 Alessandra Picollo
Alessandra Picollo- 1Institute of Biophysics, National Research Council, Genova, Italy
- 2RAISE Ecosystem, Genova, Italy
Chloride is one of the most abundant anions in the human body; it is implicated in several physiological processes such as the transmission of action potentials, transepithelial salt transport, maintenance of cellular homeostasis, regulation of osmotic pressure and intracellular pH, and synaptic transmission. The balance between the extracellular and intracellular chloride concentrations is controlled by the interplay of ion channels and transporters embedded in the cellular membranes. Vesicular members of the CLC chloride protein family (vCLCs) are chloride/proton exchangers expressed in the membrane of the intracellular organelles, where they control vesicular acidification and luminal chloride concentration. It is well known that mutations in CLCs cause bone, kidney, and lysosomal genetic diseases. However, the role of CLC exchangers in neurological disorders is only now emerging with the identification of pathogenic CLCN gene variants in patients with severe neuronal and intellectual dysfunctions. This review will provide an overview of the recent advances in understanding the role of the vesicular CLC chloride/proton exchangers in human pathophysiology.
1 Introduction
The CLC chloride transporting protein family includes four chloride channels (ClC-1, ClC-2, ClC-Ka, and ClC-Kb) expressed in the plasma membrane and five chloride/proton exchangers (from ClC-3 to ClC-7) expressed in the membrane of intracellular compartments. They are implicated in several physiological processes, including muscle contraction, transepithelial salt reabsorption, acidification of intracellular compartments, and cellular homeostasis (Jentsch and Pusch, 2018). The vCLCs can be divided into two subclasses depending on their sequence and functional similarity. ClC-3, ClC-4, and ClC-5 form the first subclass with almost 80% sequence identity. The renal ClC-5 is expressed in early endosomes, while ClC-3 and ClC-4 localize in sorting and late endosomes. ClC-6 and ClC-7, which share 45% of sequence identity, constitute the second subclass. ClC-6 resides in late endosomes, while ClC-7 localizes in lysosomes and osteoclasts, where it is implicated in bone resorption (Jentsch and Pusch, 2018). All CLCs share a similar overall dimeric architecture, with a large transmembrane domain and a cytosolic region containing two CBS (cystathionine β synthase) domains (Accardi, 2015). Recent studies have demonstrated that ClC-4 is more stable when it forms heterodimers with ClC-3 (Weinert et al., 2020).
The relevance of vCLC proteins is shown by their involvement in severe pathologies, such as neurodegeneration (ClC-3 and ClC-6) (Stobrawa et al., 2001; Polovitskaya et al., 2020), global developmental delay (ClC-3) (Duncan et al., 2021), intellectual disability combined with epilepsy, various psychiatric conditions (ClC-4) (Veeramah et al., 2013; Hu et al., 2016; Palmer et al., 2018), lysosomal storage diseases (ClC-6 and ClC-7) (Kasper, 2005; Poet et al., 2006; He et al., 2021), osteopetrosis without or with neurodegeneration (ClC-7) (Kornak et al., 2001; Kasper, 2005), and Dent’s disease (ClC-5) (Lloyd et al., 1996; Piwon et al., 2000).
With the advent of next-generation sequencing analysis, many new genetic defects are emerging. Despite the difficulty in understanding these data, it was possible to identify new pathophysiological roles for vCLCN genes, providing an extensive picture of the role of vCLCs in human physiology. This review aims to provide an overview of the similarities and differences in structural, functional, and physiological properties of vCLCs.
2 Structures
The development of cryo-EM technology allowed the resolution of the three-dimensional structures of several mammalian CLC channels and exchangers, but ClC-3, ClC-4, and ClC-5 structures are still missing. CLCs are homodimers with two identical subunits, each containing a large transmembrane domain (TMd, Figure 1A, in gray) and a cytosolic domain (CTd Figure 1A in cyan and blue). Each TMd is formed by 18 alpha helices (from helix A to R) tilted with respect to the plane of the biological membranes; some helices partially span the membranes, forming short loops within the protein. The cytosolic domain is composed of two cystathionin-β-synthase (CBS) domains containing two putative ATP binding sites. In vCLCs, each monomer contains a chloride and a proton permeation pathway. The two pathways converge toward the extracellular exit, sharing a common external gate, but they diverge toward the intracellular exit. The chloride permeation pathway is highly conserved between CLC channels and exchangers: three chloride binding sites, Sext, Scen, Sint, have been identified. Chloride ions bound to Sext and Sint are in contact with the extracellular and intracellular solutions, respectively. Conversely, the ion bound to Scen, located inside the protein, is coordinated by the side chain of the “gating glutamate” (Gluext), whose protonation/de-protonation state determines the opening or closing of the external gate, and by the side chains of a serine (Sercen) and a tyrosine (Tyrcen). The side chain of Gluext has been captured in two closed and two open configurations (Figure 1B). In the outward closed state, the side chain of Gluext occludes the extracellular chloride exit or occupies the Sext (Figure 1B, EcCLC (Dutzler et al., 2002) and hCLC-2 (Ma et al., 2023)) or the Scen (Figure 1B, CmCLC (Feng et al., 2010)). In the outward open state, the side chain of Gluext points or moves up toward the extracellular space (Figure 1B, hCLC-7 (Schrecker et al., 2020)) or toward the protein dimer interface (Figure 1B hCLC-1 (Park and MacKinnon, 2018)). The neutralization of Gluext transforms vCLC exchangers into pure chloride conductors (Picollo and Pusch, 2005; Scheel et al., 2005) and leaves CLC channels constitutively open (Traverso et al., 2003; Zuniga et al., 2004). In protein exchangers, to avoid free ion diffusion, an internal gate is necessary. Two models have been proposed. The first suggests that Cl− ions have to overcome a kinetic barrier to move from Scen to Sint or vice versa (Feng et al., 2012). In the second model, the chloride internal gate is physically constituted by the side chain of Tyrcen (Accardi et al., 2006). Indeed, the H+ translocation pathway is not well defined. In addition to Gluext, a second glutamate called “proton glutamate,” Gluint, which is closed to the cytosol and is located far away from the chloride permeation pathway, has been proposed as the internal proton acceptor. However, it is unclear how protons can travel the approximately 15 Å hydrophobic region separating Gluext from Gluint. Molecular dynamic simulations revealed the formation of transient water wires that connect the two H+ sites (Han et al., 2014; Lee et al., 2016), suggesting a water-mediated H+ transport mechanism. Unfortunately, direct functional experiments to prove the role of water wires in proton transport across CLCs are challenging. The comparison of all solved structures shows minimal local structural differences and the absence of water molecules, which could be due to a lack of sufficient experimental resolution. Indeed, NMR, biochemical, and electrophysiological studies suggest that during the transport cycle, global structural rearrangements occur (Abraham et al., 2015; Basilio et al., 2015), which may be consistent with conformational states where water wires can be formed.
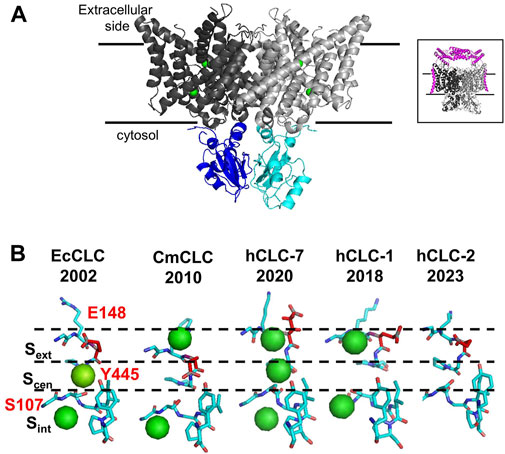
FIGURE 1. Structural properties of vCLC proteins. (A) Structure of the algae vCLC homolog CmClC, viewed within the membrane. Transmembrane domains are colored in dark and light gray and cytosolic domains are colored in blue and cyan. Green spheres correspond to chloride ions in the permeation pathway. In the inset, the structure of hClC-7 in complex with OSTM1 is shown in magenta. (B) View of the four conformations of the ion binding region captured in the corresponding protein structures solved until now. The gating glutamate is highlighted in red. Cl− ions detected by cryo-EM are shown as green spheres. [PDB ID: EcClC (1OTS), CmClC (3ORG), hClC-7 (7JM6), hClC-1 (6QVC), and hClC-2 (7xF5)].
The structure of hClC-7 in complex with its accessory protein, Ostm1 (Schrecker et al., 2020; Zhang et al., 2020) (Figure 1 A, inset), has revealed few specific structural features. First, the large highly glycosylated luminal region of Ostm1 is positioned above the luminal side of ClC-7 as a lid to protect ClC-7 from lysosomal degradation, and the unique transmembrane domain of Ostm1 interacts with ClC-7 helices facing the membrane lipid environment. Surprisingly, a phosphatidylinositol-3-phosphate (PI3P) molecule, a constituent of lysosomal membranes, has been found bound to the external surface of ClC-7 at the interface between TMd and Ctd (Schrecker et al., 2020). It has been shown that PI(3,5)P2 inhibits ClC-7 activity, but it fails in the case of the Y715C ClC-7 mutation (Leray et al., 2022), which causes lysosomal storage diseases without osteopetrosis (Nicoli et al., 2019; Leray et al., 2022). It is not known if PI3P molecules can interact with other vCLCs and modulate their transport activity.
3 Physiology and pathophysiology of vCLCs
Vesicular CLCs share common characteristics. First, they are mainly expressed in the vesicles of the endo-lysosomal pathway, in osteoclasts, in phagosomes, and in synaptic vesicles (Jentsch and Pusch, 2018). Together with the vacuolar ATPase proton pump (V-ATase), vCLCs are implicated in the regulation of the pH of intracellular organelles. Second, their biophysical properties are quite similar, but obtaining a complete functional characterization of all vCLCs took many years due to the difficulty of expressing them in plasma membranes. This limit was overcome by modification of the amino terminus or by using alternative splicing variants, as in the case of vClC-3 (Rohrbough et al., 2018) or by site-directed mutagenesis, as for vClC-7, where a leucine-based lysosomal sorting motif was identified and neutralized, partially redirecting vClC-7 to the plasma membrane (Stauber and Jentsch, 2010), or by the fusion of green fluorescence protein (GFP) to the N-terminus of vCLC-6 (Neagoe et al., 2010). vCLCs are electrogenic Cl−/H+ exchangers with a conserved stoichiometry of two chloride ions per one proton (Picollo and Pusch, 2005; Scheel et al., 2005; Graves et al., 2008; Neagoe et al., 2010; Leisle et al., 2011; Rohrbough et al., 2018). When vCLCs are expressed in the plasma membrane of Xenopus oocytes or mammalian cells, they all show strongly outwardly rectifying currents. They activate at positive membrane potentials (Figure 2), but vClC-3-ClC-4 and ClC-5 show almost instantaneous activation (Picollo and Pusch, 2005; Duncan et al., 2021) (Figure 2, left), while vClC-6 and 7 show quite slow kinetics of activation (Leisle et al., 2011; Zifarelli et al., 2022) (Figure 2, right). These outward currents correspond to the movement of chloride ions out of intracellular vesicles, coupled with an influx of protons. However, analysis of mouse models suggests that the transmembrane proton gradient of intracellular vesicles drives an increase in luminal chloride concentration, reducing the membrane potential and facilitating vesicular acidification by V-ATPase (Pressey et al., 2010; Bose et al., 2021). Mathematical model simulations (Marcoline et al., 2016, Ishida, 2013 #5753) show that an electrogenic 2Cl-/H+ exchanger allows more efficient vesicular acidification by the V-ATPase pump than a simple chloride channel.
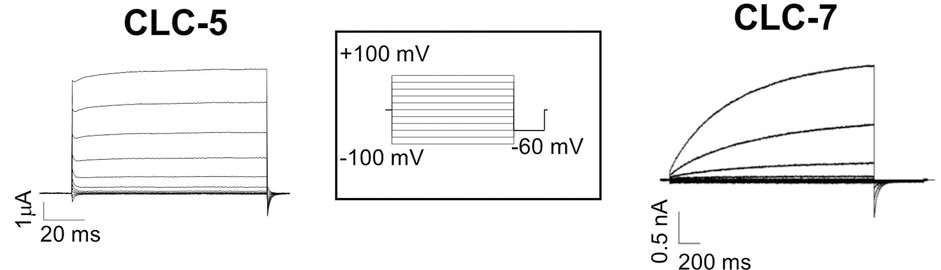
FIGURE 2. Typical recording of vCLC currents. Left two electrode voltage clamp currents recording of ClC-5 expressed in Xenopus oocytes. Right patch clamp currents recording of the plasma membrane ClC-7 construct. The stimulation voltage-clamp protocol is shown in the inset. Starting from the reversal potential, voltage steps were from −100 to 100 mV with increments of 20 mV for a duration that was protein-dependent (for 100 ms for ClC-5 and 2 s for ClC-7), and then −60 mV was applied.
Third, Clcn5, Clcn3, Clcn6, and Clcn7 knockout mice exhibit severe phenotypes resembling the severe forms of the corresponding diseases, with one exception: the Clcn4−/− mouse, which shows a milder phenotype without any neurodegeneration. In laboratory mice, the CLCN4 gene is not located on the X chromosome as in humans. An animal model with conserved human chromosome localization might be more informative and necessary for proper CLCN4 gene physiological investigation.
A common characteristic of CLCN-related disorders is a wide range of clinical symptoms, from lethal to almost asymptomatic, and a large number of pathogenic variants that cause several different functional consequences: impaired protein folding, altered protein localization, and changes in chloride and/or proton transport properties. This variety of functional alterations and clinical symptoms, combined with the limited number of patients carrying the same CLCN pathogenic variant, often represents a limit to a clear genotype-phenotype correlation.
3.1 ClC-5 and Dent’s disease
vClC-5 is expressed in the early endosomes of the epithelial cells of the proximal tube (PTCs) and in the intercalating cells of the distal nephron (Günther et al., 1998; Sakamoto et al., 1999). Although in lower abundance, ClC-5 was also found in the medullary thick ascending and thin descending limbs of Henle’s loop in the kidney (Devuyst et al., 1999). ClC-5, with the electrogenic vacuolar H+-ATPase (V-ATPase), contributes to the regulation of intra-endosomal pH and is involved in the protein uptake process in PTCs (Günther et al., 1998). Genetic defects in the CLCN5 gene cause Dent’s disease type I (Thakker, 1997; Jentsch and Pusch, 2018), a rare genetic disorder characterized by low molecular weight proteinuria, hypercalciuria, and kidney stones, mainly due to impaired endocytosis. The most common CLCN5 genetic defects are frameshift mutations that frequently result in protein misfolding and missense mutations causing loss of function (Gianesello et al., 2020). Moreover, knockout mice show altered renal endosomal acidification and reduced endocytosis in PTCs (Günther et al., 2003; Silva et al., 2003). If CLCN5 is converted in mice into a simple Cl− conductor (Clcn5unc/unc), the endosomal acidification is maintained, but the clinical phenotypes resemble those of patients affected by Dent’s disease or those developed by knockout mice (Scheinman, 1998; Piwon et al., 2000; Wang et al., 2000), suggesting that chloride conductance alone without H+ coupling is not sufficient to support tubular endocytosis.
3.2 ClC-3 and ClC-4 and heterodimers
ClC-3 and ClC-4 are broadly found in many mammalian tissues, including the brain, heart, liver, kidney, pancreas, intestine, and skeletal muscle (Kawasaki et al., 1995; Stobrawa et al., 2001); they show a similar relative abundance in the brain and muscle (Weinert et al., 2020), and both localize in the endosomes. ClC-3 has also been found in osteoclasts (Okamoto et al., 2008), and its localization and role in synaptic vesicles are still controversial (Maritzen et al., 2008; Weinert et al., 2020; Bose et al., 2021). In addition, ClC-3 is expressed in the insulin granule, where it may be implicated in insulin secretion by promoting the acidification of insulin granules (Li et al., 2009). Even if the physiological roles of ClC-3 and ClC-4 are not completely clear yet, animal models and human genetic diseases provide useful information. Knockout Clcn3 mice manifest severe neurodegeneration, leading to blindness and almost complete loss of the hippocampus in the first month of the mice’s life (Stobrawa et al., 2001; Weinert et al., 2020). A different Clcn3 mouse model, in addition, shows signs of lysosomal storage disease (Yoshikawa et al., 2002). Differently from the Clcn5unc/unc mouse model, Weinert et al. recently showed that the Clcn3unc/unc mouse does not manifest a clear phenotype. In the same study, Weinert et al. observed a significant reduction in the level of ClC-4 protein in both Clcn3−/− mice and heterozygous Clcn3+/− mice. However, in Clcn4−/− mice, they did not observe any neurodegeneration or reduction in ClC-3 protein level. To verify if the reduction of ClC-4 contributes to severe neurodegeneration in Clcn3−/− mice, they generated two genotype combinations: Clcn3−/−;Clcn4−/− and Clcn3unc;Clcn4−/−. They found that Clcn3unc/unc;Clcn4−/− mice developed more severe neurodegeneration than Clcn3−/− mice but milder than Clcn3−/−;Clcn4−/−. All this data demonstrates that ClC-4 forms stable heterodimers with ClC-3, suggesting that ClC-3 could partially compensate for the loss of ClC-4 and explaining the large variability of phenotype observed in patients carrying mutations on the CLCN4 gene. The physiological relevance of vClC-3 and vClC-4 is also emerging from the pathogenic CLCN3 and CLCN4 variants that have been found in patients affected by global developmental delay, intellectual disability, and neurodevelopmental disorders (Veeramah et al., 2013; Hu et al., 2016; Palmer et al., 2018; Duncan et al., 2021; Guo et al., 2021; Xu et al., 2021; Palmer et al., 2022). Electrophysiological analysis of these variants has revealed new biophysical mechanisms, including the gain of function and shift in voltage dependence of current gating (Duncan et al., 2021; Palmer et al., 2022).
3.3 Endolysosomal ClC-6 and ClC-7 Cl−/H+ exchangers
ClC-6 is primarily expressed in the late endosomes of the nervous system, where it partially overlaps with ClC-7 (Brandt and Jentsch, 1995; Poet et al., 2006; Neagoe et al., 2010). Clcn6−/− knockout mice showed mild neuronal lysosomal storage disease (Poet et al., 2006), resembling the phenotype observed in two patients carrying ClC-6 heterozygous missense mutations (V580M and T628R) and affected by neuronal ceroid lipofuscinosis (NCL) (Poet et al., 2006). Instead, the neutralization of the gating glutamate with alanine (E200A) is responsible for West syndrome. When the E200A mutant is heterologously expressed in vitro, it alters the autophagy lysosomal pathway (He et al., 2021) and converts ClC-6 into a pure chloride conductor (Neagoe et al., 2010). A fourth missense mutation (Y553C) was found in three unrelated children who had severe developmental delays with pronounced generalized hypotonia, respiratory insufficiency, and variable neurodegeneration (Polovitskaya et al., 2020). Electrophysiological measurements revealed that Y553C induces a drastic change in ClC-6 ion transport properties, causing a pH-dependent gain of function (stronger at luminal acidic pH) (Polovitskaya et al., 2020; Zifarelli et al., 2022). Moreover, the expression of Y553C in various cell lines exhibited an alteration in cell morphology, with the formation of enlarged vesicles (Polovitskaya et al., 2020) resembling those observed in transfected cells with the ClC-7 missense mutation, Y715C, which causes severe lysosomal storage and albinism without osteopetrosis in humans (Nicoli et al., 2019). All these observations suggest that the alteration of the endosomal-lysosomal pathway might be responsible for neurodegeneration in both cases. Instead, a decrease in the total currents has been observed in the V580M mutant, indicating a predominantly negative impact in the heterozygous variant (Zifarelli et al., 2022). For the T628R mutation, no difference in electrical activity has been detected with respect to the WT. Similar results have been reported for pathogenic ClC-4 (Palmer et al., 2022), ClC-5 (Tang et al., 2016), and ClC-7 (Leisle et al., 2011) mutants that reside either within the intracellular domain or within loops exposed to the intra/extracellular space. Classical electrophysiological in vitro approaches will likely fail to identify the pathogenic mechanisms induced by these variants.
ClC-7 is broadly expressed, and primarily, it localizes in the lysosomes (Kornak et al., 2001). Recently, it has been shown that ClC-7 also localizes in mature phagosomes where ClC-7 is important for Cl− accumulation and for phagosomal acidification (Wu et al., 2023). In the osteoclasts, through exocytosis, ClC-7 is inserted in the ruffled border of the reabsorption lacuna, a region in contact with the bone matrix and responsible for bone reabsorption (Lange et al., 2006). ClC-7 co-localizes with its accessory protein, Ostm1, to form a protein complex (Lange et al., 2006). Ostm1 shields un-glycosylated ClC-7 from degradation by lysosomal proteases (Lange et al., 2006; Schrecker et al., 2020; Zhang et al., 2020) and it is required for correct localization and function (Leisle et al., 2011). ClC-7 KO mice show severe osteopetrosis without changes in lysosomal acidification (Kornak et al., 2001; Kasper, 2005) but with a reduction in luminal [Cl−] (Weinert et al., 2010), and lysosomal storage disease, associated with retinal degeneration and severe neurodegeneration (Kornak et al., 2001; Kasper, 2005). Spontaneous Ostm1-deficient gray-lethal mice exhibit similar ClC-7 KO mice phenotype, and it mimics the severe human malignant autosomal recessive form of osteopetrosis (Chalhoub et al., 2003). Clcn7unc/unc mice, in which ClC-7 is converted into a pure chloride conductor, show a milder osteopetrosis phenotype with no changes in the fur color, but they develop lysosomal storage disease and neurodegeneration similar to Clcn7−/− mice (Weinert et al., 2010). A third mouse model, in which the transport activity of ClC-7 is abolished (Clcn7td/td), manifests severe osteopetrosis as in Clcn7−/− mice but with less severe neurodegeneration (Weinert et al., 2014). Mutations in the CLCN7 gene have been found in patients affected by autosomal recessive osteopetrosis with or without primary neurodegeneration (ARO) or by autosomal dominant osteopetrosis (ADOII) that show a quite large degree of severity, even when almost asymptomatic (Pangrazio et al., 2010; Bollerslev et al., 2013). Consistent with this heterogeneity of symptoms, electrophysiological investigations of several ClC-7 missense mutations causing diseases reveal a large variety of functional protein alterations, including impaired protein localization, complete or partial abolition of transport activity, gain of function, and accelerated kinetics of activation (Leisle et al., 2011; Nicoli et al., 2019; Di Zanni et al., 2021).
4 Conclusion
The review provides an overview of the structural similarity of vCLCs and presents recent advances in mice model investigations and functional analysis of pathogenic variants. It highlights that the variety of clinical symptoms and pathogenic functional alterations of vCLC variants represent an obstacle to identifying genotype-phenotype correlations. However, an important piece in the investigation of the entire CLC protein family is still missing: the absence of specific and powerful modulators of CLC protein activity and strategies to improve CLC protein folding, trafficking, and stability. The development of therapeutic approaches is limited by this, and therefore an effort is required to bridge the gap.
Author contributions
AP: Validation, Visualization, Writing–original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was partially funded by the European Union—NextGenerationEU (Missione 4 Componente 2, “Dalla ricerca all’impresa,” Innovation Ecosystem RAISE “Robotics and AI for Socio-economic Empowerment”, ECS00000035). In addition, this research was partially funded by the Fondazione Telethon (grant # TCP14008) granted to AP.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views and opinions expressed are those of the authors alone and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them.
References
Abraham, S. J., Cheng, R. C., Chew, T. A., Khantwal, C. M., Liu, C. W., Gong, S., et al. (2015). 13C NMR detects conformational change in the 100-kD membrane transporter ClC-ec1. J. Biomol. NMR 61 (3-4), 209–226. doi:10.1007/s10858-015-9898-7
Accardi, A. (2015). Structure and gating of CLC channels and exchangers. J. Physiol. 593 (18), 4129–4138. doi:10.1113/JP270575
Accardi, A., Lobet, S., Williams, C., Miller, C., and Dutzler, R. (2006). Synergism between halide binding and proton transport in a CLC-type exchanger. J. Mol. Biol. 362 (4), 691–699. doi:10.1016/j.jmb.2006.07.081
Basilio, D., Noack, K., Picollo, A., and Accardi, A. (2015). Conformational changes required for H(+)/Cl(-) exchange mediated by a CLC transporter. Nat. Struct. Mol. Biol. 21, 456–463. doi:10.1038/nsmb.2814
Bollerslev, J., Henriksen, K., Nielsen, M. F., Brixen, K., and Van Hul, W. (2013). Autosomal dominant osteopetrosis revisited: lessons from recent studies. Eur. J. Endocrinol. 169 (2), R39–R57. doi:10.1530/EJE-13-0136
Bose, S., He, H., and Stauber, T. (2021). Neurodegeneration upon dysfunction of endosomal/lysosomal CLC chloride transporters. Front. Cell Dev. Biol. 9, 639231. doi:10.3389/fcell.2021.639231
Brandt, S., and Jentsch, T. J. (1995). ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett. 377 (1), 15–20. doi:10.1016/0014-5793(95)01298-2
Chalhoub, N., Benachenhou, N., Rajapurohitam, V., Pata, M., Ferron, M., Frattini, A., et al. (2003). Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat. Med. 9 (4), 399–406. doi:10.1038/nm842
Devuyst, O., Christie, P. T., Courtoy, P. J., Beauwens, R., and Thakker, R. V. (1999). Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent's disease. Hum. Mol. Genet. 8 (2), 247–257. doi:10.1093/hmg/8.2.247
Di Zanni, E., Palagano, E., Lagostena, L., Strina, D., Rehman, A., Abinun, M., et al. (2021). Pathobiologic mechanisms of neurodegeneration in osteopetrosis derived from structural and functional analysis of 14 ClC-7 mutants. J. Bone Min. Res. 36 (3), 531–545. doi:10.1002/jbmr.4200
Duncan, A. R., Polovitskaya, M. M., Gaitan-Penas, H., Bertelli, S., VanNoy, G. E., Grant, P. E., et al. (2021). Unique variants in CLCN3, encoding an endosomal anion/proton exchanger, underlie a spectrum of neurodevelopmental disorders. Am. J. Hum. Genet. 108 (8), 1450–1465. doi:10.1016/j.ajhg.2021.06.003
Dutzler, R., Campbell, E. B., Cadene, M., Chait, B. T., and MacKinnon, R. (2002). X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 415 (6869), 287–294. doi:10.1038/415287a
Feng, L., Campbell, E. B., Hsiung, Y., and MacKinnon, R. (2010). Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science 330 (6004), 635–641. doi:10.1126/science.1195230
Feng, L., Campbell, E. B., and MacKinnon, R. (2012). Molecular mechanism of proton transport in CLC Cl-/H+ exchange transporters. Proc. Natl. Acad. Sci. U. S. A. 109 (29), 11699–11704. doi:10.1073/pnas.1205764109
Gianesello, L., Del Prete, D., Ceol, M., Priante, G., Calo, L. A., and Anglani, F. (2020). From protein uptake to Dent disease: an overview of the CLCN5 gene. Gene 747, 144662. doi:10.1016/j.gene.2020.144662
Graves, A. R., Curran, P. K., Smith, C. L., and Mindell, J. A. (2008). The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 453 (7196), 788–792. doi:10.1038/nature06907
Günther, W., Luchow, A., Cluzeaud, F., Vandewalle, A., and Jentsch, T. J. (1998). ClC-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. U. S. A. 95 (14), 8075–8080. doi:10.1073/pnas.95.14.8075
Günther, W., Piwon, N., and Jentsch, T. J. (2003). The ClC-5 chloride channel knock-out mouse - an animal model for Dent's disease. Pflügers Arch. 445 (4), 456–462. Epub 2002 Nov 2029. doi:10.1007/s00424-002-0950-6
Guo, Y. X., Ma, H. X., Zhang, Y. X., Chen, Z. H., and Zhai, Q. X. (2021). Whole-Exome sequencing for identifying genetic causes of intellectual developmental disorders. Int. J. Gen. Med. 14, 1275–1282. doi:10.2147/IJGM.S300775
Han, W., Cheng, R. C., Maduke, M. C., and Tajkhorshid, E. (2014). Water access points and hydration pathways in CLC H+/Cl-transporters. Proc. Natl. Acad. Sci. U. S. A. 111 (5), 1819–1824. doi:10.1073/pnas.1317890111
He, H., Cao, X., Yin, F., Wu, T., Stauber, T., and Peng, J. (2021). West syndrome caused by a chloride/proton exchange-uncoupling CLCN6 mutation related to autophagic-lysosomal dysfunction. Mol. Neurobiol. 58 (6), 2990–2999. doi:10.1007/s12035-021-02291-3
Hu, H., Haas, S. A., Chelly, J., Van Esch, H., Raynaud, M., de Brouwer, A. P., et al. (2016). X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 21 (1), 133–148. doi:10.1038/mp.2014.193
Jentsch, T. J., and Pusch, M. (2018). CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol. Rev. 98 (3), 1493–1590. doi:10.1152/physrev.00047.2017
Kasper, e.a., Planells-Cases, R., Fuhrmann, J. C., Scheel, O., Zeitz, O., Ruether, K., et al. (2005). Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. Embo J. 24, 1079–1091. doi:10.1038/sj.emboj.7600576
Kawasaki, M., Suzuki, M., Uchida, S., Sasaki, S., and Marumo, F. (1995). Stable and functional expression of the CIC-3 chloride channel in somatic cell lines. Neuron 14 (6), 1285–1291. doi:10.1016/0896-6273(95)90275-9
Kornak, U., Kasper, D., Bösl, M. R., Kaiser, E., Schweizer, M., Schulz, A., et al. (2001). Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 104 (2), 205–215. doi:10.1016/s0092-8674(01)00206-9
Lange, P. F., Wartosch, L., Jentsch, T. J., and Fuhrmann, J. C. (2006). ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature 440 (7081), 220–223. doi:10.1038/nature04535
Lee, S., Mayes, H. B., Swanson, J. M., and Voth, G. A. (2016). The origin of coupled chloride and proton transport in a Cl(-)/H(+) antiporter. J. Am. Chem. Soc. 138 (45), 14923–14930. doi:10.1021/jacs.6b06683
Leisle, L., Ludwig, C. F., Wagner, F. A., Jentsch, T. J., and Stauber, T. (2011). ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity. Embo J. 30 (11), 2140–2152. doi:10.1038/emboj.2011.137
Leray, X., Hilton, J. K., Nwangwu, K., Becerril, A., Mikusevic, V., Fitzgerald, G., et al. (2022). Tonic inhibition of the chloride/proton antiporter ClC-7 by PI(3,5)P2 is crucial for lysosomal pH maintenance. Elife 11, e74136. doi:10.7554/eLife.74136
Li, D. Q., Jing, X., Salehi, A., Collins, S. C., Hoppa, M. B., Rosengren, A. H., et al. (2009). Suppression of sulfonylurea- and glucose-induced insulin secretion in vitro and in vivo in mice lacking the chloride transport protein ClC-3. Cell Metab. 10 (4), 309–315. doi:10.1016/j.cmet.2009.08.011
Lloyd, S. E., Pearce, S. H., Fisher, S. E., Steinmeyer, K., Schwappach, B., Scheinman, S. J., et al. (1996). A common molecular basis for three inherited kidney stone diseases. Nature 379 (6564), 445–449. doi:10.1038/379445a0
Ma, T., Wang, L., Chai, A., Liu, C., Cui, W., Yuan, S., et al. (2023). Cryo-EM structures of ClC-2 chloride channel reveal the blocking mechanism of its specific inhibitor AK-42. Nat. Commun. 14 (1), 3424. doi:10.1038/s41467-023-39218-6
Marcoline, F. V., Ishida, Y., Mindell, J. A., Nayak, S., and Grabe, M. (2016). A mathematical model of osteoclast acidification during bone resorption. Bone 93, 167–180. doi:10.1016/j.bone.2016.09.007
Maritzen, T., Keating, D. J., Neagoe, I., Zdebik, A. A., and Jentsch, T. J. (2008). Role of the vesicular chloride transporter ClC-3 in neuroendocrine tissue. J. Neurosci. 28 (42), 10587–10598. doi:10.1523/JNEUROSCI.3750-08.2008
Neagoe, I., Stauber, T., Fidzinski, P., Bergsdorf, E. Y., and Jentsch, T. J. (2010). The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J. Biol. Chem. 285 (28), 21689–21697. doi:10.1074/jbc.M110.125971
Nicoli, E. R., Weston, M. R., Hackbarth, M., Becerril, A., Larson, A., Zein, W. M., et al. (2019). Lysosomal storage and albinism due to effects of a de novo CLCN7 variant on lysosomal acidification. Am. J. Hum. Genet. 104 (6), 1127–1138. doi:10.1016/j.ajhg.2019.04.008
Okamoto, F., Kajiya, H., Toh, K., Uchida, S., Yoshikawa, M., Sasaki, S., et al. (2008). Intracellular ClC-3 chloride channels promote bone resorption in vitro through organelle acidification in mouse osteoclasts. Am. J. Physiol. Cell Physiol. 294 (3), C693–C701. doi:10.1152/ajpcell.00251.2007
Palmer, E. E., Pusch, M., Picollo, A., Forwood, C., Nguyen, M. H., Suckow, V., et al. (2022). Functional and clinical studies reveal pathophysiological complexity of CLCN4-related neurodevelopmental condition. Mol. Psychiatry 28, 668–697. doi:10.1038/s41380-022-01852-9
Palmer, E. E., Stuhlmann, T., Weinert, S., Haan, E., Van Esch, H., Holvoet, M., et al. (2018). De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol. Psychiatry 23 (2), 222–230. doi:10.1038/mp.2016.135
Pangrazio, A., Pusch, M., Caldana, E., Frattini, A., Lanino, E., Tamhankar, P. M., et al. (2010). Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: report of 20 novel mutations. Hum. Mutat. 31 (1), E1071–E1080. doi:10.1002/humu.21167
Park, E., and MacKinnon, R. (2018). Structure of the CLC-1 chloride channel from Homo sapiens. Elife 7, e36629. doi:10.7554/eLife.36629
Picollo, A., and Pusch, M. (2005). Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 21 (436), 420–423. doi:10.1038/nature03720
Piwon, N., Günther, W., Schwake, M., Bösl, M. R., and Jentsch, T. J. (2000). ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature 408 (6810), 369–373. doi:10.1038/35042597
Poet, M., Kornak, U., Schweizer, M., Zdebik, A. A., Scheel, O., Hoelter, S., et al. (2006). Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. U. S. A. 103 (37), 13854–13859. doi:10.1073/pnas.0606137103
Polovitskaya, M. M., Barbini, C., Martinelli, D., Harms, F. L., Cole, F. S., Calligari, P., et al. (2020). A recurrent gain-of-function mutation in CLCN6, encoding the ClC-6 Cl(-)/H(+)-Exchanger, causes early-onset neurodegeneration. Am. J. Hum. Genet. 107 (6), 1062–1077. doi:10.1016/j.ajhg.2020.11.004
Pressey, S. N., O'Donnell, K. J., Stauber, T., Fuhrmann, J. C., Tyynela, J., Jentsch, T. J., et al. (2010). Distinct neuropathologic phenotypes after disrupting the chloride transport proteins ClC-6 or ClC-7/Ostm1. J. Neuropathol. Exp. Neurol. 69 (12), 1228–1246. doi:10.1097/NEN.0b013e3181ffe742
Rohrbough, J., Nguyen, H. N., and Lamb, F. S. (2018). Modulation of ClC-3 gating and proton/anion exchange by internal and external protons and the anion selectivity filter. J. Physiol. 596 (17), 4091–4119. doi:10.1113/JP276332
Sakamoto, H., Sado, Y., Naito, I., Kwon, T. H., Inoue, S., Endo, K., et al. (1999). Cellular and subcellular immunolocalization of ClC-5 channel in mouse kidney: colocalization with H+-ATPase. Am. J. Physiol. 277 (6), F957–F965. doi:10.1152/ajprenal.1999.277.6.F957
Scheel, O., Zdebik, A. A., Lourdel, S., and Jentsch, T. J. (2005). Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436 (7049), 424–427. doi:10.1038/nature03860
Scheinman, S. J. (1998). X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney. Int. 53 (1), 3–17. doi:10.1046/j.1523-1755.1998.00718.x
Schrecker, M., Korobenko, J., and Hite, R. K. (2020). Cryo-EM structure of the lysosomal chloride-proton exchanger CLC-7 in complex with OSTM1. Elife 9, e59555. doi:10.7554/eLife.59555
Silva, I. V., Cebotaru, V., Wang, H., Wang, X. T., Wang, S. S., Guo, G., et al. (2003). The ClC-5 knockout mouse model of Dent's disease has renal hypercalciuria and increased bone turnover. J. Bone Min. Res. 18 (4), 615–623. doi:10.1359/jbmr.2003.18.4.615
Stauber, T., and Jentsch, T. J. (2010). Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J. Biol. Chem. 285 (45), 34537–34548. doi:10.1074/jbc.M110.162545
Stobrawa, S. M., Breiderhoff, T., Takamori, S., Engel, D., Schweizer, M., Zdebik, A. A., et al. (2001). Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29 (1), 185–196. doi:10.1016/s0896-6273(01)00189-1
Tang, X., Brown, M. R., Cogal, A. G., Gauvin, D., Harris, P. C., Lieske, J. C., et al. (2016). Functional and transport analyses of CLCN5 genetic changes identified in Dent disease patients. Physiol. Rep. 4 (8), e12776. doi:10.14814/phy2.12776
Thakker, R. V. (1997). Chloride channels cough up. Nat. Genet. 17 (2), 125–127. doi:10.1038/ng1097-125
Traverso, S., Elia, L., and Pusch, M. (2003). Gating competence of constitutively open CLC-0 mutants revealed by the interaction with a small organic Inhibitor. J. Gen. Physiol. 122 (3), 295–306. doi:10.1085/jgp.200308784
Veeramah, K. R., Johnstone, L., Karafet, T. M., Wolf, D., Sprissler, R., Salogiannis, J., et al. (2013). Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 54 (7), 1270–1281. doi:10.1111/epi.12201
Wang, S. S., Devuyst, O., Courtoy, P. J., Wang, X. T., Wang, H., Wang, Y., et al. (2000). Mice lacking renal chloride channel, CLC-5, are a model for Dent's disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum. Mol. Genet. 9 (20), 2937–2945. doi:10.1093/hmg/9.20.2937
Weinert, S., Gimber, N., Deuschel, D., Stuhlmann, T., Puchkov, D., Farsi, Z., et al. (2020). Uncoupling endosomal CLC chloride/proton exchange causes severe neurodegeneration. EMBO J. 39 (9), e103358. doi:10.15252/embj.2019103358
Weinert, S., Jabs, S., Hohensee, S., Chan, W. L., Kornak, U., and Jentsch, T. J. (2014). Transport activity and presence of ClC-7/Ostm1 complex account for different cellular functions. EMBO Rep. 15 (7), 784–791. doi:10.15252/embr.201438553
Weinert, S., Jabs, S., Supanchart, C., Schweizer, M., Gimber, N., Richter, M., et al. (2010). Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl-accumulation. Science 328 (5984), 1401–1403. doi:10.1126/science.1188072
Wu, J. Z., Zeziulia, M., Kwon, W., Jentsch, T. J., Grinstein, S., and Freeman, S. A. (2023). ClC-7 drives intraphagosomal chloride accumulation to support hydrolase activity and phagosome resolution. J. Cell Biol. 222 (6), e202208155. doi:10.1083/jcb.202208155
Xu, X., Lu, F., Zhang, L., Li, H., Du, S., and Tang, J. (2021). Novel CLCN4 variant associated with syndromic X-linked intellectual disability in a Chinese girl: a case report. BMC Pediatr. 21 (1), 384. doi:10.1186/s12887-021-02860-4
Yoshikawa, M., Uchida, S., Ezaki, J., Rai, T., Hayama, A., Kobayashi, K., et al. (2002). CLC-3 deficiency leads to phenotypes similar to human neuronal ceroid lipofuscinosis. Genes cells. 7 (6), 597–605. doi:10.1046/j.1365-2443.2002.00539.x
Zhang, S., Liu, Y., Zhang, B., Zhou, J., Li, T., Liu, Z., et al. (2020). Molecular insights into the human CLC-7/Ostm1 transporter. Sci. Adv. 6 (33), eabb4747. doi:10.1126/sciadv.abb4747
Zifarelli, G., Pusch, M., and Fong, P. (2022). Altered voltage-dependence of slowly activating chloride-proton antiport by late endosomal ClC-6 explains distinct neurological disorders. J. Physiol. 600 (9), 2147–2164. doi:10.1113/JP282737
Keywords: vCLC, genetic diseases, exchangers, structures, function
Citation: Picollo A (2023) Vesicular CLC chloride/proton exchangers in health and diseases. Front. Pharmacol. 14:1295068. doi: 10.3389/fphar.2023.1295068
Received: 15 September 2023; Accepted: 16 October 2023;
Published: 07 November 2023.
Edited by:
Giovanni Zifarelli, National Research Council (CNR), ItalyReviewed by:
Anselm Zdebik, University College London, United KingdomCopyright © 2023 Picollo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Picollo, YWxlc3NhbmRyYS5waWNvbGxvQGliZi5jbnIuaXQ=