 Yujie Yang1
Yujie Yang1 Liang Zheng
Liang Zheng- 1Department of Pharmacy, The Third People’s Hospital of Chengdu, College of Medicine, Southwest Jiaotong University, Chengdu, China
- 2Department of Clinical Pharmacology, The Second Affiliated Hospital of Anhui Medical University, Hefei, China
Objectives: Codeine, a prodrug used as an opioid agonist, is metabolized to the active product morphine by CYP2D6. This study aimed to establish physiologically based pharmacokinetic (PBPK) models of codeine and morphine and explore the influence of CYP2D6 genetic polymorphisms on the pharmacokinetics of codeine and morphine.
Methods: An initial PBPK modeling of codeine in healthy adults was established using PK-Sim® software and subsequently extrapolated to CYP2D6 phenotype-related PBPK modeling based on the turnover frequency (Kcat) of CYP2D6 for different phenotype populations (UM, EM, IM, and PM). The mean fold error (MFE) and geometric mean fold error (GMFE) methods were used to compare the differences between the predicted and observed values of the pharmacokinetic parameters to evaluate the accuracy of PBPK modeling. The validated models were then used to support dose safety for different CYP2D6 phenotypes.
Results: The developed and validated CYP2D6 phenotype-related PBPK model successfully predicted codeine and morphine dispositions in different CYP2D6 phenotypes. Compared with EMs, the predicted AUC0-∞ value of morphine was 98.6% lower in PMs, 60.84% lower in IMs, and 73.43% higher in UMs. Morphine plasma exposure in IMs administered 80 mg of codeine was roughly comparable to that in EMs administered 30 mg of codeine. CYP2D6 UMs may start dose titration to achieve an optimal individual regimen and avoid a single dose of over 20 mg. Codeine should not be used in PMs for pain relief, considering its insufficient efficacy.
Conclusion: PBPK modeling can be applied to explore the dosing safety of codeine and can be helpful in predicting the effect of CYP2D6 genetic polymorphisms on drug–drug interactions (DDIs) with codeine in the future.
1 Introduction
Codeine is a relatively selective opioid agonist for the mu-opioid receptor but has a much weaker affinity than morphine (Thorn et al., 2009). Of these, approximately 70%–80% of codeine is metabolized by UDP-glucuronyl transferase (UGT) 2B7 and 2B4 to form codeine-6-glucuronide, and 5%–15% of codeine is N-demethylated by cytochrome P450 3A4 (CYP3A4) to produce demethylcodeine. A small percentage of demethylcodeine was further O-demethylated to form demethylmorphine. The most crucial metabolism of codeine occurs via cytochrome P450 2D6 (CYP2D6), and CYP2D6 demethylates 5%–10% of codeine to form the active metabolite morphine. Approximately 60% of morphine is converted to inactive morphine-3-glucuronide (M3G) and approximately 10% to active morphine-6-glucuronide (M6G) in the liver, mainly by UGT (FDA, 2023). Morphine and M6G have opioid activities, and the analgesic properties of codeine have been speculated to originate from its conversion to morphine. CYP2D6 is a crucial factor for the curative effect of codeine in vivo.
CYP2D6 is a highly polymorphic gene, with many crucial single-nucleotide polymorphisms, haplotypes, and copy number variants (Gough et al., 1993). CYP2D6 genetic polymorphisms can lead to differences in enzyme activity, directly affecting drug exposure in vivo. According to the different enzymatic activities, CYP2D6 variants can be categorized into four phenotypes as follows: extensive metabolizer (EM), intermediate metabolizer (IM), poor metabolizer (PM), and ultra-rapid metabolizer (UM) (Somogyi et al., 2007). Pharmacokinetic (PK) studies have shown significant differences in the drug exposure of the codeine metabolite morphine in populations with different CYP2D6 gene phenotypes (Linares et al., 2015). Genetic polymorphisms in CYP2D6 are associated with diminished pain relief or severe side effects. However, the Food and Drug Administration (FDA) has not published recommendations for the administration schedule adjustment of codeine in populations with different CYP2D6 phenotypes. Therefore, effective methods are needed to provide reliable evidence for the clinical application of codeine in different phenotypes.
Physiologically based pharmacokinetic (PBPK) modeling can simulate the process of drug disposal in the body based on anatomy, physiology, biochemistry, and physical chemistry, where tissues or organs are connected by blood circulation (Grimstein et al., 2019). The essential components of the PBPK model include drug- and non-drug-dependent physiologies. Non-drug-dependent physiology includes the abundance and activity of drug transporters and metabolic enzymes. With the development of PBPK software and human genomics in recent years, pharmacogenomics has provided new ideas for personalized medicine with the help of PBPK modeling. Previous studies have developed CYP2D6 phenotype-related PBPK models to explore the effect of CYP2D6 gene polymorphisms on pharmacokinetics (Grzegorzewski et al., 2022) and elucidate the extent of CYP2D6-mediated drug–drug interactions (DDIs) (Dickschen et al., 2017; Xu et al., 2021). PBPK modeling can reduce unnecessary clinical trials and maximize existing data. We used data extrapolation to improve and enrich drug use information for populations with different CYP2D6 gene phenotypes.
Several studies have used the PBPK model to explore the effects of ontogeny and genetic variation of organic cation transporter 1 or UGT2B7 on morphine disposal (Emoto et al., 2018; Schaefer et al., 2018; Sjöstedt et al., 2021). However PBPK modeling has not been established to explore the dose safety of codeine for different CYP2D6 phenotypes. Our study aimed to develop a CYP2D6 phenotype-related PBPK model of codeine and morphine and provide a scientific basis for codeine pharmacokinetics in different CYP2D6 phenotypes. This study also aimed to expand the knowledge field of CYP2D6 drug–gene interactions and DDI scenarios.
2 Methods
2.1 PBPK modeling development workflow and computer software
This study used a ‘bottom-up’ strategy to facilitate the PBPK modeling of codeine and morphine (Figure 1). First, we developed initial PBPK models of codeine and morphine without considering CYP2D6 phenotypes. Subsequently, CYP2D6 phenotype-related PBPK modeling was developed based on the initial model.

Figure 1. Schematic representation of the workflow of PBPK modeling development.
The physicochemical and ADME properties of codeine and morphine and reported PK profiles following i. v. and p. o. administration of codeine were obtained by searching PubMed, Web of Science, the MEDLINE database, or the “DrugBank” database using the keywords ‘codeine,’ ‘morphine,’ and ‘pharmacokinetics.’ The particular keywords for adults with different CYP2D6 phenotypes were ‘CYP2D6,’ ‘phenotype,’ ‘genotype,’ and ‘metabolizers.’ The collected literature was organized according to the experimental data (plasma concentration–time profile) and dosing regimen, and we excluded studies on the determination of codeine and morphine plasma concentrations using the microbial method (Supplementary Table S1).
All simulations were performed using PK-Sim® (version 11.2.0). Plasma concentration–time profile data in the published literature were obtained using the GetData Graph Digitizer (S. Fedorov, version 2.25.0.32). The observed PK parameters were obtained from the literature or calculated using non-compartment analysis by Phoenix software (version 8.1.0). Graphics were created and edited using GraphPad Prism (version 6.02).
2.2 Codeine and morphine PBPK modeling development
The PBPK model, comprising 18 tissue/organ compartments, was developed using PK-Sim® model software, where human physiological parameters were obtained using the virtual population. The physicochemical and ADME parameters of codeine and morphine are shown in Table 1. Codeine is metabolized in the liver mainly by CYP2D6 (morphine), CYP3A4 (demethylcodeine), and UGT2B7 (codeine-6-glucuronide, C6G). The relevant enzyme kinetic parameters (Km and Kcat) were optimized using parameter identification with the built-in Monte Carlo algorithm by fitting to observed PK data. Parameter identification is a means of parameter optimization to minimize the residuals between observed PK data and corresponding simulation output (codeine and morphine blood concentration) by changing the selected input parameters (Km and Kcat) in a given range. Renal clearance (CLR) combines the glomerular filtration rate (GFR), tubular secretion, and reabsorption. The total CLR was corrected according to Susanne, which met the observed value of the fraction excreted unchanged via the kidney. UGT2B7 can further metabolize morphine to form glucuronides. Liver clearance is characterized by total liver clearance, and CLR is optimized using DrugBank data. In addition, the PK-Sim® standard method was used to estimate cellular permeability, and the Rodgers and Rowland method was used for determining the partition coefficient.
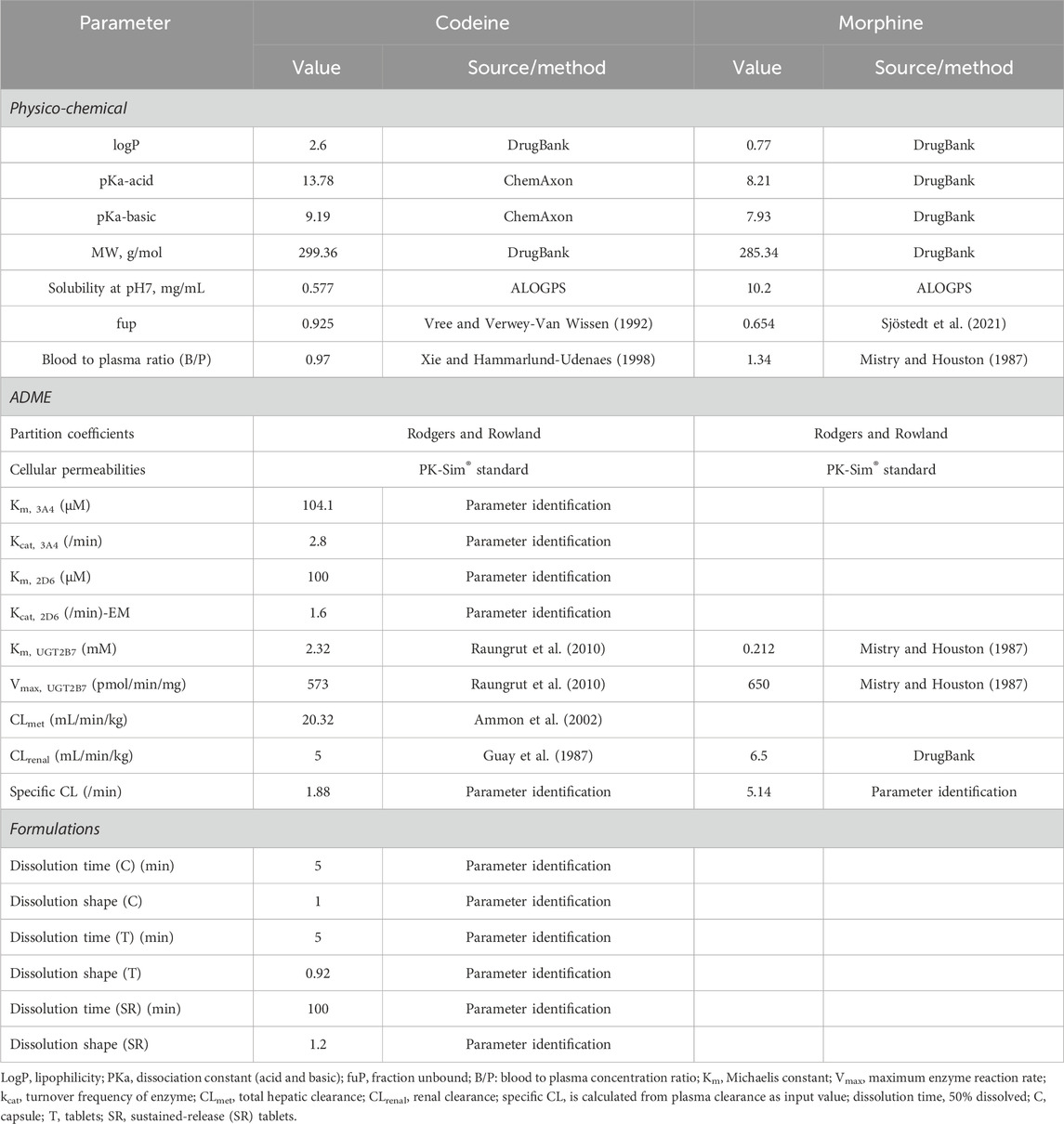
Table 1. Summary of input compound parameters of codeine and morphine.
Oral formulations included immediate-release (IR) tablets, sustained-release (SR) tablets, control-release (CR) solutions, and ordinary tablets/capsules/solutions. The IR tablet formulation was set as dissolved. The release features of the uncoated tablet/capsule and SR tablets were described using the Weibull function. The different dosage forms of codeine were calculated and converted into the corresponding administration regimens according to the content of codeine.
2.3 CYP2D6 phenotype-related PBPK modeling development
Based on the model of codeine and morphine without considering the genotype, the physicochemical properties of all drug parameters (molecular weight, log P, or Kp values) were fixed, and the genotype activity value of CYP2D6 was used to determine the phenotype of CYP2D6. CYP2D6 genotyping was conducted based on the activity score (AS) of the different CYP2D6 alleles divided into EM, IM, PM, and UM types (Supplementary Table S2). The turnover frequency (Kcat) of the EM type with an AS value of 1.5 was used as a reference (100%) and set to 1.6/min, and Kcat of other phenotypes was calculated according to the ratio of the AS value. Kcat of the CYP2D6 IM type was calculated as the median value of PM and EM and verified by clinical studies (Table 2). To avoid affecting the calculation, Kcat of the CYP2D6 PM type was not directly set to 0. The Monte Carlo method used the EM, PM, IM, and UM clinical data to optimize Kcat of CYP2D6.
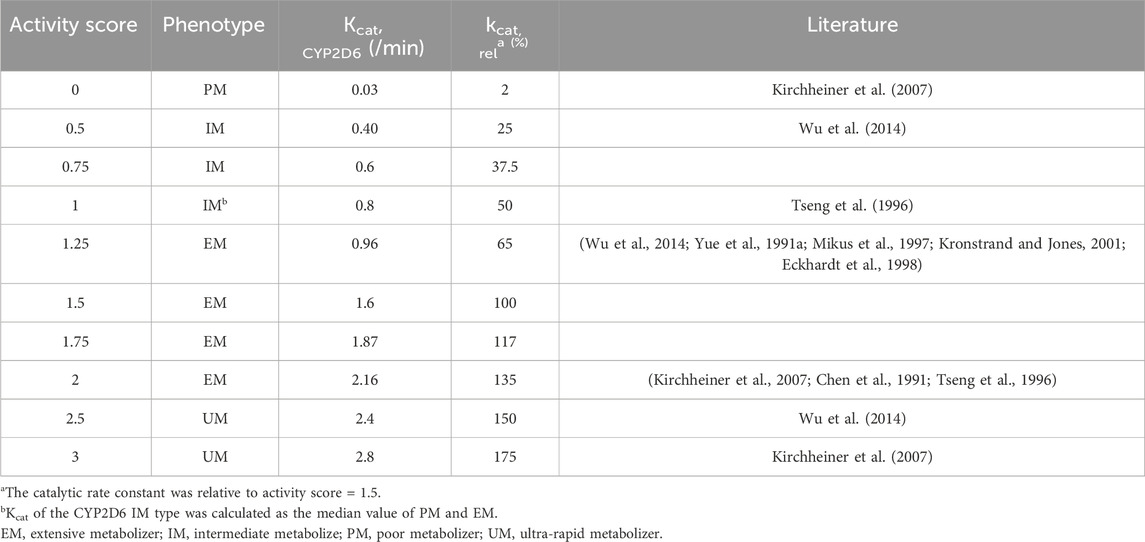
Table 2. Optimized Kcat, rel values for the different modeled CYP2D6 activity scores.
2.4 PBPK modeling evaluation
For the PBPK model evaluation, we used virtual populations generated by software with the same ethnic and demographic information as in the original literature, and the model performance was evaluated using different methods. We visually inspected the simulated and observed data following various dosing regimens. In addition, the goodness-of-fit of the plasma drug concentration and PK parameters were calculated. The PK parameters of the observed values were directly obtained from geometric or arithmetic means reported in the literature. If no PK parameters were reported, the intercepted plasma drug concentration values were used for non-compartment analysis. The mean fold error (MFE) and geometric mean fold error (GMFE) methods were used to compare the differences between the predicted and observed values of the PK parameters to evaluate the accuracy of the PBPK model. When the MFE and GMFE of PK parameters were within a 0.5- to 2-fold error (Sager et al., 2015) range, we considered the PBPK model establishment successfully.
2.5 Codeine dose simulation
We simulated codeine doses in different genotypes using genotype-dependent CYP2D6-mediated clearances of codeine. First, we analyzed the predictions of codeine and morphine in different populations with different CYP2D6 phenotypes under an administration regimen of 30 mg of codeine tablets. Then, we used the established PBPK models to simulate the situation in Asians with different CYP2D6 phenotypes after different doses. To intuitively compare different CYP2D6 phenotypes, we used the AS median value of each phenotype as a representative. All simulations were investigated in 1,000 virtual patients.
3 Results
3.1 Healthy adult PBPK modeling establishment and evaluation
First, we developed a PBPK model of codeine i. v. administration based on a typical 30-year-old European male individual with average physiological parameters. We found that using the fractions of excreted urine unchanged was consistent with the observed data obtained from the European adult population (Figure 2). We then developed the PBPK model of codeine p. o. administration based on a typical 30-year-old European and Asian male individual. Parameters were randomly optimized to obtain the best-fit parameters using the observed values from four clinical studies. The established adult codeine and morphine PBPK models are shown in Figure 2. GMFEs of AUC0-∞ and Cmax in established adult codeine, unconjugated codeine, and morphine PBPK models are shown in Supplementary Tables S3, S4.
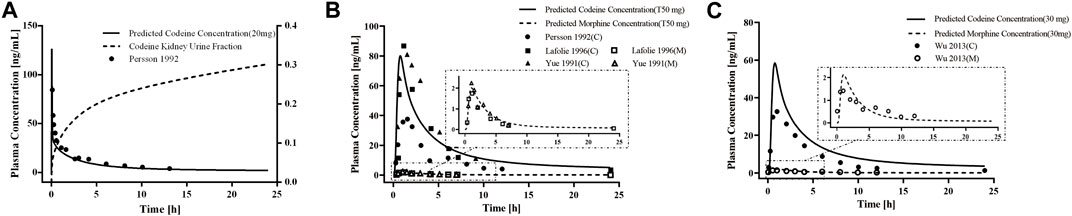
Figure 2. PBPK modeling established in healthy adults. Simulation (lines) of PK profiles for a single (A) i.v. administration of 20 mg of codeine and (B) p. o. administration of 50 mg of codeine tablet in a typical European individual, (C) p. o. administration of 30 mg of codeine tablet in a typical Asian individual. The observed concentration data were provided as the arithmetic mean values extracted from references. The solid and dotted lines represented the predicted codeine and morphine concentrations, respectively.
For the healthy adult PBPK model evaluation, 11 clinical studies following various dosing regimens were identified and used (Figure 3). For the model evaluation, models with single dosages of capsules, solutions, tablets, and sustained-release tablets and multiple dosages of tablets, sustained-release tablets, and control-release solutions were built. The goodness-of-fit diagram of codeine plasma drug concentrations showed that more than 89.11% of the predicted drug concentration values were within a 2-fold error range of the observed values, and approximately 68.09% of the predicted values were within a 1.5-fold margin of error (Figure 4). The metabolite morphine was not as well-fitted as codeine, with only 63.25% of the plasma drug concentrations within the 2-fold range. The MFE average and GMFE of all predicted PK parameters were within the 1.5-fold error range in both codeine and morphine PBPK models (Supplementary Table S4), except for the GMFE of AUC0-∞ in morphine PBPK models.
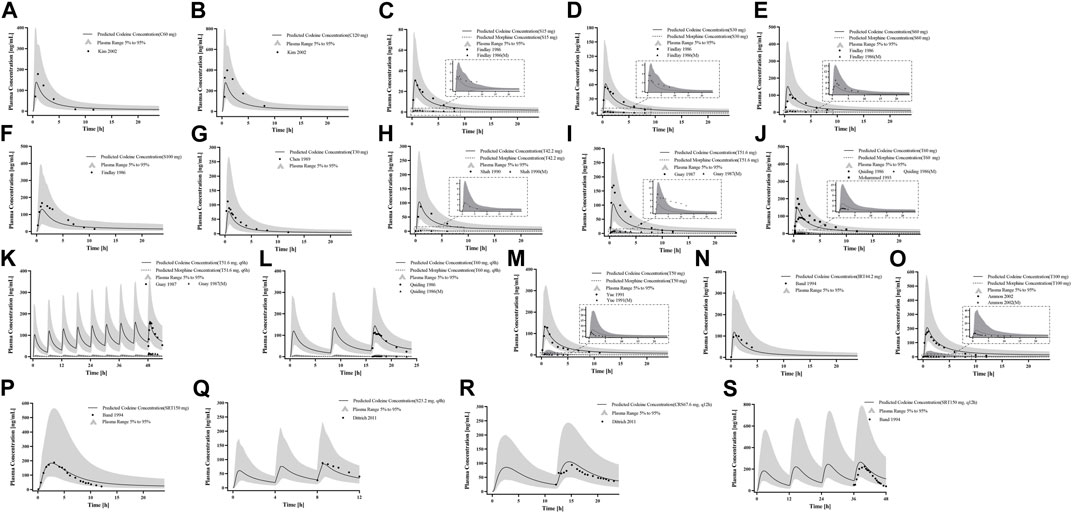
Figure 3. PK profile verification in healthy adults. (A, B) Single p. o. administration of codeine capsules in African Americans; (C–L) the p. o. administration at a dose or multiple doses of codeine in different formulations in White Americans; (M) the single p. o. administration of 50 mg codeine tablets in Asians; and (N–S) the p. o. administration at a dose or multiple doses of codeine in different formulations in Europeans. The observed concentration data were provided as the arithmetic mean values extracted from references. The solid and dotted lines represented the predicted codeine and morphine concentrations, respectively. The shaded area represented the predicted 5th to 95th percentile range.
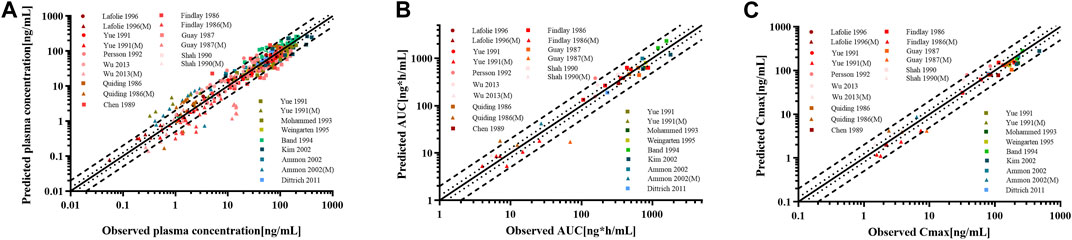
Figure 4. Goodness-of-fit plot for (A) plasma concentrations, (B) AUC0-∞, and (C) Cmax of codeine and morphine in the healthy adult population. The solid line represented the line of identity, and the bold dotted and dotted lines represented the 2-fold and 1.25-fold error ranges, respectively. The circles and squares indicated data used to PBPK modeling, building, and evaluation, and the triangle showed data about morphine.
3.2 CYP2D6 gene-related PBPK modeling establishment and evaluation
First, we predicted the PK of codeine and morphine in healthy adults with different CYP2D6 phenotypes using the physicochemical properties of the drugs along with different Kcat values of CYP2D6 (Figure 5). We found that the single AS value corresponding to the genotype could not simulate the level of morphine in vivo well; therefore, we further divided the range of AS values (Table 2). For the CYP2D6 gene-related model of codeine and morphine, eight clinical studies were used for model establishment and evaluation. We set Kcat of PM to 0.03/min according to the parameter identification. On visual inspection, the AS values of 0.5 and 1 fitted Wu et al. (2014) and Tseng et al. (1996) in IMs, and the AS values of 2.5 and 3 fitted Wu et al. (2014) and Kirchheiner et al. (2007) in UMs, respectively. As for the EMs, the AS value of 1.25 matched Wu et al. (2014), Yue et al. (1991a), Mikus et al. (1997), Kronstrand and Jones (2001), and Eckhardt et al. (1998), and the AS value of 2 matched Kirchheiner et al. (2007), Chen et al. (1991), and Tseng et al. (1996) (Figure 6).
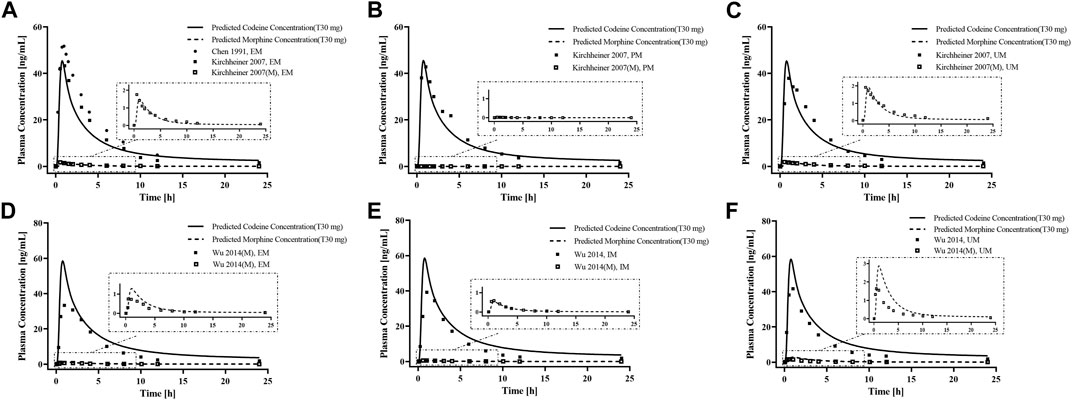
Figure 5. PBPK modeling established in healthy adults with different CYP2D6 phenotypes. Simulation (lines) of PK profiles for a single p. o. administration of 30 mg codeine tablet in a typical American individual with (A) EM type, (B) PM type, and (C) UM type, and p. o. administration of 30 mg codeine tablet in a typical Asian individual with (D) EM type, (E) IM type, and (F) UM type. The observed concentration data were provided as the arithmetic mean values extracted from references. The solid and dotted line represented the predicted codeine and morphine concentrations, respectively.
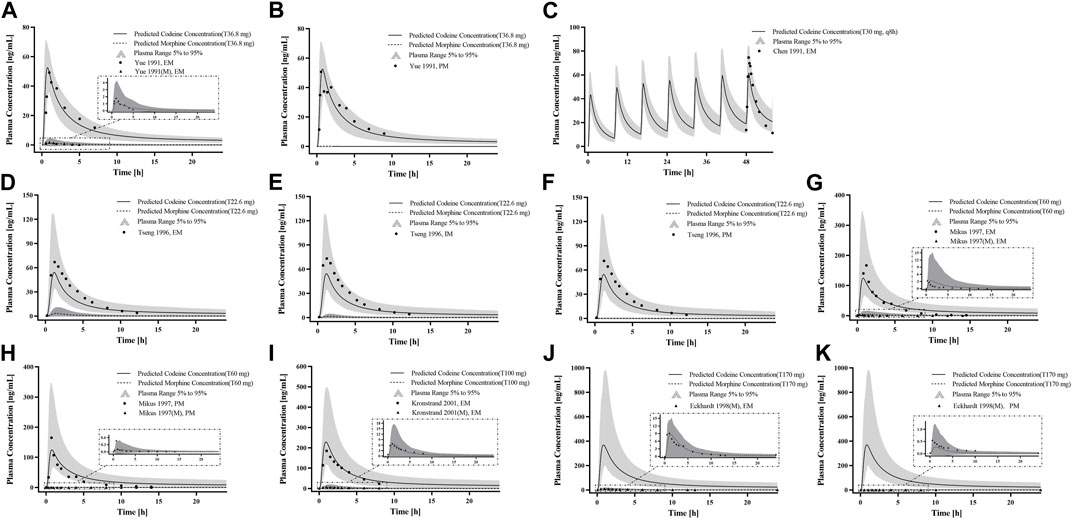
Figure 6. PK profile verification in adults with different CYP2D6 phenotypes. (A–C) p. o. administration at a dose or multiple doses of codeine tablets in Americans with EM and PM types; (D–F) the single p. o. administration of codeine tablets in Asians with EM, IM, and PM types; and (G–K) the single p. o. administration of codeine tablets in Europeans with EM and PM types. The observed concentration data were provided as the arithmetic mean values extracted from references. The solid line and dotted line represented the predicted codeine and morphine concentrations, respectively. The shaded area represented the predicted 5th to 95th percentile range.
The goodness-of-fit diagram of morphine plasma drug concentrations showed that more than 76.92% of the predicted drug concentration values were within the 2-fold error range of the observed values, and approximately 53.85% were within 1.5-fold (Figure 7). The results showed that the simulation of the metabolite morphine was better after distinguishing the CYP2D6 phenotypes. The average MFE and GMFE of AUC0-∞ and Cmax were within a 1.5-fold error range in EMs, IMs, and UMs (Supplementary Table S4). Due to the low concentration of morphine in PMs, the deviations of morphine tended to have larger GMEF values (1.45 and 1.84 for AUC0-∞ and Cmax), but this deviation was not clinically significant.
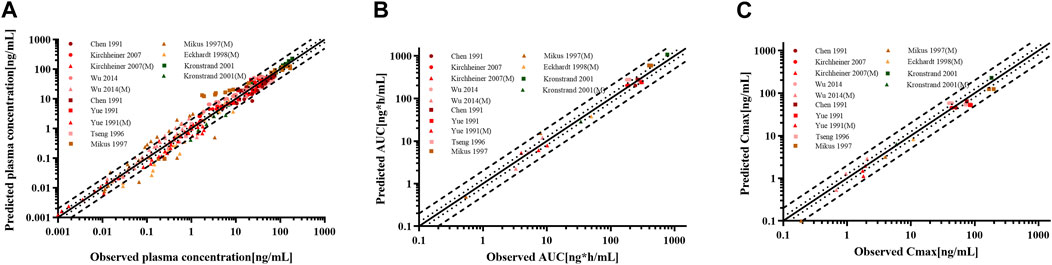
Figure 7. Goodness-of-fit plot for (A) plasma concentrations, (B) AUC0-∞, and (C) Cmax of codeine and morphine in adults with different CYP2D6 phenotypes. The solid line represented the line of identity, and the bold dotted lines and dotted lines represented the 2-fold and 1.25-fold error ranges, respectively. The circles and squares indicated data used for PBPK modeling, building, and evaluation, and the triangle showed data about morphine.
3.3 Codeine dose simulation based on CYP2D6 genetic polymorphism
Genetic polymorphisms in CYP2D6 are associated with diminished pain relief or severe side effects. Compared with EMs, the predicted AUC0-∞ of morphine was 98.6% lower in PMs, 60.84% lower in IMs, and 73.43% higher in UMs under an administration regimen of 30 mg codeine tablets (Supplementary Table S5). The results showed that the errors were controlled below 30%, except for codeine in the IMs. In addition, box–whisker and time-profile analyses were used for direct comparisons in Asians with different CYP2D6 phenotypes (Figure 8). As shown in Table 3, a dose or multiple doses of 80 mg of codeine in IMs, 1500 mg in PMs, and 20 mg in UMs had an approximately equivalent plasma exposure of morphine in EMs administered 30 mg of codeine. Notably, the simulated PM dose exceeded the daily maximum amount of codeine. Alternative analgesics are recommended for PMs who do not perform well on codeine.
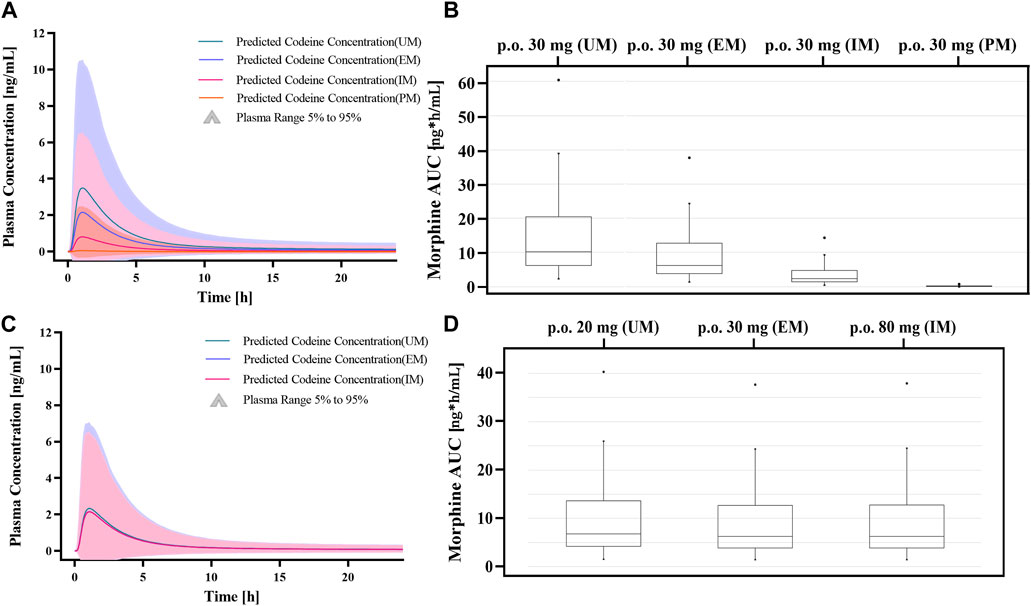
Figure 8. Dose simulations based on CYP2D6 gene-related PBPK modeling. (A, B) Simulations of morphine exposure and AUC0-∞ in Asians with different CYP2D6 phenotypes at administration of a 30 mg codeine tablet. (C, D) Simulations of morphine exposure and AUC0-∞ in Asians with different CYP2D6 phenotypes with the model-based dose.
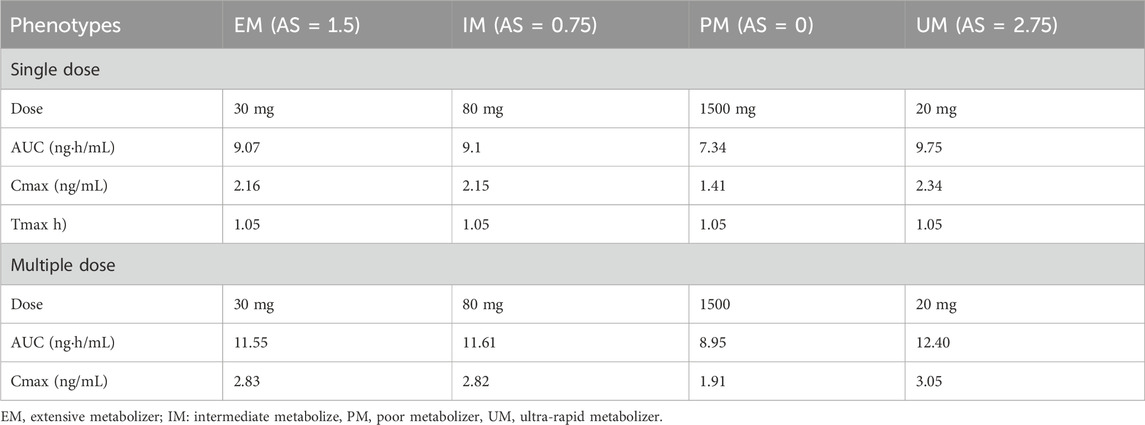
Table 3. Simulated pharmacokinetic parameters of morphine in Asians with different CYP2D6 phenotypes.
4 Discussion
Our study successfully developed a PBPK model of codeine and its active metabolite (morphine). Based on the verified basic model of codeine and morphine, the enzyme kinetic parameters of CYP2D6 with different gene phenotypes were optimized using the Monte Carlo method. The CYP2D6 gene-related PBPK modeling was successfully established using the PK data of clinical populations with different CYP2D6 gene phenotypes as observed values. The critical point in the establishment of PBPK modeling of codeine based on CYP2D6 genetic polymorphism is the change in enzyme kinetic parameters of different CYP2D6 gene phenotypes. Kcat of CYP2D6 can represent the catalytic efficiency, defined as the number of substrate molecules converted into a product at each enzyme site per unit of time. It was important to note that when we developed an initial PBPK model of codeine and morphine without considering CYP2D6 phenotypes, we set the Kcat rate of CYP2D6 to 1.6/min, precisely the median in the EMs. Therefore, the prediction of morphine in the initial PBPK modeling was poor, and the GMFE of AUC0-∞ in the morphine PBPK model was high. In addition, when we further adjusted the Kcat rate according to CYP2D6 phenotypes, we found that a single AS value corresponding to the CYP2D6 phenotypes could not accurately simulate the level of morphine in each literature work. So, we further divided the range of the AS value, where IMs included three levels of 0.5-1, EMs included four levels of 1.25–2, and UMs included two levels of 1.25–2. Notably, Kcat of CYP2D6 in the PM type was set to 0 in the previous model building (Chen et al., 2018). That is, the metabolic capacity of CYP2D6 in the PM type is entirely lost. However, previous clinical PK studies have shown that morphine generated by codeine via CYP2D6 was still exposed to CYP2D6 PMs (Kirchheiner et al., 2007). In this study, Kcat of CYP2D6 PM type was not set to 0 directly, and we set Kcat of PM to 0.03/min according to clinical PK studies of codeine and morphine in PMs (Kirchheiner et al., 2007).
In our predictions, the plasma exposure of morphine in IMs and PMs was significantly lower than that in EMs, which was consistent with observations in published studies. In our study, compared with EMs, the predicted AUC0-∞ value of morphine was 98.6% lower in PMs, 60.84% lower in IMs, and 73.43% higher in UMs. Notably, the adverse events related to morphine-related deaths were reported to be associated with ultrafast codeine metabolism (Ciszkowski et al., 2009). The FDA drug descriptions stated that the UMs converted codeine into its active metabolite, morphine, more rapidly and completely, resulting in higher serum morphine levels. Even with labeled dosage regimens or simulated dosages, UMs may have life-threatening or fatal respiratory depression or may experience signs of overdose (FDA, 2023). In addition, we found that the simulated dosage of PMs exceeded the daily maximum amount of codeine. The labeled dose of codeine may be unable to be efficiently converted to morphine. In summary, codeine should not be used by PMs for pain relief considering its insufficient efficacy; therefore, other drugs should be considered for analgesic sedation. For IMs, our predictions showed that the dose of codeine increased by a factor of 1.67 and had an approximately equivalent morphine plasma exposure as in EMs. The prevalence of CYP2D6 phenotypes varies widely. PMs and UMs have been estimated to be 5%–10% and 1%–10% for Caucasians (European and North American), 0%–19% and 3%–4% for Black people (African Americans) (Mendoza et al., 2001), respectively, and both 1% and 2% for Asians (Chinese, Japanese, and Korean) (Bradford, 2002). Additionally, UMs may be greater than 10% in certain racial/ethnic groups (that is, the Middle East) (Khalaj et al., 2019). An “a priori” and correct determination of CYP2D6 phenotypes, combined with our PBPK modeling, can promise individual predictability in drug exposure to morphine be realized.
The clinical implications of this study require further discussion. The medical indications for codeine include cough suppression and analgesia. The antitussive activity of codeine is approximately 1/4 that of morphine, and its analgesic effect is approximately 1/10 that of morphine (Beaver et al., 1978). However, the exposure of codeine after administration is much higher than that of morphine (>20 folds). Although the accurate relative contributions to in vivo activity have not been determined, evidence-based data have indicated the necessity to adjust the regimen according to the CYP2D6 genotype. The Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Dutch Pharmacogenetics Working Group (DPWG) both recommend avoiding codeine in individuals identified as CYP2D6 PMs for pain control due to significantly reduced morphine formation and insufficient pain relief (Pratt et al., 2012). This study confirms the recommendation by indicating that a markedly excessive dose of codeine is needed in PMs to obtain similar exposure to morphine. On the other hand, the DPWG recommends that no actions be required in IMs and PMs for cough prevention (Matic et al., 2022). This reflects the organization’s opinion that the antitussive activity comes primarily from the parent compound. According to the FDA’s codeine medication label, UMs may develop symptoms of overdose or potentially deadly respiratory depression, even at prescribed dosages (FDA, 2017). Although the FDA suggests that CYP2D6 UMs should not use codeine tablets, the DPWG is considered a lower dose acceptable (Matic et al., 2022). According to our simulation, a 1/3 dose reduction in UMs compared to EMs is appropriately measured by morphine formation, while the discrepancy in codeine exposure is minimal. In practice, CYP2D6 UMs may start dose titration to achieve an optimal individual regimen and avoid a single dose over 20 mg. With accumulated phenotype-based efficacy evidence, the models developed in this study can be easily used to formulate dose adjustments.
We acknowledge that this study has limitations. We did not perform pharmacodynamics simulations to determine the effects of CYP2D6 genetic polymorphisms on codeine efficacy (separately for pain control and cough prevention). Relying only on pharmacokinetic information to guide the individual administration of a drug has limitations to some extent. This study also did not address the drug evaluation at extremes of age, and we did not distinguish the difference in CYP2D6 activity between different ethnic groups in the healthy adult models. More studies are needed to explore the pharmacodynamic modeling of codeine in the future, especially the effects of genetic polymorphisms in specific populations. Our study provided a theoretical basis for exploring CYP2D6-mediated codeine interactions with other drugs. For more accurate DDI scenarios, the enzyme kinetic parameters for both codeine and inducer/inhibitors should be explored.
5 Conclusion
We developed and validated CYP2D6 phenotype-related PBPK models for codeine and its active metabolite morphine, which can predict codeine and morphine plasma concentration–time profiles. The model focused on CYP2D6 phenotype-related metabolism and has been used to explore the dosing safety of codeine for different CYP2D6 phenotypes. This study expands the knowledge of CYP2D6 drug–gene interactions and lays the foundation for future exploration of CYP2D6 DDI scenarios.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Third People’s Hospital of Chengdu. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
YY: conceptualization, funding acquisition, project administration, software, validation, and writing–original draft. XZ: data curation, validation, and writing–original draft. YW: data curation and writing–original draft. HX: validation and writing–review and editing. MX: supervision and writing–review and editing. LZ: conceptualization, funding acquisition, methodology, supervision, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Seedling Engineering project of Sichuan Science and Technology Department (no. 2022JDRC0145) and the Anhui Provincial Natural Science Foundation (no. 2208085QH279).
Acknowledgments
All authors are thankful to all members in the Open Systems Pharmacology community in GitHub.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1342515/full#supplementary-material
References
Ammon, S., Marx, C., Behrens, C., Hofmann, U., Mürdter, T., Griese, E. U., et al. (2002). Diclofenac does not interact with codeine metabolism in vivo: a study in healthy volunteers. BMC Clin. Pharmacol. 2, 2. doi:10.1186/1472-6904-2-2
Band, C. J., Band, P. R., Deschamps, M., Besner, J. G., and Coldman, A. J. (1994). Human pharmacokinetic study of immediate-release (codeine phosphate) and sustained-release (codeine Contin) codeine. J. Clin. Pharmacol. 34, 938–943. doi:10.1002/j.1552-4604.1994.tb04008.x
Beaver, W. T., Wallenstein, S. L., Rogers, A., and Houde, R. W. (1978). Analgesic studies of codeine and oxycodone in patients with cancer. I. Comparisons of oral with intramuscular codeine and of oral with intramuscular oxycodone. J. Pharmacol. Exp. Ther. 207, 92–100.
Bradford, L. D. (2002). CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 3, 229–243. doi:10.1517/14622416.3.2.229
Chen, Y., Zhou, D., Tang, W., Zhou, W., Al-Huniti, N., and Masson, E. (2018). Physiologically based pharmacokinetic modeling to evaluate the systemic exposure of gefitinib in CYP2D6 ultrarapid metabolizers and extensive metabolizers. J. Clin. Pharmacol. 58, 485–493. doi:10.1002/jcph.1036
Chen, Z. R., Reynolds, G., Bochner, F., and Somogyi, A. (1989). Direct determination of codeine-6-glucuronide in plasma and urine using solid-phase extraction and high-performance liquid chromatography with fluorescence detection. J. Chromatogr. 493, 313–324. doi:10.1016/s0378-4347(00)82737-1
Chen, Z. R., Somogyi, A. A., Reynolds, G., and Bochner, F. (1991). Disposition and metabolism of codeine after single and chronic doses in one poor and seven extensive metabolisers. Br. J. Clin. Pharmacol. 31, 381–390. doi:10.1111/j.1365-2125.1991.tb05550.x
Ciszkowski, C., Madadi, P., Phillips, M. S., Lauwers, A. E., and Koren, G. (2009). Codeine, ultrarapid-metabolism genotype, and postoperative death. N. Engl. J. Med. 361, 827–828. doi:10.1056/NEJMc0904266
Dickschen, K. J., Willmann, S., Hempel, G., and Block, M. (2017). Addressing adherence using genotype-specific PBPK modeling-impact of drug holidays on tamoxifen and endoxifen plasma levels. Front. Pharmacol. 8, 67. doi:10.3389/fphar.2017.00067
Dittrich, P., Beubler, E., Haltmeyer, K., Zimmer, A., and Roblegg, E. (2011). Pharmacokinetics of a novel liquid controlled release codeine formulation. Drug Dev. industrial Pharm. 37, 1119–1124. doi:10.3109/03639045.2011.561351
Eckhardt, K., Li, S., Ammon, S., Schänzle, G., Mikus, G., and Eichelbaum, M. (1998). Same incidence of adverse drug events after codeine administration irrespective of the genetically determined differences in morphine formation. Pain 76, 27–33. doi:10.1016/s0304-3959(98)00021-9
Emoto, C., Johnson, T. N., Neuhoff, S., Hahn, D., Vinks, A. A., and Fukuda, T. (2018). PBPK model of morphine incorporating developmental changes in hepatic OCT1 and UGT2B7 proteins to explain the variability in clearances in neonates and small infants. CPT pharmacometrics Syst. Pharmacol. 7, 464–473. doi:10.1002/psp4.12306
FDA (2017). FDA Drug Safety Communication: FDA restricts use of rescription codeine pain and cough medicines and tramadol pain medicines in children; recommends against use in breastfeeding women. Rockville, MD: US Food and Drug Administration.
Findlay, J. W., Fowle, A. S., Butz, R. F., Jones, E. C., Weatherley, B. C., Welch, R. M., et al. (1986). Comparative disposition of codeine and pholcodine in man after single oral doses. Br. J. Clin. Pharmacol. 22, 61–71. doi:10.1111/j.1365-2125.1986.tb02881.x
Gough, A. C., Smith, C. A., Howell, S. M., Wolf, C. R., Bryant, S. P., and Spurr, N. K. (1993). Localization of the CYP2D gene locus to human chromosome 22q13.1 by polymerase chain reaction, in situ hybridization, and linkage analysis. Genomics 15, 430–432. doi:10.1006/geno.1993.1082
Grimstein, M., Yang, Y., Zhang, X., Grillo, J., Huang, S. M., Zineh, I., et al. (2019). Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and drug administration's office of clinical Pharmacology. J. Pharm. Sci. 108, 21–25. doi:10.1016/j.xphs.2018.10.033
Grzegorzewski, J., Brandhorst, J., and König, M. (2022). Physiologically based pharmacokinetic (PBPK) modeling of the role of CYP2D6 polymorphism for metabolic phenotyping with dextromethorphan. Front. Pharmacol. 13, 1029073. doi:10.3389/fphar.2022.1029073
Guay, D. R., Awni, W. M., Halstenson, C. E., Findlay, J. W., Opsahl, J. A., Abraham, P. A., et al. (1987). Pharmacokinetics of codeine after single- and multiple-oral-dose administration to normal volunteers. J. Clin. Pharmacol. 27, 983–987. doi:10.1002/j.1552-4604.1987.tb05601.x
Khalaj, Z., Baratieh, Z., Nikpour, P., Khanahmad, H., Mokarian, F., Salehi, R., et al. (2019). Distribution of CYP2D6 polymorphism in the Middle eastern region. J. Res. Med. Sci. 24, 61. doi:10.4103/jrms.JRMS_1076_18
Kim, I., Barnes, A. J., Oyler, J. M., Schepers, R., Joseph, R. E., Cone, E. J., et al. (2002). Plasma and oral fluid pharmacokinetics and pharmacodynamics after oral codeine administration. Clin. Chem. 48, 1486–1496. doi:10.1093/clinchem/48.9.1486
Kirchheiner, J., Schmidt, H., Tzvetkov, M., Keulen, J. T., Lötsch, J., Roots, I., et al. (2007). Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J. 7, 257–265. doi:10.1038/sj.tpj.6500406
Knych, H. K., Baden, R. W., Gretler, S. R., and McKemie, D. S. (2019). Characterization of the in vitro CYP450 mediated metabolism of the polymorphic CYP2D6 probe drug codeine in horses. Biochem. Pharmacol. 168, 184–192. doi:10.1016/j.bcp.2019.07.005
Kronstrand, R., and Jones, A. W. (2001). Concentration ratios of codeine-to-morphine in plasma after a single oral dose (100 mg) of codeine phosphate. J. Anal. Toxicol. 25, 486–487. doi:10.1093/jat/25.6.486
Lafolie, P., Beck, O., Lin, Z., Albertioni, F., and Boréus, L. (1996). Urine and plasma pharmacokinetics of codeine in healthy volunteers: implications for drugs-of-abuse testing. J. Anal. Toxicol. 20, 541–546. doi:10.1093/jat/20.7.541
Linares, O. A., Fudin, J., Schiesser, W. E., Daly Linares, A. L., and Boston, R. C. (2015). CYP2D6 phenotype-specific codeine population pharmacokinetics. J. pain and Palliat. care Pharmacother. 29, 4–15. doi:10.3109/15360288.2014.997854
Matic, M., Nijenhuis, M., Soree, B., de Boer-Veger, N. J., Buunk, A. M., Houwink, E. J. F., et al. (2022). Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction between CYP2D6 and opioids (codeine, tramadol and oxycodone). Eur. J. Hum. Genet. 30, 1105–1113. doi:10.1038/s41431-021-00920-y
Mendoza, R., Wan, Y. J., Poland, R. E., Smith, M., Zheng, Y., Berman, N., et al. (2001). CYP2D6 polymorphism in a Mexican American population. Clin. Pharmacol. Ther. 70, 552–560. doi:10.1067/mcp.2001.120675
Mikus, G., Trausch, B., Rodewald, C., Hofmann, U., Richter, K., Gramatté, T., et al. (1997). Effect of codeine on gastrointestinal motility in relation to CYP2D6 phenotype. Clin. Pharmacol. Ther. 61, 459–466. doi:10.1016/S0009-9236(97)90196-X
Mistry, M., and Houston, J. B. (1987). Glucuronidation in vitro and in vivo. Comparison of intestinal and hepatic conjugation of morphine, naloxone, and buprenorphine. Drug metabolism Dispos. 15, 710–717.
Mohammed, S. S., Ayass, M., Mehta, P., Kedar, A., Gross, S., and Derendorf, H. (1993). Codeine disposition in sickle cell patients compared with healthy volunteers. J. Clin. Pharmacol. 33, 811–815. doi:10.1002/j.1552-4604.1993.tb01956.x
Pang, K. S., and Durk, M. R. (2010). Physiologically-based pharmacokinetic modeling for absorption, transport, metabolism and excretion. J. Pharmacokinet. pharmacodynamics 37, 591–615. doi:10.1007/s10928-010-9185-x
Persson, K., Hammarlund-Udenaes, M., Mortimer, O., and Rane, A. (1992). The postoperative pharmacokinetics of codeine. Eur. J. Clin. Pharmacol. 42, 663–666. doi:10.1007/BF00265933
Pratt, V. M., Scott, S. A., Pirmohamed, M., Esquivel, B., Kane, M. S., Kattman, B. L., et al. (2012). “Codeine therapy and CYP2D6 genotype,” in Medical genetics summaries (Bethesda (MD): National Center for Biotechnology Information).
Quiding, H., Anderson, P., Bondesson, U., Boréus, L. O., and Hynning, P. A. (1986). Plasma concentrations of codeine and its metabolite, morphine, after single and repeated oral administration. Eur. J. Clin. Pharmacol. 30, 673–677. doi:10.1007/BF00608214
Raungrut, P., Uchaipichat, V., Elliot, D. J., Janchawee, B., Somogyi, A. A., and Miners, J. O. (2010). In vitro-in vivo extrapolation predicts drug-drug interactions arising from inhibition of codeine glucuronidation by dextropropoxyphene, fluconazole, ketoconazole, and methadone in humans. J. Pharmacol. Exp. Ther. 334, 609–618. doi:10.1124/jpet.110.167916
Sager, J. E., Yu, J., Ragueneau-Majlessi, I., and Isoherranen, N. (2015). Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug metabolism Dispos. 43, 1823–1837. doi:10.1124/dmd.115.065920
Schaefer, N., Moj, D., Lehr, T., Schmidt, P. H., and Ramsthaler, F. (2018). The feasibility of physiologically based pharmacokinetic modeling in forensic medicine illustrated by the example of morphine. Int. J. Leg. Med. 132, 415–424. doi:10.1007/s00414-017-1754-8
Shah, J. C., and Mason, W. D. (1990). Plasma codeine and morphine concentrations after a single oral dose of codeine phosphate. J. Clin. Pharmacol. 30, 764–766. doi:10.1002/j.1552-4604.1990.tb03641.x
Sjöstedt, N., Neuhoff, S., and Brouwer, K. L. R. (2021). Physiologically-based pharmacokinetic model of morphine and morphine-3-glucuronide in nonalcoholic steatohepatitis. Clin. Pharmacol. Ther. 109, 676–687. doi:10.1002/cpt.2037
Somogyi, A. A., Barratt, D. T., and Coller, J. K. (2007). Pharmacogenetics of opioids. Clin. Pharmacol. Ther. 81, 429–444. doi:10.1038/sj.clpt.6100095
Thorn, C. F., Klein, T. E., and Altman, R. B. (2009). Codeine and morphine pathway. Pharmacogenetics Genomics 19, 556–558. doi:10.1097/FPC.0b013e32832e0eac
Tseng, C. Y., Wang, S. L., Lai, M. D., Lai, M. L., and Huang, J. D. (1996). Formation of morphine from codeine in Chinese subjects of different CYP2D6 genotypes. Clin. Pharmacol. Ther. 60, 177–182. doi:10.1016/S0009-9236(96)90133-2
Vree, T. B., and Verwey-Van Wissen, C. P. (1992). Pharmacokinetics and metabolism of codeine in humans. Biopharm. drug Dispos. 13, 445–460. doi:10.1002/bdd.2510130607
Weingarten, B., Wang, H. Y., and Roberts, D. M. (1995). Determination of codeine in human plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. a 696, 83–92. doi:10.1016/0021-9673(94)01212-w
Wu, X., Yuan, L., Zuo, J., Lv, J., and Guo, T. (2014). The impact of CYP2D6 polymorphisms on the pharmacokinetics of codeine and its metabolites in Mongolian Chinese subjects. Eur. J. Clin. Pharmacol. 70, 57–63. doi:10.1007/s00228-013-1573-x
Wu, X., Zhang, W., Bai, Y., Guo, T., and Gu, J. (2013). Simultaneous analysis of codeine and its active metabolites in human plasma using liquid chromatography-tandem mass spectrometry: application to a pharmacokinetic study after oral administration of codeine. J. Pharm. Biomed. analysis 78-79, 261–268. doi:10.1016/j.jpba.2013.02.027
Xie, R., and Hammarlund-Udenaes, M. (1998). Blood-brain barrier equilibration of codeine in rats studied with microdialysis. Pharm. Res. 15, 570–575. doi:10.1023/a:1011929910782
Xu, M., Zheng, L., Zeng, J., Xu, W., Jiang, X., and Wang, L. (2021). Physiologically based pharmacokinetic modeling of tramadol to inform dose adjustment and drug-drug interactions according to CYP2D6 phenotypes. Pharmacotherapy 41, 277–290. doi:10.1002/phar.2494
Yue, Q. Y., Hasselström, J., Svensson, J. O., and Säwe, J. (1991a). Pharmacokinetics of codeine and its metabolites in Caucasian healthy volunteers: comparisons between extensive and poor hydroxylators of debrisoquine. Br. J. Clin. Pharmacol. 31, 635–642. doi:10.1111/j.1365-2125.1991.tb05585.x
Keywords: codeine, physiologically based pharmacokinetic, CYP2D6, genetic polymorphism, pharmacokinetics
Citation: Yang Y, Zhang X, Wang Y, Xi H, Xu M and Zheng L (2024) Physiologically based pharmacokinetic modeling to predict the pharmacokinetics of codeine in different CYP2D6 phenotypes. Front. Pharmacol. 15:1342515. doi: 10.3389/fphar.2024.1342515
Received: 22 November 2023; Accepted: 10 April 2024;
Published: 02 May 2024.
Edited by:
Youssef Daali, University of Geneva, SwitzerlandReviewed by:
Frederic Gaspar, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandZiran Li, University of California, San Francisco, United States
Copyright © 2024 Yang, Zhang, Wang, Xi, Xu and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Zheng, emhlbmdsQGFobXUuZWR1LmNu