 Saeid Maghsoudi
Saeid Maghsoudi Rabia Shuaib
Rabia Shuaib Ben Van Bastelaere
Ben Van Bastelaere Shyamala Dakshinamurti
Shyamala Dakshinamurti- 1Department of Physiology and Pathophysiology, University of Manitoba, Winnipeg, MB, Canada
- 2Biology of Breathing Group, Children’s Hospital Research Institute of Manitoba, Winnipeg, MB, Canada
- 3Section of Neonatology, Department of Pediatrics, Health Sciences Centre, Winnipeg, MB, Canada
Adenylyl cyclases (ACs) are crucial effector enzymes that transduce divergent signals from upstream receptor pathways and are responsible for catalyzing the conversion of ATP to cAMP. The ten AC isoforms are categorized into four main groups; the class III or calcium-inhibited family of ACs comprises AC5 and AC6. These enzymes are very closely related in structure and have a paucity of selective activators or inhibitors, making it difficult to distinguish them experimentally. AC5 and AC6 are highly expressed in the heart and vasculature, as well as the spinal cord and brain; AC6 is also abundant in the lungs, kidney, and liver. However, while AC5 and AC6 have similar expression patterns with some redundant functions, they have distinct physiological roles due to differing regulation and cAMP signaling compartmentation. AC5 is critical in cardiac and vascular function; AC6 is a key effector of vasodilatory pathways in vascular myocytes and is enriched in fetal/neonatal tissues. Expression of both AC5 and AC6 decreases in heart failure; however, AC5 disruption is cardio-protective, while overexpression of AC6 rescues cardiac function in cardiac injury. This is a comprehensive review of the complex regulation of AC5 and AC6 in the cardiovascular system, highlighting overexpression and knockout studies as well as transgenic models illuminating each enzyme and focusing on post-translational modifications that regulate their cellular localization and biological functions. We also describe pharmacological challenges in the design of isoform-selective activators or inhibitors for AC5 and AC6, which may be relevant to developing new therapeutic approaches for several cardiovascular diseases.
Introduction
Over the course of life, heart health and aging have been correlated with diminished left ventricular (LV) function and weakened cardiac β-adrenergic receptor (βAR) responsiveness. The reasons for this dysfunctionality may include phenotype changes in the LV, reduced βAR density, upregulation of regulatory proteins Gαi and G-protein coupled receptor kinases, or abnormalities in the βAR signaling system (Ferrara et al., 2014). Adenylyl cyclases (ACs), as the central effector molecules for βAR signaling, play a crucial role in cardiac contractility, relaxation, and LV diastolic function (Tang et al., 2011). The failing heart has poor AC signaling and decreased LV cAMP production, leading to impaired βAR responsiveness to ligands (White et al., 1994; Roth et al., 2002). Persistent activation of the sympatho-adrenergic system in patients with congestive heart failure can also lead to unfavorable cardiac remodeling due to cardiomyocyte loss and fibrotic replacement (Zhang et al., 2013). The distribution of AC isoforms varies within cardiac tissues; ACs 2, 3, 4, 5, 6, and 7 are expressed by cardiac fibroblasts (Ostrom et al., 2003), while AC1 and AC8 are found in sinoatrial node (SAN) cells (Mattick et al., 2007; Younes et al., 2008; Robinson et al., 2021), and AC5 and AC6 are the major AC isoforms expressed in the adult ventricle (Göttle et al., 2009; Efendiev and Dessauer, 2011). AC6 also serves as the principal effector of vasodilator signaling and a regulator of membrane potential in vascular myocytes (Nelson et al., 2011). In this review, we examine the similarities and differences between ACs 5 and 6 in cardiovascular function, highlighting their divergent functional roles and opportunities for pharmacological targeting.
Overview of adenylyl cyclases
Cyclic 3′,5′-adenosine monophosphate (cAMP), a ubiquitous second messenger that mediates a variety of cellular responses, is generated by more than thousand nucleotidyl cyclase proteins classified into six groups according to the amino acid sequence of the catalytic domain (Kamenetsky et al., 2006; Seifert and Beste, 2012). The class III cyclases including eukaryotic adenylyl cyclases (ACs) are indispensable effectors of cAMP (Linder, 2006). Enumerated in order of discovery, mammalian ACs 1–9 (∼120–140 KDa) have been identified as transmembrane adenylyl cyclases (TMACs), while AC10 is a soluble enzyme. ACs are canonically regulated by heterotrimeric G-proteins, Gαi and Gαs, upon stimulation of G protein-coupled receptors (GPCRs) (Halls and Cooper, 2017). There are significant variations in the distribution and biochemical characteristics among AC isoforms, as well as distinctive chromosomal loci of individual isoforms (Vatner et al., 2013). The TMACs share a similar framework but different regulation (Sunahara et al., 1996). Despite arising from distinct transcripts, they own a similar overall structure with highly homologous active sites, making it challenging to design selective activators or inhibitors and to differentiate their functions in the organs and tissues (Rana et al., 2017). The mammalian ACs share an intracellular N-terminus, two membrane-spanning domains (TM1 and TM2), and conserved catalytic domains, C1 and C2 (each ∼40 KDa), together forming the catalytic core. The C1 and C2 cytoplasmic domains are further divided into the highly conserved catalytic regions, C1a and C2a, and the less conserved regulatory regions, C1b and C2b. Two active sites are shaped by their interface: the ATP catalytic site and forskolin (FSK) binding pocket (Figure 1A) (Schmid et al., 2014; Antoni, 2020). Crystallographic studies have revealed that the hydrophobic pocket generated by C1 and C2 catalytic subunits is the allosteric site for the interaction of FSK with ACs (Pavan et al., 2009; Bhatia et al., 2023). Despite the sequence similarity of the regulatory domains in C1–C2, various signals such as G-proteins, kinases, FSK, and Ca2+ uniquely regulate AC isoforms (Brand et al., 2013). Indeed, the C1–C2 domain, at the cytoplasmic surface of the molecule, accommodates binding sites for Gαs, Gαi, FSK, ATP, Mg2+, the regulator of G protein signaling (RGAS2), proteins associated with Myc (PAM), Snapin, Ric8a, A-kinase-anchoring protein (AKAP79), PH domain leucine-rich protein phosphatase 2 (PHLPP2), and phosphorylation and dephosphorylation sites for protein kinase A (PKA) and protein kinase C (PKC) (Sunahara et al., 1997). The crystal structures of the complex made up of the C1 cyclase homology domain (CHD) from AC5 and the C2 CHD from AC2 (VC1:IIC2) bound to FSK and GTP-S-activated Gαs served as the fundamental model for the prototype AC catalytic core (Mou et al., 2009). Binding sites for cofactors Mg2+ and Mn2+ are further located in the active site of AC and must be occupied for catalytic activity, based on structural analyses of activated VC1:IIC2 complexes coupled to substrate analogs (Sinha and Sprang, 2006; Mou et al., 2009). Forskolin, a natural diterpene, specifically binds opposite to the ATP-binding site within the catalytic core, using a mixture of hydrophobic and hydrogen interactions to approximate the two cytoplasmic domains, resulting in increased enzyme activity regardless of Gαs docking. No endogenous ligand has yet been identified for the FSK-binding site (Pavan et al., 2009; Dessauer et al., 2017; Bhatia et al., 2023).
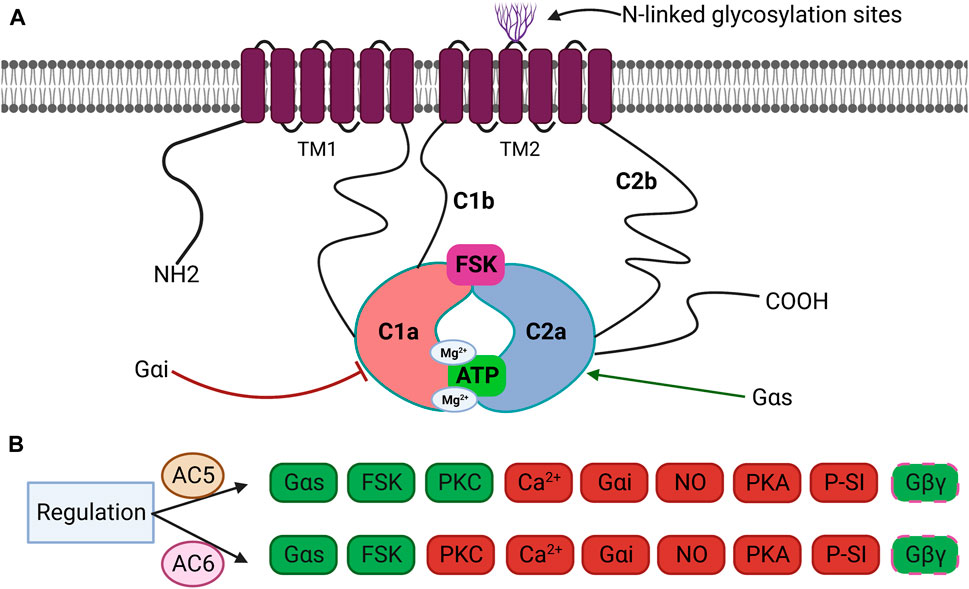
Figure 1. (A) Schematic structure of transmembrane adenylyl cyclases (TMACs). TMACs contain an intracellular N-terminus, two repetitions of six TM1 and TM2, and two cytoplasmic domains (C1 and C2) further divided into C1a and C2a. C1a–C2a forms the catalytic domain and FSK-binding site. Inhibitor Gαi binds to C1a, whereas activator Gαs binds to C2a. C1b and C2b are regulatory subdomains, and the N-terminus participates in several protein–protein interactions. Some isoforms are glycosylated on extracellular loops 5 or 6. (B) Specific regulation of AC5 and AC6 by various effectors. Both isoforms are fully activated by Gαs and FSK and partially or conditionally activated by Gβγ. AC5 is activated by PKC, whereas AC6 in inhibited by PKC. Both isoforms are inhibited by Ca2+, Gαi, NO, PKA, and P-site inhibitors (P-SI). Green: stimulation, red: inhibition, and dashed line: conditionally.
While the C1–C2 framing the catalytic core remains highly conserved among individual AC isoforms, the N-terminus is significantly varied across isoforms (Antoni, 2020). The length and composition of the N- and C-terminal domains of ACs are significantly variable outside of the non-conserved domains (Sunahara et al., 1996). The N-terminus of AC1 to AC5 spans 60 to 240 residues in length. Pharmacologically, these are the regions where type-specific regulation of ACs can occur (Halls and Cooper, 2017). The TM domains are not required for catalytic activity since C1a and C2a can be individually shaped into active enzymes by Gαs and FSK (Tang and Gilman, 1995). Removing TM domains and fusing just C1 with C2 make a small but versatile protein which can be used to replicate numerous beneficial effects of AC (Tan et al., 2019). However, the TM regions play important roles in balancing the stoichiometry of their relative catalytic domains (Sunahara et al., 1996), in regulating functional assembly and trafficking of AC (Gu et al., 2002), and may also function as receptors for extracellular signals (Seth et al., 2020).
From a regulatory standpoint, the mammalian AC isoforms are classified into four groups: Group I, ACs 1, 3, and 8, stimulated by calcium/calmodulin (Ca2+/CaM); Group II, ACs 2, 4, and 7, stimulated by Gβγ; Group III, ACs 5 and 6, inhibited by Gαi/Ca2+; and Group IV, AC9, which is partially stimulated by FSK (Ostrom et al., 2022). All isoforms are activated by Gαs while differentially regulated by Gαi, Gβγ, and protein kinases (PKA, PKC, Raf-1, and CaM kinases) (Figure 1B) (Antoni, 2020). Most cells and tissues express multiple AC isoforms; however, their varying abundance in specific tissues is noteworthy (Halls and Cooper, 2017; Ostrom et al., 2022).
Role of ACs in cardiac tissue
Key cardiac isoforms AC5 and AC6, known as the Ca2+-inhibited family of ACs, share about 91.5% sequence alignment, except for the AC6 N-terminus (aa 1–86) (Wu et al., 2017). These two isoforms have considerable similarities in their expression patterns and functions, despite their evolution at independent gene loci; the AC5 gene is on chromosome 3 at position 3q13.2–3q21, and AC6 is on chromosome 12 at position 12q12–12q13 (Haber et al., 1994). The canine (type V-a) and the rat (type V-b) forms of AC5 are in fact mRNA splice variants, co-expressed in both species (Iwami et al., 1995). Both AC5 and AC6 are non-competitively inhibited by Gαi at C1a, opposite from the docking site for Gαs at C2a (Taussig et al., 1994). Their tissue distribution and developmental mRNA expression patterns have been studied in several species, including humans (Tobise et al., 1994; Espinasse et al., 1995; Wang and Brown, 2004). AC6 is more expressed in the neonatal heart, while AC5 appears to be predominantly an adult isoform (Espinasse et al., 1995). The maximal amount of AC6 mRNA is expressed during fetal development, while steadily diminishing with age, and reaching its lowest amount in fully mature adults. On the contrary, a minimal amount of AC5 mRNA is expressed during fetal development, but progressively increasing with age, and reaching its highest point in the fully developed adult (Tobise et al., 1994; Okumura et al., 2009). In the mouse heart, the abundance of AC5 protein reaches its highest point before 1 week and then decreases to levels similar to those of mice studied at 3, 6, and 24 months of age. In the rat heart, however, the AC5 protein level peaks before 2 weeks of age and subsequently decreases by 3 months. In pigs, cardiac AC5 protein abundance is high in 1-day-old hearts and then diminishes with age, reaching its lowest content in the heart at 5–6 months (Hu et al., 2009). Feedback regulation of AC expression also varies as a function of age; PDE3 inhibition (PDE3i) has been found to alter AC expression levels in adult and pediatric subjects with dilated cardiomyopathy (DCM) and in pediatric subjects with a congenital single right ventricle. These groups demonstrated distinctive AC isoform mRNA expression patterns. Compared to the non-failing adult LV myocardium, the adult DCM myocardium exhibits upregulation of AC6; chronic PDE3i treatment enhances mRNA expression of AC5 and AC6, while all other isoforms are expressed at levels comparable to those of DCM patients not receiving PDE3i treatment. In contrast, the non-failing pediatric right ventricle expresses less AC5 than does the pediatric single ventricle; and in pediatric DCM, AC5 and AC6 remained unchanged by PDE3i (Nakano et al., 2017). Due to their structural homology and the paucity of type-specific antibodies, definitive levels of protein expression in the heart have been difficult to establish (Hu et al., 2009). Even following the deletion of AC5, 60% of immunodetectable AC5/6 was observed in cardiac myocytes, suggesting some degree of epitope overlap (Okumura et al., 2003b).
To distinguish their specific roles in cardiac function, AC5 and AC6 have been the subject of several transgenic overexpression and deletion studies. Both isoforms are highly expressed in the heart and are important negative feedback responses of cardiac rhythmicity (Cosson et al., 2019). While both isoforms regulate heart rate (HR) and contractility, AC6 appears more important at baseline cardiac function (Vatner et al., 2013). AC5 and AC6 may function distinctly in the pathogenesis of cardiac stress responses. The protein abundance of AC5 and AC6 responds oppositely to pressure overload LV hypertrophy (Okumura et al., 2003b; Sugano et al., 2011), with upregulation of AC5 and downregulation of AC6, implying that it is AC5 that plays a role in chronic pressure overload cardiomyopathy (Vatner et al., 2013). It is probable that these two isoforms also play different physiological roles in regulating cardiac function during dilated cardiomyopathy. Both AC5 and AC6 mRNAs appear decreased in dogs with pacing-induced Congestive Heart Failure (CHF) (Ishikawa et al., 1994); while in a swine model of tachypacing and severe heart failure, AC6 but not AC5 is downregulated (Ping et al., 1997). While some studies suggest AC5 overexpression improves cardiac function in transgenic mice during exercise (Esposito et al., 2008), deletion of AC5 protects the heart in most models of cardiac stress including heart failure; in contrast, AC6 overexpression has been correlated with cardioprotective effects (Wu et al., 2017; Ostrom et al., 2022). The overexpression of AC6 leads to increased LV function, improved cAMP and Ca2+ handling, and may protect the heart from pressure overload-induced systolic and diastolic dysfunction (Phan et al., 2007; Guellich et al., 2010). AC5 deletion, on the other hand, results in protecting the heart from cardiomyopathy, chronic catecholamine stress, and chronic pressure overload (Okumura et al., 2003b; Okumura et al., 2007). Isoform-specific effects are not restricted to myocardial AC5 and AC6. Cardiac-specific overexpression of AC8 in 3-month-old transgenic mice alters several pathways, resulting in elevated AC activity and cAMP-induced cardiac workload until up to 1 year of age, without excess mortality or onset of heart failure (Tarasov et al., 2022; Qu et al., 2024). However, other studies have shown cardiac overexpression of AC8 to cause early and accelerated cardiac remodeling, resulting in development of heart failure and a shortened life span, suggesting that alterations in cAMP/PKA signaling can hasten cardiac aging, in part via the glycogen-synthase-kinase 3α/β (GSK3α/β) phosphorylation pathway (Mougenot et al., 2019).
Although there remains some controversy regarding the specific regulation and roles of AC5 and AC6 isoforms in controlling cardiac function, their roles in the production of required cAMP to initiate cardiac chronotropic and inotropic responses are indispensable. It is thus important to differentiate the regulation and biological functions of AC5 versus AC6, leading the way to development of targeted treatments for different cardiac pathologies. The respective therapeutic outcomes of AC5 deletion and AC6 overexpression in the cardiovascular system are included in Figure 2.
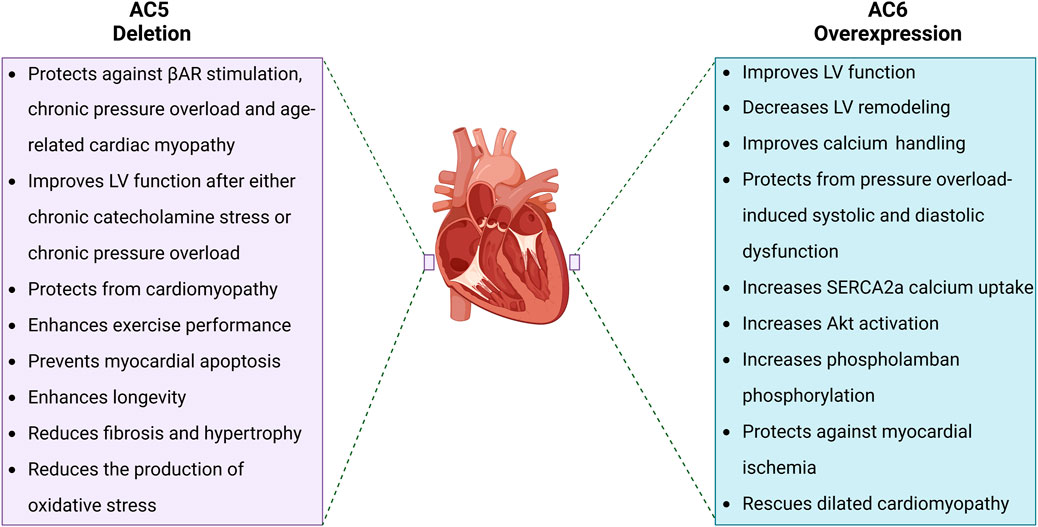
Figure 2. Pathophysiological effects of AC5 deletion and AC6 overexpression. AC5 and AC6 behave distinctly when increased or disrupted. While AC5 deletion has come with several beneficial effects for the heart such as prolonged longevity and enhanced LV function, AC6 overexpression improves cardiac cAMP generation and Ca2+ handling, resulting in improved LV function.
Regulation of AC5 and AC6 by calcium
Ca2+ serves as a pivotal regulator of AC5 and AC6, playing a crucial role in the control of cellular homeostasis and heart function regulation. The inhibition of ACs 5 and 6 by Ca2+, which is non-competitive with respect to ATP (Guillou et al., 1999), contributes to pacemaking in cardiac tissue and also sustains endothelial cell permeability. Inhibition of these cardiac AC isoforms by Ca2+, which their own cAMP generation permits to enter the cardiomyocyte, serves as a form of feedback control regulating pacemaker rhythmicity as well as the force of contraction (Mou et al., 2009). A wide range of in vitro inhibitory sensitivities are reported. Systolic [Ca2+] 1 µM is required to inhibit the activity of AC5/6, while at higher intracellular [Ca2+] (10–25 µM), the activity of all AC isoforms is inhibited (Guillou et al., 1999; Halls and Cooper, 2017). However, AC5 and AC6 inhibition by Ca2+ is biphasic, involving low- and high-affinity binding sites, so Ca2+ can inhibit AC5 and AC6 in sub- to supramicromolar concentration ranges (Steer and Levitzki, 1975; HANSKI et al., 1977; OLDHAM et al., 1984; Fagan et al., 1998; Guillou et al., 1999). The sub-micromolar [Ca2+] (0.2–0.6 µM) inhibits AC5 and AC6 in membrane preparations from different tissues, cultured cell lines, and in recombinant systems (Beazely and Watts, 2006). Mechanistically, Ca2+ antagonizes the activation of AC5 and AC6 by Mg2+ (Mou et al., 2009). Site-directed mutations of AC5 in the presence of other divalent cations indicate that the Mg2+-binding loci at the AC5 catalytic site are essential for its Ca2+-mediated inhibition (Hu et al., 2002; Mou et al., 2009). In addition to being a hub for GPCR AC interactions, lipid rafts are locations for capacitive Ca2+ entry channels; Group III AC isoforms that are localized in lipid rafts are also regulated by Ca2+ (Ostrom and Insel, 2004). Disruption of lipid rafts results in loss of capacitive Ca2+ entry and dysregulation of Ca2+-regulated AC isoforms (Dessauer et al., 2017).
Subcellular localization of AC5 and AC6
GPCR signaling pathways are type-specific for either AC5 or AC6, as determined by the colocalization of receptors and AC isoforms in lipid raft or non-raft plasma membranes (Ostrom et al., 2000). ACs 4–7 are expressed in the heart and vascular system, but among them, AC5 and AC6 are dominant subtypes in adult mammalian heart (Dessauer et al., 2017). The compartmentation of AC5 and AC6 in caveolae or membrane lipid rafts provides a key biochemical process for temporal and spatial segregation of signal transduction as well as cross-talk between signaling cascades, resulting in the compartmentalization of cAMP signaling (Thangavel et al., 2009). While native AC5/6 are found in caveolar fractions, overexpressed AC6 in rat aortic smooth muscle cells is found in non-caveolin-rich fractions, where it is functional and increases cAMP (Ostrom et al., 2002). In these cells, low levels of AC6 overexpression do not much change cAMP levels at baseline nor responses to adenosine A2b receptor challenge (Ostrom et al., 2002; Thangavel et al., 2009). In contrast, in cardiomyocytes, overexpressed AC6 localizes similarly to endogenous AC6, which is targeted to caveolae, demonstrating that GPCR and AC5/6 compartmentation to caveolin-rich membranes is cell-type dependent (Ostrom et al., 2000; Ostrom et al., 2001; Ostrom et al., 2002). Overexpression of AC6 improves βAR responses without affecting signaling of other Gαs-coupled receptors in a variety of cells, including airway smooth muscle, lung fibroblasts, and neonatal myocytes (Sunahara et al., 1996; Liu et al., 2008; Sadana and Dessauer, 2009; Dessauer et al., 2017). In vascular smooth muscle cells (VSMCs), AC3, AC5, and AC6 are the most abundant isoforms in βAR-mediated signaling, but during vasodilation, AC6 is the principal isoform involved in βAR-mediated cAMP/PKA signaling and activation of the ATP-sensitive potassium current, thus playing an important role in setting the membrane potential. Its counterpart AC5 does not have similar activity, (Nelson et al., 2011). In ventricular myocytes, isoforms AC5 and AC6 have separate subcellular compartmentalization. While AC5 is mostly found deep in transverse tubules interacting with caveolin-3 (CAV3) and phosphodiesterases (PDEs), AC6 is located in the plasma membrane outside the t-tubular area (Figure 3) (Timofeyev et al., 2013). βAR receptors are also differentially distributed; β1- and β2AR are found within t-tubules, but β1AR (comprising >70% of cardiac βAR) is found on the sarcolemma outside t-tubules (Brodde, 1991). β1AR triggers cAMP generation, which increases PKA-mediated phosphorylation of L-type Ca2+ channels as well as other regulatory proteins, greatly increasing cardiac contractility; β2AR couples with both Gαs and Gαi, resulting in a smaller increment of contractility (Zheng et al., 2005). Inactivation of Gαi by pertussis toxin enhances β1AR-mediated inotropy despite β1AR not coupling to Gαi, indicating that Gαi also inhibits the AC5/6 receptor independently (Cosson et al., 2019). Due to subcellular localization, AC5 has been connected to β2AR signaling, while AC6 is responsible for extra-tubular β1AR signaling as well as β1AR-mediated augmentation of the L-type Ca2+ channel current (ICa,L) in ventricular myocytes (Timofeyev et al., 2013) (Figure 3). Both βAR subtypes have obvious inotropic effects; dobutamine, a non-selective βAR agonist, increases cardiac contractility in both AC5KO and AC6KO mice (Tang et al., 2006; Tang et al., 2008). While β2AR overexpression enhances ventricular function and activates cell survival pathways (Milano et al., 1994), β1AR overexpression appears catastrophic, causing both cardiac hypertrophy and dilated cardiomyopathy (Engelhardt et al., 1999). The opposite effects have been reported following manipulation of their more proximal AC subtypes; AC5 disruption in t-tubules is cardioprotective (Okumura et al., 2003b), while overexpression of AC6 rescues heart function in cardiac injury (Gao et al., 2016). However, functional coupling may not only reflect localization. In AC6KO, a shift from dominant AC6 coupling to AC5 coupling occurs in the β1AR signaling cascade. This change in the AC assignment in AC6KO results in rearranged signaling compartmentalization as well as an alteration in the PDE isoform control of the cAMP pool (Cosson et al., 2019; Ostrom et al., 2022). These findings suggest some functional redundancy of AC5 or AC6 for β1AR-mediated inotropic responses. Additionally, in CHF models largely ascribed to excess β1AR activity, β2AR redistributes to the cell surface, and this loss of cAMP compartmentation correlates with heart failure (Nikolaev et al., 2010). Studies of detubulated cardiomyocytes reveal that stimulation of both β1AR and β2AR is more functionally effective at the sarcolemmal surface, rather than within t-tubules (Cros and Brette, 2013). Pharmacological inhibition studies confirm that β1AR preferentially couples with AC5 at t-tubules, while β2AR couples with AC6 when present at the cell surface, resulting in the observed AC subtype-specific effects on cardiac function and survival signaling (Tsunematsu et al., 2015).
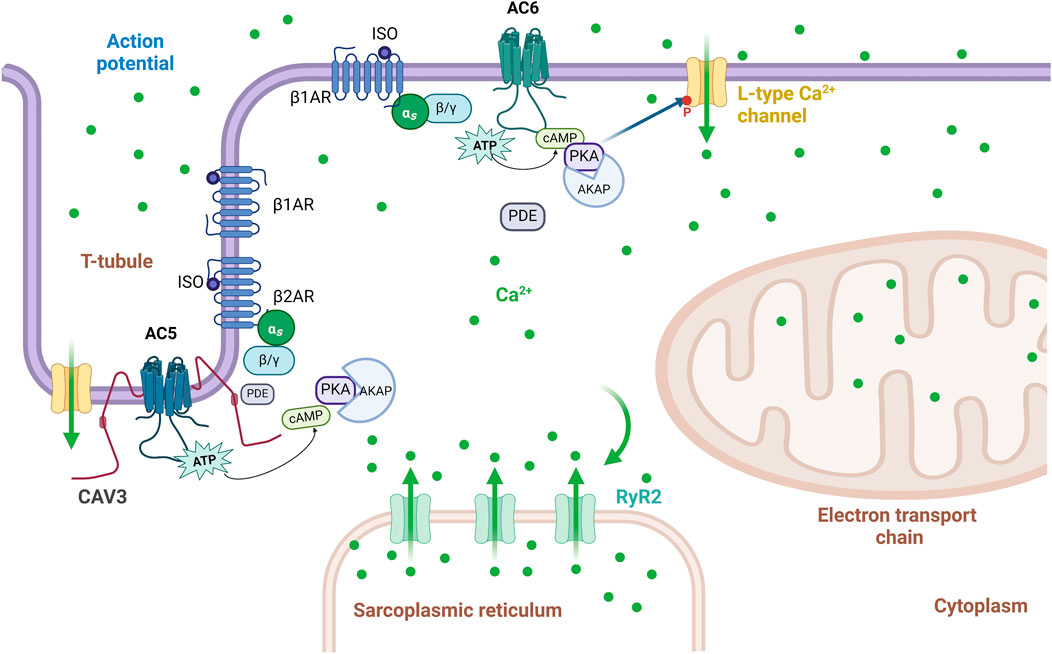
Figure 3. Schematic representation of the localization of AC5 and AC6 signaling within the t-tubule and sarcolemmal membrane. In ventricular myocytes, AC6 is localized to the plasma membrane outside of the t-tubular region, and it interacts only with β1AR signaling-mediated augmentation of the L-type Ca2+ current (ICa,L), while AC5 is localized to the membrane in t-tubular regions, and its function on ICa,L is restrained by cAMP degradation by phosphodiesterases. Adapted from (Timofeyev et al., 2013); image designed using BioRender.
Compartmentalization of cAMP signaling in the heart
While most tissues express a plethora of AC isoforms (Figure 4), the specific roles of each isoform may overlap within that cell type (Sadana and Dessauer, 2009). Functional redundancy or compensatory roles have been proposed within individual AC groups, as they are regulated or expressed in a comparable manner (Defer et al., 2000). Changes in AC activity may depend on the profile of isoforms present in a certain cell or tissue (Tao et al., 1998). Differing phenotypes of AC5 and AC6 activity suggest the maintenance of distinct pools of cAMP within cardiac myocytes, generated by each enzyme. Regulation and localization of AC5 and AC6 isoforms tailor the generation of cAMP by concurrent signals, assisting in dynamic control of the cAMP signal. The compartmentation of cAMP signaling in the heart arises, in part, from specific signaling complexes which generate distinct cell responses, with AC5 and AC6 being central effectors to the formation and maintenance of these compartments.
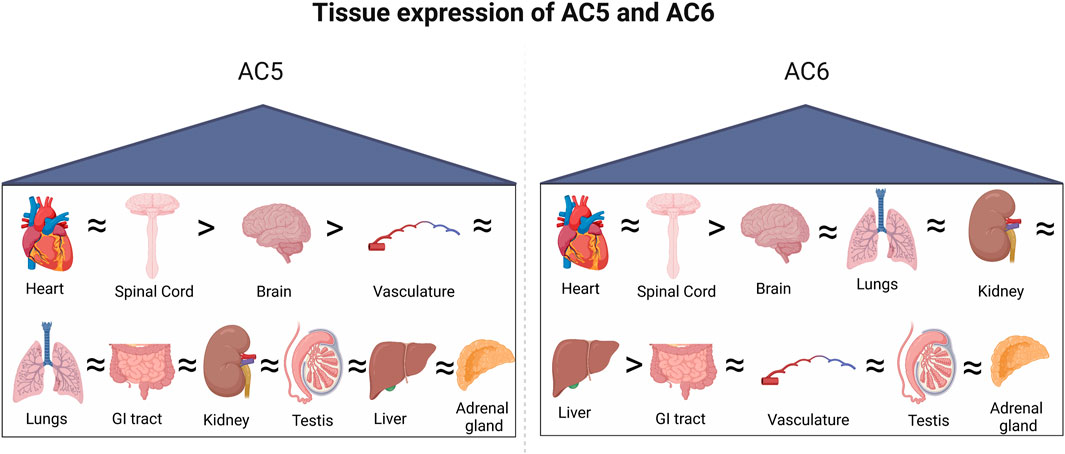
Figure 4. Tissue-specific expression of AC5 and AC6. Both isoforms are highly expressed in the heart and spinal cord, followed by the brain.
The organization of cAMP microdomains in isolated cardiac cells has been researched extensively, using various effector proteins to sort distinct cAMP pools (Patel and Gold, 2015). Several downstream effectors can regulate cAMP production or hydrolysis, including PDEs, PKA, cyclic nucleotide-gated (CNG) channels, hyperpolarization-activated cyclic nucleotide-gated exchange proteins, exchange protein directly activated by cAMP (Epac), and the Popeye domain-containing (POPDC) protein family. At the level of cAMP signal longevity, PDE isoforms, importantly PDE3/4, play pivotal roles in the discrete regulation of localized cAMP levels in neonatal cardiac myocytes (Willoughby and Cooper, 2007). PDE4 isoforms regulate cAMP signaling resulting from β1AR and β2AR stimulation in cardiac myocytes; PDE4B controls β1AR but not β2AR responses in cardiac myocytes, suggesting a localized function to control β1AR-dependent excitation–contraction coupling (Mika et al., 2014). PDE3 acts globally on cAMP in normal and heart failure models, modulating cAMP-mediated regulation of Ca2+ re-uptake in the sarcoplasmic reticulum (SR) (Calamera et al., 2022). Spatial and temporal organization of cAMP-dependent pathways is further provided by cAMP-related protein scaffolding, biasing the cAMP signal toward different end functions.
AKAP organizes AC5 and AC6 cAMP in the heart
A-kinase-anchoring proteins (AKAPs) are scaffold proteins which coordinate signaling components into multiprotein complexes, giving rise to regulation of the spatial and temporal organization and fine-tuning of cAMP signaling (Kritzer et al., 2012), ensuring correct targeting of cAMP-dependent PKA and other signaling enzymes to precise subcellular compartments (Diviani et al., 2016). AKAPs function through binding to the regulatory subunits of PKA and crucial PKA phosphorylation sites to initiate rapid and targeted coupling of the kinase to downstream effectors (Ostrom et al., 2022). AC5/6 in the heart utilizes the interactions with AKAPs to cohort with upstream and/or downstream effectors. Over 50 AKAPs are known to target PKA to various cellular locations; important cardiovascular AKAPs include AKAP15/18, AKAP79/150, Yotiao, mAKAP, AKAP-Lbc, and Gravin (Baldwin and Dessauer, 2018). The interaction of these AKAPs with AC5 or AC6 determines the localization and activity of their cAMP signaling complexes (Efendiev and Dessauer, 2011). As scaffolding effectors, AKAPs not only anchor the localization of ACs but also preserve local pools of cAMP by assembling macromolecular complexes (Baldwin and Dessauer, 2018). In cardiomyocytes, the arrangement of MAP kinase, Ca2+, and cAMP-dependent pathways is coordinated through the muscle A-kinase anchoring protein β, playing a key role in cellular hypertrophy (Kapiloff et al., 2009). AC5 binds to a specific N-terminal site on mAKAP-(245–340), resulting in impairment of AC5 activity; interruption of mAKAPβ–AC5 complexes could be targeted therapeutically to reduce cAMP generation and hypertrophy in cardiac myocytes (Bauman et al., 2006; Kapiloff et al., 2009). Both AC5 and AC6 interact with AKAP79/150 along with PKA, generating a negative feedback loop where cAMP production is impaired by PKA phosphorylation of AC5/6 (Baldwin and Dessauer, 2018).
AC5 and AC6 cAMP regulation by Epac in the heart
Another downstream cAMP effector is Epac, a family of cAMP-regulated guanine nucleotide exchange factors functioning independently of PKA as mediators of cAMP signaling. While considered less important in cardiomyocytes than PKA, Epac has high affinity to bind to cAMP, resulting in activation of small GTPases Rap1 and Rap2 (Tan et al., 2022). Epac1 and Epac2 play roles in cardiovascular Ca2+ signaling and vascular endothelial barrier formation (Lezoualc’h et al., 2016). AC5 transgenic mice with knockout of the Epac1 gene had decreased cardiac dysfunction and were less susceptible to pacing-induced atrial fibrillation after chronic isoproterenol infusion compared to controls while evincing less cardiac apoptosis and fibrosis than the AC5 transgenics, suggesting that Epac1 mediates the deleterious effects of AC5 overexpression on cardiac function and rhythmogenesis (Cai et al., 2016). On the other hand, mice lacking Epac1 are protected against pressure overload or chronic catecholamine stress-induced cardiac dysfunction (Fujita et al., 2017), reinforcing the notion that rate- and pressure-induced cardiac failure may arise from separate cAMP pools.
Advances in cAMP biosensors in live cells
Various constructs have been developed to measure real-time cAMP levels in living cells or tissues. Previous biochemical methods or radioligand tools were not able to precisely measure the spatiotemporal resolution of intercellular cAMP fluctuations; however, several fluorescent or luminescent biosensors have been introduced that can track real-time cAMP levels in living cells. These cAMP sensors have the advantage of facilitating the study of complex, compartment-specific cAMP-dependent responses (Warrier et al., 2005).
Classical methods of determining AC activity have involved detection of radiolabeled substrate ATP conversion to AMP and detection of enzyme reaction products by chromatography (Streeto and Reddy, 1967; Krishna et al., 1968); these methods can be applied to cell or tissue lysates. Loss of ATP-bound lanthanide fluorescence has been used as an indirect but specific indicator of AC ATPase activity (Spangler et al., 2008b). The terbium–norfloxacin AC activity assay measures the substrate turnover as an assessment of AC catalytic function; varying the exogenous ATP substrate concentration permits the calculation of Michaelis–Menten kinetics (Spangler et al., 2008a). In contrast, intact cell cAMP assays are cumulative measures of product formation in the absence of exogenous substrates, reflecting real-time steady state inclusive of cAMP generation, degradation, or export. Genetically encoded cAMP sensors can also be targeted to subcellular location.
Fluorescence or Förster resonance energy transfer (FRET) uses an excited donor fluorophore to transfer energy to a proximal acceptor fluorophore, which then emits a photon of a lower energy, resulting in detectable red-shift of the emitted light. FRET biosensors used to detect cAMP include PKA-, Epac-, and CNGC-based cAMP sensors. The original FRET PKA-based cAMP biosensor uses fluorescein (donor) and rhodamine (acceptor) to measure the decrease in the FRET signal due to elevation in intracellular cAMP, leading to dissociation of the regulatory subunit of PKA from its catalytic segment (Adams et al., 1991). This sensor was the first to measure microdomain cAMP levels, demonstrating a striated pattern in neonatal rat ventricular cardiomyocytes because of the interaction of PKA with AKAPs near Z-lines. These sensors have also been used to interrogate the roles of PDE3 and PDE4 in cAMP compartmentation in neonatal rat ventricular myocytes (Mongillo et al., 2004). However, the PKA-based biosensor has some limitations: a low signal-to-noise ratio, narrow dynamic range, slow response time, and need for transfection of two distinct constructs (Ponsioen et al., 2004). Epac-based cAMP biosensors address some of these issues. In these, Epac activation is determined in live cells by sandwiching Epac between cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP), taking advantage of a conformational shift generated by cAMP binding. When using cAMP-lowering agonists, FRET fully recovers, but quickly diminishes in response to cAMP-raising agents. As a result, CFP–Epac–YFP is considered a very sensitive cAMP indicator (Ponsioen et al., 2004). Epac-based cAMP biosensors have been used for monitoring Gi/o-mediated cAMP reduction after stimulation of adrenergic α2A or μ opioid receptors in a time-resolved manner without pre-stimulation with FSK or IBMX to increase endogenous cAMP levels (Storch et al., 2017), indicating detection of low levels of cAMP. The use of FRET-based sensors to visualize cAMP signaling microdomains by fluorescence microscopy has been well-reviewed (Calebiro and Maiellaro, 2014).
A newer, versatile approach for measuring cAMP after Gαs- or Gαi-mediated signaling is the cAMP difference detector in situ (cADDis) assay (schematic representation, Figure 5). This sensor uses a single fluorescent protein, the Epac cAMP-binding domain fused to circularly permuted mNeonGreen and red mMaple, resulting in a readily detectable bright green fluorescence signal; the Upward cADDis sensor manifests a rise in green fluorescence with increased cAMP, while Downward cADDis decreases fluorescence in response to increased cAMP (Tewson et al., 2018). The cADDis sensor is capable of differentiating basal cAMP from that stimulated by receptor agonism as well as cAMP changes due to Gαs or Gαi activation, when expressed in HEK293T cells (Tewson et al., 2018). Due to its robust expression in multiple cell types and ability to measure real-time cAMP kinetics in live cells over long periods of time with little loss of sensor signals, cADDis has been used in many recent studies of AC cAMP production (Baldwin et al., 2019; Bhatia et al., 2023; Cattani-Cavalieri et al., 2023; Ripoll and Von Zastrow, 2023). Other transfectable single-fluorescent protein cAMP sensors in use include fluorescent cAMP indicator (Flamindo) (Odaka et al., 2014), red fluorescent indicator for cAMP (R-FlincA) (Ohta et al., 2018), and cAMPr (Hackley et al., 2018).
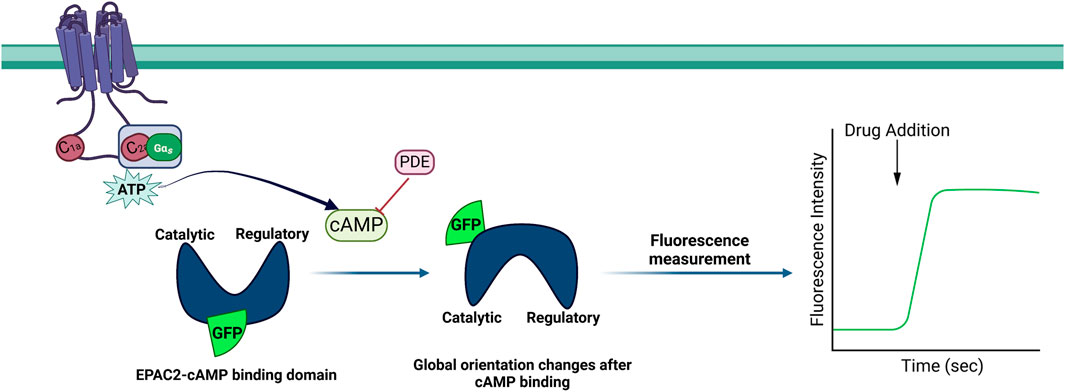
Figure 5. Schematic illustration of the cAMP difference detector in situ (cADDis) assay. The enzymatic and catalytic domains of the guanine nucleotide exchange factor Epac2 were joined to form circularly permuted green fluorescent protein (cpGFP). The binding and unbinding of the cAMP molecule cause a conformational change in Epac2.
BRET biosensors (bioluminescence resonance energy transfer) use a donor luciferase to oxidize luciferin-generating bioluminescence that excites the acceptor, emitting light at an extended wavelength (Wu and Jiang, 2022). BRET enables detection of fusion protein interactions directly, without the need of an external light source to stimulate a fluorescent energy donor (Prinz et al., 2006). This technology further addresses the problems of photobleaching, autofluorescence, and signal-to-noise ratio in FRET methods; the drawbacks include difficulty of substrate delivery and stoichiometry and potential for cytotoxicity (Wu and Jiang, 2022). BRET-based Epac1-, PKA-, and nano lantern-based cAMP sensors have been developed to investigate cAMP generation (Masri et al., 2008; Hunter et al., 2017; Valkovic et al., 2018).
All plasmid-based cAMP sensors have advantages and disadvantages, with respect to ease of introduction without toxicity, targetability of expression in specific subcellular locations, temporal and spatial resolution of cAMP detection, cAMP detection ranges, and buffering from metabolic processes or post-translational modifications in living cells or tissues. Rapid cAMP binding, instant transmission of signals, and rapid reversibility of the binding event would denote an ideal cAMP sensor (Paramonov et al., 2015). Further optimization is needed for detection of low unstimulated cAMP levels in sensitive primary cells or tissues. Available cAMP biosensors have recently been reviewed in detail (Kim et al., 2021).
Regulatory post-translational modifications of AC5 and AC6
AC5/6 phosphorylation by PKA and PKC
PKA and PKC modify AC5 and AC6 activity by phosphorylating serine (Ser) or threonine (Thr) residues. In the heart, PKA regulates metabolism, gene transcription, ion fluxes, and contraction (Colombe and Pidoux, 2021) and also acts as a feedback inhibitor for both AC5 and AC6 through inhibitory phosphorylation near the end of the C1b domain, resulting in the desensitization of AC activity (Sadana and Dessauer, 2009). Stimulation of AC6 was lost after treatment of PKA even in the presence of high concentrations of active Gαs (Chen et al., 1997). Mutational analysis indicates Ser674 is the target of phosphorylation and inhibition of AC6 by PKA in intact cells (Figure 6) (Beazely and Watts, 2006; Chen et al., 1997). AC5 possesses 14 putative PKA phosphorylation sites, including a Ser 788 that corresponds to Ser674 in AC6; however, a specific PKA phosphorylation site in AC5 has not been confirmed (Iwami et al., 1995; Chen et al., 1997).
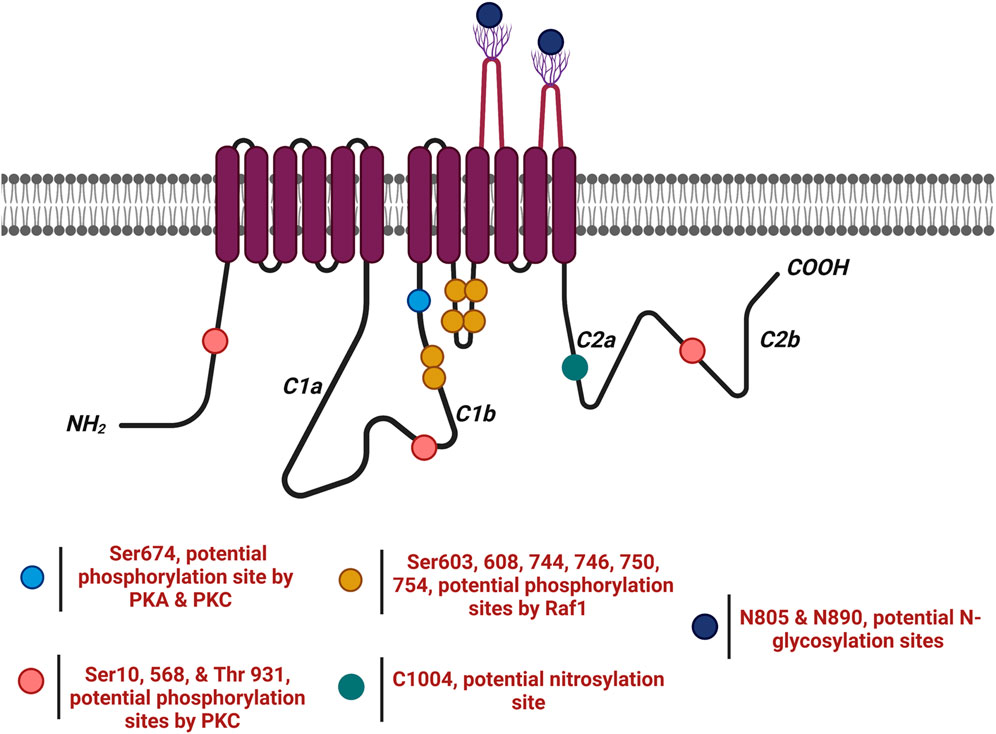
Figure 6. AC6 undergoes several post-translational modifications ( ). AC6 is phosphorylated by PKA and PKC at Ser674 (
). AC6 is phosphorylated by PKA and PKC at Ser674 ( ); phosphorylated by PKC at Ser10 and 568 and Thr931 (
); phosphorylated by PKC at Ser10 and 568 and Thr931 ( ); S-nitrosylated by NO at C1004 (
); S-nitrosylated by NO at C1004 ( ); Raf1 on Ser603, 608, 744, 746, 750, and 754 (
); Raf1 on Ser603, 608, 744, 746, 750, and 754 ( ); and glycosylated at N805 and N890 on extracellular loops 5 and 6.
); and glycosylated at N805 and N890 on extracellular loops 5 and 6.
While both AC5 and AC6 isoforms are inhibited through phosphorylation by PKA, PKC inhibits only AC6 (Kamide et al., 2015; Liu et al., 2022). Previous studies have shown that AC2, AC3, and AC5 can be stimulated by PKC, while AC6 activity is inhibited (Lai et al., 1997; Defer et al., 2000). Phosphorylation of AC5 by PKCα or PKCζ enhances basal activity as well as FSK- or Gαs-stimulated cAMP accumulation; PKC-ζ phosphorylation of AC5 results in a 20-fold increase in AC activity (Kawabe et al., 1996). In the heart, generation of phosphatidyl-inositol-3,4,5 triphosphate via hormonal or growth factor activation of phosphatidyl-inositol 3-kinase can activate PKC-ζ, thus directly activating AC5 production of cAMP (Hanoune and Defer, 2001). In contrast, PKC activators have either no effect or inhibitory effect on AC6 activity (Chen and Iyengar, 1993; Jacobowitz et al., 1993; Lai et al., 1997; Lai et al., 1999). Mutational analysis of the N-terminus of AC6 as a regulatory domain showed that elimination of residues from 1 to 86 or a single mutation of Ser10 prevents phosphorylation and inhibition of AC6 by PKC (Figure 6) (Lai et al., 1999). Subsequent studies also identified AC6 C1 Ser 568 and 674 as well as AC6 C2 Thr931 as inhibitory targets of PKC, suggesting that phosphorylation of this complex of Ser and Thr might trigger a conformational change in the catalytic core, changing AC6 catalytic activity (Lin et al., 2002).
AC5/6 regulation by glycosylation
AC6 can be glycosylated on two asparagine residues, namely, N805 and N890, on extracellular loops of TM2 (Figure 6) (Wu et al., 2001). The glycosylation of AC6 results in alteration of not only its catalytic activity but also its regulation by Gαi or by PKC. Inhibition of glycosylation by tunicamycin impairs FSK-stimulated AC6 activity, and mutation of glycosylation sites resulted in significantly lower FSK-, Mn2+-, and Mg2+-stimulated enzyme activities than did wild-type AC6, suggesting that glycosylation may be necessary for maintenance of AC6 activity (Wu et al., 2001). AC5 glycosylation has not been verified.
AC5/6 regulation by nitrosylation
Cardiac function, airway, and vascular tone, as well as regulation of immunological defense and neuronal plasticity, are regulated through nitric oxide or reactive nitrogen species (Bhatia et al., 2021). NO-mediated S-nitrosylation of AC5/AC6 plays a crucial regulatory role in physiological processes of cardiovascular function. Previous studies showed that NO, independent of its action on the guanylyl cyclase (GC) pathway, inhibits cAMP in Dictyostelium discoideum, indicating that NO can have a direct regulatory effect on ACs via modification of an AC regulatory domain (McVey et al., 1999). Agonist-stimulated cAMP accumulation is inhibited when N18TG2 neuroblastoma cells (Tao et al., 1998) or cardiac myocytes (Joe et al., 1998; Vila-Petroff et al., 1999) are treated with NO or NO donors (Tao et al., 1998). This inhibition is not dependent on the effect of Gαi (McVey et al., 1999) nor on PDE activity (Watson et al., 2001). NO or NO donors were shown to selectively decrease FSK-stimulated AC5/6 activity but not AC1 or AC2 (Hill et al., 2000), while calmodulin stimulation of AC1 is inhibited by NO (Duhe et al., 1994). NO suppression of hormonal or FSK-stimulated AC activity in neuroblastoma plasma membranes does not require CaM (McVey et al., 1999) and directly inhibits FSK-stimulated AC5 and AC6 activity (Hill et al., 2000).
The presence of caveolae, coincidentally the location of endogenous NO generation by endothelial nitric oxide synthase (eNOS), is required for NO inhibition of AC6 activity. Inhibition of AC5/6 by NO relies upon their localization in lipid rafts with caveolin signaling complexes (Ostrom et al., 2004). While lipid raft depletion with β-cyclodextrin prevented the activation of AC activity by βAR and Gαs, it has no influence on the prostanoid receptors, which are located outside of caveolin-rich microdomains and can still activate AC. Both native cardiac myocytes and pulmonary artery endothelial cells overexpressing AC6 are inhibited by the NO donor S-nitroso-N-acetylpenicillamine (SNAP), inhibiting both basal and FSK-stimulated cAMP production (Ostrom et al., 2004). This process is subject to reversal by reducing agents, indicating the involvement of cysteine residue(s) as the target for S-nitrosylation (McVey et al., 1999; Ostrom et al., 2004). The reaction between NO and superoxide (O2−) results in the production of reactive nitrogen oxygen species (RNOS) capable of altering a broader variety of biomolecules than NO itself (Ridnour et al., 2004). At higher levels of O2−, NO inactivation is followed in turn by generation of the potent and short-lived oxidant peroxynitrite (ONOO−/ONOOH), which can directly react with metal ions and thiols (Piacenza et al., 2022). ONOO− contributes more to S-nitrosylation of adjacent proteins than does NO (Maccarrone et al., 2000). It is also proposed that inhibition of AC by NO may be through S-nitrosylation caused by the reaction of another NO intermediate, nitrosonium (NO+), with cysteine residues (Tao et al., 1998). In a quantitative mass spectrometry screening investigation of modified cysteines utilizing a bioorthogonal cleavable-linker switch technique, AC6 featured among proteins identified as S-nitrosylated (Mnatsakanyan et al., 2019). AC6 inhibition due to S-nitrosylation was also demonstrated in pulmonary arteries (Sikarwar et al., 2018). Identification of AC6 cysteines susceptible to S-nitrosylation has been explored using site-directed mutagenesis of cysteines identified by bioinformatics analysis to reside within an SNO motif (Jaggupilli et al., 2018; Bhagirath et al., 2022). Mutation of cysteine 1004, located in the conserved C2 domain of AC6 near the Gαs docking position (Figure 6), decreases the basal and stimulated activity of AC6, indicating the importance of this residue in AC6 for intact catalytic activity and also susceptibility to inhibition if nitrosylated (Bhagirath et al., 2022).
AC5/6 regulation by other PTMs
AC6 may be phosphorylated through receptor tyrosine kinases (RTKs); Ser residues 603, 608, 744, 746, 750, and 754 have been implicated following RTK activation by IGF-1 or tyrosine phosphatase inhibition with sodium orthovanadate (Figure 6). Augmentation of AC6 catalytic function following RTK activity is inhibited by endogenous p74raf−1 activity, but not by inhibitors of ERK, PKC, PKA, or PI3 kinase activity (Tan et al., 2001).
Role of AC5 in cardiovascular function
In this section, we review the studies reporting the role of AC5 in cardiomyopathies induced by catecholamine stress including chronic isoproterenol stimulation, aging, and pressure overload. Upregulated AC5 mRNA expression in spontaneously hypertensive rats suggests complex regulation of LV hypertrophy in hypertension (Vatner et al., 2013). Myocardial AC5 mRNA increases from 5 to 12 weeks in spontaneously hypertensive rats, associated with the development of LV hypertrophy (Fujino et al., 2003). On the other hand, AC5KO mice have diminished sympathetic and parasympathetic responses and disrupted Ca2+-mediated cardiac regulation. Both basal and isoproterenol-stimulated AC activity are attenuated at 30%–40% in the cardiac membranes of AC5KO mice, with no compensatory increase in other AC isoforms; this reduction in AC activity does not alter cardiac function at baseline but compromises the LV inotropic response to adrenergic stimulation (Okumura et al., 2003a). In pressure overload, while there was no difference between AC5WT and AC5KO in cardiac muscle mass at baseline, AC5KO protected the heart from deleterious effects of pressure overload on LV ejection fraction, through restriction of myocardial apoptosis via upregulation of BCl-2 (Okumura et al., 2003b). Others have demonstrated enhancement in basal LV function in AC5KO as well as impairment in the responsiveness of LV to βAR stimulation (Tang et al., 2006). AC5KO mice also showed better heart function following chronic catecholamine stimulation, again through reduction in cardiac apoptosis, due to increased Bcl-2 expression and Akt signaling (Okumura et al., 2007). In contrast to the outcomes after prolonged isoproterenol infusion in AC5WT and AC5KO mice, the ejection fraction response of the LV to an abrupt isoproterenol challenge was lowered in AC5KO, which is in line with the downregulation of AC5 catalytic activity. These data indicate that AC5 impairs survival signaling after long-term catecholamine infusion, while AC5 deletion improves cardiac desensitization, suggesting a novel strategy for heart failure therapy.
Zhang et al. investigated ways in which selected Gαs-coupled receptors (GsPCRs) regulate cardiomyocyte viability by generating distinct signaling complexes governing pro-survival versus pro-death signaling. It was revealed that among the five GsPCRs studied, stimulation of β1AR and histamine-H2-receptor (H2R) has negative effects on the survival of cardiomyocytes, mediated through cAMP production by AC5 but not AC6 and via PKA activity stimulating pannexin-1 to release ATP into the extracellular space. On the other hand, activation of pro-survival GsPCRs adenosine-A2-receptor (A2R), calcitonin-gene-related-peptide-receptor (CGRPR), or relaxin-family peptide-receptor 1 (RXFP1) results in protective effects on cardiomyocyte survival as a function of cAMP generation by AC6, resulting in activation of cAMP efflux pumps. These findings indicate that selection of AC5 versus AC6 by GsPCRs determines cAMP localization which controls cAMP fate, thus altering cardiomyocyte survival (Zhang et al., 2023).
In an examination of heart rate variability during transient microgravity in parabolic flight, autonomic dysregulation became worse in AC5KO mice, while heart rate stability improved as a function of AC5 overexpression, indicating that AC5 may improve autonomic regulation (Okumura et al., 2008). It was further shown that AC5 activity is required to achieve constant responses in the low- and high-frequency ratio or normalized high frequency, two markers of sympathetic and parasympathetic activity, respectively (Bai et al., 2012). Thus, while inhibition of AC5 is beneficial for preventing cardiac myocyte myocardial apoptosis induced by excessive βAR stimulation, activation of this isoform may be advantageous in acute heart failure with low rate.
Transgenic mice overexpressing both AC5 and Gαq show efficient βAR-stimulated AC activity and cardiac contractility, but their hearts also appear pathologically fibrotic and hypertrophic (Tepe and Liggett, 1999). Microarray analysis of these hearts showed upregulation of various genes involved in pressure overload LV hypertrophy (Park et al., 2011). Examining genes relevant to ventricular hypertrophy upregulated in AC5 transgenic hearts even at baseline, transcription factor binding analysis revealed enrichment for the binding sites of nuclear factor of activated T-cells (NFAT), vital for the development and progress of cardiomyocyte hypertrophy (Park et al., 2011). NFAT binding determines the expression of cytoskeletal proteins (Schubert et al., 2003). So AC5 overexpression can mediate calcineurin–NFAT signaling involved in the development of LV hypertrophy (Park et al., 2011).
AC5KO mice also demonstrate increased physical performance, mediated through upregulation of the sirtuin-1 (SIRT1) pathway and regulating the antioxidant enzyme manganese superoxide dismutase (MnSOD) in the heart and liver of ACKO mice (Yan et al., 2012). It is proposed that SIRT1 is inhibited by AC5, leading to disruption of an interaction between SIRT1 and forkhead box O3 (FoxO3a), which eventually decreases MnSOD expression, thereby augmenting oxidative stress. The increase in MnSOD in myocytes subject to adenoviral AC5 KO is abolished by inhibition of either MEK or sirtuin, indicating MnSOD upregulation by both the mitogen-activated protein kinase kinase/extracellular signal-regulated kinases (MEK/ERK) and SIRT1/FoxO3a pathways (Chester and Watts, 2007; Lai et al., 2013). AC5KO mice are found to be resistant to cardiac stress with an enhanced median life span of roughly 30%, with protection from bone demineralization, and decreased susceptibility to fractures or aging-induced cardiomyopathy (Yan et al., 2007). The longevity, healthful aging, and stress resistance detected in AC5KO mice were correlated to diminished cAMP and PKA, resulting in activation of the Raf/MEK/ERK pathway, leading to the enhanced level of MnSOD (Yan et al., 2007). Additionally, AC5KO appears to increase NO signaling, demonstrating another mechanism by which AC5 may antagonize beneficial pathways induced by exercise (Guers et al., 2017). Two highly expressed genes encoding glutathione S-transferase (Gstk1 and Gstm) were downregulated in hypertrophic hearts, importantly because glutathione S-transferase plays the role of an antioxidant by conjugating glutathione on various substrates to protect the heart from oxidative stress (Cho et al., 2003; Yan et al., 2012).
Overall, these data place AC5 in a crucial role regulating life span and cardiac stress resistance. Figure 7 summarizes AC5 signaling in the context of cellular antioxidant imbalances in AC5WT and AC5KO. Studies delineating the role of AC5 in cardiovascular function are highlighted in Table 1.
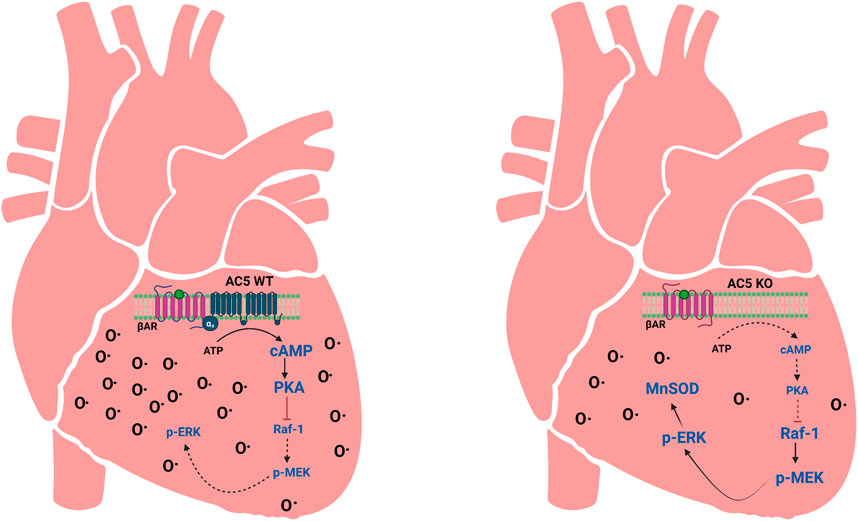
Figure 7. Depiction of AC5 signaling effects on cellular antioxidant imbalances in AC5WT and AC5KO hearts. When AC5 is disrupted (AC5KO), there is less βAR-stimulated cAMP, which decreases PKA activity. Lack of PKA leads to the activation of the Raf-1/MEK-ERK signaling pathway to increase the expression of MnSOD. This results in attenuation of oxidative stress. MnSOD antioxidant defense is not as present in AC5WT. O• indicates oxidative stress. Adapted from (Chester and Watts, 2007); image designed using BioRender.
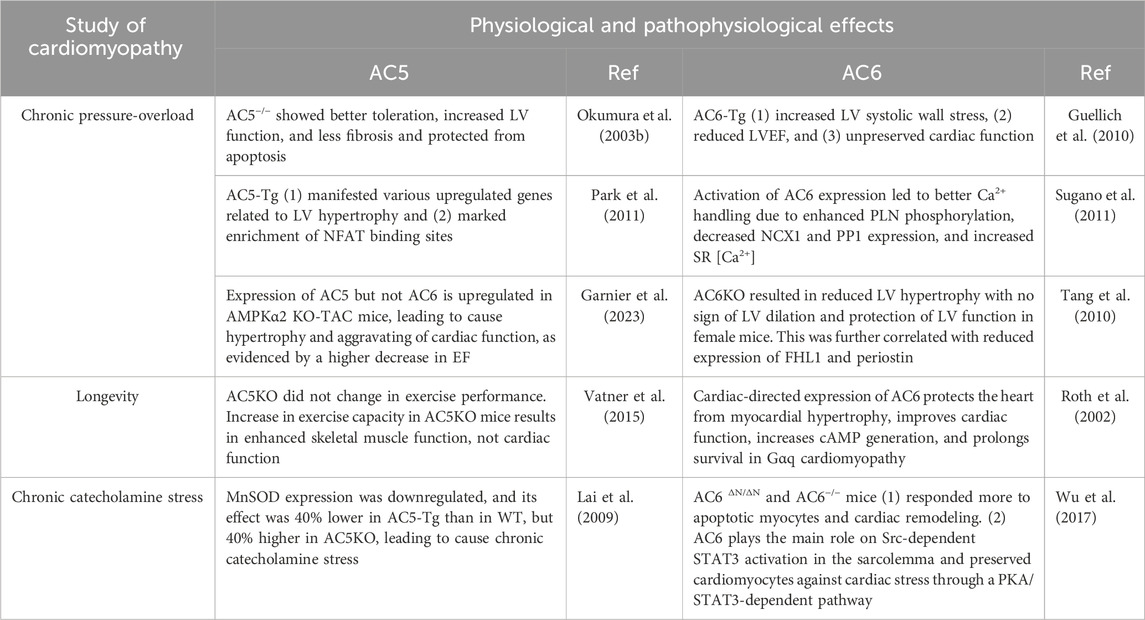
Table 1. Physiological and pathophysiological effects of AC5 and AC6.
Role of AC6 in cardiovascular function
In contrast to AC5, several studies have shown that increase in AC6 expression is beneficial for the failing heart, preserving LV contractile function and reducing dilation and dysfunction in hearts showing pressure overload (Takahashi et al., 2006; Sugano et al., 2011). The protective effect of AC6 in the heart is postulated as cAMP pathway dependence and independence (Tang et al., 2004; Gao et al., 2008; Gao et al., 2009; Gao et al., 2017). AC6 overexpression prevents cardiac hypertrophy, fibrosis, and cardiomyopathy (Roth et al., 1999; Tang et al., 2013), while cardiac-directed expression of catalytically inactive AC6 restored the detrimental effects of sustained catecholamine infusion, through diminished myocardial cAMP production (Gao et al., 2017). In addition, in ischemic cardiomyopathy, cAMP production and systolic and diastolic LV function are enhanced after activation of AC6 expression (Lai et al., 2008). AC6KO mice exhibit reduced cAMP production and have significantly higher mortality compared to AC6WT, but remain susceptible to βAR stimulation-induced cardiomyopathy, compromised electrophysiological characteristics including diminished longitudinal conduction velocity, and impaired connexin 43 phosphorylation at Ser 368, which may be part of the mechanism of ventricular dysfunction (Tang et al., 2013). Cardiac-directed AC6 overexpression with Gαq expression triggered improved βAR-stimulated AC activity, cAMP generation, and cardiac function in vivo and ex vivo, with no sign of hypertrophy and fibrosis (Roth et al., 1999), as well as improved Ca2+ handling and LV contractility manifested by ejection fraction, pressure development rate, and slope of the LV end-systolic pressure–volume relationship in aging mice (Tang et al., 2011). In 23-month-old rats, AC6 expression improved SR Ca2+ storage, while AC6 expression in 7-month-old mice did not show any difference in LV function and Ca2+ uptake (Tang et al., 2011). While cardiac troponin I (cTnI) phosphorylation diminishes with cardiac age (Jiang et al., 1993), improved LV function by AC6 is associated with phosphorylation of cTnI at Ser 23/24, which regulates thin filament function and thus contractility (Lai et al., 2008; Tang et al., 2011). While AC6 expression increases PKA activity and SR Ca2+ uptake (Tang et al., 2011), deletion of AC6 results in diminished PKA activity, phospholamban (PLB) phosphorylation, and decreased SR Ca+2-ATPase activity 2a (SERCA2) affinity toward Ca+2 in failing hearts (Takahashi et al., 2006; Tang et al., 2008). Increased AC6 content increases the expression of activating transcription factor-3, which extinguishes PLB promoter activity, resulting in reduced PLB expression (Gao et al., 2004).
Enhanced AC6 expression is also correlated with increased nuclear phospho-Akt promoting phosphorylation of Akt at Ser473 and Thr308. This process appears to be independent of PKA or βAR stimulation (Gao et al., 2008). Akt activity is reversely regulated by PH domain leucine-rich repeat protein phosphatase (PHLPP), responsible for dephosphorylation of Akt at Ser473. AC6 inhibits PHLPP activity in cardiomyocytes, bringing about high levels of Akt phosphorylation. This PHLPP suppression is, however, rescued rapidly by isoproterenol and FSK stimulation, leading to significant dephosphorylation of Akt at Ser473, but not Thr308. As PLB is an Akt target, active phospho-Akt increases PLB activity and thus improves sarcoplasmic Ca2+ cycling (Gao et al., 2009). Figure 8 gives an illustrated summary of the effects of AC5 and AC6 addition or deletion in vivo.
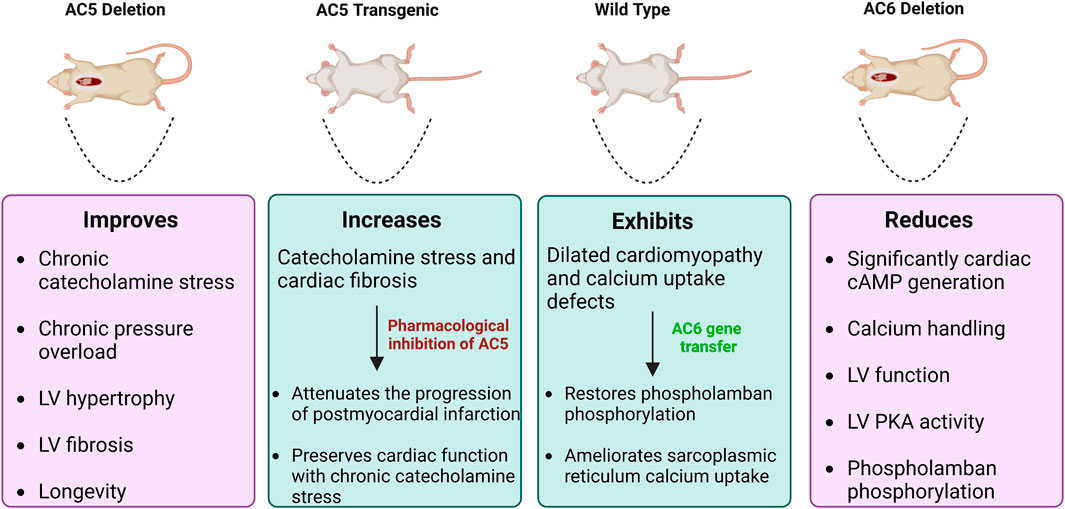
Figure 8. Effects of AC5 and AC6 overexpression or deletion on heart diseases in vivo. WT and transgenic mice subject to heart failure by catecholamine stress. Mice with deletion of AC5 demonstrate beneficial effects, whereas AC6 deletion is deleterious. AC5 transgenic mice have increased catecholamine stress and cardiac fibrosis. AC5 inhibitors rescue this condition, decreasing the progression of heart disease. Dilated cardiomyopathy in WT mice can be recovered by AC6 gene transfer.
As AC6 shows remarkable physiological benefits for the heart, while its content and function are diminished in the failing heart (Ostrom et al., 2022), there has been increased interest in the efficacy and safety of adenoviral delivery of AC6 in patients with heart failure (Hammond et al., 2016) (AC6 Clinical Trials). In a mouse model, the AC6 C1–C2 construct was shown to effectively improve cardiac dysfunction caused by prolonged βAR stimulation (Roth et al., 2002). Although AC6 transgenic mice with cardiac-directed C1–C2 expression exhibited less cAMP production, they preserved normal cardiac function through ameliorated Ca2+ handling and tolerated sustained isoproterenol infusion and pressure overload without detrimental effects on LV contractility (Gao et al., 2016; Tan et al., 2019). Preclinical analysis of AC6 gene therapy recently transitioned into clinical trials. The phase 2 results for one-time AC6 gene transfer in 56 adult patients with symptomatic heart failure (ischemic or non-ischemic) and an ejection fraction of 40% resulted in safely ameliorated LV function above standard heart failure therapy (Hammond et al., 2016). The subsequent FLOURISH trial, a double-blinded placebo-controlled, multicenter phase 3 trial of 536 patients, aims to decrease heart failure hospitalization rates and improve ejection fraction, while minimizing adverse events after intracoronary injection of the human adenovirus 5 encoding human AC6 (Ad5. hAC6) gene in patients with heart failure and diminished LV ejection fraction; patient recruitment is ongoing (Penny et al., 2018).
Studies contextualizing the AC6 role in cardiovascular function are highlighted in Table 1.
Roles of AC5 and AC6 in vasculature
AC activity in vascular cells regulates vascular reactivity, apoptosis, hypertrophy, and proliferation (Gros et al., 2006). The distribution of AC isoforms varies in the vasculature compared to the myocardium. Based on RT-PCR studies, ACs 3, 5, and 6 are expressed in the adult rat aorta (Ostrom et al., 2002), while in the perinatal period, the ductus arteriosus expresses more AC2 and 6 than does the aorta (Yokoyama et al., 2010). The pulmonary artery expresses AC6 primarily, followed by ACs 7, 9, and 3 (Sikarwar et al., 2018). In vascular myocytes, AC6 is the main AC isoform involved in βAR-mediated cAMP/PKA signaling and activation of the KATP current, important for harmonizing the membrane potential and regulating vascular tone (Nelson et al., 2011). Subjects who carry the genetic variation ADCY6 A674S have increased blood pressure, with a hyperdynamic cardiac profile that is compatible with the effect of elevated AC function (Hodges et al., 2010). Vasodilation is antagonized by Gαq-mediated signaling, in part by direct Ca2+ inhibition of AC5 and AC6 (von Hayn et al., 2010).
Changes in AC activity have been related to the development of diabetes, heart failure, and hypertension (Matsumoto et al., 2005; Hodges et al., 2010). Abnormal arterial myocyte contractility, in addition to compromised endothelium-dependent vasodilation, is an important contributing factor to vascular complications including altered myogenic tone, both in diabetic mice and in patients with diabetes (Montero et al., 2013; Sena et al., 2013; Tykocki et al., 2017; Syed et al., 2019). The contractile state of smooth muscle cells in the vessel wall determines the vascular tone, measured by a balance between the effects of vasoconstrictor and vasodilator signaling pathways, inclusive of adrenergic receptors and ACs (Shi et al., 2020). Physiological targets of ACs and downstream PKA include potassium channel phosphorylation, inducing hyperpolarization and vasodilation (Nelson et al., 2011). In resistance arteries and arterioles, the myogenic tone generated through the pulsatile stretch of the vascular wall modulates baseline smooth muscle contraction (Tykocki et al., 2017); in diabetic hyperglycemia, altered expression or function of potassium channels is linked to increased myogenic tone (Syed et al., 2019). Elevated glucose can trigger Gαs signaling (Lemaire et al., 2004) but may drive vasoconstriction through glucose-induced cAMP production via AC5, which results in the activation of an anchored PKA pool, and in turn, phosphorylates the L-type Ca2+ channel pore-forming CaV1.2 subunit at Ser 1928 (Nystoriak et al., 2017; Prada et al., 2019; Syed et al., 2019). These interactions cause potentiation of L-type Ca2+ channel activity, enhanced [Ca2+]i, and vasoconstriction (Figure 9). Supporting this, interruption of AKAP5 function in arterial myocytes prevents cAMP generation in response to either increased glucose or selective purinergic P2Y11 agonist NF546; in AKAP5-null arterial myocytes or arteries, there is no clustering of P2Y11/P2Y11-like receptors, AC5, PKA, and CaV1.2 into nanocomplexes at the plasma membrane; therefore, glucose- and NF546-induced potentiation of L-type Ca2+ channels and vasoconstriction does not occur (Prada et al., 2020). These data implicate AKAP5 and AC5 in the spatial confinement of cAMP signaling induced by elevated glucose via activation of P2Y11/P2Y11-like receptors in arterial myocytes (Prada et al., 2019; Prada et al., 2020).
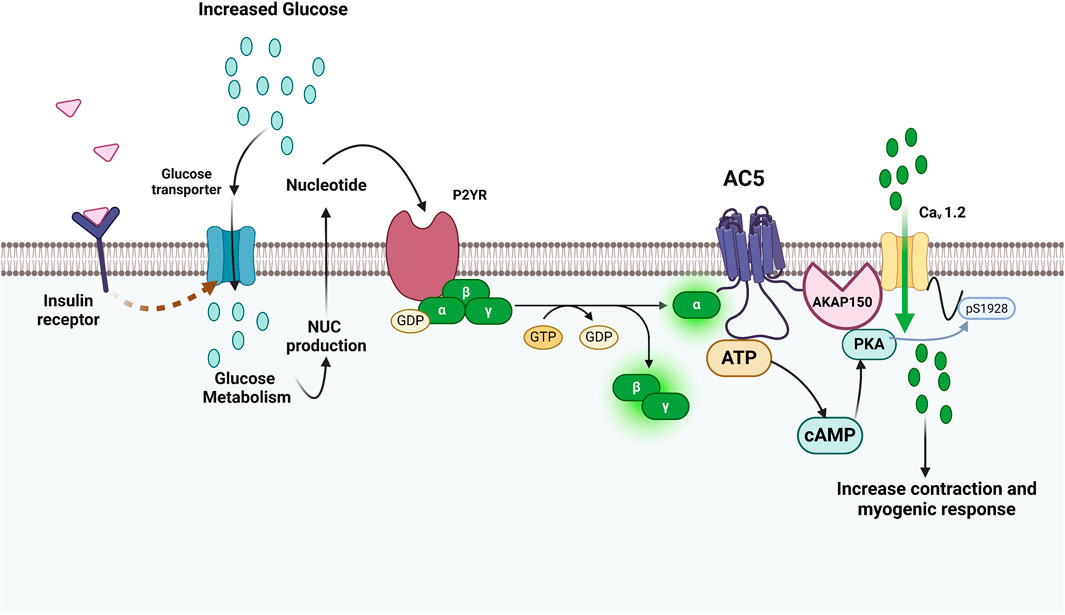
Figure 9. Involvement of AC5-mediated localized cAMP generation in diabetes and extracellular glucose activation of L-type Ca2+ channels and vasoconstriction. The illustration shows how glucose regulates L-type Ca2+ channel activity and vascular reactivity in an AC5-dependent manner. Increases in extracellular glucose can trigger P2YR linked to Gαs signaling through extracellular nucleotide signaling because of transport and metabolism. Because AC5 and CaV1.2 are close in proximity, this cAMP microdomain may stimulate a pool of AKAP150-anchored PKA that is jointly linked to CaV1.2, making it to become more phosphorylated at Ser1928 and increasing channel function. Adapted from (Syed et al., 2019); image designed using BioRender.
ACs in endothelial cells play a role in vascular permeability. Prostacyclin-mediated signaling via AC6 (but not AC5) forms part of a feedback circuit that increases endothelial barrier function. Endothelial cells overexpressing AC6 have increased prostacyclin response, reducing the permeability of the endothelial barrier. In human umbilical vein endothelial cells, adenoviral-mediated gene transfer of AC6 increased prostacyclin receptor-stimulated cAMP synthesis and concurrently decreased thrombin-stimulated increases in endothelial cell barrier function (Bundey and Insel, 2006).
Role of ACs in cardiac automaticity
The sinoatrial node (SAN) is a crescent-like shaped cluster of myocytes split by connective tissue, spread over a few millimeters (Kashou et al., 2017), located at the convergence of the superior vena cava opening and the crista terminalis in the upper wall of the right atrium. The SAN comprises coordinated actions of pacemaker cells capable of generating a cyclic electrical impulse (Kashou et al., 2017). Cardiac arrhythmia due to the abnormality of the SAN can affect annually up to 1 per 1000 adults >45 years of age (John and Kumar, 2016). The SAN is the main regulator of heart rate and is regulated by βAR signaling (Irisawa et al., 1993; Tsutsui et al., 2021). While the expression of a number of AC isoforms (types 1, 2, 3, 5, 6, 8, and 9) has been identified in rabbit SAN, AC1 and AC8, two of the Ca2+-activated AC isoforms, are predominantly distributed in atrial and SAN cells (Mattick et al., 2007; Younes et al., 2008; Robinson et al., 2021). In contrast, a recent study showed AC1 and AC6 but not AC8 expressed in SAN cells at the transcript and protein levels (Ren et al., 2022).
Ca2+ is a vital modulator of pacemaker potential via the Ca2+ clock (Maltsev and Lakatta, 2012), where the ryanodine receptor 2 (RyR2) facilitates the spontaneous release of Ca2+ from the SR, which in turn pushes Ca2+ to be released from the cytosol via the Na+–Ca2+ exchanger (Maltsev and Lakatta, 2012; Ren et al., 2022). Adrenergic control of the cardiac pacemaker current has been ascribed to AC1 (Mattick et al., 2007). AC1 mediates cAMP signaling in the SAN, in a functional microdomain with CAV3, hyperpolarization-activated cyclic nucleotide-gated 4 (HCN4), Cav1.2, and RyR2. cAMP released by AC1 leads to elevation of intracellular Ca2+ via Ca2+ channels, which triggers a positive feedback to AC1 and negative feedback to AC5/6 (Ren et al., 2022). While cardiac-specific overexpression of AC8 transgenic mice in the SAN is reported to augment the heart rate and rhythm (Moen et al., 2019), others found no differences in automaticity, basal heart rate, or isoproterenol responses in AC8-null mice compared with wild-type (Ren et al., 2022). In guinea pig atrial myocytes, sarcoplasmic reticulum type 2 inositol trisphosphate (IP3) receptors colocalize with AC8, with AC1 localized proximally. Functional AC1 and AC8 are required for the positive chronotropic effect of phenylephrine on the SAN, and activity of both AC1 and AC8 plus PKA is required for the effect of IP3 on cellular Ca2+ transients (Capel et al., 2021).
Pacemaker current is generated by hyperpolarization-activated cyclic nucleotide-gated channels (HCN). To assess the role of Ca2+ homeostasis in autonomic regulation, AC1 and AC6 were expressed in cultures of spontaneously beating neonatal rat ventricle cells co-expressing HCN2. AC1, but not AC6 expression, increased intracellular cAMP and automaticity; AC1-mediated cAMP generation was resistant to β-adrenergic blockade, but the HCN2 response to an adrenergic agonist in the presence of AC1 (but not AC6) was sensitive to Ca2+ chelation, implying that the effect of Ca2+ homeostasis on the adrenergic regulation of the pacemaker rate could be accounted by the existence of a Ca2+ sensitive AC isoform (Kryukova et al., 2012; Robinson et al., 2021). However in a study of in vivo adenoviral gene transfer of AC6 in a porcine atrioventricular node block model, the AC6 injected group developed an escape rhythm of ∼100 beats/min originating at the LV injection site, while control animals had RV escape rhythms, suggesting that biological pacemaker activity could also be triggered by AC6 (Ruhparwar et al., 2010).
AC5 and AC6 as potential drug targets for cardiovascular disease
Although βAR agonists and antagonists have therapeutic effectiveness for heart failure treatment, there are still patients who do not respond effectively; thus, heart failure is the most common cause of mortality worldwide (Capote et al., 2015). A weak cardiac response to catecholamine stimulation is a characteristic of the heart failure phenotype; hence, stimulation of the adrenergic pathway has been targeted to increase cardiac function. However, βARs can undergo downregulation after prolonged stimulation by agonists or antagonists, which leads to a reduction in their cell surface density (tachyphylaxis), modifications in subtype composition, or increase in PKA or G-protein-coupled receptor kinase (GRK) activity, resulting in uncoupling of βAR from G proteins (desensitization) (Mahmood et al., 2022). βAR desensitization also serves as a compensatory mechanism, which may disrupt cardiac function and promote arrhythmias (Ho et al., 2010).
In contrast to GPCRs, AC5 and AC6 are less likely subject to desensitization after sustained ligand exposure (Pierre et al., 2009). The AC5 and AC6 isoforms have been indirectly targeted by GPCR agonists or antagonists and PDE inhibitors due to their action as the central relay site that assembles and amplifies a wide variety of signals (Pavan et al., 2009). AC5 and AC6 have pivotal roles in cardiac disease. Despite the promising results from knockout and transgenic mice, suggesting that AC5 and AC6 can be potential drug targets, advancements to selectively and therapeutically target these isoforms have been hindered by their structural similarity. However, many positive steps in this direction are reviewed below.
AC5 and AC6 activators for the treatment of heart failure
The most potent stimulus for increasing the cardiac output is initiated by sympathetic nervous system activation through the activation of βAR and Gαs, which in turn activates AC5 or AC6 to enhance cardiac contractility and rate. In the failing heart, production of basal and stimulated cAMP is diminished, but driving cardiac cAMP levels via adrenergic agents can result in higher long-term mortality (Bristow et al., 1982; Packer et al., 1991). FSK is a well-known activator of all transmembrane ACs, but its lack of selectivity impedes its clinical application; in addition to ACs, it also targets nuclear receptors, ion channels, glucose transporters, and P-glycoprotein multidrug resistance transporters (Dessauer et al., 2017). Clinically, in patients with heart failure, forskolin decreases ventricular filling pressures and vascular resistance while increasing cardiac output, ejection fraction, and stroke volume (Bristow et al., 1984). However, novel FSK-derived compounds have been synthesized that selectively target AC5 and AC6 isoforms (Pavan et al., 2009).
To design selective AC5 and AC6 activators, maintaining hydrogen bonding of FSK C1-OH, C7-acetyl, and C9-OH has been deemed crucial (Onda et al., 2001). There has also been specific focus on modification of C6 and C7 positions, due to a large open area apparent from AC crystallographic studies (Tesmer et al., 1997), suggesting that modification on these FSK positions may increase AC activity with selectivity and/or specificity. Figure 10 illustrates the possible sites of modifications for designing novel AC5 or AC6 activators. It is important to note that many of the earlier studies which established FSK positions suitable for modification evaluated only a subset of the AC isoforms. Testing of candidate compounds on each AC isoform remains necessary to determine which site is particular to each isoform.
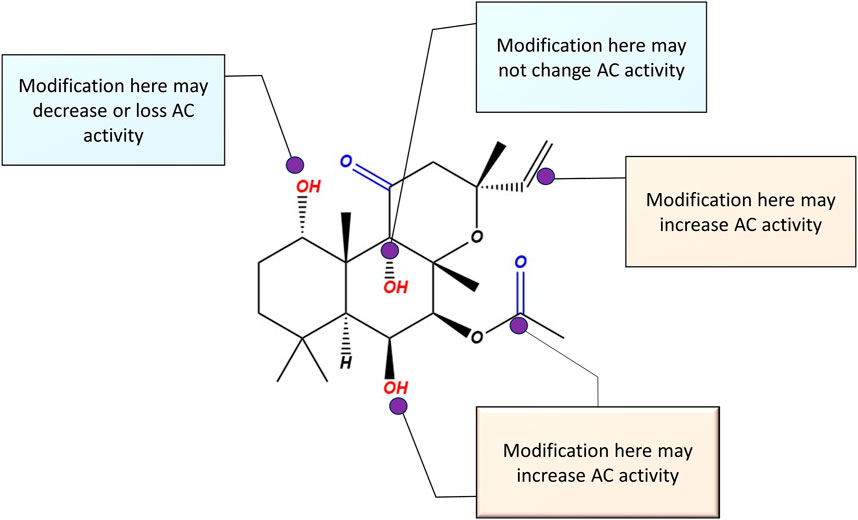
Figure 10. Possible modification sites on the parent compound forskolin molecule, for the design of selective AC5 or AC6 activators. Based on previous studies, any modification on C (1) reduces AC activity. Modifications on C (6) and (7) have the potential to enhance AC5 and AC6 activity.
The development of AC5 activator 6-[3-(dimethylamino)propionyl]FSK (NKH477, FD5, or colforsin daropate hydrochloride) (Toya et al., 1998) grabbed the attention of scientists working on AC drug discovery. Colforsin is a potent water-soluble derivative of the C6 position of FSK, initially approved in Japan for the treatment of advanced congestive heart failure due to the elevation of cAMP in cardiac tissue, resulting in the enhancement of cardiac contractility. From the standpoint of AC isoform selectivity, colforsin activates AC5 > AC2∼AC3 (Toya et al., 1998). Using the isolated perfused canine heart, the chronotropic, inotropic, coronary vasodilatory effects, and AC activity of colforsin were compared to those in catecholamines isoproterenol, dopamine, and dobutamine. All the drugs demonstrated positive chronotropic, inotropic, and coronary vasodilator effects. Colforsin cardiovascular actions were in the following order: coronary vasodilation >> positive inotropy > positive chronotropy, while isoproterenol, dopamine, and dobutamine evinced positive inotropy >> coronary vasodilation > positive chronotropy. While inotropic effects are desirable, chronotropic agents have potential arrhythmogenic effects and may impair cardiac filling; however, at doses with comparable inotropic effects, colforsin demonstrated a higher positive chronotropic effect than catecholamines or PDE inhibitors, with ventricular tachycardia triggered equally by colforsin and isoproterenol. Nevertheless, the coronary vasodilator activity of colforsin was more potent. The strong coronary vasodilator function of colforsin may therefore be applicable for the treatment of heart disease where coronary blood flow is limited and βAR-dependent signaling is also downregulated (Yoneyama et al., 2002). In a model of canine respiratory acidosis causing cardiac dysfunction, colforsin increased cardiac rate and decreased systemic vascular resistance to the same degree as did dobutamine, without attenuation by acidosis (Itami et al., 2019). Colforsin has also demonstrated pulmonary artery vasodilation (Yokochi et al., 2010), which is mediated largely through AC6 rather than AC5 (Sikarwar et al., 2018), indicating that colforsin may not be AC5-selective (Jaggupilli et al., 2018). 6-[3-(Dimethylamino)propionyl]-14–15-dihydro-FSK (FD6) is another FSK derivative, structurally similar to colforsin, but with reduced 14–15 alkyne bond, that showed AC5 and AC6 selectivity (Yokoyama et al., 2010). The lack of complete AC5 versus AC6 selectivity of colforsin and similar compounds does complicate our understanding of isoform-specific cardiac effects of AC activation, in the absence of more preclinical data. Protective or detrimental effects of individual AC manipulations on cardiac function can perhaps be best deciphered from gene overexpression or deletion studies; precise pharmacological interrogation of AC5 or AC6 activation would require further development of more selective tools.
A list of published inhibitors and FSK derivatives having AC5 and/or AC6 selectivity is depicted in Figure 11.
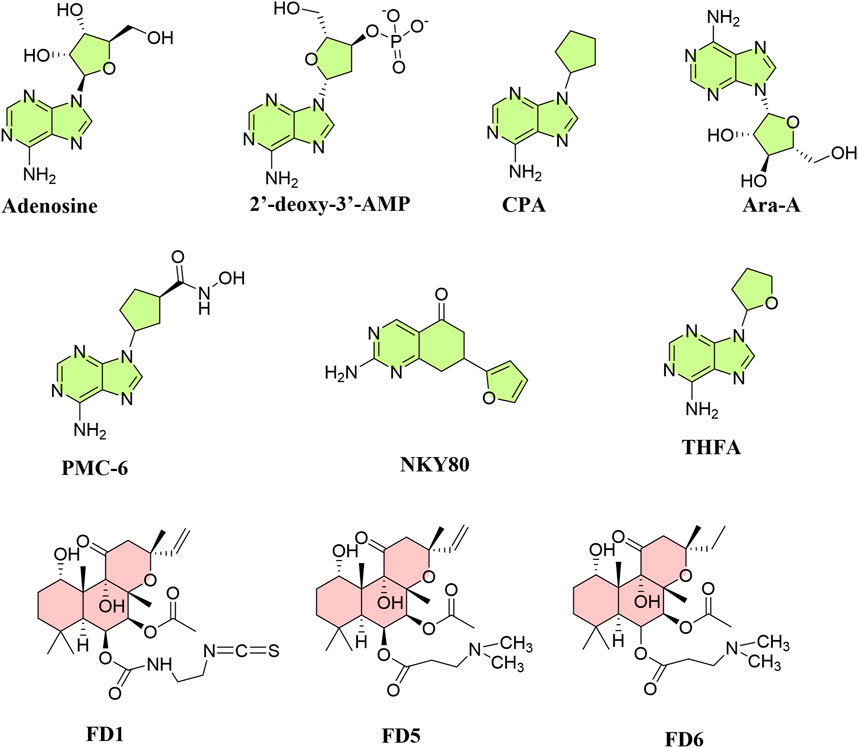
Figure 11. Structures of known inhibitors and activators targeting AC5 and AC6 allosteric or catalytic sites. Inhibitors (green) are mainly P-site inhibitors, and activators (red) are forskolin derivatives.
AC5 and AC6 inhibitors for the treatment of heart failure
As AC5 deletion appears to be protective in heart failure, there has been much research into isoform-selective AC5 inhibitors, which may be advantageous over β-blockers (Pierre et al., 2009). AC inhibitors are classified into two categories: competitive inhibitors [(M)ANT- and TNP-nucleotides] and non-competitive P-site inhibitors. Among AC5 and AC6 inhibitors discovered, 2′-d-3′-MANT-GDP has inhibitory effects on AC6 (∼8-fold) compared to AC5 (Gille et al., 2004). Among P-site inhibitors with metal chelating characteristics (PMC), PMC-6 has shown selectivity for AC5, protecting cardiac myocytes from βAR-induced apoptosis without degrading cAMP synthesis or contractility (Iwatsubo et al., 2004). P-site inhibitors that are adenosine analogs interact with ACs when PPi is present and act by obstructing the interaction of other substrates with P-sites (Dessauer and Gilman, 1997). Unlike FSK, P-site inhibitors attach to the catalytic site for substrate ATP (Tesmer et al., 2000), generating a dead-end complex with PPi (Bushfield et al., 1990). Ribose-substituted P-ligands such as 9-(cyclopentyl)-adenine (CPA) and 9-(tetrahydrofuryl)-adenine (THFA) have IC50 values in the micromolar range, selectively inhibiting AC5 more than AC3 and AC2; while 2′-deoxy-3′-AMP and 3′-AMP inhibit AC3 and AC5 more than AC2 (Onda et al., 2001; Iwatsubo et al., 2003). 2-Amino-7-(2-furanyl)-7,8-dihydro-5(6H)-quinazolinone (NKY80), derived from 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (SQ22,536), like 9-(tetrahydro-2-furyl)adenine (THFA), inhibits AC5 despite not possessing an adenine ring. NKY80, though less potent, exhibits a similar AC5 selectivity to THFA in the inhibition of AC5 catalytic activity, having a selectivity ratio of 210 between AC5 and AC2 with an IC50 of 8.3 μM for AC5, 132 μM for AC3, and 1.7 mM for AC2 when Gαs–GTPγS–forskolin is present (Onda et al., 2001).
The antiviral drug adenine 9-β-D-arabinofuranoside (Ara-A), known as vidarabine, has been found to selectively inhibit AC5; Ara-A markedly diminished AC activity in AC5 transgenic mice, but not in AC5KO, and had a minor effect in either WT or AC6 transgenic mice (Iwatsubo et al., 2012). However, another study indicated that Ara-A is also a potent AC6 inhibitor (pIC50: 5.67 and 5.34, for AC5 and AC6, respectively); indeed, SQ22,536, NKY80, and Ara-A inhibit both AC5 and AC6 without distinguishing them (Brand et al., 2013). Inhibition of AC5 by Ara-A is thought to be mediated through the MEK/ERK pathway. The Ca2+-binding protein annexin A4 (ANXA4) selectively inhibits AC5. Both ANXA4 and a peptide encompassing the ANXA4 N-terminal sequence (A4N1-22) reduced cAMP generation in AC5; ANXA4 co-immunoprecipitates with AC5 but not AC6, binding to its N-terminal domain. The peptide A4N1-22 diminishes recruitment of the L-type Ca2+ current (ICaL) and prevents action potential prolongation after catecholamine challenge, an effect similar to the loss of the β1AR signal in AC5KO models (Heinick et al., 2020). Another novel AC5 inhibitor, C90, inhibits cAMP production to FSK by 42% in WT but not in AC5KO, suggesting its selective AC5 inhibitory effect; it is five times less potent in inhibiting AC2 and AC6. C90 also produced the interesting effect of decreasing myocardial infarct size even when administered after coronary reperfusion (Zhang et al., 2018).
Conclusion
Although there have been a number of AC5 and AC6 activators/inhibitors tested in vitro, more in vivo studies are required for evaluation of the cardiac AC isoforms as drug targets and more precision of selectivity testing, given the similar structures and expressions patterns of cardiac ACs. Advances in detailed structural information (Qi et al., 2019) as well as computational homology modeling and mutational analysis of ligand-binding regions (Bhatia et al., 2023) will provide further direction to drug discovery for selective targeting of cardiovascular ACs, as will the selection of appropriate pharmacokinetic features and targeted drug delivery systems to decrease off-target adverse effects.
The prevalence of cardiovascular disease is on the rise, despite the rapid advancements in drug discovery for therapeutics addressing the alteration in the adrenergic signaling profile in the failing heart. Increased activity of AC6 has a beneficial effect on cardiac contractility, cell survival, and Ca2+ handling, while the activation of AC5 is deleterious. Differentiating the regulation of AC5 and AC6 and their signaling pathways opens up potential avenues for selective manipulation of physiologically important cAMP pools. The development of AC5/6 selective activators or inhibitors is still in its infancy, generating novel stimulators and inhibitors or taking advantage of drug repositioning; AC6 gene transfer also holds promise for heart failure treatment. Further research in these directions is clearly warranted.
Author contributions
SM: conceptualization, data curation, supervision, and writing–original draft. RS: writing–original draft. BV: writing–original draft. SD: conceptualization, funding acquisition, project administration, resources, supervision, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This project is supported by grants from Heart and Stroke Foundation (SD) (HSFC grant # G-23-0034213) and Canadian Institutes of Health Research (SD) (CIHR grant # PJT 175165).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adams, S. R., Harootunian, A. T., Buechler, Y. J., Taylor, S. S., and Tsien, R. Y. (1991). Fluorescence ratio imaging of cyclic AMP in single cells. Nature 349 (6311), 694–697. doi:10.1038/349694a0
Antoni, F. A. (2020). The chilling of adenylyl cyclase 9 and its translational potential. Cell. Signal. 70, 109589. doi:10.1016/j.cellsig.2020.109589
Bai, Y., Tsunematsu, T., Jiao, Q., Ohnuki, Y., Mototani, Y., Shiozawa, K., et al. (2012). Pharmacological stimulation of type 5 adenylyl cyclase stabilizes heart rate under both microgravity and hypergravity induced by parabolic flight. J. Pharmacol. Sci. 119 (4), 381–389. doi:10.1254/jphs.12102fp
Baldwin, T. A., and Dessauer, C. W. (2018). Function of adenylyl cyclase in heart: the AKAP connection. J. Cardiovasc. Dev. Dis. 5 (1), 2. doi:10.3390/jcdd5010002
Baldwin, T. A., Li, Y., Brand, C. S., Watts, V. J., and Dessauer, C. W. (2019). Insights into the regulatory properties of human adenylyl cyclase type 9. Mol. Pharmacol. 95 (4), 349–360. doi:10.1124/mol.118.114595
Bauman, A. L., Soughayer, J., Nguyen, B. T., Willoughby, D., Carnegie, G. K., Wong, W., et al. (2006). Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol. Cell 23 (6), 925–931. doi:10.1016/j.molcel.2006.07.025
Beazely, M. A., and Watts, V. J. (2006). Regulatory properties of adenylate cyclases type 5 and 6: a progress report. Eur. J. Pharmacol. 535 (1-3), 1–12. doi:10.1016/j.ejphar.2006.01.054
Bhagirath, A. Y., Bhatia, V., Medapati, M. R., Singh, N., Hinton, M., Chelikani, P., et al. (2022). Critical cysteines in the functional interaction of adenylyl cyclase isoform 6 with Gαs. FASEB BioAdvances 4 (3), 180–196. doi:10.1096/fba.2021-00073
Bhatia, V., Elnagary, L., and Dakshinamurti, S. (2021). Tracing the path of inhaled nitric oxide: biological consequences of protein nitrosylation. Pediatr. Pulmonol. 56 (2), 525–538. doi:10.1002/ppul.25201
Bhatia, V., Maghsoudi, S., Hinton, M., Bhagirath, A. Y., Singh, N., Jaggupilli, A., et al. (2023). Characterization of adenylyl cyclase isoform 6 residues interacting with forskolin. Biology 12 (4), 572. doi:10.3390/biology12040572
Brand, C. S., Hocker, H. J., Gorfe, A. A., Cavasotto, C. N., and Dessauer, C. W. (2013). Isoform selectivity of adenylyl cyclase inhibitors: characterization of known and novel compounds. J. Pharmacol. Exp. Ther. 347 (2), 265–275. doi:10.1124/jpet.113.208157
Bristow, M. R., Ginsburg, R., Minobe, W., Cubicciotti, R. S., Sageman, W. S., Lurie, K., et al. (1982). Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 307 (4), 205–211. doi:10.1056/NEJM198207223070401
Bristow, M. R., Ginsburg, R., Strosberg, A., Montgomery, W., and Minobe, W. (1984). Pharmacology and inotropic potential of forskolin in the human heart. J. Clin. investigation 74 (1), 212–223. doi:10.1172/JCI111404
Brodde, O. E. (1991). Beta 1- and beta 2-adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol. Rev. 43 (2), 203–242.
Bundey, R. A., and Insel, P. A. (2006). Adenylyl cyclase 6 overexpression decreases the permeability of endothelial monolayers via preferential enhancement of prostacyclin receptor function. Mol. Pharmacol. 70 (5), 1700–1707. doi:10.1124/mol.106.028035
Bushfield, M., Shoshani, I., Cifuentes, M., Stübner, D., and Johnson, R. A. (1990). Inhibition of adenylate cyclase by polyadenylate. Archives Biochem. biophysics 278 (1), 88–98. doi:10.1016/0003-9861(90)90235-q
Cai, W., Fujita, T., Hidaka, Y., Jin, H., Suita, K., Prajapati, R., et al. (2016). Disruption of Epac1 protects the heart from adenylyl cyclase type 5-mediated cardiac dysfunction. Biochem. biophysical Res. Commun. 475 (1), 1–7. doi:10.1016/j.bbrc.2016.04.123
Calamera, G., Moltzau, L. R., Levy, F. O., and Andressen, K. W. (2022). Phosphodiesterases and compartmentation of cAMP and cGMP signaling in regulation of cardiac contractility in normal and failing hearts. Int. J. Mol. Sci. 23 (4), 2145. doi:10.3390/ijms23042145
Calebiro, D., and Maiellaro, I. (2014). cAMP signaling microdomains and their observation by optical methods. Front. Cell Neurosci. 8, 350. doi:10.3389/fncel.2014.00350
Capel, R. A., Bose, S. J., Collins, T. P., Rajasundaram, S., Ayagama, T., Zaccolo, M., et al. (2021). IP3-mediated Ca2+ release regulates atrial Ca2+ transients and pacemaker function by stimulation of adenylyl cyclases. Am. J. Physiology-Heart Circulatory Physiology 320 (1), H95–H107. doi:10.1152/ajpheart.00380.2020
Capote, L. A., Perez, R. M., and Lymperopoulos, A. (2015). GPCR signaling and cardiac function. Eur. J. Pharmacol. 763, 143–148. doi:10.1016/j.ejphar.2015.05.019
Cattani-Cavalieri, I., Li, Y., Margolis, J., Bogard, A., Roosan, M. R., and Ostrom, R. S. (2023). Quantitative phosphoproteomic analysis reveals unique cAMP signaling pools emanating from AC2 and AC6 in human airway smooth muscle cells. Front. Physiology 14, 1149063. doi:10.3389/fphys.2023.1149063
Chen, J., and Iyengar, R. (1993). Inhibition of cloned adenylyl cyclases by mutant-activated Gi-alpha and specific suppression of type 2 adenylyl cyclase inhibition by phorbol ester treatment. J. Biol. Chem. 268 (17), 12253–12256. doi:10.1016/s0021-9258(18)31381-4
Chen, Y., Harry, A., Li, J., Smit, M. J., Bai, X., Magnusson, R., et al. (1997). Adenylyl cyclase 6 is selectively regulated by protein kinase A phosphorylation in a region involved in Galphas stimulation. Proc. Natl. Acad. Sci. 94 (25), 14100–14104. doi:10.1073/pnas.94.25.14100
Chester, J. A., and Watts, V. J. (2007). Adenylyl cyclase 5: a new clue in the search for the" fountain of youth"? Science's STKE 2007 (413), pe64. doi:10.1126/stke.4132007pe64
Cho, C. G., Kim, H. J., Chung, S. W., Jung, K. J., Shim, K. H., Yu, B. P., et al. (2003). Modulation of glutathione and thioredoxin systems by calorie restriction during the aging process. Exp. Gerontol. 38 (5), 539–548. doi:10.1016/s0531-5565(03)00005-6
Colombe, A.-S., and Pidoux, G. (2021). Cardiac cAMP-PKA signaling compartmentalization in myocardial infarction. Cells 10 (4), 922. doi:10.3390/cells10040922
Cosson, M.-V., Hiis, H. G., Moltzau, L. R., Levy, F. O., and Krobert, K. A. (2019). Knockout of adenylyl cyclase isoform 5 or 6 differentially modifies the β1-adrenoceptor-mediated inotropic response. J. Mol. Cell. Cardiol. 131, 132–145. doi:10.1016/j.yjmcc.2019.04.017
Cros, C., and Brette, F. (2013). Functional subcellular distribution of β1-and β2-adrenergic receptors in rat ventricular cardiac myocytes. Physiol. Rep. 1 (3), e00038. doi:10.1002/phy2.38
Defer, N., Best-Belpomme, M., and Hanoune, J. (2000). Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Physiology-Renal Physiology 279 (3), F400–F416. doi:10.1152/ajprenal.2000.279.3.F400
Dessauer, C. W., and Gilman, A. G. (1997). The catalytic mechanism of mammalian adenylyl cyclase: equilibrium binding and kinetic analysis of P-site inhibition. J. Biol. Chem. 272 (44), 27787–27795. doi:10.1074/jbc.272.44.27787
Dessauer, C. W., Watts, V. J., Ostrom, R. S., Conti, M., Dove, S., and Seifert, R. (2017). International union of basic and clinical pharmacology. CI. Structures and small molecule modulators of mammalian adenylyl cyclases. Pharmacol. Rev. 69 (2), 93–139. doi:10.1124/pr.116.013078
Diviani, D., Reggi, E., Arambasic, M., Caso, S., and Maric, D. (2016). Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochimica Biophysica Acta (BBA)-Molecular Cell Res. 1863 (7), 1926–1936. doi:10.1016/j.bbamcr.2015.11.024
Duhe, R., Nielsen, M. D., Dittman, A. H., Villacres, E. C., Choi, E.-J., and Storm, D. (1994). Oxidation of critical cysteine residues of type I adenylyl cyclase by o-iodosobenzoate or nitric oxide reversibly inhibits stimulation by calcium and calmodulin. J. Biol. Chem. 269 (10), 7290–7296. doi:10.1016/s0021-9258(17)37282-4
Efendiev, R., and Dessauer, C. W. (2011). A kinase-anchoring proteins and adenylyl cyclase in cardiovascular physiology and pathology. J. Cardiovasc. Pharmacol. 58 (4), 339–344. doi:10.1097/FJC.0b013e31821bc3f0
Engelhardt, S., Hein, L., Wiesmann, F., and Lohse, M. J. (1999). Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 96 (12), 7059–7064. doi:10.1073/pnas.96.12.7059
Espinasse, I., Iourgenko, V., Defer, N., Samson, F., Hanoune, J., and Mercadier, J.-J. (1995). Type V, but not type VI, adenylyl cyclase mRNA accumulates in the rat heart during ontogenic development. Correlation with increased global adenylyl cyclase activity. J. Mol. Cell. Cardiol. 27 (9), 1789–1795. doi:10.1016/0022-2828(95)90002-0
Esposito, G., Perrino, C., Ozaki, T., Takaoka, H., Defer, N., Petretta, M. P., et al. (2008). Increased myocardial contractility and enhanced exercise function in transgenic mice overexpressing either adenylyl cyclase 5 or 8. Basic Res. Cardiol. 103, 22–30. doi:10.1007/s00395-007-0688-6
Fagan, K. A., Mons, N., and Cooper, D. M. (1998). Dependence of the Ca2+-inhibitable adenylyl cyclase of C6–2B glioma cells on capacitative Ca2+ entry. J. Biol. Chem. 273 (15), 9297–9305. doi:10.1074/jbc.273.15.9297
Ferrara, N., Komici, K., Corbi, G., Pagano, G., Furgi, G., Rengo, C., et al. (2014). β-adrenergic receptor responsiveness in aging heart and clinical implications. Front. physiology 4, 396. doi:10.3389/fphys.2013.00396
Fujino, T., Hasebe, N., Kawabe, J.-i., Fujita, M., Fukuzawa, J., Tobise, K., et al. (2003). Effect of beta-adrenoceptor antagonist and angiotensin-converting enzyme inhibitor on hypertension-associated changes in adenylyl cyclase type V messenger RNA expression in spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 41 (5), 720–725. doi:10.1097/00005344-200305000-00008
Fujita, T., Umemura, M., Yokoyama, U., Okumura, S., and Ishikawa, Y. (2017). The role of Epac in the heart. Cell. Mol. life Sci. 74, 591–606. doi:10.1007/s00018-016-2336-5
Gao, M. H., Lai, N. C., Giamouridis, D., Kim, Y. C., Guo, T., and Hammond, H. K. (2017). Cardiac-directed expression of a catalytically inactive adenylyl cyclase 6 protects the heart from sustained β-adrenergic stimulation. PloS one 12 (8), e0181282. doi:10.1371/journal.pone.0181282
Gao, M. H., Lai, N. C., Giamouridis, D., Kim, Y. C., Tan, Z., Guo, T., et al. (2016). Cardiac-directed expression of adenylyl cyclase catalytic domain reverses cardiac dysfunction caused by sustained beta-adrenergic receptor stimulation. JACC Basic Transl. Sci. 1 (7), 617–629. doi:10.1016/j.jacbts.2016.08.004
Gao, M. H., Miyanohara, A., Feramisco, J. R., and Tang, T. (2009). Activation of PH-domain leucine-rich protein phosphatase 2 (PHLPP2) by agonist stimulation in cardiac myocytes expressing adenylyl cyclase type 6. Biochem. biophysical Res. Commun. 384 (2), 193–198. doi:10.1016/j.bbrc.2009.04.110
Gao, M. H., Tang, T., Guo, T., Miyanohara, A., Yajima, T., Pestonjamasp, K., et al. (2008). Adenylyl cyclase type VI increases Akt activity and phospholamban phosphorylation in cardiac myocytes. J. Biol. Chem. 283 (48), 33527–33535. doi:10.1074/jbc.M805825200
Gao, M. H., Tang, T., Guo, T., Sun, S. Q., Feramisco, J. R., and Hammond, H. K. (2004). Adenylyl cyclase type VI gene transfer reduces phospholamban expression in cardiac myocytes via activating transcription factor 3. J. Biol. Chem. 279 (37), 38797–38802. doi:10.1074/jbc.M405701200
Garnier, A., Leroy, J., Deloménie, C., Mateo, P., Viollet, B., Veksler, V., et al. (2023). Modulation of cardiac cAMP signaling by AMPK and its adjustments in pressure overload-induced myocardial dysfunction in rat and mouse. Plos one 18 (9), e0292015. doi:10.1371/journal.pone.0292015
Gille, A., Lushington, G. H., Mou, T.-C., Doughty, M. B., Johnson, R. A., and Seifert, R. (2004). Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J. Biol. Chem. 279 (19), 19955–19969. doi:10.1074/jbc.M312560200
Göttle, M., Geduhn, J., König, B., Gille, A., Höcherl, K., and Seifert, R. (2009). Characterization of mouse heart adenylyl cyclase. J. Pharmacol. Exp. Ther. 329 (3), 1156–1165. doi:10.1124/jpet.109.150953
Gros, R., Ding, Q., Chorazyczewski, J., Pickering, J. G., Limbird, L. E., and Feldman, R. D. (2006). Adenylyl cyclase isoform–selective regulation of vascular smooth muscle proliferation and cytoskeletal reorganization. Circulation Res. 99 (8), 845–852. doi:10.1161/01.RES.0000245189.21703.c0
Gu, C., Cali, J. J., and Cooper, D. M. (2002). Dimerization of mammalian adenylate cyclases: functional, biochemical and fluorescence resonance energy transfer (FRET) studies. Eur. J. Biochem. 269 (2), 413–421. doi:10.1046/j.0014-2956.2001.02708.x
Guellich, A., Gao, S., Hong, C., Yan, L., Wagner, T. E., Dhar, S. K., et al. (2010). Effects of cardiac overexpression of type 6 adenylyl cyclase affects on the response to chronic pressure overload. Am. J. Physiology-Heart Circulatory Physiology 299 (3), H707–H712. doi:10.1152/ajpheart.00148.2010
Guers, J. J., Zhang, J., Campbell, S. C., Oydanich, M., Vatner, D. E., and Vatner, S. F. (2017). Disruption of adenylyl cyclase type 5 mimics exercise training. Basic Res. Cardiol. 112, 59–12. doi:10.1007/s00395-017-0648-8
Guillou, J.-L., Nakata, H., and Cooper, D. M. (1999). Inhibition by calcium of mammalian adenylyl cyclases. J. Biol. Chem. 274 (50), 35539–35545. doi:10.1074/jbc.274.50.35539
Haber, N., Stengel, D., Defer, N., Roeckel, N., Mattei, M. G., and Hanoune, J. (1994). Chromosomal mapping of human adenylyl cyclase genes type III, type V and type VI. Hum. Genet. 94 (1), 69–73. doi:10.1007/BF02272844
Hackley, C. R., Mazzoni, E. O., and Blau, J. (2018). cAMPr: a single-wavelength fluorescent sensor for cyclic AMP. Sci. Signal. 11 (520), eaah3738. doi:10.1126/scisignal.aah3738
Halls, M. L., and Cooper, D. M. (2017). Adenylyl cyclase signalling complexes–Pharmacological challenges and opportunities. Pharmacol. Ther. 172, 171–180. doi:10.1016/j.pharmthera.2017.01.001
Hammond, H. K., Penny, W. F., Traverse, J. H., Henry, T. D., Watkins, M. W., Yancy, C. W., et al. (2016). Intracoronary gene transfer of adenylyl cyclase 6 in patients with heart failure: a randomized clinical trial. JAMA Cardiol. 1 (2), 163–171. doi:10.1001/jamacardio.2016.0008
Hanoune, J., and Defer, N. (2001). Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 41 (1), 145–174. doi:10.1146/annurev.pharmtox.41.1.145
Hanski, E., Sevilla, N., and Levitzki, A. (1977). The allosteric inhibition by calcium of soluble and partially purified adenylate cyclase from Turkey erythrocytes. Eur. J. Biochem. 76 (2), 513–520. doi:10.1111/j.1432-1033.1977.tb11621.x
Heinick, A., Pluteanu, F., Hermes, C., Klemme, A., Domnik, M., Husser, X., et al. (2020). Annexin A4 N-terminal peptide inhibits adenylyl cyclase 5 and limits β-adrenoceptor-mediated prolongation of cardiac action potential. FASEB J. 34 (8), 10489–10504. doi:10.1096/fj.201902094RR
Hill, J., Howlett, A., and Klein, C. (2000). Nitric oxide selectively inhibits adenylyl cyclase isoforms 5 and 6. Cell. Signal. 12 (4), 233–237. doi:10.1016/s0898-6568(99)00082-0
Ho, D., Yan, L., Iwatsubo, K., Vatner, D. E., and Vatner, S. F. (2010). Modulation of beta-adrenergic receptor signaling in heart failure and longevity: targeting adenylyl cyclase type 5. Heart Fail. Rev. 15, 495–512. doi:10.1007/s10741-010-9183-5
Hodges, G. J., Gros, R., Hegele, R. A., Van Uum, S., Shoemaker, J. K., and Feldman, R. D. (2010). Increased blood pressure and hyperdynamic cardiovascular responses in carriers of a common hyperfunctional variant of adenylyl cyclase 6. J. Pharmacol. Exp. Ther. 335 (2), 451–457. doi:10.1124/jpet.110.172700
Hu, B., Nakata, H., Gu, C., De Beer, T., and Cooper, D. M. (2002). A critical interplay between Ca2+ inhibition and activation by Mg2+ of AC5 revealed by mutants and chimeric constructs. J. Biol. Chem. 277 (36), 33139–33147. doi:10.1074/jbc.M112373200
Hu, C.-L., Chandra, R., Ge, H., Pain, J., Yan, L., Babu, G., et al. (2009). Adenylyl cyclase type 5 protein expression during cardiac development and stress. Am. J. Physiology-Heart Circulatory Physiology 297 (5), H1776–H1782. doi:10.1152/ajpheart.00050.2009
Hunter, M. R., Finlay, D. B., Macdonald, C. E., Cawston, E. E., Grimsey, N. L., and Glass, M. (2017). Real-time measurement of cannabinoid receptor-mediated cAMP signaling. Methods Enzymol. 593, 43–59. doi:10.1016/bs.mie.2017.05.001
Irisawa, H., Brown, H., and Giles, W. (1993). Cardiac pacemaking in the sinoatrial node. Physiol. Rev. 73 (1), 197–227. doi:10.1152/physrev.1993.73.1.197
Ishikawa, Y., Sorota, S., Kiuchi, K., Shannon, R., Komamura, K., Katsushika, S., et al. (1994). Downregulation of adenylylcyclase types V and VI mRNA levels in pacing-induced heart failure in dogs. J. Clin. investigation 93 (5), 2224–2229. doi:10.1172/JCI117219
Itami, T., Hanazono, K., Oyama, N., Sano, T., Makita, K., and Yamashita, K. (2019). Cardiovascular effects of intravenous colforsin in normal and acute respiratory acidosis canine models: a dose-response study. PLoS One 14 (7), e0213414. doi:10.1371/journal.pone.0213414
Iwami, G., Akanuma, M., Kawabe, J., Cannon, P. J., Homcy, C. J., and Ishikawa, Y. (1995). Multiplicity in type V adenylylcyclase: type Va and type Vb. Mol. Cell. Endocrinol. 110 (1-2), 43–47. doi:10.1016/0303-7207(95)03514-8
Iwatsubo, K., Bravo, C., Uechi, M., Baljinnyam, E., Nakamura, T., Umemura, M., et al. (2012). Prevention of heart failure in mice by an antiviral agent that inhibits type 5 cardiac adenylyl cyclase. Am. J. Physiology-Heart Circulatory Physiology 302 (12), H2622–H2628. doi:10.1152/ajpheart.00190.2012
Iwatsubo, K., Minamisawa, S., Tsunematsu, T., Nakagome, M., Toya, Y., Tomlinson, J. E., et al. (2004). Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J. Biol. Chem. 279 (39), 40938–40945. doi:10.1074/jbc.M314238200
Iwatsubo, K., Tsunematsu, T., and Ishikawa, Y. (2003). Isoform-specific regulation of adenylyl cyclase: a potential target in future pharmacotherapy. Expert Opin. Ther. targets 7 (3), 441–451. doi:10.1517/14728222.7.3.441
Jacobowitz, O., Chen, J., Premont, R., and Iyengar, R. (1993). Stimulation of specific types of Gs-stimulated adenylyl cyclases by phorbol ester treatment. J. Biol. Chem. 268 (6), 3829–3832. doi:10.1016/s0021-9258(18)53547-x
Jaggupilli, A., Dhanaraj, P., Pritchard, A., Sorensen, J. L., Dakshinamurti, S., and Chelikani, P. (2018). Study of adenylyl cyclase-GαS interactions and identification of novel AC ligands. Mol. Cell Biochem. 446 (1-2), 63–72. doi:10.1007/s11010-018-3273-4
Jiang, M. T., Moffat, M. P., and Narayanan, N. (1993). Age-related alterations in the phosphorylation of sarcoplasmic reticulum and myofibrillar proteins and diminished contractile response to isoproterenol in intact rat ventricle. Circulation Res. 72 (1), 102–111. doi:10.1161/01.res.72.1.102
Joe, E. K., Schussheim, A. E., Longrois, D., Mäki, T., Kelly, R. A., Smith, T. W., et al. (1998). Regulation of cardiac myocyte contractile function by inducible nitric oxide synthase (iNOS): mechanisms of contractile depression by nitric oxide. J. Mol. Cell. Cardiol. 30 (2), 303–315. doi:10.1006/jmcc.1997.0593
John, R. M., and Kumar, S. (2016). Sinus node and atrial arrhythmias. Circulation 133 (19), 1892–1900. doi:10.1161/CIRCULATIONAHA.116.018011
Kamenetsky, M., Middelhaufe, S., Bank, E. M., Levin, L. R., Buck, J., and Steegborn, C. (2006). Molecular details of cAMP generation in mammalian cells: a tale of two systems. J. Mol. Biol. 362 (4), 623–639. doi:10.1016/j.jmb.2006.07.045
Kamide, T., Okumura, S., Ghosh, S., Shinoda, Y., Mototani, Y., Ohnuki, Y., et al. (2015). Oscillation of cAMP and Ca2+ in cardiac myocytes: a systems biology approach. J. physiological Sci. 65 (2), 195–200. doi:10.1007/s12576-014-0354-3
Kapiloff, M. S., Piggott, L. A., Sadana, R., Li, J., Heredia, L. A., Henson, E., et al. (2009). An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J. Biol. Chem. 284 (35), 23540–23546. doi:10.1074/jbc.M109.030072
Kawabe, J.-i., Ebina, T., Toya, Y., Oka, N., Schwencke, C., Duzic, E., et al. (1996). Regulation of type V adenylyl cyclase by PMA-sensitive and-insensitive protein kinase C isoenzymes in intact cells. FEBS Lett. 384 (3), 273–276. doi:10.1016/0014-5793(96)00331-6
Kim, N., Shin, S., and Bae, S. W. (2021). cAMP biosensors based on genetically encoded fluorescent/luminescent proteins. Biosens. (Basel) 11 (2), 39. doi:10.3390/bios11020039
Krishna, G., Weiss, B., and Brodie, B. B. (1968). A simple, sensitive method for the assay of adenyl cyclase. J. Pharmacol. Exp. Ther. 163 (2), 379–385.
Kritzer, M. D., Li, J., Dodge-Kafka, K., and Kapiloff, M. S. (2012). AKAPs: the architectural underpinnings of local cAMP signaling. J. Mol. Cell Cardiol. 52 (2), 351–358. doi:10.1016/j.yjmcc.2011.05.002
Kryukova, Y. N., Protas, L., and Robinson, R. B. (2012). Ca2+-activated adenylyl cyclase 1 introduces Ca2+-dependence to beta-adrenergic stimulation of HCN2 current. J. Mol. Cell. Cardiol. 52 (6), 1233–1239. doi:10.1016/j.yjmcc.2012.03.010
Lai, H.-L., Lin, T.-H., Kao, Y.-Y., Lin, W.-J., Hwang, M.-J., and Chern, Y. (1999). The N terminus domain of type VI adenylyl cyclase mediates its inhibition by protein kinase C. Mol. Pharmacol. 56 (3), 644–650. doi:10.1124/mol.56.3.644
Lai, H.-L., Yang, T.-H., Messing, R. O., Ching, Y.-H., Lin, S.-C., and Chern, Y. (1997). Protein kinase C inhibits adenylyl cyclase type VI activity during desensitization of the A2a-adenosine receptor-mediated cAMP response. J. Biol. Chem. 272 (8), 4970–4977. doi:10.1074/jbc.272.8.4970
Lai, L., Yan, L., Gao, S., Hu, C.-L., Ge, H., Davidow, A., et al. (2013). Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of manganese superoxide dismutase via the SIRT1/FoxO3a pathway. Circulation 127 (16), 1692–1701. doi:10.1161/CIRCULATIONAHA.112.001212
Lai, L., Yan, L., Hu, C.-L., Gao, S., Iwatsubo, K., Ishikawa, Y., et al. (2009). Increased Type 5 adenylyl cyclase expression mediates chronic catecholamine stress via increases in oxidative stress and down-regulation of MnSOD. Am. Heart Assoc.
Lai, N. C., Tang, T., Gao, M. H., Saito, M., Takahashi, T., Roth, D. M., et al. (2008). Activation of cardiac adenylyl cyclase expression increases function of the failing ischemic heart in mice. J. Am. Coll. Cardiol. 51 (15), 1490–1497. doi:10.1016/j.jacc.2008.01.015
Lemaire, K., Van de Velde, S., Van Dijck, P., and Thevelein, J. M. (2004). Glucose and sucrose act as agonist and mannose as antagonist ligands of the G protein-coupled receptor Gpr1 in the yeast Saccharomyces cerevisiae. Mol. Cell 16 (2), 293–299. doi:10.1016/j.molcel.2004.10.004
Lezoualc’h, F., Fazal, L., Laudette, M., and Conte, C. (2016). Cyclic AMP sensor EPAC proteins and their role in cardiovascular function and disease. Circulation Res. 118 (5), 881–897. doi:10.1161/CIRCRESAHA.115.306529
Lin, T.-H., Lai, H.-L., Kao, Y.-Y., Sun, C.-N., Hwang, M.-J., and Chern, Y. (2002). Protein kinase C inhibits type VI adenylyl cyclase by phosphorylating the regulatory N domain and two catalytic C1 and C2 domains. J. Biol. Chem. 277 (18), 15721–15728. doi:10.1074/jbc.M111537200
Linder, J. U. (2006). Class III adenylyl cyclases: molecular mechanisms of catalysis and regulation. Cell Mol. Life Sci. 63 (15), 1736–1751. doi:10.1007/s00018-006-6072-0
Liu, X., Thangavel, M., Sun, S. Q., Kaminsky, J., Mahautmr, P., Stitham, J., et al. (2008). Adenylyl cyclase type 6 overexpression selectively enhances beta-adrenergic and prostacyclin receptor-mediated inhibition of cardiac fibroblast function because of colocalization in lipid rafts. Naunyn-Schmiedeberg's archives Pharmacol. 377, 359–369. doi:10.1007/s00210-007-0196-0
Liu, Y., Chen, J., Fontes, S. K., Bautista, E. N., and Cheng, Z. (2022). Physiological and pathological roles of protein kinase A in the heart. Cardiovasc. Res. 118 (2), 386–398. doi:10.1093/cvr/cvab008
Maccarrone, M., Bari, M., Lorenzon, T., Bisogno, T., Di Marzo, V., and Finazzi-Agro, A. (2000). Anandamide uptake by human endothelial cells and its regulation by nitric oxide. J. Biol. Chem. 275 (18), 13484–13492. doi:10.1074/jbc.275.18.13484
Mahmood, A., Ahmed, K., and Zhang, Y. (2022). β-Adrenergic receptor desensitization/down-regulation in heart failure: a friend or foe? Front. Cardiovasc. Med. 9, 925692. doi:10.3389/fcvm.2022.925692
Maltsev, V. A., and Lakatta, E. G. (2012). The funny current in the context of the coupled-clock pacemaker cell system. Heart rhythm. 9 (2), 302–307. doi:10.1016/j.hrthm.2011.09.022
Masri, B., Salahpour, A., Didriksen, M., Ghisi, V., Beaulieu, J.-M., Gainetdinov, R. R., et al. (2008). Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. 105 (36), 13656–13661. doi:10.1073/pnas.0803522105
Matsumoto, T., Wakabayashi, K., Kobayashi, T., and Kamata, K. (2005). Functional changes in adenylyl cyclases and associated decreases in relaxation responses in mesenteric arteries from diabetic rats. Am. J. Physiology-Heart Circulatory Physiology 289 (5), H2234–H2243. doi:10.1152/ajpheart.00971.2004
Mattick, P., Parrington, J., Odia, E., Simpson, A., Collins, T., and Terrar, D. (2007). Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in Guinea-pig sino-atrial node cells and modulates the if pacemaker current. J. physiology 582 (3), 1195–1203. doi:10.1113/jphysiol.2007.133439
McVey, M., Hill, J., Howlett, A., and Klein, C. (1999). Adenylyl cyclase, a coincidence detector for nitric oxide. J. Biol. Chem. 274 (27), 18887–18892. doi:10.1074/jbc.274.27.18887
Mika, D., Richter, W., Westenbroek, R. E., Catterall, W. A., and Conti, M. (2014). PDE4B mediates local feedback regulation of β₁-adrenergic cAMP signaling in a sarcolemmal compartment of cardiac myocytes. J. Cell Sci. 127 (5), 1033–1042. doi:10.1242/jcs.140251
Milano, C. A., Allen, L. F., Rockman, H. A., Dolber, P. C., McMinn, T. R., Chien, K. R., et al. (1994). Enhanced myocardial function in transgenic mice overexpressing the beta 2-adrenergic receptor. Science 264 (5158), 582–586. doi:10.1126/science.8160017
Mnatsakanyan, R., Markoutsa, S., Walbrunn, K., Roos, A., Verhelst, S. H., and Zahedi, R. P. (2019). Proteome-wide detection of S-nitrosylation targets and motifs using bioorthogonal cleavable-linker-based enrichment and switch technique. Nat. Commun. 10 (1), 2195. doi:10.1038/s41467-019-10182-4
Moen, J. M., Matt, M. G., Ramirez, C., Tarasov, K. V., Chakir, K., Tarasova, Y. S., et al. (2019). Overexpression of a neuronal type adenylyl cyclase (type 8) in sinoatrial node markedly impacts heart rate and rhythm. Front. Neurosci. 13, 615. doi:10.3389/fnins.2019.00615
Mongillo, M., McSorley, T., Evellin, S., Sood, A., Lissandron, V., Terrin, A., et al. (2004). Fluorescence resonance energy transfer–based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circulation Res. 95 (1), 67–75. doi:10.1161/01.RES.0000134629.84732.11
Montero, D., Walther, G., Pérez-Martin, A., Vicente-Salar, N., Roche, E., and Vinet, A. (2013). Vascular smooth muscle function in type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetologia 56, 2122–2133. doi:10.1007/s00125-013-2974-1
Mou, T.-C., Masada, N., Cooper, D. M., and Sprang, S. R. (2009). Structural basis for inhibition of mammalian adenylyl cyclase by calcium. Biochemistry 48 (15), 3387–3397. doi:10.1021/bi802122k
Mougenot, N., Mika, D., Czibik, G., Marcos, E., Abid, S., Houssaini, A., et al. (2019). Cardiac adenylyl cyclase overexpression precipitates and aggravates age-related myocardial dysfunction. Cardiovasc. Res. 115 (12), 1778–1790. doi:10.1093/cvr/cvy306
Nakano, S. J., Sucharov, J., Van Dusen, R., Cecil, M., Nunley, K., Wickers, S., et al. (2017). Cardiac adenylyl cyclase and phosphodiesterase expression profiles vary by age, disease, and chronic phosphodiesterase inhibitor treatment. J. cardiac Fail. 23 (1), 72–80. doi:10.1016/j.cardfail.2016.07.429
Nelson, C. P., Rainbow, R. D., Brignell, J. L., Perry, M. D., Willets, J. M., Davies, N. W., et al. (2011). Principal role of adenylyl cyclase 6 in K⁺ channel regulation and vasodilator signalling in vascular smooth muscle cells. Cardiovasc. Res. 91 (4), 694–702. doi:10.1093/cvr/cvr137
Nikolaev, V. O., Moshkov, A., Lyon, A. R., Miragoli, M., Novak, P., Paur, H., et al. (2010). Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 327 (5973), 1653–1657. doi:10.1126/science.1185988
Nystoriak, M. A., Nieves-Cintrón, M., Patriarchi, T., Buonarati, O. R., Prada, M. P., Morotti, S., et al. (2017). Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1. 2 and vasoconstriction during acute hyperglycemia and diabetes. Sci. Signal. 10 (463), eaaf9647. doi:10.1126/scisignal.aaf9647
Odaka, H., Arai, S., Inoue, T., and Kitaguchi, T. (2014). Genetically-encoded yellow fluorescent cAMP indicator with an expanded dynamic range for dual-color imaging. PloS one 9 (6), e100252. doi:10.1371/journal.pone.0100252
Ohta, Y., Furuta, T., Nagai, T., and Horikawa, K. (2018). Red fluorescent cAMP indicator with increased affinity and expanded dynamic range. Sci. Rep. 8 (1), 1866. doi:10.1038/s41598-018-20251-1
Okumura, S., Kawabe, J.-i., Yatani, A., Takagi, G., Lee, M.-C., Hong, C., et al. (2003a). Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circulation Res. 93 (4), 364–371. doi:10.1161/01.RES.0000086986.35568.63
Okumura, S., Suzuki, S., and Ishikawa, Y. (2009). New aspects for the treatment of cardiac diseases based on the diversity of functional controls on cardiac muscles: effects of targeted disruption of the type 5 adenylyl cyclase gene. J. Pharmacol. Sci. 109 (3), 354–359. doi:10.1254/jphs.08r26fm
Okumura, S., Takagi, G., Kawabe, J.-i., Yang, G., Lee, M.-C., Hong, C., et al. (2003b). Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc. Natl. Acad. Sci. 100 (17), 9986–9990. doi:10.1073/pnas.1733772100
Okumura, S., Tsunematsu, T., Bai, Y., Jiao, Q., Ono, S., Suzuki, S., et al. (2008). Type 5 adenylyl cyclase plays a major role in stabilizing heart rate in response to microgravity induced by parabolic flight. J. Appl. Physiology 105 (1), 173–179. doi:10.1152/japplphysiol.01166.2007
Okumura, S., Vatner, D. E., Kurotani, R., Bai, Y., Gao, S., Yuan, Z., et al. (2007). Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation 116 (16), 1776–1783. doi:10.1161/CIRCULATIONAHA.107.698662
Oldham, S. B., Rude, R. K., Molloy, C. T., Lipson, L. G., and Boggs, T. T. (1984). The effects of magnesium on calcium inhibition of parathyroid adenylate cyclase. Endocrinology 115 (5), 1883–1890. doi:10.1210/endo-115-5-1883
Onda, T., Hashimoto, Y., Nagai, M., Kuramochi, H., Saito, S., Yamazaki, H., et al. (2001). Type-specific regulation of adenylyl cyclase: selective pharmacological stimulation and inhibition of adenylyl cyclase isoforms. J. Biol. Chem. 276 (51), 47785–47793. doi:10.1074/jbc.M107233200
Ostrom, K. F., LaVigne, J. E., Brust, T. F., Seifert, R., Dessauer, C. W., Watts, V. J., et al. (2022). Physiological roles of mammalian transmembrane adenylyl cyclase isoforms. Physiol. Rev. 102 (2), 815–857. doi:10.1152/physrev.00013.2021
Ostrom, R. S., Bundey, R. A., and Insel, P. A. (2004). Nitric oxide inhibition of adenylyl cyclase type 6 activity is dependent upon lipid rafts and caveolin signaling complexes. J. Biol. Chem. 279 (19), 19846–19853. doi:10.1074/jbc.M313440200
Ostrom, R. S., Gregorian, C., Drenan, R. M., Xiang, Y., Regan, J. W., and Insel, P. A. (2001). Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J. Biol. Chem. 276 (45), 42063–42069. doi:10.1074/jbc.M105348200
Ostrom, R. S., and Insel, P. A. (2004). The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br. J. Pharmacol. 143 (2), 235–245. doi:10.1038/sj.bjp.0705930
Ostrom, R. S., Liu, X., Head, B. P., Gregorian, C., Seasholtz, T. M., and Insel, P. A. (2002). Localization of adenylyl cyclase isoforms and G protein-coupled receptors in vascular smooth muscle cells: expression in caveolin-rich and noncaveolin domains. Mol. Pharmacol. 62 (5), 983–992. doi:10.1124/mol.62.5.983
Ostrom, R. S., Naugle, J. E., Hase, M., Gregorian, C., Swaney, J. S., Insel, P. A., et al. (2003). Angiotensin II enhances adenylyl cyclase signaling via Ca2+/Calmodulin: gq-Gs cross-talk regulates collagen production in cardiac fibroblasts. J. Biol. Chem. 278 (27), 24461–24468. doi:10.1074/jbc.M212659200
Ostrom, R. S., Violin, J. D., Coleman, S., and Insel, P. A. (2000). Selective enhancement of beta-adrenergic receptor signaling by overexpression of adenylyl cyclase type 6: colocalization of receptor and adenylyl cyclase in caveolae of cardiac myocytes. Mol. Pharmacol. 57 (5), 1075–1079.
Packer, M., Carver, J. R., Rodeheffer, R. J., Ivanhoe, R. J., DiBianco, R., Zeldis, S. M., et al. (1991). Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N. Engl. J. Med. 325 (21), 1468–1475. doi:10.1056/NEJM199111213252103
Paramonov, V. M., Mamaeva, V., Sahlgren, C., and Rivero-Müller, A. (2015). Genetically-encoded tools for cAMP probing and modulation in living systems. Front. Pharmacol. 6, 196. doi:10.3389/fphar.2015.00196
Park, M., Park, J. Y., Lee, J. A., Tian, B., Lai, L., Iwatsubo, K., et al. (2011). Cardiac overexpression of adenylyl cyclase type 5 induces left ventricular hypertrophy potentially by activating calcineurin-NFAT signaling. Wiley Online Library.
Patel, N., and Gold, M. G. (2015). The genetically encoded tool set for investigating cAMP: more than the sum of its parts. Front. Pharmacol. 6, 164. doi:10.3389/fphar.2015.00164
Pavan, B., Biondi, C., and Dalpiaz, A. (2009). Adenylyl cyclases as innovative therapeutic goals. Drug Discov. today 14 (19-20), 982–991. doi:10.1016/j.drudis.2009.07.007
Penny, W. F., Henry, T. D., Watkins, M. W., Patel, A. N., and Hammond, H. K. (2018). Design of a Phase 3 trial of intracoronary administration of human adenovirus 5 encoding human adenylyl cyclase type 6 (RT-100) gene transfer in patients with heart failure with reduced left ventricular ejection fraction: the FLOURISH Clinical Trial. Am. Heart J. 201, 111–116. doi:10.1016/j.ahj.2018.04.005
Phan, H. M., Gao, M. H., Lai, N. C., Tang, T., and Hammond, H. K. (2007). New signaling pathways associated with increased cardiac adenylyl cyclase 6 expression: implications for possible congestive heart failure therapy. Trends Cardiovasc. Med. 17 (7), 215–221. doi:10.1016/j.tcm.2007.07.001
Piacenza, L., Zeida, A., Trujillo, M., and Radi, R. (2022). The superoxide radical switch in the biology of nitric oxide and peroxynitrite. Physiol. Rev. 102 (4), 1881–1906. doi:10.1152/physrev.00005.2022
Pierre, S., Eschenhagen, T., Geisslinger, G., and Scholich, K. (2009). Capturing adenylyl cyclases as potential drug targets. Nat. Rev. Drug Discov. 8 (4), 321–335. doi:10.1038/nrd2827
Ping, P., Anzai, T., Gao, M., and Hammond, H. (1997). Adenylyl cyclase and G protein receptor kinase expression during development of heart failure. Am. J. Physiology-Heart Circulatory Physiology 273 (2), H707–H717. doi:10.1152/ajpheart.1997.273.2.H707
Ponsioen, B., Zhao, J., Riedl, J., Zwartkruis, F., van der Krogt, G., Zaccolo, M., et al. (2004). Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 5 (12), 1176–1180. doi:10.1038/sj.embor.7400290
Prada, M. P., Syed, A. U., Buonarati, O. R., Reddy, G. R., Nystoriak, M. A., Ghosh, D., et al. (2019). A Gs-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. Elife 8, e42214. doi:10.7554/eLife.42214
Prada, M. P., Syed, A. U., Reddy, G. R., Martín-Aragón Baudel, M., Flores-Tamez, V. A., Sasse, K. C., et al. (2020). AKAP5 complex facilitates purinergic modulation of vascular L-type Ca2+ channel CaV1. 2. Nat. Commun. 11 (1), 5303. doi:10.1038/s41467-020-18947-y
Prinz, A., Diskar, M., Erlbruch, A., and Herberg, F. W. (2006). Novel, isotype-specific sensors for protein kinase A subunit interaction based on bioluminescence resonance energy transfer (BRET). Cell. Signal. 18 (10), 1616–1625. doi:10.1016/j.cellsig.2006.01.013
Qi, C., Sorrentino, S., Medalia, O., and Korkhov, V. M. (2019). The structure of a membrane adenylyl cyclase bound to an activated stimulatory G protein. Science 364 (6438), 389–394. doi:10.1126/science.aav0778
Qu, J.-H., Chakir, K., Tarasov, K. V., Riordon, D. R., Perino, M. G., Silvester, A. J., et al. (2024). Reprogramming of cardiac phosphoproteome, proteome, and transcriptome confers resilience to chronic adenylyl cyclase-driven stress. Elife 12, RP88732. doi:10.7554/eLife.88732
Rana, N., Conley, J. M., Soto-Velasquez, M., León, F., Cutler, S. J., Watts, V. J., et al. (2017). Molecular modeling evaluation of the enantiomers of a novel adenylyl cyclase 2 inhibitor. J. Chem. Inf. Model. 57 (2), 322–334. doi:10.1021/acs.jcim.6b00454
Ren, L., Thai, P. N., Gopireddy, R. R., Timofeyev, V., Ledford, H. A., Woltz, R. L., et al. (2022). Adenylyl cyclase isoform 1 contributes to sinoatrial node automaticity via functional microdomains. JCI insight 7 (22), e162602. doi:10.1172/jci.insight.162602
Ridnour, L. A., Thomas, D. D., Mancardi, D., Espey, M. G., Miranda, K. M., Paolocci, N., et al. (2004). The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putt. perspective stressful Biol. situations. doi:10.1515/BC.2004.001
Ripoll, L., and Von Zastrow, M. (2023). Spatial organization of adenylyl cyclase and its impact on dopamine signaling in neurons. bioRxiv, 2023.12.06.570478. 2023.2012. 2006.570478. doi:10.1101/2023.12.06.570478
Robinson, R. B., Dun, W., and Boyden, P. A. (2021). Autonomic modulation of sinoatrial node: role of pacemaker current and calcium sensitive adenylyl cyclase isoforms. Prog. Biophysics Mol. Biol. 166, 22–28. doi:10.1016/j.pbiomolbio.2020.08.004
Roth, D. M., Bayat, H., Drumm, J. D., Gao, M. H., Swaney, J. S., Ander, A., et al. (2002). Adenylyl cyclase increases survival in cardiomyopathy. Circulation 105 (16), 1989–1994. doi:10.1161/01.cir.0000014968.54967.d3
Roth, D. M., Gao, M. H., Lai, N. C., Drumm, J., Dalton, N., Zhou, J. Y., et al. (1999). Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 99 (24), 3099–3102. doi:10.1161/01.cir.99.24.3099
Ruhparwar, A., Kallenbach, K., Klein, G., Bara, C., Ghodsizad, A., Sigg, D. C., et al. (2010). Adenylate-cyclase VI transforms ventricular cardiomyocytes into biological pacemaker cells. Tissue Eng. Part A 16 (6), 1867–1872. doi:10.1089/ten.TEA.2009.0537
Sadana, R., and Dessauer, C. W. (2009). Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals 17 (1), 5–22. doi:10.1159/000166277
Schmid, A., Meili, D., and Salathe, M. (2014). Soluble adenylyl cyclase in health and disease. Biochimica Biophysica Acta (BBA)-Molecular Basis Dis. 1842 (12), 2584–2592. doi:10.1016/j.bbadis.2014.07.010
Schubert, W., Yang, X. Y., Yang, T. T., Factor, S. M., Lisanti, M. P., Molkentin, J. D., et al. (2003). Requirement of transcription factor NFAT in developing atrial myocardium. J. Cell Biol. 161 (5), 861–874. doi:10.1083/jcb.200301058
Seifert, R., and Beste, K. Y. (2012). Allosteric regulation of nucleotidyl cyclases: an emerging pharmacological target. Sci. Signal. 5 (240), pe37. doi:10.1126/scisignal.2003466
Sena, C. M., Pereira, A. M., and Seiça, R. (2013). Endothelial dysfunction—a major mediator of diabetic vascular disease. Biochimica Biophysica Acta (BBA)-Molecular Basis Dis. 1832 (12), 2216–2231. doi:10.1016/j.bbadis.2013.08.006
Seth, A., Finkbeiner, M., Grischin, J., and Schultz, J. E. (2020). Gsα stimulation of mammalian adenylate cyclases regulated by their hexahelical membrane anchors. Cell Signal 68, 109538. doi:10.1016/j.cellsig.2020.109538
Shi, J., Yang, Y., Cheng, A., Xu, G., and He, F. (2020). Metabolism of vascular smooth muscle cells in vascular diseases. Am. J. Physiology-Heart Circulatory Physiology 319, H613–H631. doi:10.1152/ajpheart.00220.2020
Sikarwar, A. S., Hinton, M., Santhosh, K. T., Dhanaraj, P., Talabis, M., Chelikani, P., et al. (2018). Hypoxia inhibits adenylyl cyclase catalytic activity in a porcine model of persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell Mol. Physiol. 315 (6), L933–L944. doi:10.1152/ajplung.00130.2018
Sinha, S., and Sprang, S. (2006). Structures, mechanism, regulation and evolution of class III nucleotidyl cyclases. Rev. Physiology Biochem. Pharmacol. 157, 105–140. doi:10.1007/112_0603
Spangler, C. M., Spangler, C., Gottle, M., Shen, Y., Tang, W. J., Seifert, R., et al. (2008a). A fluorimetric assay for real-time monitoring of adenylyl cyclase activity based on terbium norfloxacin. Anal. Biochem. 381 (1), 86–93. doi:10.1016/j.ab.2008.06.014
Spangler, C. M., Spangler, C., and Schaerling, M. (2008b). Luminescent lanthanide complexes as probes for the determination of enzyme activities. Ann. N. Y. Acad. Sci. 1130, 138–148. doi:10.1196/annals.1430.008
Steer, M. L., and Levitzki, A. (1975). The control of adenylate cyclase by calcium in Turkey erythrocyte ghosts. J. Biol. Chem. 250 (6), 2080–2084. doi:10.1016/s0021-9258(19)41685-2
Storch, U., Straub, J., Erdogmus, S., Gudermann, T., and Mederos y Schnitzler, M. (2017). Dynamic monitoring of G i/o-protein-mediated decreases of intracellular cAMP by FRET-based Epac sensors. Pflügers Archiv-European J. Physiology 469, 725–737. doi:10.1007/s00424-017-1975-1
Streeto, J. M., and Reddy, W. J. (1967). An assay for adenyl cyclase. Anal. Biochem. 21 (3), 416–426. doi:10.1016/0003-2697(67)90316-8
Sugano, Y., Lai, N. C., Gao, M. H., Firth, A. L., Yuan, J.X.-J., Lew, W. Y., et al. (2011). Activated expression of cardiac adenylyl cyclase 6 reduces dilation and dysfunction of the pressure-overloaded heart. Biochem. biophysical Res. Commun. 405 (3), 349–355. doi:10.1016/j.bbrc.2010.12.113
Sunahara, R. K., Dessauer, C. W., and Gilman, A. G. (1996). Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 36 (1), 461–480. doi:10.1146/annurev.pa.36.040196.002333
Sunahara, R. K., Dessauer, C. W., Whisnant, R. E., Kleuss, C., and Gilman, A. G. (1997). Interaction of Gsalpha with the cytosolic domains of mammalian adenylyl cyclase. J. Biol. Chem. 272 (35), 22265–22271. doi:10.1074/jbc.272.35.22265
Syed, A. U., Reddy, G. R., Ghosh, D., Prada, M. P., Nystoriak, M. A., Morotti, S., et al. (2019). Adenylyl cyclase 5–generated cAMP controls cerebral vascular reactivity during diabetic hyperglycemia. J. Clin. investigation 129 (8), 3140–3152. doi:10.1172/JCI124705
Takahashi, T., Tang, T., Lai, N. C., Roth, D. M., Rebolledo, B., Saito, M., et al. (2006). Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation 114 (5), 388–396. doi:10.1161/CIRCULATIONAHA.106.632513
Tan, C. M., Kelvin, D. J., Litchfield, D. W., Ferguson, S. S., and Feldman, R. D. (2001). Tyrosine kinase-mediated serine phosphorylation of adenylyl cyclase. Biochemistry 40 (6), 1702–1709. doi:10.1021/bi0015818
Tan, Y.-Q., Li, J., and Chen, H.-W. (2022). Epac, a positive or negative signaling molecule in cardiovascular diseases. Biomed. Pharmacother. 148, 112726. doi:10.1016/j.biopha.2022.112726
Tan, Z., Giamouridis, D., Lai, N. C., Kim, Y. C., Guo, T., Xia, B., et al. (2019). Cardiac-directed expression of adenylyl cyclase catalytic domain (C1C2) attenuates deleterious effects of pressure overload. Hum. Gene Ther. 30 (6), 682–692. doi:10.1089/hum.2018.176
Tang, T., Gao, M. H., Lai, N. C., Firth, A. L., Takahashi, T., Guo, T., et al. (2008). Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation 117 (1), 61–69. doi:10.1161/CIRCULATIONAHA.107.730069
Tang, T., Gao, M. H., Roth, D. M., Guo, T., and Hammond, H. K. (2004). Adenylyl cyclase type VI corrects cardiac sarcoplasmic reticulum calcium uptake defects in cardiomyopathy. Am. J. Physiology-Heart Circulatory Physiology 287 (5), H1906–H1912. doi:10.1152/ajpheart.00356.2004
Tang, T., Hammond, H. K., Firth, A., Yang, Y., Gao, M. H., Yuan, J.X.-J., et al. (2011). Adenylyl cyclase 6 improves calcium uptake and left ventricular function in aged hearts. J. Am. Coll. Cardiol. 57 (18), 1846–1855. doi:10.1016/j.jacc.2010.11.052
Tang, T., Lai, N. C., Hammond, H. K., Roth, D. M., Yang, Y., Guo, T., et al. (2010). Adenylyl cyclase 6 deletion reduces left ventricular hypertrophy, dilation, dysfunction, and fibrosis in pressure-overloaded female mice. J. Am. Coll. Cardiol. 55 (14), 1476–1486. doi:10.1016/j.jacc.2009.11.066
Tang, T., Lai, N. C., Roth, D. M., Drumm, J., Guo, T., Lee, K., et al. (2006). Adenylyl cyclase type V deletion increases basal left ventricular function and reduces left ventricular contractile responsiveness to beta-adrenergic stimulation. Basic Res. Cardiol. 101, 117–126. doi:10.1007/s00395-005-0559-y
Tang, T., Lai, N. C., Wright, A. T., Gao, M. H., Lee, P., Guo, T., et al. (2013). Adenylyl cyclase 6 deletion increases mortality during sustained β-adrenergic receptor stimulation. J. Mol. Cell. Cardiol. 60, 60–67. doi:10.1016/j.yjmcc.2013.04.005
Tang, W.-J., and Gilman, A. G. (1995). Construction of a soluble adenylyl cyclase activated by Gs alpha and forskolin. Science 268 (5218), 1769–1772. doi:10.1126/science.7792604
Tao, Y.-P., Najafi, L., Shipley, S., Howlett, A., and Klein, C. (1998). Effects of nitric oxide on adenylyl cyclase stimulation in N18TG2 neuroblastoma cells. J. Pharmacol. Exp. Ther. 286 (1), 298–304.
Tarasov, K. V., Chakir, K., Riordon, D. R., Lyashkov, A. E., Ahmet, I., Perino, M. G., et al. (2022). A remarkable adaptive paradigm of heart performance and protection emerges in response to marked cardiac-specific overexpression of ADCY8. Elife 11, e80949. doi:10.7554/eLife.80949
Taussig, R., Tang, W.-J., Hepler, J. R., and Gilman, A. G. (1994). Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J. Biol. Chem. 269 (8), 6093–6100. doi:10.1016/s0021-9258(17)37574-9
Tepe, N. M., and Liggett, S. B. (1999). Transgenic replacement of type V adenylyl cyclase identifies a critical mechanism of beta-adrenergic receptor dysfunction in the G alpha q overexpressing mouse. FEBS Lett. 458 (2), 236–240. doi:10.1016/s0014-5793(99)01147-3
Tesmer, J. J., Dessauer, C. W., Sunahara, R. K., Murray, L. D., Johnson, R. A., Gilman, A. G., et al. (2000). Molecular basis for P-site inhibition of adenylyl cyclase. Biochemistry 39 (47), 14464–14471. doi:10.1021/bi0015562
Tesmer, J. J., Sunahara, R. K., Gilman, A. G., and Sprang, S. R. (1997). Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science 278 (5345), 1907–1916. doi:10.1126/science.278.5345.1907
Tewson, P., Martinka, S., Shaner, N., Berlot, C., Quinn, A. M., and Hughes, T. (2018). Assay for detecting Gαi-mediated decreases in cAMP in living cells. SLAS Discov. Adv. Life Sci. R&D 23 (9), 898–906. doi:10.1177/2472555218786238
Thangavel, M., Liu, X., Sun, S. Q., Kaminsky, J., and Ostrom, R. S. (2009). The C1 and C2 domains target human type 6 adenylyl cyclase to lipid rafts and caveolae. Cell. Signal. 21 (2), 301–308. doi:10.1016/j.cellsig.2008.10.017
Timofeyev, V., Myers, R. E., Kim, H. J., Woltz, R. L., Sirish, P., Heiserman, J. P., et al. (2013). Adenylyl cyclase subtype–specific compartmentalization: differential regulation of L-type Ca2+ current in ventricular myocytes. Circulation Res. 112 (12), 1567–1576. doi:10.1161/CIRCRESAHA.112.300370
Tobise, K., Ishikawa, Y., Holmer, S., Im, M., Newell, J., Yoshie, H., et al. (1994). Changes in type VI adenylyl cyclase isoform expression correlate with a decreased capacity for cAMP generation in the aging ventricle. Circulation Res. 74 (4), 596–603. doi:10.1161/01.res.74.4.596
Toya, Y., Schwencke, C., and Ishikawa, Y. (1998). Forskolin derivatives with increased selectivity for cardiac adenylyl cyclase. J. Mol. Cell. Cardiol. 30 (1), 97–108. doi:10.1006/jmcc.1997.0575
Tsunematsu, T., Okumura, S., Mototani, Y., Ohnuki, Y., Jin, H., Cai, W., et al. (2015). Coupling of β1-adrenergic receptor to type 5 adenylyl cyclase and its physiological relevance in cardiac myocytes. Biochem. Biophys. Res. Commun. 458 (3), 531–535. doi:10.1016/j.bbrc.2015.01.149
Tsutsui, K., Florio, M. C., Yang, A., Wirth, A. N., Yang, D., Kim, M. S., et al. (2021). cAMP-dependent signaling restores AP firing in dormant SA node cells via enhancement of surface membrane currents and calcium coupling. Front. Physiology 12, 596832. doi:10.3389/fphys.2021.596832
Tykocki, N. R., Boerman, E. M., and Jackson, W. F. (2017). Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr. Physiol. 7 (2), 485–581. doi:10.1002/cphy.c160011
Valkovic, A. L., Leckey, M. B., Whitehead, A. R., Hossain, M. A., Inoue, A., Kocan, M., et al. (2018). Real-time examination of cAMP activity at relaxin family peptide receptors using a BRET-based biosensor. Pharmacol. Res. Perspect. 6 (5), e00432. doi:10.1002/prp2.432
Vatner, D. E., Yan, L., Lai, L., Yuan, C., Mouchiroud, L., Pachon, R. E., et al. (2015). Type 5 adenylyl cyclase disruption leads to enhanced exercise performance. Aging Cell 14 (6), 1075–1084. doi:10.1111/acel.12401
Vatner, S. F., Park, M., Yan, L., Lee, G. J., Lai, L., Iwatsubo, K., et al. (2013). Adenylyl cyclase type 5 in cardiac disease, metabolism, and aging. Am. J. Physiology-Heart Circulatory Physiology 305 (1), H1–H8. doi:10.1152/ajpheart.00080.2013
Vila-Petroff, M. G., Younes, A., Egan, J., Lakatta, E. G., and Sollott, S. J. (1999). Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circulation Res. 84 (9), 1020–1031. doi:10.1161/01.res.84.9.1020
von Hayn, K., Werthmann, R. C., Nikolaev, V. O., Hommers, L. G., Lohse, M. J., and Bunemann, M. (2010). Gq-mediated Ca2+ signals inhibit adenylyl cyclases 5/6 in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 298 (2), C324–C332. ajpcell.00197.2009 [pii]. doi:10.1152/ajpcell.00197.2009
Wang, T., and Brown, M. (2004). Differential expression of adenylyl cyclase subtypes in human cardiovascular system. Mol. Cell. Endocrinol. 223 (1-2), 55–62. doi:10.1016/j.mce.2004.05.012
Warrier, S., Belevych, A. E., Ruse, M., Eckert, R. L., Zaccolo, M., Pozzan, T., et al. (2005). Beta-adrenergic- and muscarinic receptor-induced changes in cAMP activity in adult cardiac myocytes detected with FRET-based biosensor. Am. J. Physiology-Cell Physiology 289 (2), C455–C461. doi:10.1152/ajpcell.00058.2005
Watson, E. L., Singh, J. C., Jacobson, K. L., and Ott, S. M. (2001). Nitric oxide inhibition of cAMP synthesis in parotid acini: regulation of type 5/6 adenylyl cyclase. Cell. Signal. 13 (10), 755–763. doi:10.1016/s0898-6568(01)00204-2
White, M., Roden, R., Minobe, W., Khan, M. F., Larrabee, P., Wollmering, M., et al. (1994). Age-related changes in beta-adrenergic neuroeffector systems in the human heart. Circulation 90 (3), 1225–1238. doi:10.1161/01.cir.90.3.1225
Willoughby, D., and Cooper, D. M. (2007). Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol. Rev. 87 (3), 965–1010. doi:10.1152/physrev.00049.2006
Wu, G.-C., Lai, H.-L., Lin, Y.-W., Chu, Y.-T., and Chern, Y. (2001). N-glycosylation and residues Asn805 and Asn890 are involved in the functional properties of type VI adenylyl cyclase. J. Biol. Chem. 276 (38), 35450–35457. doi:10.1074/jbc.M009704200
Wu, Y., and Jiang, T. (2022). Developments in FRET-and BRET-based biosensors. Micromachines 13 (10), 1789. doi:10.3390/mi13101789
Wu, Y.-S., Chen, C.-C., Chien, C.-L., Lai, H.-L., Jiang, S.-T., Chen, Y.-C., et al. (2017). The type VI adenylyl cyclase protects cardiomyocytes from β-adrenergic stress by a PKA/STAT3-dependent pathway. J. Biomed. Sci. 24 (1), 68–12. doi:10.1186/s12929-017-0367-3
Yan, L., Park, J. Y., Dillinger, J. G., De Lorenzo, M. S., Yuan, C., Lai, L., et al. (2012). Common mechanisms for calorie restriction and adenylyl cyclase type 5 knockout models of longevity. Aging Cell 11 (6), 1110–1120. doi:10.1111/acel.12013
Yan, L., Vatner, D. E., O'Connor, J. P., Ivessa, A., Ge, H., Chen, W., et al. (2007). Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 130 (2), 247–258. doi:10.1016/j.cell.2007.05.038
Yokochi, A., Itoh, H., Maruyama, J., Zhang, E., Jiang, B., Mitani, Y., et al. (2010). Colforsin-induced vasodilation in chronic hypoxic pulmonary hypertension in rats. J. Anesth. 24 (3), 432–440. doi:10.1007/s00540-010-0912-7
Yokoyama, U., Minamisawa, S., Katayama, A., Tang, T., Suzuki, S., Iwatsubo, K., et al. (2010). Differential regulation of vascular tone and remodeling via stimulation of type 2 and type 6 adenylyl cyclases in the ductus arteriosus. Circulation Res. 106 (12), 1882–1892. doi:10.1161/CIRCRESAHA.109.214924
Yoneyama, M., Sugiyama, A., Satoh, Y., Takahara, A., Nakamura, Y., and Hashimoto, K. (2002). Cardiovascular and adenylate cyclase stimulating effects of colforsin daropate, a water-soluble forskolin derivative, compared with those of isoproterenol, dopamine and dobutamine. Circulation J. 66 (12), 1150–1154. doi:10.1253/circj.66.1150
Younes, A., Lyashkov, A. E., Graham, D., Sheydina, A., Volkova, M. V., Mitsak, M., et al. (2008). Ca2+-stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 283 (21), 14461–14468. doi:10.1074/jbc.M707540200
Zhang, J., Levy, D., Oydanich, M., Bravo, C. A., Yoon, S., Vatner, D. E., et al. (2018). A novel adenylyl cyclase type 5 inhibitor that reduces myocardial infarct size even when administered after coronary artery reperfusion. J. Mol. Cell. Cardiol. 121, 13–15. doi:10.1016/j.yjmcc.2018.05.014
Zhang, X., Szeto, C., Gao, E., Tang, M., Jin, J., Fu, Q., et al. (2013). Cardiotoxic and cardioprotective features of chronic β-adrenergic signaling. Circulation Res. 112 (3), 498–509. doi:10.1161/CIRCRESAHA.112.273896
Zhang, Y., Chen, S., Luo, L., Greenly, S., Shi, H., Xu, J. J., et al. (2023). Role of cAMP in cardiomyocyte viability: beneficial or detrimental? Circulation Res. 133 (11), 902–923. doi:10.1161/CIRCRESAHA.123.322652
Keywords: adenylyl cyclase, G protein-coupled receptors, cyclic 3′,5′-adenosine monophosphate, signal transduction, heart disease, drug discovery
Citation: Maghsoudi S, Shuaib R, Van Bastelaere B and Dakshinamurti S (2024) Adenylyl cyclase isoforms 5 and 6 in the cardiovascular system: complex regulation and divergent roles. Front. Pharmacol. 15:1370506. doi: 10.3389/fphar.2024.1370506
Received: 14 January 2024; Accepted: 11 March 2024;
Published: 03 April 2024.
Edited by:
Ismail Laher, University of British Columbia, CanadaReviewed by:
Madeline Nieves-Cintron, University of California, Davis, United StatesCristina Ribas Fürstenau, Federal University of ABC, Brazil
Alexey V. Glukhov, University of Wisconsin-Madison, United States
Copyright © 2024 Maghsoudi, Shuaib, Van Bastelaere and Dakshinamurti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shyamala Dakshinamurti, c2h5YW1hbGEuZGFrc2hpbmFtdXJ0aUB1bWFuaXRvYmEuY2E=