 Devadoss J. Samuvel
Devadoss J. Samuvel John J. Lemasters1,2
John J. Lemasters1,2 C. James Chou
C. James Chou Zhi Zhong
Zhi Zhong- 1Departments of Drug Discovery and Biomedical Sciences, Charleston, SC, United States
- 2Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC, United States
- 3Lydex Pharmaceuticals, Mount Pleasant, SC, United States
Effective therapy for liver fibrosis is lacking. Here, we examined whether LP340, the lead candidate of a new-generation of hydrazide-based HDAC1,2,3 inhibitors (HDACi), decreases liver fibrosis. Liver fibrosis was induced by CCl4 treatment and bile duct ligation (BDL) in mice. At 6 weeks after CCl4, serum alanine aminotransferase increased, and necrotic cell death and leukocyte infiltration occurred in the liver. Tumor necrosis factor-α and myeloperoxidase markedly increased, indicating inflammation. After 6 weeks, α-smooth muscle actin (αSMA) and collagen-1 expression increased by 80% and 575%, respectively, indicating hepatic stellate cell (HSC) activation and fibrogenesis. Fibrosis detected by trichrome and Sirius-red staining occurred primarily in pericentral regions with some bridging fibrosis in liver sections. 4-Hydroxynonenal adducts (indicator of oxidative stress), profibrotic cytokine transforming growth factor-β (TGFβ), and TGFβ downstream signaling molecules phospho-Smad2/3 also markedly increased. LP340 attenuated indices of liver injury, inflammation, and fibrosis markedly. Moreover, Ski-related novel protein-N (SnoN), an endogenous inhibitor of TGFβ signaling, decreased, whereas SnoN expression suppressor microRNA-23a (miR23a) increased markedly. LP340 (0.05 mg/kg, ig., daily during the last 2 weeks of CCl4 treatment) decreased 4-hydroxynonenal adducts and miR23a production, blunted SnoN decreases, and inhibited the TGFβ/Smad signaling. By contrast, LP340 had no effect on matrix metalloproteinase-9 expression. LP340 increased histone-3 acetylation but not tubulin acetylation, indicating that LP340 inhibited Class-I but not Class-II HDAC in vivo. After BDL, focal necrosis, inflammation, ductular reactions, and portal and bridging fibrosis occurred at 2 weeks, and αSMA and collagen-1 expression increased by 256% and 560%, respectively. LP340 attenuated liver injury, ductular reactions, inflammation, and liver fibrosis. LP340 also decreased 4-hydroxynonenal adducts and miR23a production, prevented SnoN decreases, and inhibited the TGFβ/Smad signaling after BDL. In vitro, LP340 inhibited immortal human hepatic stellate cells (hTERT-HSC) activation in culture (αSMA and collagen-1 expression) as well as miR23a production, demonstrating its direct inhibitory effects on HSC. In conclusions, LP340 is a promising therapy for both portal and pericentral liver fibrosis, and it works by inhibiting oxidative stress and decreasing miR23a.
1 Introduction
Liver fibrosis is the excessive accumulation of extracellular matrix (ECM) including collagen in the liver, which occurs in patients with many chronic liver diseases (Pinzani, 1999; Bataller and Brenner, 2005). Depending on the background chronic liver disease, liver fibrosis may begin primarily around portal tracts (portal fibrosis), such as in chronic cholestatic liver disease and viral hepatitis, or occurs initially in pericentral and perisinusoidal areas (pericentral/perivenular fibrosis), such as in alcohol-associated liver disease (Pinzani, 1999; Bataller and Brenner, 2005). As hepatic fibrosis advances, bridging fibrosis is formed and ultimately cirrhosis occurs (Pinzani, 1999; Bataller and Brenner, 2005). In its advanced stage, liver cirrhosis, parenchymal cells are replaced by collagen fibers, liver architecture becomes distorted, and hepatic blood flow is disturbed, leading to portal hypertension, chronic liver failure, severe complications, and death (Crawford, 2002; Rahimi and Rockey, 2013). Liver fibrosis/cirrhosis affects >100 million people and represents one of the most common causes of death in adults in the world (Friedman, 2003; Asrani et al., 2019). Moreover, 60%–90% of cases of hepatocellular carcinoma (HCC), a highly malignant tumor, arise on a background of liver fibrosis (Scaglione et al., 2015).
Although the most effective anti-fibrotic therapies are those targeting the underlying diseases causing fibrogenesis, such as antiviral therapy for viral hepatitis and iron chelation for hemochromatosis (Powell and Kerr, 1970; Poynard et al., 2002; Hadziyannis et al., 2003), therapy for many liver diseases is still lacking, working only in certain patient populations, or incompletely effective. Moreover, onset of many liver diseases and the subsequent development of liver fibrosis is often insidious. In the U.S, majority of liver disease patients do not know that they have liver disease until advanced fibrosis occurs, which substantially delays the treatment of the underlying diseases (Scaglione et al., 2015). Despite extensive studies, FDA-approved pharmaceutical therapy for fibrosis is still unavailable. The only clinically proven treatment for cirrhosis is liver transplantation (LT), whereas the availability of LT is very low due to the severe shortage of donor livers (Meirelles Junior et al., 2015). Thus, blockade of common profibrogenic and proinflammatory pathways and/or stimulation of resolution of fibrosis represents possible alternative therapeutic approaches. No doubt, development of effective antifibrotic therapy is an urgent need for medicine.
Histone deacetylases (HDACs) are a group of epigenetic enzymes that catalyze histone deacetylation, which subsequently alters gene expression (Kuo and Allis, 1998). Histone deacetylation is also strongly associated with dysregulated expression of microRNAs, which can affect numerous biological and pathological processes (Suzuki et al., 2013; Soliman et al., 2018; Ramzan et al., 2021). In addition to deacetylating histones, HDACs also remove acetyl moieties from lysine residues on non-histone proteins, thus affecting their activation, localization, function, and degradation (Kiernan et al., 2003; Glozak et al., 2005). Eighteen isoforms of HDAC exist in mammals, belonging to 4 classes (Hammond et al., 2017; Yoon et al., 2019). HDACs regulate many biological and pathological processes, including cell death, proliferation, inflammation, fibrosis, and cancer (Shao et al., 2004; Tao et al., 2014; Li et al., 2020). In recent years, accumulated evidence suggests that HDACs stimulate fibrogenesis in several organs, such as the heart, lung, kidney, and liver (Van Beneden et al., 2013; Chen et al., 2015; Moran-Salvador and Mann, 2017; Yoon et al., 2019). As global levels of acetylation of histones H3 and H4 progressively decrease, hepatic stellate cell (HSC) activation, a key step of liver fibrosis, occurs (Chen et al., 2015). Inhibition of HDAC1-3 (Class I HDAC) suppresses HSC activation and induces apoptosis and autophagic cell death of activated HSC (Liu et al., 2013; Shaker et al., 2013). Some studies show that knockdown of HDAC4, but not HDAC5 or HDAC6 (Class II HDAC), partially hinders HSC activation through induction of microRNA-29 (Mannaerts et al., 2013). Some HDAC inhibitors (HDACi) [e.g., trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA)] have been shown to decrease fibrotic responses in vitro and in vivo (Mannaerts et al., 2010; Shaker et al., 2013; Van Beneden et al., 2013; Chen et al., 2015). However, successful clinical trials of HDACi against liver fibrosis have not been reported. Possibly, different subtypes of HDAC exert different effects on the development and resolution of fibrosis. Therefore, isozyme selective HDACi may be needed for prevention/treatment of fibrosis. Moreover, all HDACi currently on the market are for cancer therapy, and all have poor in vivo pharmacokinetics (PK), low HDAC isozyme selectivity, and long-term safety concerns regarding potential mutagenicity (Shen and Kozikowski, 2016). Therefore, more selective, potent, and safe HDACi are needed for assessment in treating liver fibrosis.
Recently, we described a new generation of hydrazide-based HDACi that inhibit HDAC1,2,3 both allosterically and competitively. These HDACi have substantially higher potency, excellent PK properties, and lower toxicity (McClure et al., 2016; Li et al., 2018). Moreover, we showed recently that LP342, one of the lead compounds of new class of HDACi, protects against hepatic ischemia/reperfusion injury (Samuvel et al., 2023). In this study, we explored whether LP340, another lead compound, decreases liver fibrosis and examined potential mechanisms of its protection.
2 Materials and methods
2.1 Materials
The sources of all chemicals, antibodies, and other reagents are listed in Supplementary Table S1 in “Supplementary Material.”
2.2 Synthesis of LP340
The molecular structure, synthesis, purification, and characterization of LP340 are described elsewhere (Jiang et al., 2022).
2.3 Animals
Liver fibrosis was induced in mice by bile duct ligation (BDL) and carbon tetrachloride (CCl4) treatment, respectively (Trams and Symons, 1957; Rehman et al., 2008; Rehman et al., 2016). For BDL, male C57Bl/6 mice (8–9 weeks, Jackson Laboratory, Bar Harbor, ME) underwent a midline abdominal incision under isoflurane (2%–3%) anesthesia. The common bile duct was located, double ligated near the liver with 6-0 nylon suture, and transected between ligatures. For sham operation, the abdomen was opened under isoflurane anesthesia and then closed without BDL. During surgery, body temperature was maintained at ∼37°C with warming lamps. Mice were gavaged with LP340 (0.05 mg/kg) or equal volume of vehicle (0.5% DMSO, 0.5% hydroxypropyl methyl cellulose) at 2 h after surgery and once daily afterwards for 2 weeks at which time the mice were euthanized. To induce liver fibrosis with CCl4, male mice were injected with CCl4 (1:3 dilution in corn oil; 1.0 µL of dilution/g mouse, i.p.) or an equal volume of corn oil once every 3 days for up to 6 weeks. Since a previous study showed that female mice are more tolerant to CCl4 hepatotoxicity (Zhang et al., 2012), female mice were treated with 1.5 µL of the CCl4 dilution/g mouse, i.p.). Both male and female mice were treated with LP340 (0.05 mg/kg, i.g.) or equal volume of vehicle once daily during the last 2 weeks of CCl4 treatment (Figure 1A). Mice were euthanized after 6 weeks of CCl4 treatment. All animals were given humane care in compliance with institutional guidelines using protocols approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina (protocol number ARC# 2018-00641).
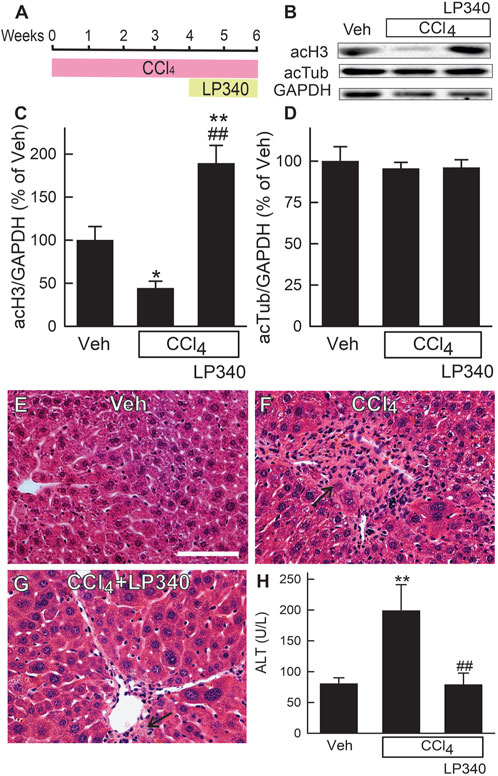
Figure 1. LP340 increases histone-3 acetylation and decreases alanine aminotransferase release and necrosis after CCl4 treatment. Male mice were injected with CCl4 (1:3 dilution in corn oil; 1.0 µL of dilution/g mouse, i.p.) or equal volume of corn oil once every 3 days for 6 weeks. LP340 (0.05 mg/kg, ig daily) or equal volume of vehicle (Veh) was administered during the last 2 weeks of CCl4 treatment. Blood and liver were collected after 6 weeks of CCl4 or corn oil treatment. (A) regimen of CCl4 and LP340 treatment. (B) representative immunoblots for acetylated histone-3 (acH3), acetylated tubulin (acTub), and housekeeping protein GAPDH; (C, D), quantification of acH3 and acTub immunoblots by densitometry. (E–G) representative images of liver histology. Bar is 50 µm. (H) serum ALT. *, p < 0.05 vs. vehicle; **, p < 0.01 vs. vehicle; ##, p < 0.01 vs. CCl4 without LP340. Data are means ± SEM (n = 3–4/group).
2.4 Culture of immortal human hepatic stellate cells
Activation of HSC is the key step in liver fibrosis. Therefore, we examined if LP340 suppresses HSC activation in vitro. Immortal human HSC (hTERT-HSC), a cell line that is often used to study the cell biology of human HSC in vitro (Schnabl et al., 2002), were cultured in DMEM high glucose medium (Supplementary Table S1) supplemented with 10% fetal bovine serum (FBS) at 5% CO2 and 37°C for 24 h to reach ∼70% confluence and then changed to the DMEM medium with 0.5% FBS and with or without LP340 (0.1 and 0.3 µM) for another 48 h. hTERT-HSC were then collected, lysed with ice-cold lysis buffer (Subedi et al., 2019), and lysates were used for immunoblotting or microRNA-23a (miR23a) measurement.
2.5 Serum alanine aminotransferase measurement
At the end of experiments, blood and liver were collected from mice under ketamine/xylazine anesthesia (90 mg/kg and 10 mg/kg, i.p.). Blood was collected by puncture of the inferior vena cava, and serum was isolated and stored at −80°C until use. Serum alanine aminotransferase (ALT) activity was determined using a commercial kit (Supplementary Table S1) according to the manufacturer’s instructions.
2.6 Histology and immunohistochemistry
After collection, part of the liver was stored at −80°C until use later, and the other part was fixed in 10% neutralized formaldehyde for 24–48 h and then embedded in paraffin. Liver sections (5 µm) were stained with hematoxylin-eosin (H&E) for histological examination. Liver sections were also stained with Mason’s trichrome staining and Sirius red/Fast green staining to reveal liver fibrosis (Kiernan, 2015; Rehman et al., 2016).
Proliferation of cholangiocytes was detected by immunohistochemical staining of cytokeratin-19 (CK19). Liver sections were deparaffinized and rehydrated, followed by antigen retrieval using antigen unmasking solution (Supplementary Table S1) according to the manufacturer’s instructions. Liver sections were pre-blocked with 3% H2O2 in distilled water and 2% bovine serum albumin (Supplementary Table S1) in 1% phosphate buffered saline with 0.1% Tween-20 (PBS-T) for 1 h at room temperature. Liver sections were then incubated with primary antibody for CK19 from rabbit (Supplementary Table S1, 1:500 dilution) at room temperature for 1 h. After washing in PBS-T 3 times (3 min each), sections were incubated with peroxidase conjugated secondary anti-rabbit antibody using a VECTASTAIN ABC kit (Supplementary Table S1). 3,3′-Diaminobenzidine (DAB) (Supplementary Table S1) was used to detect peroxidase. The sections were then counterstained with 1/5 Harris hematoxylin solution (Supplementary Table S1) for 1 min at room temperature. Liver images were acquired using a Zeiss AX10 microscope (White Plains, NY) and 10x - 40x objective lenses.
2.7 Detection of microRNA-23a in liver tissue and cell lysates
miRNAs were extracted from liver tissue (100 mg) or hTERT-HSC lysates using a miRNeasy micro-Kit (Supplementary Table S1) according to the manufacturer’s instructions. cDNAs were synthesized from 10 ng RNA using a miCURY LNA RT kit (Supplementary Table S1). Customized probes for miR23a and housekeeping gene U6 were used (Supplementary Table S1). Real-time PCR was performed using iQ™ SYBR Green Supermix (Supplementary Table S1) and a Bio-Rad CFX 96 Real time PCR System with incubations at 55°C for 5 min and 95°C for 5 min, followed by 39 cycles at 95°C for 10 s and 60°C for 1 min. The results were normalized to the expression of U6 miRNA. miR23a expression was determined by the delta-delta Ct method (Krishnasamy et al., 2016).
2.8 Immunoprecipitation of Smad4
Liver tissue was collected after 6 weeks of CCl4 treatment and homogenized in ice-cold lysis buffer (Subedi et al., 2019). The protein contents of lysates were determined using a Pierce BCA protein assay kit (Supplementary Table S1) according to the manufacturer’s instructions. Liver lysates with 500 μg protein was immunoprecipitated (IP) using a Pierce classic immunoprecipitation kit (Supplementary Table S1) with mothers against decapentaplegic homolog-4 (Smad4) antibody (5 μg, Supplementary Table S1) according to manufacturer’s instruction. Protein contents in immunoprecipitates were measured, and immunoprecipitates with equal amount of protein were loaded to each lane (Liu et al., 2015). Smad4 and Ski-related novel protein-N (SnoN) were measured by immunoblotting (IB) as described below.
2.9 Immunoblotting
Livers were collected at 2 weeks after BDL or sham-operation or 6 weeks after treatment with CCl4 or corn oil. Liver tissue was homogenized in ice-cold lysis buffer, as described above. Immunoblotting of proteins in tissue lysates, immunoprecipitates, or hTERT-HSC lysates was performed with primary antibodies specific for the proteins of interest, as described previously (Rehman et al., 2008). The sources of antibodies are shown in Supplementary Table S1. Horseradish peroxidase-conjugated secondary antibodies (Supplementary Table S1) were applied after removal of primary antibodies by washing with TBS-T solution, and detection was by chemiluminescence (Rehman et al., 2008).
2.10 Statistical analysis
Groups were compared using ANOVA plus Student-Newman-Keul’s post hoc test using p < 0.05 as the criterion of significance. Values are means ± SEM. Group sizes are described in figure legends.
3 Results
3.1 LP340 increases histone-3 but not tubulin acetylation in the liver
To determine if LP340 inhibits Class I HDAC in vivo, we assessed the acetylation status of histone-3 (substrate for Class I HDAC) in livers of male mice. After CCl4 treatment, acetylated histone-3 (acH3) decreased by 55% in vehicle-treated mice but increased ∼90% in LP340-treated mice (Figures 1B, C). By contrast, acetylation of tubulin (substrate for Class II HDAC) was not altered by CCl4 or LP340 (Figures 1B, D). These data demonstrate that LP340 inhibits Class I but not Class II HDACs in vivo.
3.2 LP340 decreases liver injury and inflammation after CCl4 treatment
In male mice, liver histology was normal after vehicle treatment (Figure 1E). After 6 weeks of CCl4 treatment, small necrotic foci with leukocyte infiltration (necro-inflammatory foci) and foci of leukocytes alone without overt necrosis developed (Figure 1F). Many necro-inflammatory foci localized in centrilobular regions, but some necro-inflammatory foci were scattering throughout the liver lobule. Cell swelling was also observed in some hepatocytes (not shown). Treatment with LP340 decreased these pathological changes (Figure 1G). ALT was measured at 6 weeks after vehicle or CCl4 treatment. In male mice, serum ALT was 80 U/L in vehicle-treated mice. After 6 weeks of CCl4 treatment, ALT increased to ∼200 U/L (Figure 1H). Treatment with LP340 during the last 2 weeks of CCl4 administration decreased serum ALT to ∼80 U/L, indicating an attenuation of CCl4-induced liver injury (Figure 1H).
Consistent with increased leukocyte infiltration in liver sections after CCl4 treatment, tumor necrosis factor-α (TNFα), a proinflammatory cytokine, increased ∼140% (Figures 2A, B). Myeloperoxidase (MPO), a marker of polymorphonuclear cell infiltration, also increased 98% after CCl4, indicating inflammation. LP340 blunted these inflammatory responses by 74% after CCl4 treatment (Figures 2A, C).
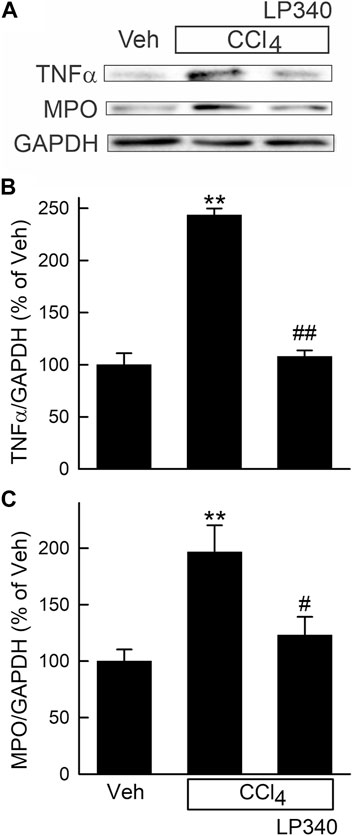
Figure 2. LP340 decreases inflammatory responses after CCl4 treatment. Male mice were treated, and livers were collected as described in Figure 1. (A) representative immunoblots for tumor necrosis factor-α (TNFα), myeloperoxidase (MPO), and housekeeping protein GAPDH. (B,C) quantification of TNFα and MPO immunoblots by densitometry. Veh, vehicle; **, p < 0.01 vs. vehicle; #, p < 0.05 vs. CCl4 without LP340; ##, p < 0.01 vs. CCl4 without LP340. Data are means ± SEM (n = 4/group).
We also examined whether LP340 protects against liver injury in female mice after CCl4 treatment. Since a previous study showed that female mice are more tolerant to CCl4 hepatotoxicity (Zhang et al., 2012), female mice were treated with a higher dose of CCl4. After 6 weeks of CCl4, serum ALT increased to 150 U/L (Supplementary Material, Supplementary Figure S1). With LP340 treatment, serum ALT was only 88 U/L. Scattered small necro-inflammatory and inflammatory foci were again observed, which LP340 also blunted (Supplementary Figure S1).
3.3 LP340 decreases liver fibrosis after CCl4 treatment
Liver fibrosis was visualized by Mason’s trichrome staining and Sirius red/Fast green staining of liver sections (Kiernan, 2015; Rehman et al., 2016). After trichrome staining, collagen stained blue (Figures 3A–C), whereas after Sirius red staining, collagen was red (Figures 3D–F). In vehicle-treated male mice, collagen staining occurred only in portal tracts and around large vessels. After CCl4 treatment, both trichrome and Sirius red staining markedly increased, primarily in centrilobular regions and around venules. Bridging fibrosis also occurred. In some portal areas, collagen staining was also slightly increased (Figures 3B, E). LP340 decreased liver fibrosis after CCl4 treatment (Figures 3C, F).
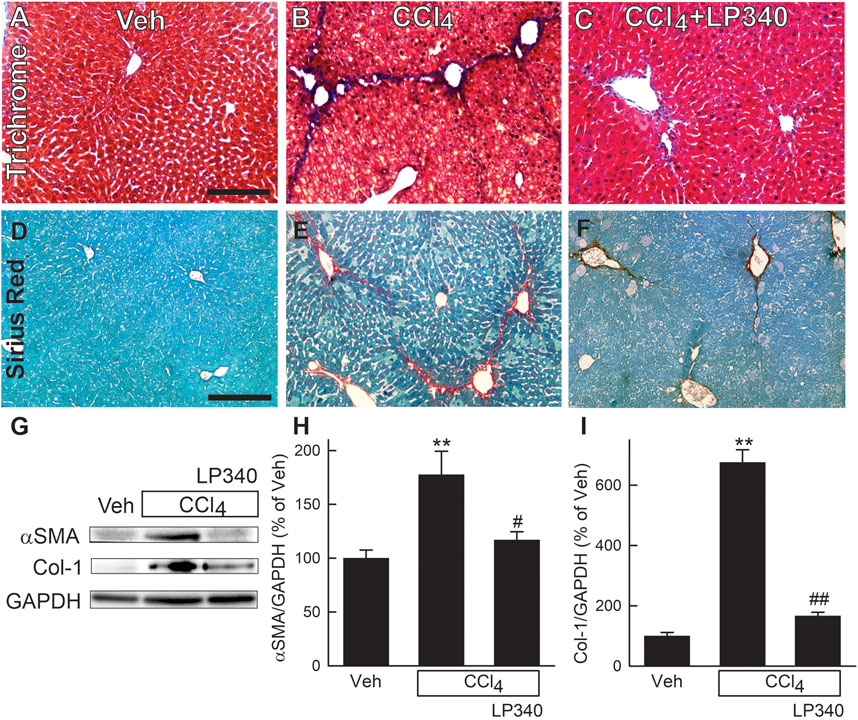
Figure 3. LP340 decreases liver fibrosis after CCl4 treatment. Male mice were treated, and livers were collected as described in Figure 1. (A–C) representative images of tichrome-stained liver sections. Bar is 100 μm; (D–F) representative images of Sirius red/Fast green-stained liver sections. Bar is 100 µm. (G) representative immunoblots for α-smooth muscle actin (αSMA), collagen-1 (Col-1), and housekeeping protein GAPDH. (H,I) quantification of αSMA and Col-1 immunoblots by densitometry. Veh, vehicle; **, p < 0.01 vs. vehicle; #, p < 0.05 vs. CCl4 without LP340; ##, p < 0.01 vs. CCl4 without LP340. Data are means ± SEM (n = 4/group).
Activation of HSC is a critical step in development of liver fibrosis. α-Smooth muscle actin (αSMA), an indicator of HSC activation, increased ∼80% after CCl4 treatment in male mice (Figures 3G, H). With LP340 treatment, αSMA increased only ∼20%. Liver fibrosis is characterized by increased formation of extracellular matrix, especially collagen. Collagen-1 expression increased 575% after CCl4 treatment. With LP340 treatment, collagen-1 expression increased only ∼70%. (Figures 3G, I).
In female mice, trichrome staining and collagen-1 expression also increased (Supplementary Material, Supplementary Figures S2A-S2E)), indicating that CCl4 treatment also caused fibrosis in females at a higher dosage. LP340 again blunted fibrosis in female mice (Supplementary Figures S2A-S2E).
3.4 LP340 decreased oxidative stress and inhibited TGFβ/Smad signaling after CCl4 treatment but did not alter matrix metalloproteinases-9 expression
CCl4 is known to cause oxidative stress, and oxidative stress can cause tissue damage and stimulate fibrosis (Bataller and Brenner, 2005; Rehman et al., 2016; Unsal et al., 2021). Therefore, we measured hepatic 4-hydroxynoneal (4-HNE) adduct formation, an indicator of lipid peroxidation, after CCl4 treatment. After vehicle treatment, weak bands of 4-HNE adducts were present (Figure 4A). After 6 weeks of CCl4 treatment, the density of 4-HNE adducts bands increased 91% (Figures 4A, B). With LP340 treatment, 4-HNE did not increase (Figures 4A, B).
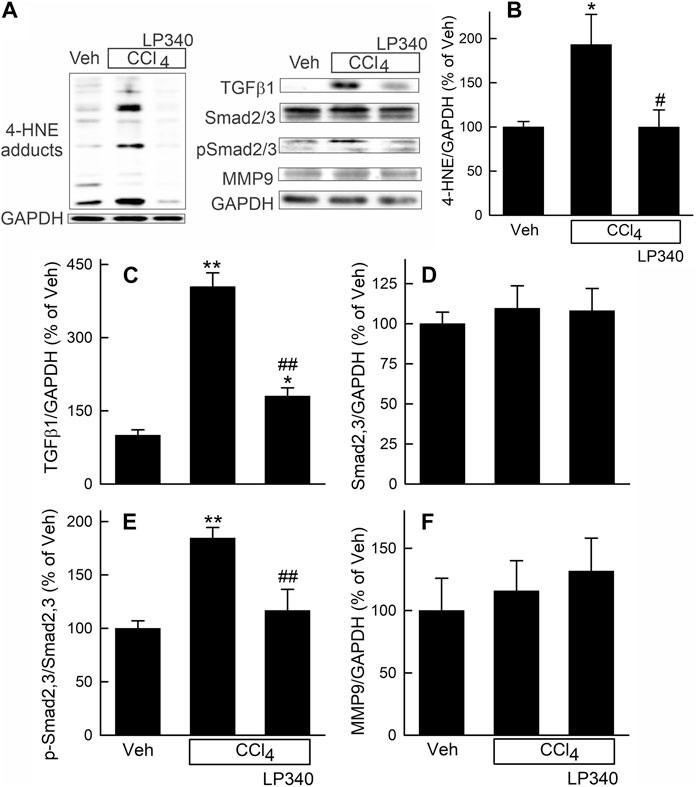
Figure 4. LP340 inhibits oxidative stress and TGFβ/Smad signaling but does not alter matrix metalloproteinase-9 expression after CCl4 treatment. Male mice were treated, and livers were collected as described in Figure 1. (A) representative immunoblots for 4-hydroxynonenal (4-HNE) adducts, transforming growth factor-β1 (TGFβ1), mothers against decapentaplegic homolog2,3 (Smad2,3), phospho-Smad2,3 (pSmad2,3), matrix metalloproteinase-9 (MMP9), and housekeeping protein GAPDH. (B–F) quantification of 4-HNE adducts, TGFβ1, Smad2,3, pSmad2,3 and MMP9 immunoblots by densitometry. Veh, vehicle; *, p < 0.05 vs. vehicle; **, p < 0.01 vs. vehicle; #, p < 0.05 vs. CCl4 without LP340; ##, p < 0.01 vs. CCl4 without LP340. Data are means ± SEM (n = 4/group).
Transforming growth factor-beta (TGFβ) is a potent profibrotic cytokine (Bataller and Brenner, 2005; Xu et al., 2016; Dewidar et al., 2019). TGFβ1 expression in liver increased ∼300% after CCl4 treatment in male mice receiving vehicle but only increased ∼80% in mice receiving LP340 (Figures 4A, C). Expression of mothers against decapentaplegic homolog-2,3 (Smad2,3), the major TGFβ downstream signaling molecules (Xu et al., 2016; Dewidar et al., 2019), was not altered by CCl4 or LP340 treatment (Figures 4A, D). By contrast, the phospho-Smad2,3 (pSmad2,3)/Smad2,3 ratio increased ∼85% after CCl4, indicating Smad2,3 activation (Figures 4A, E). LP340 blunted CCl4-induced Smad2,3 activation (Figure 4). In female mice, LP340 also blunted TGFβ1 expression after CCl4 treatment (Supplementary Material Supplementary Figure S2D, S2F).
Matrix metalloproteinases (MMPs) are proteinases that degrade the extracellular matrix, thus suppressing and reversing fibrosis (Duarte et al., 2015). In male mice, MMP9, a major MMP in the liver, was not significantly altered by CCl4 or LP340 (Figures 4A, F).
3.5 LP340 inhibited microRNA-23a but increased SnoN after CCl4 treatment
Previous studies showed that downregulation of miR23a inhibits TGFβ signaling by increasing Ski-related novel protein-N (SnoN) (Xu et al., 2018). We therefore explored if inhibition of HDAC1,2,3 alters miR23a expression. In male mice, miR23a increased 6.8-fold after CCl4 treatment, but only increased 1.6-fold in mice with LP340 treatment (Figure 5A). SnoN is a negative regulator of TGF-β/Smad signaling (Zeglinski et al., 2015). SnoN decreased by half after CCl4 treatment, which LP340 reversed (Figures 5B, C). SnoN directly interacts with Smad4, thus interfering formation of the Smad4 and pSmad2/3 complex (Wu et al., 2002; Zeglinski et al., 2015). SnoN-Smad4 interaction was examined by IP of Smad4 followed by IB for SnoN. After CCl4 treatment, SnoN co-immunoprecipitation with Smad4 decreased ∼40%, which LP340 reversed (Figures 5D, E).
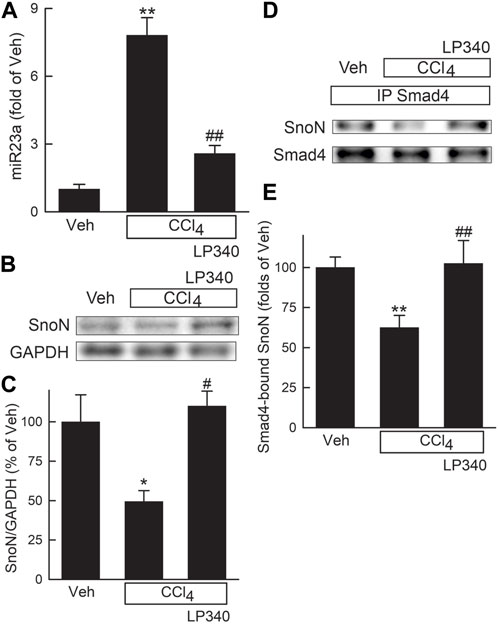
Figure 5. LP340 decreases microRNA-23a and increases SnoN expression and SnoN/Smad4 complex formation after CCl4 treatment. Male mice were treated, and livers were collected as described in Figure 1. (A) hepatic microRNA-23a (miR23a) detected by qPCR. (B) representative immunoblots of Ski-novel protein (SnoN) and housekeeping protein GAPDH. (C) quantification of SnoN. (D) representative immunoblots of Smad4-bound SnoN detected after immunoprecipitation (IP). (E) quantification of Smad4-bound SnoN. *, p < 0.05 vs. vehicle; **, p < 0.01 vs. vehicle; #, p < 0.05 vs. CCl4 without LP340; ##, p < 0.01 vs. CCl4 without LP340. Data are means ± SEM (n = 4/group).
3.6 LP340 decreases liver injury and inflammation after bile duct ligation
Hepatic cholestatic injury is typically associated with occurrence of portal fibrosis. Accordingly, we examined whether LP340 also inhibits BDL-induced cholestatic liver injury. In male mice, liver histology revealed normal liver architecture after sham-operation (Figure 6A). After BDL, numerous areas of focal necrosis developed (Figure 6B). Moreover, leukocyte infiltration increased markedly within and/or around focal necrosis, forming necro-inflammatory foci. In mice with LP340 treatment, focal necrosis areas were smaller and leukocyte infiltration within and/or around focal necrosis was markedly decreased (Figure 6C). Consistent with increased leukocyte infiltration in liver sections after BDL, proinflammatory cytokine TNFα increased by 387% (Figures 6D, E). With LP340 treatment, TNFα increased only by 60% (Figures 6D, E).
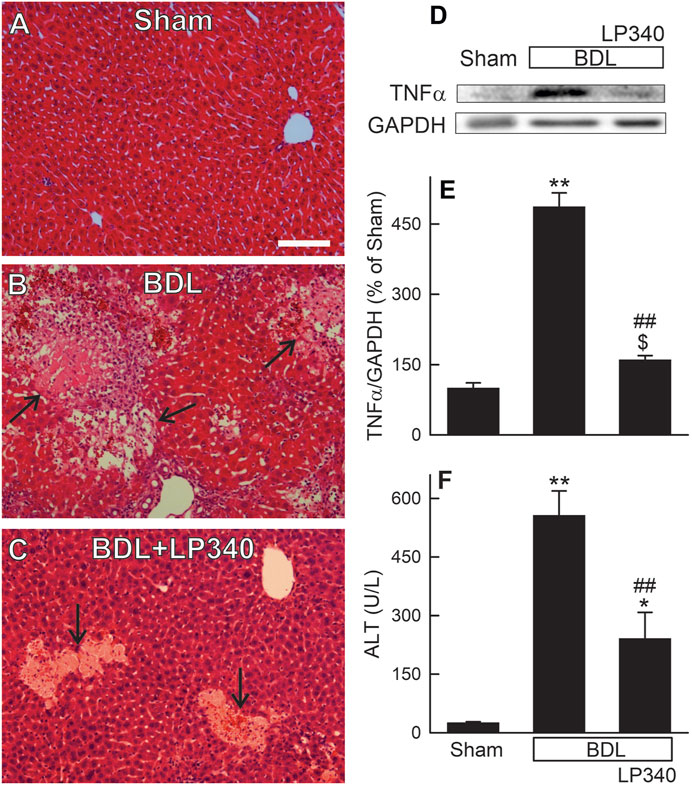
Figure 6. LP340 decreases liver injury and inflammation after bile duct ligation. Blood and livers were collected 2 weeks after BDL or sham operation (Sham). (A–C) representative images of H&E-stained liver sections. Arrows identify necro-inflammatory foci. Bar is 50 µm. (D) representative immunoblot images of TNFα and GAPDH; (E) quantification of TNFα immunoblots; (F) serum ALT; $, p = 0.052 vs. sham; *, p < 0.05 vs. sham; **, p < 0.01 vs. sham; ##, p < 0.01 vs. BDL without LP340. Data are means ± SEM (n = 3–4/group).
Serum ALT averaged 26 U/L (Figure 6F) after sham-operation. After BDL, ALT increased to ∼560 U/L at 2 weeks after BDL (Figure 6F). When mice were treated with LP340, ALT after BDL only increased to ∼240 U/L (Figure 6F). Together, these data showed that LP340 also decreased liver injury and inflammation after BDL.
3.7 LP340 decreases ductular reactions after bile duct ligation
Cholestasis typically causes a ductular reaction that is characterized by bile duct proliferation and hyperplasia. In H&E-stained liver sections from sham-operated mice, normal portal structure was observed (Figure 7A). After BDL, portal regions markedly enlarged with increased numbers of bile ducts as well as some bile ducts enlarging in size (Figure 7B). Connective tissue around portal tracts also increased (Figure 7B). Cholangiocyte proliferation was also detected by immunohistological staining for CK19, a marker of cholangiocytes (Figures 7D–F). After BDL, CK19-positive cells markedly increased, consistent with occurrence of ductular reactions (Figure 7B), which LP340 attenuated (Figures 7C, F).
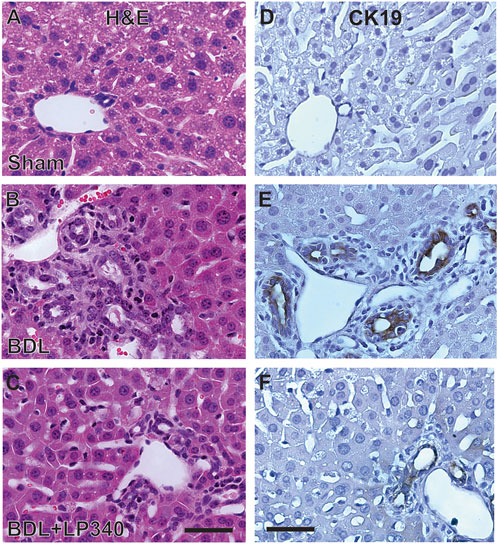
Figure 7. LP340 decreases ductular reactions after bile duct ligation. Livers were collected 2 weeks after BDL or sham operation (Sham). (A–C) representative images of liver histology after H&E staining. (D–F) representative images of immunohistological staining for CK19. Bars are 25 μm. n = 3–4/group.
3.8 LP340 decreases liver fibrosis after bile duct ligation
Liver fibrosis was revealed by trichrome and Sirius-red staining in liver sections. In sham mice, trichrome and Sirius-red labeling was confined to portal tracts and around large vessels (Figures 8A, D). After BDL in male mice, trichrome and Sirius-red staining became more intense and more widely distributed in enlarged portal areas, consistent with portal fibrosis (Figures 8B, E). Bridging fibrosis also occurred in some areas. LP340 treatment attenuated these fibrotic changes after BDL (Figures 8C, F).
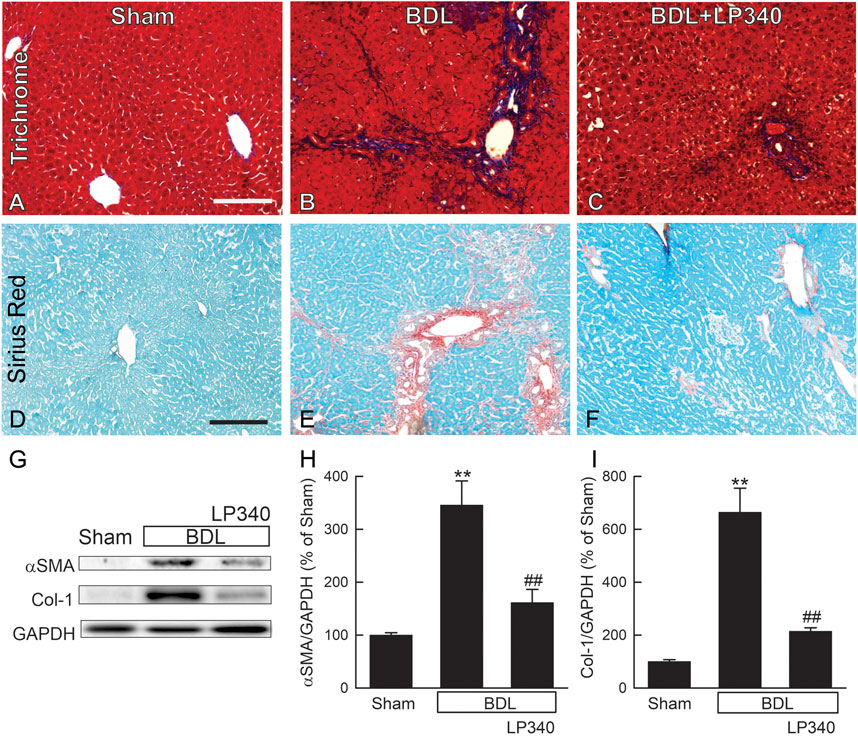
Figure 8. LP340 decreases liver fibrosis after bile duct ligation. Livers were collected 2 weeks after BDL or sham operation (Sham). (A–C) representative images of trichrome-stained liver sections. Bar is 100 µm. (D–F) representative images of Sirius red/Fast green-stained liver sections. Bar 100 µm. (G) representative immunoblots for α-smooth muscle actin (αSMA), collagen-1 (Col-1), and housekeeping protein GAPDH. (H,I) quantification of αSMA and Col-1 immunoblots by densitometry. **, p < 0.01 vs. sham; ##, p < 0.01 vs. BDL without LP340. Data are means ± SEM (n = 4/group).
αSMA expression increased ∼246% after BDL, indicating HSC activation (Figures 8G, H). Collagen-1 expression increased by ∼560% after BDL, indicating fibrogenic process (Figures 8G, I). With LP340 treatment, αSMA and collagen-1 expression increased only 62% and 114%, respectively, after BDL.
3.9 LP340 decreases oxidative stress and microRNA-23a, increases SnoN expression, and inhibits TGFβ/Smad signaling after bile duct ligation
Oxidative stress occurs in cholestatic liver injury and fibrosis (Zhong et al., 2002; Zhong et al., 2003; Heidari and Niknahad, 2019). We examined if LP340 decreases oxidative stress after BDL. In extracts of livers from sham-operated mice, Western blotting showed only weak bands of 4-HNE adducts (Figure 9A). After BDL, 4-HNE adducts increased 81%, which LP340 blocked (Figures 9A, C).
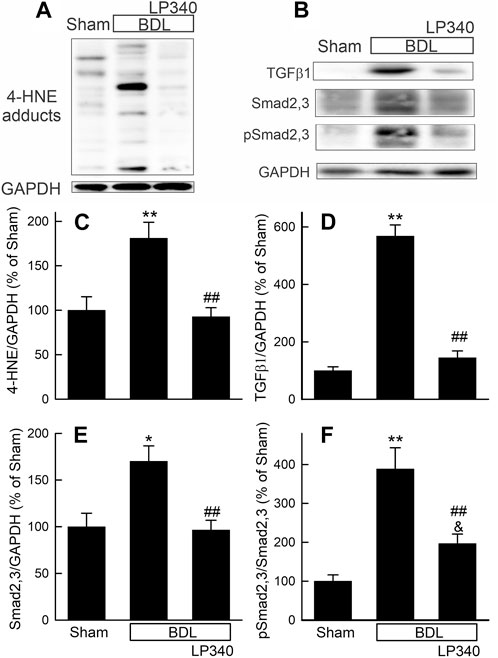
Figure 9. LP340 decreases oxidative stress and TGFβ/Smad signaling after bile duct ligation. Livers were collected 2 weeks after BDL or sham operation (Sham). (A,B) representative immunoblot for 4-HNE adducts, TGFβ1, Smad2,3, pSmad2,3 and housekeeping protein GAPDH. (C–F) quantification of 4-HNE adducts, TGFβ1, Smad2,3, pSmad2,3 immunoblots. *, p < 0.05 vs. sham; **, p < 0.01 vs. sham; &, p = 0.088 vs. sham; ##, p < 0.01 vs. BDL without LP340. Data are means ± SEM (n = 4/group).
We also examined alterations in the TGFβ/Smad pathway after BDL. Hepatic TGFβ1 expression increased ∼470% after BDL compared to sham operation (Figures 9B, D), and Smad2,3 expression increased ∼70% after BDL (Figures 9B, E). Phospho-Smad2,3 (pSmad2,3) was barely detectable in sham-operated mice and increased markedly after BDL (Figures 9B, E). The ratio of pSmad2,3 to Smad2,3 increased ∼290% after BDL, indicating Smad2,3 activation (Figure 9F). LP340 blunted both TGFβ1 and Smad2,3 expression as well as Smad2,3 activation (Figures 9B–F). After BDL, miR23a increased 3.8-fold, whereas SnoN decreased ∼44% (Figures 10A–C). LP340 blunted alterations of both miR23a and SnoN (Figure 10).
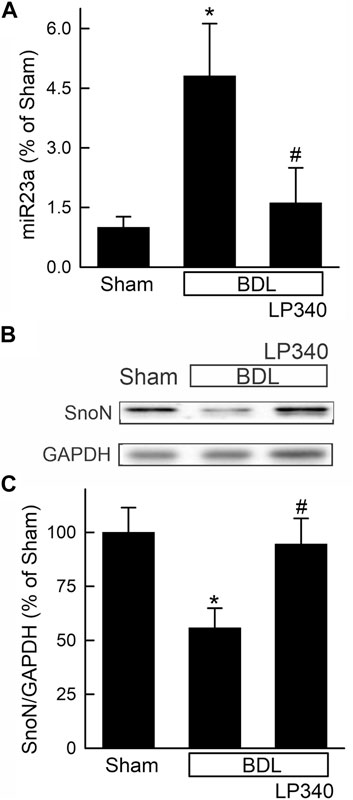
Figure 10. LP340 decreases microRNA-23a and increases SnoN after bile duct ligation. Livers were collected 2 weeks after BDL or sham operation (Sham). (A) hepatic miR23a detected by qPCR. (B) representative immunoblots for SnoN and housekeeping protein GAPDH. (C) quantification of SnoN immunoblots. *, p < 0.05 vs. sham; #, p < 0.05 vs. BDL without LP340. Data are means ± SEM (n = 4/group).
3.10 LP340 inhibits hTERT-HSC activation in vitro
HSC undergo spontaneous activation during culture. In the lysates of hTERT-HSC after 48 h culture without LP340, overt expression of αSMA and collagen-1 occurred (Figures 11A–C), indicating activation of these cells. With exposure to 0.1 and 0.3 µM LP340, αSMA expression decreased by 42% and 51%, and collagen-1 expression decreased by 58% and 66%, indicating suppression of HSC activation in a dose-dependent faction (Figures 11A–C). LP340 at 0.1 and 0.3 µM also decreased miR23a expression in hTERT-HSC by 72% and 76%, respectively (Figure 11D). These results demonstrate that LP340 directly inhibits miR23a expression and HSC activation in vitro.
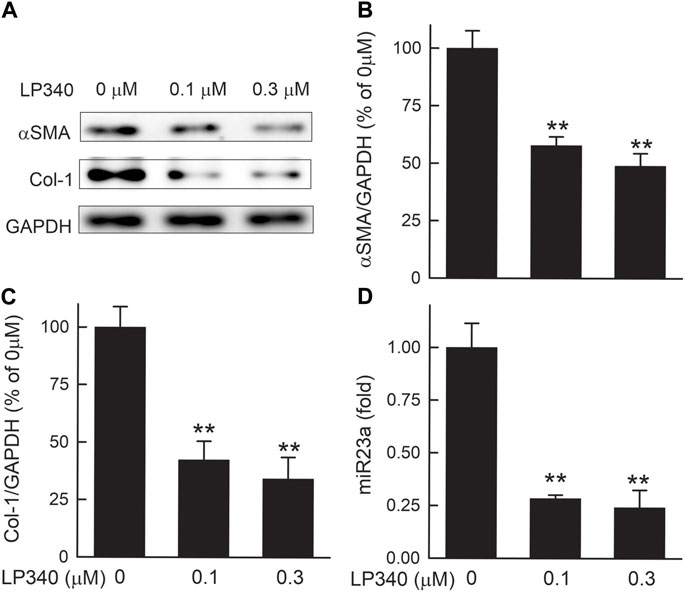
Figure 11. LP340 suppresses hTERT-HSC activation and miR340 formation in vitro. hTERT-HSC were cultured in DMEM medium with 0.5% FBS with or without LP340 (0.1 and 0.3 µM) for 48 h. (A) representative immunoblots for α-smooth muscle actin (αSMA), collagen-1 (Col-1), and housekeeping protein GAPDH. (B,C) quantification of αSMA and Col-1 immunoblots by densitometry, (D) miR23a detected by qPCR. **, p < 0.01 vs. 0 µM LP340. Data are means ± SEM (n = 3/group).
4 Discussion
4.1 LP340 protects against hepatic fibrosis in vivo
Liver fibrosis is a major medical problem with high morbidity and mortality (Friedman, 2003; Asrani et al., 2019). Importantly, insidious initiation and development of liver fibrosis substantially delay the treatment of underlying diseases. Therefore, antifibrotic treatment is much needed to decrease mortality caused by liver fibrosis (Scaglione et al., 2015). Many antifibrotic treatments targeting various fibrotic pathways have been assessed in liver fibrosis, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, interferon, pioglitazone, colchicine, farglitazar, simtuzumab, non-coding RNAs, pegbelfermin, natural herbs, and transplantation of stem cells (Friedman, 2015; Rockey et al., 2015; Trautwein et al., 2015). Unfortunately, only a very few of these treatments show modest effects in clinical trials, whereas most other treatments are ineffective (Friedman, 2015; Malaguarnera et al., 2015; Rockey et al., 2015; Trautwein et al., 2015; Zhang et al., 2023). The lack of broadly effective treatment for fibrosis continues to fuel the search for new molecular targets of fibrosis and effective drugs for prevention and therapy.
Recently, a new class of hydrazide-based HDACi targeting Class I HDAC was discovered by high-throughput screening, although the potencies of these initial hydrazide-based inhibitors was weaker than the first generation HDACi vorinostat (Wang et al., 2015). Through medicinal chemistry refinements, we developed novel HDACi with an additive inhibition mechanism via interaction with both competitive and allosteric sites of HDAC, resulting in higher potency and more specific target engagement (McClure et al., 2016; Li et al., 2018; Jiang et al., 2022). These new HDACi are highly potent (effective at low nanomolar levels) and have better isoform selectivity for HDAC1,2,3 and better PK properties than current FDA-approved HDACi on the market, especially in oral bioavailability and total systemic exposure. For example, the EC50s of LP340 for inhibiting HDAC1, 2, and 3 are 3.52, 18.14 and 0.38 nM respectively but are >10,000 nM for other HDACs (Jiang et al., 2022). LP340 can achieve oral bioavailability greater than 95%, and the maximum concentration in blood it can achieve (Cmax) is greater than 100 µM (Jiang et al., 2022).
In this study, we explored whether LP340 can be used as anti-fibrotic therapy in two mouse models of hepatic fibrosis–CCl4 and BDL, which are most widely used models for pericentral and portal fibrosis, respectively. We showed that both CCl4 and BDL caused necrosis, increased ALT release, proinflammatory cytokine formation, and leukocyte infiltration, indicating liver injury and inflammatory responses (Figure 1; Figure 2; Figure 6). LP340 blunted all these alterations. CCl4 treatment caused primarily pericentral fibrosis, whereas BDL caused mostly portal fibrosis (Figure 3; Figure 8), as expected. Bridging fibrosis occurred in each model as revealed by collagen staining with both trichrome and Sirius-red (Figure 3; Figure 8). Expression of αSMA and collagen-1 markedly increased after both CCl4 and BDL, demonstrating HSC activation and the fibrogenic process at molecular level (Figure 3; Figure 8). LP340 decreased fibrosis as revealed histologically and by molecular markers (Figure 3; Figure 8). Moreover, LP340 directly inhibited hTERT-HSC activation in vitro (Figure 11). Overall, these results suggest that LP340 not only decreases liver injury but is also a promising therapy for both pericentral and portal fibrosis. Importantly, in BDL, LP340 gavage was started right after surgery, whereas in the 6-week CCl4 model, LP340 was given only in the last 2 weeks, suggesting LP340 is effective both as a prevention and treatment strategy.
4.2 LP340 decreases oxidative stress
Liver fibrosis results from wound-healing responses to repeated injury during the progression of most chronic liver diseases (Bataller and Brenner, 2005; Zhang et al., 2023). Many cellular and molecular signals contribute to the initiation and progression of liver fibrosis, including cell death, oxidative stress, mitochondrial dysfunction, inflammatory processes, proinflammatory/profibrotic cytokines, vasoactive substances, adipokines, microRNAs, ductular reactions, and genetic factors (Gabele et al., 2003; Bataller and Brenner, 2005; Zhang et al., 2023). Chronic liver injury stimulates a multicellular response involving many hepatic resident and infiltrating cells, which release a variety of mediators to stimulate inflammatory, proliferative, and profibrogenic responses (Guo et al., 2009; Brenner et al., 2012; Higashi et al., 2017). In response to these mediators, HSC become activated and transform into myofibroblast-like cells that acquire proinflammatory, contractile, and fibrogenic properties. Activated HSCs migrate to and accumulate in sites where tissue injury occurs, producing a collagen-rich ECM to repair damaged liver tissue (Brenner et al., 2012; Higashi et al., 2017). However, dysregulated, excessive profibrogenic processes eventually lead to fibrosis/cirrhosis. By contrast, removal of activated HSCs by apoptosis and increased collagenolytic activity by upregulation of MMPs lead to resolution of fibrosis (Arthur, 2002; Bataller and Brenner, 2005).
The liver is an important site of ROS production due to its active metabolic and detoxification activities (Allameh et al., 2023). Oxidative stress develops in prevalent infectious, metabolic, drug-induced, cholestatic, autoimmune, and genetic liver diseases, such as viral hepatitis B or C, alcohol-associated liver disease, metabolic dysfunction-related steatotic liver disease, primary biliary cirrhosis, and Wilson’s disease (Allameh et al., 2023). ROS attack many biologically important macromolecules (e.g., lipids, proteins, DNA) and cause mitochondrial dysfunction, leading to cell injury and death (Hensley et al., 2000; Kim et al., 2003; Corpas and Barroso, 2013). Experimental and clinical evidence shows that oxidative stress also plays a critical role in the development of fibrosis by stimulating/amplifying inflammatory and profibrotic responses via increased expression and activation of proinflammatory/profibrotic mediators (e.g., TNFα, TGFβ, IL-1β) (Sanchez-Valle et al., 2012; Allameh et al., 2023). Oxidative stress also causes senescence in cholangiocytes and stimulates senescence-related bile ductular reactions, thus causing release of cholangiokines (e.g., TGFβ, connective tissue growth factor) that stimulate fibrosis in cholestatic liver disease (Nakanuma et al., 2015; Carpino et al., 2017; Sato et al., 2019; Cai et al., 2023). ROS and products of lipid peroxidation stimulate quiescent HSC to transdifferentiate into an activated, highly proliferative myofibroblast-like phenotype (Svegliati-Baroni et al., 2001a; Svegliati-Baroni et al., 2001b; Gandhi, 2012). Antioxidant plant polyphenols, over-expression of mitochondrial superoxide dismutase-2 (SOD2, which degrades superoxide), mitochondrial targeting antioxidant MitoQ, and activation of aldehyde dehydrogenase-2 (ALDH2, metabolically detoxifies lipid peroxidation-produced toxic aldehydes) all attenuated liver injury and fibrosis after BDL or CCl4 treatment (Zhong et al., 2002; Zhong et al., 2003; Wimborne et al., 2019).
In this study, oxidative stress occurred after CCl4 treatment and BDL, as indicated by increased 4-HNE adduct formation (Figure 4; Figure 9). LP340 blunted increases of 4-HNE, suggesting that LP340 decreases oxidative stress (Figure 12). Previous studies showed that HDACs epigenetically regulate the kelch-like ECH associated protein 1(Keap1)/nuclear factor E2-related factor 2 (Nrf2)/antioxidant pathway (Guo et al., 2015). In cultured Raw-264.7 macrophages, HDAC inhibition decreases Keap1 expression, a suppressor of Nrf2 (Wang et al., 2012). Under normal conditions, Keap1 and Nrf2 form a complex that remains localized in the cytosol. When Keap1 decreases, Keap1/Nrf2 dissociation leads to Nrf2 translocation into nuclei and then increased expression of many antioxidant proteins that contain antioxidant response elements (AREs) in their promoter regions (Kensler et al., 2007; Wang et al., 2012). TSA, a pan HDAC inhibitor, decreases infarct volume after stroke in wild-type mice, and this effect is abolished by Nrf2-deficiency (Wang et al., 2012). Consistent with an antioxidant effect of HDACi, we recently showed that LP342 decreases 4-HNE formation, increases expression of antioxidant proteins, and protects against hepatic ischemia/reperfusion injury (Samuvel et al., 2023). Therefore, LP340 likely inhibits liver injury and fibrosis, at least in part, by increasing expression of antioxidant proteins through activation of the Nrf2 pathway, thus inhibiting oxidative stress.
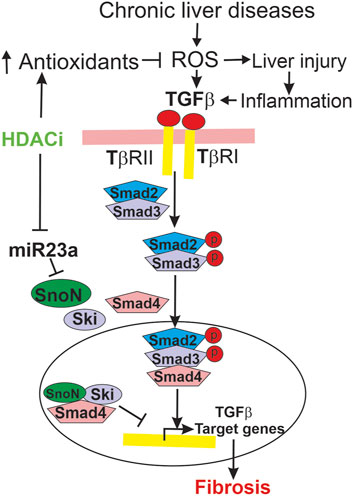
Figure 12. Mechanisms by which LP340 ameliorates liver fibrosis. Chronic liver diseases increase ROS formation, which causes cell injury and death, stimulate inflammatory responses, and increase TGFβ. Inflammatory responses also stimulate TGFβ formation. TGFβ binds to its receptors, causing phosphorylation of Smad2 and Smad3. Phosphorylated Smad2 and Smad3 form a heterocomplex with Smad4, and this complex translocates to the nucleus to upregulate transcription of TGFβ target genes, which causes HSC activation and fibrosis. SnoN and Ski, endogenous inhibitors of TGFβ/Smad signaling, compete for Smad4 with pSmad2/3 thus inhibiting pSmad2/3 nuclear translocation and TGFβ target gene transcription. Alternatively or additionally, SnoN and Ski form an inhibitory complex with Smad4 that binds to the promoter of TGFβ target genes, thus inhibiting TGFβ target gene transcription. miR23a inhibits SnoN expression. LP340 decreases oxidative stress, most likely by increasing antioxidant protein formation. Inhibition of oxidative stress would decrease liver injury and subsequent proinflammatory and profibrotic responses. Moreover, LP340 suppresses miR23a formation, which increases SnoN and its binding to Smad4, thus inhibiting TGFβ/Smad signaling-induced HSC activation and liver fibrosis.
4.3 LP340 inhibits liver fibrosis by altering microRNA-23a, SnoN, and TGFβ/Smad signaling
In addition to attenuation of liver injury and subsequent inflammatory responses, LP340 may inhibit fibrosis by epigenetically decreasing miR23a expression thus increasing SnoN, a potent suppressor of TGFβ signaling (Figure 12). TGFβ is a master profibrogenic cytokine that stimulates HSC activation, proliferation, fibrogenesis, and migration (Dewidar et al., 2019). TGFβ is produced not only by macrophages and cholangiocytes but also by activated HSC, thus acting as an autocrine/paracrine factor. Binding of TGFβ to its receptor (TβR) leads to phosphorylation of Smad2 and Smad3 (R-Smads). Phosphorylated Smad2 and Smad3 form a heterocomplex with Smad4 (co-Smad). This complex translocates to the nucleus and recruits additional transcriptional coactivators to promote transcription of TGFβ target genes, such as the gene for αSMA (Figure 12) (Derynck and Budi, 2019; Dewidar et al., 2019). HDACs enhance TGFβ-induced trans-differentiation of HSC to myofibroblasts and ECM production (Chen et al., 2015). During HSC trans-differentiation, the global acetylation of both histone H3 and H4 progressively decreases, suggesting increased HDAC activity (Qin and Han, 2010). In this study, we showed that selective and potent HDAC1,2,3 inhibitor LP340 suppresses TGFβ/Smad signaling and HSC activation in vivo (Figure 4; Figure 9).
LP340 possibly works by increasing formation of SnoN, a protein that interacts with Ski as transcriptional corepressors of Smad proteins, thus suppressing TGFβ/Smad signaling (Deheuninck and Luo, 2009; Zeglinski et al., 2015; Tecalco-Cruz et al., 2018). Ski and SnoN interact simultaneously with Smad4 (Wu et al., 2002; Deheuninck and Luo, 2009; Zeglinski et al., 2015). Binding of Ski/SnoN to Smad4 results in displacement of phospho-Smad2,3 from Smad4, thus disrupting nuclear translocation of Smad heteromeric complexes and subsequent TGFβ target gene transcription (Figure 12) (Wu et al., 2002). Another model proposes that Ski and SnoN block TGFβ signaling by forming an inhibitory complex with Smad4 that binds to the promoters of TGFβ target genes and recruits additional corepressors to inhibit TGFβ target gene expression (Figure 12) (Tecalco-Cruz et al., 2018). Other studies show that Ski and SnoN proteins also directly interact with TβRI, thus inhibiting pSmad2,3-Smad4 complex formation (Ferrand et al., 2010). Many studies have shown inhibitory effects of Ski/SnoN on fibrosis in various organs (Zeglinski et al., 2015). For example, SnoN upregulation inhibits TGFβ/Smad signaling and ameliorates renal fibrosis in diabetic rats (Liu et al., 2017). Docosahexaenoic acid, an essential n-3 polyunsaturated fatty acid, increases SnoN, thus decreasing paraquat-induced pulmonary fibrosis (Chen et al., 2013). Cpd861, a herbal compound that increases SnoN, alleviates liver fibrosis (Cai et al., 2006; Chi et al., 2018). Consistently, in this study we observed that SnoN expression markedly decreased after BDL and CCl4 treatment in association with decreased formation of SnoN/Smad4 complexes, increased TGFβ/Smad signaling, and liver fibrosis, whereas LP340 increased SnoN and inhibited hepatic fibrosis after BDL and CCl4 treatment (Figure 5; Figure 10).
LP340 increases SnoN expression likely through decreasing miR23a. miR23a plays an important role in initiation and progression of tumors (Wang et al., 2018). miR23a increases in liver fibrosis and hepatocellular carcinoma (Bao et al., 2014; Dong et al., 2019). In this study, we showed that miR23a increases after BDL and CCl4 treatment as well as in activated human HSC cell line in culture (Figure 5; Figure 10; Figure 11), which is consistent with previous reports (Dong et al., 2019). Interestingly, a previous study shows that miR23a increases TGFβ signaling by decreasing transcription of SnoN (Xu et al., 2018). Therefore, we examined the effects of inhibition of HDAC1,2,3 by LP340 on miR23a. We showed that LP340 inhibited the increase of miR23a and reversed decreases of SnoN and formation of the SnoN/Smad4 complexes after BDL and CCl4 and in cultured human HSC cell line (Figure 5; Figure 10; Figure 11).
Taken together, our findings show that LP340, the lead of a new generation of selective HDACi for HDAC1, 2 and 3, alleviates liver fibrosis either preventively or when given as therapy. LP340 inhibits Class I but not Class II HDACs in vivo. These novel HDACi not only protect against liver injury but also directly inhibit HSC activation. HDACi likely protect against liver injury and fibrosis by epigenetic upregulation of antioxidant proteins. Additionally, HDACi inhibit fibrosis by suppression of miR23a expression, thus increasing SnoN and inhibiting TGFβ signaling (Figure 12). This new generation of HDACi may represent a promising new therapy for fibrosis.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
DS: Data curation, Formal Analysis, Investigation, Methodology, Writing–original draft, Writing–review and editing. JL: Funding acquisition, Resources, Writing–review and editing. CC: Conceptualization, Funding acquisition, Investigation, Resources, Writing–original draft, Writing–review and editing. ZZ: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported, in part, by grants DK130707 and AA025379 from the National Institutes of Health. Granting agencies were not involved in the study design, collection, analysis, data interpretation, preparation of the manuscript, or other aspects of the study beyond funding. The Cell and Molecular Imaging Shared Resource was supported, in part, by NIH Grants P20 GM103542, P20 GM130457, P30 CA138313, P30 GM140964, and P30 DK123704. Shared Instrumentation Grant S10 OD018113 provided instrumentation.
Acknowledgments
The hTERT-HSC cell line was generously provided by Dr. Don Rockey at the Medical University of South Carolina. The authors thank the Cell and Molecular Imaging Shared Resource of the Hollings Cancer Center, the MUSC COBRE in Digestive and Liver Diseases, and the Digestive Disease Research Cores Center at the Medical University of South Carolina for instrumentation and technical support for microscopy in this study.
Conflict of interest
CC is the co-founder of Lydex Pharmaceuticals where LP340 was synthesized.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1386238/full#supplementary-material
References
Allameh, A., Niayesh-Mehr, R., Aliarab, A., Sebastiani, G., and Pantopoulos, K. (2023). Oxidative stress in liver pathophysiology and disease. Antioxidants (Basel) 12 (9), 1653. doi:10.3390/antiox12091653
Arthur, M. J. (2002). Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology 122 (5), 1525–1528. doi:10.1053/gast.2002.33367
Asrani, S. K., Devarbhavi, H., Eaton, J., and Kamath, P. S. (2019). Burden of liver diseases in the world. J. Hepatol. 70 (1), 151–171. doi:10.1016/j.jhep.2018.09.014
Bao, L., Zhao, J., Dai, X., Wang, Y., Ma, R., Su, Y., et al. (2014). Correlation between miR-23a and onset of hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 38 (3), 318–330. doi:10.1016/j.clinre.2013.12.002
Bataller, R., and Brenner, D. A. (2005). Liver fibrosis. J. Clin. Invest. 115 (2), 209–218. doi:10.1172/JCI24282
Brenner, D. A., Kisseleva, T., Scholten, D., Paik, Y. H., Iwaisako, K., Inokuchi, S., et al. (2012). Origin of myofibroblasts in liver fibrosis. Fibrogenes. Tissue Repair 5 (Suppl. 1), S17. doi:10.1186/1755-1536-5-S1-S17
Cai, X., Tacke, F., Guillot, A., and Liu, H. (2023). Cholangiokines: undervalued modulators in the hepatic microenvironment. Front. Immunol. 14, 1192840. doi:10.3389/fimmu.2023.1192840
Cai, Y., Shen, X. Z., Zhou, C. H., and Wang, J. Y. (2006). Abnormal expression of Smurf2 during the process of rat liver fibrosis. Chin. J. Dig. Dis. 7 (4), 237–245. doi:10.1111/j.1443-9573.2006.00275.x
Carpino, G., Pastori, D., Baratta, F., Overi, D., Labbadia, G., Polimeni, L., et al. (2017). PNPLA3 variant and portal/periportal histological pattern in patients with biopsy-proven non-alcoholic fatty liver disease: a possible role for oxidative stress. Sci. Rep. 7 (1), 15756. doi:10.1038/s41598-017-15943-z
Chen, J., Zeng, T., Zhao, X., Xiea, K., Bi, Y., Zhong, Z., et al. (2013). Docosahexaenoic acid (DHA) ameliorates paraquat-induced pulmonary fibrosis in rats possibly through up-regulation of Smad 7 and SnoN. Food Chem. Toxicol. 57, 330–337. doi:10.1016/j.fct.2013.03.045
Chen, P. J., Huang, C., Meng, X. M., and Li, J. (2015). Epigenetic modifications by histone deacetylases: biological implications and therapeutic potential in liver fibrosis. Biochimie 116, 61–69. doi:10.1016/j.biochi.2015.06.016
Chi, C., Liu, X. Y., Hou, F., Yu, X. Z., Li, C. Y., Cui, L. J., et al. (2018). Herbal compound 861 prevents hepatic fibrosis by inhibiting the TGF-β1/Smad/SnoN pathway in bile duct-ligated rats. BMC Complement. Altern. Med. 18 (1), 52. doi:10.1186/s12906-018-2119-7
Corpas, F. J., and Barroso, J. B. (2013). Nitro-oxidative stress vs oxidative or nitrosative stress in higher plants. New Phytol. 199 (3), 633–635. doi:10.1111/nph.12380
Crawford, J. M. (2002). “Liver cirrhosis,” in Pathology of the liver. 4 ed. (London, England: Churchill Livingstone), 575–619.
Deheuninck, J., and Luo, K. (2009). Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 19 (1), 47–57. doi:10.1038/cr.2008.324
Derynck, R., and Budi, E. H. (2019). Specificity, versatility, and control of TGF-β family signaling. Sci. Signal 12 (570), eaav5183. doi:10.1126/scisignal.aav5183
Dewidar, B., Meyer, C., Dooley, S., and Meindl-Beinker, A. N. (2019). TGF-Β in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells 8 (11), 1419. doi:10.3390/cells8111419
Dong, Z., Li, S., Wang, X., Si, L., Ma, R., Bao, L., et al. (2019). lncRNA GAS5 restrains CCl(4)-induced hepatic fibrosis by targeting miR-23a through the PTEN/PI3K/Akt signaling pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 316 (4), G539–G50. doi:10.1152/ajpgi.00249.2018
Duarte, S., Baber, J., Fujii, T., and Coito, A. J. (2015). Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 44-46, 147–156. doi:10.1016/j.matbio.2015.01.004
Ferrand, N., Atfi, A., and Prunier, C. (2010). The oncoprotein c-ski functions as a direct antagonist of the transforming growth factor-{beta} type I receptor. Cancer Res. 70 (21), 8457–8466. doi:10.1158/0008-5472.CAN-09-4088
Friedman, S. L. (2003). Liver fibrosis -- from bench to bedside. J. Hepatol. 38 (Suppl. 1), S38–S53. doi:10.1016/s0168-8278(02)00429-4
Friedman, S. L. (2015). Hepatic fibrosis: emerging therapies. Dig. Dis. 33 (4), 504–507. doi:10.1159/000374098
Gabele, E., Brenner, D. A., and Rippe, R. A. (2003). Liver fibrosis: signals leading to the amplification of the fibrogenic hepatic stellate cell. Front. Biosci. 8, d69–d77. doi:10.2741/887
Gandhi, C. R. (2012). Oxidative stress and hepatic stellate cells: a paradoxical relationship. Trends Cell Mol. Biol. 7, 1–10.
Glozak, M. A., Sengupta, N., Zhang, X., and Seto, E. (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23. doi:10.1016/j.gene.2005.09.010
Guo, C. J., Pan, Q., Cheng, T., Jiang, B., Chen, G. Y., and Li, D. G. (2009). Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. FEBS J. 276 (18), 5163–5176. doi:10.1111/j.1742-4658.2009.07213.x
Guo, Y., Yu, S., Zhang, C., and Kong, A. N. (2015). Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic. Biol. Med. 88 (Pt B), 337–349. doi:10.1016/j.freeradbiomed.2015.06.013
Hadziyannis, S. J., Tassopoulos, N. C., Heathcote, E. J., Chang, T. T., Kitis, G., Rizzetto, M., et al. (2003). Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N. Engl. J. Med. 348 (9), 800–807. doi:10.1056/NEJMoa021812
Hammond, C. M., Stromme, C. B., Huang, H., Patel, D. J., and Groth, A. (2017). Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 18 (3), 141–158. doi:10.1038/nrm.2016.159
Heidari, R., and Niknahad, H. (2019). The role and study of mitochondrial impairment and oxidative stress in cholestasis. Methods Mol. Biol. 1981, 117–132. doi:10.1007/978-1-4939-9420-5_8
Hensley, K., Robinson, K. A., Gabbita, S. P., Salsman, S., and Floyd, R. A. (2000). Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 28, 1456–1462. doi:10.1016/s0891-5849(00)00252-5
Higashi, T., Friedman, S. L., and Hoshida, Y. (2017). Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 121, 27–42. doi:10.1016/j.addr.2017.05.007
Jiang, Y., Xu, J., Yue, K., Huang, C., Qin, M., Chi, D., et al. (2022). Potent hydrazide-based HDAC inhibitors with a superior pharmacokinetic profile for efficient treatment of acute myeloid leukemia in vivo. J. Med. Chem. 65 (1), 285–302. doi:10.1021/acs.jmedchem.1c01472
Kensler, T. W., Wakabayashi, N., and Biswal, S. (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116. doi:10.1146/annurev.pharmtox.46.120604.141046
Kiernan, R., Bres, V., Ng, R. W., Coudart, M. P., El, M. S., Sardet, C., et al. (2003). Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 278 (4), 2758–2766. doi:10.1074/jbc.M209572200
Kim, J. S., He, L., Qian, T., and Lemasters, J. J. (2003). Role of the mitochondrial permeability transition in apoptotic and necrotic death after ischemia/reperfusion injury to hepatocytes. Curr. Mol. Med. 3 (6), 527–535. doi:10.2174/1566524033479564
Krishnasamy, Y., Ramshesh, V. K., Gooz, M., Schnellmann, R. G., Lemasters, J. J., and Zhong, Z. (2016). Ethanol and high cholesterol diet causes severe steatohepatitis and early liver fibrosis in mice. PLoS One 11 (9), e0163342. doi:10.1371/journal.pone.0163342
Kuo, M. H., and Allis, C. D. (1998). Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 20 (8), 615–626. doi:10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H
Li, G., Tian, Y., and Zhu, W. G. (2020). The roles of histone deacetylases and their inhibitors in cancer therapy. Front. Cell Dev. Biol. 8, 576946. doi:10.3389/fcell.2020.576946
Li, X., Peterson, Y. K., Inks, E. S., Himes, R. A., Li, J., Zhang, Y., et al. (2018). Class I HDAC inhibitors display different antitumor mechanism in leukemia and prostatic cancer cells depending on their p53 status. J. Med. Chem. 61 (6), 2589–2603. doi:10.1021/acs.jmedchem.8b00136
Liu, L., Shi, M., Wang, Y., Zhang, C., Su, B., Xiao, Y., et al. (2017). SnoN upregulation ameliorates renal fibrosis in diabetic nephropathy. PLoS One 12 (3), e0174471. doi:10.1371/journal.pone.0174471
Liu, Q., Rehman, H., Krishnasamy, Y., Schnellmann, R. G., Lemasters, J. J., and Zhong, Z. (2015). Improvement of liver injury and survival by JNK2 and iNOS deficiency in liver transplants from cardiac death mice. J. Hepatol. 63 (1), 68–74. doi:10.1016/j.jhep.2015.02.017
Liu, Y., Wang, Z., Wang, J., Lam, W., Kwong, S., Li, F., et al. (2013). A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-β and vascular endothelial growth factor signalling. Liver Int. 33 (4), 504–515. doi:10.1111/liv.12034
Malaguarnera, M., Motta, M., Vacante, M., Malaguarnera, G., Caraci, F., Nunnari, G., et al. (2015). Silybin-vitamin E-phospholipids complex reduces liver fibrosis in patients with chronic hepatitis C treated with pegylated interferon α and ribavirin. Am. J. Transl. Res. 7 (11), 2510–2518.
Mannaerts, I., Eysackers, N., Onyema, O. O., Van Beneden, K., Valente, S., Mai, A., et al. (2013). Class II HDAC inhibition hampers hepatic stellate cell activation by induction of microRNA-29. PLoS One 8 (1), e55786. doi:10.1371/journal.pone.0055786
Mannaerts, I., Nuytten, N. R., Rogiers, V., Vanderkerken, K., van Grunsven, L. A., and Geerts, A. (2010). Chronic administration of valproic acid inhibits activation of mouse hepatic stellate cells in vitro and in vivo. Hepatology 51 (2), 603–614. doi:10.1002/hep.23334
McClure, J. J., Zhang, C., Inks, E. S., Peterson, Y. K., Li, J., and Chou, C. J. (2016). Development of allosteric hydrazide-containing class I histone deacetylase inhibitors for use in acute myeloid leukemia. J. Med. Chem. 59 (21), 9942–9959. doi:10.1021/acs.jmedchem.6b01385
Meirelles Junior, R. F., Salvalaggio, P., Rezende, M. B., Evangelista, A. S., Guardia, B. D., Matielo, C. E., et al. (2015). Liver transplantation: history, outcomes and perspectives. Einstein (Sao Paulo) 13 (1), 149–152. doi:10.1590/S1679-45082015RW3164
Moran-Salvador, E., and Mann, J. (2017). Epigenetics and liver fibrosis. Cell Mol. Gastroenterol. Hepatol. 4 (1), 125–134. doi:10.1016/j.jcmgh.2017.04.007
Nakanuma, Y., Sasaki, M., and Harada, K. (2015). Autophagy and senescence in fibrosing cholangiopathies. J. Hepatol. 62 (4), 934–945. doi:10.1016/j.jhep.2014.11.027
Pinzani, M. (1999). Liver fibrosis. Springer Semin. Immunopathol. 21 (4), 475–490. doi:10.1007/s002810000037
Powell, L. W., and Kerr, J. F. (1970). Reversal of "cirrhosis" in idiopathic haemochromatosis following long-term intensive venesection therapy. Australas. Ann. Med. 19 (1), 54–57. doi:10.1111/imj.1970.19.1.54
Poynard, T., McHutchison, J., Manns, M., Trepo, C., Lindsay, K., Goodman, Z., et al. (2002). Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 122 (5), 1303–1313. doi:10.1053/gast.2002.33023
Qin, L., and Han, Y. P. (2010). Epigenetic repression of matrix metalloproteinases in myofibroblastic hepatic stellate cells through histone deacetylases 4: implication in tissue fibrosis. Am. J. Pathol. 177 (4), 1915–1928. doi:10.2353/ajpath.2010.100011
Rahimi, R. S., and Rockey, D. C. (2013). End-stage liver disease complications. Curr. Opin. Gastroenterol. 29 (3), 257–263. doi:10.1097/MOG.0b013e32835f43b0
Ramzan, F., Vickers, M. H., and Mithen, R. F. (2021). Epigenetics, microRNA and metabolic syndrome: a comprehensive review. Int. J. Mol. Sci. 22 (9), 5047. doi:10.3390/ijms22095047
Rehman, H., Liu, Q., Krishnasamy, Y., Shi, Z., Ramshesh, V. K., Haque, K., et al. (2016). The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 8 (1), 14–27.
Rehman, H., Ramshesh, V. K., Theruvath, T. P., Kim, I., Currin, R. T., Giri, S., et al. (2008). NIM811 (N-methyl-4-isoleucine cyclosporine), a mitochondrial permeability transition inhibitor, attenuates cholestatic liver injury but not fibrosis in mice. J. Pharmacol. Exp. Ther. 327, 699–706. doi:10.1124/jpet.108.143578
Rockey, D. C., Bell, P. D., and Hill, J. A. (2015). Fibrosis--a common pathway to organ injury and failure. N. Engl. J. Med. 372 (12), 1138–1149. doi:10.1056/NEJMra1300575
Samuvel, D. J., Krishnasamy, Y., Li, L., Lemasters, J. J., Chou, C. J., and Zhong, Z. (2023). LP342, a novel histone deacetylase inhibitor, decreases nitro-oxidative stress, mitochondrial dysfunction and hepatic ischaemia/reperfusion injury in mice. Rps. Pharm. Pharmacol. Rep. 2 (2), rqad013. doi:10.1093/rpsppr/rqad013
Sanchez-Valle, V., Chavez-Tapia, N. C., Uribe, M., and Mendez-Sanchez, N. (2012). Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr. Med. Chem. 19 (28), 4850–4860. doi:10.2174/092986712803341520
Sato, K., Marzioni, M., Meng, F., Francis, H., Glaser, S., and Alpini, G. (2019). Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology 69 (1), 420–430. doi:10.1002/hep.30150
Scaglione, S., Kliethermes, S., Cao, G., Shoham, D., Durazo, R., Luke, A., et al. (2015). The epidemiology of cirrhosis in the United States: a population-based study. J. Clin. Gastroenterol. 49 (8), 690–696. doi:10.1097/MCG.0000000000000208
Schnabl, B., Choi, Y. H., Olsen, J. C., Hagedorn, C. H., and Brenner, D. A. (2002). Immortal activated human hepatic stellate cells generated by ectopic telomerase expression. Lab. Invest. 82 (3), 323–333. doi:10.1038/labinvest.3780426
Shaker, M. E., Ghani, A., Shiha, G. E., Ibrahim, T. M., and Mehal, W. Z. (2013). Nilotinib induces apoptosis and autophagic cell death of activated hepatic stellate cells via inhibition of histone deacetylases. Biochim. Biophys. Acta 1833 (8), 1992–2003. doi:10.1016/j.bbamcr.2013.02.033
Shao, Y., Gao, Z., Marks, P. A., and Jiang, X. (2004). Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 101 (52), 18030–18035. doi:10.1073/pnas.0408345102
Shen, S., and Kozikowski, A. P. (2016). Why hydroxamates may not Be the best histone deacetylase inhibitors--what some may have forgotten or would rather forget? ChemMedChem 11 (1), 15–21. doi:10.1002/cmdc.201500486
Soliman, A. M., Das, S., Abd Ghafar, N., and Teoh, S. L. (2018). Role of MicroRNA in proliferation phase of wound healing. Front. Genet. 9, 38. doi:10.3389/fgene.2018.00038
Subedi, P., Schneider, M., Philipp, J., Azimzadeh, O., Metzger, F., Moertl, S., et al. (2019). Comparison of methods to isolate proteins from extracellular vesicles for mass spectrometry-based proteomic analyses. Anal. Biochem. 584, 113390. doi:10.1016/j.ab.2019.113390
Suzuki, H., Maruyama, R., Yamamoto, E., and Kai, M. (2013). Epigenetic alteration and microRNA dysregulation in cancer. Front. Genet. 4, 258. doi:10.3389/fgene.2013.00258
Svegliati-Baroni, G., Ridolfi, F., Di Sario, A., Saccomanno, S., Bendia, E., Benedetti, A., et al. (2001b). Intracellular signaling pathways involved in acetaldehyde-induced collagen and fibronectin gene expression in human hepatic stellate cells. Hepatology 33 (5), 1130–1140. doi:10.1053/jhep.2001.23788
Svegliati-Baroni, G., Saccomanno, S., van Goor, H., Jansen, P., Benedetti, A., and Moshage, H. (2001a). Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver 21 (1), 1–12. doi:10.1034/j.1600-0676.2001.210101.x
Tao, H., Shi, K. H., Yang, J. J., Huang, C., Zhan, H. Y., and Li, J. (2014). Histone deacetylases in cardiac fibrosis: current perspectives for therapy. Cell Signal 26 (3), 521–527. doi:10.1016/j.cellsig.2013.11.037
Tecalco-Cruz, A. C., Rios-Lopez, D. G., Vazquez-Victorio, G., Rosales-Alvarez, R. E., and Macias-Silva, M. (2018). Transcriptional cofactors Ski and SnoN are major regulators of the TGF-β/Smad signaling pathway in health and disease. Signal Transduct. Target Ther. 3, 15. doi:10.1038/s41392-018-0015-8
Trams, E. G., and Symons, A. M. (1957). Morphological and functional changes in the livers of rats after ligation or excision of the common bile duct. Am. J. Pathol. 33, 13–25.
Trautwein, C., Friedman, S. L., Schuppan, D., and Pinzani, M. (2015). Hepatic fibrosis: concept to treatment. J. Hepatol. 62 (1 Suppl. l), S15–S24. doi:10.1016/j.jhep.2015.02.039
Unsal, V., Cicek, M., and Sabancilar, I. (2021). Toxicity of carbon tetrachloride, free radicals and role of antioxidants. Rev. Environ. Health 36 (2), 279–295. doi:10.1515/reveh-2020-0048
Van Beneden, K., Mannaerts, I., Pauwels, M., Van den Branden, C., and van Grunsven, L. A. (2013). HDAC inhibitors in experimental liver and kidney fibrosis. Fibrogenes. Tissue Repair 6 (1), 1. doi:10.1186/1755-1536-6-1
Wang, B., Zhu, X., Kim, Y., Li, J., Huang, S., Saleem, S., et al. (2012). Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 52 (5), 928–936. doi:10.1016/j.freeradbiomed.2011.12.006
Wang, N., Tan, H. Y., Feng, Y. G., Zhang, C., Chen, F., and Feng, Y. (2018). microRNA-23a in human cancer: its roles, mechanisms and therapeutic relevance. Cancers (Basel) 11 (1), 7. doi:10.3390/cancers11010007
Wang, Y., Stowe, R. L., Pinello, C. E., Tian, G., Madoux, F., Li, D., et al. (2015). Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem. Biol. 22 (2), 273–284. doi:10.1016/j.chembiol.2014.12.015
Wimborne, H. J., Takemoto, K., Woster, P. M., Rockey, D. C., Lemasters, J. J., and Zhong, Z. (2019). Aldehyde dehydrogenase-2 activation by Alda-1 decreases necrosis and fibrosis after bile duct ligation in mice. Free Radic. Biol. Med. 145, 136–145. doi:10.1016/j.freeradbiomed.2019.09.026
Wu, J. W., Krawitz, A. R., Chai, J., Li, W., Zhang, F., Luo, K., et al. (2002). Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-beta signaling. Cell 111 (3), 357–367. doi:10.1016/s0092-8674(02)01006-1
Xu, F., Liu, C., Zhou, D., and Zhang, L. (2016). TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J. Histochem Cytochem 64 (3), 157–167. doi:10.1369/0022155415627681
Xu, H., Sun, F., Li, X., and Sun, L. (2018). Down-regulation of miR-23a inhibits high glucose-induced EMT and renal fibrogenesis by up-regulation of SnoN. Hum. Cell 31 (1), 22–32. doi:10.1007/s13577-017-0180-z
Yoon, S., Kang, G., and Eom, G. H. (2019). HDAC inhibitors: therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 20 (6), 1329. doi:10.3390/ijms20061329
Zeglinski, M. R., Hnatowich, M., Jassal, D. S., and Dixon, I. M. (2015). SnoN as a novel negative regulator of TGF-β/Smad signaling: a target for tailoring organ fibrosis. Am. J. Physiol. Heart Circ. Physiol. 308 (2), H75–H82. doi:10.1152/ajpheart.00453.2014
Zhang, C. Y., Liu, S., and Yang, M. (2023). Treatment of liver fibrosis: past, current, and future. World J. Hepatol. 15 (6), 755–774. doi:10.4254/wjh.v15.i6.755
Zhang, Y., Wu, L., Wang, Y., Zhang, M., Li, L., Zhu, D., et al. (2012). Protective role of estrogen-induced miRNA-29 expression in carbon tetrachloride-induced mouse liver injury. J. Biol. Chem. 287 (18), 14851–14862. doi:10.1074/jbc.M111.314922
Zhong, Z., Froh, M., Lehnert, M., Schoonhoven, R., Yang, L., Lind, H., et al. (2003). Polyphenols from Camellia sinenesis attenuate experimental cholestasis-induced liver fibrosis in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 285 (5), G1004–G1013. doi:10.1152/ajpgi.00008.2003
Zhong, Z., Froh, M., Wheeler, M. D., Smutney, O., Lehmann, T. G., and Thurman, R. G. (2002). Viral gene delivery of superoxide dismutase attenuates experimental cholestasis-induced liver fibrosis in the rat. Gene Ther. 9 (3), 183–191. doi:10.1038/sj.gt.3301638
Glossary

Keywords: histone deacetylase, liver fibrosis, microRNA-23a, oxidative stress, SnoN, TGFβ
Citation: Samuvel DJ, Lemasters JJ, Chou CJ and Zhong Z (2024) LP340, a novel histone deacetylase inhibitor, decreases liver injury and fibrosis in mice: role of oxidative stress and microRNA-23a. Front. Pharmacol. 15:1386238. doi: 10.3389/fphar.2024.1386238
Received: 14 February 2024; Accepted: 24 April 2024;
Published: 17 May 2024.
Edited by:
Wei Zhong, University of Kansas Medical Center, United StatesReviewed by:
Joy Jiang, University of California, Davis, United StatesKomal Ramani, Cedars Sinai Medical Center, United States
Copyright © 2024 Samuvel, Lemasters, Chou and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi Zhong, emhvbmdAbXVzYy5lZHU=