
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 10 April 2024
Sec. Inflammation Pharmacology
Volume 15 - 2024 | https://doi.org/10.3389/fphar.2024.1377113
This article is part of the Research Topic Recent Advances in the Pathogenesis and Potential Biomarkers of Fibrosis View all 6 articles
 Diana V. Ávila-Martínez1,2†
Diana V. Ávila-Martínez1,2† Wendy K. Mixtega-Ruiz1,3†
Wendy K. Mixtega-Ruiz1,3† José M. Hurtado-Capetillo4
José M. Hurtado-Capetillo4 Oscar Lopez-Franco1,2*
Oscar Lopez-Franco1,2* Mónica Flores-Muñoz1,2*
Mónica Flores-Muñoz1,2*The renin-angiotensin system (RAS) is an important cascade of enzymes and peptides that regulates blood pressure, volume, and electrolytes. Within this complex system of reactions, its counter-regulatory axis has attracted attention, which has been associated with the pathophysiology of inflammatory and fibrotic diseases. This review article analyzes the impact of different components of the counter-regulatory axis of the RAS on different pathologies. Of these peptides, Angiotensin-(1–7), angiotensin-(1–9) and alamandine have been evaluated in a wide variety of in vitro and in vivo studies, where not only they counteract the actions of the classical axis, but also exhibit independent anti-inflammatory and fibrotic actions when binding to specific receptors, mainly in heart, kidney, and lung. Other functional peptides are also addressed, which despite no reports associated with inflammation and fibrosis to date were found, they could represent a potential target of study. Furthermore, the association of agonists of the counter-regulatory axis is analyzed, highlighting their contribution to the modulation of the inflammatory response counteracting the development of fibrotic events. This article shows an overview of the importance of the RAS in the resolution of inflammatory and fibrotic diseases, offering an understanding of the individual components as potential treatments.
Inflammation is a physiological process used as a defense mechanism in response to tissue injury and infection. When an infection or tissue injury triggers an acute inflammatory response, blood components (proinflammatory mediators) are released to the local site where the signal begins. These signals are sent by endothelial cells, tissue-resident macrophages, and, in some tissues, mast cells, resulting in the recruitment of immune cells that orchestrate actions eliminating harmful stimulus and promoting tissue repair. If stimuli are not resolved and repaired, protein excess sets in the extracellular matrix causing the destruction of the original tissue architecture, originating fibrosis that leads to organ malfunction. The main effector cells in fibrosis, myofibroblasts, are responsible for remodeling the extracellular matrix. Increasing evidence has demonstrated that a major pathway in fibrosis generation is the transforming growth factor-β (TGF-β) and, indeed, this factor promotes a sustained fibrogenic immune cell phenotype (Frangogiannis, 2020).
Over the last years, researchers have discovered that the renin-angiotensin system (RAS), besides having an important role in the regulation of electrolyte balance, intravascular volume, and blood pressure, is also implicated in triggering inflammation and promoting tissue remodeling. Emerging evidence supports that the peptides from the newly discovered counter-regulatory arm of RAS, as part of the opposite effects to classic RAS, may also elicit anti-inflammatory and antifibrotic effects.
In this review, we describe some of the mechanisms were the counter-regulatory peptides have been found to be regulatory elements in inflammation and fibrotic processes, making them an interesting therapeutic target (Figure 1).
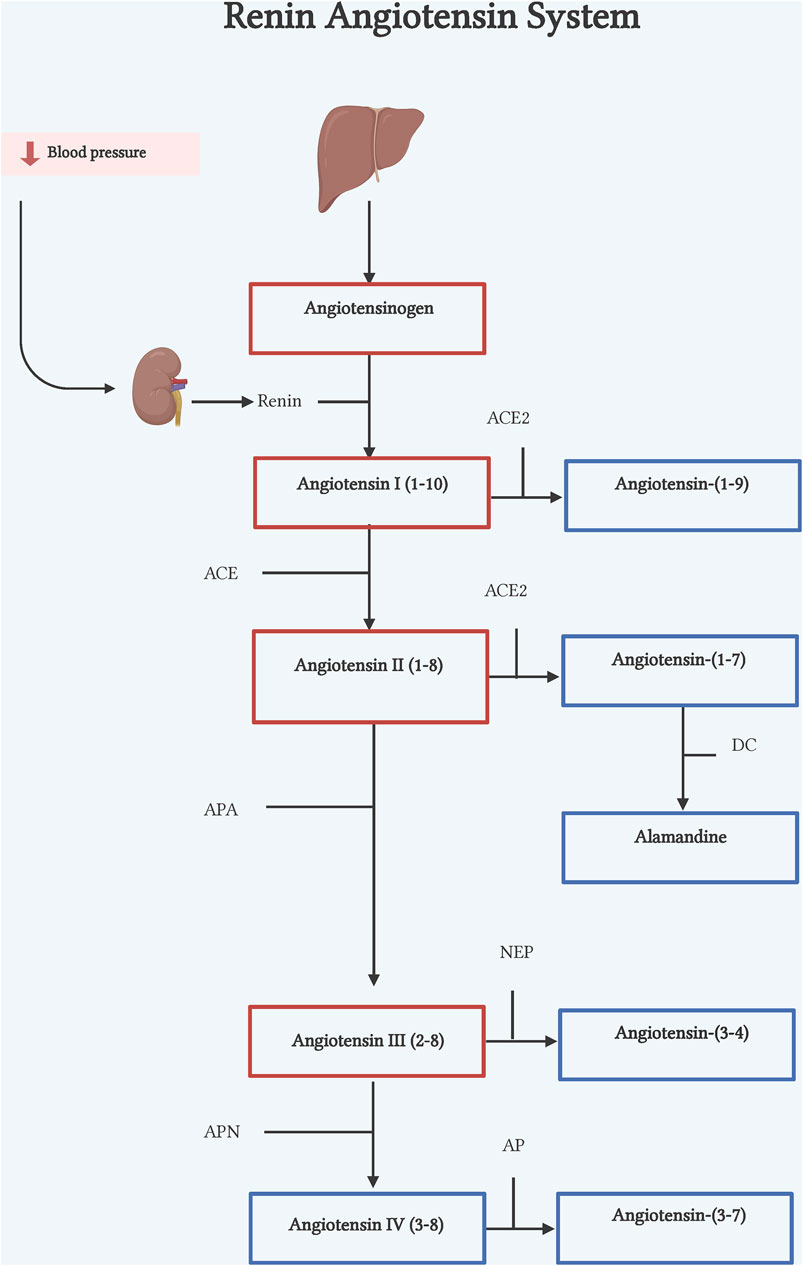
Figure 1. Renin-angiotensin system. Classic axis. Angiotensinogen (AGT), synthesized in the liver, is cleaved by renin, an enzyme synthesized by the kidneys, to form Ang I. Ang I is cleaved by ACE to form Ang II. Counter-regulatory axis. Ang I and Ang II are cleaved by ACE2 to form Ang-(1–9) and Ang-(1–7) respectively. Ang II is also cleaved by APA to form Ang III, by NEP to form Ang-(3–4), and by APN to form Ang IV. DC, decarboxylase; APA, aminopeptidase A; NEP, neutral endopeptidase; APN, aminopeptidase N; AP, aminopeptidase. Illustration was created with BioRender.com.
The initial recognition of pathogens or cell damage by tissue macrophages through pattern recognition receptors (PRRs), release a variety of cytokines, chemokines, and eicosanoids [such as tumor necrosis factor-alpha (TNF-α), interleukine (IL) 6, IL-1β, monocyte chemoattractant protein 1 (MCP-1), IL8, prostaglandins, etc.,] allowing the recruitment and transmigration of neutrophils, monocytes, and dendritic cells. Transcription nuclear factor kappa B (NF-κB) is one of the most common pathways activated by PRRs, which is responsible for inflammatory mediators’ induction by different immune cell types (Mussbacher et al., 2019). In activated M1 macrophages, NF-κB is a key pathway required for a large number of inflammatory genes including IL-1β, IL-6, IL-12, TNF-α, and cyclooxygenase-2 (Chen S. et al., 2023). Furthermore, NF-κB activation plays an important role in T cell activation and differentiation, thus, the pathway induces Th1 polarization improving the induction of cytokines and antigen presentation (Liu et al., 2017).
The transmigration mechanism is mediated by selectins (P, L, and E selectins) and integrins which facilitate adhesive interactions between leukocytes and endothelial cells of blood vessel walls, and additional chemokines and lipid chemoattractants to govern the migration of leukocyte route to inflamed tissue (Nourshargh and Alon, 2014). Inside the infection site, pathogens are opsonized, and the activated neutrophils attempt to kill the pathogens by releasing reactive oxygen species (ROS), reactive nitrogen species, proteinase 3, and cathepsin, found in the content of their granules. Consequently, surrounding tissue impairs the release of mediators that, not only contribute as inflammation signals but, also, triggers an antifibrinolytic coagulation cascade. The circulating platelets bind to exposed collagen and von Willebrand factor, activating and leading to the production of growth factors. Beyond this, other coagulation factors help to form fibrin and blood clots. (Serhan and Savill, 2005).
If the insult is eliminated, the set of signals switches from pro-inflammatory to anti-inflammatory cues such as lipoxins, resolvins, prostaglandins, and anti-inflammatory cytokines such as TGF-β or IL-10 (Levy et al., 2001; Serhan and Savill, 2005). The excess cells that had to proliferate in response to inflammation and damaged endothelial cells, now undergo apoptosis as an essential step for the clearance of resolved inflammation. During this procedure, suppression of NF-κB in neutrophils has been seen, which increases the cytotoxic effects of TNF-α (Ward et al., 1999). Apoptotic cells are phagocytized by a process called efferocytosis which includes the beginning of tissue restoration (Schmid and Brüne, 2021). The repair process involves the replacement of injury cells and components of extracellular matrix (ECM) that are beneficial for the healing process. As already mentioned, when the platelets are activated, they produce growth factors and through TGF-β action, stimulate fibroblast infiltration and its subsequent differentiation into myofibroblast, which promotes the production of collagen deposits and wound healing. In physiological conditions, ECM components are turnover and degraded by a family of proteinases termed matrix metalloproteinases (MMP) (Nathan and Ding, 2010).
Persistent tissue damage and non-resolving inflammation contribute to the development of chronic inflammation, leading to excessive deposits of ECM components generating fibrosis. Hence, fibrosis has been defined as the accumulation of ECM, mainly collagen and fibronectin, around damaged tissue. But the main question is, why is there persistent tissue damage? There are numerous reasons for that, for instance persistent infections by Helicobacter pylori (Waluga et al., 2015; Zahmatkesh et al., 2022), Schistosomes (Zhong et al., 2022), hepatitis viruses (Zhou et al., 2021), SARS-COV-2 virus (Shen et al., 2022), recurrent exposure to toxins, allergic and asthma (Hough et al., 2020), obesity, diabetes (Xiong et al., 2022) and hypertension (Schimmel et al., 2022).
The classical pathway of RAS begins with cleavage of the 10 N-terminal amino acids of angiotensinogen (ANG) by the enzyme renin, converting it into angiotensin I (Ang I) (Lu et al., 2016) (Figure 1). Renin is the active enzyme produced by juxtaglomerular cells in the kidney and released into circulation in response to sympathetic activation, low levels of sodium in renal tubules, and low pressure in afferent arterioles of the renal glomerulus (Muñoz-Durango et al., 2016). Human ANG is a glycoprotein that is composed of 485 residue amino acids. The liver is considered the main source of ANG, but other organs and cells such as the heart, kidney, brain, and leukocytes can express ANG in pathophysiological conditions (Gomez et al., 1993; Tamura et al., 1998; Kobori et al., 2007; Klimov et al., 2012). Subsequently, the angiotensin-converting enzyme (ACE), expressed in many tissues, cleaves the C-terminal dipeptide from Ang I to produce Angiotensin II (Ang II) octapeptide. Though, in vitro ACE activity also hydrolyzes other peptides such as bradykinin, N-acetyl-Ser-Asp-Lys-Pro and Angiotensin-(1–7) [Ang-(1–7)] due to its two catalytic sites (Hooper and Turner, 2003) (Figure 1).
Ang II is the most studied components of RAS, known for its principal effects as a vasoactive mediator. Ang II binds to its two main receptors, the Ang II type 1 and 2 receptor (AT1Rand AT2R, respectively). Both receptors are part of the G protein-coupled receptor family. AT1R mediates the main roles of Ang II in blood pressure regulation and body fluid homeostasis. Whereas AT2R activation is considered to have antagonist effects to AT1R. However, its complete role in different pathologies is still being studied. There are several reports describing an increase in the levels of several components of the RAS in different diseases such as hypertension, progressive nephropathic disease, diabetes, septic shock, asthma, aneurism, aging, and acute respiratory distress syndrome (Millar, et al., 1995; Kuba et al., 2006; Chawla et al., 2010; Antonucci et al., 2017), suggesting a wider effect of RAS, than merely control of blood pressure.
In the last decades, Ang II´s proinflammatory effects linked to fibrosis have been studied. Ang II is capable of up regulating many inflammatory mediators. Ang II infusion in normotensive subjects increased IL-6 plasma levels (Luther et al., 2006). Likewise, Ang II-infused mice showed an increase in the expression of cell adhesion proteins VCAM-1 and ICAM-1 on the surface of endothelial and leukocyte cells respectively, playing an important role in leukocyte adhesion (Qiu et al., 2022). Furthermore, Ang II raised IL-1β, IL-6, TNF-α levels, and macrophage infiltration in heart and vessel walls. These changes boosted TGF-β expression and collagen I/III production, promoting subsequent cardiac remodeling (Zhang et al., 2023). These effects were also observed in hypertensive patients with heart failure (Muñoz-Durango et al., 2016), suggesting that blocking inflammatory proteins could attenuate hypertensive cardiac remodeling (Wenzel et al., 2011; Qiu et al., 2022; Zhang et al., 2023). Moreover, expression of endoglin, a type III TGF-β receptor, and endothelin-1 in cardiac fibroblast (CF) were increased after Ang II treatment (Chen et al., 2004). Ang II also elicits an increase in collagen protein expression and a decrease in MMP-1 protein. These effects were abolished by AT1R and MAPp42/44 inhibitors, whereas AT2R antagonists had no effect (Chen et al., 2004). In addition, Ang II-induced TGF-β and endothelin-1 expression in CF promoted cardiac hypertrophy and renal fibrosis in vitro and in vivo (Gray et al., 1998; Sun et al., 2000; Schultz et al., 2002; Chen et al., 2004). This evidence implies that Ang II could promote profibrotic responses up-regulating TGF-β and endothelin-1 expression through the AT1R/MAPp42/44 pathway (Figure 2).
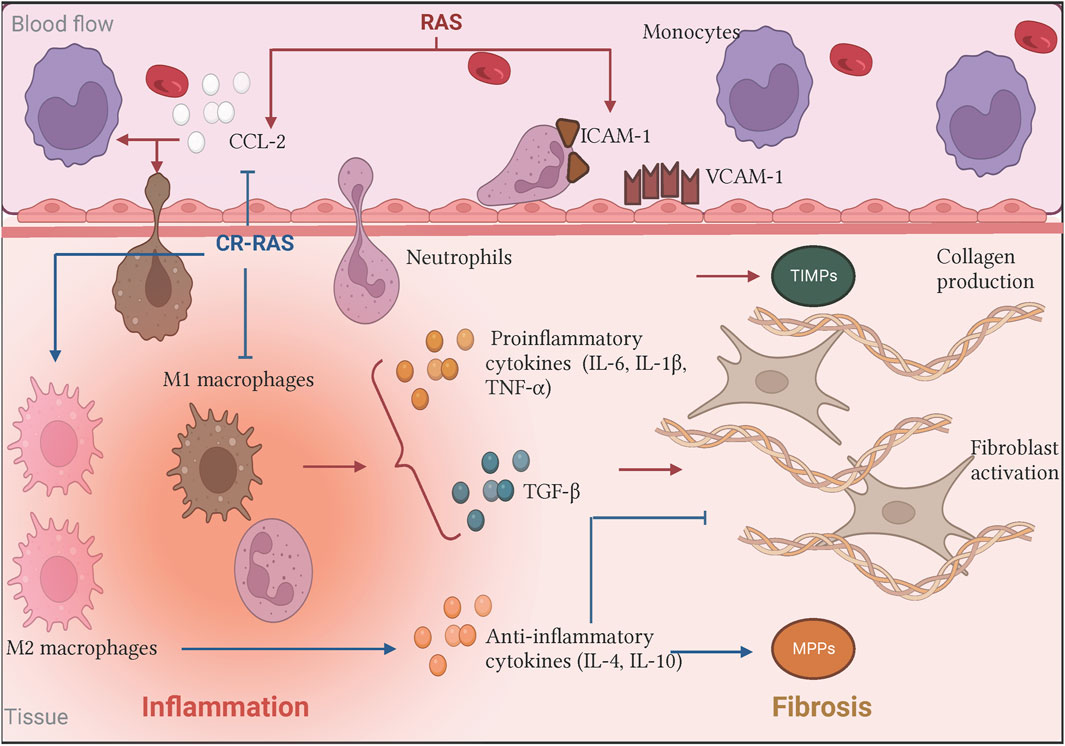
Figure 2. Biological effects of the RAS and the Counter-regulatory RAS on inflammation and fibrosis. In the classical axis, Ang II stimulus (illustrated as RAS in red bold letters) increases CCL-2 production and upregulates adhesins expression. Moreover, Ang II promotes pro-inflammatory cytokines releasing such as IL-6, which induces immunological cell recruitment and promotes its activation favoring the inflammatory process. In turn, the persistent signal gives rise to immune cells producing other mediators creating a microenvironment where the inflammation did not resolve, contributing to fibrogenesis by fibroblast activation and subsequent excessive collagen deposition. On the other hand, some of the counter-regulatory RAS (illustrated as CR-RAS in blue bold letters) have demonstrated opposite effects. Illustration was created with BioRender.com.
In addition, there is evidence of RAS overactivation in adipose tissue in obesity. It has been shown that Ang II induces insulin resistance and favors inflammation (Goossens et al., 2007). Ang II in vitro treatment increases MCP-1 secretion and proinflammatory cytokines. In contrast, adipocytes produce low levels of IL-10 after peptide stimuli, which impairs insulin secretion (Kalupahana et al., 2012). Moreover, pancreatic islet exposure to Ang II also induces cytokine production, impairs insulin secretion, and triggers β cells apoptosis. Interestingly, cytokine IL-1β blocking reduced inflammation, and restored insulin secretion, suggesting that inflammation deteriorates pancreatic islet function and could lead to end-organ damage (Sauter et al., 2015). On the other hand, scientific evidence has investigated the role of RAS in aging (Takeshita et al., 2023). Ang II has been implicated in the senescence process. The Ang II in vitro stimuli of human umbilical endothelial vein cells (HUVEC) and vascular smooth muscle cells (VSMC) induced cell growth arrest, and increased senescence-associated β-galactosidase, observing fragmented nuclei, and increase in apoptotic cells (Shan et al., 2008). Furthermore, persistent Ang II stimulation upregulates senescence-related protein expression such as p21, p16, p27, and p53 including a boost in ROS production and activation of transcription factors NF-κB and AP-1 in VSMC. These effects were inhibited by AT1R and PI3K/Akt inhibitors, suggesting that Ang II could exhibit its effect by AT1R/PI3K/Akt/p53/p21 pathway (Min et al., 2007; Li et al., 2014).
In addition to Ang II, other identified angiotensin peptides have been described as having counter effects to the classical RAS pathway. This non-classical pathway is also called the counter-regulatory arm and comprises several peptides product of Ang II and its precursor´s cleavage. The counter-regulatory RAS was identified through the concept of a local RAS based on two discoveries: first, expression of all components of the RAS in specific tissues. ACE, renin, angiotensinogen, Ang I and Ang II are expressed and synthesized in human and murine cardiomyocytes (Paul et al., 1993; Hirsch et al., 1991; Burrell et al., 2005; van Kats et al., 1998). Both the AT1R and the AT2R have also been shown to be expressed in cardiac cells (Booz and Baker, 1996). All these findings suggested that generation and consequently Ang II-mediated signaling could be initiated directly in individual tissues without the requirement for systemic, circulating RAS components. The second discovery was the identification of new components of the RAS expressed in specific tissues, predominantly the enzyme Angiotensin-converting enzyme 2 (ACE2). These findings resulted in the theory of a “local and tissue-specific” RAS. Since the recognition of this local tissue-specific RAS many studies have been described to elucidate the actions and mechanisms of their components and to attempt to segregate these actions from those mediated by circulating Ang II produced via the classical pathway. This has led to the characterization of the physiological and pathophysiological action of the RAS in different organs. Local tissue-specific RAS has been described in the heart, blood vessels, kidney, adrenal gland, nervous system, reproductive system, skin, digestive system, lymphatic and adipose tissue and in fetal development.
One of the main discoveries of the counter-regulatory RAS is the generation of Angiotensin peptide metabolites. These peptides include Ang-(1–7), angiotensin-(1–9) [Ang-(1–9)], angiotensin-(3–7) [Ang-(3–7)], angiotensin-(3–4) [Ang-(3–4)], angiotensin-(1–5) [Ang-(1–5)], angiotensin-(2–8) [Ang-(2–8)], angiotensin-(3–8) [Ang-(3–8)], also called angiotensin III (Ang III) and IV (Ang IV) respectively, and alamandine (Paz Ocaranza et al., 2020).
Ang-(1–7) is a heptapeptide discovered in 1988 in the brainstem by Santos et al. (1988). The first characterization of the peptide described it as an Ang I´s metabolite by ACE enzymatic action. However, after ACE inhibition, Ang-(1–7) was still detectable, suggesting that other enzymes could generate this peptide (Santos et al., 1988). Some years later, ACE2 was identified as an ACE homolog, that was able to hydrolyze Ang II to Ang-(1–7) (Tipnis et al., 2000) and Ang I to Ang-(1–9) (Donoghue et al., 2000). Unlike ACE, ACE2 is tissue-specific, firstly described as expressed only in heart, kidney, and testis (Donoghue et al., 2000), to then be shown to express in many other tissues, as enterocytes, reproductive cells, eyes, etc. (Hikmet et al., 2020). Subsequently, Ang I can also be converted into Ang-(1–7), via either cleavage by ACE 2, to form Ang-(1–9) which is then hydrolyzed by ACE transforming it into Ang-(1–7), or by a direct conversion by the action of prolyl endopeptidase, neutral endopeptidase or thimet oligopeptidase (Welches et al., 1993) (Figure 1). Although it has been proved that ACE2 has higher catalytic efficiency on Ang II to form Ang-(1–7), its formation at least in failing human heart, has been reported also by NEP suggesting that its pathway generation is dependent of physiological conditions (Vickers et al., 2002; Zisman et al., 2003; Rice et al., 2004). Ang-(1–7) acts through engaging the orphan Mas proto-oncogene (Santos et al., 2003). However, there is evidence that shows Ang-(1–7) can also bind to AT2R although with less specificity (Bosnyak et al., 2011).
The first described actions for Ang-(1–7) were in the cardiovascular system, where it inhibited Ang II-induced vasoconstriction (Roks et al., 1999; Jiang et al., 2014). Afterward, several studies have shown anti-inflammatory and antifibrotic effects in different pathological conditions (Figures 2, 3). There is evidence that Ang-(1–7) treatment alleviates induced inflammation by cecal ligation and puncture in murine models (Passaglia et al., 2023). Ang-(1–7) reduced expression of proinflammatory cytokines TNF-α and IL-6 while augmenting anti-inflammatory cytokines IL-4 and 10 and promoting M2 macrophage polarization. These effects were accompanied by a decrease in NF-kB phosphorylation, suggesting that Ang-(1–7) inhibits inflammation through this pathway (De Carvalho Santuchi et al., 2019; Pan et al., 2021). In another study using a E. coli-induced peritonitis, administration of Ang-(1–7) promoted the recruitment of M2 macrophages, increasing its phagocytic capacity, and increased the production of IL-10, MCP-1. These effects were inhibited in the absence of the Mas receptor, suggesting that Ang-(1–7) could contribute to the resolution of inflammation through the Mas receptor (Zaidan et al., 2022).
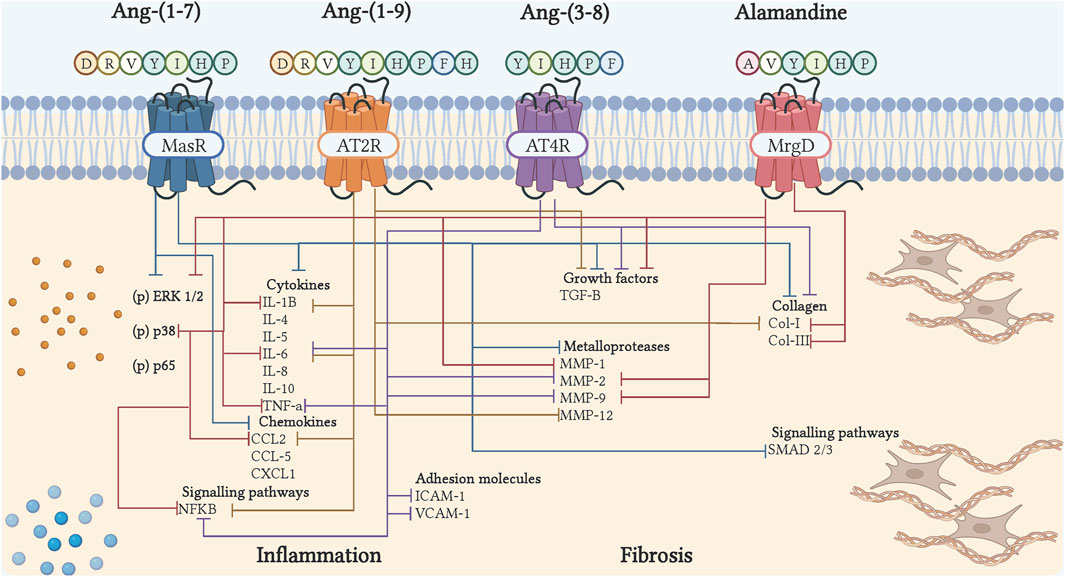
Figure 3. Cell signaling of bioactive peptides of the counter-regulatory axis in inflammation and fibrosis. The binding of peptides to their receptor induces inhibition of cytokines, chemokines, adhesion molecules, growth factors, collagen, metalloproteases, and inflammatory and fibrotic signaling pathways. The blue arrows show the actions of Ang-(1–7), through Mas; the orange arrows, showing the actions of Ang-(1–9), via AT2R; the purple arrows, show the actions of Ang-(3–8), through AT4R; the red arrows, show the actions of Alamandina, through MrgD. Illustration was created with BioRender.com.
Furthermore, the role of Ang-(1–7) during sepsis has been evaluated. The infusion of this peptide prevented septic shock in different animal models, limiting the amount of IL-6 cytokine, nitric oxide (NO) production and decreasing renal, lung and liver damage, along with IκB kinase pathway inhibition, suggesting that Ang-(1–7) ameliorate sepsis-induced organ injury likely through the inhibition of an inflammatory response (Tsai et al., 2021; Garcia et al., 2023). In the septic process, an increase in Ang II levels has been shown and is associated with renal and myocardial injury with an increase in cytokine levels (Zhu et al., 2021; Chen X. S. et al., 2023). It has been found that administration of Ang-(1–7) decreases inflammatory cells infiltration as well as the production of IL-6, IL-1β, and TNF-α in myocardial and renal tissue of septic mice. Additionally, a higher survival percentage and a decrease of Ang II levels were observed in mice that were administered with Ang-(1–7) compared to untreated mice suggesting that treatment with this peptide could attenuate the organic damage caused by sepsis (Xu et al., 2021; Zhu et al., 2021; Chen X. S. et al., 2023).
During the recent coronavirus pandemic, researchers found that there was a RAS dysregulation and ACE2 dysfunction due to SARS-COV-2 binding to ACE2 receptor in host cells, driving to acute respiratory distress syndrome. COVID-19 patients exhibit reduced Ang-(1–7) levels compared to control individuals and this phenomenon was associated with the severe form of the disease (Carpenter et al., 2022). Therefore, the peptide was also suggested as a therapy in COVID-19 ill patients confirmed with high viral load and co-morbidities such as hypertension, diabetes, heart diseases and asthma. Plasma enriched with Ang-(1–7) was transfused in combination with favipiravir treatment and, in almost all cases, the peptide transfusion improved oxygen saturation and clinical status (Onal et al., 2022). It has been shown that SARS-COV-2 infect bronchoalveolar stem cells (BACS). BACS are a lung resident stem cell population that differentiate into many types of cells, contributing to the maintenance of bronchoalveolar duct. In response to injury, BACS ratio increases and promotes tissue repair, which is impaired when viral infection. In mice, Ang-(1–7) administration increased the percentage of BACS and decreased protein expression involved in programmed cell death and inflammation process, suggesting that Ang-(1–7) peptide treatment could have a protective function in lung injury by SARS-CoV-2 (Ciechanowicz et al., 2022). In addition, Ang-(1–7)´s in vitro treatment of human pulmonary alveolar epithelial cells stimulates with SARS-COV-2 spike protein reduced production of IL-6, IL-8 and suppressed ERK1/2 and AP-1 phosphorylation, suggesting that Ang (1–7) could block SARS-COV-2-exacerbating proinflammatory response (Shen et al., 2023).
On the other hand, there is experimental evidence indicating Ang-(1–7)/Mas pathway may contribute to the attenuation of airway pathological conditions. Asthma is a chronic allergic airway disease and is characterized by airway hyperresponsiveness and pulmonary remodeling in which inflammatory response play an important role with a predominance of eosinophils, increased IL-4, IL-5, IL-13, TNF-α cytokines, and chemokines such as MCP-1 and CCL5 (Magalhães et al., 2016). Ovalbumin challenge in mice is used as an experimental asthma model, since it replicates the pathophysiological features in patients (Magalhães et al., 2015). In previous studies using this model, Ang-(1–7) treatment statistically diminished the release levels of IL-4, IL-5 cytokines and MCP-1 and CCL-5 chemokines into serum, as well as granulocyte-macrophage colony-stimulating factor. Consequently, reducing inflammatory cell infiltrate, alveolar wall thickening and airway collagen I and mucus deposition (Magalhães et al., 2015). In addition, Ang-(1–7) administration upregulated Mas expression on bronchial cells and decrease epidermal growth factor receptor (EGFR), Src kinase and ERK1/2 phosphorylation in lung tissue (El-Hashim et al., 2019). Nonetheless, it has been demonstrated that Mas deficiency aggravates chronic allergic pulmonary inflammation, suggesting that Mas is an important receptor to trigger anti-inflammatory response and could be doing it through mediation of EGFR/Src/ERK1/2 pathway on lung tissue (Magalhães et al., 2016). Additionally, ATG5 is a key protein involved in the formation of autophagosomes overexpressed in lung tissue of asthma patients. Interestingly, Ang-(1–7) elicited decreased levels of ATG5 on human bronchial epithelial and smooth muscle cells. This ATG5 deficiency suppressed inflammation and airway fibrosis, similarly to what it was observed in ovalbumin-challenged mice with Ang-(1–7) administration, suggesting another mechanism for Ang-(1–7) as an asthma treatment (Xu et al., 2023).
As already mentioned, the end result of inflammation is fibrosis. In models of acute lung injury mimicking acute respiratory distress syndrome in humans, the action of Ang-(1–7) have been examined. In this syndrome, there is a disruption of epithelial and endothelial barriers in the lung leading to severe immune reactions, hypoxic respiratory failure, pulmonary edema, and fibrosis (Chen et al., 2013). In acute lung injury, the characteristic increase in the expression of collagen I and III, TGF-β, IL-6, and Smad2/3 in the lung, is reduced after Ang-(1–7) therapy, leading to a significant improvement in oxygenation and reduction of white blood cells counts mainly in neutrophil percentage (Chen et al., 2013; Zambelli et al., 2015; Wang et al., 2022). In addition, Ang-(1–7) infusion, starting immediately after lung injury, has also been shown to prevent pulmonary vascular resistance, diminish aortic pressure, and increase ACE2 activity in lung tissue (Supé et al., 2016). Ang II-induced pulmonary fibrosis in rats triggers a cascade of proinflammatory factors and adhesion molecules that allows infiltrate cells recruitment in the lung, mediated by phosphorylation of ERK1/2/NF-κB pathway. Interestingly, Ang-(1–7) treatment in Ang II-treated animals inhibited ERK1/2/NF-κB axis, proinflammatory cascade and ameliorated lung fibrosis, effects that were reverse by Mas antagonists. By contrast, in Ang-(1–7) alone group, exhibited lung inflammation and deposition of collagen I compared to the untreated group (Meng et al., 2014). Together these data showed, at least under pathological conditions, Ang-(1–7) can protect against fibrosis development down-regulating pathways such as ERK1/2/NF-κB, which makes it a therapeutic target in fibrotic conditions (Figure 3).
Ang-(1–7) has also been studied in fibrotic renal diseases. Renal diseases are characterized by interstitial cell infiltration, NF-κB activation, increased apoptosis, oxidative stress, and fibrosis, even when patients with renal failure present complications in other organs such as heart. In previous studies in renal injury models, long-term treatment with Ang-(1–7) decreased the production of pro-inflammatory cytokines, macrophage infiltrate, caspases cleavage and blood pressure, as well as attenuating Ang II levels meliorating oxidative stress and renal fibrosis (Lu et al., 2017; Zaman and Banday, 2022). Moreover, treatment was able to increase the activity of enzymes involved in collagen degradation, and improved heart function accompanied by a reduction in cardiac fibrosis (Li et al., 2009). Interestingly, in renal failure models, Mas receptor deficiency inhibited NF-κB phosphorylation leading to reduction in MCP-1 and IL-6 cytokines and thus attenuating the fibrotic condition. Similar results were observed when wildtype experimental models were infused with short-term Ang-(1–7) treatment, suggesting that in some conditions, lack of signaling by Mas could prevent renal inflammation (Esteban et al., 2009).
One of the most studied actions of Ang-(1–7) is in cardiac remodeling. Many authors have confirmed that Ang-(1–7) has some effect in at least one of the components of cardiac remodeling. Grobe et al. demonstrated reduced myocyte hypertrophy, interstitial fibrosis and TGF-β levels in response to Ang-(1–7) in an Ang II-induced rat model of hypertension and cardiac remodeling (Grobe et al., 2007). Studies with transgenic mice overexpressing Ang-(1–7) in the heart have shown that in the presence of Ang II, hypertrophy and fibrosis of the left ventricle is reduced, as well as expression of TGF-β1 (Mercure et al., 2008). In cardiac fibrosis, exposure of Ang II-stimulated cardiac fibroblasts to Ang-(1–7) inhibited collagen synthesis and expression of endothelin-1 and leukaemia inhibitory factor (Iwata et al., 2005). It has also been described that Ang-(1–7) normalizes the decreased levels of MMP in Ang II-stimulated cardiac fibroblasts and myocytes (Pan et al., 2008). In addition, Grobe et al. (2006) showed that Ang1-(1–7) was able to prevent interstitial fibrosis by decreasing collagen deposition in the deoxycorticosterone acetate (DOCA) salt hypertensive rat model (Grobe et al., 2006). In nephrectomised mice Ang-(1–7) prevented left ventricular remodeling and diminished interstitial fibrosis by reducing the levels of TGF-β and increasing MMP2 and 9 (Li et al., 2009). Ang-(1–7) also decreased the expression levels of inflammatory cytokines and suppressed oxidative damage (Li et al., 2009) (Figure 3). It has been demonstrated that oxidative stress, vasoactive peptides, and inflammation can induce DNA damage, mitochondrial dysfunction, and protein misfolding leading to senescence and cell death (Erusalimsky, 2009). Endothelial senescence has been associated with cardiovascular diseases and atherosclerotic lesions (Mogi, 2020). Regarding Ang-(1–7), Romero et al. (2019), studied the heptapeptide role in endothelial cells senescence-induced by RAS or non-RAS mechanism. They found that Ang-(1–7) antagonized the cell senescence triggered by Ang II and IL-1β, which were blunted by A779 (Mas antagonist). The pro-inflammatory phenotype also was reduced after peptide treatment, which included a reduction in adhesines such as ICAM-1, VCAM, and IL-6 secretion. Additionally, the nuclear factor-erythroid 2-related factor 2 (Nrf2) and heme oxygenase-1 (HO-1) levels were augmented after Ang-(1–7) and inhibited when these pathways were blocked. These data suggest that Ang-(1–7) could prevent Ang II and IL-1β senescencent-induced effects by reducing oxidative stress by MasR/Nrf2/HO-1 axis (Romero et al., 2019). As mentioned above, oxidative stress and cytokine production can trigger cell senescence. Several reports on ROS dysregulation are related to the development of fibrosis diseases (Li et al., 2020). Due to that in fibrosis, there is low-grade chronic inflammation, which triggers excessive ROS production and may promote TGF-β synthesis leading to fibroblast activation with consequent ECM deposit accumulation. Hence, it could be interesting to study the pathways related to aging and fibrosis development.
Ang-(3–8), also known as Ang IV, is a hexapeptide generated through the cleavage of Ang III by aminopeptidase N. This peptide of only six amino acids long has been shown to have vasodilatory and inflammatory actions in different organs. The role of Ang-(3–8) in inflammation was described for the first time in 2005. The capacity of this peptide to activate the transcription factor NF-κB and positively regulate inflammation was demonstrated in vascular smooth muscle cells through the AT4R receptor (Esteban et al., 2005). On the contrary, studies have emerged establishing Ang-(3–8) having anti-inflammatory effects. In human endothelial cells, the capacity of Ang IV to mediate the expression of Macrophage Migration Inhibitor, a proinflammatory cytokine associated with the production of TNF-α, IL-1β and IL-6, was observed (Zhong et al., 2008). In murine macrophages, Nikolaou et al. (2014), evaluated the effects of Ang IV on the NF-κB pathway, where a lack of expression of proinflammatory genes such as ICAM-1 and TNF-α was observed, which could indicate differential effects of this peptide depending on cell lineage (Nikolaou et al., 2014). In a murine model of abdominal aortic aneurysm, treatment with Ang IV markedly reduced the infiltration of macrophages and proinflammatory cytokines (Kong et al., 2015). Furthermore, in cardiac ischemia-reperfusion (I/R) injury, Ang-(3–8) infusion managed to suppress the expression of VCAM-1, TNFα, MMP-9, and NF-κB proteins (Figure 3) (Park et al., 2016). Similar results were described in rats with cerebral hypoperfusion, where in addition to observing anti-inflammatory effects, it was discovered for the first time that this effect was induced in a dose-dependent manner (Wang et al., 2018). More recently, in mice, it was demonstrated how Ang IV was able to protect against acute myocardial infarction by inhibiting inflammation (Bai et al., 2021).
In a fibrotic context, little is still known in relation to Ang IV. However, some studies have demonstrated an interaction between this peptide and fibrotic pathologies. In a study performed in kidney cells, the capacity of Ang IV to induce the expression of plasminogen activator inhibitor-1 mRNA, a protein involved in fibrosis inhibition and progression of fibrotic events, was demonstrated (Gesualdo et al., 1999). Ang-(3–8) also induced interstitial fibrosis and cardiac deterioration in adult mouse hearts when bound to the AT1R receptor (Ainscough et al., 2009). Conversely, Ang IV dose-dependently downregulated Fox01-mediated fibrosis when bound to the AT4 receptor in a mouse model of diabetic cardiomyopathy (Zhang M. et al., 2021) (Figure 3).
Ang-(1–9) is a nine amino acid peptide that results from the metabolism of Ang I by ACE2 cleavage of the terminal amino acid (Donoghue et al., 2000). Although ACE2 is the main enzyme to form Ang-(1–9), carboxypeptidase A and cathepsin A can also produce this peptide (Jackman et al., 2002; Garabelli et al., 2008). These reports also show that formation of Ang-(1–9) from Ang I is the main pathway in cardiomyocytes (Kokkonen, et al., 1997; Garabelli et al., 2008). As already mentioned, once Ang-(1–9) is formed ACE cleaves the two last amino acids (phenylalanine and histidine) generating the active peptide Ang-(1–7). Ang I is the precursor to Ang-(1–9) through ACE2 (Figure 1). Although little is known about it, Ang-(1–9) has gained relevance as a counterregulatory active peptide. Ang-(1–9) binds mainly to the AT2R (Flores-Muñoz et al., 2011), exerting biological effects in organs such as kidney, lung and mainly heart.
Few studies have described Ang-(1–9) in an inflammatory context (Figure 2). In rats with diabetic heart disease, subcutaneous administration of this peptide normalized the levels of several proinflammatory cytokines, including TNF-α and IL-1β through the AT2R (Zheng et al., 2015). In DOCA-salt hypertensive rats, infusion of Ang-(1–9) decrease monocyte infiltration in heart, aorta and kidney tissue, reducing inflammation in these rats (Gonzalez et al., 2018). Similar results were also observed in a rat model of pulmonary hypertension, where treatment with Ang-(1–9) reduced plasma TNF-α, MCP-1, IL-1β, and IL-6 (Cha, et al., 2018). Furthermore, in prostate cancer cells Ang-(1–9) negatively regulated the expression of NF-κB1 and NF-κB2, modulating these inflammatory pathways (Domińska et al., 2020)
Within the fibrotic context Ang-(1–9) was shown to be capable of reducing cardiac fibrosis through the AT2R, by reducing the presence of type I collagen in spontaneously hypertensive rats (Flores-Munoz et al., 2012). Similarly in rats with Ang II-induced hypertension, this peptide was able to reduce the presence of collagen type I, in addition to reducing the expression of TGF-β (Ocaranza et al., 2014). These results were also observed in diabetic rats where collagen I and TGF-β mRNA expression was also decreased (Zheng et al., 2015). Adenoassociated delivery of Ang-(1–9) in mice infarcted hearts, reduced septal and perivascular fibrosis, as well as expression of MMP-12, described as promoter of fibrosis (Fattah et al., 2016). Furthermore, Fattah et al. (2016), described that Ang-(1–9) delivery, while reducing fibrosis, it also reduced acute rupture by stabilizing and thickening myocardial infarction scar, suggesting Ang-(1–9) remodeling modulation during scar evolution. Gonzalez et al. (2018), described Ang-(1–9) as having a protective role in hypertensive end-organ damage, by demonstrating reduction of collagen deposition and myofibroblast in heart, kidney and aorta when infused in DOCA-salt hypertensive rats (Gonzalez et al., 2018). Additionally, in rats with pulmonary hypertension the antifibrotic effects of Ang-(1–9) were replicated. Treatment with Ang-(1–9) in an monocrotaline induced pulmonary hypertensive model reduced pulmonary damage via the AT2R (Cha et al., 2018) (Figure 3).
Alamandine is a recently discovered heptapeptide derived from Angiotensin A, very similar to Ang-(1–7), differing only by presenting alanine instead of aspartate in the N-terminal domain (Lautner et al., 2013). Due to this similarity, alamandine has been shown to have similar actions to Ang-(1–7). However, these actions have been associated with its interaction with a different receptor, the Mas-related G protein-coupled receptor, member D (MrgD) (Lautner et al., 2013) (Figure 2). Within the inflammatory context, in vitro experiment showed that alamandine was able to dose-dependently regulate the degranulation of MMP-9 and myeloperoxidase in mouse neutrophils, suggesting an anti-inflammatory role (Da Silva et al., 2017). Later, in a murine model of cardiac dysfunction associated with sepsis, this peptide was able to prevent myocardial inflammation, by preventing the activation of ERK, JNK and P38 (Li et al., 2018). Furthermore, in mice subjected to a transverse aortic constriction procedure, it was observed that alamandine was able to decrease the expression of proinflammatory genes MCP-1, TNF-α, IL-1β and contribute to resolution with an increased expression of MRC1 and FIZZ1 (Morais Silva, 2020). Similar results were observed in a myocardial ischemia-reperfusion injury model where alamandine reduced the levels of TNF-α, IL-1β, IL-6 and NO, and protected cardiomyocytes by inhibiting the activation of NF-κB (Song et al., 2019). Therapeutic administration of this peptide decreased the number of neutrophils and M1 macrophages in a model of LPS-induced inflammation (De Carvalho Santuchi et al., 2019). Recently, alamandine was able to reduce doxorubicin-induced cardiotoxicity in rats, by counteracting the elevation of proinflammatory cytokines (Hekmat et al., 2021). In the kidney, this peptide also demonstrated anti-inflammatory actions by alleviating kidney injury by inhibiting PI3K/AK and MAPK pathways (Hu et al., 2021). Furthermore, alamandine was shown to have a protective role in a stroke model by reducing the expression of proinflammatory cytokines (TNFα, IL-1β, IL-6) (Figure 2; 3) (Gonçalves et al., 2022). Such protective effects were also observed in a collagen induced arthritis model (Ding et al., 2022).
Alamandine has also been described as having antifibrotic effects in different pathologies. Oral treatment of alamandine to spontaneously hypertensive rats resulted in decrease of collagen I, III and fibronectin expression of (Lautner et al., 2013; Liu et al., 2017). Similarly, administration of alamandine regulated vascular remodeling of ascending aorta by diminishing aortic fibrosis, collagen deposition, MMP activity and TGF-β expression in a murine model of transverse aortic contrition, as well as several important pro inflammatory genes (MCP-1, TNF-α, IL1-β) (De Souza-Neto et al., 2019). These effects are mediated through the MrgD receptor (Yang et al., 2020). In a subsequent study, when analyzing cardiac remodeling, alamandine administration reduced collagen deposition, MMP-2 and TGF-β expression in the left ventricle, induced by aortic constriction. Additionally, alamandine prevented ERK1/2 and AMPKα phosphorylation, suggesting important effects of alamandine in cardiac remodeling regulation (Morais Silva, 2020). Furthermore, Wang et al. (2023), suggested that alamandine antifibrotic effects on TGF-β activated fibroblast could be mediated by decreasing glycolysis through Parkin/CSF mitophagy (Wang et al., 2023). NF-κB and JNK pathways have been linked to alamandine actions. In a rat model of cardiac ischemia-reperfusion injury, the administration of this peptide reduced the presence of fibrotic markers, increasing the phosphorylation levels of ERK and JNK, while NF-κB decreased (Song et al., 2019). Another mechanism associated with the effects of alamandine was observed in liver and lung, where the peptide attenuated the presence of fibrosis by attenuating autophagy activated by ROS-dependent reactive oxygen species (Huang et al., 2020; Gong et al., 2022; Zhao et al., 2022). (Figure 3).
Currently, as part of the counter-regulatory RAS there are several other angiotensin peptides, but their role in inflammation or fibrosis has not been yet described. Within these, Ang III is a heptapeptide produced by Ang II cleaved in the Asp N-terminal residue, mediated by aminopeptidase A, and has been demonstrated to have high affinity to AT1R but shows selectivity to AT2R (Bosnyak et al., 2011). Ang III can induce NF-κB phosphorylation in mesangial cells and increase MAPK phosphorylation on VSMC, inducing proliferation. These effects are mainly mediated through the AT2R (Lorenzo et al., 2002; Alanazi and Clark, 2020). In addition, low concentrations of Ang III perfusion have been demonstrated to stimulate stretch-induced atrial natriuretic peptide (ANP) secretion. ANP secretion was abolished when the AT2R was blocked but not AT1R or Mas. Interestingly, such an effect was also blocked by pretreatment with PI3K, Akt or PKG inhibitor, showing that ANP secretion may be stimulated by PI3K–Akt–PKG signaling pathway (Park et al., 2013). Moreover, intrarenal treatment with Ang III augmented urine sodium excretion in rats and increased AT2R expression, effect that was abolished by an AT2R antagonist (Padia et al., 2009). In addition, Ang III treatment produced a significant reduction of diastolic blood pressure in spontaneously hypertensive rats (Matsufuji et al., 1995). However, in another study, Ang III infusion failed to increase urine sodium excretion rate and AT2R upregulation suggesting that the natriuretic effect is mediated by AT2R signaling (Padia et al., 2009; Kemp et al., 2019). These data together suggest that Ang III could have an important pathologic role in renal diseases; however, further studies are needed to understand its clinical implications.
On the other hand, Ang-(3–7) is formed by cleavage of the Pro-Phe amino acid residue from Ang IV by carboxypeptidase P (Wright et al., 2012). Early studies that evaluated Ang-(3–7) showed its pressor effect in rats (Ferreira A. J. et al., 2007). Subsequently, the role has been studied in vitro in prostate epithelial cells, observing that Ang-(3–7) incubation led to upregulation of MIK67, NF-κB gene expression, and mobility was increased (Domińska et al., 2020). Additionally, Ang-(3–7) administration reduced heart weight and cardiac hypertrophy gene markers ANP, BNP, and β-MHC. Furthermore, the peptide treatment inhibited collagen I, collagen III, and fibronectin expression in hearts’ mice and cardiac fibroblast cells. These results indicated that Ang-(3–7) could attenuate cardiac hypertrophy and prevent fibrosis development in experimental cardiac remodeling model (Zhang Y. et al., 2021).
Ang-(3–4) is one of the shorter peptides in the counter-regulatory RAS that has been shown to have some effects. It is produced from Ang III metabolism in plasma (Matsufuji et al., 1995) or by Ang-(1–7) cleavage in the kidney (Axelband et al., 2009). The first studies showing some effect of this peptide demonstrated a role as an antihypertensive, antiproliferative, and vasodilatory agent (Matsufuji et al., 1995; Matsui et al., 2005; Dias et al., 2017). In hypertensive models, it has been found that oral administration of Ang-(3–4) produced a reduction in systolic blood pressure, as well as urinary sodium excretion increased and AT1R downregulated in the renal proximal tubule of young rats (Saito et al., 1994; Luzes and Crisóstomo, 2021). Interestingly, the dipeptide depressor effect was not significant in aged mice (Matsui et al., 2003). In addition, in vitro studies show that Ang-(3–4) can also inhibit Ang II induced-human VSMC proliferation through intracellular Ca2+ suppression (Matsui et al., 2005). In a subsequent study, it was demonstrated ACE inhibition by dipeptide’s stimulus led to a reduction of Ang I-produced contraction of the aortic rings in rats (Vercruysse et al., 2008). Additionally, Ang-(3–4) effects are also investigated in overweight and undernutrition. In an overweight rat model, it was found that Ang-(3–4) treatment reduced blood pressure and hepatorenal index, suggesting that the peptide could prevent lesion formation induced by lipid deposits (Crisóstomo et al., 2022). Moreover, it has been found that in aging overweight and undernutrition juvenile rats, ACE2 levels decrease. Interestingly, after Ang-(3–4) administration, ACE2 levels are increased in overweight rats but not in rats with undernutrition conditions (Luzes and Muzi-Filho, 2021). In contrast, in a recent study, Pereira-Acácio et al. showed that a multi-deficient diet led to high blood pressure, and it could be associated with the development of hypertension (Pereira-Acácio et al., 2022). In this sense, they demonstrated that Ang-(3–4) treatment is capable of normalizing systolic blood pressure (Pereira-Acácio et al., 2022).
Finally, another recently studied peptide is Ang-(1–5). This pentapeptide is derived from Ang-(1–7) in vitro metabolism by the ACE enzyme, which hydrolyzes its Ile- His amino acid residues (Chappell et al., 1998). The most researched effects of pentapeptide are focused on the cardiovascular system. Roks et al. (1999), demonstrated that in vitro stimuli with Ang-(1–5) inhibited ACE activity from human plasma but is not capable of affecting arterial contractions (Roks et al., 1999). In addition, in isolated beating atria Ang-(1–5) stimulation induced augmented ANP secretion that was attenuated after inhibition of Mas, PI3K, Akt, and NOS, suggesting these pathways could be involved in ANP secretion stimulated by pentapeptide (Yu et al., 2016). Subsequently, the effect of Ang-(1–5) in Sprague-Dawley rats with I/R injury was evaluated. In this study, rats’ hearts were infused with low doses of Ang-(1–5) for 10 min prior to ischemic induction, further, they used AT1R, AT2R, and Mas antagonists. The Ang-(1–5) infusion ameliorated I/R-induced changes in left ventricular end-diastolic and developed pressure, cardiac infarct size and ANP secretion. These effects were abolished by the treatment with Mas antagonist, but not by AT1R or AT2R blockers. Moreover, the pentapeptide treatment also reduced pro-apoptotic proteins such as Bax, Caspase-3, and 9. However, anti-apoptotic proteins such as Bcl-2, and antioxidant enzymes such as Mn-superoxide dismutase, catalase, and HO-1 were increased. These effects were blocked when Mas was inhibited (Park, et al., 2021). Myocardial ischemic/hypoxia models are characterized by a gradual decrease in ANP secretion and changes in the profile of antioxidant enzymes and ROS production that lead to an increase in cell death (Park et al., 2021). Hence, this data together suggests that Ang-(1–5) treatment could attenuate myocardial damage in this pathology. Despite the recent evidence about the effects of these peptides, most reports are focused on their anti-hypertensive role. However, taken together all the available data, it could suggest that the upregulation of antioxidant pathways will lead to a reduction in ROS production with a consequent decrease in tissue damage to avoid chronic inflammation and fibrosis development.
Compound 21 (C21) is the first non-peptide agonist of the AT2R. This agonist was synthesized in 2004 by Yiqian Wan, and through a binding assay they demonstrated the selective affinity of C21 for AT2R. Additionally and most importantly, this new C21 had vasopressor effects in spontaneously hypertensive rats (Wan et al., 2004). The synthesis of this compound has facilitated the study of the specific effects of AT2R. Antihypertensive, proangiogenic, antifibrotic and anti-inflammatory effects in different organs have been attributed to the interaction of C21 and AT2R.
The anti-inflammatory actions of C21 with the AT2R interaction have been studied in different pathologies. In a model of acute myocardial infarction in Wistar rats, the intraperitoneal administration of C21 was able to stimulate AT2R, observing improvement in cardiac function and reduction of the scar in the post-infarction heart (Kaschina et al., 2008). In addition to these results, a null expression of p38 MAPK and p44/42 MAPK was observed. These MAPKs are traditionally involved in processes such as proliferative, apoptotic, and inflammatory. However, there is also evidence of opposite effects to those mentioned above. This association was demonstrated with evidence of anti-inflammatory actions, where C21 significantly suppressed the expression of cytokines (MCP-1, IL-1, IL-2, IL-6), so C21 contributed to the preservation of normal physiology after of myocardial infarction (Kaschina et al., 2008). Sampson et al., studied the involvement of C21 in TNF-α-induced inflammation in HUVEC, monocyte activation and in aortas of C57BL/6 mice. In vitro experiments indicated that C21 reduced TNF-α-induced expression of ICAM-1, CCL2, and IL-6 genes and proteins. In addition to reducing increased monocyte adhesion by 40% and reduced NFkB-p65 translocation from cytoplasm to the nucleus. These results were extrapolated to in vivo experiments, where treatment with C21 showed similar results (Sampson et al., 2016).
In renal pathological conditions, C21 counteracts most of the effects of the AT1R. Matavelli et al. (2011), evaluated early renal inflammation in renovascular hypertension. In Sprague-Dawley rats treated with C21 for 4 days, a reversal of the early inflammatory stage was observed, with reduction of the proinflammatory markers TNF-α, IL-6 and TGF-β1 in kidney tissue (Matavelli, et al., 2011). Another study in 2015 evaluated the involvement of C21 in a model of early diabetes in rats, where one of its main objectives was to study the effects of this compound on kidney inflammation. In this case, in addition to seeing a marked reduction in IL-6, C21 also reduced TNF-α and markers of oxidative stress (Matavelli et al., 2015). Years later, Jabber et al. (2023), demonstrated in a murine model of sepsis-induced renal inflammation that the modulation of C21-induced inflammation was mediated by the modulation of the PI3K/AKT signaling pathways (Jabber, et al., 2023).
In the lung, C21, as in the heart and kidney, proved to have anti-inflammatory potential. In experiments carried out in mice with pulmonary inflammation induced by bleomycin, the contribution of C21 as a pulmonary anti-inflammatory was confirmed for the first time, observing a reduction in cellular infiltrate in bronchoalveolar fluid (Du et al., 2009). In subsequent studies, carried out by Menk et al. (2018), it was demonstrated that, in this organ, C21 could reduce not only the cellular infiltrate, but also the expression of TNF-α and IL-6 (Menk et al., 2018). Similar results were observed in Chronic obstructive pulmonary disease (COPD) induced by cigarette smoke, showing a reduction in proinflammatory cytokines and, also, the inhibition of components of the NF-κB signaling pathway in alveolar macrophages (Mei et al., 2022). Currently, with the emergence of the COVID-19 pandemic, the study of acute respiratory distress syndrome (ARDS) has gained relevance. In this sense, Chen et al. (2024), studied the relationship of C21 in ARDS, where favorably, this compound was able to resolve the inflammatory stage, with a marked reduction of CCL-2, IL-6 through NF-κB (Chen et al., 2024).
C21 has been shown to have antifibrotic actions in various cardiac pathologies. In 2012, Rehman et al. (2012), studied the involvement of C21 in the development of hypertension and vascular damage in stroke-prone spontaneously hypertensive rats. In these rats administered with C21 for 6 weeks, a decrease in fibrotic and hypertensive events was observed, associated with the reduction of the stiffness of the mesenteric artery, the decrease in myocardial interstitial collagen type I/III and aortic oxidative stress, infiltrate of inflammatory cells and fibronectin (Rehman et al., 2012). Subsequently, in rats with myocardial infarction, in addition to observing the ability of C21 to improve arterial stiffness and reduce collagen, a decrease in the extracellular matrix metalloproteases MMP2 and MMP9 and transforming growth factor B was observed (Lauer et al., 2013). Similarly, in another study of cardiac hypertrophy induced by high salt intake, C21 was able to replicate previous results (Dopona et al., 2019). In rats with Ang II-dependent hypertension, a sustained reduction in myocardial perivascular fibrosis was observed after 1 week of intraperitoneal administration of C21 (Castoldi et al., 2016).
In in vivo and in vitro experiments carried out in ydiabetic rats and mesangial cells, Koulis et al. (2015), demonstrated that the activation of AT2R by C21 reduced the gene and protein expression of TGF-β1, CTGF, smooth muscle alpha actin and MMP-2 at the renal level. Furthermore, the effect of C21 on the extracellular matrix induced a reduction in type I and IV collagen (predominant collagen isotypes in renal fibrosis) (Koulis et al., 2015). In rats with cyclosporine-induced nephropathy, C21 reduced these same fibrotic markers, in addition to reducing glomerular and tubulo-interstitial fibrosis and macrophage infiltrate (Castoldi et al., 2016).
The presence of fibrosis in the lung can lead to decreased lung capacity, respiratory failure, and other complications such as heart problems. C21 has also been studied to treat fibrotic lung pathologies, with promising results. In a model of pulmonaryb hypertension in rats, induced by monocrotalin, the intraperitoneal administration of C21 decreased the presence of interstitial and perivascular collagen I, in addition to reducing the gene expression of profibrotic cytokines (TNF-α and IL-1β) (Bruce et al., 2015). Similarly, in a bleomycin-induced lung injury model in rats, administration of C21 at the same concentration reduced lung collagen accumulation, attenuated gene expression of fibrotic markers (Col 1, Col 3, Ctgf, Mmp12, Timp1, IL-13) (Rathinasabapathy et al., 2018). Recently, in a model of pulmonary hypertension induced by hypoxia in rats, C21 was administered intraperitoneally at two different doses. It was shown that this compound was able to reduce the presence of collagen fibers significantly at the dose of 20 mg/kg but not with the dose of 2 mg/kg (Tornling et al., 2023).
The compound AVE 0991 (AVE) was first described in 2002 by Wiemer et al. (2002). In this study they demonstrated that AVE 0991 mimicked the effects of Ang-(1–7) on endothelium, reducing the) release of nitric oxide, maintaining endothelial function, and reducing vascular injury (Wiemer et al., 2002), through the G protein-coupled receptor MAS (Pinheiro et al., 2004).
In heart diseases, AVE has been shown to induce beneficial effects for the resolution of pathologies. Zhou et al. (2019), in a murine model of transverse aortic constriction, tested the effects of AVE in combination with intraperitoneal captopril. Coadministration of these compounds completely prevented macrophage infiltration in the aortic adventitia. However, there was an accumulation of cells still present (Zhou et al., 2019). In another study of Ang II-induced hypertension in rats, AVE treatment in combination with alamandine reduced inflammatory stress related to the increase in MCP-1 (Tanrıverdi et al., 2023).
In an atherosclerosis model in knockout mice, AVE administered as treatment for 4 months, reduced the infiltration of proinflammatory cells MCP-1, IL-6, IL-12 and SAA (Jawien et al., 2012). Skiba et al. (2017), demonstrator that in this model, AVE 0991, inhibited inflammation in perivascular adipose tissue by reducing the expression of cytokines IL-1β, TNF-α, MCP-1 and CXCL10, and the differentiation of M2 macrophages to the M1 phenotype (Skiba et al., 2017).
This non-peptide agonist has also an anti-inflammatory effect in pulmonary pathologies. Rodrigues-Machado et al. (2013), evaluated the effects of AVE in a model of chronic asthma induced with ovalbumin in BALB/c mice. After administration of AVE for 28 days, this treatment was able to prevent the development of pulmonary and airway vascular remodeling, in addition to reducing the inflammatory response with a marked reduction in cytokine release (IL-5) in bronchoalveolar fluid and lung homogenates (Rodrigues-Machado et al., 2013). Subsequently, in a similar model it was observed that the anti-inflammatory effect (reduction of macrophages, MCP-1, MAPK) occurred through the MAS receptor expressed in the bronchial epithelium. Furthermore, these actions were attributed to the inhibition of the JNK signaling pathway (Hong et al., 2021).
In a fibrotic context, although little is known to date, AVE has had favorable implications in the resolution of different pathologies that develop this alteration. In a model of cardiac dysfunction in rats, induced by isoproterenol, the intraperitoneal administration of AVE attenuated the deposition of collagen in the left ventricle, in addition to reducing the presence of collagen type I, collagen III and fibronectin in the heart (Ferreira P. M. et al., 2007). In rats with myocardial infarction, AVE also attenuated the expression of collagen I and III, in addition to inhibiting the expression of TGF-β and TNF-α (Zeng et al., 2012). In a model of renal hypertension in rats, intragastric administration of AVE for 28 days was shown to be capable of reducing collagen deposition in renal tissue (Cunha et al., 2013; Cao et al., 2019). Cao et al. (2019), in a model of pulmonary fibrosis caused by LPS-induced ARDS, evaluated the effects of AVE, where they demonstrated that the application of this compound as an intraperitoneal treatment for 20 days was capable of attenuating lung injury by reducing the presence of collagen I fibers, the levels of TGF-β in bronchoalveolar fluid and plasma, the protein expression of E-cadherin and vimectin and the phosphorylation of Src kinase (Cao et al., 2019).
Peptides of the counterregulatory axis of the renin-angiotensin system are likely to play an important role in the control of inflammatory and fibrotic diseases, as supported by many in vitro and in vivo studies. Still, to date, existing studies remain limited in the clinical context.
As we have seen in this review, Ang-(1–7) is one of the most studied peptides, so there are several reports that evaluate its actions as clinical treatment. Although several of these studies are not directly in inflammatory or fibrotic diseases, it is relevant to mention them.
Ueda et al. (2000), evaluated the vascular effects of Ang-(1–7) in human forearm resistant vessels, where intra-arterial infusion of this peptide reduced the increase in blood flow caused by Ang II in normotensive patients (Ueda et al., 2000). Furthermore, in normotensive subjects, Ang-(1–7) was able to enhance the vasodilatory effects of bradykinin, reducing forearm blood flow by 10 percent (Ueda et al., 2001). Later, these same actions were evaluated in normotensive and hypertensive subjects, observing an increase in vasodilation in both conditions (Sasaki et al., 2001). More recently, in healthy normotensive postpartum women with preeclampsia, Ang-(1–7) enhanced the endothelium-dependent vasodilatory response, attenuating Ang II-mediated constriction (Stanhewicz and Alexander, 2020). The main beneficial effects described on these peptides are as vasodilators and anti-hypertensives. Hence, the improvement in blood flow could have anti-inflammatory effects by reducing the accumulation of inflammatory cells and increasing the arrival of anti-inflammatory factors, which could promote the resolution of this response. On the other hand, the reduction of inflammation could regulate the elevation of pro-fibrotic factors, which stimulate the excessive synthesis of extracellular matrix components and the proliferation of fibroblasts, reducing the progression of fibrosis.
To our knowledge, there are very few clinical trials that have evaluated the implications of Ang-(1–7) in an inflammatory context. In confirmed COVID-19 patients, the effects of plasma with Ang-(1–7) were evaluated, observing an improvement in the clinical status of the patient (Onal et al., 2022). Importantly, two independent pilot clinical trials aimed to determine the safety of human use of TXA-127 [a pharmaceutically formulated Ang-(1–7)] (Wagener et al., 2022), or Ang-(1–7) (Valle Martins et al., 2022) delivered these peptides intravenously to COVID-19 patients, showing a safe use of both molecules, and suggesting the potential clinical use as treatment for severe COVID-19.
Other peptides of the counterregulatory axis have not been evaluated in clinical studies, however the knowledge generated so far in cells and animals could lay the foundations for future research in humans focused on the development of anti-inflammatory and antifibrotic treatments.
In conclusion, the counterregulatory axis of the renin-angiotensin system has become an important object of study for more than 2 decades. The components of this axis offer a variety of effects such as antihypertensive, anti-inflammatory and antifibrotic. In the field of research, the evaluation of these components has progressed from structural observation through histological techniques to the evaluation of gene and protein expression. Several studies have described some mechanisms of action by which the components of the counterregulatory axis act in the regulation of inflammation and fibrosis. These mechanisms have focused on the evaluation of signaling pathways such as NF-κB, JAK/STAT and JNK.
Therefore, the peptides of the counterregulatory axis of the renin-angiotensin system, as well as their non-peptide analogues, offer new options for the therapy of pathologies that have inflammatory and fibrotic stages. However, several challenges remain, including:
1. There is literature that describe controversial evidence on some of the peptides regarding their anti-inflammatory effects.
2. The understanding of the mechanisms of action is still very limited, with little literature that supports the signaling pathways involved in its effects.
3. Most of the effects described so far are evaluated in in vivo or in vitro models and in numerous cases still require clinical research to demonstrate the reproducibility of their results in humans.
In future research, it is expected that by improving the understanding of the action of this axis and its relationship with the progression of diseases, these peptides can be applied clinically as treatments that help reduce the alarming response of diseases to less serious stages.
DM-A: Writing–original draft, Writing–review and editing. WM-R: Writing–original draft, Writing–review and editing. JH-C: Writing–review and editing. OL-F: Conceptualization, Funding acquisition, Writing–original draft, Writing–review and editing. MF-M: Conceptualization, Funding acquisition, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Work in the authors’ laboratory is supported by CONAHCYT-Mexico (programme grant 321869) and COVEICYDET-Mexico (programme grants: 141316, 151618), and a CONAHCYT Doctoral Training Grant PhD Studentship for DM-A, WM-R, and JG-L.
We thank Dr. Carolyn Clarke for English proofreading. We would like to acknowledge that the Figures were created with BioRender.com (agreement number: HZ26DI3FS4, YP26DI0DYW, IS26DHXW9N).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ainscough, J. F. X., Drinkhill, M. J., Sedo, A., Turner, N. A., Brooke, D. A., Balmforth, A. J., et al. (2009). Angiotensin II type-1 receptor activation in the adult heart causes blood pressure-independent hypertrophy and cardiac dysfunction. Cardiovasc. Res. 81 (3), 592–600. doi:10.1093/cvr/cvn230
Alanazi, A. Z., and Clark, M. A. (2020). Effects of angiotensin III on c-Jun N terminal kinase in Wistar and hypertensive rat vascular smooth muscle cells. Peptides 123, 170204. doi:10.1016/j.peptides.2019.170204
Antonucci, E., Gleeson, P. J., Annoni, F., Agosta, S., Orlando, S., Taccone, F. S., et al. (2017). Angiotensin II in refractory septic shock. Shock 47, 560–566. doi:10.1097/SHK.0000000000000807
Axelband, F., Dias, J., Miranda, F., Ferrão, F. M., Barros, N. M., Carmona, A. K., et al. (2009). A scrutiny of the biochemical pathways from Ang II to Ang-(3-4) in renal basolateral membranes. Regul. Pept. 158 (1–3), 47–56. doi:10.1016/j.regpep.2009.08.004
Bai, W. W., Wang, H., Gao, C. H., Liu, K. Y., Guo, B. X., Jiang, F., et al. (2021). Continuous infusion of angiotensin IV protects against acute myocardial infarction via the inhibition of inflammation and autophagy. Oxidative Med. Cell. Longev. 2021, 2860488. doi:10.1155/2021/2860488
Booz, G. W., and Baker, K. M. (1996). Role of type 1 and type 2 angiotensin receptors in angiotensin II–induced cardiomyocyte hypertrophy. Hypertension 28 (4), 635–640. doi:10.1161/01.HYP.28.4.635
Bosnyak, S., Jones, E. S., Christopoulos, A., Aguilar, M. I., Thomas, W. G., and Widdop, R. E. (2011). Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin. Sci. 121 (7), 297–303. doi:10.1042/CS20110036
Bruce, E., Shenoy, V., Rathinasabapathy, A., Espejo, A., Horowitz, A., Oswalt, A., et al. (2015). Selective activation of angiotensin AT2 receptors attenuates progression of pulmonary hypertension and inhibits cardiopulmonary fibrosis. Br. J. Pharmacol. 172 (9), 2219–2231. doi:10.1111/bph.13044
Burrell, L. M., Risvanis, J., Kubota, E., Dean, R. G., MacDonald, P. S., Lu, S., et al. (2005). Myocardial infarction increases ACE2 expression in rat and humans. Eur. Heart J. 26 (4), 369–375. doi:10.1093/eurheartj/ehi114
Cao, Y., Liu, Y., Shang, J., Yuan, Z., Ping, F., Yao, S., et al. (2019). Ang-(1-7) treatment attenuates lipopolysaccharide-induced early pulmonary fibrosis. Lab. Investig. 99 (12), 1770–1783. doi:10.1038/s41374-019-0289-7
Carpenter, R. M., Young, M. K., Petri, W. A. O., Lyons, G. R., Gilchrist, C., Carey, R. M., et al. (2022). Repressed ang 1–7 in COVID-19 is inversely associated with inflammation and coagulation. mSphere 7 (4), e0022022. doi:10.1128/msphere.00220-22
Castoldi, G., di Gioia, C. R. T., Carletti, R., Roma, F., Zerbini, G., and Stella, A. (2016). Angiotensin type-2 (AT-2)-receptor activation reduces renal fibrosis in cyclosporine nephropathy: evidence for blood pressure independent effect. Biosci. Rep. 36 (6), e00403. doi:10.1042/BSR20160278
Cha, S. A., Park, B. M., and Kim, S. H. (2018). Angiotensin-(1-9) ameliorates pulmonary arterial hypertension via angiotensin type II receptor. Korean J. Physiology Pharmacol. 22 (4), 447–456. doi:10.4196/kjpp.2018.22.4.447
Chappell, M. C., Pirro, N. T., Sykes, A., and Ferrario, C. M. (1998). Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension 31, 362–367. doi:10.1161/01.hyp.31.1.362
Chawla, T., Sharma, D., and Singh, A. (2010). Role of the renin angiotensin system in diabetic nephropathy. World J. Diabetes 1 (5), 141–145. doi:10.4239/WJD.V1.I5.141
Chen, K., Mehta, J. L., Joseph, L., and Joseph, J. (2004). Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circulation Res. 95 (12), 1167–1173. doi:10.1161/01.RES.0000150369.68826.2f
Chen, L., Gong, P., Su, Y., Meng, L., Wang, M., Gao, W., et al. (2024). Angiotensin II type 2 receptor agonist attenuates LPS-induced acute lung injury through modulating THP-1-derived macrophage reprogramming. Naunyn-Schmiedeberg’s Archives Pharmacol. 397 (1), 99–108. doi:10.1007/s00210-023-02589-0
Chen, Q., Yang, Y., Huang, Y., Pan, C., Liu, L., and Qiu, H. (2013). Angiotensin-(1-7) attenuates lung fibrosis by way of Mas receptor in acute lung injury. J. Surg. Res. 185 (2), 740–747. doi:10.1016/j.jss.2013.06.052
Chen, S., Saeed, A. F. U. H., Liu, Q., Jiang, Q., Xu, H., Xiao, G. G., et al. (2023a). Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 8, 207. doi:10.1038/s41392-023-01452-1
Chen, X. S., Cui, J. R., Meng, X. L., Wang, S. H., Wei, W., Gao, Y. L., et al. (2023b). Angiotensin-(1–7) ameliorates sepsis-induced cardiomyopathy by alleviating inflammatory response and mitochondrial damage through the NF-κB and MAPK pathways. J. Transl. Med. 21 (1), 2. doi:10.1186/s12967-022-03842-5
Ciechanowicz, A. K., Lay, W. X., Prado Paulino, J., Suchocki, E., Leszczak, S., Leszczak, C., et al. (2022). Angiotensin 1–7 stimulates proliferation of lung bronchoalveolar progenitors—implications for SARS-CoV2 infection. Cells 11 (13), 2102. doi:10.3390/cells11132102
Crisóstomo, T., Pardal, M. A. E., Herdy, S. A., Muzi-Filho, H., Mello, D. B., Takiya, C. M., et al. (2022). Liver steatosis, cardiac and renal fibrosis, and hypertension in overweight rats: angiotensin-(3–4)-sensitive hepatocardiorenal syndrome. Metab. Open 14, 100176. doi:10.1016/j.metop.2022.100176
Cunha, T. M. B., Lima, W. G., Silva, M. E., Souza Santos, R. A., Campagnole-Santos, M. J., and Alzamora, A. C. (2013). The nonpeptide ANG-(1-7) mimic AVE 0991 attenuates cardiac remodeling and improves baroreflex sensitivity in renovascular hypertensive rats. Life Sci. 92 (4–5), 266–275. doi:10.1016/j.lfs.2012.12.008
Da Silva, A. R., Lenglet, S., Carbone, F., Burger, F., Roth, A., Liberale, L., et al. (2017). Alamandine abrogates neutrophil degranulation in atherosclerotic mice. Eur. J. Clin. Investigation 47 (2), 117–128. doi:10.1111/eci.12708
De Carvalho Santuchi, M., Dutra, M. F., Vago, J. P., Lima, K. M., Galvão, I., de Souza-Neto, F. P., et al. (2019). Angiotensin-(1-7) and alamandine promote anti-inflammatory response in macrophages in vitro and in vivo. Mediat. Inflamm. 2019, 2401081. doi:10.1155/2019/2401081
De Souza-Neto, F. P., Silva, M. d. M. E., Santuchi, M. d. C., de Alcântara-Leonídio, T. C., Motta-Santos, D., Oliveira, A. C., et al. (2019). Alamandine attenuates arterial remodelling induced by transverse aortic constriction in mice. Clin. Sci. 133 (5), 629–643. doi:10.1042/CS20180547
Dias, J., Axelband, F., Lara, L. S., Muzi-Filho, H., and Vieyra, A. (2017). Is angiotensin-(3-4) (Val-Tyr), the shortest angiotensin II-derived peptide, opening new vistas on the renin-angiotensin system? JRAAS 18, 1470320316689338. doi:10.1177/1470320316689338
Ding, W., Miao, Z., Feng, X., Luo, A., Tan, W., Li, P., et al. (2022). Alamandine, a new member of the renin-angiotensin system (RAS), attenuates collagen-induced arthritis in mice via inhibiting cytokine secretion in synovial fibroblasts. Peptides 154, 170816. doi:10.1016/j.peptides.2022.170816
Domińska, K., Kowalska, K., Urbanek, K. A., Habrowska-Górczyńska, D. E., Ochędalski, T., and Piastowska Ciesielska, A. W. (2020). The impact of ang-(1-9) and ang-(3-7) on the biological properties of prostate cancer cells by modulation of inflammatory and steroidogenesis pathway genes. Int. J. Mol. Sci. 21 (17), 1–18. doi:10.3390/ijms21176227
Donoghue, M., Hsieh, F., Baronas, E., Godbout, K., Gosselin, M., Stagliano, N., et al. (2000). A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circulation Res. 87 (5), E1–E9. doi:10.1161/01.RES.87.5.e1
Dopona, E. P. B., Rocha, V. F., Furukawa, L. N. S., Oliveira, I. B., and Heimann, J. C. (2019). Myocardial hypertrophy induced by high salt consumption is prevented by angiotensin II AT2 receptor agonist. Nutr. Metabolism Cardiovasc. Dis. 29 (3), 301–305. doi:10.1016/j.numecd.2018.11.001
Du, X., Gustin, D. J., Chen, X., Duquette, J., McGee, L. R., Wang, Z., et al. (2009). Imidazo-pyrazine derivatives as potent CXCR3 antagonists. Bioorg. Med. Chem. Lett. 19 (17), 5200–5204. doi:10.1016/j.bmcl.2009.07.021
El-Hashim, A. Z., Khajah, M. A., Babyson, R. S., Renno, W. M., Ezeamuzie, C. I., Benter, I. F., et al. (2019). Ang-(1-7)/MAS1 receptor axis inhibits allergic airway inflammation via blockade of Src-mediated EGFR transactivation in a murine model of asthma. PLoS ONE 14 (11), e0224163. doi:10.1371/journal.pone.0224163
Erusalimsky, J. D. (2009). Vascular endothelial senescence: from mechanisms to pathophysiology. J. Appl. Physiology 106, 326–332. doi:10.1152/japplphysiol.91353.2008
Esteban, V., Heringer-Walther, S., Sterner-Kock, A., de Bruin, R., van den Engel, S., Wang, Y., et al. (2009). Angiotensin-(1-7) and the G protein-coupled receptor Mas are key players in renal inflammation. PLoS ONE 4 (4), e5406. doi:10.1371/journal.pone.0005406
Esteban, V., Ruperez, M., Sánchez-López, E., Rodríguez-Vita, J., Lorenzo, O., Demaegdt, H., et al. (2005). Angiotensin IV activates the nuclear transcription factor-kappaB and related proinflammatory genes in vascular smooth muscle cells. Circulation Res. 96 (9), 965–973. doi:10.1161/01.RES.0000166326.91395.74
Fattah, C., Nather, K., McCarroll, C. S., Hortigon-Vinagre, M. P., Zamora, V., Flores-Munoz, M., et al. (2016). Gene therapy with angiotensin-(1-9) preserves left ventricular systolic function after myocardial infarction. J. Am. Coll. Cardiol. 68 (24), 2652–2666. doi:10.1016/j.jacc.2016.09.946
Ferreira, A. J., Oliveira, T. L., Castro, M. C. M., Almeida, A. P., Castro, C. H., Caliari, M. V., et al. (2007a). Isoproterenol-induced impairment of heart function and remodeling are attenuated by the nonpeptide angiotensin-(1-7) analogue AVE 0991. Life Sci. 81 (11), 916–923. doi:10.1016/j.lfs.2007.07.022
Ferreira, P. M., Souza dos Santos, R. A., and Campagnole-Santos, M. J. (2007b). Angiotensin-(3-7) pressor effect at the rostral ventrolateral medulla. Regul. Pept. 141 (1–3), 168–174. doi:10.1016/j.regpep.2006.12.031
Flores-Muñoz, M., Smith, N. J., Haggerty, C., Milligan, G., and Nicklin, S. A. (2011). Angiotensin1-9 antagonises pro-hypertrophic signalling in cardiomyocytes via the angiotensin type 2 receptor. J. Physiology 589 (4), 939–951. doi:10.1113/jphysiol.2010.203075
Flores-Munoz, M., Work, L. M., Douglas, K., Denby, L., Dominiczak, A. F., Graham, D., et al. (2012). Angiotensin-(1-9) attenuates cardiac fibrosis in the stroke-prone spontaneously hypertensive rat via the angiotensin type 2 receptor. Hypertension 59 (2), 300–307. doi:10.1161/HYPERTENSIONAHA.111.177485
Frangogiannis, N. (2020). Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 217 (3), e20190103. doi:10.1084/jem.20190103
Garabelli, P. J., Modrall, J. G., Penninger, J. M., Ferrario, C. M., and Chappell, M. C. (2008). Distinct roles for angiotensin-converting enzyme 2 and carboxypeptidase A in the processing of angiotensins within the murine heart. Exp. Physiol. 93 (5), 613–621. doi:10.1113/expphysiol.2007.040246
Garcia, B., Su, F., Manicone, F., Dewachter, L., Favory, R., Khaldi, A., et al. (2023). Angiotensin 1–7 in an experimental septic shock model. Crit. Care 27 (1), 106. doi:10.1186/s13054-023-04396-8
Gesualdo, L., Ranieri, E., Monno, R., Rossiello, M. R., Colucci, M., Semeraro, N., et al. (1999). Angiotensin IV stimulates plasminogen activator inhibitor-1 expression in proximal tubular epithelial cells. Kidney Int. 56 (2), 461–470. doi:10.1046/j.1523-1755.1999.00578.x
Gomez, R. A., Norling, L. L., Wilfong, N., Isakson, P., Lynch, K. R., Hock, R., et al. (1993). Leukocytes synthesize angiotensinogen. Hypertension 21, 470–475. doi:10.1161/01.hyp.21.4.470
Gonçalves, S. C. A., Bassi, B. L. T., Kangussu, L. M., Alves, D. T., Ramos, L. K. S., Fernandes, L. F., et al. (2022). Alamandine induces neuroprotection in ischemic stroke models. Curr. Med. Chem. 29 (19), 3483–3498. doi:10.2174/0929867329666220204145730
Gong, J., Luo, M., Yong, Y., Zhong, S., and Li, P. (2022). Alamandine alleviates hypertension and renal damage via oxidative-stress attenuation in Dahl rats. Cell death Discov. 8 (1), 22. doi:10.1038/s41420-022-00822-y
Gonzalez, L., Novoa, U., Moya, J., Gabrielli, L., Jalil, J. E., García, L., et al. (2018). Angiotensin-(1-9) reduces cardiovascular and renal inflammation in experimental renin-independent hypertension. Biochem. Pharmacol. 156, 357–370. doi:10.1016/j.bcp.2018.08.045
Goossens, G. H., Jocken, J. W. E., Blaak, E. E., Schiffers, P. M., Saris, W. H. M., and van Baak, M. A. (2007). Endocrine role of the renin-angiotensin system in human adipose tissue and muscle: effect of beta-adrenergic stimulation. Hypertension 49 (3), 542–547. doi:10.1161/01.HYP.0000256091.55393.92
Gray, M. O., Long, C. S., Kalinyak, J. E., and Karliner, J. S. (1998). Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc. Res. 40 (2), 352–363. doi:10.1016/s0008-6363(98)00121-7
Grobe, J. L., Mecca, A. P., Lingis, M., Shenoy, V., Bolton, T. A., Machado, J. M., et al. (2007). Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1–7). Am. J. Physiology-Heart Circulatory Physiology 292 (2), H736–H742. doi:10.1152/ajpheart.00937.2006
Grobe, J. L., Mecca, A. P., Mao, H., and Katovich, M. J. (2006). Chronic angiotensin-(1–7) prevents cardiac fibrosis in DOCA-salt model of hypertension. Am. J. Physiology-Heart Circulatory Physiology 290 (6), H2417–H2423. doi:10.1152/ajpheart.01170.2005
Hekmat, A. S., Navabi, Z., Alipanah, H., and Javanmardi, K. (2021). Alamandine significantly reduces doxorubicin-induced cardiotoxicity in rats. Hum. Exp. Toxicol. 40 (10), 1781–1795. doi:10.1177/09603271211010896
Hikmet, F., Méar, L., Edvinsson, Å., Micke, P., Uhlén, M., and Lindskog, C. (2020). The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 16 (7), e9610. doi:10.15252/msb.20209610
Hirsch, A. T., Talsness, C. E., Schunkert, H., Paul, M., and Dzau, V. J. (1991). Tissue-specific activation of cardiac angiotensin converting enzyme in experimental heart failure. Circulation Res. 69 (2), 475–482. doi:10.1161/01.RES.69.2.475
Hong, L., Wang, Q., Chen, M., Shi, J., Guo, Y., Liu, S., et al. (2021). Mas receptor activation attenuates allergic airway inflammation via inhibiting JNK/CCL2-induced macrophage recruitment. Biomed. Pharmacother. 137, 111365. doi:10.1016/j.biopha.2021.111365
Hooper, N. M., and Turner, A. J. (2003). An ACE structure. Nat. Struct. Biol. 10 (3), 155–157. doi:10.1038/nsb0303-155
Hough, K. P., Curtiss, M. L., Blain, T. J., Liu, R. M., Trevor, J., Deshane, J. S., et al. (2020). Airway remodeling in asthma. Front. Med. 7, 191. doi:10.3389/fmed.2020.00191
Hu, W., Gao, W., Miao, J., Xu, Z., and Sun, L. (2021). Alamandine, a derivative of angiotensin-(1-7), alleviates sepsis-associated renal inflammation and apoptosis by inhibiting the PI3K/Ak and MAPK pathways. Peptides 146, 170627. doi:10.1016/j.peptides.2021.170627
Huang, Y., Li, Y., Lou, A., Wang, G. Z., Hu, Y., Zhang, Y., et al. (2020). Alamandine attenuates hepatic fibrosis by regulating autophagy induced by NOX4-dependent ROS. Clin. Sci. 134 (7), 853–869. doi:10.1042/CS20191235
Iwata, M., Cowling, R. T., Gurantz, D., Moore, C., Zhang, S., Yuan, J. X. J., et al. (2005). Angiotensin-(1–7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am. J. Physiology-Heart Circulatory Physiology 289 (6), H2356–H2363. doi:10.1152/ajpheart.00317.2005
Jabber, H., Mohammed, B., and Hadi, N. R. (2023). Investigating the renoprotective effect of C21 in male mice with sepsis via modulation of p-AKT/PI3K expression. J. Med. Life 16 (2), 203–209. doi:10.25122/jml-2022-0299
Jackman, H. L., Massad, M. G., Sekosan, M., Tan, F., Brovkovych, V., Marcic, B. M., et al. (2002). Angiotensin 1-9 and 1-7 release in human heart: role of cathepsin A. Hypertension 39 (5), 976–981. doi:10.1161/01.HYP.0000017283.67962.02
Jawien, J., Toton-Zuranska, J., Kus, K., Pawlowska, M., Olszanecki, R., and Korbut, R. (2012). The effect of AVE 0991, nebivolol and doxycycline on inflammatory mediators in an apoE-knockout mouse model of atherosclerosis. Med. Sci. Monit. 18 (10), BR389–BR393. doi:10.12659/MSM.883478
Jiang, F., Yang, J., Zhang, Y., Dong, M., Wang, S., Zhang, Q., et al. (2014). Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nat. Rev. Cardiol. 11, 413–426. doi:10.1038/nrcardio.2014.59
Kalupahana, N. S., Massiera, F., Quignard-Boulange, A., Ailhaud, G., Voy, B. H., Wasserman, D. H., et al. (2012). Overproduction of angiotensinogen from adipose tissue induces adipose inflammation, glucose intolerance, and insulin resistance. Obesity 20 (1), 48–56. doi:10.1038/oby.2011.299
Kaschina, E., Grzesiak, A., Li, J., Foryst-Ludwig, A., Timm, M., Rompe, F., et al. (2008). Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 118 (24), 2523–2532. doi:10.1161/CIRCULATIONAHA.108.784868
Kemp, B. A., Howell, N. L., Keller, S. R., Gildea, J. J., Shao, W., Navar, L. G., et al. (2019). Defective renal angiotensin III and AT2 receptor signaling in prehypertensive spontaneously hypertensive rats. J. Am. Heart Assoc. 8 (9), e012016. doi:10.1161/JAHA.119.012016
Klimov, L. O., Fedoseeva, L. A., Ryazanova, M. A., Dymshits, G. M., and Markel, A. L. (2012). Expression of renin–angiotensin system genes in brain structures of ISIAH rats with stress-induced arterial hypertension. Bull. Exp. Biol. Med. 154, 357–360. doi:10.1007/s10517-013-1950-6
Kobori, H., Nangaku, M., Navar, L. G., and Nishiyama, A. (2007). The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol. Rev. 59, 251–287. doi:10.1124/pr.59.3.3
Kokkonen, J. O., Saarinen, J., and Kovanen, P. T. (1997). Regulation of local angiotensin II formation in the human heart in the presence of interstitial fluid. Inhibition of chymase by protease inhibitors of interstitial fluid and of angiotensin-converting enzyme by Ang-(1-9) formed by heart carboxypeptidase A-like activity. Circulation 95 (6), 1455–1463. doi:10.1161/01.CIR.95.6.1455
Kong, J., Zhang, K., Meng, X., Zhang, Y., and Zhang, C. (2015). Dose-dependent bidirectional effect of angiotensin IV on abdominal aortic aneurysm via variable angiotensin receptor stimulation. Hypertension 66 (3), 617–626. doi:10.1161/HYPERTENSIONAHA.115.05482
Koulis, C., Chow, B. S. M., McKelvey, M., Steckelings, U. M., Unger, T., Thallas-Bonke, V., et al. (2015). AT2R agonist, compound 21, is Reno-protective against type 1 diabetic nephropathy. Hypertension 65 (5), 1073–1081. doi:10.1161/HYPERTENSIONAHA.115.05204
Kuba, K., Imai, Y., and Penninger, J. M. (2006). Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 6 (3), 271–276. doi:10.1016/J.COPH.2006.03.001
Lauer, D., Slavic, S., Sommerfeld, M., Thöne-Reineke, C., Sharkovska, Y., Hallberg, A., et al. (2013). Angiotensin type 2 receptor stimulation ameliorates left ventricular fibrosis and dysfunction via regulation of tissue inhibitor of matrix metalloproteinase 1/matrix metalloproteinase 9 Axis and transforming growth factor β1 in the rat heart. Hypertension 63, e60–e67. doi:10.1161/HYPERTENSIONAHA.113.02522
Lautner, R. Q., Villela, D. C., Fraga-Silva, R. A., Silva, N., Verano-Braga, T., Costa-Fraga, F., et al. (2013). Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circulation Res. 112 (8), 1104–1111. doi:10.1161/CIRCRESAHA.113.301077
Levy, B. D., Clish, C. B., Schmidt, B., Gronert, K., and Serhan, C. N. (2001). Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2, 612–619. doi:10.1038/89759
Li, P., Chen, X. R., Xu, F., Liu, C., Li, C., Liu, H., et al. (2018). Alamandine attenuates sepsis-associated cardiac dysfunction via inhibiting MAPKs signaling pathways. Life Sci. 206, 106–116. doi:10.1016/j.lfs.2018.04.010
Li, P., Guo, X., Lei, P., Shi, S., Luo, S., and Cheng, X. (2014). PI3K/Akt/uncoupling protein 2 signaling pathway may be involved in cell senescence and apoptosis induced by angiotensin II in human vascular endothelial cells. Mol. Biol. Rep. 41 (10), 6931–6937. doi:10.1007/s11033-014-3580-0
Li, X., Zhang, W., Cao, Q., Wang, Z., Zhao, M., Xu, L., et al. (2020). Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 6, 80. doi:10.1038/s41420-020-00316-9
Li, Y., Wu, J., He, Q., Shou, Z., Zhang, P., Pen, W., et al. (2009). Angiotensin (1−7) prevent heart dysfunction and left ventricular remodeling caused by renal dysfunction in 5/6 nephrectomy mice. Hypertens. Res. 32 (5), 369–374. doi:10.1038/hr.2009.25
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017). NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2, 17023. doi:10.1038/sigtrans.2017.23
Lorenzo, Ó., Ruiz-Ortega, M., Suzuki, Y., Rupérez, M., Esteban, V., Sugaya, T., et al. (2002). Angiotensin III activates nuclear transcription factor-kappaB in cultured mesangial cells mainly via AT(2) receptors: studies with AT(1) receptor-knockout mice. J. Am. Soc. Nephrol. 13 (5), 1162–1171. doi:10.1681/ASN.V1351162
Lu, H., Cassis, L. A., Kooi, C. W. V., and Daugherty, A. (2016). Structure and functions of angiotensinogen. Hypertens. Res. 39, 492–500. doi:10.1038/hr.2016.17
Lu, W., Kang, J., Hu, K., Tang, S., Zhou, X., Yu, S., et al. (2017). Angiotensin-(1-7) relieved renal injury induced by chronic intermittent hypoxia in rats by reducing inflammation, oxidative stress and fibrosis. Braz. J. Med. Biol. Res. 50 (1), e5594. doi:10.1590/1414-431X20165594
Luther, J. M., Gainer, J. V., Murphey, L. J., Yu, C., Vaughan, D. E., Morrow, J. D., et al. (2006). Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor–dependent mechanism. Hypertension 48 (6), 1050–1057. doi:10.1161/01.HYP.0000248135.97380.76
Luzes, R., Crisóstomo, T., Silva, P. A., Iack, R., de Abreu, V. G., Francischetti, E. A., et al. (2021). Angiotensin-(3–4) normalizes blood pressure, decreases Na+ and energy intake, but preserves urinary Na+ excretion in overweight hypertensive rats. Biochimica Biophysica Acta - Mol. Basis Dis. 1867 (3), 166012. doi:10.1016/j.bbadis.2020.166012
Luzes, R., Muzi-Filho, H., Pereira-Acácio, A., Crisóstomo, T., and Vieyra, A. (2021). Angiotensin-(3-4) modulates the overweight- and undernutrition-induced ACE2 downregulation in renal proximal tubule cells: implications for COVID-19? Explor. Med. 2 (2), 135–145. doi:10.37349/emed.2021.00038
Magalhães, G. S., Rodrigues-Machado, M. G., Motta-Santos, D., Alenina, N., Bader, M., Santos, R. A., et al. (2016). Chronic allergic pulmonary inflammation is aggravated in angiotensin-(1-7) Mas receptor knockout mice. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L1141–L1148. doi:10.1152/ajplung.00029.2016
Magalhães, G. S., Rodrigues-Machado, M. G., Motta-Santos, D., Silva, A. R., Caliari, M. V., Prata, L. O., et al. (2015). Angiotensin-(1-7) attenuates airway remodelling and hyperresponsiveness in a model of chronic allergic lung inflammation. Br. J. Pharmacol. 172 (9), 2330–2342. doi:10.1111/bph.13057
Matavelli, L. C., Huang, J., and Siragy, H. M. (2011). Angiotensin AT₂ receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension 57 (2), 308–313. doi:10.1161/HYPERTENSIONAHA.110.164202
Matavelli, L. C., Zatz, R., and Siragy, H. M. (2015). A nonpeptide angiotensin II type 2 receptor agonist prevents renal inflammation in early diabetes. J. Cardiovasc. Pharmacol. 65 (4), 371–376. doi:10.1097/FJC.0000000000000207
Matsufuji, H., Matsui, T., Ohshige, S., Kawasaki, T., Osajima, K., and Osajima, Y. (1995). Antihypertensive effects of angiotensin fragments in SHR. Biosci. Biotechnol. Biochem. 59 (8), 1398–1401. doi:10.1271/bbb.59.1398
Matsui, T., Hayashi, A., Tamaya, K., Matsumoto, K., Kawasaki, T., Murakami, K., et al. (2003). Depressor effect induced by dipeptide, val-tyr, in hypertensive transgenic mice is due, in part, to the suppression of human circulating renin-angiotensin system. Clin. Exp. Pharmacol. Physiology 30, 262–265. doi:10.1046/j.1440-1681.2003.03824.x
Matsui, T., Ueno, T., Tanaka, M., Oka, H., Miyamoto, T., Osajima, K., et al. (2005). Antiproliferative action of an angiotensin I-converting enzyme inhibitory peptide, val-tyr, via an L-type Ca 2+ channel inhibition in cultured vascular smooth muscle cells. Hypertens. Res. 28, 545–552. doi:10.1291/hypres.28.545
Mei, D., Liao, W., Gan, P. X. L., Tran, Q. T. N., Chan, C. C. M. Y., Heng, C. K. M., et al. (2022). Angiotensin II type-2 receptor activation in alveolar macrophages mediates protection against cigarette smoke-induced chronic obstructive pulmonary disease. Pharmacol. Res. 184, 106469. doi:10.1016/j.phrs.2022.106469
Meng, Y., Yu, C. H., Li, W., Li, T., Luo, W., Huang, S., et al. (2014). Angiotensin-converting enzyme 2/angiotensin-(1-7)/mas axis protects against lung fibrosis by inhibiting the MAPK/NF-κB pathway. Am. J. Respir. Cell Mol. Biol. 50 (4), 723–736. doi:10.1165/rcmb.2012-0451OC
Menk, M., Graw, J. A., von Haefen, C., Steinkraus, H., Lachmann, B., Spies, C. D., et al. (2018). Angiotensin II type 2 receptor agonist compound 21 attenuates pulmonary inflammation in a model of acute lung injury. J. Inflamm. Res. 11, 169–178. doi:10.2147/JIR.S160573
Mercure, C., Yogi, A., Callera, G. E., Aranha, A. B., Bader, M., Ferreira, A. J., et al. (2008). Angiotensin(1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circulation Res. 103 (11), 1319–1326. doi:10.1161/CIRCRESAHA.108.184911
Millar, E. A., Nally, J. E., and Thomson, N. C. (1995). Angiotensin II potentiates methacholine-induced bronchoconstriction in human airway both in vitro and in vivo. Eur. Respir. J. 8 (11), 1838–1841. doi:10.1183/09031936.95.08111838
Min, L. J., Mogi, M., Iwanami, J., Li, J. M., Sakata, A., Fujita, T., et al. (2007). Cross-talk between aldosterone and angiotensin II in vascular smooth muscle cell senescence. Cardiovasc. Res. 76 (3), 506–516. doi:10.1016/j.cardiores.2007.07.008
Mogi, M. (2020). Effect of renin–angiotensin system on senescence. Geriatrics Gerontology Int. 20, 520–525. doi:10.1111/ggi.13927
Muñoz-Durango, N., Fuentes, C. A., Castillo, A. E., González-Gómez, L. M., Vecchiola, A., Fardella, C. E., et al. (2016). Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int. J. Mol. Sci. 17, 797. doi:10.3390/ijms17070797
Mussbacher, M., Salzmann, M., Brostjan, C., Hoesel, B., Schoergenhofer, C., Datler, H., et al. (2019). Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front. Immunol. 10, 85. doi:10.3389/fimmu.2019.00085
Nathan, C., and Ding, A. (2010). Nonresolving inflammation. Cell 140, 871–882. doi:10.1016/j.cell.2010.02.029
Nikolaou, A., Stijlemans, B., Laoui, D., Schouppe, E., Tran, H. T. T., Tourwé, D., et al. (2014). Presence and regulation of insulin-regulated aminopeptidase in mouse macrophages. JRAAS - J. Renin-Angiotensin-Aldosterone Syst. 15 (4), 466–479. doi:10.1177/1470320313507621
Nourshargh, S., and Alon, R. (2014). Leukocyte migration into inflamed tissues. Immunity 41, 694–707. doi:10.1016/j.immuni.2014.10.008
Ocaranza, M. P., Moya, J., Barrientos, V., Alzamora, R., Hevia, D., Morales, C., et al. (2014). Angiotensin-(1-9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis. J. Hypertens. 32 (4), 771–783. doi:10.1097/HJH.0000000000000094
Onal, H., Ergun, N. U., Arslan, B., Topuz, S., Semerci, S. Y., Ugurel, O. M., et al. (2022). Angiotensin (1−7) peptide replacement therapy with plasma transfusion in COVID-19. Transfus. Apher. Sci. 61 (4), 103418. doi:10.1016/j.transci.2022.103418
Padia, S. H., Kemp, B. A., Howell, N. L., Gildea, J. J., Keller, S. R., and Carey, R. M. (2009). Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension 53 (2), 338–343. doi:10.1161/HYPERTENSIONAHA.108.124198
Pan, C., Wen, C., and Lin, C. (2008). Interplay of angiotensin II and angiotensin(1–7) in the regulation of matrix metalloproteinases of human cardiocytes. Exp. Physiol. 93 (5), 599–612. doi:10.1113/expphysiol.2007.041830
Pan, H., Huang, W., Wang, Z., Ren, F., Luo, L., Zhou, J., et al. (2021). The ACE2-ang-(1‑7)-mas Axis modulates M1/M2 macrophage polarization to relieve CLP-induced inflammation via TLR4-mediated NF-кb and MAPK pathways. J. Inflamm. Res. 14, 2045–2060. doi:10.2147/JIR.S307801
Park, B. M., Cha, S. A., Lee, S. H., and Kim, S. H. (2016). Angiotensin IV protects cardiac reperfusion injury by inhibiting apoptosis and inflammation via AT4R in rats. Peptides 79, 66–74. doi:10.1016/j.peptides.2016.03.017
Park, B. M., Li, W., and Kim, S. H. (2021). Cardio-protective effects of angiotensin-(1-5) via mas receptor in rats against ischemic-perfusion injury. Peptides 139, 170516. doi:10.1016/j.peptides.2021.170516
Park, B. M., Oh, Y. B., Gao, S., Cha, S. A., Kang, K. P., and Kim, S. H. (2013). Angiotensin III stimulates high stretch-induced ANP secretion via angiotensin type 2 receptor. Peptides 42, 131–137. doi:10.1016/j.peptides.2013.01.018
Passaglia, P., Silva, H. B., de Jesus, A. A., Filho, M. A. M., Trajano, I. P., Batalhão, M. E., et al. (2023). Angiotensin-(1−7) improves tail skin heat loss and increases the survival of rats with polymicrobial sepsis. Peptides 167, 171042. doi:10.1016/j.peptides.2023.171042
Paul, M., Wagner, J., and Dzau, V. J. (1993). Gene expression of the renin-angiotensin system in human tissues. Quantitative analysis by the polymerase chain reaction. J. Clin. Investigation 91 (5), 2058–2064. doi:10.1172/JCI116428
Paz Ocaranza, M., Riquelme, J. A., García, L., Jalil, J. E., Chiong, M., Santos, R. A. S., et al. (2020). Counter-regulatory renin–angiotensin system in cardiovascular disease’, Nature Reviews Cardiology. Nat. Res. 17, 116–129. doi:10.1038/s41569-019-0244-8
Pereira-Acácio, A., Veloso-Santos, J. P. M., Nossar, L. F., Costa-Sarmento, G., Muzi-Filho, H., and Vieyra, A. (2022). Angiotensin-(3–4) normalizes the elevated arterial blood pressure and abnormal Na+/energy handling associated with chronic undernutrition by counteracting the effects mediated by type 1 angiotensin II receptors. PLoS ONE 17, e0273385. doi:10.1371/journal.pone.0273385
Pinheiro, S. V. B., Simões e Silva, A. C., Sampaio, W. O., de Paula, R. D., Mendes, E. P., Bontempo, E. D., et al. (2004). Nonpeptide AVE 0991 is an angiotensin-(1-7) receptor mas agonist in the mouse kidney. Hypertension 44 (4), 490–496. doi:10.1161/01.HYP.0000141438.64887.42
Qiu, Z. Y., Yu, W. J., Bai, J., and Lin, Q. Y. (2022). Blocking VCAM-1 ameliorates hypertensive cardiac remodeling by impeding macrophage infiltration. Front. Pharmacol. 13, 1058268. doi:10.3389/fphar.2022.1058268
Rathinasabapathy, A., Horowitz, A., Horton, K., Kumar, A., Gladson, S., Unger, T., et al. (2018). The selective angiotensin II type 2 receptor agonist, compound 21, attenuates the progression of lung fibrosis and pulmonary hypertension in an experimental model of bleomycin-induced lung injury. Front. Physiology 9, 180. doi:10.3389/fphys.2018.00180
Rehman, A., Leibowitz, A., Yamamoto, N., Rautureau, Y., Paradis, P., and Schiffrin, E. L. (2012). Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 59 (2), 291–299. doi:10.1161/HYPERTENSIONAHA.111.180158
Rice, G. I., Thomas, D. A., Grant, P. J., Turner, A. J., and Hooper, N. M. (2004). Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 383, 45–51. doi:10.1042/BJ20040634
Rodrigues-Machado, M. G., Magalhães, G. S., Cardoso, J. A., Kangussu, L. M., Murari, A., Caliari, M. V., et al. (2013). AVE 0991, a non-peptide mimic of angiotensin-(1-7) effects, attenuates pulmonary remodelling in a model of chronic asthma. Br. J. Pharmacol. 170 (4), 835–846. doi:10.1111/bph.12318
Roks, A. J. M., van Geel, P. P., Pinto, Y. M., Buikema, H., Henning, R. H., de Zeeuw, D., et al. (1999). Angiotensin-(1-7) is a modulator of the human renin-angiotensin system. Hypertension 34, 296–301. doi:10.1161/01.hyp.34.2.296
Romero, A., San Hipólito-Luengo, Á., Villalobos, L. A., Vallejo, S., Valencia, I., Michalska, P., et al. (2019). The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell 18 (3), e12913. doi:10.1111/acel.12913
Saito, Y., Wanezaki, K., Kawato, A., and Imayasu, S. (1994). Antihypertensive effects of peptide in sake and its by products on spontaneously hypertensive rats. Biosci. Biotechnol. Biochem. 58 (5), 812–816. doi:10.1271/bbb.58.812
Sampson, A. K., Irvine, J. C., Shihata, W. A., Dragoljevic, D., Lumsden, N., Huet, O., et al. (2016). Compound 21, a selective agonist of angiotensin AT2 receptors, prevents endothelial inflammation and leukocyte adhesion in vitro and in vivo. Br. J. Pharmacol. 173 (4), 729–740. doi:10.1111/bph.13063
Santos, R. A. S., Brosnihan, K. B., Chappell, M. C., Pesquero, J., Chernicky, C. L., Greene, L. J., et al. (1988). Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension 11, I153–I157. doi:10.1161/01.hyp.11.2_pt_2.i153
Santos, R. A. S., Simoes e Silva, A. C., Maric, C., Silva, D. M. R., Machado, R. P., de Buhr, I., et al. (2003). Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. U. S. A. 100, 8258–8263. doi:10.1073/pnas.1432869100
Sasaki, S., Higashi, Y., Nakagawa, K., Matsuura, H., Kajiyama, G., and Oshima, T. (2001). Effects of angiotensin-(1-7) on forearm circulation in normotensive subjects and patients with essential hypertension. Hypertension 38 (1), 90–94. doi:10.1161/01.HYP.38.1.90
Sauter, N. S., Thienel, C., Plutino, Y., Kampe, K., Dror, E., Traub, S., et al. (2015). Angiotensin II induces interleukin-1β-mediated islet inflammation and β-cell dysfunction independently of vasoconstrictive effects. Diabetes 64 (4), 1273–1283. doi:10.2337/DB14-1282
Schimmel, K., Ichimura, K., Reddy, S., Haddad, F., and Spiekerkoetter, E. (2022). Cardiac fibrosis in the pressure overloaded left and right ventricle as a therapeutic target. Front. Cardiovasc. Med. 9, 886553. doi:10.3389/fcvm.2022.886553
Schmid, T., and Brüne, B. (2021). Prostanoids and resolution of inflammation – beyond the lipid-mediator class switch. Front. Immunol. 12, 714042. doi:10.3389/fimmu.2021.714042
Schultz, J. E. J., Witt, S. A., Glascock, B. J., Nieman, M. L., Reiser, P. J., Nix, S. L., et al. (2002). TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Investigation 109 (6), 787–796. doi:10.1172/JCI14190
Serhan, C. N., and Savill, J. (2005). Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197. doi:10.1038/ni1276
Shan, H. Y., Bai, X. J., and Chen, X. M. (2008). Apoptosis is involved in the senescence of endothelial cells induced by angiotensin II. Cell Biol. Int. 32 (2), 264–270. doi:10.1016/j.cellbi.2007.09.003
Shen, H., Zhang, N., Liu, Y., Yang, X., He, Y., Li, Q., et al. (2022). The interaction between pulmonary fibrosis and COVID-19 and the application of related anti-fibrotic drugs. Front. Pharmacol. 12, 805535. doi:10.3389/fphar.2021.805535
Shen, Y. L., Hsieh, Y. A., Hu, P. W., Lo, P. C., Hsiao, Y. H., Ko, H. K., et al. (2023). Angiotensin-(1–7) attenuates SARS-CoV2 spike protein-induced interleukin-6 and interleukin-8 production in alveolar epithelial cells through activation of Mas receptor. J. Microbiol. Immunol. Infect. 56, 1147–1157. doi:10.1016/j.jmii.2023.09.003
Skiba, D. S., Nosalski, R., Mikolajczyk, T. P., Siedlinski, M., Rios, F. J., Montezano, A. C., et al. (2017). Anti-atherosclerotic effect of the angiotensin 1–7 mimetic AVE0991 is mediated by inhibition of perivascular and plaque inflammation in early atherosclerosis. Br. J. Pharmacol. 174 (22), 4055–4069. doi:10.1111/bph.13685
Song, X.-D., Feng, J.-P., and Yang, R.-X. (2019). Alamandine protects rat from myocardial ischemia-reperfusion injury by activating JNK and inhibiting NF-κB. Eur. Rev. Med. Pharmacol. Sci. 23 (15), 6718–6726. doi:10.26355/eurrev_201908_18563
Stanhewicz, A. E., and Alexander, L. M. (2020). Local angiotensin-(1–7) administration improves microvascular endothelial function in women who have had preeclampsia. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 318 (1), R148–R155. doi:10.1152/ajpregu.00221.2019
Sun, Y., Zhang, J., Zhang, J. Q., and Ramires, F. J. (2000). Local angiotensin II and transforming growth factor-beta1 in renal fibrosis of rats. Hypertension 35 (5), 1078–1084. doi:10.1161/01.HYP.35.5.1078
Supé, S., Kohse, F., Gembardt, F., Kuebler, W. M., and Walther, T. (2016). Therapeutic time window for angiotensin-(1-7) in acute lung injury. Br. J. Pharmacol. 173 (10), 1618–1628. doi:10.1111/bph.13462
Takeshita, H., Yamamoto, K., Mogi, M., Nozato, S., and Rakugi, H. (2023). Is the anti-aging effect of ACE2 due to its role in the renin-angiotensin system? findings from a comparison of the aging phenotypes of ACE2-deficient, Tsukuba hypertensive, and Mas-deficient mice. Hypertens. Res. 46, 1210–1220. doi:10.1038/s41440-023-01189-y
Tamura, K., Umemura, S., Nyui, N., Hibi, K., Ishigami, T., Kihara, M., et al. (1998). Activation of angiotensinogen gene in cardiac myocytes by angiotensin II and mechanical stretch. Am. J. Physiol. 275, R1–R9. doi:10.1152/ajpregu.1998.275.1.R1
Tanrıverdi, L. H., Özhan, O., Ulu, A., Yıldız, A., Ateş, B., Vardı, N., et al. (2023). Activation of the Mas receptors by AVE0991 and MrgD receptor using alamandine to limit the deleterious effects of Ang II-induced hypertension. Fundam. Clin. Pharmacol. 37 (1), 60–74. doi:10.1111/fcp.12829
Tipnis, S. R., Hooper, N. M., Hyde, R., Karran, E., Christie, G., and Turner, A. J. (2000). A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 275 (43), 33238–33243. doi:10.1074/jbc.M002615200
Tornling, G., Batta, R., Salvail, D., Raud, J., and Denton, C. P. (2023). Effects of the oral angiotensin II type 2 receptor agonist C21 in sugen-hypoxia induced pulmonary hypertension in rats. Int. J. Mol. Sci. 24 (8), 7478. doi:10.3390/ijms24087478
Tsai, H. J., Shih, C. C., Chang, K. Y., Liao, M. H., Liaw, W. J., Wu, C. C., et al. (2021). Angiotensin-(1–7) treatment blocks lipopolysaccharide-induced organ damage, platelet dysfunction, and IL-6 and nitric oxide production in rats. Sci. Rep. 11 (1), 610. doi:10.1038/s41598-020-79902-x
Ueda, S., Masumori-Maemoto, S., Ashino, K., Nagahara, T., Gotoh, E., Umemura, S., et al. (2000). Angiotensin-(1-7) attenuates vasoconstriction evoked by angiotensin II but not by noradrenaline in man. Hypertension 35 (4), 998–1001. doi:10.1161/01.HYP.35.4.998
Ueda, S., Masumori-Maemoto, S., Wada, A., Ishii, M., Brosnihan, K. B., and Umemura, S. (2001). Angiotensin(1–7) potentiates bradykinin-induced vasodilatation in man. J. Hypertens. 19 (11), 2001–2009. doi:10.1097/00004872-200111000-00010
Valle Martins, A. L., Annoni, F., da Silva, F. A., Bolais-Ramos, L., de Oliveira, G. C., dos Santos, A. H., et al. (2022). Angiotensin 1-7 in severe COVID-19 patients: a phase 1 clinical trial. medRxiv. doi:10.1101/2022.09.15.22279897
van Kats, J. P., Danser, A. H., van Meegen, J. R., Sassen, L. M., Verdouw, P. D., and Schalekamp, M. A. (1998). Angiotensin production by the heart: a quantitative study in pigs with the use of radiolabeled angiotensin infusions. Circulation 98 (1), 73–81. doi:10.1161/01.cir.98.1.73
Vercruysse, L., Morel, N., Van Camp, J., Szust, J., and Smagghe, G. (2008). Antihypertensive mechanism of the dipeptide Val-Tyr in rat aorta. Peptides 29 (2), 261–267. doi:10.1016/j.peptides.2007.09.023
Vickers, C., Hales, P., Kaushik, V., Dick, L., Gavin, J., Tang, J., et al. (2002). Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 277 (17), 14838–14843. doi:10.1074/jbc.M200581200
Wagener, G., Goldklang, M. P., Gerber, A., Elisman, K., Eiseman, K. A., Fonseca, L. D., et al. (2022). A randomized, placebo-controlled, double-blinded pilot study of angiotensin 1–7 (TXA-127) for the treatment of severe COVID-19. Crit. Care 26, 229. doi:10.1186/s13054-022-04096-9
Waluga, M., Kukla, M., Żorniak, M., Bacik, A., and Kotulski, R. (2015). From the stomach to other organs: Helicobacter pylori and the liver. World J. Hepatology 7 (18), 2136–2146. doi:10.4254/WJH.V7.I18.2136
Wan, Y., Wallinder, C., Johansson, B., Holm, M., Mahalingam, A. K., Wu, X., et al. (2004). First reported nonpeptide AT1 receptor agonist (L-162,313) acts as an AT2 receptor agonist in vivo. J. Med. Chem. 47 (6), 1536–1546. doi:10.1021/jm031031i
Wang, L., Jiang, T., Yang, Y., Mao, J., Wang, Q., Yu, R., et al. (2022). Angiotensin-(1–7) alleviates acute lung injury by activating the Mas receptor in neutrophil. Ann. Transl. Med. 10 (24), 1395. doi:10.21037/atm-22-6193
Wang, Q. G., Xue, X., Yang, Y., Gong, P. Y., Jiang, T., and Zhang, Y. D. (2018). Angiotensin IV suppresses inflammation in the brains of rats with chronic cerebral hypoperfusion. JRAAS 19 (3), 1470320318799587. doi:10.1177/1470320318799587
Wang, W., Zhang, Y., Huang, W., Yuan, Y., Hong, Q., Xie, Z., et al. (2023). Alamandine/MrgD axis prevents TGF-β1-mediated fibroblast activation via regulation of aerobic glycolysis and mitophagy. J. Transl. Med. 21 (1), 24. doi:10.1186/s12967-022-03837-2
Ward, C., Chilvers, E. R., Lawson, M. F., Pryde, J. G., Fujihara, S., Farrow, S. N., et al. (1999). NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J. Biol. Chem. 274, 4309–4318. doi:10.1074/jbc.274.7.4309
Welches, W. R., Bridget Brosnihan, K., and Ferrario, C. M. (1993). A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: angiotensin converting enzyme, prolyl endopeptidase and neutral endopeptidase 24.11. Life Sci. 52 (18), 1461–1480. doi:10.1016/0024-3205(93)90108-F
Wenzel, P., Knorr, M., Kossmann, S., Stratmann, J., Hausding, M., Schuhmacher, S., et al. (2011). Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 124, 1370–1381. doi:10.1161/CIRCULATIONAHA.111.034470
Wiemer, G., Dobrucki, L. W., Louka, F. R., Malinski, T., and Heitsch, H. (2002). AVE 0991, a nonpeptide mimic of the effects of angiotensin-(1-7) on the endothelium. Hypertension 40 (6), 847–852. doi:10.1161/01.HYP.0000037979.53963.8F
Wright, J. W., Mizutani, S., and Harding, J. W. (2012). Focus on brain angiotensin III and aminopeptidase A in the control of hypertension. Int. J. Hypertens. 2012, 124758. doi:10.1155/2012/124758
Xiong, S., Tan, J., Wang, Y., He, J., Hu, F., Wu, X., et al. (2022). Fibrosis in fat: from other diseases to Crohn’s disease. Front. Immunol. 13, 935275. doi:10.3389/fimmu.2022.935275
Xu, H., An, X., Tian, J., Fu, M., Wang, Q., Li, C., et al. (2021). Angiotensin-(1-7) protects against sepsis-associated left ventricular dysfunction induced by lipopolysaccharide. Peptides 144, 170612. doi:10.1016/j.peptides.2021.170612
Xu, J., Yu, Z., and Liu, X. (2023). Angiotensin-(1–7) suppresses airway inflammation and airway remodeling via inhibiting ATG5 in allergic asthma. BMC Pulm. Med. 23 (1), 422. doi:10.1186/s12890-023-02719-7
Yang, C., Wu, X., Shen, Y., Liu, C., Kong, X., and Li, P. (2020). Alamandine attenuates angiotensin II-induced vascular fibrosis via inhibiting p38 MAPK pathway. Eur. J. Pharmacol. 883, 173384. doi:10.1016/j.ejphar.2020.173384
Yu, L., Yuan, K., Phuong, H. T. A., Park, B. M., and Kim, S. H. (2016). Angiotensin-(1-5), an active mediator of renin-angiotensin system, stimulates ANP secretion via Mas receptor. Peptides 86, 33–41. doi:10.1016/j.peptides.2016.09.009
Zahmatkesh, M. E., Jahanbakhsh, M., Hoseini, N., Shegefti, S., Peymani, A., Dabin, H., et al. (2022). Effects of exosomes derived from Helicobacter pylori outer membrane vesicle-infected hepatocytes on hepatic stellate cell activation and liver fibrosis induction. Front. Cell. Infect. Microbiol. 12, 857570. doi:10.3389/fcimb.2022.857570
Zaidan, I., Tavares, L. P., Sugimoto, M. A., Lima, K. M., Negreiros-Lima, G. L., Teixeira, L. C., et al. (2022). Angiotensin-(1-7)/MasR axis promotes migration of monocytes/macrophages with a regulatory phenotype to perform phagocytosis and efferocytosis. JCI Insight 7, e147819. doi:10.1172/jci.insight.147819
Zaman, A., and Banday, A. A. (2022). Angiotensin (1-7) protects against renal ischemia-reperfusion injury via regulating expression of NRF2 and microRNAs in Fisher 344 rats. Am. J. Physiology - Ren. Physiology 323 (1), F33–F47. doi:10.1152/ajprenal.00283.2021
Zambelli, V., Bellani, G., Borsa, R., Pozzi, F., Grassi, A., Scanziani, M., et al. (2015). Angiotensin-(1-7) improves oxygenation, while reducing cellular infiltrate and fibrosis in experimental Acute Respiratory Distress Syndrome. Intensive Care Med. Exp. 3 (1), 44–17. doi:10.1186/s40635-015-0044-3
Zeng, W. T., Chen, W. y., Leng, X. y., Tang, L. l., Sun, X. t., Li, C. l., et al. (2012). Impairment of cardiac function and remodeling induced by myocardial infarction in rats are attenuated by the nonpeptide angiotensin-(1-7) analog AVE 0991. Cardiovasc. Ther. 30 (3), 152–161. doi:10.1111/j.1755-5922.2010.00255.x
Zhang, M., Sui, W., Xing, Y., Cheng, J., Cheng, C., Xue, F., et al. (2021a). Angiotensin IV attenuates diabetic cardiomyopathy via suppressing FoxO1-induced excessive autophagy, apoptosis and fibrosis. Theranostics 11 (18), 8624–8639. doi:10.7150/THNO.48561
Zhang, Y., Shang, Z., and Liu, A. (2021b). Angiotensin-(3–7) alleviates isoprenaline-induced cardiac remodeling via attenuating cAMP-PKA and PI3K/Akt signaling pathways. Amino Acids 53 (10), 1533–1543. doi:10.1007/s00726-021-03074-9
Zhang, Y. L., Bai, J., Yu, W. J., Lin, Q. Y., and Li, H. H. (2023). CD11b mediates hypertensive cardiac remodeling by regulating macrophage infiltration and polarization. J. Adv. Res. 55, 17–31. doi:10.1016/J.JARE.2023.02.010
Zhao, K., Xu, T., Mao, Y., Wu, X., Hua, D., Sheng, Y., et al. (2022). Alamandine alleviated heart failure and fibrosis in myocardial infarction mice. Biol. Direct 17 (1), 25. doi:10.1186/s13062-022-00338-6
Zheng, H., Pu, S. Y., Fan, X. F., Li, X. S., Zhang, Y., Yuan, J., et al. (2015). Treatment with angiotensin-(1-9) alleviates the cardiomyopathy in streptozotocin-induced diabetic rats. Biochem. Pharmacol. 95 (1), 38–45. doi:10.1016/j.bcp.2015.03.009
Zhong, H., Gui, X., Hou, L., Lv, R., and Jin, Y. (2022). From inflammation to fibrosis: novel insights into the roles of high mobility group protein box 1 in schistosome-induced liver damage. Pathogens 11 (3), 289. doi:10.3390/PATHOGENS11030289
Zhong, J. C., Yu, X. Y., Lin, Q. X., Li, X. H., Huang, X. Z., Xiao, D. Z., et al. (2008). Enhanced angiotensin converting enzyme 2 regulates the insulin/Akt signalling pathway by blockade of macrophage migration inhibitory factor expression. Br. J. Pharmacol. 153 (1), 66–74. doi:10.1038/sj.bjp.0707482
Zhou, H., Yan, Z. H., Yuan, Y., Xing, C., and Jiang, N. (2021). The role of exosomes in viral hepatitis and its associated liver diseases. Front. Med. 8, 782485. doi:10.3389/fmed.2021.782485
Zhou, Z., Peters, A. M., Wang, S., Janda, A., Chen, J., Zhou, P., et al. (2019). Reversal of aortic enlargement induced by increased biomechanical forces requires AT1R inhibition in conjunction with AT2R activation. Arteriosclerosis, Thrombosis, Vasc. Biol. 39 (3), 459–466. doi:10.1161/ATVBAHA.118.312158
Zhu, Y., Xu, D., Deng, F., Yan, Y., Li, J., Zhang, C., et al. (2021). Angiotensin (1-7) attenuates sepsis-induced acute kidney injury by regulating the NF-κB pathway. Front. Pharmacol. 12, 601909. doi:10.3389/fphar.2021.601909
Zisman, L. S., Keller, R. S., Weaver, B., Lin, Q., Speth, R., Bristow, M. R., et al. (2003). Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme homologue ACE2. Circulation 108 (14), 1707–1712. doi:10.1161/01.CIR.0000094734.67990.99
Keywords: inflammation, fibrosis, counter-regulatory RAS, angiotensin type 2 receptor, Mas receptor
Citation: Ávila-Martínez DV, Mixtega-Ruiz WK, Hurtado-Capetillo JM, Lopez-Franco O and Flores-Muñoz M (2024) Counter-regulatory RAS peptides: new therapy targets for inflammation and fibrotic diseases?. Front. Pharmacol. 15:1377113. doi: 10.3389/fphar.2024.1377113
Received: 26 January 2024; Accepted: 18 March 2024;
Published: 10 April 2024.
Edited by:
Yassine Sassi, Fralin Biomedical Research Institute, United StatesReviewed by:
Francisco O. Silva, University of Texas Southwestern Medical Center, United StatesCopyright © 2024 Ávila-Martínez, Mixtega-Ruiz, Hurtado-Capetillo, Lopez-Franco and Flores-Muñoz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mónica Flores-Muñoz, bW9mbG9yZXNAdXYubXg=; Oscar Lopez-Franco, b3NjYXJsb3BlejAxQHV2Lm14
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.