 Mohamed Tawengi
Mohamed Tawengi Yazan Al-Dali
Yazan Al-Dali Abdelaziz Tawengi
Abdelaziz Tawengi Ibrahim F. Benter
Ibrahim F. Benter Saghir Akhtar
Saghir Akhtar- 1College of Medicine, QU Health, Qatar University, Doha, Qatar
- 2Faculty of Pharmacy, Final International University, Kyrenia, Cyprus
Epidermal growth factor receptor (EGFR), which is referred to as ErbB1/HER1, is the prototype of the EGFR family of receptor tyrosine kinases which also comprises ErbB2 (Neu, HER2), ErbB3 (HER3), and ErbB4 (HER4). EGFR, along with other ErbBs, is expressed in the kidney tubules and is physiologically involved in nephrogenesis and tissue repair, mainly following acute kidney injury. However, its sustained activation is linked to several kidney pathologies, including diabetic nephropathy, hypertensive nephropathy, glomerulonephritis, chronic kidney disease, and renal fibrosis. This review aims to provide a summary of the recent findings regarding the consequences of EGFR activation in several key renal pathologies. We also discuss the potential interplay between EGFR and the reno-protective angiotensin-(1–7) (Ang-(1–7), a heptapeptide member of the renin-angiotensin-aldosterone system that counter-regulates the actions of angiotensin II. Ang-(1–7)-mediated inhibition of EGFR transactivation might represent a potential mechanism of action for its renoprotection. Our review suggests that there is a significant body of evidence supporting the potential inhibition of EGFR/ErbB, and/or administration of Ang-(1–7), as potential novel therapeutic strategies in the treatment of renal pathologies. Thus, EGFR inhibitors such as Gefitinib and Erlinotib that have an acceptable safety profile and have been clinically used in cancer chemotherapy since their FDA approval in the early 2000s, might be considered for repurposing in the treatment of renal pathologies.
1 Introduction
Epidermal growth factor receptor (EGFR), also called ErbB1/HER1, is the prototype of the four-membered EGFR receptor tyrosine kinases family that also comprises ErbB-2 (Neu, HER2), ErbB-3 (HER3), and ErbB-4 (HER4) (Kumagai et al., 2021; Sharifi et al., 2021; Shraim et al., 2021; Shaban et al., 2023; Popović et al., 2024) (see also Figure 1). Through their regulation by several different receptor ligands and ability to form multiple homo- and heterodimer combinations, signalling via the ErbB family members yields a diverse array of signalling outputs that regulate key cellular functions (Lemmon and Schlessinger, 2010; Wee and Wang, 2017; Shaban et al., 2023; Popović et al., 2024). All members of the EGFR family have an N-terminal extracellular ligand-binding domain, a cytoplasmic tyrosine kinase catalytic domain linked by a single transmembrane helix, and a C-terminal tail with multiple phosphorylation sites. Although they share a general architecture, the extracellular domain of each receptor is quite different, resulting in variability of ligand specificity and several biological responses (Tang et al., 2013a; Shaban et al., 2023; Popović et al., 2024). In contrast, a delicate conformation of the tyrosine kinase domain makes up the intracellular domain of the different ErbB receptors. The kinase domain of ErbB3 is an exception containing substitutions of important amino acids rendering it without kinase activity (Normanno et al., 2005).
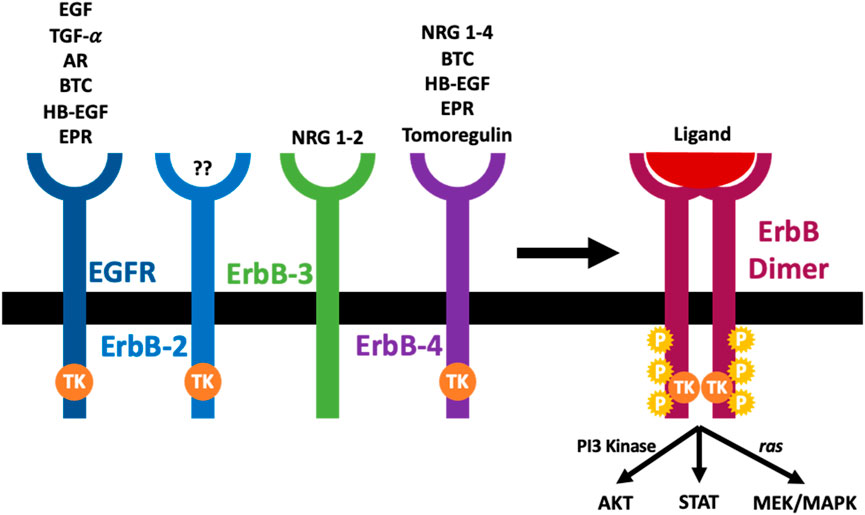
Figure 1. The arrangement and cognate ligands of the ErbB family of receptors. An ErbB receptor is composed of an extracellular domain that binds specific ligands, a transcellular component, and an intracellular domain with a tyrosine kinase activity (TK). The ErbB-2 is not known to have any ligand, while the ErbB-3 is known to lack kinase activity. Once a ligand binds the extracellular domain of ErbB receptors, it induces dimerization of the receptor (homodimerization or heterodimerization), tyrosine kinase activation (TK), trans-phosphorylation (P) and initiation of downstream signaling cascades (see main text for further details).
The EGFR family can interact with various ligands (see Figure 1). Based on their ability to bind to the different ErbB receptors, the epidermal growth factor (EGF)-related growth factors are split into three different groups. The first group has EGF, transforming growth factor-α (TGF-α), and amphiregulin (AR), and they bind the EGF receptor; the second group is made up of several growth factors which can bind both EGFR and ErbB4, and they include betacellulin (BTC), heparin-binding growth factor (HB-EGF) and epiregulin (EPR). The final group comprises tomoregulin and the neuregulins (NRGs) or heregulins (HRG) which are split into two subclasses depending on their affinity to ErbB-3 and ErbB-4 (NRG-1 and NRG-2) or merely ErbB-4 (NRG-3 and NRG-4). All EGF-ligand family members are produced as membrane-tethered precursors that can be cleaved by specific metalloproteases to shed the soluble bioactive form from the cell surface (Harris et al., 2003; Rayego-Mateos et al., 2018a, 2018b; Shraim et al., 2021; Shaban et al., 2023; Popović et al., 2024). Until now, no ligand has been identified with a high affinity for ErbB-2 (Normanno et al., 2005). EGFR activation without direct ligand-stimulated interaction with the receptor ectodomain is known as transactivation (indirect activation). Several ‘transactivating” molecules exist, and they are generally classified into activators of G protein-coupled receptors (e.g., Angiotensin II (Ang II) and endothelin 1 (ET-1), cytokines (e.g., TNFA, TWEAK, and other TNF receptor family proteins), growth factors (e.g., TGFB and other proteins, including TPA and LPA), integrins, ion channels, and other physical stimuli (Rayego-Mateos et al., 2018b; Shraim et al., 2021; Shaban et al., 2023; Popović et al., 2024).
Under unstimulated conditions, EGFR is in a state of auto-inhibition and is unable to dimerize on the plasma membrane. Physiological EGFR signaling involves two structurally coupled allosteric activation events across the membrane: an extracellular ligand-dependent extracellular domain dimerization followed by an intracellular dimer-dependent tyrosine kinase activation. Ultimately, ligand binding promotes receptor dimerization, which regulates a chain of structural readjustments which get transported to the intracellular domain to allow for the formation of asymmetric dimers between two juxtaposed catalytic domains (Sigismund et al., 2018; Tsai and Nussinov, 2019). Downstream effects include the activation of several signaling pathways including the extracellular signal-regulated kinase (ERK1/2) pathway, the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway, and the phosphoinositide-3-kinase (PI3K)/AKT (protein kinase B) pathway. Indeed, EGFR stimulation activates a cascade of nuclear signaling that controls the function of different transcription factors including c-JUN, c-FOS, c-MYC, and NF-κB that control gene transcription. The net effects of such molecular events are associated with multiple functions including cell survival, proliferation, differentiation, and migration (Tang et al., 2013a; Rayego-Mateos et al., 2018a; 2018b; Shaban et al., 2023; Popović et al., 2024).
In the kidney, all four EGFR/ErbB receptors, as well as their ligands are expressed along the length of the nephron (see also Figure 2). The receptors are mostly localized at the distal tubular cells, the connecting tubules, and the collecting ducts, and most of the ligands for these receptors are formed locally in the kidneys (Staruschenko et al., 2013) implying a possible local paracrine-autocrine function. Recent evidence suggests that kidney EGFR expression and activation after injury might also be dependent on gender (Harris, 2021).
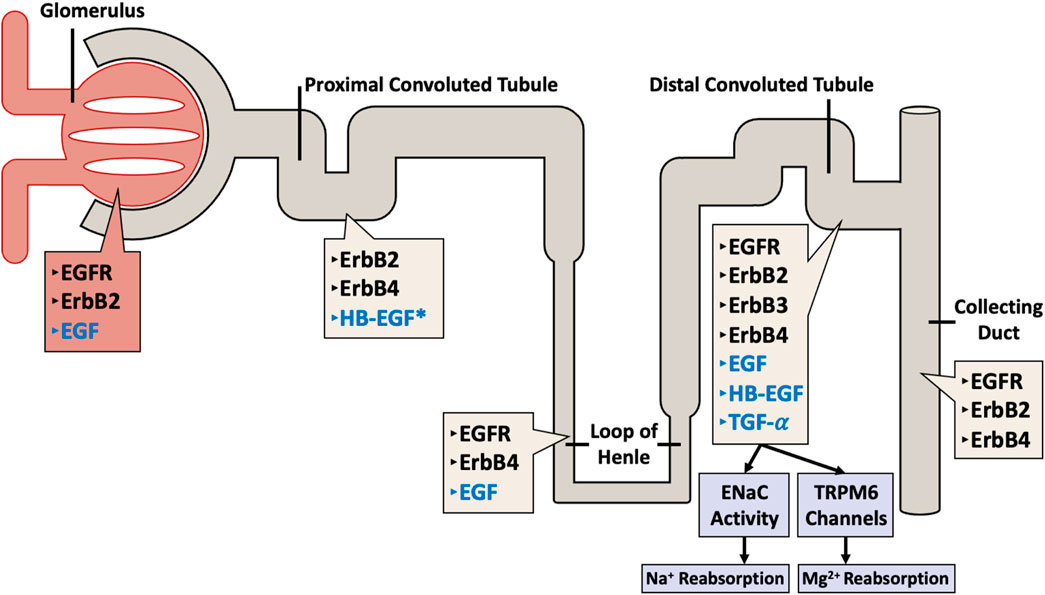
Figure 2. Distribution of ErbB family of receptors (in black text) with their corresponding ligands (in blue text) throughout the nephron. The different receptors and their ligands are differentially expressed along the various segments of the nephron (including the glomerulus, proximal convoluted tubule, loop of Henle, distal convoluted tubule, and the collecting duct) in a human adult kidney. Note the exception: HB-EGF expression data is from rat kidneys (asterisked).
The EGFR/ErbB pathway plays an important physiological or beneficial role in kidney development and function. Indeed, EGFR knockout mice suffer from impaired epithelial cell development in multiple organs, including the kidney (Threadgill et al., 1995; Sibilia et al., 2007). However, limited in vivo functional information can be obtained from knockout (KO) mice of any of the four EGFR/ErbB receptors because of the perinatal lethality of EGFR deficient mice, with embryos typically only surviving a few weeks due to, amongst others, impaired cardiovascular, skin, neuronal and gastrointestinal development (for reviews see also Sibilia et al., 2007; Sanchez-Soria and Camenisch, 2010; Shraim et al., 2021). However, knockouts of their ligands or conditional and/or site-specific (e.g., podocyte-specific) deletion of EGFR family of receptors as well as pharmacological inhibition studies have revealed that these RTKs play a detrimental role in the development and progression of different renal pathologies (Harskamp et al., 2016; Rayego-Mateos et al., 2018b; Melenhorst et al., 2008) (to be discussed further below). Thus, this potential dual (or “Jekyl and Hyde”) role of EGFR family members in the kidneys, whereby there are necessary/beneficial for development and tissue repair, but sustained overactivity is detrimental in mediating pathologies, is akin to that observed in the cardiovascular system (Akhtar and Benter, 2013; Shraim et al., 2021).
2 Glomerulonephritis
Glomerulonephritis (GN) is an inflammation within the glomerulus and other parts of the kidney resulting from a variety of immune-mediated disorders or infections (Chadban and Atkins, 2005; Satoskar et al., 2020; Pesce et al., 2021; Zhan et al., 2024). The incidence rate of GN worldwide is estimated to be between 0.2 and 2.5/100,000/year (McGrogan et al., 2011), with the prevalence being 306/100,000 in the US (Wetmore et al., 2016). EGFR ligands and receptor signaling have been implicated in the development of several forms of GN (see Table 1 for summary). One of these disorders in GN is the Rapid Progressive Glomerulonephritis (RPGN) which is the formation of crescent-shaped necrotizing lesions within the glomeruli with accumulation of CD4+ T cells and macrophages as well as proliferation of intrinsic glomerular cells, resulting in a swift decline of renal function. Increased EGFR signalling is known to promote glomerular damage and renal failure in rapidly progressive crescentic glomerulonephritis (Bollée et al., 2011). The detrimental role of EGFR in GN is supported by a study in a type 2 diabetes mouse model where EGFR inhibition with Erlotinib (a selective, clinically used EGFR inhibitor) resulted in decreased albuminuria and glomerulosclerosis, attenuated podocyte loss, and reduced renal fibrosis (Li et al., 2018).
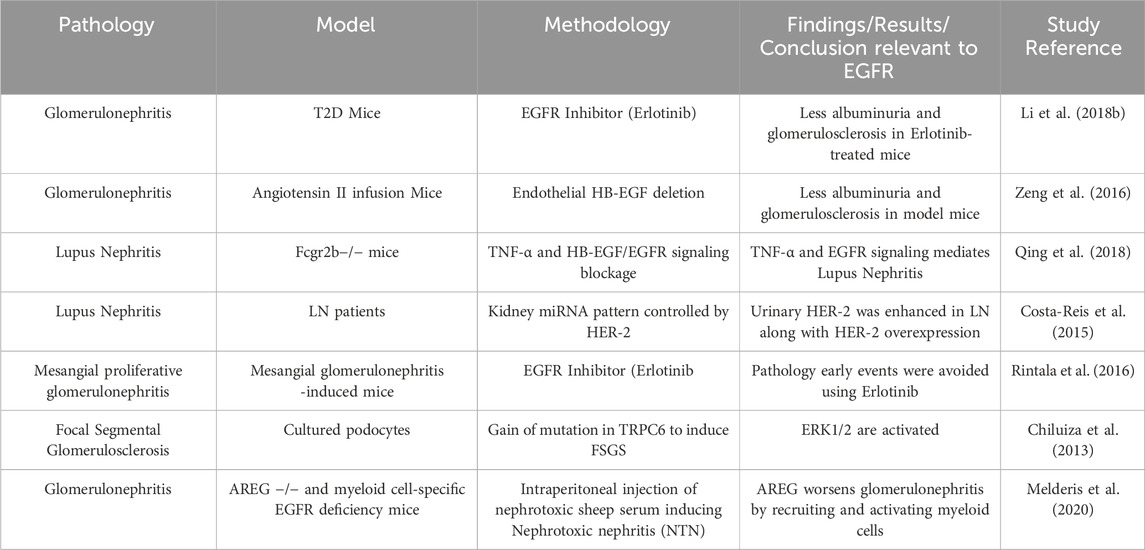
Table 1. Summary of selected studies highlighting the role of EGFR signalling in glomerulonephritis and associated pathologies.
Other studies have also implicated EGFR ligands in mediating GN pathology (see also Table 1). For example, several studies have shown that HB-EGF, a ligand for EGFR, appears to be a key player in mediating pathological changes in the glomerulus resulting in glomerulonephritis (Feng et al., 2000; Tang et al., 2013a; Tang et al., 2013b; Hénique et al., 2013). Thus, HB-EGF ligand-activated EGFR signalling appears critical in mediating GN. Interestingly, a recent study showed that a subset of EGF-deficient mice developed crescentic glomerulonephritis due to upregulated HB-EGF/EGFR activation in glomeruli (Zeid et al., 2022) implying also that physiological levels of EGF, as well as HB-EGF, act as reno-protective agents against the development of glomerulonephritis.
Additionally, it has been shown that Ang II-mediated shedding of EGF-like ligands such as HB-EGF and TGF-alpha, can transactivate EGFR signalling in the kidney that leads to several renal pathologies (Lautrette et al., 2005; Chen et al., 2006; Zeng et al., 2016). For example, deletion of endothelial HB-EGF in mice reduced Ang II infusion-related renal damage by decreasing albuminuria, glomerulosclerosis, and podocyte injury, whilst maintaining the integrity of the endothelium (Zeng et al., 2016). These studies imply important crosstalk between the Renin-Angiotensin-Aldosterone system (RAAS) of which Ang II is the main component, and EGFR signalling (see also discussion on EGFR/ErbB and Ang-(1–7) crosstalk below in Section 9).
To further study the role of EGFR ligands in glomerular pathology, a recent study investigated the role of amphiregulin (AREG), an EGF-like ligand that can possess both pro and anti-inflammatory properties (Melderis et al., 2020). In a mouse model of glomerulonephritis, it was found that upregulation of AREG plays an important proinflammatory role, by enhancing myeloid cell responses and directing the macrophages toward a pro-inflammatory M1 phenotype whereas AREG knockout mice displayed considerable improvement in disease. Importantly, in the myeloid cells, the selective deletion of EGFR signaling was enough to be protective against nephritis (Melderis et al., 2020) (See also Table 1). These authors also found a strong upregulation of renal AREG expression in human crescentic glomerulonephritis (Melderis et al., 2020) and thereby suggesting that AREG/EGFR axis might represent a new therapeutic target for patients with acute glomerulonephritis.
Another disorder that could lead to progressive renal dysfunction is Lupus Nephritis (LN). A recent study suggested that permanent kidney injury occurs due to iRhom2/ADAM17-dependent TNF-α and EGFR signaling through activation of the inactive rhomboid two (iRhom2), the regulator of disintegrin and metalloprotease 17 (ADAM17) (Qing et al., 2018). In addition, urinary HER-2/ErbB2, that is involved in mesangial cell proliferation, was increased in LN and correlated with disease activity (Costa-Reis et al., 2015). Thus, a potentially novel strategy to reduce cell proliferation and damage in LN could be via blockade of both EGFR and ErbB2 signalling.
Amongst the spectrum of glomerular diseases, mesangial proliferative glomerulonephritis (MPGN) is another disorder that could result in end-stage renal disease. Treatment of mice bearing Thy1.1-antibody-induced MPGN with Erlotinib, a selective EGFR inhibitor, significantly attenuated the glomerular inflammatory response, mesangial cell proliferation, matrix accumulation and further, preserved renal function (Rintala et al., 2016). These results suggested that EGFR inhibition could effectively prevent the initial or early-phase pathological events involved in the development of MPGN and thus, may represent a potential therapeutic option in halting the progression of GN. Supportive of the role of EGFR signaling in glomerulonephritis is the finding that extracellular signal-regulated kinases 1/2 (ERK1/2), which are downstream signalling effectors for EGFR, are also activated in focal segmental glomerulosclerosis that was induced by a gain of mutation in the transient receptor potential C6 (TRPC6) (Chiluiza et al., 2013). Thus, EGFR activation and subsequent signalling via ERK1/2 may be a critical pathway leading to glomerular pathology.
Additionally, there is emerging evidence that the pro-inflammatory transcription factor, NF-κB, plays a key role in the pathogenesis of renal inflammation including GN (Sanz et al., 2010; Song et al., 2019; Lin and Hu, 2022). NF-κB activation and subsequent transcription of multiple proinflammatory genes has been reported in glomerular podocytes, mesangial, tubular, and endothelial cells in acute renal injury or upon exposure to inflammatory stimuli in vivo and in vitro (Guijarro and Egido, 2001; Sanz et al., 2010; Song et al., 2019). Reports also suggest that NF-κB is a downstream effector of both Ang II/Ang II- type 1 (AT1) receptor signalling axis as well as EGFR signalling and that EGFR inhibitors can attenuate NF-κB signalling as well as the associated inflammation (Mezzano et al., 2003; Akhtar et al., 2015; Benter et al., 2015; Cong et al., 2022). Thus, it is tempting to speculate that Ang II-mediated shedding of EGF-like ligands, subsequent transactivation of EGFR and activation of the pro-inflammatory NF-κB could be a likely mechanism in the development of GN–though this requires further study and verification in vivo.
In summary, the above studies provide emerging evidence for upregulated EGFR signalling, either via direct ligand-mediated activation or via Ang-II-mediated transactivation, and the possible involvement of downstream effectors, such as ERK1/2 and/or NF-κB, to be critical in the development of GN (for a summary see also Table 1).
3 Diabetic nephropathy
Diabetic nephropathy (DN) is the leading cause of end-stage renal failure worldwide (Duran-Salgado and Rubio-Guerra, 2014; Li et al., 2022; Nomura et al., 2022). In particular, one-third of all type 1 diabetes (T1D) patients along with around 25% of type 2 diabetic (T2D) patients are affected (Duran-Salgado and Rubio-Guerra, 2014). Chronic or prolonged intermittent hyperglycemia associated with diabetes mellitus can lead to a general dysfunction in the kidney (e.g., in mesangial cells, podocytes, endothelial and tubular epithelial cells) through multiple pathological mechanisms including excess production of reactive oxidative species (ROS) or reactive nitrogen species (RNS), and downstream activation of several signaling networks including EGFR (for a recent review, see also Sheng et al., 2021). DN arises from the subsequent dysfunction in renal cell growth/proliferation, angiogenesis, and/or apoptosis and clinically manifests with symptoms of proteinuria together with morphological changes or remodeling in the glomerulus and interstitium (Sheng et al., 2021). Its complex pathogenesis may also involve low grade inflammation as well as hemodynamic and metabolic disturbances (Sheng et al., 2021).
Several reports have now shown that renal EGFRs are involved in DN (for a summary, see Table 2). Upregulation of EGFR, and other ErbBs, has been reported in conditions of hyperglycemia and/or diabetes (Konishi and Berk, 2003; Benter et al., 2005a; Benter et al., 2005b; Saad et al., 2005; Portik-Dobos et al., 2006; Uttarwar et al., 2011; Akhtar et al., 2012; Akhtar et al., 2013; Akhtar et al., 2015; Benter et al., 2015; Li et al., 2015; Akhtar et al., 2019). Inhibiting EGFR or its downstream signaling is protective against the development of DN and as such might be exploited as a potential new therapeutic strategy in its treatment. In a comprehensive study, Li et al. tested if blocking the activation of EGFR with Erlotinib could influence the development of DN in an accelerated T2D mouse model (Li et al., 2018b). Compared to controls, the Erlotinib-treated eNOS deficient db/db mice had markedly decreased albuminuria, glomerulosclerosis, podocyte loss, and fibrosis (Li et al., 2018a). In addition, the intervention reduced the infiltration of the immune cells such as the macrophages and T-lymphocytes, decreased the renal oxidative stress, and macrophage and T-lymphocyte infiltration, as well as fewer proinflammatory cytokine levels. Interestingly, Erlotinib also exerted anti-diabetic actions in this mouse model. Thus, apart from the kidneys, EGFR inhibition was also reported to preserve pancreatic function and enhance insulin levels in eNOS deficient db/db mice. Erlotinib also decreased fasting blood glucose levels along with increased glucose tolerance, insulin sensitivity and circulating levels of the adipokine, adiponectin-that has reno-protective and insulin sensitizing actions (Li et al., 2018b). Thus, this dual action upon EGFR inhibition might be particularly useful in DN. However, it should be noted that EGFR inhibition or deletion does not always lead to a lowering of blood glucose levels as observed in several other animal models (Benter et al., 2005a; Benter et al., 2005b; Akhtar et al., 2013; 2019; Benter et al., 2015; Xu et al., 2017; Li et al., 2021) despite a couple of case studies in human lung cancer patients have pointed to anti-diabetic effects with Erlotinib (Costa and Huberman, 2006; Brooks, 2012). The potentially beneficial action of EGFR inhibitors in diabetes, and specifically in DN, requires further confirmation and validation in large scale clinical studies in human subjects.
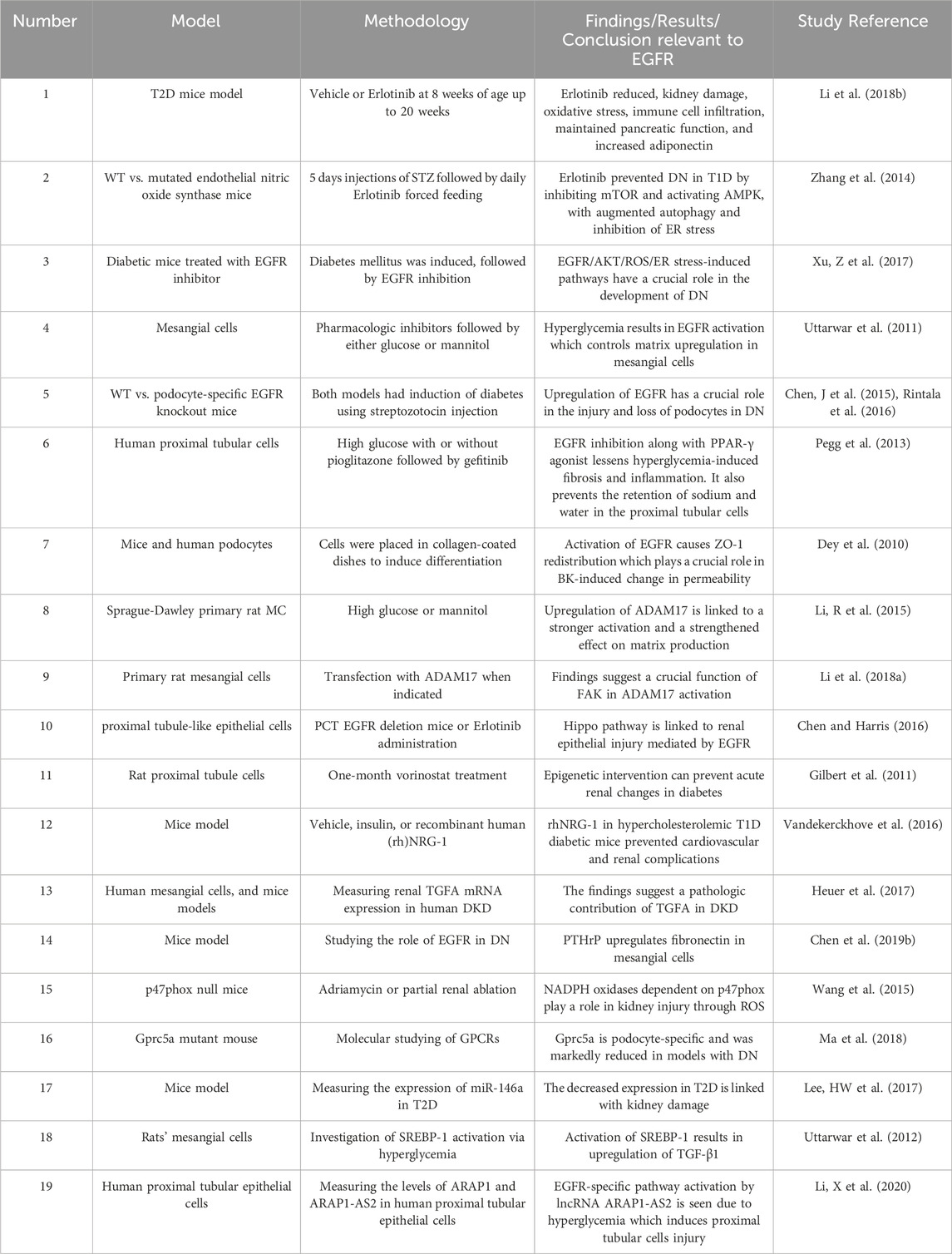
Table 2. Summary of selected studies highlighting the role of EGFR signalling in diabetic nephropathy.
In another study, Zhang et al. (2014) showed beneficial effects of Erlotinib treatment on the progression of DN in mouse models of T1D through attenuation of the albumin/creatinine ratio, reduced histological glomerular injury, reduced renal expression of connective tissue growth factor, and collagens I and IV. Furthermore, mice treated with Erlotinib had indications of enhanced renal autophagy which reduces the endoplasmic reticulum (ER) stress and thus renal tissue injury. Furthermore, treated mice also had proof of AMP-activated protein kinase activation which inhibits the mammalian target of rapamycin (mTOR) pathway, an important factor in the development of DN and an inhibitor of autophagy (Zhang et al., 2014). These findings were further supported by Xu et al. (2017) who showed that oxidative stress and ER stress were ameliorated following treatment with AG1478, a specific inhibitor of EGFR phosphorylation, in the same mouse model. In addition, pro-fibrotic genes such as TGF-β and collagen IV were shown to be decreased by AG1478. The intervention also reduced oxidative and ER stress that results from high glucose levels in renal mesangial SV40 cells. The results showed that EGFR/AKT/ROS/ER stress signaling is crucial in the development of DN (Xu et al., 2017).
Several mechanistic studies have aimed to provide insight into the likely underlying mechanisms by which activation of ErbBs and/or shedding of their ligands might lead to DN in vitro and in vivo. Indeed, several of the EGF-like ligands that can activate multiple members of the EGFR family of receptors are now thought to be involved in the pathogenesis of DN. For example, in mesangial cells exposed to hyperglycemia, general inhibition of matrix metalloproteases, MMPs, (known to be involved in EGF-like ligand shedding) or specifically of HB-EGF ligand inhibited the activation of both, EGFR and downstream AKT signaling (Uttarwar et al., 2011; Uttarwar et al., 2012). Another ligand that plays a role in multiple kidney pathologies, including DN, is TGF-α. Antibody-mediated blockade of TGF-α reduced prolonged EGFR signaling in renal tubular and mesangial cells (Heuer et al., 2017). This study implicated TGF-α as a pharmacological target that can play a role in treating kidney diseases. Thus, it appears that targeting the upstream mediators of EGFR transactivation (e.g., MMP-mediated HB-EGF shedding) or its direct ligands such as TGF-α could also represent a viable therapeutic approach for managing DN.
Another set of enzymes involved in the shedding of EGF-like ligands in the kidney are the family of ADAMs (a disintegrin and metalloproteases). Of these, ADAM 17 has been implicated in several renal pathologies including DN. For example, ADAM17-mediated shedding of epiregulin has been implicated in the beneficial actions of bradykinin (BK), whose levels are typically elevated upon clinical treatment of chronic kidney disease with angiotensin-converting enzyme inhibitors. BK decreases mouse podocyte permeability by acting on the tight junction protein zonula occludens-1 (ZO-1) (Dey et al., 2010). These actions of BK2 receptor appear to involve ADAM-17-dependent EGFR transactivation. By inhibiting the EGFR receptor, the ZO-1 rearrangement and BK-induced decrease in the permeability of podocytes was attenuated (Dey et al., 2010). In addition, ADAM17 mediates hyperglycemia-induced matrix production in mesangial cells (Li et al., 2015; Li et al., 2018a). Interestingly, this study showed that ADAM17 upregulation in conditions of hyperglycemia might be self-augmented via an EGFR/ADAM17 signaling loop (Li et al., 2015) and further implicated ADAM17 as a target in treating diabetic kidney disease (Li et al., 2015). In summary, the above studies provide evidence that ADAMs, in addition to EGFR, are potential therapeutic targets in DN and possibly other chronic kidney pathologies.
Interestingly, in contrast to other EGFR ligands discussed above, NRG-1 (neuregulin-1) has been reported to be reno-protective in a model of type 1 diabetes bearing a mild form of nephropathy with significant cardiovascular dysfunction (Vandekerckhove et al., 2016). Recombinant human neuregulin-1 (rhNRG-1) treatment provoked a systemic activation of both ErbB2 and ErbB4 receptors in the model hearts and kidneys. In addition to the multiple cardiovascular benefits, rhNRG-1 significantly reduced albuminuria, neutrophil gelatinase-associated lipocalin, and glomerular fibrosis. This ligand also inhibited the synthesis of collagen of mesangial cells in vitro, although it showed no effect on Ang II-induced vasoconstriction of glomerular arterioles (Vandekerckhove et al., 2016). The authors conceded that their model of nephropathy did not mimic the full hallmarks of the disease and thus, for a better understanding of the role of NRGs in DN, it would be necessary to confirm these findings in better models of DN that more closely resemble the characteristics of human DN.
Apart from increased production of EGF-like ligands in DN, it was found that in the glomeruli and tubules of patients with diabetic kidney disease, the parathyroid hormone-related protein (PTHrP) is also upregulated. Additionally, reactive oxygen species derived from nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) facilitate the activation of PTHrP (1–34)-induced Src kinase which activates the EGFR (Chen et al., 2019a). The subsequent sequelae result in excessive protein synthesis of fibronectin. PTHrP’s role in fibronectin upregulation adds to the efforts dedicated to treating glomerular sclerosis (Chen et al., 2019b). The main generators of reactive oxygen species (ROS) in the glomerulus (P47phox, NOX1, and NOX2) have now been implicated in the pathogenesis of many kidney diseases including DN. P47phox, that regulates the formation as well as the function of NOX1 and NOX2, has been shown to exacerbate DN (Sheng et al., 2021). Indeed, P47phox deletion safeguarded mice from developing glomerulosclerosis and albuminuria (Wang et al., 2015). These results showed that P47phox and its dependent NADPH oxidases could be potential targets to attenuate fibrotic disease through antioxidant therapy (Wang et al., 2015). Oxidative stress pathways were also implicated in a study on podocyte-specific EGFR knockout diabetic mice which exhibited markedly decreased albuminuria and podocyte loss when compared to their wild-type littermates (Chen et al., 2015). The study also showed that podocyte-specific EGFR knockout mice had reduced TGF-β1 expression, Smad2/3 phosphorylation, and glomerular fibronectin deposition. The alterations seen in the wild-type mice were attenuated by treatment with antioxidants or by preventing EGFR expression or activity. Therefore, it is likely that NOX-mediated EGFR signaling is essential in the downstream activation of pathways that facilitate podocyte injury and loss in DN (Chen et al., 2015).
A more recent study by Li et al. 2021 showed that selective podocyte EGFR deletion led to relative podocyte preservation and marked reduction in albuminuria and glomerulosclerosis, renal proinflammatory cytokine/chemokine expression, and decreased profibrotic and fibrotic components in nos3 −/−; db/db mice. Podocyte EGFR deletion led to decreased podocyte expression of the autophagy-regulator, rubicon ((RUN domain and cysteine-rich domain containing Beclin one-interacting protein) and was associated with increased podocyte autophagy activity. Therefore, activation of EGFR signaling in podocytes contributes to progression of DN at least in part by increasing rubicon expression resulting in subsequent attenuation of autophagy and podocyte injury.
Pegg et al. (2013) provided further evidence which demonstrated that using EGFR inhibitor with a peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist attenuates the fibrosis and inflammation resulting from hyperglycemia. In addition, it also counteracts the activation of the channels and transporters that play a role in the retention of sodium and water in human proximal tubular cells. These data shows that blocking EGFR, apart from limiting the tubulointerstitial pathology in DN, also reduces the retention of sodium and water noted in diabetics. This effect is further induced by the PPAR-γ activators. (Pegg et al., 2013).
A novel mechanism was postulated by Chen and Harris (2016), who showed that upregulation of Yes-associated protein (YAP) that is seen in diabetic patients which is EGFR-dependent is crucial in the upregulation and expression of the profibrotic factor CTGF and amphiregulin. For the first time, these findings demonstrated the interaction between EGFR signaling and the Hippo pathway which could be a crucial step in the pathogenesis of DN (Chen and Harris, 2016). Thus, further studies are necessary to evaluate whether targeting of the Hippo pathway alone or in combination with EGFR signaling could more effectively slow the progression of DN in patients.
The acetylation status of histone proteins can alter EGF-EGFR signaling in renal tubule cells. Gilbert et al. evaluated the effects of vorinostat, a histone deacetylase (HDAC) inhibitor, in attenuating the enlargement of the kidneys in diabetics with a focus on the EGF interaction with EGFR (Gilbert et al., 2011). In diabetic mice treated with vorinostat, the results showed a reduction in the proliferation of the tubular epithelial cells. Treatment with vorinostat daily for a month reduced the glomerular hypertrophy and renal growth, which the authors claimed was most likely due to downregulation of EGFR following blockade of epigentic changes in histone acetylation (Gilbert et al., 2011). These data suggest that modulation of epigenetic changes might prevent early renal changes in DN.
Several other therapeutic targets have been proposed for diabetic nephropathy. Among these is a novel molecular target, Gprc5a, which is only located within the glomerular podocytes and plays an essential role in healthy and diseased kidneys. In a link to EGFR, the stable overexpression of Gprc5a in glomerular podocytes was proven to inhibit EGFR activation (Ma et al., 2018). In addition to other effects like blunting the downstream signaling to the TGF-β pathway, it was concluded that this receptor can serve as a key therapeutic target in treating DN. However, this could be challenging as the drugability assessment showed several challenges that require dedicated investigation (Ma et al., 2018).
MicroRNAs (miRNAs), small non-coding RNAs that control gene expression by regulating target messenger RNA translation, have been implicated in several kidney diseases (Mahtal et al., 2022). As miRNAs are critical regulators of cellular homeostasis, their dysregulation is a crucial component of cell and organ injury (Mahtal et al., 2022). Of particular interest is microRNA-146a (miR-146a) that may regulate expression of ErbBs. The expression of this miRNA is increased in different cells in homeostatic conditions as it has a crucial anti-inflammatory role in myeloid cells. A study showed that the expression of this molecule in the glomeruli is decreased in patients with type 2 diabetes, which is linked with an increase in albuminuria and glomerular damage (Lee et al., 2017). miR-146a deficient mice were found to have faster progression of glomerular disease and albuminuria following induction of hyperglycemia using streptozotocin. In addition, the targets of miR-146a, Notch-1, and ErbB4 were found to be highly upregulated in the glomeruli of diabetic mice and patients (Lee et al., 2017). Consequently, it was shown that using an inhibitor of all ErbB kinase receptors significantly reduced the diabetic glomerular injury and albuminuria in wild-type as well as miR-146a deficient animal models (Lee et al., 2017). Also at the genetic level, ARAP1 is a gene associated with an increased risk for type 2 diabetes. This gene controls the internalization and ubiquitination of membrane receptors. In a recently published study, the antisense long non-coding RNA of this gene (ARAP1-AS2) and the gene itself were investigated to see their effect on the ubiquitination of EGFR in DN (Li et al., 2020). The results of the study showed that ARAP1-AS2 has the ability to interact with ARAP1. This interaction plays a crucial part in the hyperglycemia-induced injury and fibrosis of the proximal tubules by retaining chronic activation of the EGFR/TGF-β/Smad3 pathway (Li et al., 2020). The results showed that ARAP-AS2 and ARAP1 could be potential targets to treat DN.
Another transcription factor that was proven to have a role in DN is the sterol-responsive element-binding protein (SREBP)-1. A study showed that EGFR/PI3K/RhoA signaling is essential for hyperglycemia-induced SREBP-1 activation which then results in TGF- β1 upregulation (Uttarwar et al., 2012). The findings provided further evidence to support the importance of EGFR in DN and presented SREBP-1 as a potential target in treating DN (Uttarwar et al., 2011; Uttarwar et al., 2012).
Finally, diabetes-induced vascular remodeling and hemodynamic changes are also thought to contribute significantly to the development of renal pathologies including DN (Sheng et al., 2021). Chronic hyperglycemia-induced glomerular hyperfiltration arises from asymmetric alterations in arteriolar resistance with greater dilation of the efferent versus afferent arterioles that ultimately leads to increased transcapillary hydrostatic pressure (Sheng et al., 2021). This eventually leads to albuminuria. Many molecules, in particular the RAAS octapeptide, Ang Ⅱ, have been implicated in causing hyperfiltration/hyperperfusion. (Cooper, 2001; Wolf, 2004; Forbes et al., 2007; Sheng et al., 2021). Since Ang II mediates transactivation of EGFR, it has been suggested that inhibition of EGFR can reduce Ang Ⅱ-mediated hemodynamic alterations (Krishna et al., 2007; Akhtar et al., 2012; Shraim et al., 2021).
There is a growing body of evidence to suggest that EGFR and related family members are key mediators of diabetes-induced vascular dysfunction (for a recent review see Shraim et al., 2021). Indeed, several in vivo studies have now shown that treatment with EGFR inhibitors, mostly without correcting blood sugar levels, can markedly normalize the altered vasoconstrictor and vasodilator responses typically observed in the diabetic vasculature including in the renal artery (Benter et al., 2005a; Benter et al., 2005b; Yousif et al., 2005; Benter et al., 2009a; Akhtar et al., 2012; Akhtar et al., 2013; Schreier et al., 2014; Akhtar et al., 2019). Mechanistically, upregulation of EGFR signaling appears to be a key early vascular change induced by diabetes as EGFR inhibition was able to correct more that 90% of all diabetes-induced gene expressions in the diabetic vasculature (Benter et al., 2009b). Hyperglycemia-induced upregulation of EGFR receptors likely includes other family members especially ErbB2 (Akhtar et al., 2013; Akhtar et al., 2015; Benter et al., 2015). Use of selective inhibitors of EGFR and/other ErbBs were able to reverse multiple diabetes-induced signaling changes including oxidative stress signaling, inflammatory markers, nitric oxide (NO) synthase (eNOS) activity and NO production in the vasculature (Benter et al., 2015; Kassan et al., 2015). In addition to EGFR/ErbBs mediating diabetes-induced vascular dysfunction, EGFR has also been directly linked to diabetes-induced vascular remodeling (Akhtar et al., 2019). Chronic EGFR inhibition resulted in a significant reduction in diabetes-induced blood vessel intima and media thickening and correction in vascular hyper-responsiveness through EGFR-ERK1/2-ROCK dependent pathway (Akhtar et al., 2019). Thus, therapeutic targeting of one or more EGFR/ErbB receptors might be useful in correcting diabetes-induced renal vascular dysfunction as well as for reversing the underlying morphological changes and alterations in key renal cell signaling cascades typically associated with DN.
In summary, the above studies suggest that a complex cascade of signaling networks involving multiple signaling effectors both upstream and downstream of EGFR appear to be involved in the development of DN through modulation of hemodynamic, inflammatory, fibrosis, and oxidative stress pathways amongst others. Importantly, inhibition of EGFR seems sufficient to correct many of the hyperglycemia/diabetes-induced hemodynamic, morphological, and renal cell signaling changes associated with DN.
4 Hypertensive nephropathy
After diabetes, hypertension is believed to be the second or third commonest cause of chronic kidney disease as well as renal replacement therapy and poses a significant burden on healthcare globally (Carriazo et al., 2020; Costantino et al., 2021; Wang et al., 2024). Despite the strong association of hypertension with nephropathy, hypertensive nephropathy does not have a clear enough definition to allow diagnosis other than by exclusion (Carriazo et al., 2020). In the absence of uniform criteria to accurately diagnose hypertensive nephropathy, the prevalence of chronic kidney disease resulting from hypertension is therefore difficult to quantify with any precision (Udani et al., 2011; Carriazo et al., 2020). Thus, there appears to be an important need to come up with a clear definition of hypertensive nephropathy with a comprehensive set of criteria to be able to exclude other nephropathies (Carriazo et al., 2020).
The etiology of hypertensive nephropathy appears complex, but key pathological features involve arteriolar nephrosclerosis, a vascular disease characterized by progressive intimal thickening of small arterioles due to persistent elevation in blood pressure, followed by renal parenchymal damage and ischemic/hypoxia-mediated glomerulosclerosis (Freedman and Cohen, 2016; Costantino et al., 2021). Renal microvasculature dysfunction and the associated local ischemia/hypoxia culminates in signaling changes leading to renal inflammation and fibrosis. Additionally, chronic elevated systemic blood pressure causes renal tubular cell injury as well as podocyte effacement and loss, leading to disruption of the renal filtration barrier and microalbuminuria (Costantino et al., 2021), though clinical symptoms often appear much later than the underlying cell signaling and histopathological changes (Sun, 2019; Kao and Huang, 2021).
Several key signaling effectors including RAAS peptides may be involved (Sun, 2019), but recent evidence implicates EGFR as a key player in the pathology of hypertensive nephropathy (Benter et al., 2009a; Tang et al., 2013b; Staruschenko et al., 2013) ((for a summary of selected studies see also Table 3)). We and others have shown that in animal models of hypertension, expression of EGFR is elevated in kidneys (François et al., 2004; Benter et al., 2009a) including in the renal vasculature, such as the media of afferent and efferent renal arterioles (Swaminathan et al., 1996; Ying and Sanders, 2005). In deoxycortisone acetate (DOCA) salt-induced hypertensive mice, early treatment with a selective inhibitor of EGFR was able to prevent changes in more than 97% of the approximately 2,400 genes upregulated in the hypertensive kidney (Benter et al., 2009a). EGFR inhibition with AG1478 concomitantly reduced proteinuria, blood pressure, and histological changes indicative of kidney damage in this mouse model, (Benter et al., 2009a), implying that EGFR is a key early initiator step in the development of hypertension-induced renal dysfunction in this model. Further, it suggested that many of the upregulated genes associated with DOCA–salt hypertensive kidneys are likely downstream of EGFR signaling and thus, highlighting this important receptor tyrosine kinase as a potentially viable pharmacological drug target. In support of this notion, gefitinib -a clinically used EGFR inhibitor-was also reported to counter the progression of glomerular and vascular fibrosis in this model. In addition, inhibition of EGFR also reduced vasoconstriction and improved blood pressure in models of hypertension (Benter et al., 2009a; Tang et al., 2013b). It is known that EGFR expression is elevated in the vascular smooth muscle cells from hypertensive rats and in atherosclerotic plaques that may have an impact on hypertension (Rayego-Mateos et al., 2018b). Thus, further studies are needed to ascertain the relative contribution of EGFR in mediating hypertension versus its direct effects on kidney damage per se.
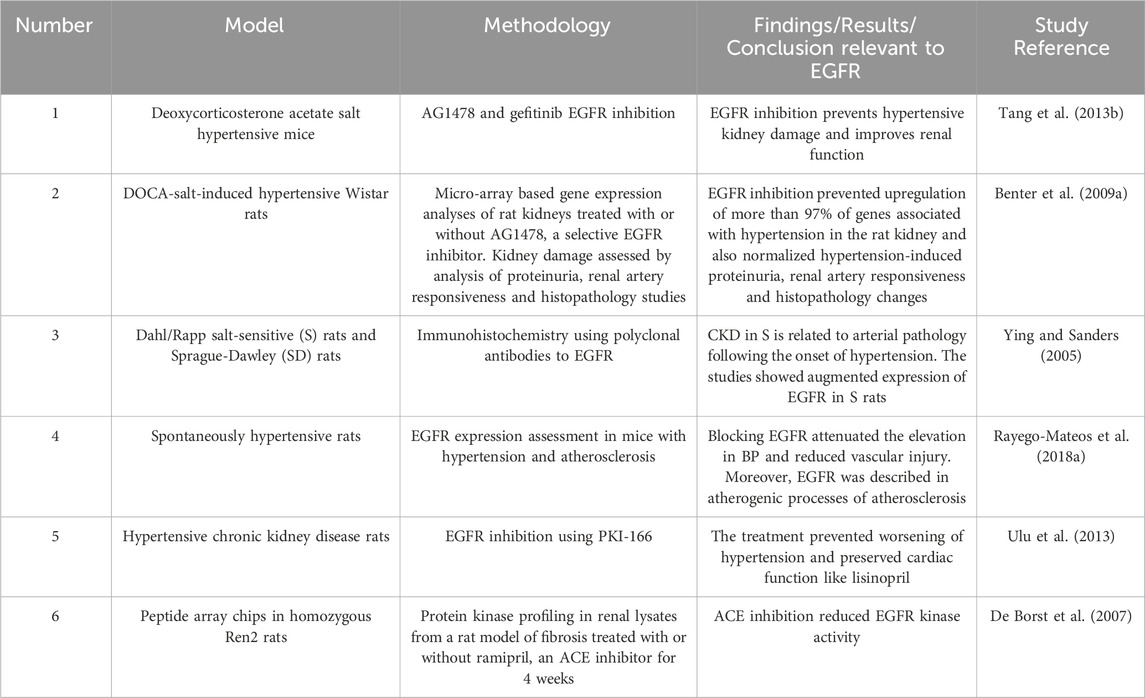
Table 3. Summary of selected studies highlighting the role of EGFR signalling in hypertensive nephropathy.
5 Acute kidney injury
Acute kidney injury (AKI) is the abrupt reduction in kidney function in a few hours, which includes structural injury and functional impairment (Makris and Spanou, 2016; Nourie et al., 2024). Even though there is scarcity of data, it is believed that the prevalence of AKI goes up to a little over 13 million cases per year (Ponce and Balbi, 2016). The prevalence is estimated to range from <1% to 66%, where this variability is explained by the inconsistent use of standardized AKI classification criteria (Hoste et al., 2018). AKI can develop in different clinical situations including sepsis, ischemia, and nephrotoxicity. Animal studies of AKI have illustrated that following an AKI, the kidney has the capacity of recovering from the insult. The regeneration process includes the dedifferentiation, proliferation, and migration and ends with redifferentiation into mature tubular cells. It is thought that this regeneration process occurs through the affected renal tubular epithelial cells (RTECs) undergoing differentiation to a mesenchymal stage by reactivating pathways which are common to the early development of the kidneys (Cirio et al., 2014). Different cellular molecules like vimentin and neural cell adhesion molecule 1 (NCAM1) which are no longer expressed after the metanephric mesenchyme stage and kidney maturation are reexpressed during the recovery phase in renal tubular cells. Other molecules such as Pax-2 are markedly expressed in the surviving tubular epithelium although it is not found in adult kidneys. This indicates the presence of cells with immature characteristics. Similarly, another marker of dedifferentiation, Sox9 was elevated up to 30 days following an injury. Moreover, nestin, which is an intermediate filament was proposed as an indicator of dedifferentiation (Andrianova et al., 2019). These findings suggest that renal regeneration and recovery after acute injury share lots of similarities with the early development of the kidneys. During development, dedifferentiation, high proliferation, and regulation by different growth factors such as the EGFR ligands are noted on the renal mesenchymal cells. Experiments showed that the EGFR ligands EGF, TGF-α, epiregulin, and HB-EGF, are upregulated in metanephric structures and play a role in tubulogenesis and breaching (Zeng et al., 2009). Thus, EGFR signaling is thought to be a major regulator of renal development, the proliferative capability of mature proximal tubule cells and in mediating recovery from acute kidney injury (for a summary of selected studies see also Table 4). Increased phosphorylation of the EGFR was noted in the proximal tubular cells in different experimental models of AKI, including hypoperfusion/reperfusion, aminoglycoside toxicity, and administration of folic acid (He et al., 2013; Gao et al., 2020). Further, EGFR/FOXM1 signaling is known to induce proliferative repair after kidney injury (Chang-Panesso et al., 2019).
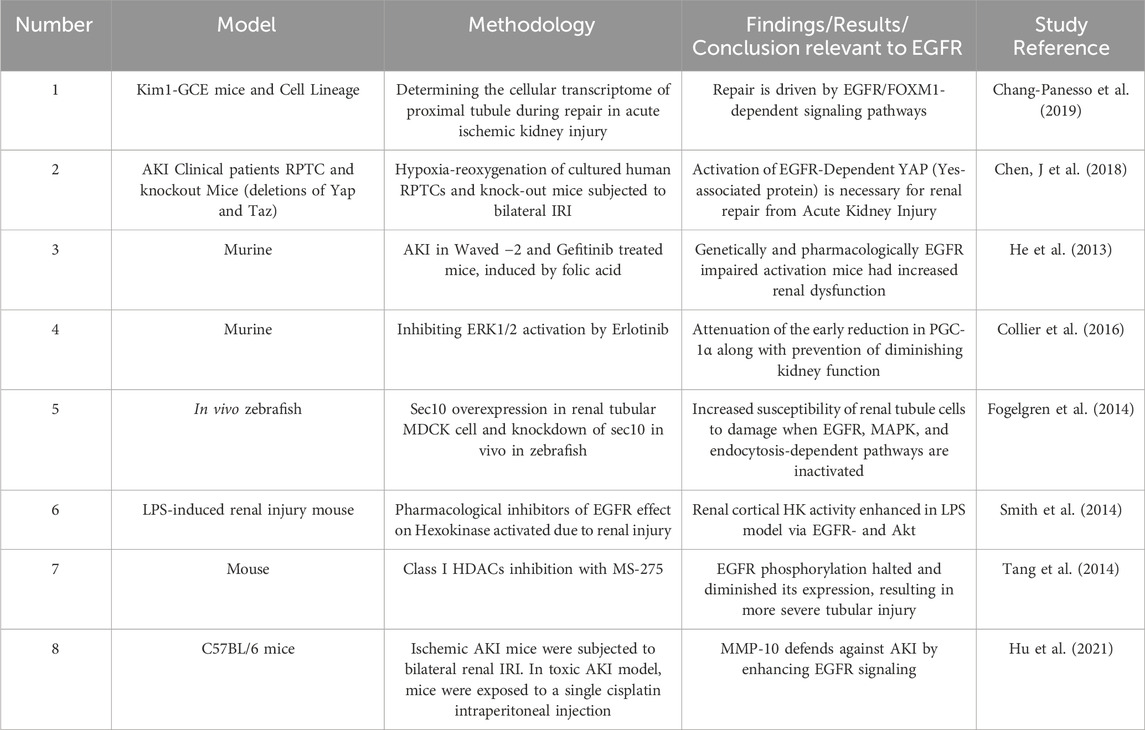
Table 4. Summary of selected studies highlighting the role of EGFR signalling in acute kidney injury (AKI).
While growing evidence shows that renal recovery from AKI is a result of dedifferentiation and proliferation of surviving tubular epithelial cells, two pathways seem to have a crucial role in the proliferation and differentiation process, namely, the EGF receptor (EGFR) and the Hippo signaling pathway (Harris, 2021). Several articles have proven that the activation of EGFR in renal proximal tubule epithelial cells (RPTCs) had a major role in preventing ischemia-reperfusion injury (IRI). It was proven that EGFR-Dependent YAP (Yes-associated protein) upregulation is necessary for renal recovery from AKI (Chen, J et al., 2018). These results come in line with another study that supported the evidence that activation of EGFR is critical for the onset of dedifferentiation and proliferation of the renal tubular cells after inducing acute kidney injury by folic acid (He et al., 2013). This was further supported by another model which provided genetic and pharmacologic proof that activation of EGFR in the proximal tubular cells is crucial in the recovery from AKI, by generating mice with EGFR mutation in RPTCs (Chen et al., 2012). Ischemia-reperfusion induced AKI resulted in significant activation of EGFR in control mice, unlike the genetically mutated (i.e., knockout) mice or the wild-type which were treated by Erlotinib which had no functional EGFR. It was noted that kidney injury was reduced in the control mice compared to the knockout and Erlotinib-treated mice which had chronic dilation of the proximal tubules, epithelial simplification, as well as cast formation. In addition, the proliferation of the renal cells was slowed because of decreased ERK and AKT signaling (Chen et al., 2012).
Regarding the precise signaling mechanisms in the kidney, phosphorylation of EGFR was noted as an upstream activator of ERK1/2 (extracellular signal-regulated kinase), through negative regulation of PGC-1α transcription in normal physiological conditions. However, under the abnormal/pathological state of renal ischemia/reperfusion-induced AKI in mice, the activation of ERK1/2 resulted in a decrease in PGC-1α transcription and a decline in kidney function as indicated by serum creatinine level. Through inhibition of EGFR-ERK1/2, Erlinotib treatment halted the initial reduction in PGC-1α and stopped the decline in kidney function (Collier et al., 2016). Furthermore, a study in renal tubular MDCK cells showed that the mechanism by which the overexpression of exocyst Sec10, which is a highly conserved eight-protein complex that regulates protein trafficking, increases phosphorylation of ERK and healing from oxidative injury is EGFR, MAPK, and endocytosis dependent (Fogelgren et al., 2014), thereby highlighting the potential use of exocyst Sec10 as a therapeutic target in AKI therapy.
Many previous reports have indicated that glycolysis provides an alternative way of providing energy following tubular injury, which under normal conditions, uses oxidative phosphorylation for ATP generation with low glycolytic capacity. To investigate if glycolysis occurs in AKI, LPS-induced renal injury in a mouse model indicated a particular acute increase in hexokinase (HK) function. This activation was halted by pharmacological inhibition of EGFR, showing that LPS increases HK function in the renal cortex occurs via EGFR-dependent mechanisms (Smith et al., 2014).
Although the fact that histone deacetylases (HDACs) activation is crucial for the proliferation of renal epithelial cells and the development of the kidney, it remained unclear whether they play a role following an AKI. To shed light on their significance, a study inhibited class I HDACs which showed suppression of EGFR phosphorylation as well as a reduction in its expression, suggesting that this class protects the kidney, helps in regaining kidney function and is necessary for renal regeneration after acute kidney injury (Tang et al., 2014).
Matrix metalloproteinase-10 is a zinc-dependent endopeptidase that plays a role in various biological processes. It is upregulated in the tubular epithelium in models of AKI provoked by ischemia, reperfusion, or pharmacologically through cisplatin. The increased expression of exogenous MMP-10 improved AKI, while the knockdown of endogenous MMP-10 worsened AKT. Both findings resulted from activating and inhibiting EGFR signaling pathways respectively, that modulated the downstream AKT and extracellular signal-regulated kinase-1 and 2 (ERK1/2) signaling (Hu et al., 2021).
Another molecule is Lysophosphatidic acid (LPA) which raises the production of platelet-derived growth factor-B (PDGFB) and connective tissue growth factor (CTGF) by proximal tubular (PT) cells via LPA2 receptor-Gqα-αvβ6- activation of transforming growth factor-β1 (TGFB1) mediated by integrin. These secretions increase after ischemia-reperfusion injury to the kidneys, paving the way for kidney fibrosis (Geng et al., 2021). Recent findings prove that LPA1 receptor signaling via EGFR-ERK1/2-activator protein-1 conjoins with LPA2-dependent TGFB1 signaling and increases PDGFB/CTGF formation and secretion by PT cells (Geng et al., 2021). Therefore, it is proposed that antagonists of the TGFB1 receptor, LPA1, and LPA2 inhibitors can be used in AKI to prevent the development of CKD (Geng et al., 2021) (see also Table 4).
In summary, consistent with its ‘Jekyl and Hyde’ role, EGFR signaling network is needed for recovery and repair of kidneys following AKI but prolonged EGFR activation can lead to unwanted pathologies such as renal fibrogenesis post-AKI (Tang et al., 2013a; Tang et al., 2013b).
6 Chronic kidney disease and its progression from AKI
Chronic kidney disease (CKD) is a description of many different diseases that disturb the kidneys’ structure and function (Levey and Coresh, 2012; Yeh et al., 2024). CKD is diagnosed when there is evidence of kidney damage or reduced function based on the glomerular filtration rate (below 60 mL/min) for at least 3 months, regardless of the clinical diagnosis. The incidence of CKD goes up to 200 per million per year in many countries. The average survival in the USA is 3–5 years, and the prevalence is approximately 1800 cases per million. The higher prevalence is noted in countries with high survival like Japan and Taiwan, with around 2,400 cases per million. CKD is most commonly caused by diabetes, and the disease has some genetic and environmental factors that contribute to its development (Levey and Coresh, 2012).
Multiple research articles have linked EGFR with CKD (for a summary of selected studies see also Table 5). Activation of EGFR results in several cellular responses that depend on the ligand that is bound to it. A major ligand of interest in relevance to CKD is CTGF/CCN2 which when bound to EGFR controls renal inflammation, cellular growth, and fibrosis. In addition, other ligands are also linked to kidney disease, including Ang II, TGF-β, and cytokines, including members of the TNF superfamily (Rayego-Mateos et al., 2018b). Regarding the therapeutic approach, CTGF is being considered as a target because of its pro-inflammatory and fibrotic properties, however, further evidence is required to recognize the consequence of its binding to EGFR. Nevertheless, it is proven that EGFR and some of its ligands could be targeted in treating CKD (Rayego-Mateos et al., 2018b).
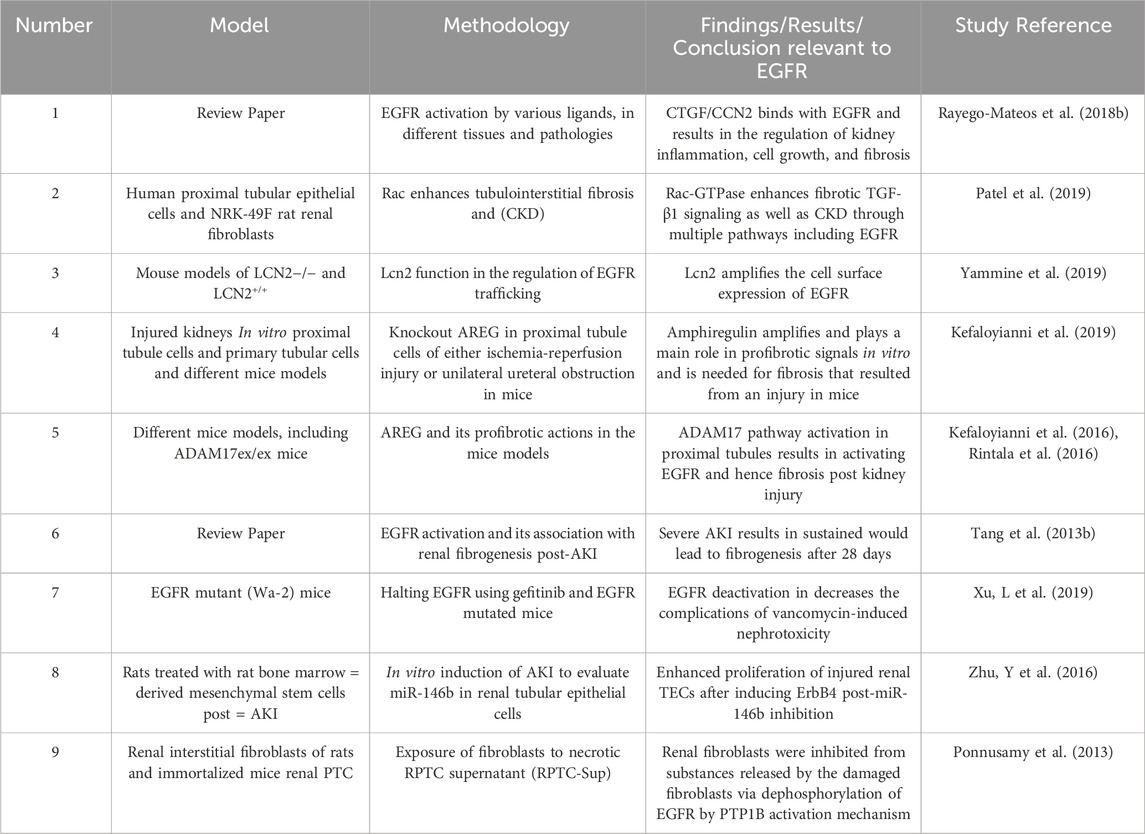
Table 5. Summary of selected studies highlighting the role of EGFR signalling in Chronic Kidney Disease.
In addition, recent evidence has shown that EGFR activation was also linked to Rac-GTPases which are important regulators of cytoskeletal remodeling (Patel et al., 2019). The deregulation of Rac-GTPases is a main contributor to numerous pathologies. In the context of CKD, Rac-GTPase activates the EGFR, p53, and Hippo/YAZ/TAZ pathways which results in TGF- β1 signaling that promotes fibrosis and CKD. Inhibition of the Rac pathway inhibition was shown to suppress renal free radical damage and maladaptive recovery which made it a new target in treating CKD (Patel et al., 2019).
Another cellular molecule that is linked to EGFR activation and is observed in several pathologies is lipocalin-2 (Lcn2). It was reported that this molecule causes an increased expression of EGFR on the cell surface which is needed for TGF α-induced EGFR salvaging to the plasma membrane and constant activation (Yammine et al., 2019). Lcn2 does this function by inhibiting the lysosomal degradation of the intracellular domain of EGFR. The inhibition of Lcn2 gene activation prevents EGFR recycling and expression in a trial model of CKD. The inhibition resulted in a significant reduction of kidney damage which identifies Lcn2 as a major target to offset the damaging outcomes of EGFR signaling in CKD (Yammine et al., 2019).
Although research has shown that the activity of EGFR is important in the acute phase of recovery in AKI (day 1–2 after injury), the constant activation beyond this phase of days to weeks after injury was found to be associated with kidney fibrosis in experimental models of bilateral ischemic AKI and unilateral ureteral obstruction (UUO). To identify which ligands are responsible for the chronic activation of EGFR and consequent fibrosis, it was found that amphiregulin, a low-affinity ligand for EGFR whose expression is increased following an AKI, drives, amplifies, and integrates profibrotic signals in the proximal tubule cells and is crucial and adequate for AKI-induced fibrosis in mice in vivo (Kefaloyianni et al., 2019).
These findings also correlate with another study which reported that ADAM17 pathway upregulation and activation in the proximal tubule results in constant activation of EGFR and fibrosis following different types of kidney injury including ischemia and ureteral obstruction. This occurs through different cellular mechanisms including the EGFR ligand substrate pro-amphiregulin and EGFR-TNFα interaction (Kefaloyianni et al., 2016; Harris, 2021). Similar studies supported this proposal, of which Waved-2 (Wa-2) mice with EGFR reduced activity were compared to normal mice following ischemic renal injury as an exposure. The results showed that severe kidney damage causes prolonged activation of EGFR which is essential for the recovery of renal tubular cells in the initial phase (the first 2 days), but this sustained EGFR activation would lead to fibrogenesis after 28 days (Tang et al., 2013a).
To study the mechanism of the progression of AKI to CKD induced by vancomycin, a model of a mutated EGFR (Wa-2) mice and Gefitinib were used to deactivate EGFR. It was found that AKI caused by vancomycin was significantly reduced in waved-2 mice in the acute phase. While the latter stage ameliorated the renal fibrosis and inflammation. Hence, the study highlighted the significance of EGFR deactivation in decreasing the complications of vancomycin-induced nephrotoxicity (Xu et al., 2019). To further investigate the role of drug-induced AKI, cisplatin-induced acute kidney injury mice treated with rat bone marrow mesenchymal stem cells have been studied. It was found that the expression of microRNA-146b (miR-146b) was increased in AKI rats as opposed to healthy counterparts. In addition, the expression lessened after mesenchymal stem cell therapy. The use of cisplatin in vitro augmented the expression of miR-146b renal TECs. This all comes into significance when it was identified that ErbB4 is targeted by miR-146b, its inhibition resulted in an increase in the expression of ErbB4, causing an enhancement in the proliferation of injured renal TECs. Furthermore, this restoration process via rMSCs can be controlled by downregulating ErbB4. Thus, indicating that inhibition of miR-146b is a potential therapeutic option for acute kidney injury (Zhu et al., 2016). In line with this, a study illustrated that products released by injured epithelial cells have the ability to halt renal fibroblasts through dephosphorylation of EGFR by a mechanism involving activation of PTP1B. Given the importance of peritubular fibroblasts in preserving the structural and functional integrity of the kidneys, their inactivation through damage via any insult will impair the process of regeneration of renal epithelium following an AKI (Ponnusamy et al., 2013).
7 Renal transplant
Renal transplant is the management option for end-stage renal disease because of its survival benefits in the short and long term compared to dialysis (Kälble et al., 2005; Goto et al., 2024; Jørgensen et al., 2024). The incidence of CKD that is treated by transplantation is limited depending on the difference in the underlying disease rates and the availability of government-sponsored treatment (Levey and Coresh, 2012). The transplanted kidney survival rates are 95% when taken from a living donor and 89% from a deceased donor during the first year after the transplant. A successful renal transplant is influenced by allograft rejection and serious infections (Shams et al., 2017; Sawinski and Blumberg, 2019). Proper preventative measures and prophylaxis are essential to guarantee successful transplantation.
To date, the mechanisms of renal allograft rejection are not fully understood. However, polymorphisms in EGFR may be implicated. For example, Kim et al. 2020 showed that specific single nucleotide polymorphisms (SNPs) displayed a significant association with acute renal allograft rejection in Korean patients. The study involved 347 patients who received their first renal transplant following end-stage renal disease, out of which 63 had acute renal allograft rejection. Among the results, it was found that the SNP (rs11568835) in the epidermal growth factor gene had a significant association with the susceptibility to allograft rejection. In addition, (rs1050171) SNP in the EGFR showed a significant association with susceptibility to allograft rejection as well (Kim et al., 2020).
An additional study looked at the expression of genes using records of allograft biopsies and blood, and it comprised patients experiencing rejection as well as patients with steady kidney function. Based on the biopsy samples, the researchers found out that PIK3R2 and EGFR are the major signaling molecules in inflammatory-immune pathways (Meng et al., 2018). Other studies have proposed the use of TGF-β1, AGT, and EGFR mRNA levels in urine samples of KTP as timely indicators of chronic allograft nephropathy (CAN) development (Mas et al., 2007a; Mas et al., 2007b). However, more research is needed to verify the clinical importance of these findings (See also Table 6 for a summary).

Table 6. Summary of selected studies highlighting the role of EGFR signalling in Renal transplant.
8 Renal fibrosis
The common result in end-stage renal disease is renal fibrosis (chronic kidney disease), where renal fibroblasts abnormally activate and grow, along with overproduction of extracellular matrix proteins (Huang et al., 2023; Nørregaard et al., 2023). Around half of the adults above age 70% and 10% of the world’s population develop the outcomes CKD and renal fibrosis, with the incidence increasing worldwide (Huang et al., 2023; Nørregaard et al., 2023). After AKI, tissue repair is able to return the integrity of the damaged tissue only when the injury is mild. However, if the injury was severe, it could lead to renal fibrosis that is known to occur via sustained EGFR activation has been linked to the development of renal fibrosis (Cao et al., 2023; Tang, et al., 2013a; Xu et al., 2019).
Expression of EGFR was elevated in interstitial myofibroblasts in human and mouse fibrotic kidneys and its selective deletion in the fibroblast/pericyte cells inhibited interstitial fibrosis resulting from unilateral ureteral obstruction, ischemia or nephrotoxins (Cao et al., 2023). Additionally, increased HER2 expression in unilateral ureteral obstruction-induced renal fibrosis, type 1 and type 2 diabetic nephropathy, and in kidney biopsies from patients with renal fibrosis has also been reported (Li et al., 2019).
Furthermore, in a mice model study, the progression of chronic kidney disease in rats with remnant kidneys was found to be attenuated using Erlotinib (Yamamoto et al., 2018). An EGFR mimotope (that prevents downstream signaling) alleviated renal fibrosis in the murine unilateral ureteral obstruction model (Yang et al., 2019). Renal fibrosis in Ang II-stimulated mice was also prevented using a new EGFR inhibitor (Skibba et al., 2016). Similarly, supportive of the crucial role that EGFR plays in renal fibrosis induced by Ang II, Ang II-treated SV40 mesangial cells in vitro were administered novel small molecules or short hairpin RNA as inhibitors of EGFR, that resulted in preventing the accumulation of connective tissue growth factor (CTGF) and collagen IV, known fibrotic markers (Qian et al., 2016). Consistent with this, it was demonstrated in vivo that proteinase-activated receptor-2 (PAR2) activation in kidney tubular epithelial cells transactivates the EGF and TGFβ receptors. This in turn resulted in Smad2 activation and production of the pro-fibrotic factors and CTGF, all of which contribute to renal fibrosis (Chung et al., 2013).
Supported by murine model evidence, it was concluded that tissue fibrosis resulting from aldosterone depends on ROS-induced EGFR/ERK activation, thus making EGFR a possible point for controlling renal fibrosis (Sheng et al., 2016). Likewise, continuous activation of EGFR by expressing the EGFR ligand human heparin-binding EGF-like growth factor (hHB-EGF) in the proximal tubule epithelium of a mouse model kidney gave rise to spontaneous, progressive renal tubulointerstitial fibrosis (Overstreet et al., 2017). To study the methods of albumin-induced renal fibrosis, it was observed that EGFR has a significant role in albumin-induced fibrotic events, as inhibition of γ-secretase activity played the major role, depending on ERK-MAPK (Slattery et al., 2013). The ligands responsible for this sustained EGFR activation were suggested to be Src mediated, which is needed in the stimulation of renal interstitial fibroblasts and gene expression of numerous profibrogenic cytokines, such as TNF-b1 (TGF-b1) (Tang et al., 2013b). To evaluate the role of Src, PP1 (a selective inhibitor of Src) was introduced into cultured renal interstitial fibroblasts (NRK-49F). PPI treatment inhibited stimulation of TGF-β1 signaling, activation of EGFR and STAT3, and decreased amount of renal epithelial cells arrested at the G2/M phase of the cell cycle after ureteral obstruction (Yan et al., 2016). To support this, it was shown that renal fibrosis was decreased when Fyn (a member of the Src family of kinases) deficiency was implemented by the inhibition of phospho-STAT3 (Seo et al., 2016). Another reason for this sustained EGFR activation could be the excessive expression of connective tissue growth factor (CCN2) in fibrotic areas. It was shown that upon administration of CCN2, the fibrotic response in the kidney of a murine model was diminished by EGFR blockade, indicating that blocking EGFR could be a promising treatment option to halt the profibrotic effects of CCN2 in renal pathologies (Rayego-Mateos et al., 2018a, 2018b).
HB-EGF, another ligand for EGFR, is likely also involved in renal fibrosis. Reduced renal fibrosis was noticed when endothelial HB-EGF expression was deleted, by diminishing Ang II infusion-related renal damage (Zeng et al., 2016). It is well known that ErbB4 expression increases in CKD. A study reported that a novel long non-coding RNA (lncRNA) called Erbb4-IR is accountable for renal fibrosis that is mediated by TGF-β/Smad3, making it a selective therapeutic target for CKD (Feng et al., 2018). Supporting this, it was also found that by suppressing miR-29b, renal fibrosis was enhanced in T2DN by Erbb4-IR through a Smad3-dependent lncRNA (Sun et al., 2018).
It has been implicated that oxidative stress could lead to CKD. While studying the possible underlying mechanisms for this, it was found that CKD-bearing mice given Tempol (a Superoxide Dismutase-Mimetic) exhibited reduced renal fibrosis along with decreased signaling via TGF-ß/Smad3, EGFR and MAPK pathways (Ding et al., 2015). Furthermore, exploring other possible mechanisms, it was found that EGFR activation, p53, and ROS are all needed in the initiation of renal fibrotic genes by TGF-β1, confirming the role of EGFR in renal fibrosis (Samarakoon et al., 2013).
Studying the precise EGFR signaling cascades, it was proven that mice lacking ADAM17 in smooth muscle cells had transient protective effects from renal fibrosis (Shen et al., 2017). These results come in line with those that indicated that the hepatic stellate cell (HSC) transactivation of Ang II-induced EGFR is mediated through ADAM17, which could lead to the progression of liver fibrosis (Oikawa et al., 2014). Histone deacetylases classes (HDAC) 1 and 2 were comprehensively investigated for their part in the stimulation of fibroblasts and the pathogenesis of numerous chronic diseases, including renal fibrosis (Marchion and Münster, 2007; Liu et al., 2013; Wang et al., 2022). Furthermore, it was found that renal fibrosis improved along with attenuation of renal fibroblast activation by manipulating TGB-beta and EGFR signaling through blocking class I histone deacetylase (Liu et al., 2013). Blocking Sirtuin-1 and 2 (Class III HDAC) prevented renal interstitial fibroblast activation and halted renal interstitial fibrosis in obstructive nephropathy through inhibition of EGFR and PDGFRβ signaling (Ponnusamy et al., 2014). A recent study of a unilateral ureteral obstruction (UUO) mouse, the expression of an E3 ubiquitin ligase called HUWE1 and EGFR was examined. Importantly, the overexpression of HUWE1 decreased the expression of EGFR, by physically interacting with the receptor and promoting its ubiquitination and degradation, hence regulating the expression of EGFR, and protecting against kidney injury (Zhu et al., 2020).
Inhibiting AREG, a ligand of EGFR, by AREG-targeting Self-Assembled-Micelle inhibitory RNA (SAMiRNA-AREG) in animal models of renal fibrosis resulted in decreased expression of fibrotic markers, including α-smooth muscle actin, fibronectin, α1(I) collagen, and α1(III) collagen. Remarkably, reduced renal fibrosis was accompanied by decreased EGFR phosphorylation (Son et al., 2021). The aforementioned studies are therefore suggestive of an important role of EGFR in mediating renal fibrosis and its selective inhibiton might represent a viable therapeutic strategy (for a summary of selected studies see also Table 7).
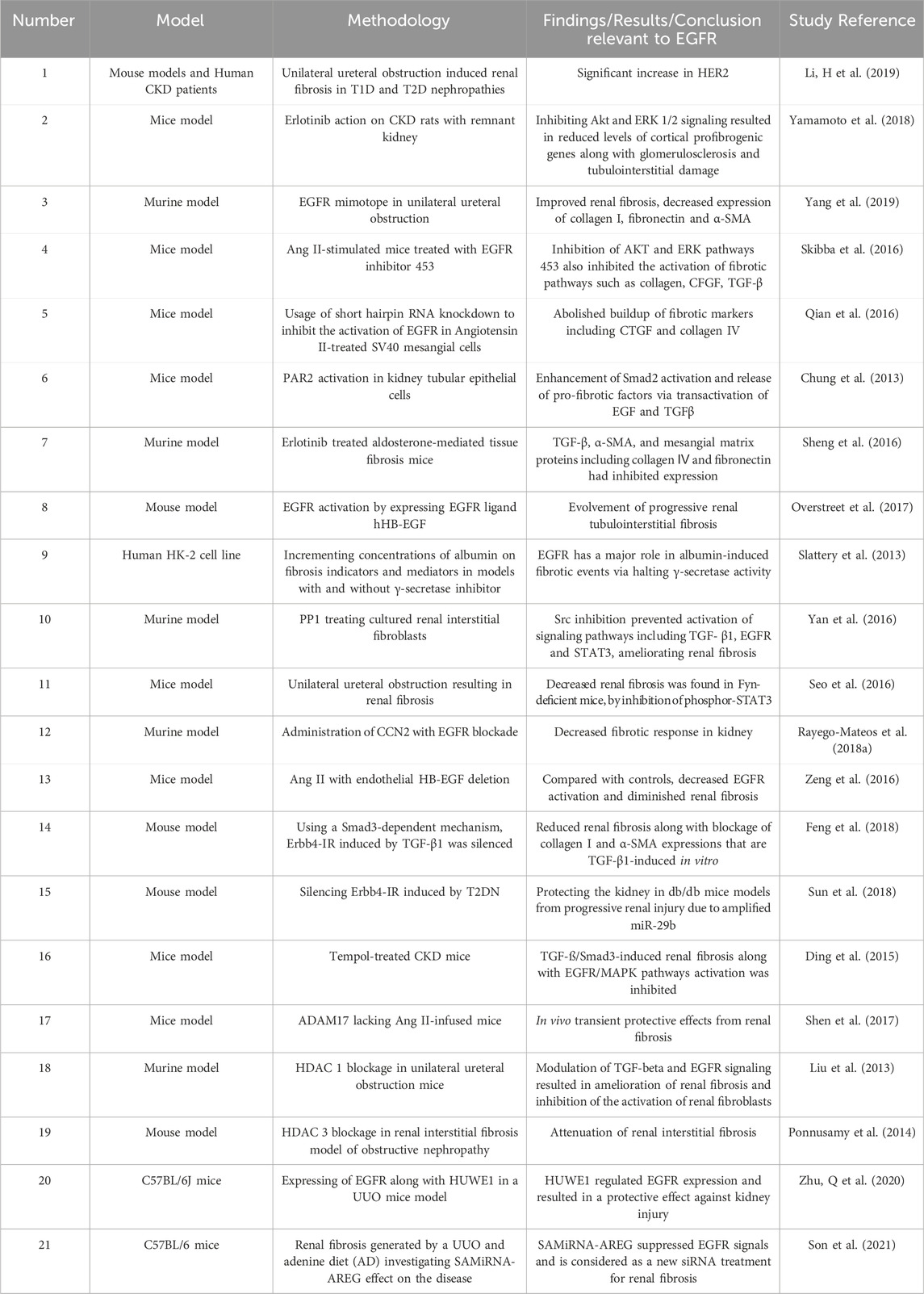
Table 7. Summary of selected studies highlighting the role of EGFR signalling in renal fibrosis.
9 Potential cross talk between EGFR/ErbB family of receptors and Ang-(1–7) in renal pathologies
The renin-angiotensin-aldosterone system (RAAS) plays a key role in regulating cardiovascular homeostasis. For many years Ang II which is a pro-hypertensive peptide that is known to have inflammatory and fibrotic effects was regarded as the only active member of the RAS. However recent studies have shown that Angiotensin-(1–7) [Ang-(1–7)] is also an active part of RAAS and produces effects that are opposite to those of Ang II (Benter et al., 1993; Benter et al., 1995a; Benter et al., 1995b). Today, it is well accepted that RAAS has two opposing arms with their relative balance determining the precise physiological and pathological consequences of RAAS (Benter et al., 2006; Benter et al., 2007; Ferrario et al., 2010; Ferrario et al., 2005; Li et al., 2017; Bader et al., 2024; Ye et al., 2004).
Activation of the ACE-Ang II-AT1 receptor arm produces pro-inflammatory, pro-oxidative stress, and pro-fibrotic signals leading to cardiovascular disorders. These pathophysiological effects are known, in part, to occur via Ang II mediated transactivation of EGFR and/or other ErbBs (Lautrette et al., 2005; Akhtar et al., 2012; Akhtar et al., 2015; Forrester et al., 2018; Akhtar et al., 2020; Shraim et al., 2021). Overactivity of this arm has been implicated in various pathologies, including hypertension, cardiovascular diseases, diabetes-induced complications, respiratory and renal diseases (Ferrario et al., 2010; Ferrario and Varagic, 2010; Re RN, 2018; Paz Ocaranza et al., 2020). This over activity can be mitigated by RAAS inhibitors such as ACE inhibitors (ACEIs), AT1R blockers (ARBs), and mineralocortocoid receptor inhibitors (MCRIs), commonly used in managing hypertension, heart failure, cardiac myopathies, and delaying the progression of cardiac/renal pathologies (Arendse et al., 2019) as well as by EGFR inhibitors (Qian et al., 2016; Rintala et al., 2016).
In the counter-regulating arm, ACE2 converts Ang II to Ang-(1–7), which acts via its G-protein coupled Mas receptor (MasR) (Kostenis et al., 2005). Ang-(1–7) inhibits ACE activity and decreases Ang II production whereas MasR is known to heterodimerize with AT1 receptor and inhibit actions of Ang II (Kostenis et al., 2005; Galandrin et al., 2016; Teixeira et al., 2017). Activation of the ACE2-Ang-(1–7)-MasR arm leads to vasodilation, anti-proliferative, anti-inflammatory, anti-oxidative stress, and anti-fibrotic effects (Benter et al., 1993; Benter et al., 1995a; Benter et al., 1995b; Dhaunsi et al., 2010; Ferrario et al., 2010). Indeed, Benter and colleagues were the first to report that Ang-(1–7) behaves differently from Ang II and produces vasodilatory effects by activating its own receptor in a pithed rat model in 1993 that was later confirmed in animal models of hypertension (Benter et al., 1993; Benter et al., 1995a; Benter et al., 1995b). In human subjects, Ang-(1–7) produces vasodilation in the coronary, splanchic, and renal circulations (Schindler et al., 2007; Medina and Arnold, 2019). Numerous studies have shown that ACE2-Ang-(1–7)-MasR activation is protective in various disease states that include renal, cardiovascular, respiratory, and diabetes-induced complications (Ferrario et al., 2005; Benter et al., 2006; Benter et al., 2007; Benter et al., 2008; Ferrario et al., 2010; Ferrario and Varagic, 2010; El-Hashim et al., 2012; Benter et al., 2015; Cao et al., 2019; Santos et al., 2019; Magalhães et al., 2020).
The kidney is a key site for Ang-(1–7) production and ACE2 protein, which is expressed in glomerular endothelial cells, podocytes and proximal tubular endothelial cells, and has been reported to be decreased in kidneys from diabetic and hypertensive mice (Crackower et al., 2002; Tikellis et al., 2003; Mizuiri and Ohashi, 2015). Furthermore, deletion or suppression of the ACE2 gene is associated with renal injury, dysfunction, and fibrosis (Ortiz-Melo and Gurley, 2016) and more severe nephropathy in response to high blood pressure (Liu et al., 2017). Conversely, treatment with exogenous recombinant ACE2 or transgenic overexpression of ACE2 has been demonstrated to limit kidney injury (Ortiz-Melo and Gurley, 2016). Overexpression of human ACE2 in a model of kidney injury showed a protective phenotype (Wang et al., 2015). Furthermore, administration of Ang-(1–7) has been shown to reduce renal resistance and improve renal vascular function in rats (Benter et al., 2008). In rats with diabetic nephropathy, Ang-(1–7) has been shown to reduce proteinuria and improve kidney function (Benter et al., 2007; Kim et al., 2015; Shi et al., 2015). In hypertensive kidney disease, Ang-(1–7) plays a protective role independent of its effects on blood pressure (Benter et al., 2006; Chen et al, 2019). Ang-(1–7) also protects against drug (Adriamycin)-induced nephropathy and renal ischemia-reperfusion injury in animal models (Silveira et al., 2013; Zaman and Banday, 2022). In humans, levels of Ang-(1–7) are reduced in patients with chronic kidney disease, and supplementation with Ang-(1–7) may improve kidney function in these patients (Chen and Harris, 2016).
Ang II activation of the AT1R contributes to proximal tubule brush border injury and remodeling, in part, due to enhanced mTOR signaling which promotes tubulointerstitial fibrosis (Whaley-Connell et al., 2011). Studies in models of diabetic and polycystic kidney disease indicate that targeting reductions in mTOR activity attenuates the progression of tubulointerstitial fibrosis (Whaley-Connell et al., 2011). AT1R antagonist telmisartan was shown to reduce the phosphorylation of EGFR and ERBB2 in human esophageal adenocarcinoma cells and inhibit cell proliferation and tumor growth, inducing cell cycle arrest by regulating cell cycle-related molecules via the AMPK/mTOR pathway (Fujihara et al., 2017). It has been shown that ACE inhibitors and ARBs increase Ang-(1–7) formation and Ang-(1–7) receptor antagonists attenuate their cardiovascular benefits indicating that at least part of the beneficial effects of ACEIs and ARBs are due to Ang-(1–7) activation (Ishiyama et al., 2004; Benter et al., 2011; Santos et al., 2019). Recently it was shown that Ang-(1–7) suppresses mTOR signaling, mitigates oxidative stress and protects human proximal tubular cells from endoplasmic reticulum stress, mitochondrial dysfunction and apoptosis (Farooqui and Banday, 2024). Although some opposing views exist (Zimmerman and Burns, 2012), the majority of studies provide increasing evidence for the renoprotective effects of ACE2/Ang-(1–7)/Mas receptor pathway that most likely occurs, at least partly, through counteracting the actions of ACE/Ang II/AT1 arm of RAAS and EGFR signalling.
So how might Ang-(1–7) precisely function in ameliorating renal pathologies? Oxidative stress and inflammation are well-known drivers of kidney injury, and reducing these factors is a key strategy in the prevention and treatment of kidney disease. The potential benefits of Ang-(1–7) in the regulation of kidney function are believed to be due to its ability to reduce oxidative stress, inflammation and the ensuing fibrosis in the kidney, as well as to its direct effects on renal blood flow and function (Benter et al., 2008; Lu et al., 2017; Yousif et al., 2012; Simões e Silva et al., 2013). Ang-(1–7) has been shown to have direct anti-inflammatory and antioxidant effects in the kidney (Benter et al., 2008; Shi et al., 2015). In a study by Lu et al. (2017), Ang-(1–7) was shown to reduce oxidative stress and inflammation in an experimental model of renal injury. Additionally, administration of Ang-(1–7) has been shown to improve renal vascular function and to improve kidney function in rats (Benter et al., 2007; Van Twist et al., 2014; Kim et al., 2015; Shi et al., 2015). These effects are believed to be due to the ability of Ang-(1–7) to reduce renal resistance, promote vasodilation, and increase blood flow to the kidney-characteristics that are actually exacerbated by EGFR signaling. Therefore, could the beneficial effects of ACE2/Ang-(1–7)/Mas receptor pathway in renal pathologies be somehow mediated by inhibition of EGFR? Indeed, there now appears to be significant evidence for cross-talk between Ang-(1–7) and EGFR. We were the first to show that Ang-(1–7) inhibits EGFR transactivation in vitro and in vivo (Akhtar et al., 2012). More recently, it was shown that Ang-(1–7) also inhibits transactivation to ErbB2, ErbB3 and ErbB4 receptors from the EGFR family of receptor tyrosine kinases and might act as a novel “pan-ErbB inhibitor (Akhtar et al., 2015). Furthermore, it was shown in the diabetic vasculature that Ang-(1–7)-mediated inhibition of EGFR receptor family occurred via a src-dependent pathway (Akhtar et al., 2012; Akhtar et al., 2015). As to whether this occurs in the diabetic kidney pathologies remains unknown. However, in a study by Qin et al. (2015), administration of Ang-(1–7) reduced EGFR expression and activity in the kidney in rats with experimental glomerulonephritis supporting the notion that Ang-(1–7) is an inhibitor of EGFR and that this might represent a potential mechanism by which it exerts its beneficial renal effects.
Overall, the potential interplay between EGFR and Ang-(1–7) in the kidney is likely to be complex and multifaceted relationship depending on the site and duration of EGFR actions. Thus, further research is needed to fully understand the mechanisms involved. However, the current evidence suggests that both EGFR and Ang-(1–7) are important effectors in the regulation of kidney function, and that the interplay between these two systems may play a key role in the prevention and treatment of kidney disease.
10 Concluding remarks
Our review paper highlights the role of epidermal growth factor receptor (EGFR) and its downstream signaling in seven different kidney pathologies: glomerulonephritis, diabetic nephropathy, hypertensive nephropathy, acute kidney injury, Chronic kidney disease, real transplant, and renal fibrosis. EGFR signaling appears to have a dual role in kidney physiology, repair and pathology. Although EGFR activation is crucial for kidney physiology and repair such as following acute kidney injury through mechanisms involving dedifferentiation, migration and proliferation of tubular cells, its sustained activation has been linked to renal fibrosis via activation of myofibroblasts. The overall impact of EGFR activation on kidney health depends on the duration and site of activation. Where overactivity of EGFR family members has been demonstrated, pre-clinical studies with inhibitors of EGFR family of RTKs as potential therapeutic agents have shown promising results in some kidney pathologies. Indeed, small molecule EGFR inhibitors such as Gefitinib and Erlinotib have been clinically used in cancer chemotherapy since their FDA approval in the early 2000s (Karachaliou et al., 2019; Abourehab et al., 2021; Singh et al., 2023; Zubair and Bandyopadhyay, 2023). They are deemed to have an acceptable safety profile with diarrhea and skin rashes as the most common side effects but these can be readily managed with conventional approaches (Hirsh, 2011; Aw et al., 2018). Relatively more recently, large molecule, monoclonal antibody-based inhibitors of EGFR family of receptors, such Cetuximab and Panitumumab, have also been introduced clinically for the treatment of several cancers (Yamaoka et al., 2017). The clinical effectiveness and seemingly acceptable safety and toxicological profile of both large and small molecule inhibitors suggests that the repurposing of these FDA-approved anticancer EGFR inhibitors for renal pathologies might be a clinically feasible therapeutic approach-though this requires further clinical evaluation including monitoring the possible development of drug resistance (Cooper et al., 2022). Thus, it is clear that further research is necessary to fully understand the role of these RTKs, including their interplay with members of the RAAS such as Ang-(1–7) as well as the potential use of specific EGFR inhibitors, in renal pathologies in the clinical setting.
Author contributions
MT: Writing–original draft, Writing–review and editing, Formal Analysis. YA-D: Writing–original draft, Writing–review and editing. AT: Writing–original draft, Writing–review and editing. IB: Writing–original draft, Writing–review and editing. SA: Writing–original draft, Writing–review and editing, Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Funding for research in the laboratory of SA was provided by Qatar University grant QUCG-CMED-22/23-540.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abourehab, M. A. S., Alqahtani, A. M., Youssif, B. G. M., and Gouda, A. M. (2021). Globally approved EGFR inhibitors: insights into their syntheses, target kinases, biological activities, receptor interactions, and metabolism. Molecules 26 (21), 6677. doi:10.3390/molecules26216677
Akhtar, S., and Benter, I. F. (2013). The role of epidermal growth factor receptor in diabetes-induced cardiac dysfunction. Bioimpacts 3 (1), 5–9. doi:10.5681/bi.2013.008
Akhtar, S., Benter, I. F., Danjuma, M. I., Doi, S. A. R., Hasan, S. S., and Habib, A. M. (2020). Pharmacotherapy in COVID-19 patients: a review of ACE2-raising drugs and their clinical safety. J. Drug Target 28 (7-8), 683–699. doi:10.1080/1061186X.2020.1797754
Akhtar, S., Chandrasekhar, B., Attur, S., Dhaunsi, G. S., Yousif, M. H., and Benter, I. F. (2015). Transactivation of ErbB family of receptor tyrosine kinases is inhibited by angiotensin-(1-7) via its mas receptor. PLoS One 10 (11), e0141657. doi:10.1371/journal.pone.0141657
Akhtar, S., Chandrasekhar, B., Yousif, M. H., Renno, W., Benter, I. F., and El-Hashim, A. Z. (2019). Chronic administration of nano-sized PAMAM dendrimers in vivo inhibits EGFR-ERK1/2-ROCK signaling pathway and attenuates diabetes-induced vascular remodeling and dysfunction. Nanomedicine 18, 78–89. doi:10.1016/j.nano.2019.02.012
Akhtar, S., Yousif, M. H., Dhaunsi, G. S., Chandrasekhar, B., Al-Farsi, O., and Benter, I. F. (2012). Angiotensin-(1-7) inhibits epidermal growth factor receptor transactivation via a Mas receptor-dependent pathway. Br. J. Pharmacol. 165 (5), 1390–1400. doi:10.1111/j.1476-5381.2011.01613.x
Akhtar, S., Yousif, M. H., Dhaunsi, G. S., Sarkhouh, F., Chandrasekhar, B., Attur, S., et al. (2013). Activation of ErbB2 and downstream signalling via rho kinases and ERK1/2 contributes to diabetes-induced vascular dysfunction. PLoS One 8 (6), e67813. doi:10.1371/journal.pone.0067813
Andrianova, N. V., Buyan, M. I., Zorova, L. D., Pevzner, I. B., Popkov, V. A., Babenko, V. A., et al. (2019). Kidney cells regeneration: dedifferentiation of tubular epithelium, resident stem cells and possible niches for renal progenitors. Int. J. Mol. Sci. 20 (24), 6326. doi:10.3390/ijms20246326
Arendse, L. B., Danser, A. H. J., Poglitsch, M., Touyz, R. M., Burnett, J. C., Llorens-Cortes, C., et al. (2019). Novel therapeutic approaches targeting the renin-angiotensin system and associated peptides in hypertension and heart failure. Pharmacol. Rev. 71 (4), 539–570. doi:10.1124/pr.118.017129
Aw, D. C., Tan, E. H., Chin, T. M., Lim, H. L., Lee, H. Y., and Soo, R. A. (2018). Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J. Clin. Oncol. 14 (1), 23–31. doi:10.1111/ajco.12687
Bader, M., Steckelings, U. M., Alenina, N., Santos, R. A. S., and Ferrario, C. M. (2024). Alternative renin-angiotensin system. Hypertension 81 (5), 964–976. doi:10.1161/HYPERTENSIONAHA.123.21364
Benter, I. F., Benboubetra, M., Hollins, A. J., Yousif, M. H., Canatan, H., and Akhtar, S. (2009b). Early inhibition of EGFR signaling prevents diabetes-induced up-regulation of multiple gene pathways in the mesenteric vasculature. Vasc. Pharmacol. 51 (4), 236–245. doi:10.1016/j.vph.2009.06.008
Benter, I. F., Canatan, H., Benboubetra, M., Yousif, M. H., and Akhtar, S. (2009a). Global upregulation of gene expression associated with renal dysfunction in DOCA-salt-induced hypertensive rats occurs via signaling cascades involving epidermal growth factor receptor: a microarray analysis. Vasc. Pharmacol. 51 (2-3), 101–109. doi:10.1016/j.vph.2009.04.004
Benter, I. F., Diz, D. I., and Ferrario, C. M. (1993). Cardiovascular actions of angiotensin(1-7). Peptides 14 (4), 679–684. doi:10.1016/0196-9781(93)90097-z
Benter, I. F., Diz, D. I., and Ferrario, C. M. (1995b). Pressor and reflex sensitivity is altered in spontaneously hypertensive rats treated with angiotensin-(1-7). Hypertension 26 (6 Pt 2), 1138–1144. doi:10.1161/01.hyp.26.6.1138
Benter, I. F., Ferrario, C. M., Morris, M., and Diz, D. I. (1995a). Antihypertensive actions of angiotensin-(1-7) in spontaneously hypertensive rats. Am. J. Physiol. 269 (1 Pt 2), H313–H319. doi:10.1152/ajpheart.1995.269.1.H313
Benter, I. F., Sarkhou, F., Al-Khaldi, A. T., Chandrasekhar, B., Attur, S., Dhaunsi, G. S., et al. (2015). The dual targeting of EGFR and ErbB2 with the inhibitor Lapatinib corrects high glucose-induced apoptosis and vascular dysfunction by opposing multiple diabetes-induced signaling changes. J. Drug Target 23 (6), 506–518. doi:10.3109/1061186X.2015.1057150
Benter, I. F., Yousif, M. H., Al-Saleh, F. M., Raghupathy, R., Chappell, M. C., and Diz, D. I. (2011). Angiotensin-(1-7) blockade attenuates captopril- or hydralazine-induced cardiovascular protection in spontaneously hypertensive rats treated with NG-nitro-L-arginine methyl ester. J. Cardiovasc Pharmacol. 57 (5), 559–567. doi:10.1097/FJC.0b013e31821324b6
Benter, I. F., Yousif, M. H., Anim, J. T., Cojocel, C., and Diz, D. I. (2006). Angiotensin-(1-7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am. J. Physiol. Heart Circ. Physiol. 290 (2), H684–H691. doi:10.1152/ajpheart.00632.2005
Benter, I. F., Yousif, M. H., Cojocel, C., Al-Maghrebi, M., and Diz, D. I. (2007). Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 292 (1), H666–H672. doi:10.1152/ajpheart.00372.2006
Benter, I. F., Yousif, M. H., Dhaunsi, G. S., Kaur, J., Chappell, M. C., and Diz, D. I. (2008). Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am. J. Nephrol. 28 (1), 25–33. doi:10.1159/000108758
Benter, I. F., Yousif, M. H., Griffiths, S. M., Benboubetra, M., and Akhtar, S. (2005a). Epidermal growth factor receptor tyrosine kinase-mediated signalling contributes to diabetes-induced vascular dysfunction in the mesenteric bed. Br. J. Pharmacol. 145 (6), 829–836. doi:10.1038/sj.bjp.0706238
Benter, I. F., Yousif, M. H., Hollins, A. J., Griffiths, S. M., and Akhtar, S. (2005b). Diabetes-induced renal vascular dysfunction is normalized by inhibition of epidermal growth factor receptor tyrosine kinase. J. Vasc. Res. 42 (4), 284–291. doi:10.1159/000085904
Bollée, G., Flamant, M., Schordan, S., Fligny, C., Rumpel, E., Milon, M., et al. (2011). Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat. Med. 17 (10), 1242–1250. doi:10.1038/nm.2491
Brooks, M. B. (2012). Erlotinib appears to produce prolonged remission of insulin-requiring type 2 diabetes associated with metabolic syndrome and chronic kidney disease. Br. J. Diabetes and Vasc. Dis. 12 (2), 87–90. doi:10.1177/1474651412442694
Cao, S., Pan, Y., Terker, A. S., Arroyo Ornelas, J. P., Wang, Y., Tang, J., et al. (2023). Epidermal growth factor receptor activation is essential for kidney fibrosis development. Nat. Commun. 14 (1), 7357. doi:10.1038/s41467-023-43226-x
Cao, Y., Liu, Y., Shang, J., Yuan, Z., Ping, F., Yao, S., et al. (2019). Ang-(1-7) treatment attenuates lipopolysaccharide-induced early pulmonary fibrosis. Lab. Invest 99 (12), 1770–1783. doi:10.1038/s41374-019-0289-7
Carriazo, S., Vanessa Perez-Gomez, M., and Ortiz, A. (2020). Hypertensive nephropathy: a major roadblock hindering the advance of precision nephrology. Clin. Kidney J. 13 (4), 504–509. doi:10.1093/ckj/sfaa162
Chadban, S. J., and Atkins, R. C. (2005). Glomerulonephritis. Lancet. 365 (9473), 1797–1806. doi:10.1016/S0140-6736(05)66583-X
Chang-Panesso, M., Kadyrov, F. F., Lalli, M., Wu, H., Ikeda, S., Kefaloyianni, E., et al. (2019). FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J. Clin. Invest 129 (12), 5501–5517. doi:10.1172/JCI125519
Chen, H. M., Dai, J. J., Zhu, R., Peng, F. F., Wu, S. Z., Yu, H., et al. (2019a). Parathyroid hormone-related protein induces fibronectin up-regulation in rat mesangial cells through reactive oxygen species/Src/EGFR signaling. Biosci. Rep. 39 (4), BSR20182293. doi:10.1042/BSR20182293
Chen, J., Chen, J. K., and Harris, R. C. (2012). Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int. 82 (1), 45–52. doi:10.1038/ki.2012.43
Chen, J., Chen, J. K., and Harris, R. C. (2015). EGF receptor deletion in podocytes attenuates diabetic nephropathy. J. Am. Soc. Nephrol. 26 (5), 1115–1125. doi:10.1681/ASN.2014020192
Chen, J., Chen, J. K., Neilson, E. G., and Harris, R. C. (2006). Role of EGF receptor activation in angiotensin II-induced renal epithelial cell hypertrophy. J. Am. Soc. Nephrol. 17 (6), 1615–1623. doi:10.1681/ASN.2005111163
Chen, J., and Harris, R. C. (2016). Interaction of the EGF receptor and the Hippo pathway in the diabetic kidney. J. Am. Soc. Nephrol. 27 (6), 1689–1700. doi:10.1681/ASN.2015040415
Chen, J., You, H., Li, Y., Xu, Y., He, Q., and Harris, R. C. (2018). EGF receptor-dependent YAP activation is important for renal recovery from AKI. J. Am. Soc. Nephrol. 29 (9), 2372–2385. doi:10.1681/ASN.2017121272
Chen, Y., Zhao, W., Liu, C., Meng, W., Zhao, T., Bhattacharya, S. K., et al. (2019b). Molecular and cellular effect of angiotensin 1-7 on hypertensive kidney disease. Am. J. Hypertens. 32 (5), 460–467. doi:10.1093/ajh/hpz009
Chiluiza, D., Krishna, S., Schumacher, V. A., and Schlöndorff, J. (2013). Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J. Biol. Chem. 288 (25), 18407–18420. doi:10.1074/jbc.M113.463059
Chung, H., Ramachandran, R., Hollenberg, M. D., and Muruve, D. A. (2013). Proteinase-activated receptor-2 transactivation of epidermal growth factor receptor and transforming growth factor-β receptor signaling pathways contributes to renal fibrosis. J. Biol. Chem. 288 (52), 37319–37331. doi:10.1074/jbc.M113.492793
Cirio, M. C., de Groh, E. D., de Caestecker, M. P., Davidson, A. J., and Hukriede, N. A. (2014). Kidney regeneration: common themes from the embryo to the adult. Pediatr. Nephrol. 29 (4), 553–564. doi:10.1007/s00467-013-2597-2
Collier, J. B., Whitaker, R. M., Eblen, S. T., and Schnellmann, R. G. (2016). Rapid renal regulation of peroxisome proliferator-activated receptor γ coactivator-1α by extracellular signal-regulated kinase 1/2 in physiological and pathological conditions. J. Biol. Chem. 291 (52), 26850–26859. doi:10.1074/jbc.M116.754762
Cong, C., Yuan, X., Hu, Y., Chen, W., Wang, Y., and Tao, L. (2022). Catalpol alleviates Ang II-induced renal injury through NF-κB pathway and TGF-β1/smads pathway. J. Cardiovasc Pharmacol. 79 (1), e116–e121. doi:10.1097/FJC.0000000000001148
Cooper, A. J., Sequist, L. V., and Lin, J. J. (2022). Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat. Rev. Clin. Oncol. 19 (8), 499–514. doi:10.1038/s41571-022-00639-9
Cooper, M. E. (2001). Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia 44 (11), 1957–1972. doi:10.1007/s001250100000
Costa, D. B., and Huberman, M. S. (2006). Improvement of type 2 diabetes in a lung cancer patient treated with Erlotinib. Diabetes Care 29 (7), 1711. doi:10.2337/dc06-0558
Costantino, V. V., Gil Lorenzo, A. F., Bocanegra, V., and Vallés, P. G. (2021). Molecular mechanisms of hypertensive nephropathy: renoprotective effect of losartan through Hsp70. Cells 10 (11), 3146. doi:10.3390/cells10113146
Costa-Reis, P., Russo, P. A., Zhang, Z., Colonna, L., Maurer, K., Gallucci, S., et al. (2015). The role of MicroRNAs and human epidermal growth factor receptor 2 in proliferative lupus nephritis. Arthritis Rheumatol. 67 (9), 2415–2426. doi:10.1002/art.39219
Crackower, M. A., Sarao, R., Oudit, G. Y., Yagil, C., Kozieradzki, I., Scanga, S. E., et al. (2002). Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417 (6891), 822–828. doi:10.1038/nature00786
de Borst, M. H., Diks, S. H., Bolbrinker, J., Schellings, M. W., van Dalen, M. B. A., Peppelenbosch, M. P., et al. (2007). Profiling of the renal kinome: a novel tool to identify protein kinases involved in angiotensin II-dependent hypertensive renal damage. Am. J. Physiol. Ren. Physiol. 293 (1), F428–F437. doi:10.1152/ajprenal.00367.2006
Dey, M., Baldys, A., Sumter, D. B., Göoz, P., Luttrell, L. M., Raymond, J. R., et al. (2010). Bradykinin decreases podocyte permeability through ADAM17-dependent epidermal growth factor receptor activation and zonula occludens-1 rearrangement. J. Pharmacol. Exp. Ther. 334 (3), 775–783. doi:10.1124/jpet.110.168054
Ding, W., Wang, B., Zhang, M., and Gu, Y. (2015). Tempol, a Superoxide dismutase-mimetic drug, ameliorates progression of renal disease in CKD mice. Cell Physiol. Biochem. 36 (6), 2170–2182. doi:10.1159/000430183
Dhaunsi, G. S., Yousif, M. H., Akhtar, S., and Chappell, M. C. (2010). Angiotensin-(1-7) prevents diabetes-induced attenuation in PPAR-gamma and catalase activities. Eur. J. Pharmacol. 638 (1-3), 108–114. doi:10.1016/j.ejphar.2010.04.030
Duran-Salgado, M. B., and Rubio-Guerra, A. F. (2014). Diabetic nephropathy and inflammation. World J. Diabetes 5 (3), 393–398. doi:10.4239/wjd.v5.i3.393
El-Hashim, A. Z., Renno, W. M., Raghupathy, R., Abduo, H. T., Akhtar, S., and Benter, I. F. (2012). Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-κB-dependent pathways. Br. J. Pharmacol. 166 (6), 1964–1976. doi:10.1111/j.1476-5381.2012.01905.x
Farooqui, Z., and Banday, A. A. (2024). Angiotensin 1-7 exerts antioxidant effects, suppresses Mammalian Target of Rapamycin (mTOR) signaling, and inhibits apoptosis in renal proximal tubular cells. Peptides 172, 171136. doi:10.1016/j.peptides.2023.171136
Feng, L., Garcia, G. E., Yang, Y., Xia, Y., Gabbai, F. B., Peterson, O. W., et al. (2000). Heparin-binding EGF-like growth factor contributes to reduced glomerular filtration rate during glomerulonephritis in rats. J. Clin. Invest 105 (3), 341–350. doi:10.1172/JCI2869
Feng, M., Tang, P. M., Huang, X. R., Sun, S. F., You, Y. K., Xiao, J., et al. (2018). TGF-Β mediates renal fibrosis via the smad3-erbb4-IR long noncoding RNA Axis. Mol. Ther. 26 (1), 148–161. doi:10.1016/j.ymthe.2017.09.024
Ferrario, C. M., Ahmad, S., Joyner, J., and Varagic, J. (2010). Advances in the renin angiotensin system focus on angiotensin-converting enzyme 2 and angiotensin-(1-7). Adv. Pharmacol. 59, 197–233. doi:10.1016/S1054-3589(10)59007-0
Ferrario, C. M., Trask, A. J., and Jessup, J. A. (2005). Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1-7) in regulation of cardiovascular function. Am. J. Physiol. Heart Circ. Physiol. 289 (6), H2281–H2290. doi:10.1152/ajpheart.00618.2005
Ferrario, C. M., and Varagic, J. (2010). The ANG-(1-7)/ACE2/mas axis in the regulation of nephron function. Am. J. Physiol. Ren. Physiol. 298 (6), F1297–F1305. doi:10.1152/ajprenal.00110.2010
Fogelgren, B., Zuo, X., Buonato, J. M., Vasilyev, A., Baek, J. I., Choi, S. Y., et al. (2014). Exocyst Sec10 protects renal tubule cells from injury by EGFR/MAPK activation and effects on endocytosis. Am. J. Physiol. Ren. Physiol. 307 (12), F1334–F1341. doi:10.1152/ajprenal.00032.2014
Forbes, J. M., Fukami, K., and Cooper, M. E. (2007). Diabetic nephropathy: where hemodynamics meets metabolism. Exp. Clin. Endocrinol. Diabetes 115 (2), 69–84. doi:10.1055/s-2007-949721
Forrester, S. J., Booz, G. W., Sigmund, C. D., Coffman, T. M., Kawai, T., Rizzo, V., et al. (2018). Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev. 98 (3), 1627–1738. doi:10.1152/physrev.00038.2017
François, H., Placier, S., Flamant, M., Tharaux, P. L., Chansel, D., Dussaule, J. C., et al. (2004). Prevention of renal vascular and glomerular fibrosis by epidermal growth factor receptor inhibition. FASEB J. 18 (7), 926–928. doi:10.1096/fj.03-0702fje
Freedman, B. I., and Cohen, A. H. (2016). Hypertension-attributed nephropathy: what's in a name? Nat. Rev. Nephrol. 12 (1), 27–36. doi:10.1038/nrneph.2015.172
Fujihara, S., Morishita, A., Ogawa, K., Tadokoro, T., Chiyo, T., Kato, K., et al. (2017). The angiotensin II type 1 receptor antagonist telmisartan inhibits cell proliferation and tumor growth of esophageal adenocarcinoma via the AMPKα/mTOR pathway in vitro and in vivo. Oncotarget 8 (5), 8536–8549. doi:10.18632/oncotarget.14345
Galandrin, S., Denis, C., Boularan, C., Marie, J., M'Kadmi, C., Pilette, C., et al. (2016). Cardioprotective angiotensin-(1-7) peptide acts as a natural-biased ligand at the angiotensin II type 1 receptor. Hypertension 68 (6), 1365–1374. doi:10.1161/HYPERTENSIONAHA.116.08118
Gao, L., Zhong, X., Jin, J., Li, J., and Meng, X. M. (2020). Potential targeted therapy and diagnosis based on novel insight into growth factors, receptors, and downstream effectors in acute kidney injury and acute kidney injury-chronic kidney disease progression. Signal Transduct. Target Ther. 5 (1), 9. doi:10.1038/s41392-020-0106-1
Geng, H., Lan, R., Liu, Y., Chen, W., Wu, M., Saikumar, P., et al. (2021). Proximal tubule LPA1 and LPA2 receptors use divergent signaling pathways to additively increase profibrotic cytokine secretion. Am. J. Physiol. Ren. Physiol. 320 (3), F359–F374. doi:10.1152/ajprenal.00494.2020
Gilbert, R. E., Huang, Q., Thai, K., Advani, S. L., Lee, K., Yuen, D. A., et al. (2011). Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 79 (12), 1312–1321. doi:10.1038/ki.2011.39
Goto, S., Fujii, H., Mieno, M., Yagisawa, T., Abe, M., Nitta, K., et al. (2024). Survival benefit of living donor kidney transplantation in patients on hemodialysis. Clin. Exp. Nephrol. 28 (2), 165–174. doi:10.1007/s10157-023-02417-y
Guijarro, C., and Egido, J. (2001). Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int. 59 (2), 415–424. doi:10.1046/j.1523-1755.2001.059002415.x
Harris, R. C. (2021). The epidermal growth factor receptor axis and kidney fibrosis. Curr. Opin. Nephrol. Hypertens. 30 (3), 275–279. doi:10.1097/MNH.0000000000000696
Harris, R. C., Chung, E., and Coffey, R. J. (2003). EGF receptor ligands. Exp. Cell Res. 284 (1), 2–13. doi:10.1016/s0014-4827(02)00105-2
Harskamp, L. R., Gansevoort, R. T., van Goor, H., and Meijer, E. (2016). The epidermal growth factor receptor pathway in chronic kidney diseases. Nat. Rev. Nephrol. 12 (8), 496–506. doi:10.1038/nrneph.2016.91
He, S., Liu, N., Bayliss, G., and Zhuang, S. (2013). EGFR activity is required for renal tubular cell dedifferentiation and proliferation in a murine model of folic acid-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 304 (4), F356–F366. doi:10.1152/ajprenal.00553.2012
Hénique, C., Fligny, C., and Tharaux, P. L. (2013). L22. Crescent formation: unraveling local mediators that break glomerular epithelial cell tolerance to immune injury. Presse Med. 42 (4 Pt 2), 565–568. doi:10.1016/j.lpm.2013.01.021
Heuer, J. G., Harlan, S. M., Yang, D. D., Jaqua, D. L., Boyles, J. S., Wilson, J. M., et al. (2017). Role of TGF-alpha in the progression of diabetic kidney disease. Am. J. Physiol. Ren. Physiol. 312 (6), F951–F962. doi:10.1152/ajprenal.00443.2016
Hirsh, V. (2011). Managing treatment-related adverse events associated with egfr tyrosine kinase inhibitors in advanced non-small-cell lung cancer. Curr. Oncol. 18 (3), 126–138. doi:10.3747/co.v18i3.877
Hoste, E. A. J., Kellum, J. A., Selby, N. M., Zarbock, A., Palevsky, P. M., Bagshaw, S. M., et al. (2018). Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 14 (10), 607–625. doi:10.1038/s41581-018-0052-0
Hu, C., Zuo, Y., Ren, Q., Sun, X., Zhou, S., Liao, J., et al. (2021). Matrix metalloproteinase-10 protects against acute kidney injury by augmenting epidermal growth factor receptor signaling. Cell Death Dis. 12 (1), 70. doi:10.1038/s41419-020-03301-3
Huang, R., Fu, P., and Ma, L. (2023). Kidney fibrosis: from mechanisms to therapeutic medicines. Sig Transduct. Target Ther. 8, 129. doi:10.1038/s41392-023-01379-7
Ishiyama, Y., Gallagher, P. E., Averill, D. B., Tallant, E. A., Brosnihan, K. B., and Ferrario, C. M. (2004). Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 43 (5), 970–976. doi:10.1161/01.HYP.0000124667.34652.1a
Jørgensen, I. F., Muse, V. P., Aguayo-Orozco, A., Brunak, S., and Sørensen, S. S. (2024). Stratification of kidney transplant recipients into five subgroups based on temporal disease trajectories. Transpl. Direct 10 (2), e1576. doi:10.1097/TXD.0000000000001576
Kälble, T., Lucan, M., Nicita, G., Sells, R., Burgos Revilla, F. J., Wiesel, M., et al. (2005). EAU guidelines on renal transplantation. Eur. Urol. 47 (2), 156–166. doi:10.1016/j.eururo.2004.02.009
Kao, T. W., and Huang, C. C. (2021). Blood pressure management and renal protection: revisiting hypertensive nephropathy. J. Chin. Med. Assoc. 84 (10), 911–916. doi:10.1097/JCMA.0000000000000600
Karachaliou, N., Fernandez-Bruno, M., Bracht, J. W. P., and Rosell, R. (2019). EGFR first- and second-generation TKIs-there is still place for them in EGFR-mutant NSCLC patients. Transl. Cancer Res. 8 (Suppl. 1), S23–S47. doi:10.21037/tcr.2018.10.06
Kassan, M., Ait-Aissa, K., Ali, M., Trebak, M., and Matrougui, K. (2015). Augmented EGF receptor tyrosine kinase activity impairs vascular function by NADPH oxidase-dependent mechanism in type 2 diabetic mouse. Biochim. Biophys. Acta 1853 (10 Pt A), 2404–2410. doi:10.1016/j.bbamcr.2015.05.032
Kefaloyianni, E., Keerthi Raja, M. R., Schumacher, J., Muthu, M. L., Krishnadoss, V., Waikar, S. S., et al. (2019). Proximal tubule-derived amphiregulin amplifies and integrates profibrotic EGF receptor signals in kidney fibrosis. J. Am. Soc. Nephrol. 30 (12), 2370–2383. doi:10.1681/ASN.2019030321
Kefaloyianni, E., Muthu, M. L., Kaeppler, J., Sun, X., Sabbisetti, V., Chalaris, A., et al. (2016). ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight 1 (13), e87023. doi:10.1172/jci.insight.87023
Kim, B. W., Kim, S. K., Heo, K. W., Bae, K. B., Jeong, K. H., Lee, S. H., et al. (2020). Association between epidermal growth factor (EGF) and EGF receptor gene polymorphisms and end-stage renal disease and acute renal allograft rejection in a Korean population. Ren. Fail 42 (1), 98–106. doi:10.1080/0886022X.2019.1710535
Kim, C. S., Kim, I. J., Bae, E. H., Ma, S. K., Lee, J., and Kim, S. W. (2015). Angiotensin-(1-7) attenuates kidney injury due to obstructive nephropathy in rats. PLoS One 10 (11), e0142664. doi:10.1371/journal.pone.0142664
Konishi, A., and Berk, B. C. (2003). Epidermal growth factor receptor transactivation is regulated by glucose in vascular smooth muscle cells. J. Biol. Chem. 278 (37), 35049–35056. doi:10.1074/jbc.M304913200
Kostenis, E., Milligan, G., Christopoulos, A., Sanchez-Ferrer, C. F., Heringer-Walther, S., Sexton, P. M., et al. (2005). G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 111 (14), 1806–1813. doi:10.1161/01.CIR.0000160867.23556.7D
Krishna, S. B., Alfonso, L. F., Thekkumkara, T. J., Abbruscato, T. J., and Bhat, G. J. (2007). Angiotensin II induces phosphorylation of glucose-regulated protein-75 in WB rat liver cells. Arch. Biochem. Biophys. 457 (1), 16–28. doi:10.1016/j.abb.2006.10.011
Kumagai, S., Koyama, S., and Nishikawa, H. (2021). Antitumour immunity regulated by aberrant ERBB family signalling. Nat. Rev. Cancer 21 (3), 181–197. doi:10.1038/s41568-020-00322-0
Lautrette, A., Li, S., Alili, R., Sunnarborg, S. W., Burtin, M., Lee, D. C., et al. (2005). Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat. Med. 11 (8), 867–874. doi:10.1038/nm1275
Lee, H. W., Khan, S. Q., Khaliqdina, S., Altintas, M. M., Grahammer, F., Zhao, J. L., et al. (2017). Absence of miR-146a in podocytes increases risk of diabetic glomerulopathy via up-regulation of ErbB4 and notch-1. J. Biol. Chem. 292 (2), 732–747. doi:10.1074/jbc.M116.753822
Lemmon, M. A., and Schlessinger, J. (2010). Cell signaling by receptor tyrosine kinases. Cell 141 (7), 1117–1134. doi:10.1016/j.cell.2010.06.011
Levey, A. S., and Coresh, J. (2012). Chronic kidney disease. Lancet 379 (9811), 165–180. doi:10.1016/S0140-6736(11)60178-5
Li, H., Shao, F., Qian, B., Sun, Y., Huang, Z., Ding, Z., et al. (2019). Upregulation of HER2 in tubular epithelial cell drives fibroblast activation and renal fibrosis. Kidney Int. 96 (3), 674–688. doi:10.1016/j.kint.2019.04.012
Li, R., Uttarwar, L., Gao, B., Charbonneau, M., Shi, Y., Chan, J. S. D., et al. (2015). High glucose up-regulates ADAM17 through HIF-1α in mesangial cells. J. Biol. Chem. 290 (35), 21603–21614. doi:10.1074/jbc.M115.651604
Li, R., Wang, T., Walia, K., Gao, B., and Krepinsky, J. C. (2018a). Regulation of profibrotic responses by ADAM17 activation in high glucose requires its C-terminus and FAK. J. Cell Sci. 131 (4), jcs208629. doi:10.1242/jcs.208629
Li, S., Xie, H., Shi, Y., and Liu, H. (2022). Prevalence of diabetic nephropathy in the diabetes mellitus population: a protocol for systematic review and meta-analysis. Med. Baltim. 101 (42), e31232. doi:10.1097/MD.0000000000031232
Li, X., Ma, T. K., Wen, S., Li, L. L., Xu, L., Zhu, X. W., et al. (2020). LncRNA ARAP1-AS2 promotes high glucose-induced human proximal tubular cell injury via persistent transactivation of the EGFR by interacting with ARAP1. J. Cell Mol. Med. 24 (22), 12994–13009. doi:10.1111/jcmm.15897
Li, X. C., Zhang, J., and Zhuo, J. L. (2017). The vasoprotective axes of the renin-angiotensin system: physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol. Res. 125 (Pt A), 21–38. doi:10.1016/j.phrs.2017.06.005
Li, Y., Pan, Y., Cao, S., Sasaki, K., Wang, Y., Niu, A., et al. (2021). Podocyte EGFR inhibits autophagy through upregulation of rubicon in type 2 diabetic nephropathy. Diabetes 70 (2), 562–576. doi:10.2337/db20-0660
Li, Z., Li, Y., Overstreet, J. M., Chung, S., Niu, A., Fan, X., et al. (2018b). Inhibition of epidermal growth factor receptor activation is associated with improved diabetic nephropathy and insulin resistance in type 2 diabetes. Diabetes 67 (9), 1847–1857. doi:10.2337/db17-1513
Lin, L., and Hu, K. (2022). Annexin A2 and kidney diseases. Front. Cell Dev. Biol. 10, 974381. doi:10.3389/fcell.2022.974381
Liu, N., He, S., Ma, L., Ponnusamy, M., Tang, J., Tolbert, E., et al. (2013). Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS One 8 (1), e54001. doi:10.1371/journal.pone.0054001
Liu, Z., Huang, X. R., Chen, H. Y., Fung, E., Liu, J., and Lan, H. Y. (2017). Deletion of angiotensin-converting enzyme-2 promotes hypertensive nephropathy by targeting Smad7 for ubiquitin degradation. Hypertension 70 (4), 822–830. doi:10.1161/HYPERTENSIONAHA.117.09600
Lu, W., Kang, J., Hu, K., Tang, S., Zhou, X., Yu, S., et al. (2017). Angiotensin-(1-7) relieved renal injury induced by chronic intermittent hypoxia in rats by reducing inflammation, oxidative stress and fibrosis. Braz. J. Med. Biol. Res. 50 (1), e5594. doi:10.1590/1414-431X20165594
Ma, X., Schwarz, A., Sevilla, S. Z., Levin, A., Hultenby, K., Wernerson, A., et al. (2018). Depletion of Gprc5a promotes development of diabetic nephropathy. J. Am. Soc. Nephrol. 29 (6), 1679–1689. doi:10.1681/ASN.2017101135
Magalhães, G. S., Gregório, J. F., Ramos, K. E., Cançado-Ribeiro, A. T. P., Baroni, I. F., Barcelos, L. S., et al. (2020). Treatment with inhaled formulation of angiotensin-(1-7) reverses inflammation and pulmonary remodeling in a model of chronic asthma. Immunobiology 225 (3), 151957. doi:10.1016/j.imbio.2020.151957
Mahtal, N., Lenoir, O., Tinel, C., Anglicheau, D., and Tharaux, P. L. (2022). MicroRNAs in kidney injury and disease. Nat. Rev. Nephrol. 18 (10), 643–662. doi:10.1038/s41581-022-00608-6
Makris, K., and Spanou, L. (2016). Acute kidney injury: definition, pathophysiology and clinical phenotypes. Clin. Biochem. Rev. 37 (2), 85–98.
Marchion, D., and Münster, P. (2007). Development of histone deacetylase inhibitors for cancer treatment. Expert Rev. Anticancer Ther. 7 (4), 583–598. doi:10.1586/14737140.7.4.583
Mas, V., Maluf, D., Archer, K., Yanek, K., Mas, L., King, A., et al. (2007a). Establishing the molecular pathways involved in chronic allograft nephropathy for testing new noninvasive diagnostic markers. Transplantation 83 (4), 448–457. doi:10.1097/01.tp.0000251373.17997.9a
Mas, V. R., Mas, L. A., Archer, K. J., Yanek, K., King, A. L., Gibney, E. M., et al. (2007b). Evaluation of gene panel mRNAs in urine samples of kidney transplant recipients as a non-invasive tool of graft function. Mol. Med. 13 (5-6), 315–324. doi:10.2119/2007–00017.Mas
McGrogan, A., Franssen, C. F., and de Vries, C. S. (2011). The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol. Dial. Transpl. 26 (2), 414–430. doi:10.1093/ndt/gfq665
Medina, D., and Arnold, A. C. (2019). Angiotensin-(1-7): translational avenues in cardiovascular control. Am. J. Hypertens. 32 (12), 1133–1142. doi:10.1093/ajh/hpz146
Melderis, S., Hagenstein, J., Warkotsch, M. T., Dang, J., Herrnstadt, G. R., Niehus, C. B., et al. (2020). Amphiregulin aggravates glomerulonephritis via recruitment and activation of myeloid cells. J. Am. Soc. Nephrol. 31 (9), 1996–2012. doi:10.1681/ASN.2019111215
Melenhorst, W. B., Mulder, G. M., Xi, Q., Hoenderop, J. G. J., Kimura, K., Eguchi, S., et al. (2008). Epidermal growth factor receptor signaling in the kidney: key roles in physiology and disease. Hypertension 52 (6), 987–993. doi:10.1161/HYPERTENSIONAHA.108.113860
Meng, H., Liang, Y., Hao, J., and Lu, J. (2018). Comparison of rejection-specific genes in peripheral blood and allograft biopsy from kidney transplant. Transpl. Proc. 50 (1), 115–123. doi:10.1016/j.transproceed.2017.11.022
Mezzano, S., Droguett, A., Burgos, M. E., Ardiles, L. G., Flores, C. A., Aros, C. A., et al. (2003). Renin-angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int. Suppl. 64 (86), S64–S70. doi:10.1046/j.1523-1755.64.s86.12.x
Mizuiri, S., and Ohashi, Y. (2015). ACE and ACE2 in kidney disease. World J. Nephrol. 4 (1), 74–82. doi:10.5527/wjn.v4.i1.74
Nomura, H., Kuruppu, S., and Rajapakse, N. W. (2022). Stimulation of angiotensin converting enzyme 2: a novel treatment strategy for diabetic nephropathy. Front. Physiol. 12, 813012. doi:10.3389/fphys.2021.813012
Normanno, N., Bianco, C., Strizzi, L., Mancino, M., Maiello, M. R., De Luca, A., et al. (2005). The ErbB receptors and their ligands in cancer: an overview. Curr. Drug Targets 6 (3), 243–257. doi:10.2174/1389450053765879
Nørregaard, R., Mutsaers, H. A. M., Frøkiær, J., and Kwon, T. H. (2023). Obstructive nephropathy and molecular pathophysiology of renal interstitial fibrosis. Physiol. Rev. 103 (4), 2827–2872. doi:10.1152/physrev.00027.2022
Nourie, N., Ghaleb, R., Lefaucheur, C., and Louis, K. (2024). Toward precision medicine: exploring the landscape of biomarkers in acute kidney injury. Biomolecules 14 (1), 82. doi:10.3390/biom14010082
Oikawa, H., Maesawa, C., Tatemichi, Y., Nishinari, Y., Nishiya, M., Mizugai, H., et al. (2014). A disintegrin and metalloproteinase 17 (ADAM17) mediates epidermal growth factor receptor transactivation by angiotensin II on hepatic stellate cells. Life Sci. 97 (2), 137–144. doi:10.1016/j.lfs.2013.12.028
Ortiz-Melo, D. I., and Gurley, S. B. (2016). Angiotensin converting enzyme 2 and the kidney. Curr. Opin. Nephrol. Hypertens. 25 (1), 59–66. doi:10.1097/MNH.0000000000000182
Overstreet, J. M., Wang, Y., Wang, X., Niu, A., Gewin, L. S., Yao, B., et al. (2017). Selective activation of epidermal growth factor receptor in renal proximal tubule induces tubulointerstitial fibrosis. FASEB J. 31 (10), 4407–4421. doi:10.1096/fj.201601359RR
Patel, S., Tang, J., Overstreet, J. M., Anorga, S., Lian, F., Arnouk, A., et al. (2019). Rac-GTPase promotes fibrotic TGF-β1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways. FASEB J. 33 (9), 9797–9810. doi:10.1096/fj.201802489RR
Paz Ocaranza, M., Riquelme, J. A., García, L., Jalil, J. E., Chiong, M., Santos, R. A. S., et al. (2020). Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 17 (2), 116–129. doi:10.1038/s41569-019-0244-8
Pegg, K., Zhang, J., Pollock, C., and Saad, S. (2013). Combined effects of PPAR γ agonists and epidermal growth factor receptor inhibitors in human proximal tubule cells. PPAR Res. 2013, 982462. doi:10.1155/2013/982462
Pesce, F., Stea, E. D., Rossini, M., Fiorentino, M., Piancone, F., Infante, B., et al. (2021). Glomerulonephritis in AKI: from pathogenesis to therapeutic intervention. Front. Med. (Lausanne) 7, 582272. doi:10.3389/fmed.2020.582272
Ponnusamy, M., Ma, L., and Zhuang, S. (2013). Necrotic renal epithelial cell inhibits renal interstitial fibroblast activation: role of protein tyrosine phosphatase 1B. Am. J. Physiol. Ren. Physiol. 304 (6), F698–F709. doi:10.1152/ajprenal.00564.2012
Ponnusamy, M., Zhou, X., Yan, Y., Tang, J., Tolbert, E., Zhao, T. C., et al. (2014). Blocking sirtuin 1 and 2 inhibits renal interstitial fibroblast activation and attenuates renal interstitial fibrosis in obstructive nephropathy. J. Pharmacol. Exp. Ther. 350 (2), 243–256. doi:10.1124/jpet.113.212076
Popović, L., Wintgens, J. P., Wu, Y., Brankatschk, B., Menninger, S., Degenhart, C., et al. (2024). Profiling of ERBB receptors and downstream pathways reveals selectivity and hidden properties of ERBB4 antagonists. iScience 27 (2), 108839. doi:10.1016/j.isci.2024.108839
Portik-Dobos, V., Harris, A. K., Song, W., Hutchinson, J., Johnson, M. H., Imig, J. D., et al. (2006). Endothelin antagonism prevents early EGFR transactivation but not increased matrix metalloproteinase activity in diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290 (2), R435–R441. doi:10.1152/ajpregu.00300.2005
Qian, Y., Peng, K., Qiu, C., Skibba, M., Huang, Y., Xu, Z., et al. (2016). Novel epidermal growth factor receptor inhibitor attenuates angiotensin II-induced kidney fibrosis. J. Pharmacol. Exp. Ther. 356 (1), 32–42. doi:10.1124/jpet.115.228080
Qing, X., Chinenov, Y., Redecha, P., Madaio, M., Roelofs, J. J., Farber, G., et al. (2018). iRhom2 promotes lupus nephritis through TNF-α and EGFR signaling. J. Clin. Invest 128 (4), 1397–1412. doi:10.1172/JCI97650
Rayego-Mateos, S., Morgado-Pascual, J. L., Rodrigues-Diez, R. R., Rodrigues-Diez, R., Falke, L. L., Mezzano, S., et al. (2018a). Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J. Pathol. 244 (2), 227–241. doi:10.1002/path.5007
Rayego-Mateos, S., Rodrigues-Diez, R., Morgado-Pascual, J. L., Valentijn, F., Valdivielso, J. M., Goldschmeding, R., et al. (2018b). Role of epidermal growth factor receptor (EGFR) and its ligands in kidney inflammation and damage. Mediat. Inflamm. 2018, 8739473. doi:10.1155/2018/8739473
Re, R. N. (2018). Role of intracellular angiotensin II. Am. J. Physiol. Heart Circ. Physiol. 314 (4), H766–H771. doi:10.1152/ajpheart.00632.2017
Rintala, J. M., Savikko, J., Rintala, S. E., Palin, N., and Koskinen, P. K. (2016). Epidermal growth factor receptor inhibition with erlotinib ameliorates anti-Thy 1.1-induced experimental glomerulonephritis. J. Nephrol. 29 (3), 359–365. doi:10.1007/s40620-015-0233-x
Saad, S., Stevens, V. A., Wassef, L., Poronnik, P., Kelly, D. J., Gilbert, R. E., et al. (2005). High glucose transactivates the EGF receptor and up-regulates serum glucocorticoid kinase in the proximal tubule. Kidney Int. 68 (3), 985–997. doi:10.1111/j.1523-1755.2005.00492.x
Samarakoon, R., Overstreet, J. M., and Higgins, P. J. (2013). TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal 25 (1), 264–268. doi:10.1016/j.cellsig.2012.10.003
Sanchez-Soria, P., and Camenisch, T. D. (2010). ErbB signaling in cardiac development and disease. Semin. Cell Dev. Biol. 21 (9), 929–935. doi:10.1016/j.semcdb.2010.09.011
Santos, R. A. S., Oudit, G. Y., Verano-Braga, T., Canta, G., Steckelings, U. M., and Bader, M. (2019). The renin-angiotensin system: going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 316 (5), H958–H970. doi:10.1152/ajpheart.00723.2018
Sanz, A. B., Sanchez-Niño, M. D., Ramos, A. M., Moreno, J. A., Santamaria, B., Ruiz-Ortega, M., et al. (2010). NF-kappaB in renal inflammation. J. Am. Soc. Nephrol. 21 (8), 1254–1262. doi:10.1681/ASN.2010020218
Satoskar, A. A., Parikh, S. V., and Nadasdy, T. (2020). Epidemiology, pathogenesis, treatment and outcomes of infection-associated glomerulonephritis. Nat. Rev. Nephrol. 16 (1), 32–50. doi:10.1038/s41581-019-0178-8
Sawinski, D., and Blumberg, E. A. (2019). Infection in renal transplant recipients. Chronic Kidney Dis. Dialysis, Transplant. 621–638.e6. doi:10.1016/B978-0-323-52978-5.00040-9
Schindler, C., Bramlage, P., Kirch, W., and Ferrario, C. M. (2007). Role of the vasodilator peptide angiotensin-(1-7) in cardiovascular drug therapy. Vasc. Health Risk Manag. 3 (1), 125–137.
Schreier, B., Gekle, M., and Grossmann, C. (2014). Role of epidermal growth factor receptor in vascular structure and function. Curr. Opin. Nephrol. Hypertens. 23 (2), 113–121. doi:10.1097/01.mnh.0000441152.62943.29
Seo, H. Y., Jeon, J. H., Jung, Y. A., Jung, G. S., Lee, E. J., Choi, Y. K., et al. (2016). Fyn deficiency attenuates renal fibrosis by inhibition of phospho-STAT3. Kidney Int. 90 (6), 1285–1297. doi:10.1016/j.kint.2016.06.038
Shaban, N., Kamashev, D., Emelianova, A., and Buzdin, A. (2023). Targeted inhibitors of EGFR: structure, biology, biomarkers, and clinical applications. Cells 13 (1), 47. doi:10.3390/cells13010047
Shams, S. F., Eidgahi, E. S., Lotfi, Z., Khaledi, A., Shakeri, S., Sheikhi, M., et al. (2017). Urinary tract infections in kidney transplant recipients 1st year after transplantation. J. Res. Med. Sci. 22, 20. doi:10.4103/1735-1995.200274
Sharifi, J., Khirehgesh, M. R., Safari, F., and Akbari, B. (2021). EGFR and anti-EGFR nanobodies: review and update. J. Drug Target 29 (4), 387–402. doi:10.1080/1061186X.2020.1853756
Shen, M., Morton, J., Davidge, S. T., and Kassiri, Z. (2017). Loss of smooth muscle cell disintegrin and metalloproteinase 17 transiently suppresses angiotensin II-induced hypertension and end-organ damage. J. Mol. Cell Cardiol. 103, 11–21. doi:10.1016/j.yjmcc.2016.12.001
Sheng, L., Bayliss, G., and Zhuang, S. (2021). Epidermal growth factor receptor: a potential therapeutic target for diabetic kidney disease. Front. Pharmacol. 11, 598910. doi:10.3389/fphar.2020.598910
Sheng, L., Yang, M., Ding, W., Zhang, M., Niu, J., Qiao, Z., et al. (2016). Epidermal growth factor receptor signaling mediates aldosterone-induced profibrotic responses in kidney. Exp. Cell Res. 346 (1), 99–110. doi:10.1016/j.yexcr.2016.06.009
Shi, Y., Lo, C. S., Padda, R., Abdo, S., Chenier, I., Filep, J. G., et al. (2015). Angiotensin-(1-7) prevents systemic hypertension, attenuates oxidative stress and tubulointerstitial fibrosis, and normalizes renal angiotensin-converting enzyme 2 and Mas receptor expression in diabetic mice. Clin. Sci. (Lond). 128 (10), 649–663. doi:10.1042/CS20140329
Shraim, B. A., Moursi, M. O., Benter, I. F., Habib, A. M., and Akhtar, S. (2021). The role of epidermal growth factor receptor family of receptor tyrosine kinases in mediating diabetes-induced cardiovascular complications. Front. Pharmacol. 12, 701390. doi:10.3389/fphar.2021.701390
Sibilia, M., Kroismayr, R., Lichtenberger, B. M., Natarajan, A., Hecking, M., and Holcmann, M. (2007). The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 75 (9), 770–787. doi:10.1111/j.1432-0436.2007.00238.x
Sigismund, S., Avanzato, D., and Lanzetti, L. (2018). Emerging functions of the EGFR in cancer. Mol. Oncol. 12 (1), 3–20. doi:10.1002/1878-0261.12155
Silveira, K. D., Barroso, L. C., Vieira, A. T., Cisalpino, D., Lima, C. X., Bader, M., et al. (2013). Beneficial effects of the activation of the angiotensin-(1-7) MAS receptor in a murine model of adriamycin-induced nephropathy. PLoS One 8 (6), e66082. doi:10.1371/journal.pone.0066082
Simões e Silva, A. C., Silveira, K. D., Ferreira, A. J., and Teixeira, M. M. (2013). ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 169 (3), 477–492. doi:10.1111/bph.12159
Singh, S., Sadhukhan, S., and Sonawane, A. (2023). 20 years since the approval of first EGFR-TKI, gefitinib: insight and foresight. Biochim. Biophys. Acta Rev. Cancer 1878 (6), 188967. doi:10.1016/j.bbcan.2023.188967
Skibba, M., Qian, Y., Bao, Y., Lan, J., Peng, K., Zhao, Y., et al. (2016). New EGFR inhibitor, 453, prevents renal fibrosis in angiotensin II-stimulated mice. Eur. J. Pharmacol. 789, 421–430. doi:10.1016/j.ejphar.2016.08.009
Slattery, C., Jang, Y., Kruger, W. A., Hryciw, D. H., Lee, A., and Poronnik, P. (2013). γ-Secretase inhibition promotes fibrotic effects of albumin in proximal tubular epithelial cells. Br. J. Pharmacol. 169 (6), 1239–1251. doi:10.1111/bph.12214
Smith, J. A., Stallons, L. J., and Schnellmann, R. G. (2014). Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 307 (4), F435–F444. doi:10.1152/ajprenal.00271.2014
Son, S. S., Hwang, S., Park, J. H., Ko, Y., Yun, S. I., Lee, J. H., et al. (2021). In vivo silencing of amphiregulin by a novel effective Self-Assembled-Micelle inhibitory RNA ameliorates renal fibrosis via inhibition of EGFR signals. Sci. Rep. 11 (1), 2191. doi:10.1038/s41598-021-81726-2
Song, N., Thaiss, F., and Guo, L. (2019). NFκB and kidney injury. Front. Immunol. 10, 815. doi:10.3389/fimmu.2019.00815
Staruschenko, A., Palygin, O., Ilatovskaya, D. V., and Pavlov, T. S. (2013). Epidermal growth factors in the kidney and relationship to hypertension. Am. J. Physiol. Ren. Physiol. 305 (1), F12–F20. doi:10.1152/ajprenal.00112.2013
Sun, H. J. (2019). Current opinion for hypertension in renal fibrosis. Adv. Exp. Med. Biol. 1165, 37–47. doi:10.1007/978-981-13-8871-2_3
Sun, S. F., Tang, P. M. K., Feng, M., Xiao, J., Huang, X. R., Li, P., et al. (2018). Novel lncRNA erbb4-IR promotes diabetic kidney injury in db/db mice by targeting miR-29b. Diabetes 67 (4), 731–744. doi:10.2337/db17-0816
Swaminathan, N., Vincent, M., Sassard, J., and Sambhi, M. P. (1996). Elevated epidermal growth factor receptor levels in hypertensive Lyon rat kidney and aorta. Clin. Exp. Pharmacol. Physiol. 23 (9), 793–796. doi:10.1111/j.1440-1681.1996.tb01181.x
Tang, J., Liu, N., Tolbert, E., Ponnusamy, M., Ma, L., Gong, R., et al. (2013b). Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am. J. Pathol. 183 (1), 160–172. doi:10.1016/j.ajpath.2013.04.005
Tang, J., Liu, N., and Zhuang, S. (2013a). Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 83 (5), 804–810. doi:10.1038/ki.2012.435
Tang, J., Yan, Y., Zhao, T. C., Gong, R., Bayliss, G., Yan, H., et al. (2014). Class I HDAC activity is required for renal protection and regeneration after acute kidney injury. Am. J. Physiol. Ren. Physiol. 307 (3), F303–F316. doi:10.1152/ajprenal.00102.2014
Teixeira, L. B., Parreiras-E-Silva, L. T., Bruder-Nascimento, T., Duarte, D. A., Simões, S. C., Costa, R. M., et al. (2017). Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 7 (1), 11903. doi:10.1038/s41598-017-12074-3
Threadgill, D. W., Dlugosz, A. A., Hansen, L. A., Tennenbaum, T., Lichti, U., Yee, D., et al. (1995). Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 269 (5221), 230–234. doi:10.1126/science.7618084
Tikellis, C., Johnston, C. I., Forbes, J. M., Burns, W. C., Burrell, L. M., Risvanis, J., et al. (2003). Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension 41 (3), 392–397. doi:10.1161/01.HYP.0000060689.38912.CB
Tsai, C. J., and Nussinov, R. (2019). Emerging allosteric mechanism of EGFR activation in physiological and pathological contexts. Biophys. J. 117 (1), 5–13. doi:10.1016/j.bpj.2019.05.021
Udani, S., Lazich, I., and Bakris, G. L. (2011). Epidemiology of hypertensive kidney disease. Nat. Rev. Nephrol. 7 (1), 11–21. doi:10.1038/nrneph.2010.154
Ulu, N., Mulder, G. M., Vavrinec, P., Landheer, S. W., Duman-Dalkilic, B., Gurdal, H., et al. (2013). Epidermal growth factor receptor inhibitor PKI-166 governs cardiovascular protection without beneficial effects on the kidney in hypertensive 5/6 nephrectomized rats. J. Pharmacol. Exp. Ther. 345 (3), 393–403. doi:10.1124/jpet.113.203497
Uttarwar, L., Gao, B., Ingram, A. J., and Krepinsky, J. C. (2012). SREBP-1 activation by glucose mediates TGF-β upregulation in mesangial cells. Am. J. Physiol. Ren. Physiol. 302 (3), F329–F341. doi:10.1152/ajprenal.00136.2011
Uttarwar, L., Peng, F., Wu, D., Kumar, S., Gao, B., Ingram, A. J., et al. (2011). HB-EGF release mediates glucose-induced activation of the epidermal growth factor receptor in mesangial cells. Am. J. Physiol. Ren. Physiol. 300 (4), F921–F931. doi:10.1152/ajprenal.00436.2010
Vandekerckhove, L., Vermeulen, Z., Liu, Z. Z., Boimvaser, S., Patzak, A., Segers, V. F. M., et al. (2016). Neuregulin-1 attenuates development of nephropathy in a type 1 diabetes mouse model with high cardiovascular risk. Am. J. Physiol. Endocrinol. Metab. 310 (7), E495–E504. doi:10.1152/ajpendo.00432.2015
Van Twist, D. J., Houben, A. J., De Haan, M. W., Mostard, G. J., De Leeuw, P. W., and Kroon, A. A. (2014). Angiotensin-(1-7)-induced renal vasodilation is reduced in human kidneys with renal artery stenosis. J. Hypertens. 32 (12), 2428–2432. doi:10.1097/HJH.0000000000000351
Wang, H., Chen, X., Su, Y., Paueksakon, P., Hu, W., Zhang, M. Z., et al. (2015). p47(phox) contributes to albuminuria and kidney fibrosis in mice. Kidney Int. 87 (5), 948–962. doi:10.1038/ki.2014.386
Wang, J., Li, J., Zhang, X., Zhang, M., Hu, X., and Yin, H. (2022). Molecular mechanisms of histone deacetylases and inhibitors in renal fibrosis progression. Front. Mol. Biosci. 9, 986405. doi:10.3389/fmolb.2022.986405
Wang, L., Wang, J., Zhang, Y., and Zhang, H. (2024). Current perspectives and trends of the research on hypertensive nephropathy: a bibliometric analysis from 2000 to 2023. Ren. Fail 46 (1), 2310122. doi:10.1080/0886022X.2024.2310122
Wee, P., and Wang, Z. (2017). Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) 9 (5), 52. doi:10.3390/cancers9050052
Wetmore, J. B., Guo, H., Liu, J., Collins, A. J., and Gilbertson, D. T. (2016). The incidence, prevalence, and outcomes of glomerulonephritis derived from a large retrospective analysis. Kidney Int. 90 (4), 853–860. doi:10.1016/j.kint.2016.04.026
Whaley-Connell, A., Habibi, J., Panfili, Z., Hayden, M. R., Bagree, S., Nistala, R., et al. (2011). Angiotensin II activation of mTOR results in tubulointerstitial fibrosis through loss of N-cadherin. Am. J. Nephrol. 34 (2), 115–125. doi:10.1159/000329327
Wolf, G. (2004). New insights into the pathophysiology of diabetic nephropathy: from haemodynamics to molecular pathology. Eur. J. Clin. Invest 34 (12), 785–796. doi:10.1111/j.1365-2362.2004.01429.x
Xu, L., Li, X., Zhang, F., Wu, L., Dong, Z., and Zhang, D. (2019). EGFR drives the progression of AKI to CKD through HIPK2 overexpression. Theranostics 9 (9), 2712–2726. doi:10.7150/thno.31424
Xu, Z., Zhao, Y., Zhong, P., Wang, J., Weng, Q., Qian, Y., et al. (2017). EGFR inhibition attenuates diabetic nephropathy through decreasing ROS and endoplasmic reticulum stress. Oncotarget 8 (20), 32655–32667. doi:10.18632/oncotarget.15948
Yamamoto, Y., Iyoda, M., Tachibana, S., Matsumoto, K., Wada, Y., Suzuki, T., et al. (2018). Erlotinib attenuates the progression of chronic kidney disease in rats with remnant kidney. Nephrol. Dial. Transpl. 33 (4), 598–606. doi:10.1093/ndt/gfx264
Yamaoka, T., Ohba, M., and Ohmori, T. (2017). Molecular-targeted therapies for epidermal growth factor receptor and its resistance mechanisms. Int. J. Mol. Sci. 18 (11), 2420. doi:10.3390/ijms18112420
Yammine, L., Zablocki, A., Baron, W., Terzi, F., and Gallazzini, M. (2019). Lipocalin-2 regulates epidermal growth factor receptor intracellular trafficking. Cell Rep. 29 (7), 2067–2077. doi:10.1016/j.celrep.2019.10.015
Yan, Y., Ma, L., Zhou, X., Ponnusamy, M., Tang, J., Zhuang, M. A., et al. (2016). Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. 89 (1), 68–81. doi:10.1038/ki.2015.293
Yang, L., Yuan, H., Yu, Y., Yu, N., Ling, L., Niu, J., et al. (2019). Epidermal growth factor receptor mimotope alleviates renal fibrosis in murine unilateral ureteral obstruction model. Clin. Immunol. 205, 57–64. doi:10.1016/j.clim.2019.05.014
Ye, M., Wysocki, J., Naaz, P., Salabat, M. R., LaPointe, M. S., and Batlle, D. (2004). Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice: a renoprotective combination? Hypertension 43 (5), 1120–1125. doi:10.1161/01.HYP.0000126192.27644.76
Yeh, T. H., Tu, K. C., Wang, H. Y., and Chen, J. Y. (2024). From acute to chronic: unraveling the pathophysiological mechanisms of the progression from acute kidney injury to acute kidney disease to chronic kidney disease. Int. J. Mol. Sci. 25 (3), 1755. doi:10.3390/ijms25031755
Ying, W. Z., and Sanders, P. W. (2005). Enhanced expression of EGF receptor in a model of salt-sensitive hypertension. Am. J. Physiol. Ren. Physiol. 289 (2), F314–F321. doi:10.1152/ajprenal.00003.2005
Yousif, M. H., Benter, I. F., and Akhtar, S. (2005). The role of tyrosine kinase-mediated pathways in diabetes-induced alterations in responsiveness of rat carotid artery. Auton. Autacoid Pharmacol. 25 (2), 69–78. doi:10.1111/j.1474-8673.2004.00333.x
Yousif, M. H., Dhaunsi, G. S., Makki, B. M., Qabazard, B. A., Akhtar, S., and Benter, I. F. (2012). Characterization of Angiotensin-(1-7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacol. Res. 66 (3), 269–275. doi:10.1016/j.phrs.2012.05.001
Zaman, A., and Banday, A. A. (2022). Angiotensin (1-7) protects against renal ischemia-reperfusion injury via regulating expression of NRF2 and microRNAs in Fisher 344 rats. Am. J. Physiol. Ren. Physiol. 323 (1), F33–F47. doi:10.1152/ajprenal.00283.2021
Zeid, A. M., Lamontagne, J. O., Zhang, H., and Marneros, A. G. (2022). Epidermal growth factor deficiency predisposes to progressive renal disease. FASEB J. 36 (5), e22286. doi:10.1096/fj.202101837R
Zeng, F., Kloepfer, L. A., Finney, C., Diedrich, A., and Harris, R. C. (2016). Specific endothelial heparin-binding EGF-like growth factor deletion ameliorates renal injury induced by chronic angiotensin II infusion. Am. J. Physiol. Ren. Physiol. 311 (4), F695–F707. doi:10.1152/ajprenal.00377.2015
Zeng, F., Singh, A. B., and Harris, R. C. (2009). The role of the EGF family of ligands and receptors in renal development, physiology and pathophysiology. Exp. Cell Res. 315 (4), 602–610. doi:10.1016/j.yexcr.2008.08.005
Zhan, H. Q., Zhang, X., Chen, X. L., Cheng, L., and Wang, X. (2024). Application of nanotechnology in the treatment of glomerulonephritis: current status and future perspectives. J. Nanobiotechnology 22 (1), 9. doi:10.1186/s12951-023-02257-8
Zhang, M. Z., Wang, Y., Paueksakon, P., and Harris, R. C. (2014). Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes 63 (6), 2063–2072. doi:10.2337/db13-1279
Zhu, Q., Dong, H., Bukhari, A. A., Zhao, A., Li, M., Sun, Y., et al. (2020). HUWE1 promotes EGFR ubiquitination and degradation to protect against renal tubulointerstitial fibrosis. FASEB J. 34 (3), 4591–4601. doi:10.1096/fj.201902751R
Zhu, Y., Yu, J., Yin, L., Zhou, Y., Sun, Z., Jia, H., et al. (2016). MicroRNA-146b, a sensitive indicator of mesenchymal stem cell repair of acute renal injury. Stem Cells Transl. Med. 5 (10), 1406–1415. doi:10.5966/sctm.2015-0355
Zimmerman, D., and Burns, K. D. (2012). Angiotensin-(1-7) in kidney disease: a review of the controversies. Clin. Sci. (Lond). 123 (6), 333–346. doi:10.1042/CS20120111
Keywords: epidermal growth factor receptor, ErbB, diabetic nephropathy, hypertensive nephropathy, glomerulonephritis, chronic kidney disease, renal fibrosis, acute kidney injury (AKI)
Citation: Tawengi M, Al-Dali Y, Tawengi A, Benter IF and Akhtar S (2024) Targeting the epidermal growth factor receptor (EGFR/ErbB) for the potential treatment of renal pathologies. Front. Pharmacol. 15:1394997. doi: 10.3389/fphar.2024.1394997
Received: 02 March 2024; Accepted: 29 July 2024;
Published: 21 August 2024.
Edited by:
Muthuvel Jayachandran, Mayo Clinic, United StatesReviewed by:
Takashi Kato, Yasuda Women’s University, JapanKiyoshi Mori, Shizuoka Graduate University of Public Health, Japan
Copyright © 2024 Tawengi, Al-Dali, Tawengi, Benter and Akhtar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saghir Akhtar, cy5ha2h0YXJAcXUuZWR1LnFh; c2FfcGhhcm1hQGhvdG1haWwuY28udWs=
†These authors have contributed equally to this work and share first authorship