 Sisi Lin
Sisi Lin Yutao Lou
Yutao Lou Rui Hao1
Rui Hao1 Meihua Bao
Meihua Bao Wu Yi
Wu Yi Yiwen Zhang
Yiwen Zhang- 1Department of Pharmacy, Zhejiang Provincial People’s Hospital, People’s Hospital of Hangzhou Medical College, Hangzhou, China
- 2Hunan Key Laboratory of the Research and Development of Novel Pharmaceutical Preparations, School of Pharmaceutical Science, Changsha Medical University, Changsha, China
- 3Key Laboratory of Endocrine Gland Diseases of Zhejiang Province, Hangzhou, China
- 4Zhejiang Provincial Clinical Research Center for Malignant Tumor, Hangzhou, China
Purpose: The aim of this study was to evaluate the bioequivalence of two formulations of rupatadine (10-mg tablets) under fasting and fed conditions in healthy Chinese subjects.
Methods: A total of 72 subjects were randomly assigned to the fasting cohort (n = 36) and fed cohort (n = 36). Each cohort includes four single-dose observation periods and 7-day washout intervals. Blood samples were collected at several timepoints for up to 72 h post-dose. The plasma concentration of rupatadine and the major active metabolites (desloratadine and 3-hydroxydesloratadine) were analyzed by a validated HPLC–MS/MS method. The non-compartmental analysis method was employed to determine the pharmacokinetic parameters. Based on the within-subject standard deviation of the reference formulation, a reference-scaled average bioequivalence or average bioequivalence method was used to evaluate the bioequivalence of the two formulations.
Results: For the fasting status, the reference-scaled average bioequivalence method was used to evaluate the bioequivalence of the maximum observed rupatadine concentration (Cmax; subject standard deviation > 0.294), while the average bioequivalence method was used to evaluate the bioequivalence of the area under the rupatadine concentration–time curve from time 0 to the last detectable concentration (AUC0-t) and from time 0 to infinity (AUC0-∞). The geometric mean ratio (GMR) of the test/reference for Cmax was 95.91%, and the upper bound of the 95% confidence interval was 95.91%. For AUC0-t and AUC0-∞ comparisons, the GMR and 90% confidence interval (CI) were 98.76% (93.88%–103.90%) and 98.71% (93.93%–103.75%), respectively. For the fed status, the subject standard deviation values of Cmax, AUC0-t, and AUC0-∞ were all <0.294; therefore, the average bioequivalence method was used. The GMR and 90% CI for Cmax, AUC0-t, and AUC0-∞ were 101.19% (91.64%–111.74%), 98.80% (94.47%–103.33%), and 98.63% (94.42%–103.03%), respectively. The two-sided 90% CI of the GMR for primary pharmacokinetic endpoints of desloratadine and 3-hydroxydesloratadine was also within 80%–125% for each cohort. These results met the bioequivalence criteria for highly variable drugs. All adverse events (AEs) were mild and transient.
Conclusion: The test drug rupatadine fumarate showed a similar safety profile to the reference drug Wystamm® (J. Uriach y Compañía, S.A., Spain), and its pharmacokinetic bioequivalence was confirmed in healthy Chinese subjects based on fasting and postprandial status.
Clinical trial registration:: http://www.chinadrugtrials.org.cn/index.html, identifier CTR20213217
1 Introduction
Allergic rhinitis is a common clinical condition in otorhinolaryngology. Epidemiological studies showed that the global incidence of the disease is 10%–40% in adults and 2%–25% in children (Asher et al., 2006; Hoyte and Nelson, 2018). In China, the incidence of allergic rhinitis fluctuates between 8% and 25% in adults and between 10% and 22% in children (Zhang et al., 2009; Wang et al., 2016; Pang et al., 2022). Urticaria is also a common skin condition characterized by wind clumps, angioedema, or both (Zuberbier et al., 2014). It manifests as acute and chronic urticaria, affecting approximately 20% and 5% of the general population, respectively; moreover, these rates are increasing annually (Fine and Bernstein, 2016; Antia et al., 2018; Maurer, Zuberbier, and Metz, 2022). In addition to elevating medical expenses, allergic rhinitis and urticaria impose a heavy economic burden on the society; they also exert a detrimental effect on the ability of patients to work, sleep, interact socially, and even manage their emotions. These effects have a significant negative impact on quality of life and are associated with a significant healthcare burden. Therefore, the control of the disease progression is clinically significant for improving the quality of life, daily activities, and wellbeing of patients.
Rupatadine, a second-generation H1 antihistamine, was initially approved for the treatment of allergic rhinitis and urticaria by the European Medicines Agency in 2002 (Valero et al., 2009). Rupatadine exhibits anti-allergic and anti-inflammatory characteristics, with good safety features. This is shown by the inhibition of mast cell degranulation due to immunological or non-immune system stimulation and inhibition of mediators involved in the inflammatory response (Mullol et al., 2008). This agent has demonstrated clinical benefits in relieving symptoms of nasal airway, rhinocnesmus, and obstruction due to a runny nose (Picado, 2006; Fantin et al., 2008; Mullol et al., 2019; Muñoz-Cano et al., 2019). Moreover, it alleviates clinical symptoms in patients with urticaria (Hide, et al., 2019a). As a prodrug, rupatadine has a rapid onset of action and is extensively metabolized by P450 3A4, yielding desloratadine and 3-hydroxydesloratadine as the major active metabolites (Valero et al., 2009). The within-subject variabilities for rupatadine in the maximum observed rupatadine concentration (Cmax) (coefficient of variation [CV]: −38.8%) and area under curve (AUC; CV: −33.9%) are considerable (Merlos et al., 1997; Picado, 2006). According to the U.S. Food and Drug Administration and the National Medical Products Administration of China (NMPA) guidelines on bioequivalence studies, the reference-scaled average bioequivalence (RSABE) approach is recommended for evaluating the bioequivalence of highly variable drugs (U.S. Food and Drug Administration, 2001; The NMPA, Center for Drug Evaluation, 2019).
Rupatadine fumarate tablet (10-mg tablets, T) is a generic drug, developed by Haisco Pharmaceutical Group Co., Ltd. (Meishan, China) and launched in China in 2014. Its active ingredient, dosage form, specifications, indications, route of administration, and dosage are consistent with those of the reference drug Wystamm® (10-mg tablets, R). The generic drugs have lower costs than original products, providing a potential method to overcome the economic burden on patients. Bioequivalence (BE) studies comparing generic to innovator products are required for marketing a new generic product by the NMPA of China. A systemically active generic drug is considered to be bioequivalent to the reference drug if the rate and extent of absorption of the two products do not show any significant difference, which is assessed by conducting BE studies in human subjects to compare their pharmacokinetic characteristics. The purpose of our study was to 1) evaluate the safety and PK parameters of rupatadine and its active metabolites and 2) compare the bioequivalence of two rupatadine fumarate (10-mg tablets) formulations acquired from different suppliers.
2 Methods
2.1 Subjects
The study was performed at the Zhejiang Provincial People’s Hospital-Phase I Clinical Research Center (Hangzhou, China) from December 2021 to March 2022. All subjects were informed regarding the study and provided written informed consent prior to their participation. The inclusion criteria are as follows: healthy male and female subjects aged 18–45 years; weight ≥50 kg for male subjects and ≥45 kg for female subjects; body mass index 19.0–26.0 kg/m2; and full understanding of the informed consent, test content, process, and possible adverse events (AEs).
The exclusion criteria included the following: a history/presence of any clinically relevant condition or disease; clinically significant abnormal physical examination, electrocardiogram, laboratory, or viral serology tests; HBV Ag, HCV Ab, HIV Ab, and TP Ab tests have clinical significance; have special dietary requirements or a history of dysphagia or lactose intolerance; allergic constitution or known allergy to the study drug; cannot tolerate venipuncture or a history of needle sickness and blood sickness; have had a special diet or strenuous exercise within 2 weeks prior screening, which will affect the drug ADME prediction; a history/presence of alcohol and/or smoking and/or drug abuse; treatment with a drug that affects liver drug enzymes within 4 weeks or other drugs within 1 week prior to enrollment, received live vaccination within 3 months or plan to be vaccinated during the trial; a history of participation in other drug clinical trials, or blood loss > 450 mL within 3 months; and subjects deemed unsuitable for participation by investigators. Pregnant or lactating women were also excluded.
2.2 Study design
The protocol, amendments, and informed consent forms were approved by the Ethics Committee of the Zhejiang Provincial People’s Hospital. The study was conducted in accordance with the Declaration of Helsinki (World Medical Association, 2013), principle of Good Clinical Practice (NMPA, National Health Commission of the People’s Republic of China, 2020), and Chinese laws and regulations.
According to the NMPA guidelines (NMPA, Center for Drug Evaluation, 2019), this study was an open-label, randomized, two-treatment, full-replicated, four-period, two-sequence crossover design that was conducted in two cohorts under fasting and fed conditions. The trial featured a screening period, four treatment periods, a washout period of 7 days after each treatment period, and a follow-up period following the last treatment (Figure 1). Healthy subjects were randomly assigned to sequences of RTRT or TRTR by a randomized block design using SAS version 9.4 software. In each treatment period, all subjects received a single oral administration of 10 mg of rupatadine fumarate (specification: 10 mg; lot number: 210703; Haisco Pharmaceutical Co., Ltd.) or Wystamm® (specification: 10 mg, lot number: P003; J. Uriach y Compañía, S.A.). In the fasting cohort, subjects received rupatadine following an overnight fasting period (≥10 h). After a 7-day washout period, the subjects received orally the same dose of another formulation of rupatadine following a sequence of RTRT or TRTR. In the fed cohort, subjects consumed a high-fat breakfast (containing 528.3 calories of fat, 274.8 calories of carbohydrate, and 180.4 calories of protein) within 30 min before dosing. Investigational drugs were administered using 240 mL of water under supervision by a qualified pharmacist. Subjects were not allowed to drink additional water for 1 h before and after treatment. Food intake was strictly controlled, and standardized lunch and dinner were provided approximately 4 and 10 h post-administration, respectively.
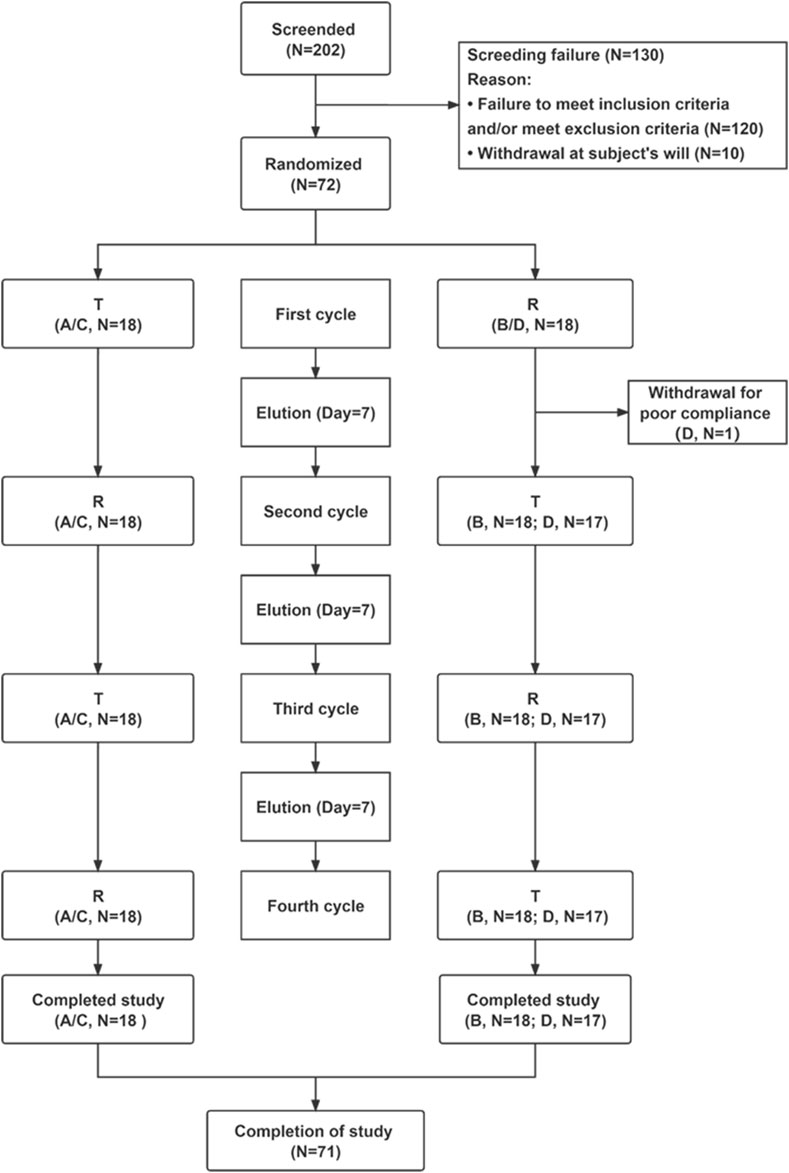
Figure 1. Flow chart of the study. Abbreviations: T, test; R, rupatadine; N, number of subjects. Notes: reference indicates Wystamm®; A and B represent groups of the fasting status; and C and D represent groups of the postprandial status.
2.3 Sample collection and analysis
Blood samples (4 mL) were collected before treatment (≤30 min) at 10 min, 20 min, 30 min, and 45 min and at 1, 1.25, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 12, 24, 48, and 72 h post-dose in K2-ethylenediaminetetraacetic acid anticoagulation tubes by direct venipuncture. After sample collection, plasma was separated by centrifugation (1,700 g × 10 min, 4°C) and stored in a −70°C freezer for 2 h until transfer to the analysis department. The plasma concentrations of rupatadine, desloratadine, and 3-hydroxydesloratadine were analyzed using a validated liquid chromatography–tandem mass spectrometry method (Sun et al., 2015). The quantitative range of the standard curve was 0.05–10.0 ng/mL for rupatadine and 0.025–5.00 ng/mL for desloratadine and 3-hydroxydesloratadine.
2.4 Pharmacokinetic analysis
This study evaluated the bioequivalence of rupatadine fumarate, a generic chemical drug of Wystamm®, using PK parameters as endpoints. The main PK parameters were Cmax and area under the rupatadine concentration–time curve from time 0 to the last detectable concentration (AUC0-t) and from time 0 to infinity (AUC0-∞). Secondary PK parameters included the time of the maximum measured plasma concentration (Tmax), elimination half-life (t1/2), and terminal rate constant (λz). Both the main and secondary PK parameters were calculated using the non-compartmental analysis model with Phoenix® WinNonlin 8.3 (Certara, Princeton, New Jersey).
2.5 Safety evaluation
Safety was evaluated at the screening period, treatment period, and follow-up period. The evaluation included AE monitoring throughout the study period, physical examination, vital signs (i.e., temperature, blood pressure, and pulse rate), clinical laboratory tests (i.e., blood routine, urine routine, blood biochemistry, and coagulation), 12-lead electrocardiogram, and pregnancy screening (female subjects only). All AEs were recorded immediately by the clinical research physician, and the relationship with the study drug and severity were evaluated with reference to the Common Terminology Criteria for the Evaluation of Adverse Events (version 5.0). In the safety analysis set, AEs were coded using the Preferred Terminology of the International Medical Terminology Dictionary (MedDRA version 24.1) and summarized according to the systematic organ classification.
2.6 Sample size and statistical analysis
According to a previous study (Solans et al., 2007), the total CV for Cmax was 56.9% and 59.8%, respectively, and the total CV for AUC0-t was 62.9% and 50.8%, after a single administration of rupatadine tablets under fasting and fed conditions, respectively. Assuming no influence of food intake on the PK parameters of rupatadine, intra-individual CV for Cmax and AUC0-t was estimated to be 38.8% and 33.9%, respectively, based on a 90% confidence interval (CI) for fasting versus fed conditions. Therefore, the intra-individual CV of AUC0-t, AUC0-∞, and Cmax for the reference drug Wystamm® in this study was overestimated to be 38%. Assuming that the geometric mean ratio (GMR) of the two formulations ranges from 0.95 to 1.05, with a one-sided test type I error probability α = 0.05 (two-sided total of 0.10), it was estimated that a minimum of 34 subjects would be required for the equivalence test method of the four-cycle crossover trial. This sample size would guarantee that the 90% CI for the GMR of the main PK parameters (AUC0-t, AUC0-∞, and Cmax) for the test and reference drugs with >80% power was between 80.0% and 125.0%. Considering the possibility of dropout, it was planned to recruit 36 subjects (18 subjects per administration group). Therefore, it was decided that the final sample size for the fasting status and postprandial status study would be 72 subjects.
Following the natural logarithmic transformation of the main PK parameters (Cmax, AUC0-t, and AUC0-∞), the fixed-effect analysis of variance (ANOVA) model was used to analyze statistical significance. Statistical analysis was performed using a two-sided test and 90% Cl to evaluate the bioequivalence between the test drug rupatadine fumarate and the reference drug Wystamm®. Prior to the evaluation of bioequivalence, the within-subject CV (CVWR) was calculated for each PK parameter of Wystamm®. If the CVWR was <30% for the main PK parameters (i.e., Cmax, AUC0-t, and AUC0-∞), the PK bioequivalence evaluation was conducted using the average bioequivalence (ABE) method (U.S. Food and Drug Administration, 2001). A 90% CI of the GMR for the main PK parameters between the predefined intervals of 80.0%–125.0% denoted PK bioequivalence for the relevant reference product. If CVWR was ≥30% for the main PK parameters, the RSABE method was performed for bioequivalence evaluation (Davit et al., 2012). In this case, PK bioequivalence was concluded if the upper limit of the one-sided 95% CI for
3 Results
3.1 Subject demographics and baseline characteristics
Of the 202 subjects initially screened in this study, 130 did not meet the inclusion criteria and were excluded (Figure 1). The remaining 72 subjects were randomly divided into four groups (18 subjects per group): group A or group B (fasting status) and group C or group D (postprandial status). Only one subject in group D dropped out of the study due to poor adherence, while all other subjects completed the study. Demographic features and baseline clinical characteristics (listed in Table 1) were comparable between the treatment groups. There were no significant differences in the demographic characteristics between the two sequences for the fasting and fed conditions.
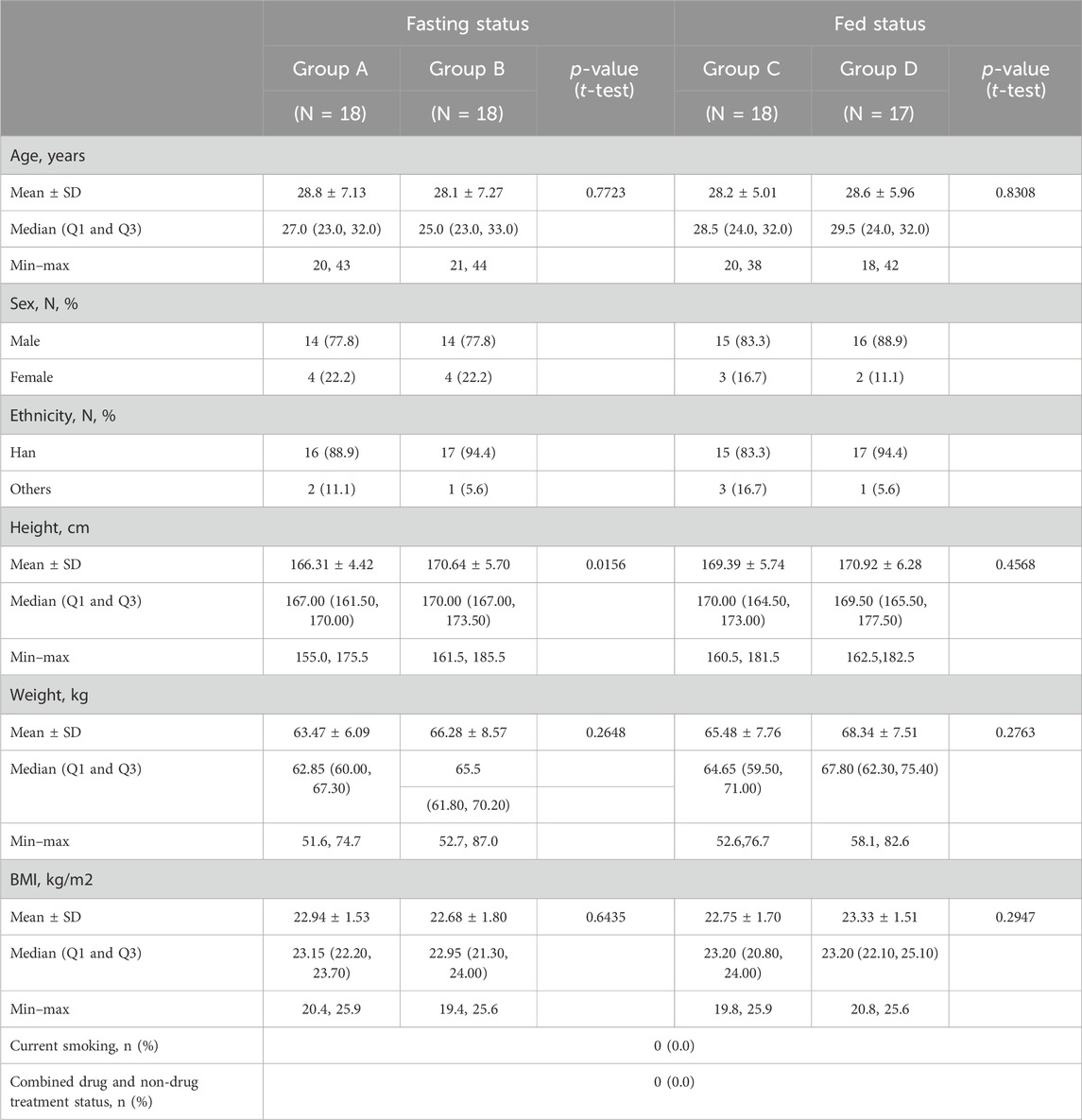
Table 1. Subject demographics and baseline characteristics by treatment sequence.
3.2 PK properties
The PK parameters for the fasting and postprandial conditions are summarized in Table 2. The mean ± standard deviation (SD) plasma concentration–time curves of rupatadine (T vs. R) (Figures 2A, B) and those of desloratadine and 3-hydroxydesloratadine (Figure 2C–F) were drawn after four periods of administration under fasting and fed conditions.
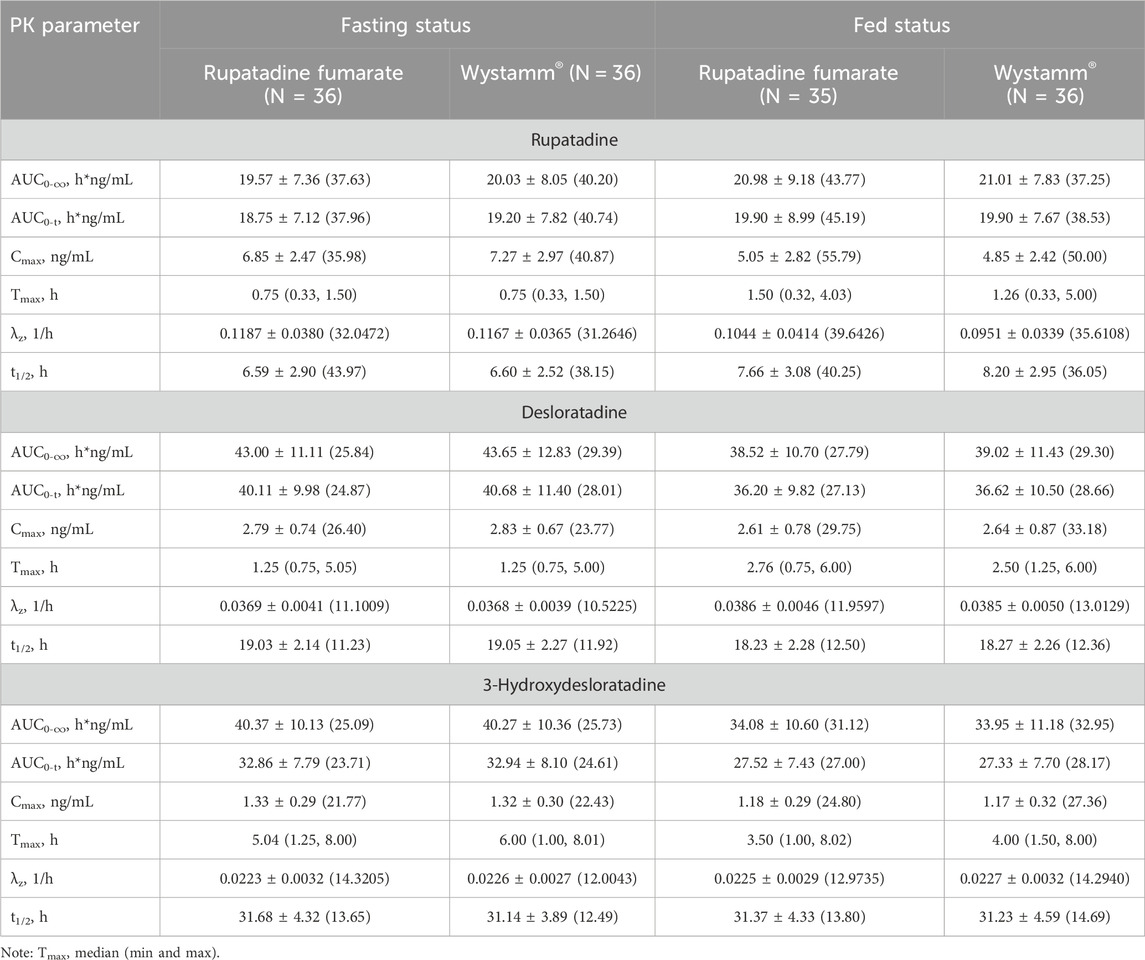
Table 2. Pharmacokinetic parameters (mean ± SD) after the administration of rupatadine fumarate and Wystamm® under fasting and fed conditions.
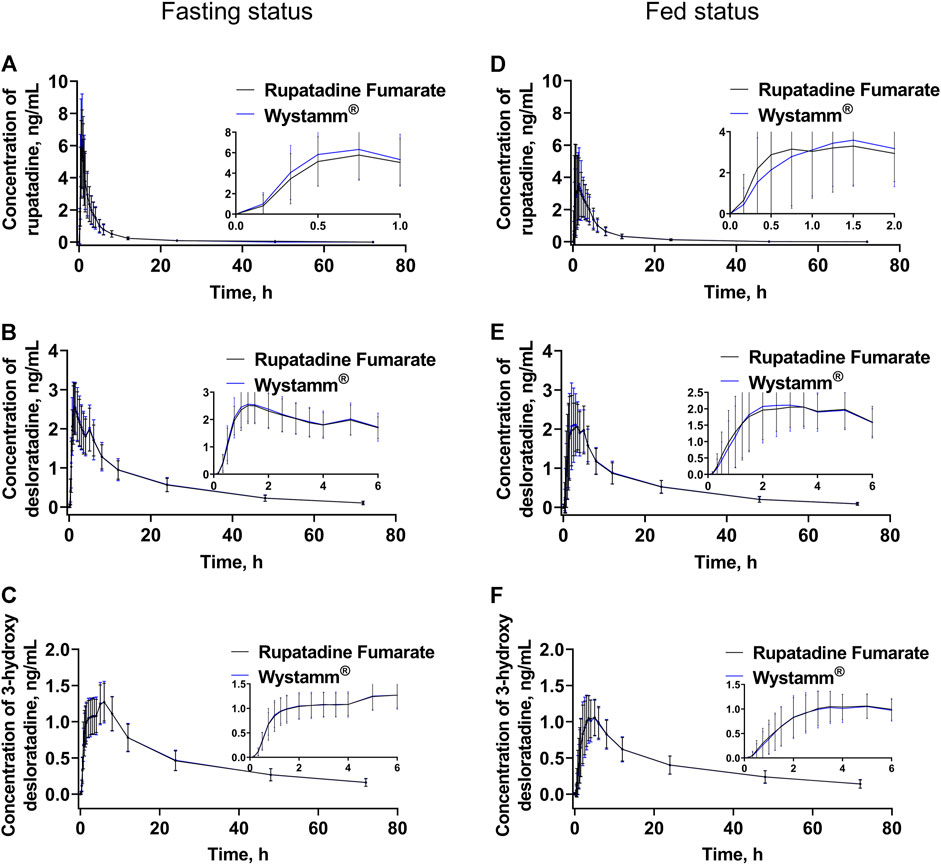
Figure 2. Plasma concentration–time curves. The mean (±SD) plasma concentration–time curves of rupatadine (A), desloratadine (B), and 3-hydroxydesloratadine (C) after a single oral administration of 10 mg rupatadine fumarate or Wystamm® under the fasting status. The mean (±SD) plasma concentration–time curves of rupatadine (D), desloratadine (E), and 3-hydroxydesloratadine (F) after a single oral administration of 10 mg rupatadine fumarate or Wystamm® under the postprandial status. Abbreviations: SD, standard deviation.
In the fasting condition, the mean ± SD (CV%) of the AUC0-t values for T and R were 18.75 ± 7.12 h*ng/mL (37.96%) and 19.20 ± 7.82 h*ng/mL (40.74%), respectively; the AUC0-∞ values were 19.57 ± 7.36 h*ng/mL (37.63%) and 20.03 ± 8.05 h*ng/mL (40.20%), respectively; and the Cmax values were 6.85 ± 2.47 ng/mL (35.98%) and 7.27 ± 2.97 ng/mL (40.87%), respectively. The median Tmax was 0.75 for both medications. The mean ± SD (CV%) of the λz values for T and R were 0.1187 ± 0.0380 1/h (32.0472%) and 0.1167 ± 0.0365 1/h (31.2646%), respectively, and the t1/2 values were 6.59 ± 2.90 h (43.97%) and 6.60 ± 2.52 h (38.15%), respectively. The period, sequence, and formulation factors may affect the equivalence of the T formulation and R formulation in the bioequivalence study. The ANOVA results showed that a significant period effect was observed in the Cmax (p = 0.0307), AUC0-t (p = 0.0226), and AUC0-∞ (p = 0.0255) values of rupatadine. However, the bioequivalence of statistical differences in the administration period could still be recognized; there was no significant difference in Cmax, AUC0-t, and AUC0-∞ between the drug formulations and drug sequences (p > 0.05) (Table 3).
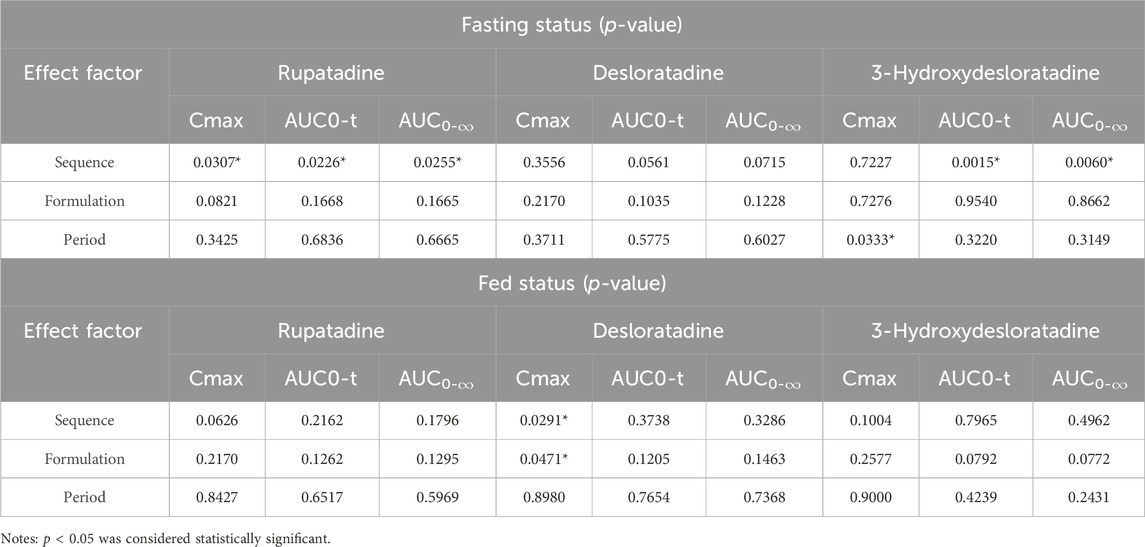
Table 3. Results of variance analysis of the main PK parameters after logarithmic transformation.
In the fed condition, Cmax, AUC0-t, and AUC0-∞ of subjects in the four periods were included in the PK parameter set of rupatadine, in addition to the parameters for the first period of the subject in group D who withdrew early from the study. The mean ± SD (CV%) of the AUC0-t values for T and R were 19.90 ± 8.99 h*ng/mL (45.19%) and 19.90 ± 7.67 h*ng/mL (38.53%), respectively; the AUC0-∞ values were 20.98 ± 9.18 h*ng/mL (43.77%) and 21.01 ± 7.83 h*ng/mL (37.25%), respectively; and the Cmax values were 5.05 ± 2.82 ng/mL (55.79%) and 4.85 ± 2.42 ng/mL (50.00%), respectively. The median Tmax values for T and R were 1.50 and 1.26 h, respectively. The mean ± SD (CV%) of the λz values for T and R were 0.1044 ± 0.0414 1/h (39.6426%) and 0.0951 ± 0.0339 1/h (35.6108%), respectively, and the t1/2 values were 7.66 ± 3.08 h (40.25%) and 8.20 ± 2.95 h (36.05%), respectively.
3.3 Bioequivalence evaluation
The results of the bioequivalence evaluation between T and R under the fasting status and fed status are shown in Table 4.
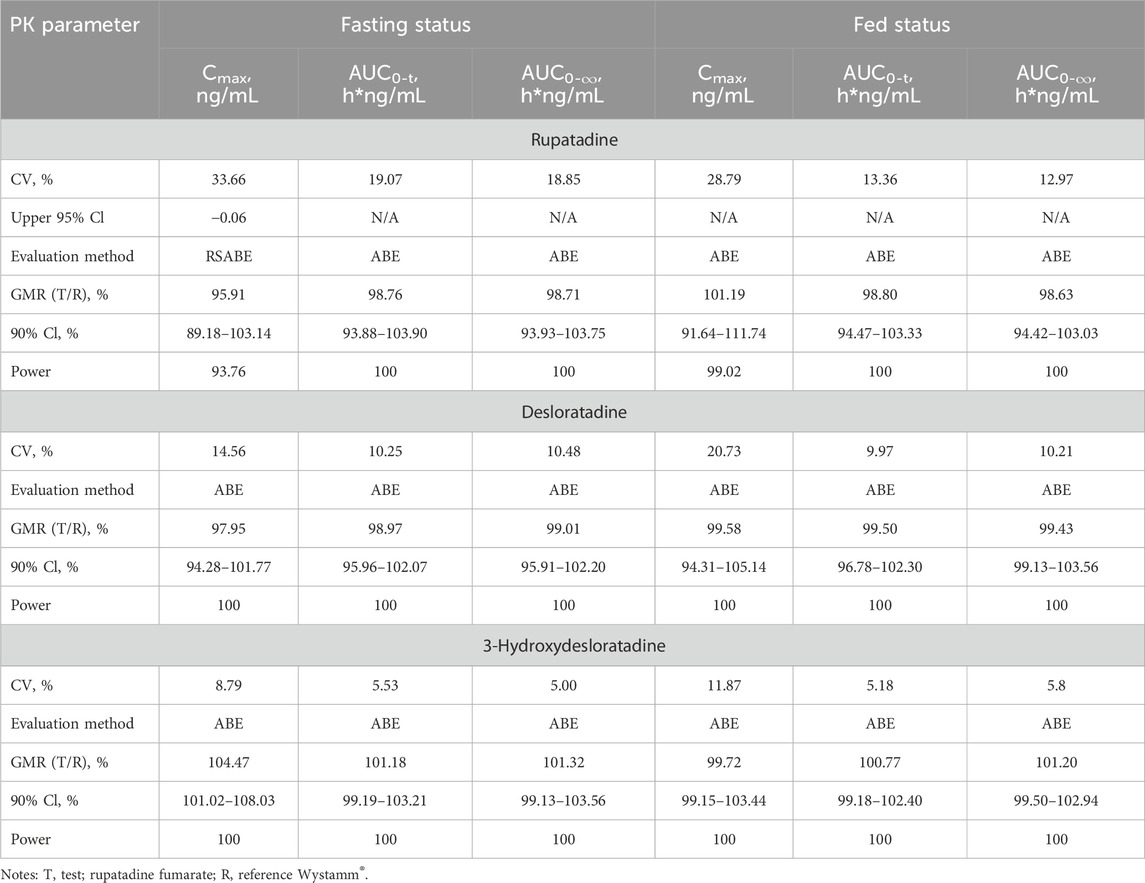
Table 4. Bioequivalence statistics for PK parameters of rupatadine fumarate and Wystamm® under the fasting and fed status.
In the fasting status, 36 subjects completed the four-period study, and all PK parameters were calculated. Since the CVWR value for Cmax was >30%, the RSABE method was employed for equivalence evaluation. The T/R GMR (power) for Cmax was 95.91% (93.76%), falling within the predefined interval of 80%–125%. Of note, the upper limit of the one-sided 95% CI was −0.06 < 0, indicating that the test drug rupatadine fumarate was bioequivalent to the reference drug Wystamm® based on the endpoint Cmax. Furthermore, since the CVWRs values for AUC0-t and AUC0-∞ were below the threshold value (i.e., <30%), the ABE method was employed to evaluate the PK bioequivalence. The 90% CIs (power) of GMR were 93.88%–103.90% (100%) and 93.93%–103.75% (100%), respectively, both falling within the predefined interval (80%–125%) and, therefore, meeting the bioequivalence criteria for ABE.
In the fed condition, one subject dropped out after receiving the first single dose of the R formulation. Thus, the PK parameters of the T formulation were calculated in 35 subjects. Since the CVWRs values for Cmax, AUC0-t, and AUC0-∞ were <30%, the bioequivalence evaluation was conducted using the ABE method. The 90% CIs (power) of the GMR for these PK parameters were 91.64%–111.74% (99.02%), 94.47%–103.33% (100%), and 94.47%–103.33% (100%), respectively, all falling within the predefined interval of 80%–125%; the upper limit of the two-sided 95% CI was <0. These results indicate that the test drug rupatadine fumarate was bioequivalent to the reference drug Wystamm®.
According to the guidelines, the GMR for PK parameter endpoints of the major metabolites was not used as a basis to determine the bioequivalence of T and R formulations; notably, all values were within the predefined interval of 80%–125%. In summary, T and R were bioequivalent under both the fasting and fed conditions (Figure 3).
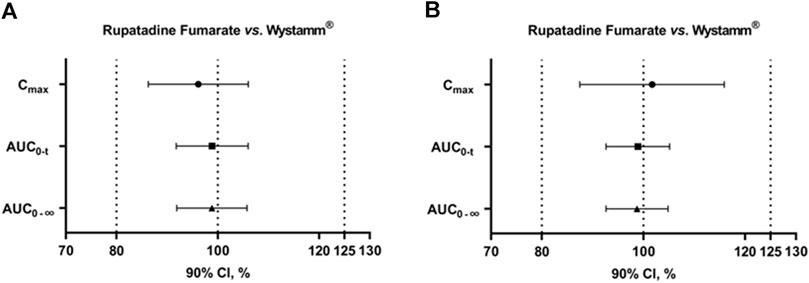
Figure 3. 90% CIs of AUC0-t, Cmax, and AUC0-∞ for rupatadine fumarate and Wystamm® under the fasting status (A) and postprandial status (B). Abbreviations: AUC0-t, area under the rupatadine concentration–time curve from time 0 to the last detectable concentration; AUC0-∞, area under the rupatadine concentration–time curve from time 0 to infinity; CI, confidence interval; and Cmax, maximum observed rupatadine concentration.
3.4 Safety evaluation
A summary of AEs by the systematic organ classification and preferred terminology for T and R under the fasting and postprandial conditions is presented in Table 5. Overall, both drugs exhibited a good safety profile in healthy Chinese subjects.
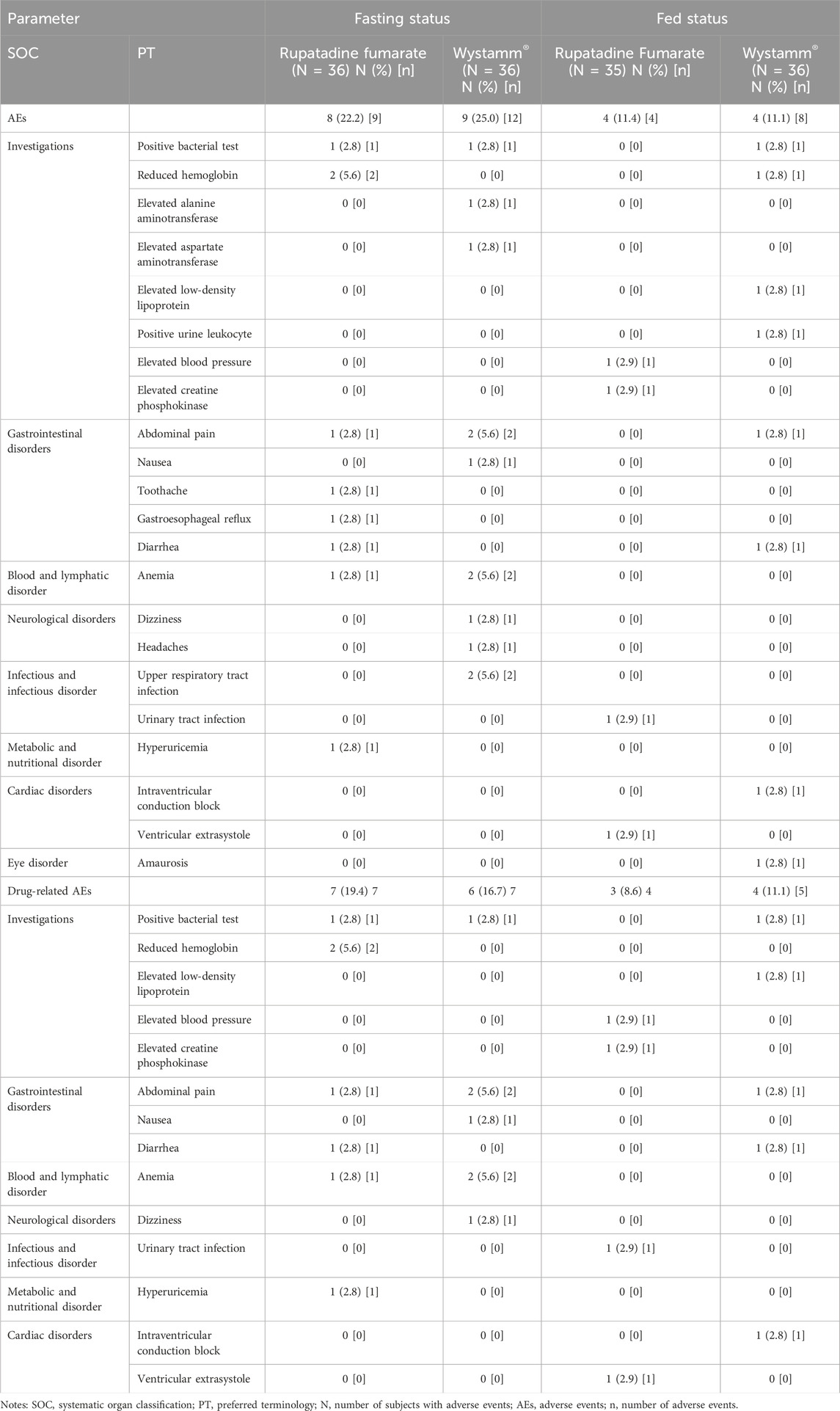
Table 5. Summary of AEs under fasting and fed condition.
In the fasting status, seven (19.4%) and six (16.7%) subjects who received the T and R formulation, respectively, experienced drug-related treatment-emergent AEs (TEAEs). In the postprandial status, three (8.6%) and four (11.1%) subjects who received the T and R formulation, respectively, experienced TEAEs. The incidence of TEAEs was similar, and the severity of all AEs was grade 1; in addition, there were no severe AEs and AEs leading to subject withdrawal or death. Frequent TEAEs between the T and R groups were a positive bacterial test (2.8% vs. 2.8%, respectively), reduced hemoglobin (5.6% vs. 0.0%, respectively), abdominal pain (2.8% vs. 5.6%, respectively), anemia (2.8% vs. 5.6%, respectively), and upper respiratory tract infection (0.0% vs. 5.6%, respectively).
4 Discussion
Allergic rhinitis and CIU are chronic disorders that are associated with increased morbidity and thus have a major impact on the quality of life. Therefore, the control of the progression of anaphylactic disease is clinically significant. Oral antihistamines are the major pharmacological treatment. Rupatadine is a second-generation H1-receptor antagonist antihistamine and has potent PAF antagonist activity, the efficacy and safety of which have been demonstrated in clinical trials and clinical applications. However, the high cost of innovative products imposes a financial burden on patients. Thus, the exploitation of generic rupatadine extends the range of oral agents available for the treatment of allergic disorders.
The present trial was a phase Ⅰ, single-dose, randomized, open-label, four-period, crossover study on healthy Chinese subjects. It was designed to evaluate the bioequivalence and safety between the test drug rupatadine fumarate and the reference drug Wystamm®. The study involved 36 subjects under the fasting status and another 36 subjects under the fed status. One subject withdrew after the first administration under the fed status, and the remaining 71 subjects received the investigational drug (i.e., rupatadine fumarate or Wystamm®). The results revealed that the PK parameters (i.e., Cmax, AUC0-t, AUC0–∞, Tmax, t1/2, and λz) were similar between T and R. PK parameters were also similar for desloratadine and 3-hydroxydesloratadine. All 90% CIs for the GMR of Cmax, AUC0-t, and AUC0-∞ were within the standard prespecified range (i.e., 80%–125%), indicating that T and R were bioequivalent.
The PK profiles of rupatadine in healthy subjects after a single dose of 10 mg under the fasting status have been well-established (Solans et al., 2008; Mullol et al., 2015; Sun et al., 2015). Rupatadine is rapidly absorbed within 45 min to 1 h after oral administration in adults, with a Cmax of 2.3 ng/mL (Mullol et al., 2015). PK parameters, including Cmax, AUC0-t, and AUC0-∞, were consistent with those reported in previous studies, in which healthy Chinese subjects received rupatadine fumarate tablets (Zhuhai Kinhoo Pharmaceutical Co., Ltd., Zhuhai, China) (Sun et al., 2015). The Cmax, Tmax, and t1/2 values of two rupatadine formulations in our study were generally consistent with those reported by Solans et al. (2008), demonstrating rapid absorption and elimination. However, the AUC (AUC0-t and AUC0-∞) in the present study was noticeably higher, indicating higher bioavailability and exposure level. Ethnic differences may be one of the factors responsible for this discrepancy. The majority of participants in the comparative study were European, whereas all subjects in our study were Chinese. The smaller body size and lower levels of cytochrome P450, family 3, and subfamily A (CYP3A) enzymes in Chinese compared with European individuals (two inter-ethnic physiological characteristics) mean a higher exposure to rupatadine (Ramamoorthy et al., 2015). In addition, the AUC0-∞ and t1/2 values of rupatadine were lower than those of desloratadine and 3-hydroxydesloratadine, indicating that both active metabolites may exert a lasting effect.
Consistent with a previous study, in this investigation, the effect of food on the PK parameters of rupatadine was mainly reflected in t1/2 and Tmax, which were prolonged in both the T and R groups (Solans et al., 2007). In fact, the PK profiles of rupatadine shifted to the right under the postprandial status; thus, it is highly probable that gastric emptying is responsible for the increase in t1/2 and Tmax. For both T and R, food consumption increased the AUC but decreased the Cmax of rupatadine, while the Cmax and AUC of desloratadine and 3-hydroxydesloratadine were decreased. This evidence indicated that food could accelerate the rate of rupatadine absorption, as reflected by an increase in AUC; nevertheless, it could slow it down, as reflected by a delay in Tmax and a decrease in Cmax, which affected the formation of metabolites desloratadine and 3-hydroxydesloratadine. However, our primary objective was to estimate bioequivalence rather than the food effect; hence, the effect of food on the PK profile was imprecise in these two cohorts. In conclusion, despite the discrepancies in Cmax and AUC compared with previous data, these differences did not affect the bioequivalence between T and R.
AEs were evaluated for both treatment groups in two separate cohorts; all AEs were TEAEs and rated as mild. The TEAEs included ventricular extrasystole, anemia, dizziness, headache, abdominal pain, positive bacterial tests, presence of albumin and red blood cells in urine, and elevated levels of creatine phosphokinase, alanine aminotransferase, and aspartate aminotransferase in blood, all of which have a low incidence rate. Headache was the most commonly reported AE in this study, with one and three cases occurring under the fasting status and fed status, respectively. However, a low rate of narcolepsy, which was frequently reported in European, Korean, Japanese, and Brazilian populations, was recorded in this study (Gimenez-Arnau et al., 2007; Mion et al., 2009; Hide, et al., 2019b; Won et al., 2021). Furthermore, there was no difference in the incidence of TEAEs between the T and R groups (p > 0.05). Therefore, we concluded that the test drug rupatadine fumarate and reference drug Wystamm® have an equivalent safety profile.
Several limitations are present in this study. The recommended dose of two investigational drugs (10 mg bid) was used in this clinical trial, and dose titration was not selected. Further studies are warranted to verify the dose relationship between the investigational drugs and AEs. In addition, in this trial, the sample size was small, and physiological differences were limited. Although the requirements of a bioequivalence trial were met, this investigation is unable to provide a comprehensive evaluation of the PK profiles and safety of those drugs. Lastly, adolescents and children were not recruited in the trial; thus, the safety and effectiveness of the drugs in these populations should be further evaluated.
5 Conclusion
The results of the study confirmed that rupatadine fumarate 10 mg was bioequivalent to the reference drug Wystamm® in healthy Chinese subjects under the fasting status and postprandial status. The two investigational drugs were generally well-tolerated and safe. These findings based on Cmax, AUC0-t, and AUC0-∞ in this clinical trial indicated that the test drug rupatadine fumarate could be an alternative to the reference drug Wystamm® in China, thereby improving accessibility and reducing drug-related costs.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Zhejiang Provincial People’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
SL: conceptualization, writing–original draft, and writing–review and editing. YL: writing–original draft and writing–review and editing. RH: conceptualization and writing–original draft. YS: conceptualization and writing–original draft. JY: conceptualization and writing–original draft. LF: conceptualization and writing–original draft. MB: writing–review and editing. WY: supervision and writing–review and editing. YZ: writing–review and editing and Supervision.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by the Zhejiang Medical and Health Science and Technology Project (Project: 2022496567), Zhejiang Provincial Department of Education General Research Project (Project: Y202146122), Medical and Health Research Program of Zhejiang Province (Grant Nos: 2021KY040 and 2022KY069), and Zhejiang Provincial Program for the Cultivation of New Heath Talents to YZ.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1328142/full#supplementary-material
References
Antia, C., Baquerizo, K., Korman, A., Bernstein, J. A., and Alikhan, A. (2018). Urticaria: a comprehensive review: epidemiology, diagnosis, and work-up. J. Am. Acad. Dermatol. 79 (4), 599–614. doi:10.1016/j.jaad.2018.01.020
Asher, M. I., Montefort, S., Björkstén, B., Lai, C. K., Strachan, D. P., Weiland, S. K., et al. (2006). Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet 368 (9537), 733–743. doi:10.1016/S0140-6736(06)69283-0
Davit, B. M., Chen, M. L., Conner, D. P., Haidar, S. H., Kim, S., Lee, C. H., et al. (2012). Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration. AAPS J. 14 (4), 915–924. doi:10.1208/s12248-012-9406-x
Fantin, S., Maspero, J., Bisbal, C., Agache, I., Donado, E., Borja, J., et al. (2008). A 12-week placebo-controlled study of rupatadine 10 mg once daily compared with cetirizine 10 mg once daily, in the treatment of persistent allergic rhinitis. Allergy 63 (7), 924–931. doi:10.1111/j.1398-9995.2008.01668.x
Fine, L. M., and Bernstein, J. A. (2016). Guideline of chronic urticaria beyond. Allergy Asthma Immunol. Res. 8 (5), 396–403. doi:10.4168/aair.2016.8.5.396
Gimenez-Arnau, A., Pujol, R. M., Ianosi, S., Kaszuba, A., Malbran, A., Poop, G., et al. (2007). Rupatadine in the treatment of chronic idiopathic urticaria: a double-blind, randomized, placebo-controlled multicentre study. Allergy 62 (5), 539–546. doi:10.1111/j.1398-9995.2007.01330.x
Hide, M., Suzuki, T., Tanaka, A., and Aoki, H. (2019a). Efficacy and safety of rupatadine in Japanese adult and adolescent patients with chronic spontaneous urticaria: a double-blind, randomized, multicenter, placebo-controlled clinical trial. Allergol. Int. 68 (1), 59–67. doi:10.1016/j.alit.2018.06.002
Hide, M., Suzuki, T., Tanaka, A., and Aoki, H. (2019b). Long-term safety and efficacy of rupatadine in Japanese patients with itching due to chronic spontaneous urticaria, dermatitis, or pruritus: a 12-month, multicenter, open-label clinical trial. J. Dermatol. Sci. 94 (3), 339–345. doi:10.1016/j.jdermsci.2019.05.008
Hoyte, F. C. L., and Nelson, H. S. (2018). Recent advances in allergic rhinitis. F1000 Res. 7, F1000 Faculty Rev-1333. doi:10.12688/f1000research.15367.1
Maurer, M., Zuberbier, T., and Metz, M. (2022). The classification, pathogenesis, diagnostic workup, and management of urticaria: an update. Handb. Exp. Pharmacol. 268, 117–133. doi:10.1007/164_2021_506
Merlos, M., Giral, M., Balsa, D., Ferrando, R., Queralt, M., Puigdemont, A., et al. (1997). Rupatadine, a new potent, orally active dual antagonist of histamine and platelet-activating factor (PAF). J. Pharmacol. Exp. Ther. 280 (1), 114–121.
Mion, O. deG., Campos, R. A., Antila, M., Rapoport, P. B., Rosario, N., de Mello Junior, J. F., et al. (2009). Futura study: evaluation of efficacy and safety of rupatadine fumarate in the treatment of persistent allergic rhinitis. Braz. J. Otorhinolaryngol. 75 (5), 673–679. doi:10.1590/s1808-86942009000500011
Mullol, J., Bousquet, J., Bachert, C., Canonica, G. W., Giménez-Arnau, A., Kowalski, M. L., et al. (2015). Update on rupatadine in the management of allergic disorders. Allergy 70, 1–24. Suppl 100. doi:10.1111/all.12531
Mullol, J., Bousquet, J., Bachert, C., Canonica, W. G., Gimenez-Arnau, A., Kowalski, M. L., et al. (2008). Rupatadine in allergic rhinitis and chronic urticaria. Allergy 63, 5–28. Suppl 87. doi:10.1111/j.1398-9995.2008.01640.x
Mullol, J., Izquierdo, I., Okubo, K., Canonica, G. W., Bousquet, J., and Valero, A. (2019). Clinically relevant effect of rupatadine 20 mg and 10 mg in seasonal allergic rhinitis: a pooled responder analysis. Clin. Transl. Allergy 9, 50. doi:10.1186/s13601-019-0293-4
Muñoz-Cano, R. M., Casas-Saucedo, R., Valero Santiago, A., Bobolea, I., Ribó, P., and Mullol, J. (2019). Platelet-Activating factor (PAF) in allergic rhinitis: clinical and therapeutic implications. J. Clin. Med. 8 (9), 1338. doi:10.3390/jcm8091338
National Medical Products Administration, National Health Commission of the People’s Republic of China (2020) Good clinical Practice.
Pang, K., Li, G., Li, M., Zhang, L., Fu, Q., Liu, K., et al. (2022). Prevalence and risk factors for allergic rhinitis in China: a systematic review and meta-analysis. Evid. Based. Complement. Altern. Med. 2022, 7165627. doi:10.1155/2022/7165627
Picado, C. (2006). Rupatadine: pharmacological profile and its use in the treatment of allergic disorders. Expert. Opin. Pharmacother. 7 (14), 1989–2001. doi:10.1517/14656566.7.14.1989
Ramamoorthy, A., Pacanowski, M. A., Bull, J., and Zhang, L. (2015). Racial/ethnic differences in drug disposition and response: review of recently approved drugs. Clin. Pharmacol. Ther. 97 (3), 263–273. doi:10.1002/cpt.61
Solans, A., Carbó, M. L., Peña, J., Nadal, T., Izquierdo, I., and Merlos, M. (2007). Influence of food on the oral bioavailability of rupatadine tablets in healthy volunteers: a single-dose, randomized, open-label, two-way crossover study. Clin. Ther. 29 (5), 900–908. doi:10.1016/j.clinthera.2007.05.004
Solans, A., Izquierdo, I., Donado, E., Antonijoan, R., Peña, J., Nadal, T., et al. (2008). Pharmacokinetic and safety profile of rupatadine when coadministered with azithromycin at steady-state levels: a randomized, open-label, two-way, crossover, Phase I study. Clin. Ther. 30 (9), 1639–1650. doi:10.1016/j.clinthera.2008.09.002
Sun, C., Li, Q., Pan, L., Liu, B., Gu, P., Zhang, J., et al. (2015). Development of a highly sensitive LC-MS/MS method for simultaneous determination of rupatadine and its two active metabolites in human plasma: application to a clinical pharmacokinetic study. J. Pharm. Biomed. Anal. 111, 163–168. doi:10.1016/j.jpba.2015.03.025
The National Medical Products Administration of China, Center for Drug Evaluation (2019) Guideline for bioavailability and bioequivalence studies of generic drug products.
The U.S. Food and Drug Administration (2001) Guidance for industry—statistical approaches to establishing bioequivalence rockville.
Valero, A., de la Torre, F., Castillo, J. A., Rivas, P., del Cuvillo, A., Antépara, I., et al. (2009). Safety of rupatadine administered over a period of 1 year in the treatment of persistent allergic rhinitis: a multicentre, open-label study in Spain. Drug Saf. 32 (1), 33–42. doi:10.2165/00002018-200932010-00003
Wang, X. D., Zheng, M., Lou, H. F., Wang, C. S., Zhang, Y., Bo, M. Y., et al. (2016). An increased prevalence of self-reported allergic rhinitis in major Chinese cities from 2005 to 2011. Allergy 71 (8), 1170–1180. doi:10.1111/all.12874
Won, T. B., Kim, H. G., Kim, J. W., Kim, J. K., Kim, Y. H., Kim, S. W., et al. (2021). Efficacy and safety of rupatadine fumarate in the treatment of perennial allergic rhinitis: a multicenter, double-blinded, randomized, placebo-controlled, bridging study in Koreans. Asian. pac. J. Allergy Immunol. doi:10.12932/AP-201220-1019
World Medical Association (2013). World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 310 (20), 2191–2194. doi:10.1001/jama.2013.281053
Zhang, L., Han, D., Huang, D., Wu, Y., Dong, Z., Xu, G., et al. (2009). Prevalence of self-reported allergic rhinitis in eleven major cities in China. Int. Arch. Allergy Immunol. 149 (1), 47–57. doi:10.1159/000176306
Keywords: rupatadine fumarate, pharmacokinetics, bioequivalence, reference-scaled average bioequivalence, safety
Citation: Lin S, Lou Y, Hao R, Shao Y, Yu J, Fang L, Bao M, Yi W and Zhang Y (2024) A single-dose, randomized, open-label, four-period, crossover equivalence trial comparing the clinical similarity of the proposed biosimilar rupatadine fumarate to reference Wystamm® in healthy Chinese subjects. Front. Pharmacol. 15:1328142. doi: 10.3389/fphar.2024.1328142
Received: 26 October 2023; Accepted: 22 April 2024;
Published: 17 May 2024.
Edited by:
Md Saquib Hasnain, Palamau Institute of Pharmacy, IndiaReviewed by:
Syed Anees Ahmed, East Carolina University, United StatesIndrashis Podder, College of Medicine & Sagore Dutta Hospital, India
Copyright © 2024 Lin, Lou, Hao, Shao, Yu, Fang, Bao, Yi and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiwen Zhang, emhhbmd5aXdlbkBobWMuZWR1LmNu; Wu Yi, d3V5aTEwMDAwQDEyNi5jb20=
†These authors have contributed equally to this work