 Dayma Llanes1§Robert Rennert1†§Paul Jänicke1
Dayma Llanes1§Robert Rennert1†§Paul Jänicke1 Ibrahim Morgan1†Leslie Reguera1,2
Ibrahim Morgan1†Leslie Reguera1,2 Daniel G. Rivera1,2*
Daniel G. Rivera1,2* Manuel G. Ricardo1,2*‡
Manuel G. Ricardo1,2*‡ Ludger A. Wessjohann1*†
Ludger A. Wessjohann1*†- 1Department of Bioorganic Chemistry, Leibniz Institute of Plant Biochemistry, Halle (Saale), Germany
- 2Laboratory of Synthetic and Biomolecular Chemistry, Faculty of Chemistry, University of Havana, Havana, Cuba
Peptide-drug conjugates (PDCs) have recently gained significant attention for the targeted delivery of anticancer therapeutics, mainly due to their cost-effective and chemically defined production and lower antigenicity compared to ADCs, among other benefits. In this study, we designed and synthesized novel PDCs by conjugating new thiol-functionalized tubulysin analogs (tubugis) to bombesin, a peptide ligand with a relevant role in cancer research. Two tubulysin analogs bearing ready-for-conjugation thiol groups were prepared by an on-resin multicomponent peptide synthesis strategy and subsequently tested for their stand-alone in vitro anti-proliferative activity against human cancer cells, which resulted in IC50 values in the nanomolar range. In addition, various fluorescently labeled [K5]-bombesin(6–14) peptides, non-lipidated and lipidated with fatty acid chains of variable length, were also synthesized using the versatile multicomponent chemistry. These bombesin derivatives were tested for their gastrin-related peptide receptor (GRPR)-mediated internalization into cancer cells using flow cytometry, proving that the lipid tail (especially C14) enhances the cell internalization. Using the tubugi toxins and bombesin peptides, three different bombesin-tubugi conjugates were synthesized with different cleavage propensity and lipophilicity. Preliminary in vitro experiments revealed that, depending on the linker and the presence of a lipid tail, these novel PDCs possess good to potent anticancer activity and moderate selectivity for GRPR-overexpressing cancer cells.
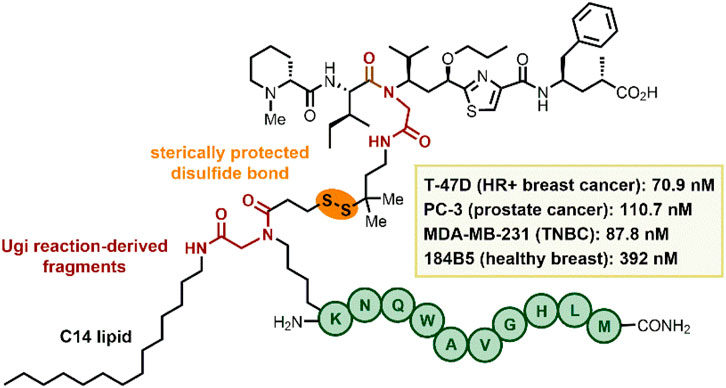
GRAPHICAL ABSTRACT | Table of Content Graphic.
Introduction
Peptide-drug conjugates (PDCs) have emerged in recent years and increasingly become a focus in targeted anticancer drug research (He et al., 2019; Hoppenz et al., 2020). Employing a homing peptide as a cancer cell-targeting entity offers significant benefits over more complex biomolecules such as monoclonal antibodies. These benefits include a well-defined, homogenous and synthetically flexible chemical structure as well as reduced immunogenicity and a more efficient distribution in solid tumors (Hoppenz et al., 2020; Cooper et al., 2021). Additionally, the cost-effective and reproducible methods for synthesizing peptides also make PDCs particularly attractive from both regulatory and manufacturing perspectives. Importantly, many targeting peptides derived from natural peptide ligands exhibit a high affinity and selectivity for cancer-associated cell surface receptors, often G protein-coupled receptors (GPCRs), which makes them promising candidates for targeted tumor therapy approaches (Dorsam and Gutkind, 2007).
The bombesin peptide (Bn) is a 14-residue neuropeptide originally isolated from the Bombina bombina frog (Figure 1A), and its associated receptor family holds significant therapeutic potential in cancer treatment (Anastasi et al., 1972; Faintuch et al., 2008; Chen et al., 2014). The gastrin-releasing peptide receptor (GRPR), in humans the primary and most relevant Bn receptor, is of particular interest due to its overexpression in several prevalent tumors, including those of breast, prostate, lung, and pancreas (Worm et al., 2020). Currently, a series of synthetic Bn analogs comprising simplified sequences are being clinically studied, especially for their application in radionuclide-based conjugates for tumor imaging (Figure 1B). Abd-Elgaliel et al. (2008), Ohki-Hamazaki et al. (2005), Begum et al. (2016), Mansi et al. (2021) While the majority of breakthroughs in this area focuses on diagnostics, Bn analogs also show considerable promise in drug delivery therapies (Anastasi et al., 1972; Faintuch et al., 2008; Chen et al., 2014).
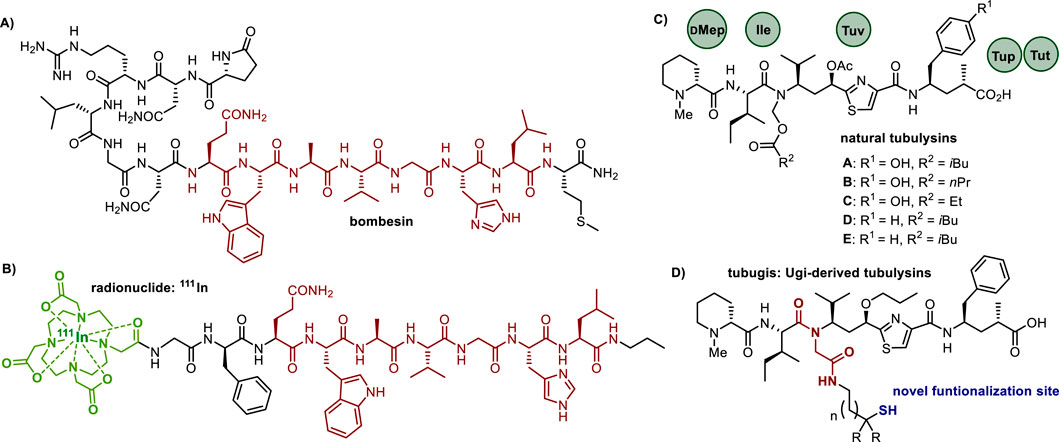
Figure 1. Structures of (A) the natural bombesin peptide, showing in red the core sequence Bn (Chen et al., 2014; Worm et al., 2020; Abd-Elgaliel et al., 2008; Ohki-Hamazaki et al., 2005; Begum et al., 2016; Mansi et al., 2021; Steinmetz et al., 2013) compelling for high receptor affinity. (B) a synthetic bombesin analog used in radionuclide-peptide conjugates. (C) natural tubulysins, highlighting their diverse N,O-acetal moieties at the tertiary amide and substitutions of the C-terminal residue. (D) synthetic Ugi-reaction derived tubulysin analogs (tubugis) functionalized with a thiol at a new position.
A very crucial aspect in designing an effective PDC is the selection of the payload component, with a prime requirement of having a very potent cytotoxicity. In this context, tubulysins are outstanding due to their cell growth-inhibiting effects in nano to picomolar concentrations (Steinmetz et al., 2013). As depicted in Figure 1C, tubulysins are tetrapeptides composed of unusual residues such as N-methyl-D-pipecolic acid (Mep), L-isoleucine (Ile), tubuvaline (Tuv), and either tubuphenylalanine (Tup) or tubutyrosine (Tut).
Despite their potent anti-proliferative activity, tubulysins have been less used in PDC development than other antimitotics, which can be attributed to their complex syntheses and the absence of residues amenable for functionalization with cleavable, ideally traceless linkers (Cooper et al., 2021; Dorsam and Gutkind, 2007; Worm et al., 2020). Early attempts of tubulysin conjugations relied on the linkages of the C-terminal carboxylic acid to entities like vitamins, polymers, antibodies and peptides, mostly via a difficult to cleave amide or a hydrazide moiety (Murray et al., 2015; Kufka et al., 2019; Kaake et al., 2018; Chen et al., 2017; Cohen et al., 2014). Other research groups focused on the insertion of an amino group at Tup’s phenyl moiety or the functionalization of Tuv’s thiazole ring (Parker et al., 2017; Xu et al., 2021). However, the derivatization of the N-methyl-D-pipecolic acid into a self-immolating quaternary ammonium linker constitutes the most recurrent alternative, owing to the improved pharmacokinetic and therapeutic efficacy of these conjugates (Rodrigues and Bernardes, 2016; Staben et al., 2016).
Our research is directed at devising suitable directing molecules and linkages for the targeted delivery of potent tubulysin analogs referred to as tubugis (Figure 1D). These analogs were originally obtained by fragment coupling using the Ugi four-component reaction (Ugi-4CR) and they not only exhibit cytotoxic activities in the picomolar concentration range but also provide higher stability under physiological conditions (Pando et al., 2011). While most of the known tubulysin analogs bear a small methyl, ethyl, or propyl N-substituent at the tertiary amide (Parker et al., 2017; Leverett et al., 2016), the Ugi-derived N-substituent of tubugis replaces the N,O-acetal with a retro amide residue incorporating alkyl moieties just like the natural products. Since previous studies have shown that the simplification of the N-alkyl motif is well accepted to keep a potent cytotoxicity (Sani et al., 2017), we hypothesized that further Ugi-4CR derivatizations could be done, seeking to expand the conjugation possibilities of this relevant payload family. Herein we describe for the first time the functionalization of the Ugi-derived N-substituent with a functional group suitable for conjugation to a targeting molecule, specifically a thiol capable of forming a stable conjugate with a homing peptide. Because disulfide bond formation is a successful strategy in the conjugation to targeting biomolecules (Danial and Postma, 2017; Bernardes et al., 2012), we sought to produce PDCs by combining bombesin peptides with tubugis via disulfide linkages, and subsequently evaluate their anticancer potential.
Results and discussion
Synthesis of thiol-functionalized tubugis
The building blocks required for the synthesis of tubulysin derivatives can be purchased commercially or synthesized on gram-scale using standard protocols (Murray et al., 2015; Wang et al., 2019; Shankar et al., 2013; Wipf et al., 2004). However, the assembly of some of these blocks using solution-phase protocols can be challenging due to the steric hindrance of some of them, especially in the coupling step leading to the tertiary amide formation (Peltier et al., 2006; Balasubramanian et al., 2009). We recently developed a solid-phase protocol that enables the construction and multicomponent derivatization of the tubugi skeleton, including the combinatorial diversification of the Ugi-derived N-substituent (Ricardo et al., 2024). We applied this protocol to synthesize tubugi derivatives with N-substituents bearing a variety of functional groups suitable for conjugation, such as carboxylic acids, amines, and alcohols. However, their introduction led to a significant drop in the anti-proliferative activity, thus limiting their use as PDC payloads. As shown in Scheme 1, here we extend this strategy to generate novel tubugis functionalized with a thiol group at the N-substituent, thus paving the way for the subsequent conjugation to a homing peptide without overly reducing the payloads activity.
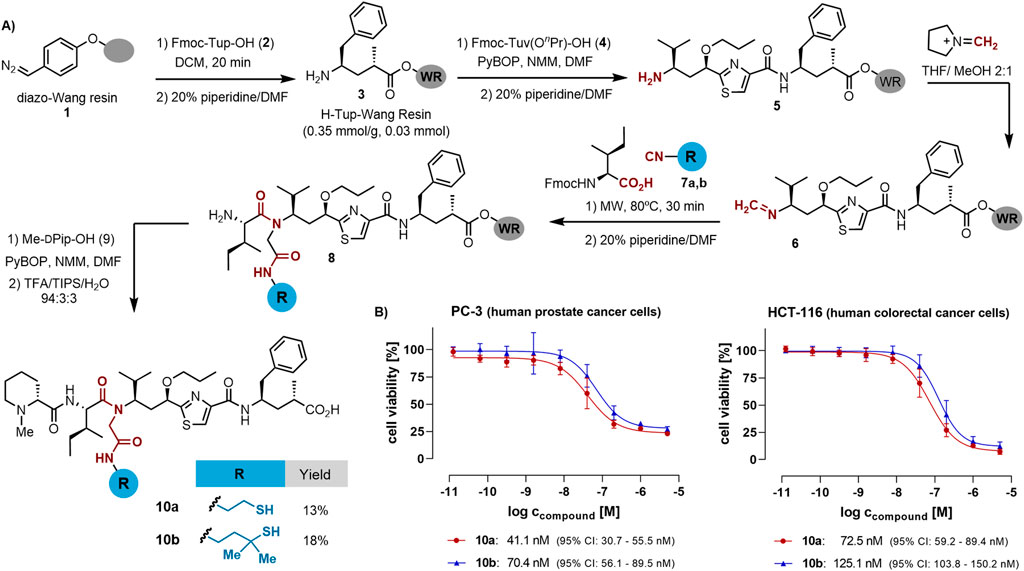
Scheme 1. (A) Solid-phase synthesis of tubugi analogs functionalized with a thiol group at the internal amide N-substituent. WR = Wang resin. (B) Evaluation of the anti-proliferative activity: tubugi effects on cell viability, as determined by a resazurin assay for human prostate cancer PC-3 cells and human colon cancer HCT-116 cells treated for 48 h with tubugi 10a (red dots and line) and tubugi 10b (blue triangles and line). For control and data normalization, cells were routinely treated in parallel with 0.5% (v/v) DMSO (negative control), and as a positive cytotoxicity control with 100 µM digitonin (for data normalization set to 0% cell viability), both in standard growth medium.
The synthetic strategy for tubugi analogs begins with the preparation of the diazo resin 1 following a reported protocol (Bhalay and Dunstan, 1998; Lévesque et al., 2017). The resin can be stored at −20°C until further use. A portion of this resin equivalent to a synthesis scale of 0.05 mmol was used to quantitatively incorporate the Tup amino acid 2 without the need to use an excess of this valuable building block. After incorporation of the Tuv residue 4 using standard coupling protocol, the dipeptide was deprotected for the two-step multicomponent protocol. The first step involved an organocatalytic transimination reaction that allowed the quantitative formation of the resin-linked imine 6, which was subjected to the Ugi-4CR by treatment with thiol-functionalized isocyanides 7 and the Fmoc-protected Ile under microwave irradiation (Rivera et al., 2021; Ricardo et al., 2018). The two isocyanides were synthesized with a thiol moiety protected with an acid-labile group (see the Supplementary Material), bearing geminal methyl groups at the α position or not. Because the final payloads were designed to form a disulfide bond as a linkage to the targeting peptide, the α-substitution was chosen to influence the conjugate stability. The two geminal methyls are known to form rather stable disulfide bonds that only get cleaved in the presence of high concentrations of free thiols like glutathione or similar reductants found under the reducing conditions in cancer cells and solid tumors. After the on-resin Ugi-4CR, N-methyl-D-pipecolic acid 9 was coupled and the resulting tubugis 10a and 10b were obtained in good crude purity after acidic cleavage from the resin. A final HPLC purification step rendered the pure thiol-functionalized tubugis in good isolated yields considering the number of synthetic steps implemented in the solid-phase protocol.
Evaluation of anticancer activity
Tubulysin analogs 10a and 10b were tested for anti-proliferative activity on the human prostate cancer cell line PC-3 and the colon cancer cell line HCT-116. As illustrated in Scheme 1B, both 10a and 10b inhibited the proliferation of the cancer cell lines under investigation with IC50 values in the low nanomolar range. Several remarks regarding the activity of the compounds were noted: i) the non-methylated analog 10a showed a 2-fold lower anti-proliferative activity toward the two cell lines than the α-di-methylated analog 10b (Scheme 1B); ii) the prostate cancer PC-3 cells were found to be slightly more sensitive towards both tubugi analogs than the colorectal cancer HCT-116 cells, and iii) the compounds’ effect on PC-3 cells seems to be rather cytostatic (lower curve plateau does not reach the 0% level of cell viability), whereas the effect on HCT-116 cells appears to be cytotoxic, reaching nearly 0% cell viability.
Synthesis and derivatization of bombesin peptides
To design bombesin targeting peptides suitable for PDC applications, structural modifications are required to enable the conjugation without compromising the peptide’s receptor binding and activation. In this context, it has been described that a N-terminally truncated bombesin analog [Bn (Faintuch et al., 2008; Chen et al., 2014; Worm et al., 2020; Abd-Elgaliel et al., 2008; Ohki-Hamazaki et al., 2005; Begum et al., 2016; Mansi et al., 2021; Steinmetz et al., 2013; Bouchard et al., 2014): Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met] mimics the full-length bombesin’s ability to activate bombesin receptors, particularly the human bombesin-binding gastrin-releasing peptide receptor (GRPR) (Begum et al., 2016). This study also proved that adding a Lys residue at the N-terminus of this truncated sequence permits the subsequent derivatization at the Lys ɛ-amino group without affecting the internalization capacity. Thus, by tagging this Lys residue with a fluorescein label, significant agonist activity and high levels of internalization were observed, particularly in prostate cancer cells (Begum et al., 2016).
To validate this [K5]-Bn(6–14) sequence as a targeting peptide, we synthesized various truncated bombesins and compared their internalization in the prostate cancer PC-3 cells by conducting a flow cytometry analysis. Our approach was influenced by similar studies on other targeting peptides, e.g., the human pancreatic polypeptide (hPP), (Mäde et al., 2014), for which enhanced cell internalization was achieved by increasing the hydrophobicity through attachment of a fatty acid. Given that multicomponent reactions are powerful approaches for the one-pot incorporation of various functionalities, we relied on a multicomponent strategy for the on-resin derivatization of the bombesin analogs with a lipid and a fluorescent label simultaneously. To minimize a potential negative impact of a large hydrophobic label (e.g., fluorescein) on the ligand-receptor binding and cell internalization, we chose to incorporate the considerably smaller 7-nitro-benzofurazan (NBD) fluorophore.
As depicted in Scheme 2A, the truncated sequence Bn(6–14) 11 was assembled on a Tentagel resin (0.05 mmol scale), followed by the incorporation of Boc-Lys(Fmoc)-OH using the standard SPPS methodology to afford the resin-bound [K5]-Bn(6–14) 12. The non-lipidated analog 13 was obtained in 32% overall yield after a final reaction of NBD-Cl with the ɛ-amino group of Lys and subsequent standard cleavage with TFA/TIPS/H2O (94:3:3). For the incorporation of both the fatty acid and the fluorescent label, we employed an on-resin Ugi-4CR comprising the aminocatalytic transimination approach followed by reaction of the resulting imine with various fatty acids and isocyanide 14, bearing an orthogonally (Alloc) protected amino group (Rivera et al., 2021; Ricardo et al., 2018). The parallel incorporation of four fatty acids of different chain lengths (C14, C16, C18, and C20) followed by Alloc deprotection, incorporation of the NBD label and cleavage from the resin led to bombesin analogs 15a, b, c, and d in good overall yield after HPLC purification (see the Supplementary Material).
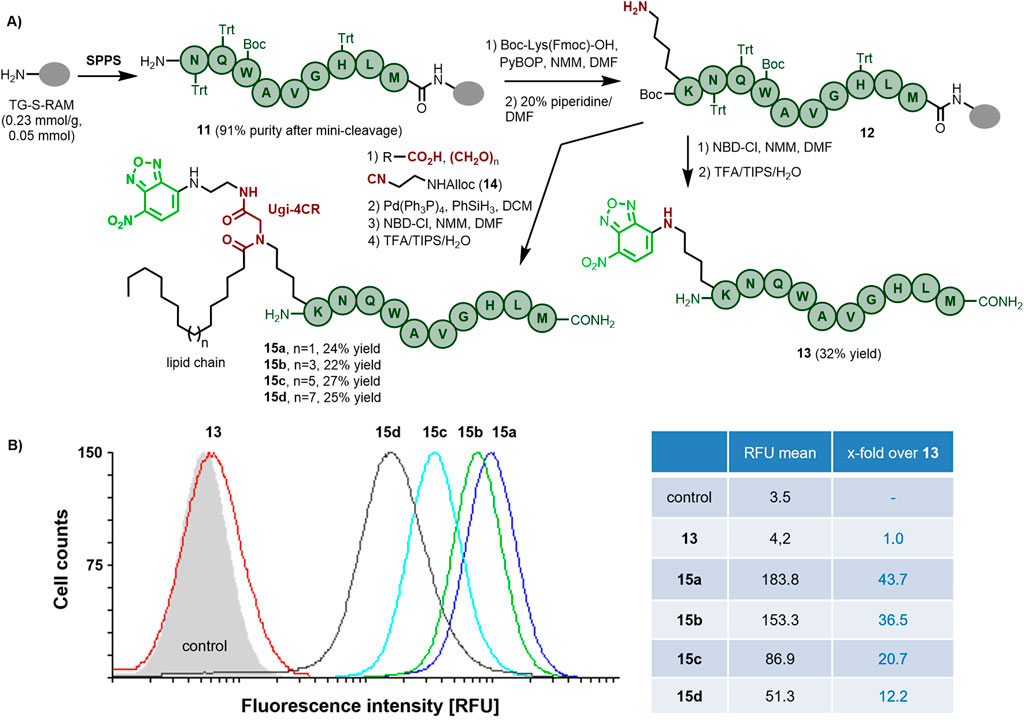
Scheme 2. (A) Synthesis of fluorescently-labeled bombesin derivatives incorporating lipid chains of different lengths. (B) Flow cytometry analysis of the internalization of the bombesin derivatives 13 (reference without lipid tail), 15a, 15 b, 15c and 15d in human prostate cancer PC-3 cells. The cells were treated for 2 h (for other time points see the SI, Supplementary Figure S46) with 5 µM concentrations of the peptides at 37 °C.
Internalization study of bombesin peptides
The cellular internalization of the NBD-labeled bombesin derivatives without (13) and with lipid tails of varied lengths C14-C20 (15a, 15b, 15c, and 15d) into GRPR-expressing PC-3 prostate cancer cells were semi-quantified by flow cytometry. Trypan blue quenching of outer cell surface fluorescence was done immediately before and while measuring, aiming to inspect as much as possible truly intracellular, hence internalized fluorescence of the labeled bombesin analogs (Rennert et al., 2009). The results of the flow cytometry measurements are illustrated in Scheme 2B and Supplementary Figure S46. As shown, the non-lipidated bombesin analog 13 showed only a poor cell internalization within the 2 h of treatment, as indicated by the just slightly elevated fluorescence count (RFU 4.2 compared to 3.5 of untreated control cells, this base value is due to their autofluorescence) caused by the intracellularly located peptide. On the other hand, all lipidated analogs were internalized better than the non-lipidated one, even within a short treatment time of only 2 h. Interestingly, a reverse correlation between the alkyl chain length and the internalization potency was observed, making 15a (C14 chain) the most efficiently internalized bombesin analog with a 43.7-fold higher intracellular fluorescence compared to 13, followed by 15b (C16), 15c (C18), and 15d (C20) with decreasing 36.5-fold, 20.7-fold, and 12.2-fold higher intracellular fluorescence as compared to 13. Previously, other groups have reported a similar trend for membrane-active lipopeptides, where C6–C12 acyl chains improved the lipopeptide interaction with the cell membranes better than longer and shorter lipid tails (Toniolo et al., 1996). As summarized in Supplementary Figure S46, the time dependency of the bombesin analogs’ cellular internalization was investigated as well, indicating a fast cell internalization process for all lipidated bombesins, with substantial internalization already within the first minute of treatment, reaching the majority of internalized ligand after just 1 hour, and with little increase to a maximum within the second hour. Such a fast cell internalization kinetics is typical for several G protein-coupled receptor peptide ligands. For example, the data of another GPCR-ligand system, the neuropeptide Y Y1 receptor (NPY1R), also shows the GPCR rapidly internalizing the peptide ligand NPY or agonistic analogs within minutes. (Gicquiaux et al., 2002; Babilon et al., 2013). However, for the GRPR receptor, at least in our hands, fast internalization is only achieved with the lipidated peptide analogs, and not with the non-lipidated ligand 13.
Synthesis of bombesin-tubugi conjugates via a disulfide linkage
Disulfide bond formation is widely used in the field of peptide derivatization, especially in dimerization processes and side chain crosslinking involving Cys residues (Danial and Postma, 2017; Deng et al., 2020). However, the formation of asymmetric disulfide bonds is of greater synthetic challenge and often requires thiol exchange in electrophilic disulfides, typically using either Elman’s reagent or 2-pyridyl disulfides (Bernardes et al., 2012; Pillow et al., 2016). As shown in Scheme 3A, the bombesin peptide was functionalized at the Lys side chain using 2-pyridyl disulfide containing a carboxylic acid (Beck et al., 2017) derived from 3-mercaptopropionic acid. The integrity of the 2-pyridyl disulfide component remained unaffected by the acidic conditions used during cleavage from the resin, leading to the activated bombesin analog 18 in 82% overall yield and 87% crude purity without the need for HPLC purification. Because of the superior internalization performance of the lipidated bombesin in cancer cells, i.e., derivative 15a with a C14 chain, we aimed to functionalize the truncated Bn with both a 2-pyridyl disulfide linker and a C14 lipid chain. For this purpose, lipidic isocyanide 17, paraformaldehyde and linker 16 were simultaneously incorporated at the Lys side chain using the Ugi-4CR (Scheme 3A), yielding bombesin derivative 19 in 17% isolated yield after RP-HPLC purification.
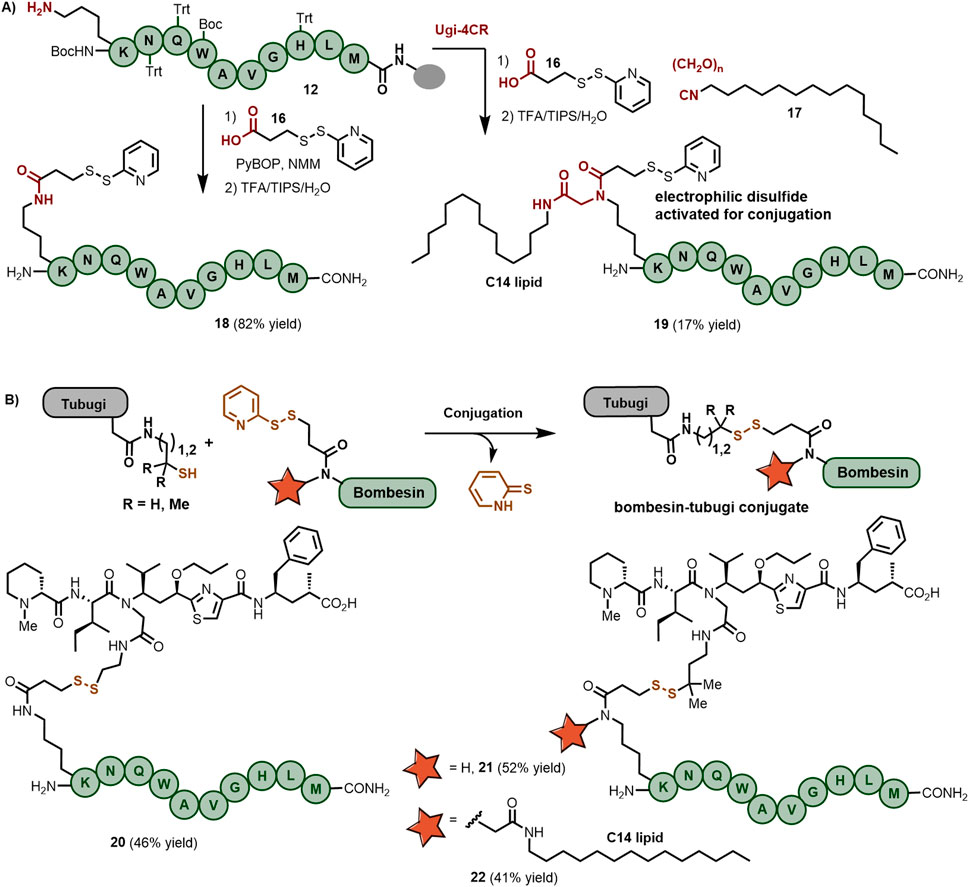
Scheme 3. (A) On-resin synthesis of 2-pyridyl disulfide-functionalized bombesin peptides either containing a C14 lipid chain or not. (B) Bombesin-tubugi conjugates produced by disulfide bond formation between activated bombesin and thiol-functionalized tubugis.
Integrating the activated disulfide into the bombesin peptide streamlined the solution-phase coupling with the thiol-functionalized tubugi. As depicted in Scheme 3B, bombesin analog 18 was ligated to the thiol-tubugi 10a in MeOH/DIPEA, with full consumption of starting materials after 2 h, leading to bombesin-tubugi conjugate 20 in 46% yield after purification. Following a similar conjugation protocol, conjugate 21 was produced in 52% isolated yield, while the preparation of the lipidated bombesin-tubugi conjugate required 12 h to reach complete consumption of the starting materials, ultimately giving 22 in 41% isolated yield.
Anti-proliferation study of bombesin-tubugi conjugates
The bombesin-tubugi conjugates 20, 21, and 22 were tested on human cell lines representing various receptor expression levels of GRPR, a human receptor with a high affinity for bombesin. We sought to investigate the correlation between the anti-proliferative efficacy of the conjugates and the GRPR expression levels of the cells, hypothesizing a better anti-proliferative effect in the cells with the higher expression level. For this purpose, a panel of human, primarily cancer, cell lines was initially selected to encompass a broad spectrum of GRPR expression levels. The selection process was based on differences in mRNA expression levels of GRPR as documented in the publicly available RNA-Seq database, GRCh38.p12 (O’Leary et al., 2024). Differential expression visualization was performed using the Genevestigator® gene expression analysis tool (Hruz et al., 2008), aiming to cover a wide range of GRPR expression levels. Accordingly, the following cell lines were selected for testing: T-47D (HR+ breast cancer), MDA-MB-231 (triple-negative breast cancer, TNBC), PC-3 (prostate cancer), MCF-10A (non-cancer, “healthy” breast epithelium), and HCT-116 (colorectal cancer). Of notice, T-47D has been reported with relatively high, MDA-MB-231 and PC-3 with medium, and HCT-116 and MCF-10A with relatively low GRPR expression levels (decreasing in the order of that list).
The actual GRPR expression levels of these cell lines were checked by conducting RT-qPCR, as described in the SI. Indeed, as shown in Figure 2A, the five cell lines ranked in the proposed order described above. The cell viability and proliferation assays were carried out by using a resazurin-based fluorometric assay. The cells were exposed to the bombesin conjugates 20, 21, and 22 for initial 6 h and 48 h, and regardless of the initial incubation time, the cells were then allowed to grow until 72 h in sum were finalized, after which the cell viability was determined (see the SI).
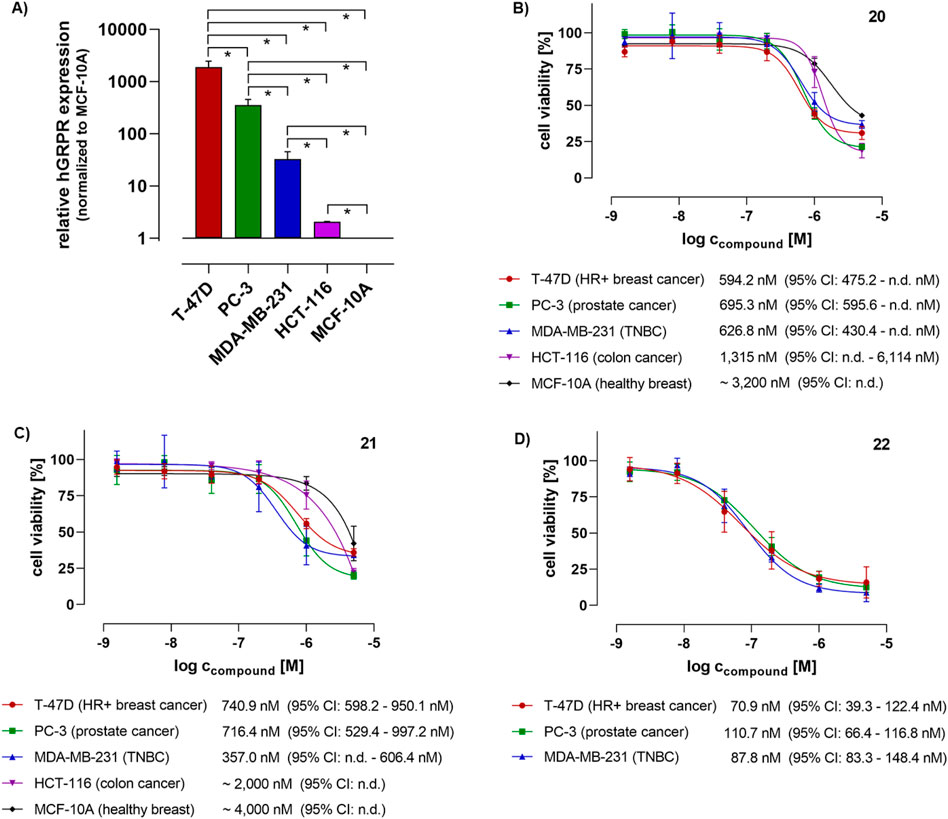
Figure 2. (A) Relative human GRPR (mRNA) expression levels in the cell lines used for the flow cytometry and cell viability studies, as detected by using RT-qPCR (n = 3). Data were analyzed by conducting the ΔΔCt methodology using GAPDH as a reference gene and normalized to the GRPR expression level of non-cancerous MCF-10A breast cells as a “healthy” cell reference (set to 1.0). Statistical significance was tested by using an unpaired t-test (*p < 0.05). (B–D) Cell viability of human cancer (T-47D, PC-3, MDA-MB-231, and HCT-116) and non-cancer (MCF-10A) cell lines after 6 h initial treatment with the bombesin-tubugi conjugates 20 (normal disulfide), 21 (hindered disulfide), and 22 (hindered disulfide and lipidated), medium exchange and subsequent cell cultivation until finalization of 72 h, and subsequent measurement by using fluorometric resazurin assay read-out. For control and data normalization, cells were routinely treated in parallel with 0.5% (v/v) DMSO (negative control), and as a positive cytotoxicity control with 100 µM digitonin (for data normalization set to 0% cell viability), both in standard growth medium. Each data point was determined with 3-4 biological replicates, each with technical triplicates. The indicated concentrations are IC50 values as calculated based on the non-linear regression dose-response curves as drawn by using GraphPad Prism 8 software. Error bars indicate the 95% confidence interval.
The results of the cell viability assays indicating the anti-proliferative and cytotoxic effects of the bombesin-tubugi conjugates are shown in Figures 2B–D (conjugate 20–22, respectively) for the initial 6 h treatment, and in the SI (Supplementary Figures S7, S48, S49) for the 48 h treatment. Because of the rather short metabolic half-lives of peptides in vivo, usually minutes up to just a few hours, the 6 h initial treatment was thought to be of higher relevance for potential anticancer therapeutics. The 48 h treatment was intended to show a proposed maximum effect of the sample. As illustrated in Figure 2B, after only 6 h initial cell treatment, conjugate 20 showed an anticancer activity in the high nanomolar range, as proven by an IC50 ∼ 600 nM in T-47D breast cancer cells, and just slightly higher IC50 values in PC-3 prostate and MDA-MB-231 TNBC cancer cells. Furthermore, IC50 values approximately 2-fold and 5.5-fold higher were detected in the GRPR-lower expressing HCT-116 (IC50 ∼ 1.3 µM) and GRPR-lowest expressing, healthy MCF-10A cells (IC50 ∼ 3.2 µM), respectively. Although the anticancer activity of conjugate 20 is not remarkable, the data illustrate a direct correlation between the cells’ GRPR expression levels and the anti-proliferative efficacy. The results of conjugate 21 after 6 h initial treatment (as shown in Figure 1C) are quite comparable to those described above for conjugate 20. The IC50 values of 21 are slightly higher in the different cell lines than for 20, except for the MDA-MB-231 cell line. In this latter cell line, conjugate 21 showed an IC50 ∼ 360 nM, whereas the IC50s in the higher expressing T-47D and PC-3 cancer cells were determined to be 741 nM and 716 nM, respectively. However, compared to these values, IC50 values of 21 in the lower and lowest GRPR-expressing HCT-116 cancer and MCF-10A healthy breast cells were approximately 2.5- and 5.5-fold higher, respectively. Compared to the lowest IC50 of 21 in MDA-MB-231 (∼360 nm), the IC50s of the conjugate in HCT-116 and MCF-10A were even ∼10-fold higher.
Interestingly, in the case of both 20 and 21 treatments for the initial 6 h, a significant difference in IC50s between the three highest and medium GRPR-expressing cell lines - i.e., T-47D, PC-3, and MDA-MB-231 – was not detected, which is in contrast to what was expected. Seemingly, there might be a certain GRPR expression level that allows a maximum targeting effect of the PDC, here IC50 ∼ 600–700 nM after 6 h initial treatment, that is already reached at least with the medium GRPR expression level of MDA-MB-231 cells. This could imply that even far higher GRPR levels would not have a substantial benefit for the targeting and thus selective cytotoxic effect.
After the cell treatment for 48 h with both conjugates 20 and 21, all IC50 values were measured in the nanomolar range, as well as those against the lower GRPR-expressing cell lines HCT-116 and MCF-10A. The reason for this could be the time that cells have to import the conjugates via GRPR-mediated endocytosis in multiple receptor internalization and recycling cycles. This could also be the reason for the lack of a clear correlation between GRPR expression levels and the conjugates’ anti-proliferative impact after 48 h initial incubation, as indicated by a difference of the lowest (in T-47D; IC50 ∼ 200 nM) and highest IC50 (in HCT-116; IC50 ∼ 570 nM) of at most ∼2.5-fold. This has, of course, consequences for the cancer cell selectivity of the bombesin conjugates, and should be kept in mind in future developments of bombesin-toxin conjugates. Considering the high similarity of the conjugates 20 and 21 in terms of chemical structure, in vitro cytotoxicity (IC50 values) and selectivity, the intended stabilization of the disulfide bridge by the dimethylation in 21 seems to have only a very modest effect on the PDC efficacy, at least in vitro.
As illustrated in Figure 1D (see also Supplementary Figure S49) the C14-lipidated conjugate 22 was 4- to 10-fold more potent in three cancer cells overexpressing the GRPR than the non-lipidated conjugates 20 and 21, respectively, with IC50 values in the range of ∼70–110 nM. We also conducted a direct comparison of conjugate 22 and tubugi 10b with an initial treatment of the cells for 6 h, as it makes sense to compare a PDC with its free payload. Besides the cancer cells, both compounds were tested (Supplementary Figure S50) in another healthy breast cell line, 184B5, which has a GRPR (mRNA) expression level that is comparable to that of MCF-10A, according to the Genevestigator® gene expression analysis tool (Hruz et al., 2008). As summarized in Table 1, the bombesin conjugate 22 exhibited a higher cytotoxicity than free tubugi 10b in the three cancer cells, but compound 10b also proved to be three times more toxic than conjugate 22 in the 184B5 healthy cells. In this case, the lack of selectivity of tubugi 10b among the cell lines tested is expected, given that this small compound can have a similar internalization efficiency in all cells after a short initial treatment of 6 h. It is also noteworthy the selectivity of conjugate 22, which exhibited a 3.5- to 5.5-fold higher cytotoxicity for cells overexpressing the GRPR receptor (Figure 1D) than for healthy 184B5 cells (Supplementary Figure S50). While this level of selectivity is not yet satisfactory for therapeutic applications, it is a clear indication of what can be achieved using this strategy.
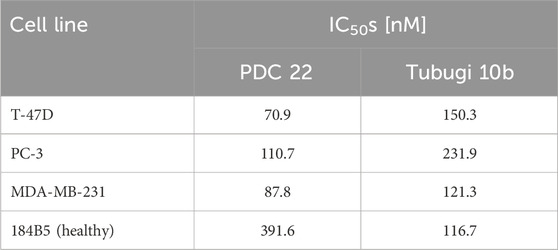
Table 1. Comparison of the anti-proliferative activity of bombesin-tubugi conjugate 22 and its free tubugi payload 10b in cancer and healthy cells after 6 h of initial treatmment.
Study limitations
While this study represents the first report on the development of bombesin-derived PDCs incorporating tubulysin analogs, we acknowledge that our approach has some limitations derived from both the type of receptor targeted and the methodology employed (Heh et al., 2023). For example, the initial cytotoxicity evaluation of the tubulysin analogs 10a and 10b was conducted using only the PC-3 and HCT-116 cells, with 48 h of initial treatment, as part of a general screening program in our laboratory. Accordingly, these results have limited comparability to the data obtained for the PDCs 20, 21 and 22, for which additional cancer cells and an initial treatment of 6 h were employed (as recommended for PDC evaluation). Fortunately, we were able to compare PDC 22 and its parent unconjugated payload 10b using 6 h of initial treatment and the same cell lines, which provided valuable information regarding the selectivity achieved by the bombesin PDC compared to the free toxin (discussed above). A further limitation is the lack of sufficient data concerning the role of lipidation on the PDC behavior. In addition to addressing PDC disadvantages such as poor metabolic stability and rapid renal clearance, lipidation also increases the PDC’s interaction with the cell membrane. The impact of this can be beneficial or detrimental, depending on whether it increases the PDC concentration on the cell membrane where the receptors are localized or also boosts an undesired, non-receptor-mediated internalization process. We did a comparative internalization analysis with the labeled bombesin peptide bearing various lipid chain lengths or no lipid chain, but assessing the internalization behavior of the final PDCs on different cell lines is further required.
Another limitation can be associated with the use of GRPR as target, since its level of overexpression in specific cancer cells might be insufficient to achieve a suitable selectivity index. The GRPR is known to be overexpressed in various cancer cells (D’Onofrio et al., 2023; Belge et al., 2024; Zhang et al., 2024), which was corroborated by our RT-qPCR data, but - like other GPCRs - it is not absent in healthy cells and tissues. It is also known that agonistic GPCR activation results in a subsequent desensitization and downregulation of the receptor, albeit with a certain delay. This also applies to the GRPR targeted by our PDCs (Benya et al., 1994). Furthermore, we don’t have data demonstrating the agonistic behavior of our PDCs or the lipidated bombesins used in the internalization study. The flow cytometry analysis in PC-3 cells of the bombesin derivatives (Scheme 2) indicated a fast and efficient cellular internalization of the lipidated bombesins, but other internalization mechanims cannot be ruled out. Overall, as expected, lipidated derivatives showed better internalization and thus higher bioactivity (lower IC50s). Indeed, new experiments focus on assessing both the capacity of our PDCs to functionally activate GRPR and the effectiveness of a receptor-mediated payload delivery into the cancer cells. This will help either to confirm or exclude alternative mechanisms of internalization for the lipidated PDC.
Conclusion
A novel solid-phase methodology was implemented for the synthesis of tubulysin analogs, known as tubugis, functionalized with ready-to-conjugate thiol groups. The on-resin protocol incorporates a key Ugi multicomponent reaction that allows the simultaneous assembly of the tubulysin peptide skeleton and the incorporation of the thiol-functionalized amide N-substituent. In parallel, cell internalization studies conducted with a fluorescently labeled bombesin peptide (active truncated sequence) proved that the peptide lipidation increases the homing peptide internalization, with a C14 chain showing the best cell internalization. As a result, a Ugi multicomponent approach was employed for the on-resin functionalization of the truncated bombesin with both a 2-pyridyl-activated disulfide and a C-14 lipid tail. Both lipidated and non-lipidated bombesin peptides were conjugated to the thiol-tubugi payloads and the resulting conjugates were evaluated for their anti-proliferative activity in a variety of cancer cells expressing GRPR (bombesin receptor) at different levels. The lipidated bombesin-tubugi 22 showed the best anti-proliferative activity and a good selectivity for cancer cells, although its selectivity is not sufficient to be considered for therapeutic applications. The present results do not yet allow for the establishment of a suitable correlation between the receptor expression levels and the cytotoxic activity of the PDCs. There appears to be an early saturation, with higher expressen not having additional benefits. Apparently, our data reflect a saturation of the receptor-mediated PDC internalization, causing higher GRPR expression not having additional benefit. However, the data suggest that the lipidation plays an important role in the PDC behavior, and the possibility of non-specific internalization of the lipidated bombesin and its conjugate cannot be ruled out, thereby circumventing receptor-mediated uptake and reducing selectivity. Further studies are required to get deeper insights into the mechanism of action of (lipidated) bombesin peptides as targeting molecules, and the potential of this conjugate class in targeted anticancer therapy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required in accordance with the local legislation and institutional requirements because only commercially available established in vitro cell lines were used.
Author contributions
DL: Data curation, Formal Analysis, Investigation, Writing–original draft, Writing–review and editing. RR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Supervision, Validation, Writing–original draft, Writing–review and editing. PJ: Formal Analysis, Investigation, Writing–original draft, Writing–review and editing. IM: Data curation, Formal Analysis, Investigation, Writing–original draft, Writing–review and editing. LR: Data curation, Formal Analysis, Investigation, Writing–original draft, Writing–review and editing. DR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. MR: Conceptualization, Formal Analysis, Investigation, Methodology, Validation, Writing–original draft, Writing–review and editing. LW: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Public funding for research was provided by the Alexander von Humboldt Foundation for an Experienced Researcher fellowship to DR and LR, by the BMBF (AZ_01DN18063) and by the DAAD, Germany, in scope of the GLACIER project (DAAD 57592717).
Acknowledgments
We are grateful to the Alexander von Humboldt Foundation for an Experienced Researcher fellowship to DR and LR. We acknowledge financial support from BMBF (AZ_01DN18063) and DAAD, Germany, in scope of the GLACIER project (DAAD 57592717).
Conflict of interest
LW is a patent holder of the tubugis anticancer compounds.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1408091/full#supplementary-material
References
Abd-Elgaliel, W. R., Gallazzi, F., Garrison, J. C., Rold, T. L., Sieckman, G. L., Figueroa, S. D., et al. (2008). Design, synthesis, and biological evaluation of an antagonist-bombesin analogue as targeting vector. Bioconjug Chem. 19, 2040–2048. doi:10.1021/bc800290c
Anastasi, A., Erspamer, V., and Bucci, M. (1972). Isolation and amino acid sequences of alytesin and bombesin, two analogous active tetradecapeptides from the skin of European discoglossid frogs. Arch. Biochem. Biophys. 148, 443–446. doi:10.1016/0003-9861(72)90162-2
Babilon, S., Mörl, K., and Beck-Sickinger, A. G. (2013). Towards improved receptor targeting: anterograde transport, internalization and postendocytic trafficking of neuropeptide y receptors. Biol. Chem. 394, 921–936. doi:10.1515/hsz-2013-0123
Balasubramanian, R., Raghavan, B., Begaye, A., Sackett, D. L., and Fecik, R. A. (2009). Total synthesis and biological evaluation of tubulysin U, tubulysin V, and their analogues. J. Med. Chem. 52, 238–240. doi:10.1021/jm8013579
Beck, A., Goetsch, L., Dumontet, C., and Corvaïa, N. (2017). Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 16, 315–337. doi:10.1038/nrd.2016.268
Begum, A. A., Moyle, P. M., and Toth, I. (2016). Investigation of bombesin peptide as a targeting ligand for the gastrin releasing peptide (GRP) receptor. Bioorg Med. Chem. 24, 5834–5841. doi:10.1016/j.bmc.2016.09.039
Belge, B. G., Bilgin, C., Orscelik, A., Burkett, B. J., Thorpe, M. P., Johnson, D. R., et al. (2024). Detection rate of gastrin-releasing peptide receptor (GRPr) targeted tracers for positron emission tomography (PET) imaging in primary prostate cancer: a systematic review and meta-analysis. Ann. Nucl. Med. 38, 865–876. doi:10.1007/s12149-024-01978-6
Benya, R. V., Fathi, Z., Kusui, T., Pradhan, T., Battey, J. F., and Jensen, R. T. (1994). Gastrin-releasing peptide receptor-induced internalization, down-regulation, desensitization, and growth: possible role for cyclic AMP. Mol. Pharmacol. 46 (2), 235–245.
Bernardes, G. J. L., Casi, G., Trüssel, S., Hartmann, I., Schwager, K., Scheuermann, J., et al. (2012). A traceless vascular-targeting antibody-drug conjugate for cancer therapy. Angew. Chem. - Int. Ed. 51 (51), 941–944. doi:10.1002/anie.201106527
Bhalay, G., and Dunstan, A. R. (1998). Facile solid phase synthesis of an activated diazo linker. Tetrahedron Lett. 39, 7803–7806. doi:10.1016/S0040-4039(98)01706-7
Bouchard, H., Viskov, C., and Garcia-Echeverria, C. (2014). Antibody – drug conjugates — a new wave of cancer drugs. Bioorg Med. Chem. Lett. 24, 5357–5363. doi:10.1016/j.bmcl.2014.10.021
Chen, H., Lin, Z., Arnst, K. E., Miller, D. D., and Li, W. (2017). Tubulin inhibitor-based antibody-drug conjugates for cancer therapy. Molecules 22, 1281. doi:10.3390/molecules22081281
Chen, H., Wan, S., Zhu, F., Wang, C., Cui, S., Dua, C., et al. (2014). A fast tumor-targeting near-infrared fluorescent probe based on bombesin analog for in vivo tumor imaging. Contrast Media Mol. Imaging 9, 122–134. doi:10.1002/cmmi.1545
Cohen, R., Vugts, D. J., Visser, G. W. M., Walsum, M. S., Bolijn, M., Spiga, M., et al. (2014). Development of novel ADCs: conjugation of tubulysin analogues to trastuzumab monitored by dual radiolabeling. Cancer Res. 74, 5700–5710. doi:10.1158/0008-5472.CAN-14-1141
Cooper, B. M., Iegre, J., O’Donovan, D. H., Ölwegård Halvarsson, M., and Spring, D. R. (2021). Peptides as a platform for targeted therapeutics for cancer: peptide-drug conjugates (PDCs). Chem. Soc. Rev. 50, 1480–1494. doi:10.1039/d0cs00556h
Danial, M., and Postma, A. (2017). Disulfide conjugation chemistry: a mixed blessing for therapeutic drug delivery? Ther. Deliv. 8, 359–362. doi:10.4155/tde-2017-0003
Deng, Z., Hu, J., and Liu, S. (2020). Disulfide-based self-immolative linkers and functional bioconjugates for biological applications. Macromol. Rapid Commun. 41, 1900531. doi:10.1002/marc.201900531
D’Onofrio, A., Engelbrecht, S., Läppchen, T., Rominger, A., and Gourni, E. (2023). GRPR-targeting radiotheranostics for breast cancer management. Front. Med. (Lausanne) 10, 1250799. doi:10.3389/fmed.2023.1250799
Dorsam, R. T., and Gutkind, J. S. (2007). G-protein-coupled receptors and cancer. Nat. Rev. Cancer 7, 79–94. doi:10.1038/nrc2069
Faintuch, B. L., Teodoro, R., Duatti, A., Muramoto, E., Faintuch, S., and Smith, C. J. (2008). Radiolabeled bombesin analogs for prostate cancer diagnosis: preclinical studies. Nucl. Med. Biol. 35, 401–411. doi:10.1016/j.nucmedbio.2008.02.005
Gicquiaux, H., Lecat, S., Gaire, M., Dieterlen, A., Mély, Y., Takeda, K., et al. (2002). Rapid internalization and recycling of the human neuropeptide Y Y1 receptor. J. Biol. Chem. 277, 6645–6655. doi:10.1074/jbc.M107224200
He, R., Finan, B., Mayer, J. P., and Dimarchi, R. D. (2019). Peptide conjugates with small molecules designed to enhance efficacy and safety. Molecules 24, 1855. doi:10.3390/molecules24101855
Heh, E., Allen, J., Ramirez, F., Lovasz, D., Fernandez, L., Hogg, T., et al. (2023). Peptide drug conjugates and their role in cancer therapy. Int. J. Mol. Sci. 24, 829–844. doi:10.3390/ijms24010829
Hoppenz, P., Els-Heindl, S., and Beck-Sickinger, A. G. (2020). Peptide-drug conjugates and their targets in advanced cancer therapies. Front. Chem. 8, 571. doi:10.3389/fchem.2020.00571
Hruz, T., Laule, O., Szabo, G., Wessendorp, F., Bleuler, S., Oertle, L., et al. (2008). Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes. Adv. Bioinforma. 2008, 420747. doi:10.1155/2008/420747
Kaake, M., Srinivasarao, M., and Low, P. S. (2018). Targeted tubulysin B hydrazide conjugate for the treatment of luteinizing hormone-releasing hormone receptor-positive cancers. Bioconjugate Chem. 29, 2208–2214. doi:10.1021/acs.bioconjchem.8b00164
Kufka, R., Rennert, R., Kalu, G. N., Weber, L., Richter, W., and Wessjohann, L. A. (2019). Synthesis of a tubugi-1-toxin conjugate by a modulizable disulfide linker system with a neuropeptide Y analogue showing selectivity for hY1R-overexpressing tumor cells. Beilstein J. Org. Chem. 15, 96–105. doi:10.3762/bjoc.15.11
Leverett, C. A., Sukuru, S. C. K., Vetelino, B. C., Musto, S., Parris, K., Loganzo, F., et al. (2016). Design, synthesis, and cytotoxic evaluation of novel tubulysin analogues as ADC payloads. ACS Med. Chem. Lett. 7, 999–1004. doi:10.1021/acsmedchemlett.6b00274
Lévesque, É., Laporte, S. T., and Charette, A. B. (2017). Continuous flow synthesis and purification of aryldiazomethanes through hydrazone fragmentation. Angew. Chem. Int. Ed. 56, 837–841. doi:10.1002/anie.201608444
Mäde, V., Bellmann-Sickert, K., Kaiser, A., Meiler, J., and Beck-Sickinger, A. G. (2014). Position and length of fatty acids strongly affect receptor selectivity pattern of human pancreatic polypeptide analogues. ChemMedChem 9 (9), 2463–2474. doi:10.1002/cmdc.201402235
Mansi, R., Nock, B. A., Dalm, S. U., Busstra, M. B., van Weerden, W. M., and Maina, T. (2021). Radiolabeled bombesin analogs. Cancers (Basel) 13, 5766. doi:10.3390/cancers13225766
Murray, B. C., Peterson, M. T., and Fecik, R. A. (2015). Chemistry and biology of tubulysins: antimitotic tetrapeptides with activity against drug resistant cancers. Nat. Prod. Rep. 32, 654–662. doi:10.1039/c4np00036f
Ohki-Hamazaki, H., Iwabuchi, M., and Maekawa, F. (2005). Development and function of bombesin-like peptides and their receptors. Int. J. Dev. Biol. 49, 293–300. doi:10.1387/ijdb.041954ho
O’Leary, N. A., Cox, X., Holmes, J. B., Anderson, W. R., Falk, R., Hem, V., et al. (2024). Exploring and retrieving sequence and metadata for species across the tree of life with NCBI Datasets. Sci Data. 11 (1), 732. doi:10.1038/s41597-024-03571-y
Pando, O., Stark, S., Denkert, A., Porzel, A., Preusentanz, R., and Wessjohann, L. A. (2011). The multiple multicomponent approach to natural product mimics: tubugis, N-substituted anticancer peptides with picomolar activity. J. Am. Chem. Soc. 133, 7692–7695. doi:10.1021/ja2022027
Parker, J. S., McCormick, M., Anderson, D. W., Maltman, B. A., Gingipalli, L., and Toader, D. (2017). The development and scale-up of an antibody drug conjugate tubulysin payload. Org. Process Res. Dev. 21, 1602–1609. doi:10.1021/acs.oprd.7b00232
Peltier, H. M., Mcmahon, J. P., Patterson, A. W., and Ellman, J. A. (2006). The total synthesis of tubulysin D. JACS 128, 16018–16019. doi:10.1021/ja067177z
Pillow, T. H., Sadowsky, J. D., Zhang, D., Yu, S. F., Del Rosario, G., Xu, K., et al. (2016). Decoupling stability and release in disulfide bonds with antibody-small molecule conjugates. Chem. Sci. 8, 366–370. doi:10.1039/C6SC01831A
Rennert, R., Neundorf, I., and Beck-Sickinger, A. G. (2009). Synthesis and application of peptides as drug carriers. Methods Mol. Biol. 535, 389–403. doi:10.1007/978-1-59745-557-2_22
Ricardo, M. G., Llanes, D., Rennert, R., Jänicke, P., Rivera, D. G., and Wessjohann, L. A. (2024). Improved access to potent anticancer tubulysins and linker-functionalized payloads via an all-on-resin strategy. Chem. Eur. J. 30, e202401943. doi:10.1002/chem.202401943
Ricardo, M. G., Marrero, J. F., Valdes, O., Rivera, D. G., and Wessjohann, L. A. (2018). A peptide backbone stapling strategy enabled by the multicomponent incorporation of amide N-substituents. Chem. - A Eur. J. 25, 769–774. doi:10.1002/chem.201805318
Rivera, D. G., Ricardo, M. G., Vasco, A. V., Wessjohann, L. A., and Van der Eycken, E. V. (2021). On-resin multicomponent protocols for biopolymer assembly and derivatization. Nat. Protoc. 16, 561–578. doi:10.1038/s41596-020-00445-6
Rodrigues, T., and Bernardes, G. J. L. (2016). Antibody-Drug Conjugates: the missing link. Nat. Chem. 8, 1088–1090. doi:10.1038/nchem.2685
Sani, M., Lazzari, P., Folini, M., Spiga, M., Zuco, V., Cesare, M. D., et al. (2017). Synthesis and superpotent anticancer activity of tubulysins carrying non-hydrolysable N-substituents on tubuvaline. Chem-Eur J. 23, 5842–5850. doi:10.1002/chem.201700874
Shankar, S. P., Jagodzinska, M., Malpezzi, L., Paolo, L., Manca, I., Greig, I. R., et al. (2013). Synthesis and structure–activity relationship studies of novel tubulysin U analogues – effect on cytotoxicity of structural variations in the tubuvaline fragment. Org. Biomol. Chem. 11, 2273–2287. doi:10.1039/C3OB27111K
Staben, L. R., Koenig, S. G., Lehar, S. M., Vandlen, R., Zhang, D., Chuh, J., et al. (2016). Targeted drug delivery through the traceless release of tertiary and heteroaryl amines from antibody – drug conjugates. Nat. Chem. 8, 1112–1119. doi:10.1038/nchem.2635
Steinmetz, H., Glaser, N., Herdtweck, E., Sasse, F., Reichenbach, H., Höfle, G., et al. (2013). Isolation, crystal and solution structure determination, and biosynthesis of tubulysins - powerful inhibitors of tubulin polymerization from myxobacteria. Angew. Chem. Int. Ed, 1213–1222. doi:10.1002/asia.201300051
Toniolo, C., Crisma, M., Formaggio, F., Peggion, C., Monaco, V., Goulard, C., et al. (1996). Effect of Nα-acyl chain length on the membrane-modifying properties of synthetic analogs of the lipopeptaibol trichogin GA IV. J. Am. Chem. Soc. 118, 4952–4958. doi:10.1021/ja954081o
Wang, X. M., Liu, Y. W., Wang, Q. E., Zhou, Z., Si, C. M., and Wei, B. G. (2019). A divergent method to key unit of tubulysin V through one-pot diastereoselective Mannich process of N,O-acetal with ketone. Tetrahedron 75, 260–268. doi:10.1016/j.tet.2018.11.053
Wipf, P., Takada, T., and Rishel, M. J. (2004). Synthesis of the tubuvaline-tubuphenylalanine (Tuv-Tup) fragment of tubulysin. Org. Lett. 6, 4057–4060. doi:10.1021/ol048252i
Worm, D. J., Els-Heindl, S., and Beck-Sickinger, A. G. (2020). Targeting of peptide-binding receptors on cancer cells with peptide-drug conjugates. Pept. Sci. 112, e24171. doi:10.1002/pep2.24171
Xu, X., Fan, M., Qi, J., and Yao, L. (2021). Design, synthesis, and antitumor activity evaluation of pretubulysin analogs. Chem. Biol. Drug Des. 98, 341–351. doi:10.1111/cbdd.13852
Keywords: anticancer agents, peptide-drug conjugates (PDCs), tubulysin therapy, lipopeptides, gastrin-releasing peptide (GRP) receptor, bombesin (BBN), bombesin receptor
Citation: Llanes D, Rennert R, Jänicke P, Morgan I, Reguera L, Rivera DG, Ricardo MG and Wessjohann LA (2024) Development of bombesin-tubulysin conjugates using multicomponent chemistry to functionalize both the payload and the homing peptide. Front. Pharmacol. 15:1408091. doi: 10.3389/fphar.2024.1408091
Received: 27 March 2024; Accepted: 18 October 2024;
Published: 12 November 2024.
Edited by:
Norbert Sewald, Bielefeld University, GermanyReviewed by:
Fernando Formaggio, University of Padua, ItalyMarc Nazare, Leibniz-Institut für Molekulare Pharmakologie (FMP), Germany
Copyright © 2024 Llanes, Rennert, Jänicke, Morgan, Reguera, Rivera, Ricardo and Wessjohann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel G. Rivera, ZGdyQGZxLnVoLmN1; Manuel G. Ricardo, bWFudWVsLmdhcmNpYXJpY2FyZG9AbXBpa2cubXBnLmRl; Ludger A. Wessjohann, d2Vzc2pvaGFubkBpcGItaGFsbGUuZGU=
†ORCID ID: Robert Rennert, orcid.org/0000-0003-3665-6696; Ibrahim Morgan, orcid.org/0000-0002-4032-2691; Ludger A. Wessjohann, orcid.org/0000-0003-2060-8235
‡Present address: Manuel G. Ricardo, Department of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Potsdam, Germany
§These authors have contributed equally to this work