
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 01 May 2024
Sec. Experimental Pharmacology and Drug Discovery
Volume 15 - 2024 | https://doi.org/10.3389/fphar.2024.1385598
This article is part of the Research Topic The Magic Bullet Evolution in Tumor Therapy: Novel Drug Conjugates for Targeted Delivery View all 9 articles
 Ioanna Liatsou1
Ioanna Liatsou1 Betelhem Assefa1Wathsala Liyanage2Sharmane Surasinghe3Zora Nováková4
Betelhem Assefa1Wathsala Liyanage2Sharmane Surasinghe3Zora Nováková4 Cyril Bařinka4Kathleen Gabrielson5,6
Cyril Bařinka4Kathleen Gabrielson5,6 Venu Raman1,6,7,8
Venu Raman1,6,7,8 Dmitri Artemov1,6
Dmitri Artemov1,6 Sudath Hapuarachchige1,6*
Sudath Hapuarachchige1,6*Prostate cancer (PC) is an aggressive cancer that can progress rapidly and eventually become castrate-resistant prostate cancer (CRPC). Stage IV metastatic castrate-resistant prostate cancer (mCRPC) is an incurable late-stage cancer type with a low 5-year overall survival rate. Targeted therapeutics such as antibody-drug conjugates (ADCs) based on high-affinity monoclonal antibodies and potent drugs conjugated via smart linkers are being developed for PC management. Conjugating further with in vitro or in vivo imaging agents, ADCs can be used as antibody-theranostic conjugates (ATCs) for diagnostic and image-guided drug delivery. In this study, we have developed a novel ATC for PSMA (+) PC therapy utilizing (a) anti-PSMA 5D3 mAb, (b) Aurora A kinase inhibitor, MLN8237, and (c) for the first time using tetrazine (Tz) and trans-cyclooctene (TCO) click chemistry-based conjugation linker (CC linker) in ADC development. The resulting 5D3(CC-MLN8237)3.2 was labeled with suitable fluorophores for in vitro and in vivo imaging. The products were characterized by SDS-PAGE, MALDI-TOF, and DLS and evaluated in vitro by optical imaging, flow cytometry, and WST-8 assay for cytotoxicity in PSMA (+/−) cells. Therapeutic efficacy was determined in human PC xenograft mouse models following a designed treatment schedule. After the treatment study animals were euthanized, and toxicological studies, complete blood count (CBC), blood clinical chemistry analysis, and H&E staining of vital organs were conducted to determine side effects and systemic toxicities. The IC50 values of 5D3(CC-MLN8237)3.2-AF488 in PSMA (+) PC3-PIP and PMSA (−) PC3-Flu cells are 8.17 nM and 161.9 nM, respectively. Pure MLN8237 shows 736.9 nM and 873.4 nM IC50 values for PC3-PIP and PC3-Flu cells, respectively. In vivo study in human xenograft mouse models confirmed high therapeutic efficacy of 5D3(CC-MLN8237)3.2-CF750 with significant control of PSMA (+) tumor growth with minimal systemic toxicity in the treated group compared to PSMA (−) treated and untreated groups. Approximately 70% of PSMA (+) PC3-PIP tumors did not exceed the threshold of the tumor size in the surrogate Kaplan-Meyer analysis. The novel ATC successfully controlled the growth of PSMA (+) tumors in preclinical settings with minimal systemic toxicities. The therapeutic efficacy and favorable safety profile of novel 5D3(CC-MLN8237)3.2 ATC demonstrates their potential use as a theranostic against aggressive PC.
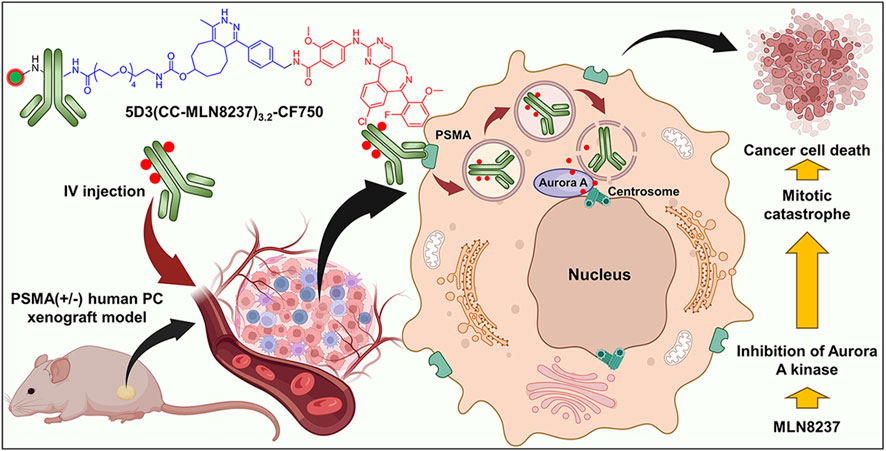
Prostate cancer (PC) is estimated to account for almost 29% of all diagnosed cancer cases and 12% of cancer deaths in men in the United States (Siegel et al., 2023; Siegel et al., 2024). Radical prostatectomy, chemotherapy, radiotherapy, immunotherapy, and androgen/hormone deprivation therapy (ADT) are the standard of care at different stages of PC disease (Lindner et al., 2010; Lorente et al., 2016; Perera et al., 2016; Mottet et al., 2017; Evans, 2018). However, almost all PCs eventually become castrate-resistant prostate cancer (CRPC), which can progress rapidly and metastasize to stage IV metastatic castrate-resistant prostate cancer (mCRPC) (Sartor and de Bono, 2018; Einstein et al., 2021). Despite the continued progress in the treatment landscape of mCRPC in the last few decades from palliative care to taxane chemotherapy (e.g., docetaxel, cabazitaxel), sipuleucel-T immunotherapy, bone-specific radionuclide radium-223, androgen receptor directed therapies (e.g., abiraterone, enzalutamide), and poly (ADP-ribose) polymerase inhibitors, presently, there are no curative treatments for mCRPC (Antonarakis et al., 2020). Moreover, these existing therapies may be associated with poor tolerability and toxicity to healthy cells (Antonarakis et al., 2020). An increasing volume of clinical data proves the limitation of chemotherapeutics due to the lack of target-specificity, lack of efficacy in metastatic PC therapy, and induction of severe systemic toxicities in patients (Diamantis and Banerji, 2016; Rosenfeld et al., 2020; Vazquez et al., 2021; Wang et al., 2022). The off-targeted accumulation of these cytotoxic agents in healthy organs and tissues results in severe systemic toxicities and adverse side effects in PC patients. To overcome these issues, the development of image-guided and targeted drug delivery systems that enable early diagnosis of the disease and treatment are critically important to treat this disease effectively (Hapuarachchige and Artemov, 2020).
Image guidance is an essential approach in the early development of novel therapeutics and drug delivery systems. In the preclinical settings, imaging is used for several purposes including the non-invasive determination of pharmacokinetics, biodistribution, tumor uptake, off-target accumulation, controlled-release, and treatment response (Ojha et al., 2015). The use of image-guided drug delivery in clinics tremendously supports diagnostic imaging to recognize the targeted biomarkers, determine the position, size, and stage of cancer, and design the treatment plan in personalized medicine (Arranz and Ripoll, 2015; Hapuarachchige and Artemov, 2020). Hence, image-guided drug delivery systems are called theranostics when the delivery platform provides both diagnostic information and therapy (Herrmann et al., 2017).
Developing antibody-drug conjugates (ADCs) is a continuously thriving field for targeted drug delivery. An ADC consists of a target-specific antibody and cytotoxic agents conjugated through a linker. (Drago et al., 2021). Their target-specificity enhances therapeutic efficacy with minimal exposure to healthy tissue, expanding the therapeutic window of ADCs (Marei et al., 2022). Developing antibodies with high specificity and binding affinity on targeted cells, designing novel linkers for targeted-specific controlled release, and novel potent chemotherapeutics that can specifically kill the cancer cells vastly support the development of new ADCs for cancer therapy (Bakhtiar, 2016). Currently, following PC-specific antigens, (a) prostate-specific membrane antigen, PSMA, (b) trophoblast cell surface antigen-2, TROP-2, (c) six-transmembrane epithelial antigen of prostate-1, STEAP-1, (d) tissue factor, TF, (e) delta-like protein 3, DLL-3, (f) B7-H3 family of proteins, B7-H3, and (g) human epidermal growth factor receptor 2, HER2 are being studied as targets for the development of ADCs for PC therapy (Rosellini et al., 2021; Sardinha et al., 2023). Among these biomarkers, PSMA is one of the most well-known and clinically validated biomarkers that is expressed 1000-fold more on PC tumors than normal prostatic tissue (Ghosh and Heston, 2004; Jeitner et al., 2022). PSMA is internalized via clathrin-coated pits and subsequent endocytosis (clathrin-mediated endocytosis) upon ligand binding. Targeting PSMA is expected to result in high in vitro and in vivo image quality and cellular uptake by tumor cells due to these internalization characteristics that lead to enhanced tumor uptake and retention (Jeitner et al., 2022). Moreover, its overexpression in more than 70% of patients with mCRPC, in addition to its extracellular domain that can be targeted by antibodies and a motif that results in the internalization of bound agents, is what makes PSMA an attractive biomarker for diagnostic and therapeutic targeting (Donin and Reiter, 2018).
Recently, two PSMA-targeted ADCs (MLN2704 and PSMA-MMAE) have been used in clinical trials (Milowsky et al., 2016; Petrylak et al., 2020; Mjaess et al., 2023). However, both drugs were discontinued as they didn’t meet their primary endpoint due to the narrow therapeutic window, neurotoxicity effects, treatment-related adverse events, and lack of efficacy and safety in a significant percentage of patients (Galsky et al., 2008; Milowsky et al., 2016; Petrylak et al., 2020). The reported trial results of ADCs in PCs are indicative of the need for the development of novel, effective, and high-safety profile theranostics to treat advanced PCs. As of 2023, seven ADCs for PC are in phase I/II ongoing clinical trials (Sardinha et al., 2023). We have recently developed an anti-PSMA 5D3 monoclonal antibody, which can specifically bind to the extracellular domain of PSMA with sub-nanomolar affinity, and successfully used it as a therapeutic platform (Nováková et al., 2017; Huang et al., 2020). The unique characteristics of anti-PSMA 5D3 mAb include high target-specificity, enhanced binding affinity, fast internalization, and localization at the centrosome.
Aurora A kinase is a key mitotic regulator in the assembly of spindles, maturation of centrosomes, chromosomal segregation, and cytokinesis (Barr and Gergely, 2007; Görgün et al., 2010; Venkatakrishnan et al., 2015; Yan et al., 2016). The deregulation of Aurora A kinase is shown to induce mitotic arrest followed by apoptosis as well as severe mitotic abnormalities that result in selective lethality for many types of solid and hematological malignancies (Du and Hannon, 2004; Harrington et al., 2004; Cheung et al., 2009; Perez Fidalgo et al., 2009; Moore et al., 2010; Malumbres and Perez de Castro, 2014). Because of its significant role in mitotic progression and tumor proliferation (Cheung et al., 2009), Aurora A kinase inhibition would be expected to have an antitumor effect across a broad range of human tumors (Zhou et al., 2022). Alisertib (MLN8237) is a selective small molecular Aurora-A kinase inhibitor that is investigated for treatment as a single agent or in combination with other agents across a range of solid and hematologic malignancies (Manfredi et al., 2011; Venkatakrishnan et al., 2015; Tayyar et al., 2017). The high antitumor activity of MLN8237 through disruption of mitotic progression (Hoar et al., 2007; Nováková et al., 2017) can provide significant advantages as a drug for the development of the anti-PSMA 5D3-based ADC that targets PSMA (+) PC cells. In this study, we used Aurora A kinase inhibitor, MLN8237, for the first time as a conjugated chemotherapeutic using a novel click chemistry-based conjugation chemistry (CC linker) in ADC development with anti-PSMA 5D3 mAb. The resulting 5D3(CC-MLN8237)3.2 were labeled with imaging agents for image-guided evaluation of 5D3(CC-MLN8237)3.2 antibody-theranostic conjugates (ATCs). 5D3(CC-MLN8237)3.2 ATCs were fully characterized and evaluated in vitro for cytotoxicity and cellular uptake in PSMA (+/−) cells. In vivo, therapeutic efficacy was determined in bilateral/dual tumor human PC subcutaneous xenograft mouse models (Figure 1). At the end of the therapeutic study, animals were euthanized and evaluated for systemic toxicities by CBC study, clinical chemistry analyses of blood, and hematoxylin and eosin (H&E) staining.
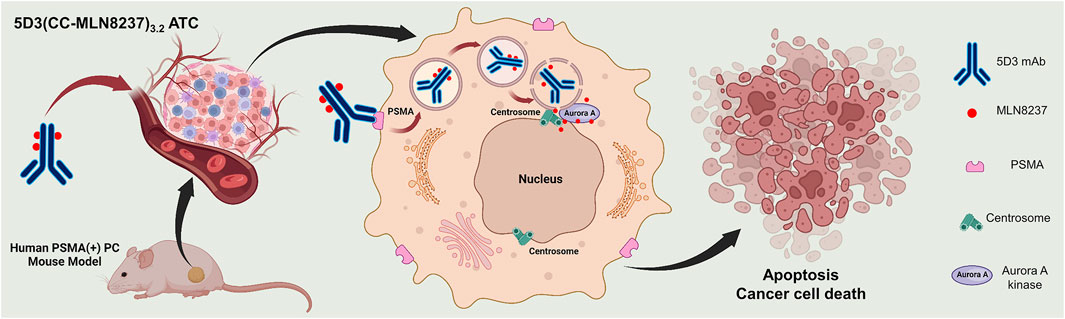
Figure 1. Schematic representation of the therapeutic approach using 5D3(CC-MLN8237)3.2 ATC in a PSMA (+) prostate cancer cell.
We used PSMA (+) PC3-PIP and PSMA (−) PC3-Flu PC cells in this study. Cells were kindly provided by Dr. Martin Pomper’s lab in the Johns Hopkins University School of Medicine. Cells were grown in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin−streptomycin. In addition, the medium for PC3-PIP was supplemented with puromycin (20 μg/mL). Cells were maintained at 37°C in a humidified incubator with a 5% CO2 atmosphere. Cells were tested for mycoplasma and confirmed to be free of contamination. These cells were used for in vitro imaging, cytotoxicity studies, flow cytometric analysis, and for the preparation of mouse models for in vivo imaging, therapeutic studies, and toxicological analyses.
5D3 mAb was produced following the protocol as previously described and stored in 0.02% NaN3/phosphate-buffered saline (PBS) for long-term storage (Nováková et al., 2017). The NaN3 was removed by buffer exchange using 30 kDa MWCO ultrafiltration centrifugal filter units purchased from Sigma-Aldrich, Inc. before use. Methyltetrazine amine.HCl and trans-cyclooctene PEG4-NHS (TCO-PEG4-NHS) were purchased from Click Chemistry Tools, Scottsdale, AZ, United States. The Aurora A kinase inhibitor drug, MLN8237 (Alisertib), was purchased from MCE (MedChemExpress) LLC, NJ, United States NHS esters of fluorophores, AlexaFluor 488, AlexaFluor 555, and CF750 dyes were purchased from MilliporeSigma Inc. and Biotium Inc., United States, respectively. Organic solvents and HPLC-grade water were purchased from Sigma-Aldrich and FisherScientific, Inc., respectively. Dulbecco’s phosphate-buffered saline (DPBS), and BupH phosphate-buffered-saline were purchased from Thermo Fisher, Inc. All chemicals, reagents, and solvents were used without further purification unless otherwise stated.
The syntheses were designed and conducted as shown in the Figure 2. MLN8237 (25 mg, 48.2 µmol), 1.2 equivalent of methytetrazine amine.HCl (13.7 mg, 57.8 µmoles), and 1.2 equivalent of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 9.0 mg, 57.8 µmoles) were taken into a micro-scale reaction vial and dissolved in dimethylformamide (DMF, 2.0 mL). The reaction mixture was treated with 1.0 µL of N,N-diisopropylethylamine (DIPEA) and stirred at room temperature for 12 h. The organic layer was evaporated using a rotavapor, and the crude solid product was purified by the HPLC-PDI system (Shimadzu LC-AD HPLC-PDI) equipped with a C18 reverse-phase column using a 10%–90% gradient acetonitrile/water with 0.1% Trifluoroacetic acid (TFA) mobile phase (Supplementary Figure S1). The desired pure product MLN8237-Tz was isolated as a pink-red solid (20 mg, yield 67%). The structure was confirmed by 1H NMR (Supplementary Figure S2, Bruker Avance III 500 MHz NMR spectrometer) and HPLC-MS (Agilent Infinity Lab LC/MSD XT system) equipped with a C18 reverse-phase column and a high-speed, sensitive single quadrupole mass spectrometer (Supplementary Figure S3). 1H NMR (500 MHz, CDCl3): δ 8.60 (s, 1H), 8.56 (d, J = 8 Hz, 2H), 8.32 (m, 2H), 8.25 (d, J = 9 Hz, 2H), 7.79 (bs, 1H), 7.73 (d, J = 10 Hz, 2H), 7.58 (d, J = 5 Hz, 2H), 7.44 (m, 1H), 7.40 (bs, 1H), 6.77 (bs, 2H), 4.80 (s, 2H), 3.99 (s, 3H), 3.08 (s, 3H), 1.24 (s, 3H). HPLC-MS (m/z): [M+H]+ calcd for C38H29ClFN9O3 702.2, found 702.3.
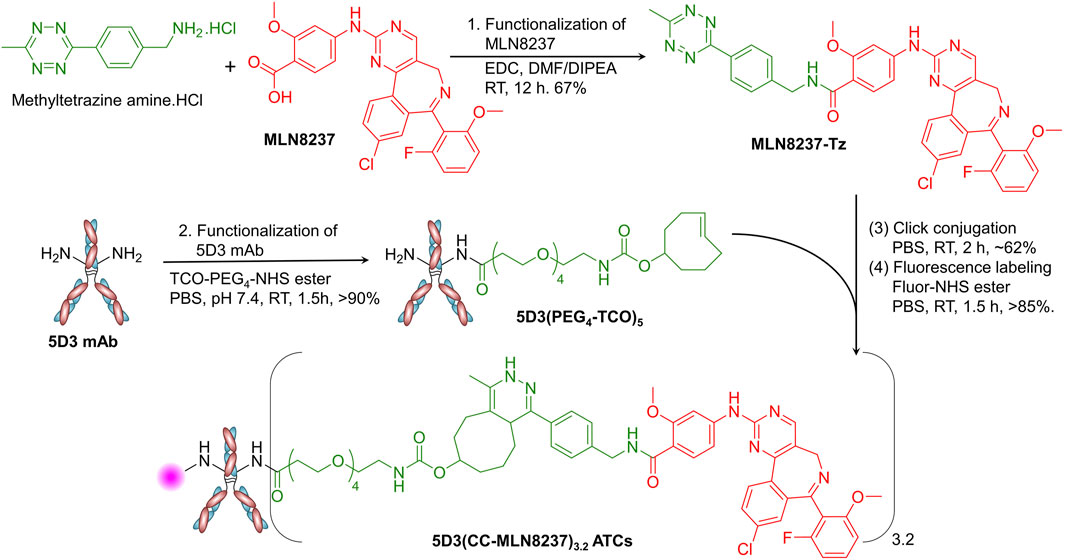
Figure 2. Synthesis of the 5D3(CC-MLN8237)3.2 ATCs. In the synthesis of 5D3(CC-MLN8237)3.2 ATC, first, (1) MLN8237 was functionalized with methyltetrazine using EDC-activated conjugation chemistry in DMF/DIPEA (Yield: 67%). (2) 5D3 mAb was functionalized with pegylated TCO using TCO-PEG4-NHS ester in PBS (Yield: >90%). (3) 5D3(PEG4-TCO)5 and MLN8237-Tz were conjugated by catalyst-free strain-promoted click reaction in PBS (Yield: ∼62%). (4) The product, 5D3(CC-MLN8237)3.2, was subsequently labeled with fluorophores for in vitro and in vivo imaging using NHS ester of fluorophores in PBS (Yield: >85%).
5D3 mAb (20 mg, 0.133 µmol) in PBS (1.0 mL) was treated with 10 equivalent of TCO-PEG4-NHS ester (0.35 mg, 1.33 µmol in 10 µL of anhydrous DMSO) and stirred for 1.5 h at room temperature (Figure 2). Unreacted reagents and byproducts were removed by ultrafiltration using 10 kDa MWCO (4 mL) centrifugal filter units, and the product was further purified by size exclusion column (SEC) chromatography using an ÄKTA go protein purification system (Cytiva Life Sciences, MA, United States). The pure product, 5D3(PEG4-TCO)5 was isolated with over 90% yield and it was characterized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), matrix assisted desorption/ionization-time of flight (MALDI-TOF), and dynamic light scattering (DLS).
5D3(PEG4-TCO)5 (10.0 mg, 66.67 nmoles in 1.0 mL of PBS) was taken into a 1.5 mL Eppendorf tube and treated with MLN8237-Tz (2.34 mg, 33.33 µmole in 20 µL of anhydrous DMSO). The mixture was vortexed and stirred gently using a tube rotator for 3 h at room temperature. Unreacted reagents and byproducts were removed by ultrafiltration using 10 kDa MWCO (4 mL) centrifugal filter units, and the product was further purified by SEC chromatography to obtain 5D3(CC-MLN8237)3.2 with approximately 62% yield. For fluorescent labeling, the product (2–5 mg/mL) was treated with NHS-ester form of Alexa Fluor 488, Alexa Fluor 555, or CF750 (5 mol equiv. in 10 µL of anhydrous DMSO) and stirred at room temperature in PBS for 1.5 h. The reaction mixture was purified by ultrafiltration (10 kDa MWCO) followed by SEC chromatography as described earlier to obtain pure final products, fluorescent 5D3(CC-MLN8237)3.2 ATCs (Figure 3A) with over 85% yield. The degree of fluorescence labeling (DOL) was calculated following the manufacturer’s protocol (DOL = 1–2). All ATC samples were stored in 1X PBS at 4°C.
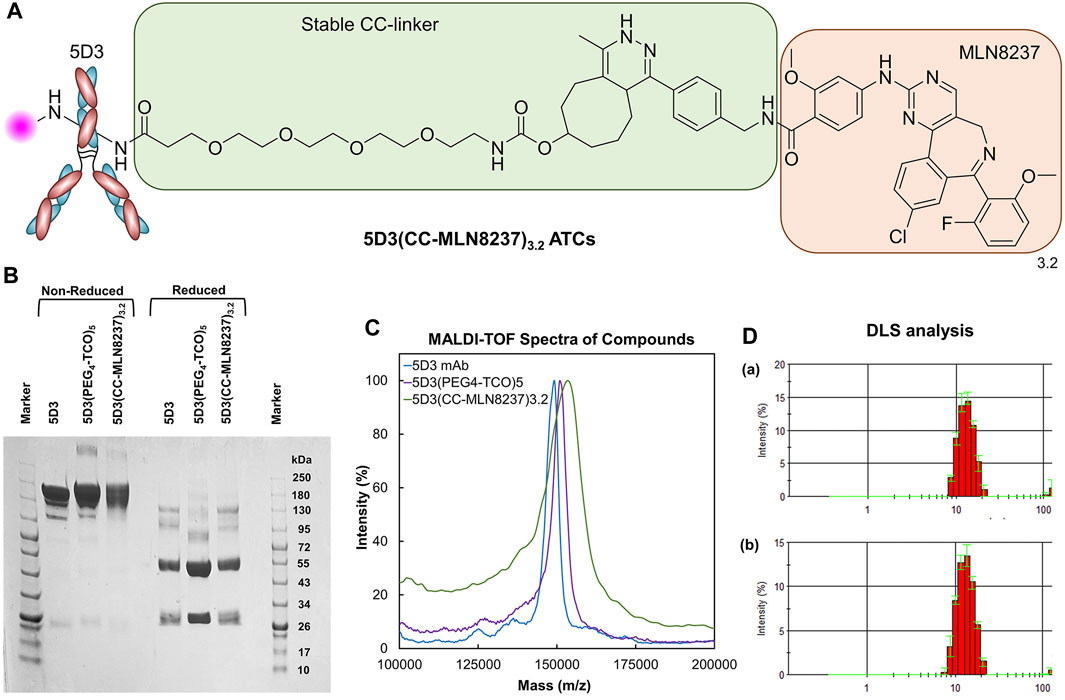
Figure 3. Structure and Characterization of 5D3(CC-MLN8237)3.2. (A) The structure of 5D3(CC-MLN8237)3.2 ATCs. (B) SDS-PAGE analysis of 5D3 mAb, intermediate 5D3(PEG4-TCO)5, and 5D3(CC-MLN8237)3.2 under non-reducing and reducing conditions. (C) MALDI-TOF spectra of 5D3 mAb, intermediate 5D3(PEG4-TCO)5, and 5D3(CC-MLN8237)3.2. (D) Dynamic light scattering analysis (DLS) and size distribution of (a) 5D3 mAb (12.7 nm) and (b) 5D3(CC-MLN8237)3.2 (13.4 nm).
Unconjugated 5D3 mAb, 5D3(PEG4-TCO)5, and 5D3(CC-MLN8237)3.2 were analyzed by SDS-PAGE under reducing and non-reducing conditions following standard lab procedures (Figure 3B). A 4%–20% Mini-Protean TGX gel (Bio-Rad Laboratories Inc., United States) was used, and a color protein standard broad-range (10–250 kDa) molecular weight marker (New England Biolabs Inc., MA, United States) was co-run to estimate molecular weights of proteins. Protein samples (3–4 μg/mL) were heated for 10 min at 70°C in NuPAGE sample reducing agent and non-reducing sample buffer (ThermoFisher Scientific, MA, United States). Samples were subsequently loaded into the wells in a volume of 10 μL and ran on Mini-PROTEAN Tetra Vertical Electrophoresis Cell (Bio-Rad Laboratories Inc., United States). Gels were stained with Coomassie Brilliant Blue R-250 Staining Solution (Bio-Rad laboratories, United States) and imaged using the Chemidoc Touch Gel Imaging System.
For the MALDI-TOF analysis, a 1.0 µL sample (5 mg/mL in H2O) was spotted on a targeted plate, and 1.0 µL of sinapinic acid (10 mg/mL in 1:1 H2O:CH3CN, 0.1% TFA) was co-spotted after drying in air. MALDI-TOF spectra were acquired after the spot was completely dry in the air on a Voyager DE-STR MALDI-TOF mass spectrometer (Mass Spectrometry and Proteomics Facility, JHU School of Medicine). The degree of conjugation (DOC) of PEG4-TCO and drug-to-antibody ratio (DAR) were calculated based on the change of molecular weights measured by MALDI-TOF (Figure 3C). The DOC for PEG4-TCO and the DAR were 5 and 3.2, respectively.
Cells grown to approximately 90% confluency were harvested by trypsinization. Twelve cell samples in 200 µL of media (0.2 million cells/sample) were treated with 5D3-AF488 or 5D3(CC-MLN8237)3.2-AF488 (20 μg/mL) and incubated at 37°C for 1.5 h. Unbound mAb and ATCs were washed using DPBS and the cells were then fixed by 4% paraformaldehyde (PFA). Isolated cell samples were filtered and analyzed by SONY SH800 flow cytometer (Supplementary Figure S4). Data was analyzed and fluorescence histograms were generated by FlowJo software (BD Biosciences, United States). The hydrodynamic diameter of free 5D3 mAb and 5D3(CC-MLN8237)3.2 (1.0 mg/mL) was determined by DLS (Figure 3D) using a Nano-ZS90 Zetasizer (Malvern Instruments, UK).
PSMA (+) PC3-PIP or PSMA (−) PC3-Flu cells (0.2×106 cells/chamber in 500 µL of media) were seeded in 4-well chamber slides (Nunc Lab-Tek, Thermo Scientific) and grown to ∼90% confluency (24–48 h). Cells were then fed with fresh media containing 5D3-AF555 or 5D3(CC-MLN8237)3.2-AF555 (20 μg/mL) and incubated at 37°C for 1.5 h. Cells were washed twice with 1X DPBS and fixed using 4% PFA in PBS on ice for 10 min. After washing once with DPBS, cells were permeabilized with 0.1% Triton X-100 for 5 min and then blocked with 1.0% BSA/10% goat serum/0.3 M glycine in 0.1% PBS-Tween for 1 h. After a quick wash with 1X DPBS, cells were treated with centrosome marker, recombinant anti-pericentrin Alexa Fluor® 488 antibody (Abcam, Inc, 1:500 dilution), and anti-α-tubulin Alexa Fluor® 647 (Abcam, Inc. 1:250 dilution) for overnight at 4°C. Cell nuclei were counterstained with Hoechst 33342 (10 μg/mL in H2O at room temperature for 10 min). After washing with 1X DPBS twice, the slides with cells were wet mounted. Cells were imaged using a Zeiss LSM880-Airyscan FAST super-resolution single-point, laser scanning confocal microscope. Images were processed using Zeiss Zen software.
The cytotoxicity of 5D3(CC-MLN8237)3.2-AF488 was determined by treating PC3-PIP and PC3-Flu (control) cells in 96-well plates in RPMI 1640 culture media in the presence of 5D3-(CC-MLN8237)3.2-AF488 over a series of concentrations. Cells were seeded in a 96-well plate (2000 cells/well) and grown for 24 h to achieve 30%–40% confluency. Then, cells were treated with increasing concentrations of 5D3(CC-MLN8237)3.2-AF488 or pure MLN8237 equivalent to the drug concentration in 5D3-(CC-MLN8237)3.2-AF555 (1.0 ng/mL-1.0 mg/mL by 10-fold increment). After the incubation for 72 h, IC50 values of 5D3(CC-MLN8237)3.2-AF488 ATC and corresponding free MLN8237 were determined using the WST-8 assay (CCK-8 assay, Dojindo Molecular Technologies), following the manufacturer’s protocol. Briefly, media in each well were replaced by 100 μL of the fresh media. Then, cells in each well were treated with 10 μL of WST-8 reagent and incubated at 37°C for 3 h. During incubation, WST-8 tetrazolium salt is reduced by dehydrogenase in living cells, forming a yellow formazan dye with λmax at 450 nm. After 3 h, the absorbance in wells was measured at 450 nm using a BioTek Epoch microplate spectrophotometer (Agilent Technologies). The concentration of the formazan dye in the media produced by dehydrogenases is directly proportional to the density of viable cells per well. The cell viability of treated cells was normalized to readings in untreated control cells with 100% viability. Data were fitted, and IC50 values for free MLN8237 and 5D3(CC-MLN8237)3.2-AF488 were calculated using Prism 9 software (GraphPad, San Diego CA).
Healthy, four-to-six weeks-old, athymic, nude male mice were purchased from Charles River Laboratories (United States) and maintained in the animal facility in Miller Research Building (B-level) of the Johns Hopkins School of Medicine (at 25°C/12 h light-dark cycle/food and water ad libitum). The bilateral PSMA (+/−) dual-tumor subcutaneous xenograft mouse models were prepared for the in vivo experiments. Mice were inoculated on the right and left flanks with PC3-PIP and PC3-Flu cells (2 × 106 cells in 50 μL of 1:1 RPMI 1640/Matrigel). The treatment started when tumor size was approximately 0.5 cm, a volume of approximately 65 mm3 (after 7–10 days). Tumor volumes were calculated using the formula (L×W2)π/6, where L is the longest diameter (the major axis), and W is the tumor width, measured perpendicular to the major axis.
All animal experiments were conducted strictly following the animal protocol approved by the Animal Care and Use Committee (ACUC) at the Johns Hopkins University School of Medicine.
We measured the tumor sizes using a digital caliper every other day to make sure not to exceed the tumor sizes subcutaneous xenograft tumor size above 2 cm in any direction. Animals were euthanized immediately when the tumor size exceeded the 2 cm limit or the tumor volume reached the 2000 mm3 limit following the approved protocol. All mice were sacrificed at the end of the treatment period by 3% isoflurane-assisted cervical dislocation.
Mice were randomly selected into treatment and control groups (n = 10). The first dose of 5D3(CC-MLN8237)3.2-CF750 (120 µL of 5.0 mg/kg in PBS) was injected through the tail vein. In vivo optical images were taken using Xenogen IVIS in vivo small animal imaging system using 745 nm as excitation wavelength (λex) and collecting the fluorescence signals at emission wavelength (λemi) 820 nm on day 4. Tumor sizes were measured every other day during the treatment period using a digital caliper and tumor volumes were calculated using the formula (L×W2)π/6. The second dose was administered on day 14 of the treatment period and measuring tumor sizes was continued for up to 21 days. Relative tumor volume (Vt/V0) was calculated and plotted against time (days) based on the tumor volume at day t (Vt) with respect to the tumor volume at day 0. We have also conducted a biodistribution study by ex vivo optical imaging of tumor tissues and selected vital organs extracted from treated mice after 96 h of drug administration.
Daily clinical observations and body weight measurements were conducted every other day as a part of a toxicological assessment during the treatment period. After the 21-days treatment period, we investigated possible in vivo toxicities of 5D3(CC-MLN8237)3.2-CF750 by (a) determining the complete blood count (CBC) and (b) analyzing the blood clinical chemistry profiles related to liver and kidney toxicities. Blood collections by cardiocentesis were performed at the time of euthanasia of mice in both treatment and control groups under sterile conditions in a biosafety cabinet. The total serum protein (T-Pro), blood urea nitrogen (BUN), alkaline phosphatase (ALP), creatinine (Cre), and alanine transaminase (ALT) levels were measured using Spotchem EZ vet veterinary chemistry analyzer (Scil Animal Care Company, Gurnee, Illinois, United States). Red blood cells (RBC), white blood cells (WBC) and platelet (PLT) contents were counted using the scil Vet abc Plus + hematology analyzer (Scil Animal Care Company, Gurnee, Illinois, United States). All aqueous solutions were prepared with ultrapure water. Following blood collections, tissues of tumors and vital organs, brain, lungs, heart, liver, kidneys, and spleen, were extracted and stained by H&E for pathological evaluation. Tissue samples were preserved in 10% neutral buffered formalin, later processed, and embedded in paraffin. H&E staining was performed on 4 μm sections following a standard protocol by the Reference Histology Laboratory, Department of Pathology, The Johns Hopkins Hospital.
The cell viability study was performed in triplicate independent experiments with triplicates per plate for statistical analyses. The one-way analysis of variance (ANOVA) was used for the omnibus F-test. In in vivo experiments, the statistical analysis (t-test) between treated and untreated groups was performed using JMP 12.1.0 Statistical Discovery from SAS. A p-value of less than 0.05 was considered significant (*p < 0.05). Survival curves in Kaplan−Meier analysis use the time when the tumor has reached over 2000 mm3 increase in the volume relative to the initial tumor volume as the surrogate endpoint to develop the Kaplan−Meier graph.
5D3(CC-MLN8237)3.2 ATCs are composed of an anti-PSMA 5D3 mAb conjugated with Aurora A kinase inhibitor (MLN8237) via a non-cleavable TCO-Tz-based click linker (Figure 2). This click linker is stable in the circulatory system because the drug was conjugated through an amide/peptide bond to the linker. Proteolysis of amide/peptide bonds is naturally catalyzed by intracellular enzymes predominantly in lysosomes (Yang et al., 2023). The concentration of proteolytic enzymes in the circulatory system is extremely low. Hence, the MLN8237 conjugated by the amide bond is stable in the circulatory system. The TCO- and Tz-functionalized 5D3(PEG4-TCO)5 and MLN8237-Tz undergo rapid, catalyst-free click chemistry. This reaction occurs with a kinetic rate ranging from 2 × 103 to 1 × 106 M−1s−1 under physiological conditions, driven by inverse electron demand Diels–Alder (IEDDA) conjugation chemistry (Blackman et al., 2008). Remarkably, this process produces only non-toxic N2 as a byproduct, without any cross-reactions with other functional groups. Additionally, pegylation of the linker enhances water solubility and biocompatibility of the resulting conjugate (Veronese and Mero, 2008). These advantages position our approach favorably compared to the common uncleavable heterobifunctional MCC linker chemistry for antibody-drug conjugation. 5D3(CC-MLN8237)3.2 was labeled with suitable fluorophores for in vitro or in vivo imaging. We conjugated only 1-2 fluorophores per antibody. Based on our previous studies and experience, these small molecular fluorophores (Molecular Weight <1.0 kDa) have not made any contributions or interference to cytotoxicity, therapeutic efficacy, internalization, or tumor uptake of the drug delivery system.
The SDS-PAGE analysis was conducted for 5D3(CC-MLN8237)3.2 parallel with starting 5D3 mAb and activated 5D3(PEG4-TCO)5 under both non-reduced and reduced conditions (Figure 3B). We observed the high purity of activated 5D3(PEG4-TCO)5 and 5D3(CC-MLN8237)3.2, same as starting 5D3 mAb, and all biomolecules revealed expected molecular weight of 150 kDa in non-reduced conditions. The standard pattern of bands was also observed under reduced condition, showing a heavy chain band at ∼50 kDa and a light chain band at ∼25 kDa. 5D3(CC-MLN8237)3.2 had slightly decreased gel mobility relative to the unmodified 5D3 mAb; however, it is not pronounced since the conjugated drug, MLN8237 is a small molecule. Under reduced condition, the heavy chain had lower mobility, therefore, we suggest that the drug is conjugated to the heavy chain.
MALDI-TOF spectra of 5D3 mAb, 5D3(PEG4-TCO)5, and 5D3(CC-MLN8237)3.2 were obtained using sinapinic acid-based matrix (Figure 3C). The TCO functionalization (DOF) and DAR were calculated based on changes in the molecular weights after each conjugation step. Based on MALDI-TOF calculation, the average DOF of PEG4-TCO groups is 5, and DAR is 3.2 in 5D3(CC-MLN8237), which are optimal levels to maintain the solubility in aqueous media, binding affinity, and pharmacokinetics. Our previous studies showed that DAR less than four including hydrophobic drug conjugation does not significantly increase lipophilicity (logP <5) and often exhibits optimal solubility in aqueous media studies (Huang et al., 2020). As a proof-of-concept study, we conjugated 3.2 drugs per antibody and used direct amine conjugation chemistry to minimize the mAb exposure in reaction conditions at reduced temperatures.
The flow cytometry study was conducted to determine any significant changes in the binding affinity of 5D3 after drug conjugation and fluorescence labeling in PSMA (+) PC3-PIP cells and compared with PSMA (−) PC3-Flu cells. The results in Supplementary Figure S4 show no significant difference in binding affinity between 5D3 mAb (i) and 5D3(CC-MLN8237)3.2-AF488 (ii) in PSMA (+) cells. This result confirms the preservation of 5D3 immunoreactivity and specificity to PSMA (+) PC3-PIP cells upon drug conjugation and fluorescent labeling. There was no 5D3(CC-MLN8237)3.2-AF488 affinity to PSMA (−) PC3-Flu cells (Supplementary Figure S4 (iii)/(iv)). The hydrodynamic diameters of 5D3 mAb and 5D3(CC-MLN8237)3.2 were 12.7 and 13.4 nm, respectively. There is an insignificant increase in hydrodynamic diameter after conjugation (Figure 3D); however, it does not affect the binding affinity and internalization kinetics, as shown in the in vitro study results below.
PSMA (+) PC3-PIP cells were labeled with fluorescent 5D3-AF555 and 5D3(CC-MLN8237)3.2-AF555 conjugates and incubated for internalization and localization in the subcellular components. Centrosomes of fixed cells were labeled with anti-pericentrin Alexa Fluor-488, a centrosome marker, and high-resolution confocal fluorescence images were acquired as shown in Figure 4. We observed the internalization of 5D3-AF555 and localization in the cytoplasm in PSMA (+) PC3-PIP cells (Figure 4A) and higher cellular uptake compared to PSMA (−) PC3-Flu cells (Supplementary Figure S5). Figure 4B and Supplementary Figure S6 show the super-resolution fluorescence image of a dividing PC3-PIP cell at telophase, visualizing the colocalization of 5D3-AF555 and anti-pericentrin-Alexa Fluor-488 marker, proving that 5D3 localizes in the proximity of centrosomes immediately after internalization. As shown in Figure 4C, 5D3(CC-MLN8237)3.2-AF555 also internalizes faster and localizes at the centrosomes, the same as free 5D3 mAb. Specific uptake of 5D3(CC-MLN8237)3.2-AF555 was not observed in PSMA (−) PC3-Flu cells (Figure 4D). We have previously quantitatively analyzed the cell surface binding and cellular uptake of 5D3 mAb, Fab fragments of 5D3 and their conjugates in PC3-PIP and PC3-Flu cells; we have not observed a significant change of internalization rate of 5D3 antibody upon drug conjugation (Hapuarachchige et al., 2020).
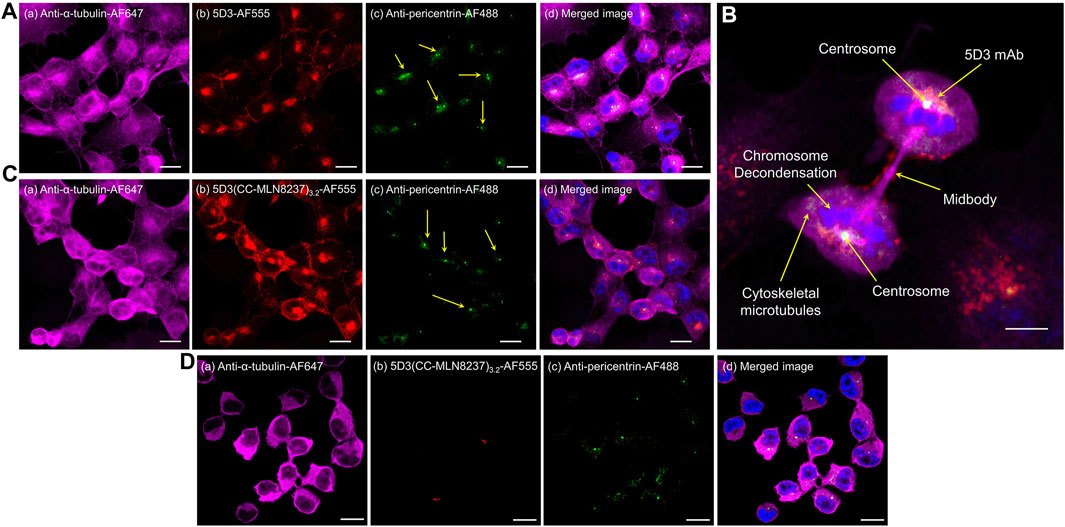
Figure 4. Super-resolution confocal fluorescence images of PSMA (+) PC3-PIP or PSMA (−) PC3-Flu cells. Panel (A) PSMA (+) cells treated with anti-α-tubulin-Alexa Fluor-647 (a), 5D3-AF555 (b), and anti-pericentrin Alexa Fluor-488 (c), with channel merged image with the blue channel for Hoechst 33342 nuclei counterstaining (d). Panel (B) Telophase of PSMA (+) mitotic cell showing the localization of 5D3 mAb at the centrosome. Cells treated with anti-α-tubulin-Alexa Fluor-647, 5D3(CC-MLN8237)3.2-AF555, anti-pericentrin Alexa Fluor-488, and Hoechst 33342 for nuclei counterstaining. Panel (C) PSMA (+) cells treated with anti-α-tubulin-Alexa Fluor-647 (a), 5D3(CC-MLN8237)3.2-AF555 (b), and anti-pericentrin Alexa Fluor-488 (c), with channel merged image with the blue channel for Hoechst 33342 nuclei counterstaining (d). Panel (D) PSMA (−) cells treated with anti-α-tubulin-Alexa Fluor-647 (a), 5D3(CC-MLN8237)3.2-AF555 (b), and anti-pericentrin Alexa Fluor-488 (c), with channel merged image with the blue channel for Hoechst 33342 nuclei counterstaining (d). (Scale bar: 10 μm, Magnification: ×63).
The in vitro cytotoxicity of 5D3(CC-MLN8237)3.2-AF488 on PSMA (+) PC3-PIP cells was evaluated using PSMA (−) PC3-Flu cells and free MLN8237 drug as controls. The concentration of MLN8237 in 5D3(CC-MLN8237)3.2-AF488 and free MLN8237 was used to directly compare IC50 values (Figure 5). To directly compare cytotoxicity, the dose was normalized for MLN8237 concentrations in the nanomolar range. As shown in Figure 5A, we observed a significantly high cytotoxicity by 5D3(CC-MLN8237)3.2-AF488 in PSMA (+) PC3-PIP cells (IC50 = 8.17 nM) compared to the cytotoxicity in PSMA (−) PC3-Flu cells (IC50 = 161.9 nM). PC3-PIP and PC3-Flu cells treated with the equivalent concentration of free MLN8237 show IC50 values of 736.9 and 873.4 nM, respectively (Figure 5B). These results suggest that the cytotoxicity of 5D3(CC-MLN8237)3.2 was target-mediated by the anti-PSMA antibody.
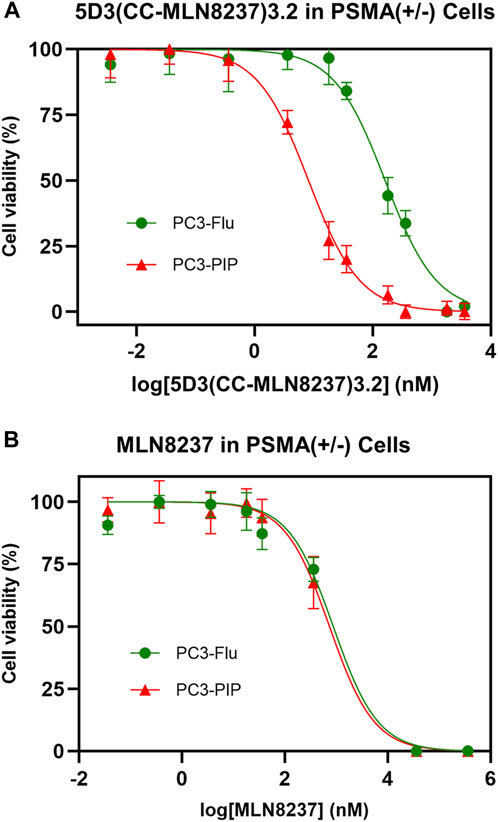
Figure 5. Effect of 5D3(CC-MLN8237)3.2-AF488 and free MLN8237 on PSMA (+/−) cell viability. (A). Cytotoxicity of 5D3(CC-MLN8237)3.2-AF488 in PSMA (+) PC3-PIP cells (IC50 = 8.17 nM) and PSMA (−) PC3-Flu cells (IC50 = 161.9 nM). (B). Cytotoxicity of free MLN8237 in PSMA (+) PC3-PIP cells (IC50 = 736.9 nM) and PSMA (−) PC3-Flu cells (IC50 = 873.4 nM). The cell viability was assessed with the WST-8 assay after 72 h of treatment with 5D3(CC-MLN8237)3.2-AF488 or equivalent free MLN8237 drug. The study was performed in triplicate per plate, and triplicate independent experiments, 5D3(CC-MLN8237)3.2-AF488 is 19.8 fold more efficacious in PSMA (+) cells than in PSMA (−) cells and 90.1 fold more efficacious than free MLN8237 drug in PSMA (+) cells. *p < 0.05.
The in vivo therapeutic efficacy studies of 5D3(CC-MLN8237)3.2-CF750 were conducted in PSMA (+/−) bilateral/dual human PC subcutaneous tumor xenograft mouse models following the therapeutic schedule shown in Figure 6A. In this study, two groups of mice (n = 10) with both PSMA (+) and PSMA (−) tumors were treated with 5.0 mg/kg dose of 5D3(CC-MLN8237)3.2-CF750 on day 1. We have administered two doses (5.0 mg/kg per dose) on day 1 and day 14 in this proof-of-concept study based on our previous dose establishment of in vivo therapeutic studies of 5D3 mAb-ADCs. Tumor sizes were measured every other day by a caliper, and the second dose was administered on day 14. In vivo optical images were taken at day 1, 3, and 4 post-injection time points using a Xenogen IVIS small animal imaging system, shown in Figure 6B. A higher tumor uptake of 5D3(CC-MLN8237)3.2-CF750 in PSMA (+) tumors was exhibited, proving the tumor specificity of the ATC compared to PSMA (−) tumors. We observed signals in the lower abdominal area mainly due to the bladder, urine contamination, or autofluorescence from feces. PC3-PIP tumor uptake of 5D3(CC-MLN8237)3.2-CF750 is significantly higher than its uptake in PC3-Flu tumors as shown in the ex vivo biodistribution study. There was high liver uptake, which was not detected in whole body imaging due to the limitation of fluorescent signal penetration through the body tissues. The tumor measurement was continued for a total of 21 days. Remarkably, PSMA (+) tumor growth ceased during the 5D3(CC-MLN8237)3.2-CF750 treatment period (Figure 6C). In contrast, PSMA (+) and PMSA (−) tumors in untreated groups and PSMA (−) tumors in the treated group grew rapidly during the treatment period.
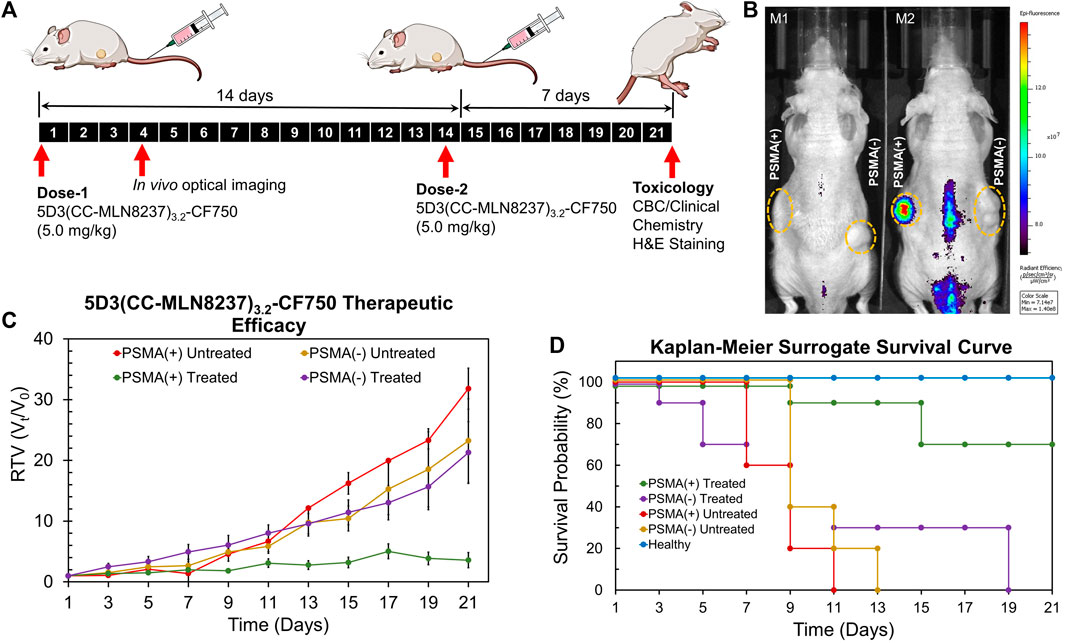
Figure 6. Imaging and therapeutic study in animal models. (A) Treatment schedules. Mice were inoculated with PSMA (+) PC3-PIP and PSMA (−) PC3-Flu cells. Two doses of 5D3(CC-MLN8237)3.2-CF750 (5.0 mg/kg) were given on day 1 and day 14 and tumor sizes were measured every other day for 21 days. (B) IVIS Xenogen images (λex:745 nm/λemi: 820 nm) of tumor-bearing mice (M1: untreated mouse and M2: treated mouse) on day 4 showed high tumor uptake of 5D3(CC-MLN8237)3.2-CF750 by PSMA (+) tumor (left flank) in treated mouse. (C) Change of relative tumor volume (Vt/V0) against the time of treatment period. The graph show the significant control of PSMA (+) tumor growth. (D) Kaplan-Meier surrogate survival curves showing higher overall survival of mice bearing PSMA (+) tumors and treated by 5D3(CC-MLN8237)3.2-CF750 (green, 70%) compared to PSMA (−) tumors in treated mice (purple, 0% on day 19), untreated mice with PSMA (+) tumors (red, 0% on day 11), and untreated mice with PSMA (−) tumors (yellow, 0% on day 13), and untreated healthy mice (blue, 100% during the treatment period). n = 10, *p < 0.05.
The Kaplan–Meier surrogate survival results for untreated mice and mice treated with 5D3(CC-MLN8237)3.2-CF750 are shown in Figure 6D. We used 2000 mm3 tumor size limit for the surrogate survival criterion. The results demonstrated that the 5.0 mg/kg treatment increased the survival of mice with PSMA (+) tumors (*p < 0.05), and 70% of PSMA (+) treated tumors did not reach the threshold level of tumor sizes during the treatment. In the treated group, 1 mouse died on day 9, and the tumors of the two mice became ulcerous and started to bleed. These two mice were euthanized according to the protocol on day 15. All mice in treated and untreated groups with PSMA (−) tumors and untreated PSMA (+) tumors exceeded the threshold tumor volume of 2000 mm3 before day 19.
Since Aurora A kinase is an intracellular enzyme, MLN8237 is effective upon intracellular drug release. MLN8237 is conjugated with the stable TCO-Tz linker via an α-peptide type amide bond. Amide bonds are hydrolysable by lysosomal enzymes such as proteases, amidases and cutinases (Rajagopalan and Kroutil, 2011; Mahesh et al., 2018) and it is also cleavable in acidic conditions (Dhavale et al., 2018). Therefore, we suggest that MLN8237 is released as free drug without traces of the linker by breaking this bond by the combination of enzymatic cleavage and acidic hydrolysis. We observed a significantly slower PSMA (+) tumor growth in treated mice (*p < 0.05) compared to the PSMA (−) tumors and PSMA (+) and PSMA (−) tumors in untreated mice from day 9 onward. PSMA (−) tumor growth was slower in both untreated and treated groups; however, this effect was insignificant. The overall efficacy of treatment was higher in PSMA (+) tumors and exhibited significantly (*p < 0.05) higher therapeutic efficacy in PSMA (+) tumors compared to PSMA (−) tumors.
To determine the toxicological clinical chemistry profile and CBC, we measured the levels of total T-Pro, BUN, ALP, Cre, and ALT levels, and RBC, WBC, and PLT counts in the blood. The results show a decrease in T-Pro and slight increase in Cre levels but these values were similar to healthy controls. The WBC and PLT counts in the blood of mice treated with 5D3(CC-MLN8237)3.2-CF750 was slightly decreased (Figure 7) likely due to consumption due to inflammation of the cancer and its necrosis. There is evidence of high liver uptake of the ATC in ex vivo optical imaging. However, all clinical chemistry and CBC changes are within the range of standard healthy levels. It is possible that ATC are not internalized into liver cells and show no significant liver toxicity since no liver enzymes were elevated. Supplementary Figure S8 shows the plot of the change in body weights of mice during the treatment period. No loss in body weight was observed in all mice of treated, untreated, and healthy groups. Overall, the toxicological study results are consistent with H&E staining of major organs isolated from mice following treatment. The H&E histological staining results show more necrosis in the PSMA (+) PC3-PIP tumor, less necrosis in the PSMA (−) PC3-Flu tumor, and no necrosis or other toxicological damage in the mice liver, kidneys, lungs, and spleen of 5D3(CC-MLN8237)3.2-CF750 treated group (Figure 8). Slides were reviewed in a blinded manner by a veterinary pathologist.
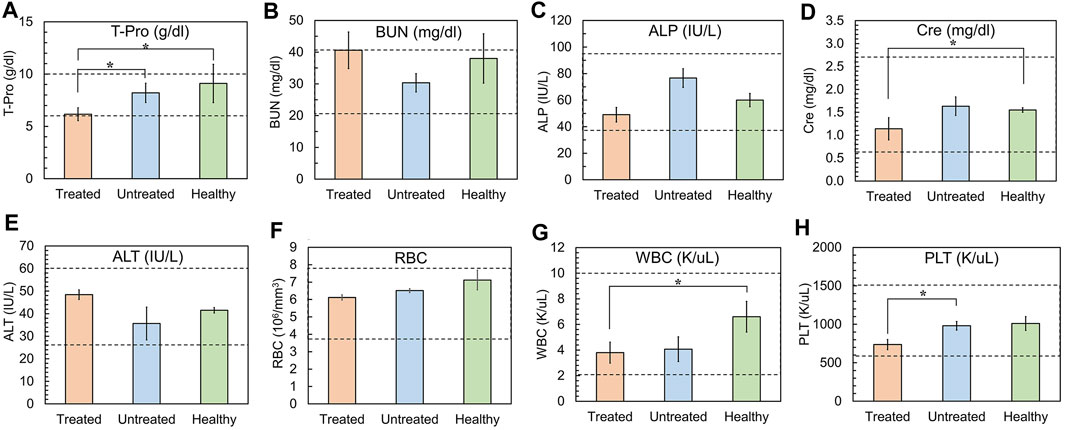
Figure 7. Analyses of clinical chemistry profile and, RBC, WBC, and PLT counts. (A) Total serum protein (T-Pro, normal range: 6.0–10.0 g/dL), (B) Blood Urea Nitrogen (BUN, normal range: 20–40 mg/dL), (C) Alkaline Phosphatase (ALP, normal range: 35–96 IU/L), (D) Creatinine (Cre, normal range: 0.60–2.72 mg/dL), (E) Alanine Transaminase (ALT, normal range: 25–60 U/L), (F) Red Blood cell Count (RBC, normal range: 3.8-7.9 ×106/µL, (G) White Blood cell Count (WBC, normal range: 2–10 K/μL) (H) Platelet Count (PLT, normal range: 600–1500 K/μL). Gray area: normal range of analytes in healthy mice. (Plotted with standard error bars, n = 5, *p < 0.05).
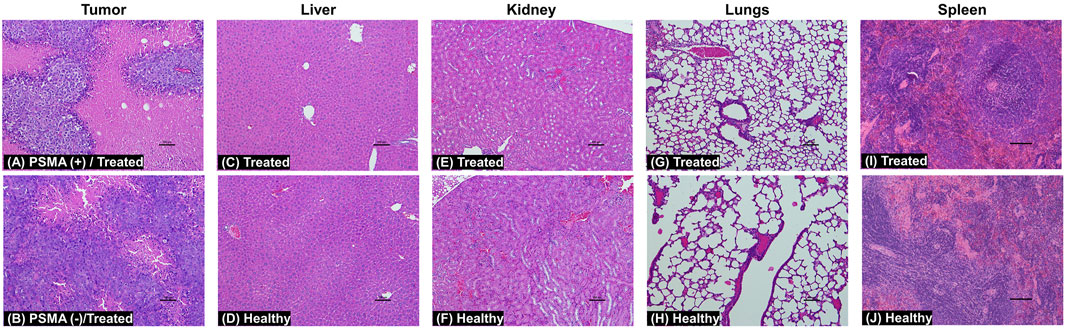
Figure 8. H&E staining images of tumors, liver, kidneys, lungs, and spleen at day 21. Treated mice were administered with a 5.0 mg/kg dose of 5D3(CC-MLN8237)3.2-CF750 or saline. Tissue sections reveal extensive necrosis in the PSMA (+) PC3-PIP tumors (A) and much less necrosis in the PSMA (−) PC3-Flu tumors (B) in treated mice. There was no toxicological damage in the liver (C, D), kidney (E, F), lung (G, H), and spleen (I, J) tissues. (n = 4, magnification: ×10, scale bar: 100 µm).
We have developed 5D3(CC-MLN8237)3.2, novel PSMA (+) PC targeting ATCs using anti-PSMA 5D3 mAb and Aurora A kinase inhibitor, MLN8237, conjugated via a TCO-Tz click chemistry-based stable linker (CC linker). The use of Aurora A kinase as a conjugated payload, and TCO-Tz click chemistry linker is demonstrated for the first time in the ADC development. 5D3(CC-MLN8237)3.2 were labeled with visible light or NIR fluorophores to integrate theranostic properties and to evaluate ATCs by in vitro and in vivo optical imaging. 5D3(CC-MLN8237)3.2 ATCs exhibited unaltered characteristics of 5D3 mAb in binding affinity, internalization, and subcellular localization. 5D3(CC-MLN8237)3.2-CF750 exhibited an enhanced therapeutic efficacy in vivo with minimal side effects and systemic toxicities in preclinical human PC xenograft mouse models compared to previously developed 5D3-based ADCs. The results indicate that 5D3(CC-MLN8237)3.2 ATCs could be valuable theranostics with potential benefit to patients with PSMA (+) PCs. To further prove these points, we are expecting to extend the therapeutic and toxicological studies to prostate cancer PDX models in the near future.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was approved by the Animal Care and Use Committee (ACUC) at the Johns Hopkins University School of Medicine. The study was conducted in accordance with the local legislation and institutional requirements.
IL: Data curation, Formal Analysis, Resources, Validation, Writing–review and editing, Methodology. BA: Data curation, Visualization, Writing–review and editing, Formal Analysis. WL: Formal Analysis, Validation, Writing–review and editing, Data curation. SS: Data curation, Formal Analysis, Writing–review and editing. ZN: Formal Analysis, Validation, Writing–review and editing, Methodology. CB: Formal Analysis, Resources, Validation, Writing–review and editing, Methodology. KG: Formal Analysis, Validation, Visualization, Writing–review and editing, Resources. VR: Validation, Writing–review and editing. DA: Formal Analysis, Methodology, Validation, Visualization, Writing–review and editing. SH: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was mainly supported by the Emerson Collective Cancer Research Fund (2021 Emerson Collective 139601) and was partially supported by the Congressionally Directed Medical Research Programs in the Department of Defense of United States (CDMRP/DOD) (W81XWH-20-1-0429/PC190040). This work was also in part supported by the Czech Academy of Sciences (RVO: 86652036) and the Ministry of Education, Youth, and Sports of the Czech Republic (LUAUS23254).
We thank Gary Cromwell and Xiaoju Yang for the technical assistance given for cell culture and animal studies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1385598/full#supplementary-material
Antonarakis, E. S., Piulats, J. M., Gross-Goupil, M., Goh, J., Ojamaa, K., Hoimes, C. J., et al. (2020). Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II KEYNOTE-199 study. J. Clin. Oncol. 38 (5), 395–405. doi:10.1200/JCO.19.01638
Arranz, A., and Ripoll, J. (2015). Advances in optical imaging for pharmacological studies. Front. Pharmacol. 6, 189. doi:10.3389/fphar.2015.00189
Bakhtiar, R. (2016). Antibody drug conjugates. Biotechnol. Lett. 38, 1655–1664. doi:10.1007/s10529-016-2160-x
Barr, A. R., and Gergely, F. (2007). Aurora-A: the maker and breaker of spindle poles. J. Cell Sci. 120 (17), 2987–2996. doi:10.1242/jcs.013136
Blackman, M. L., Royzen, M., and Fox, J. M. (2008). Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 130 (41), 13518–13519. doi:10.1021/ja8053805
Cheung, C. H., Coumar, M. S., Hsieh, H. P., and Chang, J. Y. (2009). Aurora kinase inhibitors in preclinical and clinical testing. Expert Opin. Investig. Drugs 18 (4), 379–398. doi:10.1517/13543780902806392
Dhavale, R. P., Waifalkar, P. P., Sharma, A., Dhavale, R. P., Sahoo, S. C., Kollu, P., et al. (2018). Monolayer grafting of aminosilane on magnetic nanoparticles: an efficient approach for targeted drug delivery system. J. Colloid Interface Sci. 529, 415–425. doi:10.1016/j.jcis.2018.06.006
Diamantis, N., and Banerji, U. (2016). Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 114 (4), 362–367. doi:10.1038/bjc.2015.435
Donin, N. M., and Reiter, R. E. (2018). Why targeting PSMA is a game changer in the management of prostate cancer. J. Nucl. Med. 59 (2), 177–182. doi:10.2967/jnumed.117.191874
Drago, J. Z., Modi, S., and Chandarlapaty, S. (2021). Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 18 (6), 327–344. doi:10.1038/s41571-021-00470-8
Du, J., and Hannon, G. J. (2004). Suppression of p160ROCK bypasses cell cycle arrest after Aurora-A/STK15 depletion. Proc. Natl. Acad. Sci. U. S. A. 101 (24), 8975–8980. doi:10.1073/pnas.0308484101
Einstein, D. J., Arai, S., Calagua, C., Xie, F., Voznesensky, O., Capaldo, B. J., et al. (2021). Metastatic castration-resistant prostate cancer remains dependent on oncogenic drivers found in primary tumors. JCO Precis. Oncol. 5, 1514–1522. doi:10.1200/PO.21.00059
Evans, A. J. (2018). Treatment effects in prostate cancer. Mod. Pathol. 31, 110–121. doi:10.1038/modpathol.2017.158
Galsky, M. D., Eisenberger, M., Moore-Cooper, S., Kelly, W. K., Slovin, S. F., DeLaCruz, A., et al. (2008). Phase I trial of the prostate-specific membrane antigen-directed immunoconjugate MLN2704 in patients with progressive metastatic castration-resistant prostate cancer. J. Clin. Oncol. 26 (13), 2147–2154. doi:10.1200/JCO.2007.15.0532
Ghosh, A., and Heston, W. D. (2004). Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J. Cell Biochem. 91 (3), 528–539. doi:10.1002/jcb.10661
Görgün, G., Calabrese, E., Hideshima, T., Ecsedy, J., Perrone, G., Mani, M., et al. (2010). A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood, J. Am. Soc. Hematol. 115 (25), 5202–5213. doi:10.1182/blood-2009-12-259523
Hapuarachchige, S., and Artemov, D. (2020). Theranostic pretargeting drug delivery and imaging platforms in cancer precision medicine. Front. Oncol. 10, 1131. doi:10.3389/fonc.2020.01131
Hapuarachchige, S., Huang, C. T., Donnelly, M. C., Barinka, C., Lupold, S. E., Pomper, M. G., et al. (2020). Cellular delivery of bioorthogonal pretargeting therapeutics in PSMA-positive prostate cancer. Mol. Pharm. 17 (1), 98–108. doi:10.1021/acs.molpharmaceut.9b00788
Harrington, E. A., Bebbington, D., Moore, J., Rasmussen, R. K., Ajose-Adeogun, A. O., Nakayama, T., et al. (2004). VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 10 (3), 262–267. doi:10.1038/nm1003
Herrmann, K., Larson, S. M., and Weber, W. A. (2017). Theranostic concepts: more than just a fashion trend—introduction and overview. J. Nucl. Med. 58 (2), 1S–2S. doi:10.2967/jnumed.117.199570
Hoar, K., Chakravarty, A., Rabino, C., Wysong, D., Bowman, D., Roy, N., et al. (2007). MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol. Cell Biol. 27 (12), 4513–4525. doi:10.1128/MCB.02364-06
Huang, C. T., Guo, X., Barinka, C., Lupold, S. E., Pomper, M. G., Gabrielson, K., et al. (2020). Development of 5D3-DM1: a novel anti-prostate-specific membrane antigen antibody-drug conjugate for PSMA-positive prostate cancer therapy. Mol. Pharm. 17 (9), 3392–3402. doi:10.1021/acs.molpharmaceut.0c00457
Jeitner, T. M., Babich, J. W., and Kelly, J. M. (2022). Advances in PSMA theranostics. Transl. Oncol. 22, 101450. doi:10.1016/j.tranon.2022.101450
Lindner, U., Lawrentschuk, N., Weersink, R. A., Davidson, S. R., Raz, O., Hlasny, E., et al. (2010). Focal laser ablation for prostate cancer followed by radical prostatectomy: validation of focal therapy and imaging accuracy. Eur. Urol. 57 (6), 1111–1114. doi:10.1016/j.eururo.2010.03.008
Lorente, D., Fizazi, K., Sweeney, C., and de Bono, J. S. (2016). Optimal treatment sequence for metastatic castration-resistant prostate cancer. Eur. Urol. Focus 2 (5), 488–498. doi:10.1016/j.euf.2016.10.008
Mahesh, S., Tang, K. C., and Raj, M. (2018). Amide bond activation of biological molecules. Molecules 23, 2615. doi:10.3390/molecules23102615
Malumbres, M., and Perez de Castro, I. (2014). Aurora kinase A inhibitors: promising agents in antitumoral therapy. Expert Opin. Ther. Targets 18 (12), 1377–1393. doi:10.1517/14728222.2014.956085
Manfredi, M. G., Ecsedy, J. A., Chakravarty, A., Silverman, L., Zhang, M., Hoar, K. M., et al. (2011). Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res. 17 (24), 7614–7624. doi:10.1158/1078-0432.CCR-11-1536
Marei, H. E., Cenciarelli, C., and Hasan, A. (2022). Potential of antibody-drug conjugates (ADCs) for cancer therapy. Cancer Cell Int. 22 (1), 255. doi:10.1186/s12935-022-02679-8
Milowsky, M. I., Galsky, M. D., Morris, M. J., Crona, D. J., George, D. J., Dreicer, R., et al. (2016). Phase 1/2 multiple ascending dose trial of the prostate-specific membrane antigen-targeted antibody drug conjugate MLN2704 in metastatic castration-resistant prostate cancer. Urol. Oncol. 34 (12), 530 e515–e530. doi:10.1016/j.urolonc.2016.07.005
Mjaess, G., Aoun, F., Rassy, E., Diamand, R., Albisinni, S., and Roumeguère, T. (2023). Antibody-drug conjugates in prostate cancer: where are we? Clin. Genitourin. Cancer 21 (1), 171–174. doi:10.1016/j.clgc.2022.07.009
Moore, A. S., Blagg, J., Linardopoulos, S., and Pearson, A. D. (2010). Aurora kinase inhibitors: novel small molecules with promising activity in acute myeloid and Philadelphia-positive leukemias. Leukemia 24 (4), 671–678. doi:10.1038/leu.2010.15
Mottet, N., Bellmunt, J., Bolla, M., Briers, E., Cumberbatch, M. G., De Santis, M., et al. (2017). EAU-ESTRO-SIOG guidelines on prostate cancer. Part 1: screening, diagnosis, and local treatment with curative intent. Eur. Urol. 71 (4), 618–629. doi:10.1016/j.eururo.2016.08.003
Nováková, Z., Foss, C. A., Copeland, B. T., Morath, V., Baranová, P., Havlínová, B., et al. (2017). Novel monoclonal antibodies recognizing human prostate-specific membrane antigen (PSMA) as research and theranostic tools. Prostate 77 (7), 749–764. doi:10.1002/pros.23311
Ojha, T., Rizzo, L., Storm, G., Kiessling, F., and Lammers, T. (2015). Image-guided drug delivery: preclinical applications and clinical translation. Expert Opin. Drug Deliv. 12 (8), 1203–1207. doi:10.1517/17425247.2015.1059420
Perera, M., Krishnananthan, N., Lindner, U., and Lawrentschuk, N. (2016). An update on focal therapy for prostate cancer. Nat. Rev. Urol. 13 (11), 641–653. doi:10.1038/nrurol.2016.177
Perez Fidalgo, J. A., Roda, D., Rosello, S., Rodriguez-Braun, E., and Cervantes, A. (2009). Aurora kinase inhibitors: a new class of drugs targeting the regulatory mitotic system. Clin. Transl. Oncol. 11 (12), 787–798. doi:10.1007/s12094-009-0447-2
Petrylak, D. P., Vogelzang, N. J., Chatta, K., Fleming, M. T., Smith, D. C., Appleman, L. J., et al. (2020). PSMA ADC monotherapy in patients with progressive metastatic castration-resistant prostate cancer following abiraterone and/or enzalutamide: efficacy and safety in open-label single-arm phase 2 study. Prostate 80 (1), 99–108. doi:10.1002/pros.23922
Rajagopalan, A., and Kroutil, W. (2011). Biocatalytic reactions: selected highlights. Mater. Today 14 (4), 144–152. doi:10.1016/s1369-7021(11)70086-4
Rosellini, M., Santoni, M., Mollica, V., Rizzo, A., Cimadamore, A., Scarpelli, M., et al. (2021). Treating prostate cancer by antibody-drug conjugates. Int. J. Mol. Sci. 22 (4), 1551. doi:10.3390/ijms22041551
Rosenfeld, L., Sananes, A., Zur, Y., Cohen, S., Dhara, K., Gelkop, S., et al. (2020). Nanobodies targeting prostate-specific membrane antigen for the imaging and therapy of prostate cancer. J. Med. Chem. 63 (14), 7601–7615. doi:10.1021/acs.jmedchem.0c00418
Sardinha, M., Palma Dos Reis, A. F., Barreira, J. V., Fontes Sousa, M., Pacey, S., and Luz, R. (2023). Antibody-drug conjugates in prostate cancer: a systematic review. Cureus 15 (2), e34490. doi:10.7759/cureus.34490
Sartor, O., and de Bono, J. S. (2018). Metastatic prostate cancer. N. Engl. J. Med. 378 (7), 645–657. doi:10.1056/NEJMra1701695
Siegel, R. L., Giaquinto, A. N., and Jemal, A. (2024). Cancer statistics, 2024. CA Cancer J. Clin. 74 (1), 12–49. doi:10.3322/caac.21820
Siegel, R. L., Miller, K. D., Wagle, N. S., and Jemal, A. (2023). Cancer statistics, 2023. CA Cancer J. Clin. 73 (1), 17–48. doi:10.3322/caac.21763
Tayyar, Y., Jubair, L., Fallaha, S., and McMillan, N. A. J. (2017). Critical risk-benefit assessment of the novel anti-cancer aurora a kinase inhibitor alisertib (MLN8237): a comprehensive review of the clinical data. Crit. Rev. Oncol. Hematol. 119, 59–65. doi:10.1016/j.critrevonc.2017.09.006
Vazquez, R., Civenni, G., Kokanovic, A., Shinde, D., Cantergiani, J., Marchetti, M., et al. (2021). Efficacy of novel bromodomain and extraterminal inhibitors in combination with chemotherapy for castration-resistant prostate cancer. Eur. Urol. Oncol. 4 (3), 437–446. doi:10.1016/j.euo.2019.07.013
Venkatakrishnan, K., Zhou, X., Ecsedy, J., Mould, D. R., Liu, H., Danaee, H., et al. (2015). Dose selection for the investigational anticancer agent alisertib (MLN8237): pharmacokinetics, pharmacodynamics, and exposure-safety relationships. J. Clin. Pharmacol. 55 (3), 336–347. doi:10.1002/jcph.410
Veronese, F. M., and Mero, A. (2008). The impact of PEGylation on biological therapies. BioDrugs 22 (5), 315–329. doi:10.2165/00063030-200822050-00004
Wang, X., Xuetao, X., Wu, M., Wu, P., Sheng, Z., Liu, W., et al. (2022). Inhibitory effect of roburic acid in combination with docetaxel on human prostate cancer cells. J. Enzyme Inhib. Med. Chem. 37 (1), 542–553. doi:10.1080/14756366.2021.2018684
Yan, M., Wang, C., He, B., Yang, M., Tong, M., Long, Z., et al. (2016). Aurora-A kinase: a potent oncogene and target for cancer therapy. Med. Res. Rev. 36 (6), 1036–1079. doi:10.1002/med.21399
Yang, N., Matthew, M. A., and Yao, C. (2023). Roles of cysteine proteases in biology and pathogenesis of parasites. Microorganisms 11 (6), 1397. doi:10.3390/microorganisms11061397
Keywords: prostate cancer, PSMA -prostate-specific membrane antigen, image-guided drug delivery, targeted therapy, theranostics, Aurora A kinase inhibition, MLN8237, antibody-theranostic conjugates
Citation: Liatsou I, Assefa B, Liyanage W, Surasinghe S, Nováková Z, Bařinka C, Gabrielson K, Raman V, Artemov D and Hapuarachchige S (2024) Development and therapeutic evaluation of 5D3(CC-MLN8237)3.2 antibody-theranostic conjugates for PSMA-positive prostate cancer therapy. Front. Pharmacol. 15:1385598. doi: 10.3389/fphar.2024.1385598
Received: 13 February 2024; Accepted: 15 April 2024;
Published: 01 May 2024.
Edited by:
Silvia Gazzola, University of Insubria, ItalyReviewed by:
Hancheng Cai, Mayo Clinic Florida, United StatesCopyright © 2024 Liatsou, Assefa, Liyanage, Surasinghe, Nováková, Bařinka, Gabrielson, Raman, Artemov and Hapuarachchige. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sudath Hapuarachchige, c2hhcHVhcjFAamguZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.