteins suppress p53-mediated response to DNA damage. J. Biol. Chem. 280 (26), 24315–24321. doi:10.1074/jbc.M412237200
Yan, B., Claxton, D., Huang, S., and Qiu, Y. (2020). AML chemoresistance: the role of mutant TP53 subclonal expansion and therapy strategy. Exp. Hematol. 87, 13–19. doi:10.1016/j.exphem.2020.06.003
Yang, M., Pan, Z., Huang, K., Busche, G., Liu, H., Gohring, G., et al. (2022). A unique role of p53 haploinsufficiency or loss in the development of acute myeloid leukemia with FLT3-ITD mutation. Leukemia 36 (3), 675–686. doi:10.1038/s41375-021-01452-6
Yilmaz, M., Kantarjian, H., and Ravandi, F. (2021). Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J. 11 (6), 123. doi:10.1038/s41408-021-00514-3
Yilmaz, M., Kantarjian, H., Short, N. J., Reville, P., Konopleva, M., Kadia, T., et al. (2022). Hypomethylating agent and venetoclax with FLT3 inhibitor "triplet" therapy in older/unfit patients with FLT3 mutated AML. Blood Cancer J. 12 (5), 77. doi:10.1038/s41408-022-00670-0
Young, A. L., Tong, R. S., Birmann, B. M., and Druley, T. E. (2019). Clonal hematopoiesis and risk of acute myeloid leukemia. Haematologica 104 (12), 2410–2417. doi:10.3324/haematol.2018.215269
Zeidan, A. M., Ando, K., Rauzy, O., Turgut, M., Wang, M. C., Cairoli, R., et al. (2024). Sabatolimab plus hypomethylating agents in previously untreated patients with higher-risk myelodysplastic syndromes (STIMULUS-MDS1): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Haematol. 11 (1), e38–e50. doi:10.1016/S2352-3026(23)00333-2
Zeidan, A. M., DeAngelo, D. J., Palmer, J., Seet, C. S., Tallman, M. S., Wei, X., et al. (2022). Phase 1 study of anti-CD47 monoclonal antibody CC-90002 in patients with relapsed/refractory acute myeloid leukemia and high-risk myelodysplastic syndromes. Ann. Hematol. 101 (3), 557–569. doi:10.1007/s00277-021-04734-2
Zhang, J., Sun, W., Kong, X., Zhang, Y., Yang, H. J., Ren, C., et al. (2019). Mutant p53 antagonizes p63/p73-mediated tumor suppression via Notch1. Proc. Natl. Acad. Sci. U. S. A. 116 (48), 24259–24267. doi:10.1073/pnas.1913919116
Zhang, Q., Riley-Gillis, B., Han, L., Jia, Y., Lodi, A., Zhang, H., et al. (2022). Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct. Target Ther. 7 (1), 51. doi:10.1038/s41392-021-00870-3
Zhao, D., Zarif, M., Zhou, Q., Capo-Chichi, J. M., Schuh, A., Minden, M. D., et al. (2023). TP53 mutations in AML patients are associated with dismal clinical outcome irrespective of frontline induction regimen and allogeneic hematopoietic cell transplantation. Cancers (Basel) 15 (12), 3210. doi:10.3390/cancers15123210
Zhou, T., Ke, Z., Ma, Q., Xiang, J., Gao, M., Huang, Y., et al. (2023). Molecular mechanism of CCDC106 regulating the p53-Mdm2/MdmX signaling axis. Sci. Rep. 13 (1), 21892. doi:10.1038/s41598-023-47808-z
Zhou, Y., Takacs, G. P., Lamba, J. K., Vulpe, C., and Cogle, C. R. (2020). Functional dependency analysis identifies potential druggable targets in acute myeloid leukemia. Cancers (Basel) 12 (12), 3710. doi:10.3390/cancers12123710
Keywords: AML, TP53 mutations, drug resistance, immunosuppression, metabolic rewiring, therapeutic approaches
Citation: Chomczyk M, Gazzola L, Dash S, Firmanty P, George BS, Mohanty V, Abbas HA and Baran N (2024) Impact of p53-associated acute myeloid leukemia hallmarks on metabolism and the immune environment. Front. Pharmacol. 15:1409210. doi: 10.3389/fphar.2024.1409210
Received: 16 April 2024; Accepted: 08 July 2024;
Published: 05 August 2024.
Edited by:
Adrian Bogdan Tigu, University of Medicine and Pharmacy Iuliu Hatieganu, RomaniaReviewed by:
Gábor Barna, Semmelweis University, HungaryCopyright © 2024 Chomczyk, Gazzola, Dash, Firmanty, George, Mohanty, Abbas and Baran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalia Baran, bmJhcmFuQGloaXQud2F3LnBs, bmJhcmFuQG1kYW5kZXJzb24ub3Jn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.