 Ifraz Ahmad
Ifraz Ahmad Warda Parveen
Warda Parveen Shah Noor
Shah Noor Zahoor Udin3
Zahoor Udin3 Riaz Ullah
Riaz Ullah Hamid Ali
Hamid Ali
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 26 March 2024
Sec. Experimental Pharmacology and Drug Discovery
Volume 15 - 2024 | https://doi.org/10.3389/fphar.2024.1332184
This article is part of the Research Topic What does Experimental Pharmacology and Drug Discovery look like in 2035? View all 8 articles
Tyrosinase (TYR) inhibitors are very significant as they inhibit enzyme tyrosinase activity, and its inhibition is vital for skin care, anticancer medication, and antibrowning of fruits and vegetables. This work presents a novel and economical route for the preparation of new synthetic tyrosinase inhibitors using amlodipine (4). The novel conjugates 6 (a–o) were designed, synthesized, and characterized by spectroscopic analyses, including Fourier transform infrared and low- and high-resolution mass spectroscopy. The purified compound 4 was refluxed with various aldehydes and ketones 5 (a–o) for 5–8 h in methanol at 60°C–90°C. This research modified the drug in a step-by-step manner to develop therapeutic properties as a tyrosinase inhibitor. The structures of synthesized ligands 6 (a–o) were established based on spectral and analytical data. The synthesized compounds 6 (a–o) were screened against tyrosinase enzyme. Kojic acid was taken as standard. All the prepared compounds 6 (a–o) have good inhibition potential against the enzyme tyrosinase. Compounds 6o, 6b, 6f, and 6k depicted excellent antityrosinase activity. Compound 6k, with an IC50 value of 5.34 ± 0.58 µM, is as potent as the standard kojic acid (IC50 6.04 ± 0.11 µM), standing out among all synthesized compounds 6 (a–o). The in silico studies of the conjugates 6 (a–o) were evaluated via PatchDock. Compound 6k showed a binding affinity score of 8,999 and an atomic contact energy (ACE) value of −219.66 kcal/mol. The structure–activity relationship illustrated that the presence of dihydropyridine nuclei and some activating groups at the ortho and para positions of the benzylideneimine moiety is the main factor for good tyrosinase activity. The compound 6k could be used as a lead compound for drug modification as a tyrosinase inhibitor for skin care, anticancer medication, and antibrowning for fruits and vegetables.
Tyrosinase (TYR), a binuclear copper-containing enzyme belonging to the oxidoreductase class of enzymes that catalyze oxidation, is a primary target for the treatment of over-pigmentation disorder and associated skin problems because it directly contributes to the synthesis of melanin (Roulier et al., 2020). Protein tyrosine kinase (PTK) is one of the many cellular proteins that have been identified as an anticancer study target as a result of the quick advancement of life science research (e.g., DNA, tubulin, and essential enzymes) (Mongre et al., 2021). PTK is a group of tyrosine kinase-active proteins that contribute to the control of several physiological and biochemical processes, including cell growth, differentiation, and death (Wang et al., 2019). PTK expression may be abnormal, which may promote tumor neovascularization, invasion, metastasis, and resistance to therapy (Drake et al., 2014; Knosel et al., 2014). As a result, PTK has evolved into an important target for antitumor drug research (Jiao et al., 2018). One of the biggest issues facing food companies is enzymatic browning, particularly for fruits, vegetables, and seafood items. Enzymatic and nonenzymatic oxidation are the two processes that cause browning. Producing poisonous and antinutritional substances could further diminish the nutritional value and safety of food (Atasever et al., 2013; Aydin et al., 2015; Arnold and Gramza-Michalowska, 2022). Tyrosinase enzymatic oxidation of phenols, which results in the degradation of plant-based foods and beverages, can cause degradation of important amino acids, inhibits proteolytic and glycolytic enzymes, and, therefore, reduces digestibility and nutrition and releases hazardous chemicals (Qian et al., 2020). Tyrosinase and its polyphenolic substrates combine with oxygen to cause the enzymatic browning of fruits, vegetables, and beverages after brushing, peeling, and crushing operations that cause cell structures to break down (Moon et al., 2020). This enzyme occurs naturally in a wide variety of organisms, including bacteria, fungi, higher plants (including mushrooms, bananas, apples, pears, potatoes, avocados, and peaches), and animals (Yuan et al., 2019). Tyrosinase and phenolic substrate concentrations, oxygen availability, pH, temperature, and other factors all affect how quickly an object will turn brown through enzymatic means (Panis and Rompel, 2022). Food additives like reducing agents and enzyme inhibitors can be used to reduce browning.
Hydroquinone-based tyrosinase inhibitors utilized for skin disorders are linked to a range of unfavorable effects, including contact dermatitis, irritation, transitory erythema, burning, prickling feeling, leukoderma, chestnut patches on the nails, hypochromia, and ochronosis, and they are potentially carcinogenic to mammalian cells (Bilovol et al., 2021). The natural substance arbutin, a prodrug of hydroquinone, decreases or prevents melanin formation by blocking tyrosinase (Boo, 2021). Though chemically unstable, natural forms of arbutin have the ability to produce hydroquinone, which is catabolized into benzene metabolites, which may be hazardous to bone marrow (Zhou et al., 2013). Due to its cancer-causing properties and instability during storage, the use of kojic acid in cosmetics has been restricted (Masub and Khachemoune, 2020). The degradation of L-ascorbic acid is rapid and sensitive to heat (Yin et al., 2022). Ellagic acid is insoluble and, therefore, poorly bioavailable (Arulmozhi et al., 2013), and the melanogenic path remains undecided for tranexamic acid (Tse and Hui, 2013).
In brief, tyrosinase inhibitors, besides being used to treat specific dermatological conditions linked to melanin hyperpigmentation, play a vital role in the cosmetics industry due to their ability to brighten skin and reduce sunburn-related pigmentation. Tyrosinase inhibitors also find applications in the food and beverage industry and as anticancer medication. Developing effective tyrosinase inhibitors is important in order to address these concerns. We have synthesized de novo conjugates of amlodipine that have a dihydropyridine-type heterocyclic ring structure and a benzylideneimine moiety, which are very effective as tyrosinase inhibitors, via facile and routine methods.
The novel compounds 6 (a–o) were designed rationally via in silico studies and synthesized, characterized, and evaluated for their tyrosinase inhibition potential.
Tyrosinase (EC 1.14.18.1), also known as polyphenol oxidase, is a type of multi-functional binuclear copper ion metalloenzyme that is abundantly present in animals, plants, and microbes. It is crucial for several physiological and pathological functions (Casanola-Martin et al., 2014). The primary enzyme in the manufacture of melanin is TYR. A number of pigmented skin illnesses, including malignant melanoma, can develop as a result of high levels of TYR activity that result in melanin buildup (Buitrago et al., 2016).
Dihydropyridine-based tyrosinase inhibitors were reported by (Bhosle et al., 2018), as given in Scheme 1 structure (3). Amlodipine (4), which is commercially available in the form of amlodipine besylate (1) with dihydropyridine nuclei, is a commonly prescribed medication for hypertension and angina. It belongs to a class of drugs called calcium channel blockers, which work by relaxing and widening blood vessels to improve blood flow and reduce cardiac workload. Amlodipine (4) is known for its long-lasting effects, with a half-life of approximately 30 h, making it convenient for once-daily dosing. It is a well-tolerated drug with few side effects, making it a reliable choice for patients with cardiovascular diseases (Tang et al., 2016). Overall, amlodipine plays a significant role in managing hypertension and improving heart health (Zhou et al., 2022). Over the past 2 decades, it has remained under study for several types of biological activities. Based on its active dihydropyridine structure, we analyzed it for the design of novel biologically effective tyrosinase inhibitors.
Benzylideneimine-type tyrosinase inhibitors were reported by Wang et al. (2019) as given in structure (2) in Scheme 1. Bae et al. (2012) and Lima et al. (2013) also described benzylideneimine-containing tyrosinase inhibitors. In this research, we synthesized novel tyrosinase inhibitors by improving amlodipine chemically based on the proposition that dihydropyridine and benzylideneimine play a role in antityrosinase activity. Hence, the benzylideneimine moiety with different substituents on the benzyl ring has been introduced to the original structure of amlodipine via chemical reactions. In this way, 15 novel compounds were synthesized. The rational designing of these novel amlodipine conjugates 6 (a–o) as tyrosinase inhibitors is given below in Scheme 1.
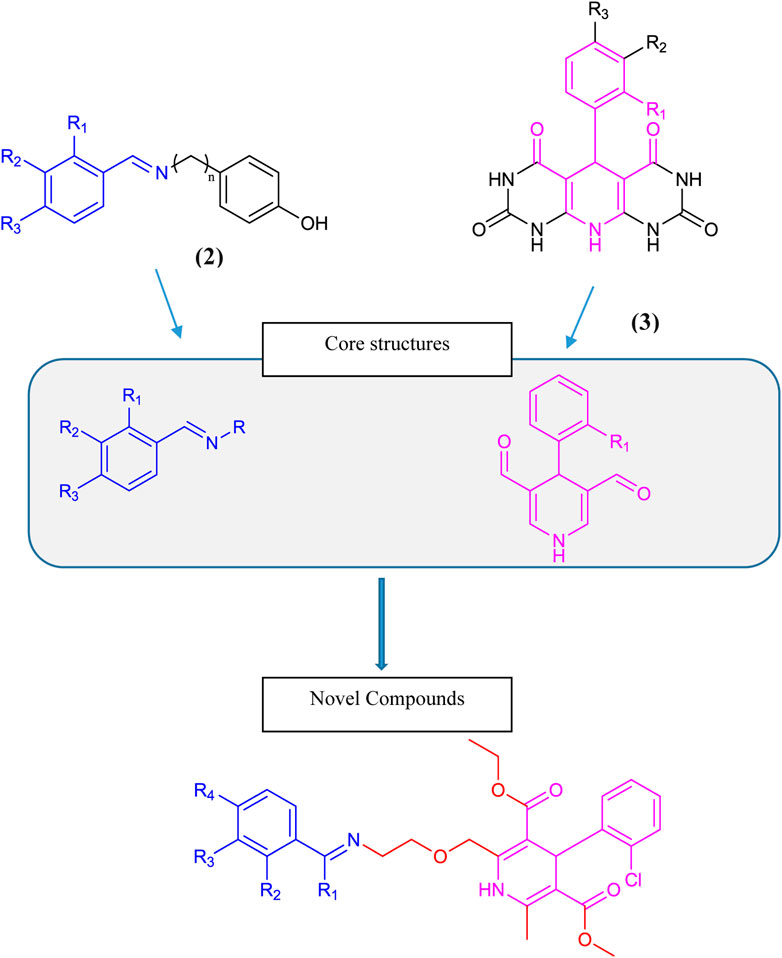
Scheme 1. Rational design of conjugates 6 (a–o).
The crystal structure of the tyrosinase protein was retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB ID 2y9x). The coordinate files were subjected to Discovery Studio 4.5 Visualizer for pre-docking receptor preparation by removing water molecules and adding hydrogen atoms. Ligands 6 (a–o) were docked with tyrosinase (2y9x) by PatchDock. PatchDock is a molecular docking tool aimed at finding docking transformations that generate good molecular shape complementarity based on shape complementarity principles (Schneidman-Duhovny et al., 2005). The input files comprise the receptor protein and ligand in PDB format.
The PatchDock server offers multiple solutions. “Solution 1” was selected as it surrounded the most significant residues as a binding pocket for docking analyses assigned in the crystal structure of the tyrosinase receptor (2y9x) (Silva et al., 2017; Nazir et al., 2022). The docked structures were analyzed via Discovery Studio 4.5 Visualizer.
The binding affinity scores and ACEs of the docked ligand complexes were assessed. The hydrogen bonding and hydrophobic contacts of each ligand were evaluated within the binding pocket of the receptor protein. The conformations of the ligands that demonstrated the highest biological activities are explained in Supplementary Table S1 and Figures 1–3, which show their favorable interactions in the binding pouches. We examined the docked complexes of ligands (6 (a–o) and kojic acid to qualitatively estimate their biological activities (IC50) and identify the molecular basis for these effects.
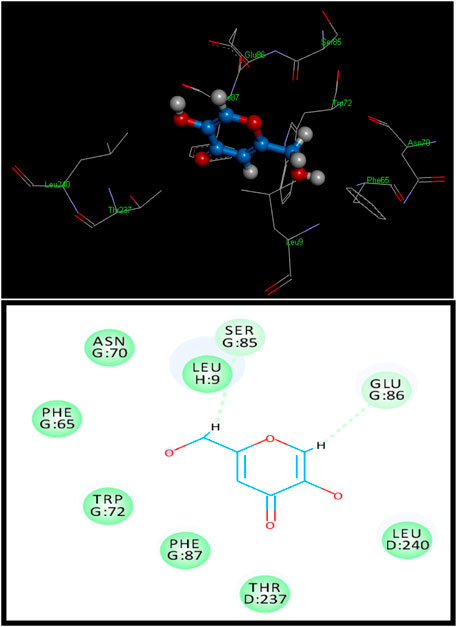
Figure 1. 3D (above) and 2D (below) binding site contacts of standard kojic acid with a tyrosinase binding pocket.
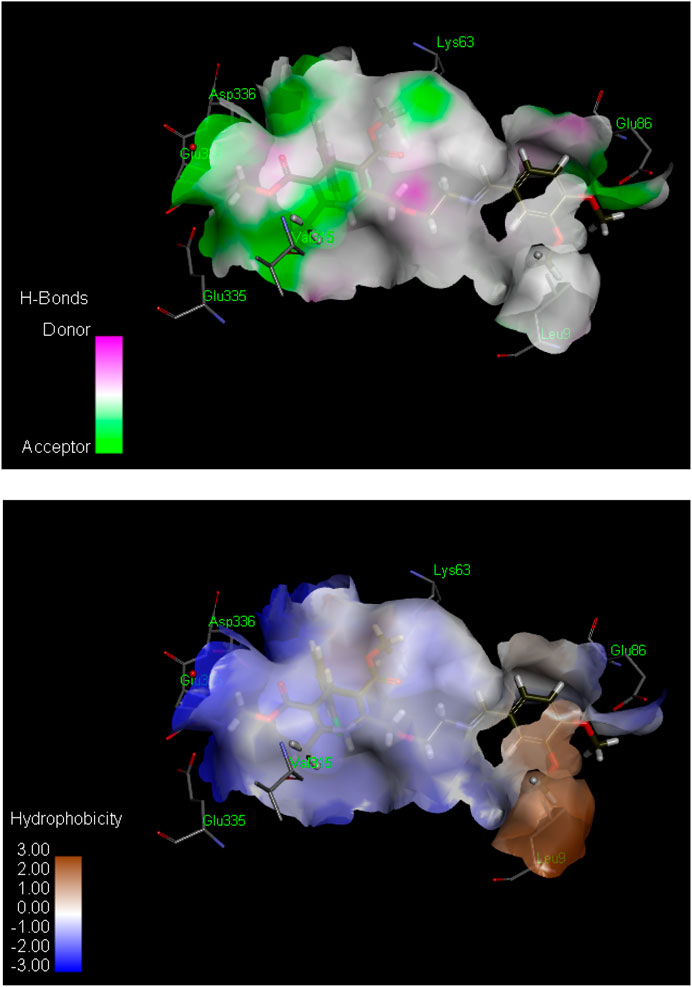
Figure 2. Hydrogen bond (above) and hydrophobic interaction (below) analyses of the most active inhibitor, 6k. The structural elements (atoms and torsions) that contribute favorably to the overall binding energy are colored blue, those that contribute unfavorably are colored pink, and neutral elements are colored white.
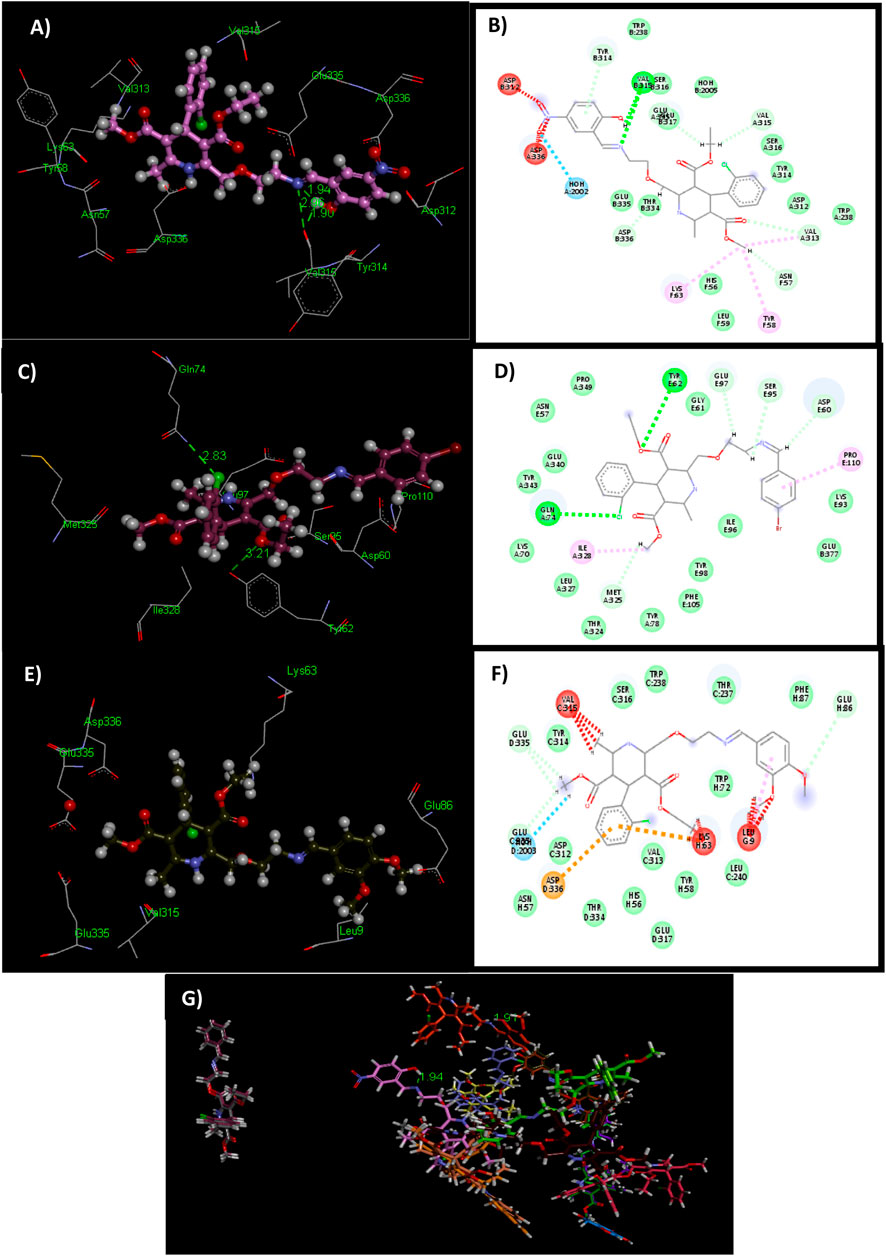
Figure 3. 3D and 2D structures of docked ligands 6o (A, B), 6f (C, D), and 6k (E, F). Illustrating unfavorable protrusion (red), H–bond (light green), pi–alkyl (light pink), pi–cation (brown), and amide pi–stack (pink) interactions within a binding pocket of the tyrosinase protein (PDB ID: 2y9x). (G) Overlap of bound conformations of kojic acid (blue) with ligands 6d (gray), 6a (yellow), 6e (mauve), 6o (pale pink), 6n (green), 6i (brown), 6f (magenta), 6k (olive green), 6c (mustard), 6m (purple), 6h (pink), 6g (bright green), 6h (beige), 6j (orange), and 6b (chocolate brown).
All of the chemicals and solvents were purchased from Sigma-Aldrich and Alfa Aesar and utilized directly for experimentation. The melting points of the compounds were determined by using the Gallenkamp melting point apparatus via the open capillary method. Fourier transform infrared (FTIR) spectra were recorded on a Bruker alpha 2 FTIR spectrophotometer with a spectral range of 4,000–400 cm−1. Low-resolution mass spectra (LRMS) were obtained using an ESI LC TOF 6224 (Agilent) under the positive ion mode. High-resolution mass spectra (HSMS) were obtained on a 9.4 T Bruker FT-ICR-MS spectrometer in MeOH: MeCN (1: 1). Thin-layer chromatography (TLC) was determined on silica gel plates (Analtech 02521), using the solvent systems EtOAc:hexanes (8:2) and CHCl3:CH3OH (1:1).
Amlodipine besylate (1) was used as the starting material. It was then processed to obtain a purified form 4 by mixing it with a combination of dichloromethane and water in a 1:1 ratio. Following this step, the mixture underwent basification at a pH of 11. Subsequently, an extraction process using dichloromethane as the solvent was conducted. The final purification of the extracted substance was achieved through preparative TLC. The purified extract was dried and analyzed for physical and chemical characterization.
Fifteen novel amlodipine conjugates 6 (a–o) were synthesized by the conventional imine formation reaction. All the compounds were synthesized by the one-pot reaction of various aldehydes and ketones 5 (a–o) with compound 4 in ethanol under reflux. Amlodipine (4) and substituted aldehydes and ketones 5(a–o) were refluxed using ethanol and a catalytic amount of CH3COOH for approximately 5–8 h. Imines were obtained as sole products (Scheme 2). The completion of the reaction was observed by TLC. The solvent system was chloroform and methanol in a ratio of 9.9:0.1. Then, the reaction mixture was quenched by aqueous NaHCO3 solution (1 M). Next, the organic product was extracted using DCM (3 × 10 mL). Anhydrous MgSO4 was used to dry the combined organic extract. Later, the solid product was filtered. After evaporation, preparative TLC was used for purification. Chloroform and methanol (90:10) were used as a solvent system. The purified organic product was dried over silica gel beads in desiccators for a week. Finally, the dried compound was collected for further analysis (Mocanu and Cernătescu, 2008). Characterization of synthesized compounds 6(a–o) was done via FTIR, high-resolution mass spectroscopy (HRMS), and LRMS.
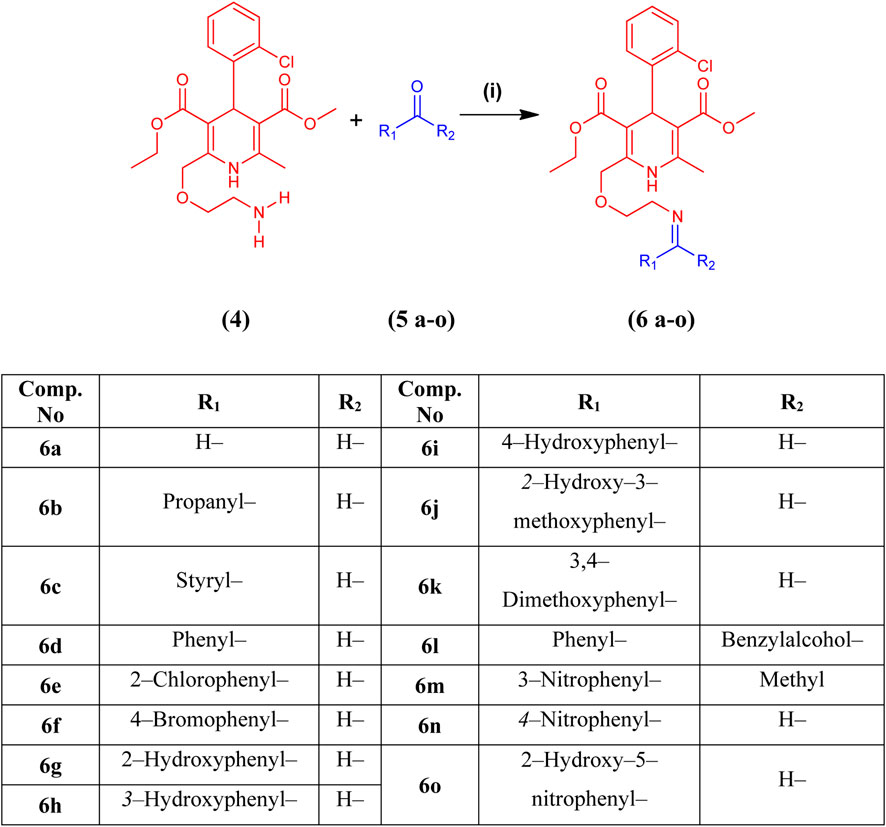
Scheme 2. Synthesis of amlodipine derivatives 6 (a–o) from amlodipine (4) and aldehydes or ketones 5 (a–o). Reagents and conditions: (i) CH3CH2OH and CH3COOH, reflux for 5–8 h at 60°C–90°C.
Tyrosinase assay materials (100 μL) were mixed with 100 mM phosphate buffer (60 μL), pH 6.8, 10 μL (5 units) of mushroom tyrosinase enzyme, and 10 μL (0.5 mM) of the test substance (Lee, 2002) in a 96-well-plate. The assay contents were pre-incubated for 5 min at 37°C. Then, 20 μL of L-dopamine (10 mM) was added as a substrate, the contents were mixed, and the solution was incubated for a period of 30 min. The absorbance was recorded at 490 nm by using a Synergy HT BioTek 96-well plate reader.
The enzyme inhibition (%) was determined by the given formula
Inhibition (%) = 100 − (Abs of the test sample/Abs of control × 100)
The IC50 values (the concentration of a drug or inhibitor needed to inhibit a biological process or response by 50%) of compounds were calculated using EZ-Fit Enzyme Kinetics Software (Perrella Scientific Inc. Amherst, USA) (Asadzadeh et al., 2016; Shao et al., 2018; Wang et al., 2019).
To assess the tyrosinase inhibition potential, compounds 6 (a–o) and standard kojic acid were docked against the tyrosinase enzyme via the software, as explained above. To obtain a qualitative estimation and to identify the molecular origin of the analyzed biological activities (IC50), the docked complexes of ligands (6 (a–o) and kojic acid) were investigated. The initial assessment of the docked complexes of tyrosinase (6 (a–o) and kojic acid) disclosed that 6k and 6o have superior docking scores than the standard, kojic acid.
Standard kojic acid was also docked with tyrosinase for comparison and had a score of 2,530 with an ACE value of −68.92 kcal/mol (Figure 1). Kojic acid has shown hydrogen bonding with Tyr128. The length of the hydrogen bond was 3.35 Å, and hydrophobic contact potential with pocket amino acids Leu9, Asn70, Phe65, Trp72, Phe87, Thr237, and Leu240 was identified (Supplementary Table S1).
The ligand 6k showed the highest IC50 value (Figure 2; Table 1) and exhibited excellent binding affinity with a score of 8,999 and an ACE value of −219.66 kcal/mol. No hydrogen bond was observed between 6k and the binding pocket (PDB ID 2y9x). Visual examination of the ligand 6k depicted the potential for hydrophobic interactions with pocket amino acid residues Ser316, Trp238, Thr237, Phe37, Trp72, Leu240, Tyr58, Val313, Glu317, His56, Thr334, Asn57, Asp312, and Tyr314. These interactions can contribute significantly to comparatively superior binding affinity. A pi–alkyl interaction can also be visualized between Leu9 and the 3-hydroxy-4-methoxyphenyl species (Supplementary Table S1).
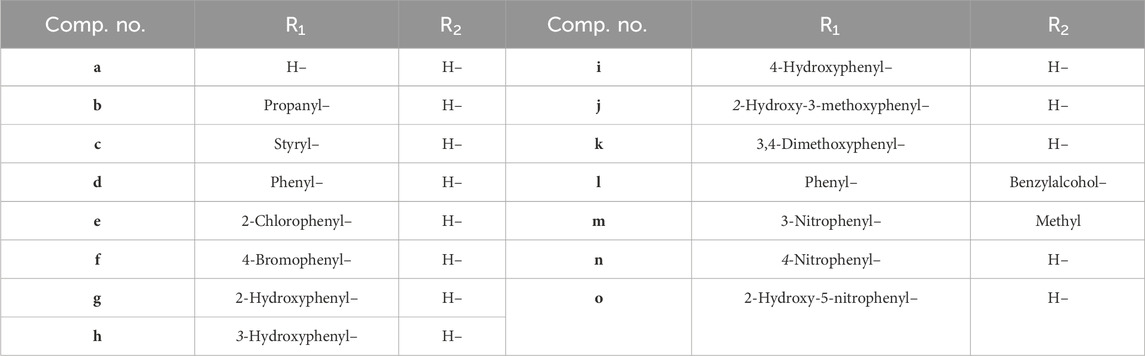
Table 1. Compounds (a-o) having different substituents with respect to R1 and R2 shown below.
Study of the contacts between ligand 6o and the tyrosinase pocket discloses the binding affinity as a score of 8,908 and an ACE of −152.73 kcal/mol. Ligand 6o exhibited two hydrogen bond interactions among the carbonyl group of amino acid Val315 and the –C=N (1.90 Å) and –OH (2.96 Å) groups of ligand 6o. However, ligand 6o showed hydrophobic interaction with Trp238, Ser316, Ser316, Glu317, HOH 2005, Tyr314, Leu59, Trp238, Asp312, His56, Thr334, and Glu335 amino acid residues. The pi–alkyl interaction can also be visualized between Tyr58 and the methyl formate directly attached to the dihydropyridine ring moiety (Supplementary Table S1).
Similarly, ligand 6f and the tyrosinase pocket had a binding affinity score of 8,758 and an ACE value of −146.96 kcal/mol. Two hydrogen bonds were observed between the amino group directly attached to the carbonyl of Gln74 and the 2-chlorophenyl group (2.83 Å). Another hydrogen bond was observed between the 4-hydroxyphenyl group of Tyr62 and the oxygen of the methyl formate moiety (3.21 Å). In contrast, ligand 6f depicted hydrophobic contact potential with Tyr343, Glu340, Asn57, Pro349, Gly61, Lys93, Ile96, Glu377, Tyr98, Phe105, Tyr78, Thr324, Leu327, and Lys70 amino acid residues. The pi–alkyl interaction can also be visualized between Pro110 and the 4-hydroxyphenyl moiety (Supplementary Table S1).
The structures of the title compounds 6 (a–o) were confirmed by FTIR, LRMS, and HRMS, which revealed that the results were in accordance with the proposed and calculated values and testify to our synthetic approach. The overall results are given in Supplementary Material Section 2.
A series of novel amlodipine conjugates (6 a–o) were analyzed for their tyrosinase inhibition activity. All the compounds were found to be active against the enzyme tyrosinase. The strength of the tyrosinase inhibition activity of compounds 6 (a–o) decreases as follows: 6k > 6o > 6b > 6f > 6e > 6c > 6n > 6a > 6g > 6l > 6h > 6d > 6j > 6i > 6m (Table 2).
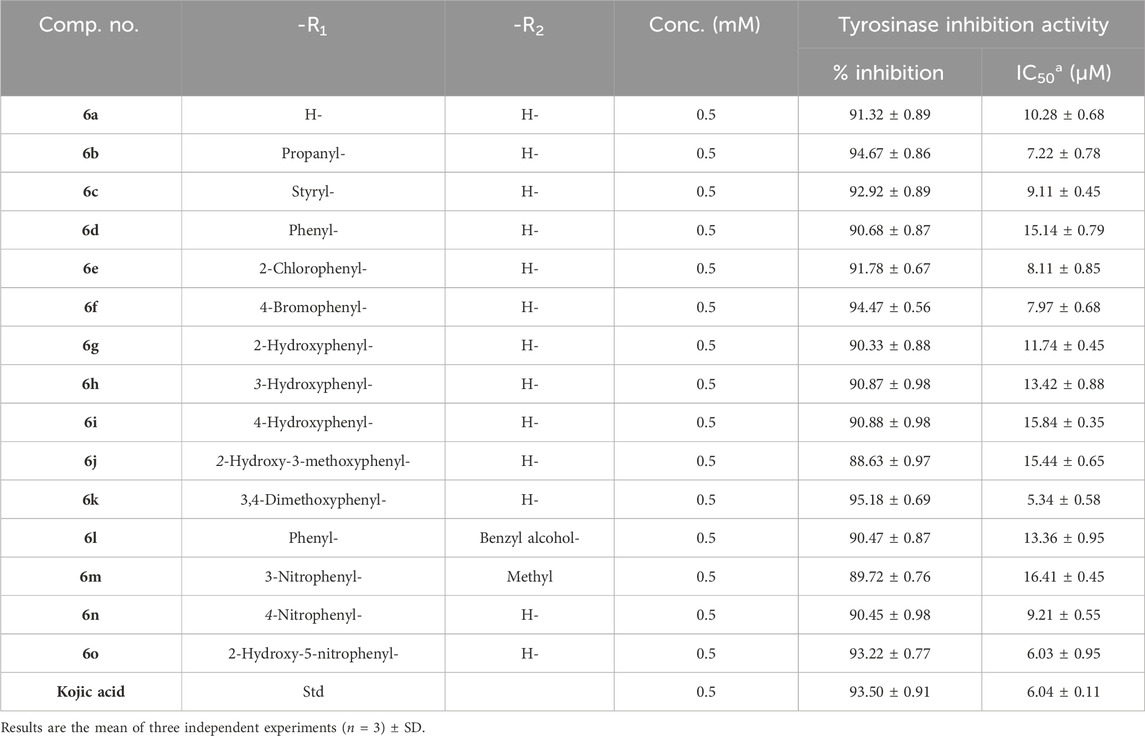
Table 2. Determination of IC50 values of tyrosinase inhibition activity of the amlodipine conjugates (6a–o).
The results revealed that compound 6k is the most potent compound of series 6 (a–o), having an IC50 of 5.34 ± 0.58 µM, which is as potent as the standard kojic acid, with an IC50 of 6.04 ± 0.11 µM. Three other members of series 6 (a–o) also showed good inhibition; for example, 6o had an IC50 of 6.03 ± 0.95 µM, 6b had an IC50 of 7.22 ± 0.78 µM, and 6f had an IC50 of 7.97 ± 0.68 µM.
The structure–activity relationship revealed two main core structures in the synthesized compounds 6 (a–o) that are responsible for antityrosinase activity, the pyridine ring and the benzylideneimine moiety (Scheme 3). The dihydropyridine nuclei in the molecule are the main cause of the antityrosinase activity (Bhosle et al., 2018). Therefore, 6b (IC50 7.22 ± 0.78 µM) has the third highest value of tyrosinase inhibition among compounds 6 (a–o) due to the presence of two dihydropyridine rings. Electron-withdrawing substituents of the benzylideneimine moiety of compounds 6 (d–o) contribute to the change in the antityrosinase activity. The electron-donating substituents at the ortho or para positions or the electron-withdrawing groups at the meta position enhance the tyrosinase inhibition activity. It is noteworthy that the substituent position and number on the benzylideneimine aromatic ring of compounds 6 (d–o) are important for inhibition activity, as the hydroxyl group at the ortho position enhances antityrosinase activity (Lima et al., 2013). A methoxy substituent at the para position of benzylideneimine is very important to enhance the tyrosinase inhibition activity (Bae et al., 2012). Hence, compound 6k gives the best value of tyrosinase inhibition among 6 (a–o). The order of activity for ortho-, meta-, and para-hydroxy substituted benzylideneimine is o–OH > p–OH > m–OH, as given by the compounds 6g (IC50 11.74 ± 0.45 µM) > 6h (IC50 15.84 ± 0.35 µM) > (6i IC50 13.42 ± 0.88 µM). Among the nitro-substituted benzylideneimine, the para nitros have a lower IC50 than the meta nitro-substituted, as 6n (IC50 9.21 ± 0.55 µM) has a smaller value of tyrosinase inhibition than 6o (6.03 ± 0.95 µM). Non-substituted benzylideneimine-containing compounds give poor inhibition, as shown by compounds 6d (IC50 15.14 ± 0.79 µM) and 6l (IC50 13.36 ± 0.95 µM). However, it is difficult to establish the SAR of the conjugates 6 (a–o), probably due to multiple factors, as size, shape, electronegativity, and polarizability of the ligand play a role in enzyme inhibition.
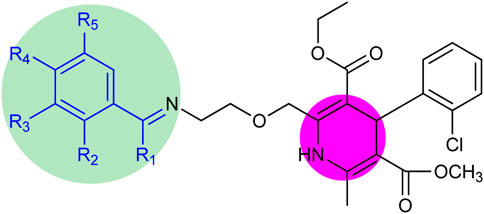
Scheme 3. Core structure of compounds 6 (a–o) showing the benzylideneimine group in the green-shaded region and dihydropyridine in the pink. R1 = H, CH3 and R2, R3, R4, R5 = H, CH3O, CH3CH2O, OH, NO2, Br, Cl.
A series of novel amlodipine conjugates 6 (a–o) were designed, synthesized, and characterized by spectroscopic analyses, including FTIR, LRMS, and HRMS. The purified ligand 4 was refluxed with various aldehydes and ketones 6 (a–o) for 5–8 h in ethanol at 60°C–90°C. The research work was progressively completed so that the drug could be altered to develop therapeutic properties as a tyrosinase inhibitor. The structures of the prepared derivatives 6 (a–o) were established on the basis of analytical and spectral data. The synthesized conjugates 6 (a–o) were evaluated for antityrosinase activity against mushroom tyrosinase enzyme, for which kojic acid was taken as standard. The biological activity assay revealed that all the compounds have inhibition potential against the enzyme tyrosinase. Compounds 6o, 6b, 6f, and 6k depicted excellent antityrosinase activity. Compound 6k has the highest tyrosinase inhibition activity with an IC50 value of 5.34 ± 0.58 µM, which is as potent as the standard kojic acid (IC50 6.04 ± 0.11 µM). From the structure–activity relationship, it could be inferred that the existence of pyridine nuclei and activating groups at the ortho and para positions of the benzylideneimine moiety is the main factor for tyrosinase activity. The presence of a methoxy substituent at the para position of benzylideneimine was the main cause of the outstanding activity of ligand 6k. The molecular docking studies depicted that the conjugates 6 (a–o) showed a much higher docking score than the standard kojic acid. The compound 6k has an excellent binding affinity with a score of 8,999 and an ACE value of −219.66 kcal/mol. Three other compounds, 6o (6.03 ± 0.95 µM), 6b (7.22 ± 0.78 µM), and 6f (7.97 ± 0.68 µM), also have remarkable activity. Furthermore, high binding affinities toward tyrosinase were exhibited by the compounds 6o (IC50 6.03 ± 0.95 µM), 6b (IC50 7.22 ± 0.78 µM), and 6f (IC50 7.97 ± 0.68 µM) with binding affinity scores of 8,908, 8,894, and 8,758 and ACE values of −152.73, −156.85, and −146.96 kcal/mol, respectively. Compounds 6o and 6f have two hydrogen bonding interactions with tyrosinase pocket amino acids. Compound 6b, which does not have benzylideneimine in its structure, gives excellent activity due to the presence of two pyridine rings.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
IA: Conceptualization, Investigation, Writing–original draft. WP: Investigation, Writing–original draft, Formal Analysis, Methodology. SN: Data curation, Software, Validation, Writing–review and editing. ZU: Validation, Writing–review and editing, Formal Analysis, Visualization. AA: Formal Analysis, Validation, Software, Writing–original draft. IA: Writing–original draft, Conceptualization, Data curation, Methodology. RU: Conceptualization, Funding acquisition, Resources, Writing–review and editing. HA: Writing–review and editing, Project administration, Supervision.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The research work was supported by Researchers Supporting Project number (RSP2024R110) at King Saud University, Riyadh, Saudi Arabia.
The authors wish to thank Researchers Supporting Project Number (RSP2024R110) at King Saud University Riyadh Saudi Arabia for financial support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1332184/full#supplementary-material
Arnold, M., and Gramza-Michałowska, A. (2022). Enzymatic browning in apple products and its inhibition treatments: a comprehensive review. Compr. Rev. Food Sci. Food Saf. 21 (6), 5038–5076. doi:10.1111/1541-4337.13059
Arulmozhi, V., Pandian, K., and Mirunalini, S. (2013). Ellagic acid encapsulated chitosan nanoparticles for drug delivery system in human oral cancer cell line (KB). Colloids Surf. B Biointerfaces 110, 313–320. doi:10.1016/j.colsurfb.2013.03.039
Asadzadeh, A., Pourfarzam, M., Yaghmaei, P., Fassihi, A., and Afshin, F. (2016). In vitro and in silico studies of the inhibitory effects of some novel kojic acid derivatives on tyrosinase enzyme. Iran. J. Basic Med. Sci. 19, 132–144.
Atasever, A., Ozdemir, H., Gulcin, I., and Irfan Kufrevioglu, O. (2013). One-step purification of lactoperoxidase from bovine milk by affinity chromatography. Food Chem. 136 (2), 864–870. doi:10.1016/j.foodchem.2012.08.072
Aydın, B., Gülçın, İ., and Alwasel, S. (2015). Purification and characterization of polyphenol oxidase from hemşin apple (malus communisL.). Int. J. Food Prop. 18 (12), 2735–2745. doi:10.1080/10942912.2015.1012725
Bae, S. J., Ha, Y. M., Park, Y. J., Park, J. Y., Song, Y. M., Ha, T. K., et al. (2012). Design, synthesis, and evaluation of (E)-N-substituted benzylidene-aniline derivatives as tyrosinase inhibitors. Eur. J. Med. Chem. 57, 383–390. doi:10.1016/j.ejmech.2012.09.026
Bhosle, M. R., Khillare, L. D., Mali, J. R., Sarkate, A. P., Lokwani, D. K., and Tiwari, S. V. (2018). DIPEAc promoted one-pot synthesis of dihydropyrido[2,3-d:6,5-d]dipyrimidinetetraone and pyrimido[4,5-d]pyrimidine derivatives as potent tyrosinase inhibitors and anticancer agents: in vitro screening, molecular docking and ADMET predictions. New J. Chem. 42 (23), 18621–18632. doi:10.1039/c8nj04622k
Bilovol, A., Tkachenko, S., Berehova, A., Havrylyuk, O., and Manhusheva, V. (2021). Сosmetology: textbook for the 2nd-year dentistry students. (English medium).
Boo, Y. C. (2021). Arbutin as a skin depigmenting agent with antimelanogenic and antioxidant properties. Antioxidants 10 (7), 1129. doi:10.3390/antiox10071129
Buitrago, E., Hardré, R., Haudecoeur, R., Jamet, H., Belle, C., Boumendjel, A., et al. (2016). Are human tyrosinase and related proteins suitable targets for melanoma therapy? Curr. Top. Med. Chem. 16 (27), 3033–3047. doi:10.2174/1568026616666160216160112
Casanola-Martin, G. M., Le-Thi-Thu, H., Marrero-Ponce, Y., Castillo-Garit, J. A., Torrens, F., Rescigno, A., et al. (2014). Tyrosinase enzyme: 1. An overview on a pharmacological target. Curr. Top. Med. Chem. 14 (12), 1494–1501. doi:10.2174/1568026614666140523121427
Chen, X., Liao, Y., Long, D., Yu, T., Shen, F., and Lin, X. (2017). The Cdc2/Cdk1 inhibitor, purvalanol A, enhances the cytotoxic effects of taxol through Op18/stathmin in non-small cell lung cancer cells in vitro. Int. J. Mol. Med. 40 (1), 235–242. doi:10.3892/ijmm.2017.2989
da Silva, A. P., Silva, N. F., Andrade, E. H. A., Gratieri, T., Setzer, W. N., Maia, J. G. S., et al. (2017). Tyrosinase inhibitory activity, molecular docking studies and antioxidant potential of chemotypes of Lippia origanoides (Verbenaceae) essential oils. Plos One 12 (5), e0175598. doi:10.1371/journal.pone.0175598
Drake, J. M., Lee, J. K., and Witte, O. N. (2014). Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol. Cell Biol. 34 (10), 1722–1732. doi:10.1128/MCB.01592-13
Fares, H., DiNicolantonio, J. J., O’Keefe, J. H., and Lavie, C. J. (2016). Amlodipine in hypertension: a first-line agent with efficacy for improving blood pressure and patient outcomes. Open Heart 3 (2), e000473. doi:10.1136/openhrt-2016-000473
Guo, Z., Wang, Y., He, B., Zhou, J., and Chiarla, C. (2022). Linc00312 single nucleotide polymorphism as biomarker for chemoradiotherapy induced hematotoxicity in nasopharyngeal carcinoma patients. Dis. Markers 2022, 6707821. doi:10.1155/2022/6707821
Huang, H., Liu, L., Wang, J., Zhou, Y., Hu, H., Ye, X., et al. (2022). Aggregation caused quenching to aggregation induced emission transformation: a precise tuning based on BN-doped polycyclic aromatic hydrocarbons toward subcellular organelle specific imaging. Chem. Sci. 13 (11), 3129–3139. doi:10.1039/D2SC00380E
Jiao, Q., Bi, L., Ren, Y., Song, S., Wang, Q., and Wang, Y. S. (2018). Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 17 (1), 36–12. doi:10.1186/s12943-018-0801-5
Knosel, T., Kampmann, E., Kirchner, T., and Altendorf-Hofmann, A. (2014). Tyrosine kinases in soft tissue tumors. Dermatol. Pathol. 35 (2), 198–201. doi:10.1007/s00292-014-1958-3
Lee, H. –S. (2002). Tyrosinase inhibitors of Pulsatilla cernua root-derived materials. J. Agric. Food Chem. 50 (6), 1400–1403. doi:10.1021/jf011230f
Lima, L. L., Lima, R. M., Da Silva, A. F., Carmo, A. M. R. D., Da Silva, A. D., and Raposo, N. R. B. (2013). Azastilbene analogs as tyrosinase inhibitors: new molecules with depigmenting potential. Sci. World J. 2013, 274643–274647. doi:10.1155/2013/274643
Masub, N., and Khachemoune, A. (2020). Cosmetic skin lightening use and side effects. J. Dermatological Treat. 33 (3), 1287–1292. doi:10.1080/09546634.2020.1845597
Mocanu, A. M., and Cernătescu, C. (2008). Metal complexes of 3-chloro-4-hydrazino--carbonyl- methoxy-benzenesulfondibuthylamide. Chem. Industry Chem. Eng. Q. 14 (1), 1–4. doi:10.2298/ciceq0801001m
Mongre, R. K., Mishra, C. B., Shukla, A. K., Prakash, A., Jung, S., Ashraf-Uz-Zaman, M., et al. (2021). Emerging importance of tyrosine kinase inhibitors against cancer: quo vadis to cure? Int. J. Mol. Sci. 22 (21), 11659. doi:10.3390/ijms222111659
Moon, K. M., Kwon, E. B., Lee, B., and Kim, C. Y. (2020). Recent trends in controlling the enzymatic browning of fruit and vegetable products. Mol. Basel, Switz. 25 (12), 2754. doi:10.3390/molecules25122754
Nazir, Y., Rafique, H., Roshan, S., Shamas, S., Ashraf, Z., Rafiq, M., et al. (2022). Molecular docking, synthesis, and tyrosinase inhibition activity of acetophenone amide: potential inhibitor of melanogenesis. BioMed Res. Int. 2022, 1040693. doi:10.1155/2022/1040693
Pan, L., Feng, F., Wu, J., Fan, S., Han, J., Wang, S., et al. (2022). Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol. Res. 181, 106270. doi:10.1016/j.phrs.2022.106270
Panis, F., and Rompel, A. (2022). The novel role of tyrosinase enzymes in the storage of globally significant amounts of carbon in wetland ecosystems. Environ. Sci. Technol. 56 (17), 11952–11968. doi:10.1021/acs.est.2c03770
Qian, W., Liu, W., Zhu, D., Cao, Y., Tang, A., Gong, G., et al. (2020). Natural skin whitening compounds for the treatment of melanogenesis (Review). Exp. Ther. Med. 20, 173–185. doi:10.3892/etm.2020.8687
Qiu, H., Chen, X., Luo, Z., Zhao, L., Zhang, T., Yang, N., et al. (2018). Inhibition of endogenous hydrogen sulfide production exacerbates the inflammatory response during urine-derived sepsis-induced kidney injury. Exp. Ther. Med. 16 (4), 2851–2858. doi:10.3892/etm.2018.6520
Roulier, B., Pérès, B., and Haudecoeur, R. (2020). Advances in the design of genuine human tyrosinase inhibitors for targeting melanogenesis and related pigmentations. J. Med. Chem. 63 (22), 13428–13443. doi:10.1021/acs.jmedchem.0c00994
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R., and Wolfson, H. J. (2005). Patch Dock and Symm Dock: servers for rigid and symmetric docking. Nucleic Acids Res. 33 (Suppl. 2), W363–W367. doi:10.1093/nar/gki481
Shao, L., Wang, X., Chen, K., Dong, X., Kong, L., Zhao, D., et al. (2018). Novel hydroxypyridinone derivatives containing an oxime ether moiety: synthesis, inhibition on mushroom tyrosinase and application in anti-browning of fresh-cut apples. Food Chem. 242, 174–181. doi:10.1016/j.foodchem.2017.09.054
Sun, Q., Tao, Q., Ming, T., Tang, S., Zhao, H., Liu, M., et al. (2023). Berberine is a suppressor of Hedgehog signaling cascade in colorectal cancer. Phytomedicine 114, 154792. doi:10.1016/j.phymed.2023.154792
Tang, L., El-Din, T. M., Swanson Tamer, M. G., Swanson, T. M., Pryde, D. C., Scheuer, T., et al. (2016). Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs. Nature 537, 117–121. doi:10.1038/nature19102
Tse, T. W., and Hui, E. (2013). Tranexamic acid: an important adjuvant in the treatment of melasma. J. Cosmet Dermatology 12, 57–66. doi:10.1111/jocd.12026
Wang, H., Yang, T., Wu, J., Chen, D., and Wang, W. (2023). Unveiling the mystery of SUMO-activating enzyme subunit 1: a groundbreaking biomarker in the early detection and advancement of hepatocellular carcinoma. Transplant. Proc. 55 (4), 945–951. doi:10.1016/j.transproceed.2023.03.042
Wang, Y., Du, Y., Liu, Z., Fu, Y., Sheng, X., and Wang, S. (2019). The synthesis of imine compounds with polyphenol structure and their tyrosinase inhibitory activity. Scientific Workshop on Chemical and Agricultural Sciences (SWCAS 2019), 116–122.
Yi, Z., Yi, Z., Huang, K., Cao, Y., Xiao, C., Li, Y., et al. (2018). Propofol attenuates mast cell degranulation via inhibiting the miR-221/PI3K/Akt/Ca2+ pathway. Exp. Ther. Med. 16 (2), 1426–1432. doi:10.3892/etm.2018.6317
Yin, X., Chen, K., Cheng, H., Chen, X., Feng, S., Song, Y., et al. (2022). Chemical stability of ascorbic acid integrated into commercial products: a review on bioactivity and delivery technology. Antioxidants 11 (1), 153. doi:10.3390/antiox11010153
Yu, T., Xu, B., Bao, M., Gao, Y., Zhang, Q., Zhang, X., et al. (2022). Identification of potential biomarkers and pathways associated with carotid atherosclerotic plaques in type 2 diabetes mellitus: a transcriptomics study. Front. Endocrinol. 13, 981100. doi:10.3389/fendo.2022.981100
Yuan, Y., Jin, W., Nazir, Y., Fercher, C., Blaskovich, M. A., Cooper, M. A., et al. (2019). Tyrosinase inhibitors as potential antibacterial agents. Eur. J. Med. Chem. 187, 111892. doi:10.1016/j.ejmech.2019.111892
Zhao, J., Liu, Y., Zhu, L., Li, J., Liu, Y., Luo, J., et al. (2023). Tumor cell membrane-coated continuous electrochemical sensor for GLUT1 inhibitor screening. J. Pharm. Analysis 13 (6), 673–682. doi:10.1016/j.jpha.2023.04.015
Zhou, L., Liu, Y., Sun, H., Li, H., Zhang, Z., and Hao, P. (2022). Usefulness of enzyme-free and enzyme-resistant detection of complement component 5 to evaluate acute myocardial infarction. Sensors Actuators B Chem. 369, 132315. doi:10.1016/j.snb.2022.132315
Keywords: tyrosinase inhibitors, tyrosine kinase, skin care, antibrowning, anticancer, amlodipine, dihydropyridine, benzylideneimine
Citation: Ahmad I, Parveen W, Noor S, Udin Z, Ali A, Ali I, Ullah R and Ali H (2024) Design and synthesis of novel dihydropyridine- and benzylideneimine-based tyrosinase inhibitors. Front. Pharmacol. 15:1332184. doi: 10.3389/fphar.2024.1332184
Received: 02 November 2023; Accepted: 04 January 2024;
Published: 26 March 2024.
Edited by:
Mohamed Abdo Rizk, Mansoura University, EgyptReviewed by:
Mizaton Hazizul Hasan, Universiti Teknologi MARA, MalaysiaCopyright © 2024 Ahmad, Parveen, Noor, Udin, Ali, Ali, Ullah and Ali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hamid Ali, aGFtaWRwY21kQHlhaG9vLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.