 Ni Zhou1,2,3
Ni Zhou1,2,3 Wenqun Li
Wenqun Li Bikui Zhang
Bikui Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 08 December 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1292088
This article is part of the Research Topic Metallodrugs in Cancer Therapy: Past, Present and New Strategies View all 5 articles
Doxorubicin is a widely used anticancer drug in clinical practice for the treatment of various human tumors. However, its administration is associated with cardiotoxicity. Administration of doxorubicin with low side effects for cancer treatment and prevention are, accordingly, urgently required. The human body harbors various endogenous metal ions that exert substantial influences. Consequently, extensive research has been conducted over several decades to investigate the potential of targeting endogenous metal ions to mitigate doxorubicin’s side effects and impede tumor progression. In recent years, there has been a growing body of research indicating the potential efficacy of metal ion-associated therapeutic strategies in inhibiting doxorubicin-induced cardiotoxicity (DIC). These strategies offer a combination of favorable safety profiles and potential clinical utility. Alterations in intracellular levels of metal ions have been found to either facilitate or mitigate the development of DIC. For instance, ferroptosis, a cellular death mechanism, and metal ions such as copper, zinc, and calcium have been identified as significant contributors to DIC. This understanding can contribute to advancements in cancer treatment and provide valuable insights for mitigating the cardiotoxic effects of other therapeutic drugs. Furthermore, potential therapeutic strategies have been investigated to alleviate DIC in clinical settings. The ultimate goal is to improve the efficacy and safety of Dox and offer valuable insights for future research in this field.
Doxorubicin (Dox), the first anthracycline used in clinical therapy, is a widely used class of antitumor antibiotics, commonly employed in the treatment of childhood leukemia, breast cancer, lymphoma, and sarcoma, among other conditions. Unfortunately, the clinical application of Dox in anticancer therapy is greatly limited by various systemic adverse reactions, which can lead to acute and chronic cardiomyopathy and, in severe cases, congestive heart failure, and the cardiotoxicity is considered irreversible (Ewer and Lippman, 2005). DIC is classified as acute and chronic, the clinical dose of Dox is 50–60 mg once every 3–4 weeks or 20–30 mg weekly for 3 weeks, repeated after 2–3 weeks of withdrawal, with a maximum cumulative dose of 550 mg/m2 (Li and Hill, 2014). Children’s dosage is about half that of an adult. Up to 25% of patients using Dox may experience DIC (Swain et al., 2003).
Dox inhibits the growth and proliferation of tumor cells by inhibiting topoisomerase II (Top2β), inserting into the double helix structure of DNA to unravel the double strand and inhibiting DNA and RNA synthesis (Kitakata et al., 2022). However, the exact mechanism of DIC is not fully deciphered, and it is only certain that oxidative stress and apoptosis play a key role in DIC. Numerous studies have identified various potential pathogenic mechanisms of DIC, including transcriptional dysregulation, calcium dysregulation, oxidative stress, DNA damage, nitric oxide release, inflammatory mediates, mitochondrial dysfunction, accumulation of iron in mitochondria, and dysregulation of autophagy due to the inhibition of Top2β (Renu et al., 2018). Therefore, it is important and advantageous to gain an understanding of the pathological factors that contribute to the onset or progression of DIC.
The human body needs trace elements such as iron, copper, zinc, calcium, manganese, etc., to maintain health and normal function of the system. These essential metal elements play a crucial role within cells, as many catalytic enzymes and proteins involved in biochemical processes heavily depend on them (Zoroddu et al., 2019). Disorders in iron metabolism can result in significant tissue degeneration, impairment of organ function, and potential development of cancer, ferroptosis activation may be a potential strategy to overcome mechanisms of resistance to conventional cancer therapy (Zhao et al., 2022). In mice mesothelioma models, reducing bioavailable copper through the use of D-pen, tetrathiomolybdate (TM), or trientine also reduced tumor growth and hindered tumor blood vessel formation (Ishida et al., 2013). Zinc can bind to Tau-specific amino acids to accelerate deposition, induce Tau abnormal phosphorylation, amplify its cytotoxicity, and aggravate cognitive impairment in Alzheimer’s patients (Hu et al., 2017). Calcium is chelated through the sarcoplasmic/endoplasmic Ca2+-ATPase (SERCa2a) after its release from the Ryanodine receptors (RyR2), and changes in these proteins in the circulation occur as the heart ages, leading to arrhythmias (Hamilton and Terentyev, 2019).
The disturbance of metal ion balance in cardiomyocyte of DIC patients is associated with oxidative stress and mitochondrial dysfunction, indicating the significant role of metal ions in the pathogenesis of DIC (Sheibani et al., 2022). In heart tissues affected by DIC, it is possible to observe alterations in the regulation of metal ions, including upregulation, downregulation, or dysregulation. In 2022, Abe et al. found that Dox accumulated in mitochondria by intercalating into mitochondrial DNA and decreased the abundance of 5′-aminolevulinate synthase 1, the rate-limiting enzyme in this process, thereby resulting in iron overload and ferroptosis in mitochondria in cultured cardiomyocytes (Abe et al., 2022). Clinical trials have been conducted in experimental animal models of DIC, using supplementation, chelation, or modulation of metal ions for treatment.
In the following section, we have reviewed the various roles of metal ions in the pathological mechanism of DIC and have discussed the roles of iron, copper, zinc, and calcium in triggering oxidative stress, mitochondrial dysfunction, DNA damage, among other effects.
Ferroptosis is a newly discovered mode of regulatory cell death (RCD) by Stockwell et al., in 2002. Unlike other RCD modes. Ferroptosis does not show apoptotic features such as nuclear rupture, DNA ladder, and caspase-3 activation (Dolma et al., 2003), instead, it relies on Fe2+ and reactive oxygen species (ROS), along with mitochondrial dysfunction. This dysfunction is observed as mitochondrial outer membrane rupture, reduction or disappearance of mitochondrial ridges and shrinkage of the mitochondrial membrane (Friedmann Angeli et al., 2014). By focusing on the three primary features of ferroptosis, namely, dependence on iron, disrupted lipid metabolism, and excessive production of ROS (Jiang et al., 2021), new therapeutic targets can be pinpointed and more precise drugs can be developed. These are illustrated in Figure 1.
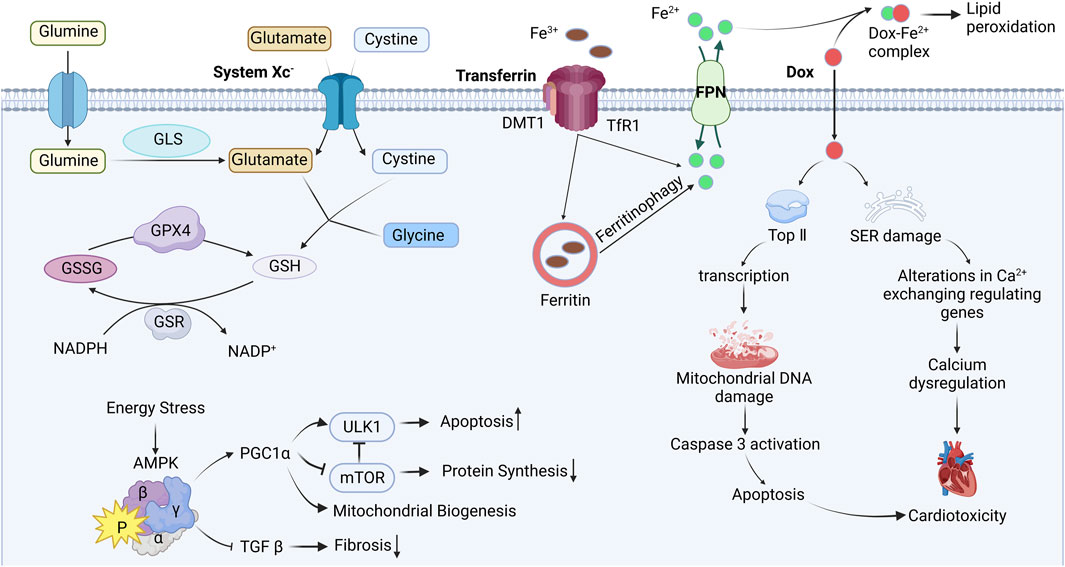
FIGURE 1. Mechanism of ferroptosis induced by Doxorubicin. Interference with iron metabolism after Dox administration, leads to DNA damage and endoplasmic reticulum stress, impairing mitochondrial function, ultimately resulting in cardiac toxicity. Blunt-ended lines indicate inhibition while arrows indicate promotion; DOX Doxorubicin; Top Ⅱ topoisomerase Ⅱ; SER sarcoplasmic (endoplasmic) reticulum; FPN ferroportin; DMT1 divalent metal-ion transporter 1; TfR1 transferrin receptor 1; GLS glutaminase; GSH glutathione; GSSG glutathione disulfide; GPX4 glutathione peroxidase 4; GSR glutathione reductase; NADPH nicotinamide adenine dinucleotide phosphate; AMPK AMP-activated protein kinase; PGC1α peroxisome proliferator–activated receptor-gamma coactivator 1α; TGF β transforming growth factor β; ULK1 Unc-51 like autophagy activating kinase; mTOR mammalian target of rapamycin. Image from biorender.
There are two main sources of dietary iron absorption: inorganic iron and heme iron (Uzel and Conrad, 1998). In a study conducted by Panjrath, G. S. et al., two groups of animals were fed either iron-rich food or ordinary food, and then treated with Dox and normal saline, respectively. The results showed that animals fed iron-rich food exhibited more severe DIC compared to those fed ordinary food. Additionally, feeding iron-rich foods alone did not cause cardiac damage, suggesting that iron accumulation and its bioavailability in the body may serve as crucial independent predictors of DIC sensitivity (Panjrath et al., 2007). The deposition of iron in cardiomyocyte can result in oxidative stress, mitochondrial damage and dysfunction, which further worsens DIC.
Disruption of the redox equilibrium is a core pathological mechanism in many heart diseases. The conversion of Fe3+ to Fe2+ is a reversible redox process, but electron transfer may result in the production of excessive ROS when iron is overloaded in cardiomyocyte. Normally, there is not enough free iron in the body to bind with Dox, the problem is that Dox carries a positive charge which enhances its affinity for iron and promotes the formation of the Dox-Fe complex. This complex alters the distribution and metabolism of iron and further combines with free oxygen in the body, resulting in cardiotoxicity (Gutteridge, 1984). Furthermore, iron and copper ions promote the binding of Dox to DNA, leading to increased cytotoxicity. A study demonstrated that 5-fluorouracil can upregulate iron levels in animals and cause iron accumulation in cell experiments, resulting in fluorescence quenching and increasing lipid peroxide levels (Li et al., 2022a). In addition, a study by He et al. revealed that pretreatment with Epigallocatechin-3-gallate can reduce iron deposition, inhibit oxidative stress by activating AMPKα2 (He et al., 2021). Recent studies have also shown that cardiac dysfunction caused by ferroptosis can be inhibited by iron complexing agents and antioxidants such as Ferrostatin-1 (Baba et al., 2018), Dexrazoxane (Weiss et al., 1999), Deferoxamine (Menon and Kim, 2022), Deferiprone (El-Ammar el et al., 2011), and Vitamin E (Berthiaume et al., 2005). These studies demonstrate that iron overload exacerbates Dox-induced oxidative stress and cardiomyocyte death. Interestingly, the use of antioxidants alone is insufficient to protect against DIC (Mukhopadhyay et al., 2009).
Cardiomyocyte contain a large number of mitochondria (Sheibani et al., 2022). However, mitochondria are also prone to iron overload, leading to lipid peroxidation on their membranes. Li et al. discovered that injection of endotoxin into mice increases the expression of nuclear receptor coactivator 4, facilitating the transfer of Fe3+ to mitochondria, leading to mitochondrial iron overload, triggering oxidative stress (Li et al., 2020b). On the other hand, Chang H.C. et al. suggested that overexpression of the mitochondrial iron efflux pump or the application of specifically targeted iron chelating agents within mitochondria could decrease mitochondrial iron levels (Chang et al., 2016). For instance, MitoTEMPO, a mitochondria-targeting antioxidant, can be easily integrated into the mitochondria (Dikalova et al., 2010). Experimental results have demonstrated that mitochondrial oxidative damage is the primary mechanism of heart injury caused by ferroptosis. Interestingly, the non-targeted antioxidant TEMPO does not exhibit any cardiac protective effects (Fang et al., 2019). Similarly, Ichikawa et al. confirmed that overexpression of the mitochondrial transporter ABCB8 promotes iron efflux, and prevents DIC in vivo and in vitro (Ichikawa et al., 2014). This suggests that reducing the iron levels in cardiomyocyte mitochondria can reverse the cardiotoxicity caused by Dox in cancer therapy.
The enzyme selenate Gpx4 plays a crucial role in regulating ferroptosis by minimizing lipid peroxidation. It accomplishes this by converting reduced glutathione (GSH) into glutathione disulfide (GSSG) and utilizing hydrogen ions to effectively diminish the presence of hydrogen peroxide and lipid peroxidation, subsequently reducing the accumulation of ROS (Forcina and Dixon, 2019). Several studies have reported that Dox can attenuate the levels of Gpx4. Protein arginine methyltransferase 4 can inhibit the Nrf2/Gpx4 to enhance ferroptosis, then mitigate DIC (Wang et al., 2022c). Moreover, the overexpression of Gpx4 or the introduction of ferrous ion chelators specifically targeting mitochondria have demonstrated the ability to mitigate DIC (Tadokoro et al., 2020).
GSH, a tripeptide comprising glutamine, cysteine and glycine, exists in cells in either its oxidized form, GSSG, or reduced form, GSH. GSH/GSSG is influenced by redox reaction, and maintaining an optimal ratio is vital for combating oxidative stress (Anderson, 1998). GSH catalytic detoxification of various electrophilic compounds and peroxides, facilitating the elimination of ROS through the, a process mediated by glutathione S-transferase and Gpx enzymes (Townsend et al., 2003). In a study conducted by Sun et al. observed that the protein expression of Gpx4 and SLC7A11, as well as the GSH/GSSG, were diminished upon Herceptin treatment (Sun et al., 2022). Furthermore, the intervention of Herceptin resulted in an increase in cellular and mitochondrial iron levels (Sun et al., 2022).
System Xc-is a heterodimer reverse transport system for input cysteine and output glutamate, consisting of two subunits SLC3A2 and SLC7A11. Among them, SLC7A11 can promote cystine uptake and glutathione biosynthesis, maintain redox balance in vivo, and inhibit ferroptosis (Kim et al., 2020). In addition, the expression level of SLC7A11 is usually positively correlated with the activity of the reverse transporter (Lewerenz et al., 2013). Activation of transcription factor 3 (ATF3), a common stress receptor, inhibits SLC7A11 expression, thereby exacerbating elastin-induced lipid peroxidation and ferroptosis (Wang et al., 2020a). Repression of the SLC7A11/GSH/GPX4 axis triggers ferroptosis of vascular smooth muscle cells to promote vascular calcification under chronic kidney disease conditions (Ye et al., 2022).
Nuclear factor erythroid 2-related factor 2 (Nrf2) possesses redox activity, when activated, iit enhances the overall detoxification and clearance of harmful substances in cells. Keap1 serves as an adapter protein for the CUL3-based E3 ubiquitin ligase and, under normal circumstances, binds to cytoplasmic Nrf2, promoting its ubiquitination and degradation. Hence, Keap1 functions as a negative inhibitor of Nrf2, a proposition that has also been experimentally supported through gene-deficient mice. Tertbutylhydroquinone (tBHQ) is an Nrf2 activator (Dinkova-Kostova and Talalay, 2008), the research demonstrates that tBHQ can induce Nrf2 overexpression, resulting in the upregulation of SLC7A11 expression in Hela or human corneal endothelial cells (Guha and Roy, 2021). In transgenic mice models, it has been proven that sequestration of Keap1 has been found to protect the heart from doxorubicin-induced ferroptosis (Hou et al., 2021). The ubiquitin E3 ligase TRIM21 interacts with p62 and ubiquitinates it, negatively regulating the p62-KEAP1-Nrf2 antioxidant pathway (Refaie et al., 2021).
Adenosine monophosphate-activated protein kinase (AMPK) is a macromolecular protein complex composed of three subunits: one catalytic site α and two regulatory sites β and γ. The AMPK pathway is closely associated with key mechanisms of DIC, including oxidative stress, mitochondrial damage, dysregulation of autophagy, increased apoptosis, and fibrosis. Dox inhibits the activation of other signals such as Akt and mitogen activated protein kinase (MAPK) through the AMPK pathway, leading to DNA damage. Activation of AMPK has several effects: firstly, it inhibits the autophagy activating protein ULK1, promoting autophagy (Kim et al., 2011); secondly, it inhibits the TGFβ pathway, which decreases myocardial fibrosis (Lin et al., 2015); thirdly, it inhibits the mTOR signal, reducing apoptosis (Ren et al., 2016). Various drugs can activate the AMPK signal, including metformin, statins, resveratrol, thiazolidinedione, AICAR (Acadesine), and specific AMPK agonists. These drugs are expected to have clinical applications in resisting DIC.
Copper, an essential trace element in basic physiological activities of the human body, is involved in numerous key biological processes, including free radical scavenging, iron metabolism, connective tissue synthesis, immunity and cell signal transduction (Tainer et al., 1983; Banci and Bertini, 2013; Apresova et al., 2014; Bhuvanasundar et al., 2014). Additionally, several important “cuproproteins” in the human body, such as cytochrome C oxidase (COX), NADH dehydrogenase-2, Cu/Zn-superoxide dismutase (SOD1) and tyrosinase, rely on copper as a catalytic cofactor to fulfill their roles (Denoyer et al., 2015). Furthermore, the high redox activity of copper allows it to easily transition between the two valence states, and as a result, the generated electrons can promote the formation of ROS (Purchase, 2013).
In patients with osteosarcoma and leukemia, endogenous copper levels are higher compared to normal cells and tissues, and patients with more aggressive typically exhibit elevated serum copper levels (Fisher et al., 1976). Copper has been shown to promote angiogenesis, tumor growth, and metastasis (Gupte and Mumper, 2009). The body regulates copper concentrations through processes of absorption, excretion and bioavailability. Copper obtained from food sources (organic copper) undergoes processing by the liver, which controls its distribution in serum and tissues, excretes excess copper into bile (Angelova et al., 2011); on the other hand, copper (inorganic copper) present in drinking water or from copper supplements is absorbed by the intestinal mucosa and directly enters the free copper pool in the blood, bypassing the liver. This route likely allows copper to cross the blood-brain barrier (BBB) (Brewer, 2009). Based on Figure 2, three main approaches can be identified for addressing copper overload.
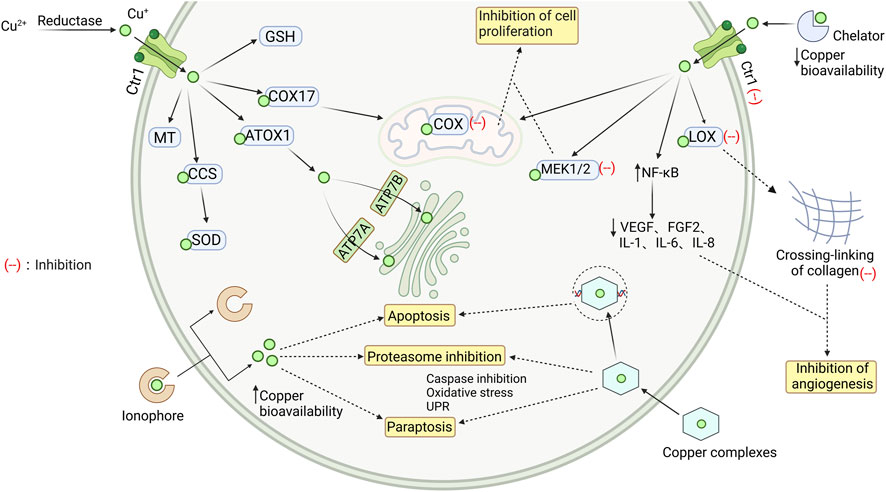
FIGURE 2. Intracellular transport of copper and the mechanism of chelating agents, ionophores, and complexes. The Cu2+ is reduced to Cu+, which then enters myocardial cells and binds to different proteins to maintain a steady state of copper ions in the cytoplasm. When there is an overload of copper ions, they can be removed by copper chelators, copper ionophores, and copper coordination compounds, which have a protective effect by clearing free copper ions. GSH glutathione; COX17 cytochrome c oxidase copper chaperone; ATOX1 antioxidant protein; CCS copper chaperone for SOD1; SOD Cu/Zn-superoxide dismutase; MT metallothionein; Ctr1 copper transporter 1; LOX recombinant lysyl oxidase; COX cytochrome c oxidase; MEK1/2 mitogen-activated protein kinase kinase; NF-κB nuclear factor kappa-B; VEGF vascular endothelial growth factor; FGF2 human basic fibroblast growth factor 2; ATP7A/B copper transporting ATPase A/B. Image from biorender.
Copper deficiency may not necessarily worsen the cardiac toxicity induced by Dox (Fischer et al., 1993). It was found that copper-deficient rats may compensate for Dox-induced oxidative stress in the heart by improving the activity of Cu, Zn-superoxide dismutase and glutathione S-transfers (Fischer et al., 1992). However, Mizutani, H. et al. measured the levels of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG), a marker of DNA damage, and found that Dox can cause DNA damage in the presence of Cu2+ or cytochrome P450 reductase. Curiously, the extent of Cu2+-mediated DNA damage, including the formation of 8-oxodG, is far greater than the extent of DNA damage mediated by cytochrome P450 reductase (Mizutani et al., 2003). Therefore, people are striving to maintain stable levels of copper within cells when using Dox. This is currently being done through three approaches: chelation, ion carriers, and coordination compounds.
The use of copper chelators to reduce the concentration and bioavailability of copper in vivo is an effective method. By forming complexes with copper, chelators help remove it from the body and inhibit the formation of cancer blood vessels, thereby preventing further progression of the disease.
Several common copper chelators, such as D-pen, trientine, ALXN 1840, or TM, have been shown to delay the spread of cancer by inhibiting angiogenesis in various animal models. This effect has been demonstrated in animal models of rat gliosarcoma (Brem et al., 1990), mice hepatocellular carcinoma (Yoshii et al., 2001), head and neck squamous cell carcinoma (Cox et al., 2001) and mesothelioma (Crowe et al., 2013). D-PEN, for example, can inhibit the activity of lysine oxidase and disrupt collagen crosslinking. Consequently, it affects the expression of endothelial growth factor, leading to the delayed progression of glioblastoma multiforme in vivo (Mammoto et al., 2013). Animal experiments have shown that treatment with trientine at doses ranging from approximately 34–68 mg/kg/day for 6–8 weeks resulted in increased cardiac output, increased left ventricular pressure, as well as improvements in left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF). TM-induced copper deficiency inhibits angiogenesis by activating the transcription factor NF-κB, thereby reducing the secretion of angiogenic factors (VEGF, FGF2) and interleukins (IL-1a, IL-6, IL-8) (Pan et al., 2003). ALXN1840 acts by forming a tripartite complex with copper and albumin, which reduces the levels of non-ceruloplasmin-bound copper in the bloodstream. It specifically targets intracellular copper in the liver and increases the copper excretion through bile (Komatsu et al., 2000).
Copper ionophores are a class of fat-soluble molecules that reversibly bind copper ions and can induce apoptosis in copper-containing cells. Unlike copper chelators, which remove copper from the body, copper ionophores redistribute the distribution of copper ions within cells. By doing so, they exploit the cytotoxicity of copper to specifically target and eliminate cancer cells.
Three different compounds—Cu2+(gtsm) (a bis(thiosemicarbazone) analog), clioquinol (a hydroxyquinoline analog) and disulfiram (a dithiocarbamate analog)—have been studied for their ability to inhibit proteasomal chymotrypsin-like activity, thereby preventing the release of copper (Chen et al., 2007). These compounds have demonstrated anticancer effects both in vitro and in rodent models (Cater et al., 2013). Cu2+(gtsm), when used in combination with its ligands, exhibited enhanced anti-proliferative activity against tumor cells compared to the ligands alone (Anjum et al., 2019). It is worth noting that the growth of tumors in an anoxic microenvironment diminishes the anti-proliferative efficacy of most bis(thiosemicarbazone) compounds and their copper complexes, compared to an oxygen-rich environment (Anjum et al., 2019). Clioquinol and disulfiram have undergone extensive clinical trials, the results have shown that these compounds can indeed transport copper into human cells and exert selective cytotoxicity in tumor cells. However, the underlying reasons for this selectivity have not been identified (Johansson, 1992; Schimmer et al., 2012). Studies have demonstrated that the anticancer activity of clioquinol is intensified with higher extracellular copper levels, which can be counteracted by the formation of copper complexes with a chelating agent called TM (Cater and Haupt, 2011). Li et al.'s study showed that the disulfiram-copper complex exerts anti-tumor activity in nasopharyngeal carcinoma cells through the ROS/MAPK pathway and the p53-mediated ferroptosis pathway. Additionally, this complex can inhibit α-SMA expression, inactivate cancer-related fibroblasts (Li et al., 2020c).
The interest in finding alternative metal complexes for supplementary treatment has been growing, considering the widespread clinical use of platinum-based drugs such as cisplatin, carboplatin, and oxaliplatin. This is driven by the aim of reducing the side effects associated with platinum-containing chemotherapy (Marzano et al., 2009). Copper has been shown to have potential to enhance efficacy, reduce side effects, and bypass drug resistance (Marzano et al., 2009). A study conducted by Pivetta evaluated the combined effects of three copper coordination compounds containing one or two 1,10-phenanthroline molecules in combination with cisplatin. The results indicated a significant synergistic anti-tumor effect on cell proliferation. This combination therapy holds promise for enhancing the effectiveness of existing treatments (Pivetta et al., 2015). Additionally, the combination of copper and tin binary complexes has been investigated, demonstrating superior anticancer activity compared to monometallic complexes (Chauhan et al., 2007). CuSn2 has been particularly effective in inducing apoptotic cell death in various cancer cell lines in vitro. Notably, CuSn2 exhibits a significantly higher maximum tolerated dose than cisplatin while causing fewer toxic side effects. Interestingly, there is no evidence of liver, kidney, or brain toxicity associated with CuSn2 at equivalent doses (Zaidi et al., 2014).
In conclusion, the increasing research on copper complexes as complementary chemotherapy agents presents an encouraging avenue for cancer treatment. Novel copper complexes, such as the binuclear Cu2+ complex targeting DNA, combinations with existing drugs like cisplatin, and heterologous copper-tin complexes, hold promise for future cancer therapies. Further studies are warranted to fully explore the efficacy and safety profiles of these complexes and their potential for clinical translation.
Zinc is the second most abundant metal element in the human body after iron. The majority of enzymatic reactions in the human body rely on zinc, making it indispensable for normal physiological functions and overall health. Zinc is found in various tissues and organs, but its content in the heart is minimal, accounting for 0.4% of the total zinc content (King et al., 2000). With age and the presence of certain pathological conditions, the level of zinc in the body gradually decreases, leading to an increased risk of complications (Bayır et al., 2013). Decreased zinc ion concentration in peripheral blood has been suggested as an independent risk factor for predicting coronary heart disease, especially in elderly patients, non-smokers, and postmenopausal women (Meng et al., 2021).
The [Zn2+]i in Dox-treated H9c2 cells is significantly higher compared to the control group, indicating the presence of endoplasmic reticulum (ER) stress as evidenced by elevated levels of ER stress markers, such as GRP78 and CHOP/Gadd15 (Olgar et al., 2018). Interestingly, directly increasing [Zn2+]i through a zinc ion carrier induced a significant increase in these markers, while directly inducing ER stress did not alter the levels of zinc ion transport proteins (Olgar et al., 2018). Disruption of Zn2+ homeostasis in cells can lead to mitochondrial and endoplasmic reticulum stress, causing disruption of normal ER/mitochondrial crosstalk and mitochondrial autophagy, resulting in metabolic dysfunction (Dabravolski et al., 2022). Therefore, controlling intracellular zinc levels through zinc ion transporters may be an important approach in the treatment of DIC.
Zinc homeostasis is regulated by multiple proteins at various levels. Zinc ion transporters are the most significant among these regulators, which can be classified into two families: ZnTs (SLC30) and ZIPs (SLC39) (Kambe et al., 2021), as shown in Figure 3. The ZIPs family increases zinc ion concentration in the cytoplasm by promoting extracellular uptake or release from subcellular organelles. Conversely, the ZnTs family reduces zinc ion concentration in the cytoplasm. Imbalances or changes in the expression and localization of these zinc ion transporters can lead to zinc ion imbalances, resulting in various severe pathophysiological stimuli within cells (Tuncay et al., 2019). The expression of zinc ion transporters ZIP7 and ZnT7 at the protein and RNA levels is relatively low in mammalian heart tissues. ZIP7 is widely expressed in human and mice tissues, while ZnT7 is abundant in the liver and small intestine (Kirschke and Huang, 2003; Huang et al., 2005).
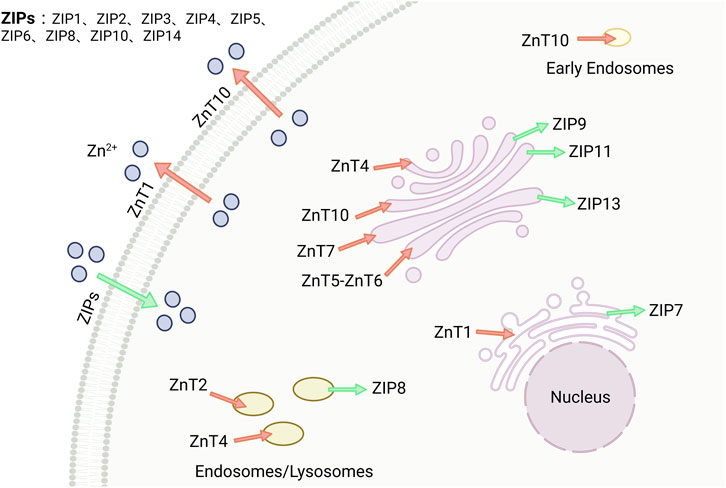
FIGURE 3. Zinc is maintained by two families, ZnTs and ZIPs. There are two families of zinc transport proteins that maintain the stability of zinc ions within the body. ZIPs (SLC39A) increase the concentration of zinc in the cytoplasm, while ZnTs (SLC30A) decrease the concentration of zinc in the cytoplasm. Image from biorender.
Kirschke et al. and Turan et al. analyzed confocal images of cardiomyocyte using immunofluorescence microscopy and Huygens software, they found that ZIP7 was primarily located in the sarcoplasmic(endoplasmic) reticulum [S(E)R], with a small portion in the Golgi apparatus, but not in the nucleus(Kirschke and Huang, 2003; Turan, 2019). Similarly, Tuncay et al. examined ZIP7 and ZnT7 colocalization in S(E)R preparations isolated from rat cardiomyocyte and observed the same localization pattern (Tuncay et al., 2017). They also discovered that hyperglycemia-induced intracellular redistribution of free zinc led to an increased cytoplasmic [Zn2+]i and decreased [Zn2+]i in S(E)R through activation of CK2α-associated ZIP7 phosphorylation (Tuncay et al., 2017).
Metallothionein (MT) is a low-molecular-weight protein rich in mercaptans, which functions as a scavenger of free radicals (Wang et al., 2001), It contains a high concentration of cysteine, which effectively protects cells and tissues from oxidative damage (Kang, 1999). There are four subtypes of MT: MT-I and MT-II are present in all tissues and organs of mammals, MT-III is unique to brain tissues, and MT-IV is found only in certain tissues containing lamellar squamous epithelial cells (Meloni et al., 2006). Cardio-specific overexpression of MT and catalase significantly inhibited acute (Kang et al., 1997) and chronic (Sun et al., 2001) cardiotoxicity induced by Dox, proved by characteristic histopathological, ultrastructural changes, and dysfunction. Cardiomyocyte apoptosis induced by Dox was significantly reduced in mice with cardiac-specific overexpression of MT (Kang et al., 2000). What’s more, Zinc-MT strongly inhibited lipid peroxidation when present during incubation (Thomas et al., 1986).
Researchers have made many attempts and innovations to reduce the cardiac side effects during chemotherapy. For example, Y. Zhang et al. found zinc-selenium tea can effectively alleviate the extent of myocardial fiber disarray, rupture, and inflammatory cell infiltration induced by nonylphenol (Zhang et al., 2023b); although green tea has a similar effect, not as pronounced as zinc-selenium tea (Zhang et al., 2023b). P.K. Badkoobeh found that the antioxidant nano-zinc oxide (nZnO) has a cellular protective effect for Dox-induced male gonadal toxicity (Badkoobeh et al., 2013). Zinc finger protein 260 (Zfp260), also known as phenylephrine-induced complex-1 (PEX1), is an effector of α-1-adrenergic signaling in cardiac hypertrophy. It has been found that overexpression of Zfp260 can upregulate anti-apoptotic genes and reduce doxorubicin-induced apoptosis in primary cardiomyocyte (Li et al., 2022b). Researchers also evaluated the cardioprotective effects of zinc taurine solid dispersion in SD rats. After giving Dox treatment, zinc taurine was found to alleviate the decrease in blood pressure and left ventricular pressure caused by Dox (Wang et al., 2015). It also reduced serum Zn2+ and albumin levels and inhibited cardiomyocyte apoptosis (Wang et al., 2015). In addition, R. Wu et al. found that Zn2+-Curcumin supplementation significantly attenuates Dox-induced zinc imbalance, improves Dox-induced cardiac dysfunction, reduces myocardial injury (Wu et al., 2019).
Calcium is a vital trace element that plays various roles in the human body, both structurally and functionally. The concentration of intracellular and extracellular calcium is strictly regulated through processes such as intestinal absorption, renal reabsorption, and bone exchange, which are controlled by a group of interacting hormones, including parathyroid hormone, parathyroid hormone-related peptides, and key receptors (Matikainen et al., 2021), These mechanisms ensure that blood calcium concentrations and systemic calcium ion balance are maintained within a narrow range (Matikainen et al., 2021). Calcium exists in two forms in the body: as an inactive binding form and as the highly active bivalent cation Ca2+.
In cardiomyocyte, calcium is predominantly found in the SR. According to Figure 4, under normal physiological conditions, the release of calcium ions from the SR generates ROS. However, under the influence of Dox, the release of Ca2+ increases, leading to apoptosis of cardiomyocyte. On the other hand, chelating agents of Ca2+ inhibit ROS production and cardiomyocyte apoptosis (Kalivendi et al., 2005). Disruption of Ca2+ balance triggers activation of the ER stress response. Maintaining Ca2+ homeostasis requires a highly integrated and complex system, which includes the plasma membrane Na+/Ca2+ exchanger (NCX), a system with low affinity but a strong capability to transport Ca2+. The plasma membrane Ca2+ pump (PMCA) and the S(E)R calcium ATPase (SERCA) have a high affinity for Ca2+ but poor capacity (Krebs et al., 2015). Similarly, in the case of DIC, ER stress can also induce cardiomyocyte apoptosis, further exacerbating the condition (Lakshmanan et al., 2013). Furthermore, during the metabolism of Dox, the toxic metabolite DOXOL is produced, which inhibits sodium/calcium exchange channels (Fu et al., 1990).

FIGURE 4. Calcium depends on the transport of different proteins between cytoplasm and organelles, and calcium overload promotes apoptosis. Ca2+ is pumped into the cytoplasm by L-Type channels and CaMKII. It then influences the calcium ion concentration in organelles such as mitochondria and endoplasmic reticulum through proteins like RyR, SERCA, and NCX. When there is calcium overload in cells, it results in endoplasmic reticulum stress and mitochondrial energy imbalance. This triggers the activation of apoptosis-related proteins, ultimately leading to myocardial disease. PMCA plasma membrane Ca2+-ATPases; CaMKII calcium/calmodulin-dependent protein kinase II; ATP adenosine triphosphate; SERCA sarcoendoplasmic reticulum calcium ATPase; RyR ryanodine receptor; NCX sodium (Na)-calcium exchanger; SR sarcoplasmic reticulum; mPTP mitochondrial permeability transition pore. Image from biorender.
In a rabbit model with DIC, Arai et al. found that reduced expression levels of the SR mRNA gene responsible for calcium transport are the main cause of disrupt calcium homeostasis (Arai et al., 1998). Therefore, several studies have identified targets related to ER stress, aiming to expand the possibilities for preventing and treating DIC. For instance, it has been observed that Stim1 expression is downregulated in Dox-treated mice cardiomyocyte. Conversely, cardio-specific deficiency of Stim1 worsens Dox-induced cardiac dysfunction and cardiomyocyte apoptosis, while cardio-specific overexpression of Stim1 alleviates these phenomenon (Zhu et al., 2021); Additionally, downregulation of Stim1 enhances Dox-induced ER stress in cardiomyocyte, whereas overexpression of Stim1 inhibits the activation of molecular markers of ER stress (Zhu et al., 2021).
Dox also causes calcium overload in mitochondria. On one hand, Dox treatment can disrupt mitochondrial energy synthesis, and induce mutations and defects in mitochondrial DNA (Sheibani et al., 2022); On the other hand, elevated intracellular calcium concentration can also lead to mitochondrial dysfunction and apoptosis (Mitry and Edwards, 2016). To maintain the flow of Ca2+ across the mitochondrial membrane, Ca2+ enters the mitochondria through a specific non-adenosine triphosphate-dependent single transporter and then exits through Na+ carriers or Ca2+/H+ reverse transporters in the heart and liver, respectively (Mughal et al., 2018). Interestingly, calcium directly affects enzymes that stimulate the tricarboxylic acid cycle and the electron transport chain, resulting in increased oxidative phosphorylation (Ivannikov and Macleod, 2013). Mitochondria, in turn, regulate cellular calcium signaling by sequestering and buffering cytoplasmic calcium.
Calcium channel blockers can be used to prevent cellular death to alleviate calcium overload. Nicorandil, an indirect calcium channel blocker, can inhibit oxidative stress-induced apoptosis by opening mitochondrial KATP channels or activating NO/cGMP-dependent pathways (Nishikawa et al., 2006). Studies have shown that Nicorandil can preserve phosphocreatine and adenine nucleotide levels by restoring mitochondria’s oxidative phosphorylation ability and creatine kinase activity (Ahmed and El-Maraghy, 2013). Diltiazem (DIL), another calcium channel blocker, has been reported to downregulate the expression of the mitochondrial transporter ABCB1 gene in the human breast cancer cell line MCF-7 when used in combination with Dox. DIL treatment can reverse the resistance of breast cancer cells to Dox and has a protective effect against DIC (Al-Malky et al., 2019).
There are three overall types of calcium sensitizer agents (Pollesello et al., 2016). Type I calcium sensitizers increase the affinity of Ca2+ to troponin C either through direct action or conformational changes, enhancing myocardial contractility. Their mechanism of action is central, such as Pimobendan, CGP-48506, and Oxyphenamone. Type II calcium sensitizers act directly on cardiac myofilaments, promoting the regulation of actin and myosin, which increases the sensitivity of myocardial fibers to Ca2+ but does not affect the affinity of troponin C to Ca2+. Their mechanism of action is downstream, such as Levosimendan (LEVO). Type III calcium sensitizer directly acts on the troponin C and is considered a downstream mechanism. Moreover, it exhibits myocontractile effects by activating the actomyosin ATPase, even in the absence of Ca2+, such as EMD57033. While LEVO is one of the well-studied calcium sensitizer and vasodilator. Studies have shown that LEVO can activate the cAMP-PKA-PLN axis, reducing calcium overload in cardiomyocyte, and alleviating DIC (Efentakis et al., 2020). Additionally, LEVO can inhibit the activation of PTEN, promote the expression of P-Akt, decrease cell apoptosis, and repair cardiac function damage caused by Dox (Li et al., 2020a).
All of the metals mentioned above, although the amount needed by the human body is very small, can cause signs of disease when absent. Abnormal transport of metal ions has been observed in patients with cardiomyopathy caused by continuous use of Dox in cancer treatment and in rodents with cumulative dosing of Dox to model cardiomyopathy. Previous literature suggests that overload and deposition of endogenous metal ions are part of the mechanism of DIC. An effective metal chelator should have the ability to cross the BBB, be specific to a single metal ion, and not interfere with the normal physiological metabolism of metal ions (Wang et al., 2020b). At present, metal chelators that satisfy these three conditions have not yet been identified for clinical treatment of DIC. Ferroptosis currently has only one clinical treatment option, which is Dexrazoxane. Studies have shown that Ferrostatin 1 can inhibit ferroptosis more than Dexrazoxane in mice (Fang et al., 2019). Interestingly, researchers have pointed out that the in vivo therapeutic function of Ferrostatin 1 is weaker than in vitro due to its instability in plasma. To tackle this, a soluble Ferrostatin analog named UAMC-3203 has been developed. UAMC-3203 is a more stable and effective inhibitor of ferroptosis (Jin et al., 2022a).
Currently, we have discovered various chemical drugs, natural compounds or traditional Chinese medicine formulations that exhibit different mechanisms in combating DIC. Examples of chemical compounds include empagliflozin (Quagliariello et al., 2021), sulforaphane (Wang et al., 2022b), dexmedetomidine (Wang et al., 2022d). Natural products such as puerarin (Huang et al., 2022), isoquercitrin (Luo et al., 2023), icariside II (Gao et al., 2023) have also shown efficacy. Additionally, traditional Chinese medicine compound formulations like qing-xin-jie-yu granule (Zhang et al., 2023a), kaixin san (Cao et al., 2023), and qishenyiqi drops (Wu et al., 2023) have demonstrated the ability to inhibit ferroptosis and maintain intracellular iron balance. Furthermore, supplementation with Ginkgolide B (Gao et al., 2016), vitamins B and D (Awad et al., 2021), Epalastat analogue NARI-29 (Syamprasad et al., 2023), and d-Limonene complexed with cyclodextrin (Durço et al., 2023) have been found to alleviate DIC through calcium-related pathways. Taurine zinc solid dispersions (Wang et al., 2015), Zn(ii)-curcumin supplementation (Wu et al., 2019), magnolol and honokiol complex (Aktay et al., 2023) have been shown to improve the abnormal state of Zn2+ during DIC. Interestingly, cannabidiol has the ability to restore normal calcium and zinc levels simultaneously, thereby playing a protective role for the heart (Fouad et al., 2013).
In addition to reducing DIC, metal ion overload also has certain cardiotoxic effects. For example, a series of previous meta-analysis studies conducted by Wang’s team found that an increase in dietary heme iron intake by 1 mg per day led to a 7% increase in the risk of heart disease (Fang et al., 2015). Furthermore, hereditary hemochromatosis, an autosomal recessive genetic disease characterized by large deposits of Fe3+ in multiple organs. According to a study by the Mayo Clinic in the United States, approximately one-third of hemochromatosis patients die from heart disease (Olson et al., 1987). Moreover, women with hemochromatosis have a lower risk of cardiovascular events than men, possibly due to persistent iron deficiency during women’s periods (Gaenzer et al., 2002). Similarly, uncombined or free copper is toxic, as almost all copper in the human body must be combined with copper proteins. Acute copper poisoning can lead to abdominal pain, nausea and vomiting, liver necrosis, kidney damage, and more. In terms of the cardiovascular system, it can lead to atherosclerosis. Excessive zinc or calcium in adults can also increase the risk of coronary heart disease and arteriosclerosis. As summarized in Table 1, Dox promotes apoptosis and mortality rates, induces myocardial diseases. However, when administered with metal ion inhibitors through various mechanisms, it exhibits significant cardiac protective effects. Metal ions have characteristics of a “double-edged sword,” so it is important to avoid excessive adjustment of their concentration.
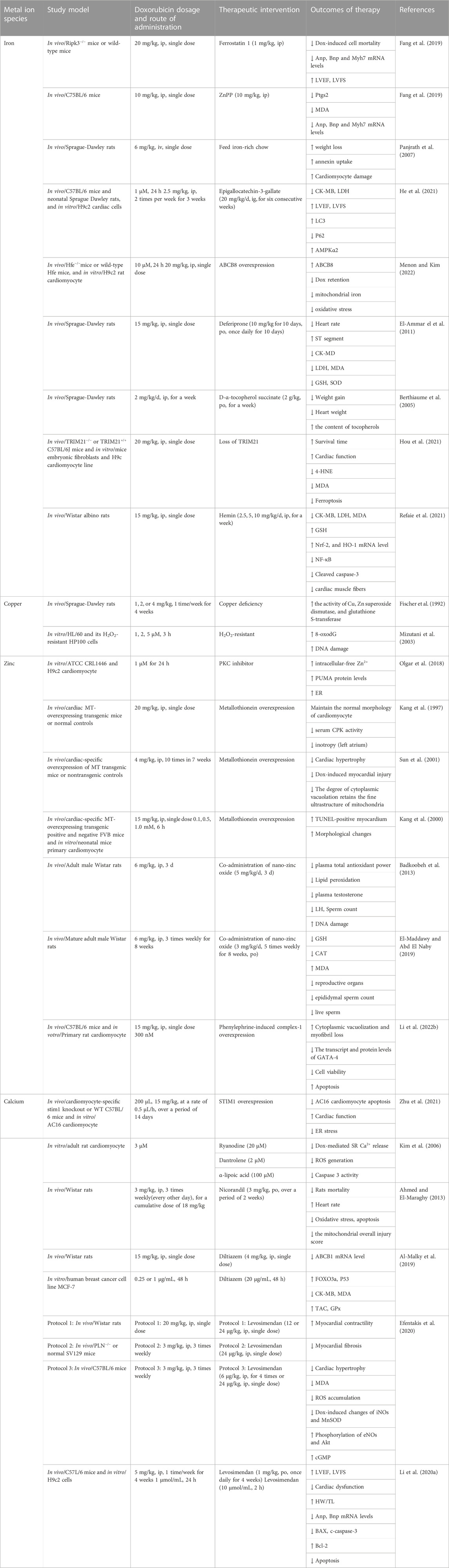
TABLE 1. Potential therapeutic interventions in Dox-induced cardiotoxicity in vivo/vitro.
Despite its side and adverse effects, Dox remains a key drug in many cancer treatments. The mechanism of DIC is influenced by many factors. Depending on the mechanism, different strategies can be developed to prevent or reduce the adverse cardiotoxicity of Dox. Previous studies have shown that ADAR2 overexpression attenuates DIC by enhancing cardiac function and reducing apoptosis (Wu et al., 2022). Similarly, DIC can be alleviated by activating some classical signaling pathways such as cAMP/PKA/SIRT1 (Hu et al., 2020), or AKT/SIRT3/SOD2 (Liu et al., 2022). Recently, some potential DIC therapeutic targets have been newly discovered, such as PDE10A (Chen et al., 2023), Sestrin 2 (Wang et al., 2022a), FAM134B (Qu et al., 2022), TFEB (Chen et al., 2022), etc., which play a cardioprotective role in reducing apoptosis, improving cardiac function, reducing oxidative stress and endoplasmic reticulum stress, and promoting autophagy. In addition, some non-coding RNAs have been shown to play important roles in DIC, such as miR-128-3p (Zhao et al., 2019), miR-451 (Li et al., 2019), miR-152 (Zhang et al., 2021), miR-125b (Jin et al., 2022b), etc. Recently, there are also newly discovered drugs with evidence of reducing DIC, such as Di’ao Xinxuekang capsule (Li et al., 2022c), tanshinone I (Jiang et al., 2022), glycyrrhetinic acid (Cheng et al., 2022), and Berberine (Wang et al., 2023), etc.
This paper discusses various metal ion chelators, ionic carriers, metal complexes, and natural chemical products that can reduce ROS production by regulating endogenous metal ion homeostasis, reducing oxidative stress and mitochondrial dysfunction, and thereby alleviating the cardiac toxicity of Dox. In future studies, it is hoped that by further improving the structure and deficiencies of these drugs, they will not only be effective at the animal and cellular level, but can also be used clinically through drug clinical trials to prevent or treat DIC.
NZ: Investigation, Visualization, Writing–original draft. SW: Investigation, Methodology, Writing–review and editing. TS: Resources, Writing–review and editing. SX: Supervision, Writing–review and editing. JL: Investigation, Writing–review and editing. WL: Conceptualization, Funding acquisition, Writing–review and editing. BZ: Funding acquisition, Writing–review and editing, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants of the National Natural Scientific Foundation of China (Nos 82173911 and 81973406), Fundamental Research Funds for the Central Universities of Central South University (No. 2023ZZTS0891), Hunan Provincial Natural Scientific Foundation (Nos 2023JJ30761 and 2023JJ30802), Scientific Research Project of Hunan Provincial Health and Family Planning Commission (No. 202113050843), and Research Project established by Chinese Pharmaceutical Association Hospital Pharmacy department (No. CPA-Z05-ZC-2021-002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abe, K., Ikeda, M., Ide, T., Tadokoro, T., Miyamoto, H. D., Furusawa, S., et al. (2022). Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci. Signal 15, eabn8017. doi:10.1126/scisignal.abn8017
Ahmed, L. A., and El-Maraghy, S. A. (2013). Nicorandil ameliorates mitochondrial dysfunction in doxorubicin-induced heart failure in rats: possible mechanism of cardioprotection. Biochem. Pharmacol. 86, 1301–1310. doi:10.1016/j.bcp.2013.07.005
Aktay, I., Bitirim, C. V., Olgar, Y., Durak, A., Tuncay, E., Billur, D., et al. (2023). Cardioprotective role of a magnolol and honokiol complex in the prevention of doxorubicin-mediated cardiotoxicity in adult rats. Mol. Cell Biochem. doi:10.1007/s11010-023-04728-w
Al-Malky, H. S., Osman, A. M., Damanhouri, Z. A., Alkreathy, H. M., Al Aama, J. Y., Ramadan, W. S., et al. (2019). Modulation of doxorubicin-induced expression of the multidrug resistance gene in breast cancer cells by diltiazem and protection against cardiotoxicity in experimental animals. Cancer Cell Int. 19, 191. doi:10.1186/s12935-019-0912-0
Anderson, M. E. (1998). Glutathione: an overview of biosynthesis and modulation. Chem. Biol. Interact. 111-112, 1–14. doi:10.1016/s0009-2797(97)00146-4
Angelova, M., Asenova, S., Nedkova, V., and Kolevakolarova, R. (2011). Copper in the human organism. Trakia J. Sci.
Anjum, R., Palanimuthu, D., Kalinowski, D. S., Lewis, W., Park, K. C., Kovacevic, Z., et al. (2019). Synthesis, characterization, and in vitro anticancer activity of copper and zinc bis(thiosemicarbazone) complexes. Inorg. Chem. 58, 13709–13723. doi:10.1021/acs.inorgchem.9b01281
Apresova, M. A., Efremova, I. E., Babayants, A. A., and Cheknev, S. B. (2014). γ-Globulin fraction proteins and their metal complexes with copper cations in induction of IL-8 production. Bull. Exp. Biol. Med. 156, 823–825. doi:10.1007/s10517-014-2460-x
Arai, M., Tomaru, K., Takizawa, T., Sekiguchi, K., Yokoyama, T., Suzuki, T., et al. (1998). Sarcoplasmic reticulum genes are selectively down-regulated in cardiomyopathy produced by doxorubicin in rabbits. J. Mol. Cell Cardiol. 30, 243–254. doi:10.1006/jmcc.1997.0588
Awad, H. H., El-Derany, M. O., Mantawy, E. M., Michel, H. E., El-Naa, M. M., Salah El-Din, R. A., et al. (2021). Comparative study on beneficial effects of vitamins B and D in attenuating doxorubicin induced cardiotoxicity in rats: emphasis on calcium homeostasis. Biomed. Pharmacother. 140, 111679. doi:10.1016/j.biopha.2021.111679
Baba, Y., Higa, J. K., Shimada, B. K., Horiuchi, K. M., Suhara, T., Kobayashi, M., et al. (2018). Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 314, H659–H668. doi:10.1152/ajpheart.00452.2017
Badkoobeh, P., Parivar, K., Badkoobeh, P. K., Kalantar, S. M., Hosseini, S. D., and Salabat, A. (2013). Effect of nano-zinc oxide on doxorubicin-induced oxidative stress and sperm disorders in adult male Wistar rats. Iran. J. Reproductive Med. 11, 355–364.
Banci, L., and Bertini, I. (2013). Metallomics and the cell: some definitions and general comments. Met. Ions Life Sci. 12, 1–13. doi:10.1007/978-94-007-5561-1_1
BayıR, A., Kara, H., KıYıCı, A., OztüRK, B., and AkyüREK, F. (2013). Levels of selenium, zinc, copper, and cardiac troponin I in serum of patients with acute coronary syndrome. Biol. Trace Elem. Res. 154, 352–356. doi:10.1007/s12011-013-9754-0
Berthiaume, J. M., Oliveira, P. J., Fariss, M. W., and Wallace, K. B. (2005). Dietary vitamin E decreases doxorubicin-induced oxidative stress without preventing mitochondrial dysfunction. Cardiovasc Toxicol. 5, 257–267. doi:10.1385/ct:5:3:257
Bhuvanasundar, R., John, A., Sulochana, K. N., Coral, K., Deepa, P. R., and Umashankar, V. (2014). A molecular model of human Lysyl Oxidase (LOX) with optimal copper orientation in the catalytic cavity for induced fit docking studies with potential modulators. Bioinformation 10, 406–412. doi:10.6026/97320630010406
Brem, S., Tsanaclis, A. M., and Zagzag, D. (1990). Anticopper treatment inhibits pseudopodial protrusion and the invasive spread of 9L gliosarcoma cells in the rat brain. Neurosurgery 26, 391–396. doi:10.1097/00006123-199003000-00003
Brewer, G. J. (2009). The risks of copper toxicity contributing to cognitive decline in the aging population and to Alzheimer's disease. J. Am. Coll. Nutr. 28, 238–242. doi:10.1080/07315724.2009.10719777
Cao, Y., Li, M., Gu, L., Zhao, X., Zhou, A., Miao, Y., et al. (2023). Chinese traditional formula Kaixin San suppressed ferroptosis of hippocampal neurons and cardiomyocytes in mice with paradoxical sleep deprivation. J. Ethnopharmacol. 304, 116034. doi:10.1016/j.jep.2022.116034
Cater, M. A., and Haupt, Y. (2011). Clioquinol induces cytoplasmic clearance of the X-linked inhibitor of apoptosis protein (XIAP): therapeutic indication for prostate cancer. Biochem. J. 436, 481–491. doi:10.1042/BJ20110123
Cater, M. A., Pearson, H. B., Wolyniec, K., Klaver, P., Bilandzic, M., Paterson, B. M., et al. (2013). Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem. Biol. 8, 1621–1631. doi:10.1021/cb400198p
Chang, H. C., Wu, R., Shang, M., Sato, T., Chen, C., Shapiro, J. S., et al. (2016). Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 8, 247–267. doi:10.15252/emmm.201505748
Chauhan, M., Banerjee, K., and Arjmand, F. (2007). DNA binding studies of novel Copper(II) complexes containing L-tryptophan as chiral auxiliary: in vitro antitumor activity of Cu-Sn2 complex in human neuroblastoma cells. Inorg. Chem. 46, 3072–3082. doi:10.1021/ic061753a
Chen, D., Cui, Q. C., Yang, H., Barrea, R. A., Sarkar, F. H., Sheng, S., et al. (2007). Clioquinol, a therapeutic agent for Alzheimer's disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 67, 1636–1644. doi:10.1158/0008-5472.CAN-06-3546
Chen, D., Yu, W., Zhong, C., Hong, Q., Huang, G., Que, D., et al. (2022). Elabela ameliorates doxorubicin-induced cardiotoxicity by promoting autophagic flux through TFEB pathway. Pharmacol. Res. 178, 106186. doi:10.1016/j.phrs.2022.106186
Cheng, Y., Wu, X., Nie, X., Wu, Y., Zhang, C., Lee, S. M., et al. (2022). Natural compound glycyrrhetinic acid protects against doxorubicin-induced cardiotoxicity by activating the Nrf2/HO-1 signaling pathway. Phytomedicine 106, 154407. doi:10.1016/j.phymed.2022.154407
Chen, S., Chen, J., Du, W., Mickelsen, D. M., Shi, H., Yu, H., et al. (2023). PDE10A inactivation prevents doxorubicin-induced cardiotoxicity and tumor growth. Circ. Res. 133, 138–157. doi:10.1161/CIRCRESAHA.122.322264
Cox, C., Teknos, T. N., Barrios, M., Brewer, G. J., Dick, R. D., and Merajver, S. D. (2001). The role of copper suppression as an antiangiogenic strategy in head and neck squamous cell carcinoma. Laryngoscope 111, 696–701. doi:10.1097/00005537-200104000-00024
Crowe, A., Jackaman, C., Beddoes, K. M., Ricciardo, B., and Nelson, D. J. (2013). Rapid copper acquisition by developing murine mesothelioma: decreasing bioavailable copper slows tumor growth, normalizes vessels and promotes T cell infiltration. PLoS One 8, e73684. doi:10.1371/journal.pone.0073684
Dabravolski, S. A., Sadykhov, N. K., Kartuesov, A. G., Borisov, E. E., Sukhorukov, V. N., and Orekhov, A. N. (2022). Interplay between Zn(2+) homeostasis and mitochondrial functions in cardiovascular diseases and heart ageing. Int. J. Mol. Sci. 23, 6890. doi:10.3390/ijms23136890
Denoyer, D., Masaldan, S., La Fontaine, S., and Cater, M. A. (2015). Targeting copper in cancer therapy: 'Copper that Cancer. Metallomics 7, 1459–1476. doi:10.1039/c5mt00149h
Dikalova, A. E., Bikineyeva, A. T., Budzyn, K., Nazarewicz, R. R., Mccann, L., Lewis, W., et al. (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116. doi:10.1161/CIRCRESAHA.109.214601
Dinkova-Kostova, A. T., and Talalay, P. (2008). Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 52 (1), S128–S138. doi:10.1002/mnfr.200700195
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. doi:10.1016/s1535-6108(03)00050-3
DurçO, A. O., Souza, D. S., Rhana, P., Costa, A. D., Marques, L. P., Santos, L., et al. (2023). d-Limonene complexed with cyclodextrin attenuates cardiac arrhythmias in an experimental model of doxorubicin-induced cardiotoxicity: possible involvement of calcium/calmodulin-dependent protein kinase type II. Toxicol. Appl. Pharmacol. 474, 116609. doi:10.1016/j.taap.2023.116609
Efentakis, P., Varela, A., Chavdoula, E., Sigala, F., Sanoudou, D., Tenta, R., et al. (2020). Levosimendan prevents doxorubicin-induced cardiotoxicity in time- and dose-dependent manner: implications for inotropy. Cardiovasc Res. 116, 576–591. doi:10.1093/cvr/cvz163
El-Ammar, S. M., Said, S. A., Suddek, G. M., and El-Damarawy, S. L. (2011). Amelioration of doxorubicin-induced cardiotoxicity by deferiprone in rats. Can. J. Physiol. Pharmacol. 89, 269–276. doi:10.1139/y11-020
El-Maddawy, Z. K., and Abd El Naby, W. S. H. (2019). Protective effects of zinc oxide nanoparticles against doxorubicin induced testicular toxicity and DNA damage in male rats. Toxicol. Res. (Camb) 8, 654–662. doi:10.1039/c9tx00052f
Ewer, M. S., and Lippman, S. M. (2005). Type II chemotherapy-related cardiac dysfunction: time to recognize a new entity. J. Clin. Oncol. 23, 2900–2902. doi:10.1200/JCO.2005.05.827
Fang, X., An, P., Wang, H., Wang, X., Shen, X., Li, X., et al. (2015). Dietary intake of heme iron and risk of cardiovascular disease: a dose-response meta-analysis of prospective cohort studies. Nutr. Metab. Cardiovasc Dis. 25, 24–35. doi:10.1016/j.numecd.2014.09.002
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 116, 2672–2680. doi:10.1073/pnas.1821022116
Fischer, J. G., Tackett, R. L., Howerth, E. W., and Johnson, M. A. (1992). Copper deficient rat heart can compensate for doxorubicin-induced oxidant stress. Biol. Trace Elem. Res. 37, 233–251. doi:10.1007/bf02783798
Fischer, J. G., Tackett, R. L., Howerth, E. W., and Johnson, M. A. (1993). Copper deficient rat heart can compensate for doxorubicin-induced oxidant stress. Biol. Trace Elem. Res. 37, 233–251. doi:10.1007/BF02783798
Fisher, G. L., Byers, V. S., Shifrine, M., and Levin, A. S. (1976). Copper and zinc levels in serum from human patients with sarcomas. Cancer 37, 356–363. doi:10.1002/1097-0142(197601)37:1<356::aid-cncr2820370146>3.0.co;2-w
Forcina, G. C., and Dixon, S. J. (2019). GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 19, e1800311. doi:10.1002/pmic.201800311
Fouad, A. A., Albuali, W. H., Al-Mulhim, A. S., and Jresat, I. (2013). Cardioprotective effect of cannabidiol in rats exposed to doxorubicin toxicity. Environ. Toxicol. Pharmacol. 36, 347–357. doi:10.1016/j.etap.2013.04.018
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi:10.1038/ncb3064
Fu, L. X., Waagstein, F., and Hjalmarson, A. (1990). A new insight into adriamycin-induced cardiotoxicity. Int. J. Cardiol. 29, 15–20. doi:10.1016/0167-5273(90)90267-9
Gaenzer, H., Marschang, P., Sturm, W., Neumayr, G., Vogel, W., Patsch, J., et al. (2002). Association between increased iron stores and impaired endothelial function in patients with hereditary hemochromatosis. J. Am. Coll. Cardiol. 40, 2189–2194. doi:10.1016/s0735-1097(02)02611-6
Gao, J., Chen, T., Zhao, D., Zheng, J., and Liu, Z. (2016). Ginkgolide B exerts cardioprotective properties against doxorubicin-induced cardiotoxicity by regulating reactive oxygen species, Akt and calcium signaling pathways in vitro and in vivo. PLoS One 11, e0168219. doi:10.1371/journal.pone.0168219
Gao, J., Ma, C., Xia, D., Chen, N., Zhang, J., Xu, F., et al. (2023). Icariside II preconditioning evokes robust neuroprotection against ischaemic stroke, by targeting Nrf2 and the OXPHOS/NF-κB/ferroptosis pathway. Br. J. Pharmacol. 180, 308–329. doi:10.1111/bph.15961
Guha, S., and Roy, S. (2021). Enhanced expression of SLC4A11 by tert-Butylhydroquinone is mediated by direct binding of Nrf2 to the promoter of SLC4A11. Free Radic. Biol. Med. 167, 299–306. doi:10.1016/j.freeradbiomed.2021.03.006
Gupte, A., and Mumper, R. J. (2009). Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 35, 32–46. doi:10.1016/j.ctrv.2008.07.004
Gutteridge, J. M. (1984). Lipid peroxidation and possible hydroxyl radical formation stimulated by the self-reduction of a doxorubicin-iron (III) complex. Biochem. Pharmacol. 33, 1725–1728. doi:10.1016/0006-2952(84)90340-x
Hamilton, S., and Terentyev, D. (2019). Altered intracellular calcium homeostasis and arrhythmogenesis in the aged heart. Int. J. Mol. Sci. 20, 2386. doi:10.3390/ijms20102386
He, H., Wang, L., Qiao, Y., Yang, B., Yin, D., and He, M. (2021). Epigallocatechin-3-gallate pretreatment alleviates doxorubicin-induced ferroptosis and cardiotoxicity by upregulating AMPKα2 and activating adaptive autophagy. Redox Biol. 48, 102185. doi:10.1016/j.redox.2021.102185
Hou, K., Shen, J., Yan, J., Zhai, C., Zhang, J., Pan, J. A., et al. (2021). Loss of TRIM21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. EBioMedicine 69, 103456. doi:10.1016/j.ebiom.2021.103456
Hu, J. Y., Zhang, D. L., Liu, X. L., Li, X. S., Cheng, X. Q., Chen, J., et al. (2017). Pathological concentration of zinc dramatically accelerates abnormal aggregation of full-length human Tau and thereby significantly increases Tau toxicity in neuronal cells. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 414–427. doi:10.1016/j.bbadis.2016.11.022
Huang, L., Kirschke, C. P., Zhang, Y., and Yu, Y. Y. (2005). The ZIP7 gene (Slc39a7) encodes a zinc transporter involved in zinc homeostasis of the Golgi apparatus. J. Biol. Chem. 280, 15456–15463. doi:10.1074/jbc.M412188200
Huang, Y., Wu, H., Hu, Y., Zhou, C., Wu, J., Wu, Y., et al. (2022). Puerarin attenuates oxidative stress and ferroptosis via AMPK/PGC1α/Nrf2 pathway after subarachnoid hemorrhage in rats. Antioxidants (Basel) 11, 1259. doi:10.3390/antiox11071259
Hu, C., Zhang, X., Song, P., Yuan, Y. P., Kong, C. Y., Wu, H. M., et al. (2020). Meteorin-like protein attenuates doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1 pathway. Redox Biol. 37, 101747. doi:10.1016/j.redox.2020.101747
Ichikawa, Y., Ghanefar, M., Bayeva, M., Wu, R., Khechaduri, A., Naga Prasad, S. V., et al. (2014). Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 124, 617–630. doi:10.1172/JCI72931
Ishida, S., Andreux, P., Poitry-Yamate, C., Auwerx, J., and Hanahan, D. (2013). Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. U. S. A. 110, 19507–19512. doi:10.1073/pnas.1318431110
Ivannikov, M. V., and Macleod, G. T. (2013). Mitochondrial free Ca2⁺ levels and their effects on energy metabolism in Drosophila motor nerve terminals. Biophys. J. 104, 2353–2361. doi:10.1016/j.bpj.2013.03.064
Jiang, Q., Chen, X., Tian, X., Zhang, J., Xue, S., Jiang, Y., et al. (2022). Tanshinone I inhibits doxorubicin-induced cardiotoxicity by regulating Nrf2 signaling pathway. Phytomedicine 106, 154439. doi:10.1016/j.phymed.2022.154439
Jiang, X., Stockwell, B. R., and Conrad, M. (2021). Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282. doi:10.1038/s41580-020-00324-8
Jin, T., He, Q., Cheng, C., Li, H., Liang, L., Zhang, G., et al. (2022a). UAMC-3203 or/and deferoxamine improve post-resuscitation myocardial dysfunction through suppressing ferroptosis in a rat model of cardiac arrest. Shock 57, 344–350. doi:10.1097/SHK.0000000000001869
Jin, X., Yu, W., and Ye, P. (2022b). MiR-125b enhances doxorubicin-induced cardiotoxicity by suppressing the nucleus-cytoplasmic translocation of YAP via targeting STARD13. Environ. Toxicol. 37, 730–740. doi:10.1002/tox.23438
Johansson, B. (1992). A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr. Scand. Suppl. 369, 15–26. doi:10.1111/j.1600-0447.1992.tb03310.x
Kalivendi, S. V., Konorev, E. A., Cunningham, S., Vanamala, S. K., Kaji, E. H., Joseph, J., et al. (2005). Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochem. J. 389, 527–539. doi:10.1042/BJ20050285
Kambe, T., Taylor, K. M., and Fu, D. (2021). Zinc transporters and their functional integration in mammalian cells. J. Biol. Chem. 296, 100320. doi:10.1016/j.jbc.2021.100320
Kang, Y. J. (1999). The antioxidant function of metallothionein in the heart. Proc. Soc. Exp. Biol. Med. 222, 263–273. doi:10.1046/j.1525-1373.1999.d01-143.x
Kang, Y. J., Chen, Y., Yu, A., Voss-Mccowan, M., and Epstein, P. N. (1997). Overexpression of metallothionein in the heart of transgenic mice suppresses doxorubicin cardiotoxicity. J. Clin. Invest. 100, 1501–1506. doi:10.1172/JCI119672
Kang, Y. J., Zhou, Z. X., Wang, G. W., Buridi, A., and Klein, J. B. (2000). Suppression by metallothionein of doxorubicin-induced cardiomyocyte apoptosis through inhibition of p38 mitogen-activated protein kinases. J. Biol. Chem. 275, 13690–13698. doi:10.1074/jbc.275.18.13690
Kim, D. H., Kim, W. D., Kim, S. K., Moon, D. H., and Lee, S. J. (2020). TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 11, 406. doi:10.1038/s41419-020-2618-6
Kim, S. Y., Kim, S. J., Kim, B. J., Rah, S. Y., Chung, S. M., Im, M. J., et al. (2006). Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp. Mol. Med. 38, 535–545. doi:10.1038/emm.2006.63
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi:10.1038/ncb2152
King, J. C., Shames, D. M., and Woodhouse, L. R. (2000). Zinc homeostasis in humans. J. Nutr. 130, 1360S–6s. doi:10.1093/jn/130.5.1360S
Kirschke, C. P., and Huang, L. (2003). ZnT7, a novel mammalian zinc transporter, accumulates zinc in the Golgi apparatus. J. Biol. Chem. 278, 4096–4102. doi:10.1074/jbc.M207644200
Kitakata, H., Endo, J., Ikura, H., Moriyama, H., Shirakawa, K., Katsumata, Y., et al. (2022). Therapeutic targets for DOX-induced cardiomyopathy: role of apoptosis vs. Ferroptosis. Int. J. Mol. Sci. 23, 1414. doi:10.3390/ijms23031414
Komatsu, Y., Sadakata, I., Ogra, Y., and Suzuki, K. T. (2000). Excretion of copper complexed with thiomolybdate into the bile and blood in LEC rats. Chem. Biol. Interact. 124, 217–231. doi:10.1016/s0009-2797(99)00159-3
Krebs, J., Agellon, L. B., and Michalak, M. (2015). Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 460, 114–121. doi:10.1016/j.bbrc.2015.02.004
Lakshmanan, A. P., Harima, M., Suzuki, K., Soetikno, V., Nagata, M., Nakamura, T., et al. (2013). The hyperglycemia stimulated myocardial endoplasmic reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: a differential role of unfolded protein response (UPR) signaling proteins. Int. J. Biochem. Cell Biol. 45, 438–447. doi:10.1016/j.biocel.2012.09.017
Lewerenz, J., Hewett, S. J., Huang, Y., Lambros, M., Gout, P. W., Kalivas, P. W., et al. (2013). The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 18, 522–555. doi:10.1089/ars.2011.4391
Li, D. L., and Hill, J. A. (2014). Cardiomyocyte autophagy and cancer chemotherapy. J. Mol. Cell Cardiol. 71, 54–61. doi:10.1016/j.yjmcc.2013.11.007
Li, L. L., Wei, L., Zhang, N., Wei, W. Y., Hu, C., Deng, W., et al. (2020a). Levosimendan protects against doxorubicin-induced cardiotoxicity by regulating the PTEN/Akt pathway. Biomed. Res. Int. 2020, 8593617. doi:10.1155/2020/8593617
Li, D., Song, C., Zhang, J., and Zhao, X. (2022a). ROS and iron homeostasis dependent ferroptosis play a vital role in 5-Fluorouracil induced cardiotoxicity in vitro and in vivo. Toxicology 468, 153113. doi:10.1016/j.tox.2022.153113
Li, J., Wan, W., Chen, T., Tong, S., Jiang, X., and Liu, W. (2019). miR-451 silencing inhibited doxorubicin exposure-induced cardiotoxicity in mice. Biomed. Res. Int. 2019, 1528278. doi:10.1155/2019/1528278
Li, N., Wang, W., Zhou, H., Wu, Q., Duan, M., Liu, C., et al. (2020b). Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 160, 303–318. doi:10.1016/j.freeradbiomed.2020.08.009
Lin, H., Li, N., He, H., Ying, Y., Sunkara, S., Luo, L., et al. (2015). AMPK inhibits the stimulatory effects of TGF-β on smad2/3 activity, cell migration, and epithelial-to-mesenchymal transition. Mol. Pharmacol. 88, 1062–1071. doi:10.1124/mol.115.099549
Liu, X., Li, D., Pi, W., Wang, B., Xu, S., Yu, L., et al. (2022). LCZ696 protects against doxorubicin-induced cardiotoxicity by inhibiting ferroptosis via AKT/SIRT3/SOD2 signaling pathway activation. Int. Immunopharmacol. 113, 109379. doi:10.1016/j.intimp.2022.109379
Li, W., Xie, H., Hu, H., Huang, J., and Chen, S. (2022b). PEX1 is a mediator of α1-adrenergic signaling attenuating doxorubicin-induced cardiotoxicity. J. Biochem. Mol. Toxicol. 36, e23196. doi:10.1002/jbt.23196
Li, X., Liang, J., Qin, A., Wang, T., Liu, S., Li, W., et al. (2022c). Protective effect of Di'ao Xinxuekang capsule against doxorubicin-induced chronic cardiotoxicity. J. Ethnopharmacol. 287, 114943. doi:10.1016/j.jep.2021.114943
Li, Y., Chen, F., Chen, J., Chan, S., He, Y., Liu, W., et al. (2020c). Disulfiram/copper induces antitumor activity against both nasopharyngeal cancer cells and cancer-associated fibroblasts through ROS/MAPK and ferroptosis pathways. Cancers (Basel) 12, 138. doi:10.3390/cancers12010138
Luo, X., Gong, Y., Jiang, Q., Wang, Q., Li, S., and Liu, L. (2023). Isoquercitrin promotes ferroptosis and oxidative stress in nasopharyngeal carcinoma via the AMPK/NF-κB pathway. J. Biochem. Mol. Toxicol., e23542. doi:10.1002/jbt.23542
Mammoto, T., Jiang, A., Jiang, E., Panigrahy, D., Kieran, M. W., and Mammoto, A. (2013). Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am. J. Pathol. 183, 1293–1305. doi:10.1016/j.ajpath.2013.06.026
Marzano, C., Pellei, M., Tisato, F., and Santini, C. (2009). Copper complexes as anticancer agents. Anticancer Agents Med. Chem. 9, 185–211. doi:10.2174/187152009787313837
Matikainen, N., Pekkarinen, T., Ryhanen, E. M., and Schalin-Jantti, C. (2021). Physiology of calcium homeostasis: an overview. Endocrinol. Metab. Clin. North Am. 50, 575–590. doi:10.1016/j.ecl.2021.07.005
Meloni, G., Zovo, K., Kazantseva, J., Palumaa, P., and VasáK, M. (2006). Organization and assembly of metal-thiolate clusters in epithelium-specific metallothionein-4. J. Biol. Chem. 281, 14588–14595. doi:10.1074/jbc.M601724200
Meng, H., Wang, Y., Zhou, F., Ruan, J., Duan, M., Wang, X., et al. (2021). Reduced serum zinc ion concentration is associated with coronary heart disease. Biol. Trace Elem. Res. 199, 4109–4118. doi:10.1007/s12011-020-02551-8
Menon, A. V., and Kim, J. (2022). Iron promotes cardiac doxorubicin retention and toxicity through downregulation of the mitochondrial exporter ABCB8. Front. Pharmacol. 13, 817951. doi:10.3389/fphar.2022.817951
Mitry, M. A., and Edwards, J. G. (2016). Doxorubicin induced heart failure: phenotype and molecular mechanisms. Int. J. Cardiol. Heart Vasc. 10, 17–24. doi:10.1016/j.ijcha.2015.11.004
Mizutani, H., Oikawa, S., Hiraku, Y., Murata, M., Kojima, M., and Kawanishi, S. (2003). Distinct mechanisms of site-specific oxidative DNA damage by doxorubicin in the presence of copper(II) and NADPH-cytochrome P450 reductase. Cancer Sci. 94, 686–691. doi:10.1111/j.1349-7006.2003.tb01503.x
Mughal, W., Martens, M., Field, J., Chapman, D., Huang, J., Rattan, S., et al. (2018). Myocardin regulates mitochondrial calcium homeostasis and prevents permeability transition. Cell Death Differ. 25, 1732–1748. doi:10.1038/s41418-018-0073-z
Mukhopadhyay, P., Rajesh, M., BáTKAI, S., Kashiwaya, Y., Haskó, G., Liaudet, L., et al. (2009). Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 296, H1466–H1483. doi:10.1152/ajpheart.00795.2008
Nishikawa, S., Tatsumi, T., Shiraishi, J., Matsunaga, S., Takeda, M., Mano, A., et al. (2006). Nicorandil regulates Bcl-2 family proteins and protects cardiac myocytes against hypoxia-induced apoptosis. J. Mol. Cell Cardiol. 40, 510–519. doi:10.1016/j.yjmcc.2006.01.020
Olgar, Y., Durak, A., Tuncay, E., Bitirim, C. V., Ozcinar, E., Inan, M. B., et al. (2018). Increased free Zn(2+) correlates induction of sarco(endo)plasmic reticulum stress via altered expression levels of Zn(2+) -transporters in heart failure. J. Cell Mol. Med. 22, 1944–1956. doi:10.1111/jcmm.13480
Olson, L. J., Edwards, W. D., Mccall, J. T., Ilstrup, D. M., and Gersh, B. J. (1987). Cardiac iron deposition in idiopathic hemochromatosis: histologic and analytic assessment of 14 hearts from autopsy. J. Am. Coll. Cardiol. 10, 1239–1243. doi:10.1016/s0735-1097(87)80124-9
Panjrath, G. S., Patel, V., Valdiviezo, C. I., Narula, N., Narula, J., and Jain, D. (2007). Potentiation of Doxorubicin cardiotoxicity by iron loading in a rodent model. J. Am. Coll. Cardiol. 49, 2457–2464. doi:10.1016/j.jacc.2007.02.060
Pan, Q., Bao, L. W., and Merajver, S. D. (2003). Tetrathiomolybdate inhibits angiogenesis and metastasis through suppression of the NFkappaB signaling cascade. Mol. Cancer Res. 1, 701–706.
Pivetta, T., Lallai, V., Valletta, E., Trudu, F., Isaia, F., Perra, D., et al. (2015). Mixed copper-platinum complex formation could explain synergistic antiproliferative effect exhibited by binary mixtures of cisplatin and copper-1,10-phenanthroline compounds: an ESI-MS study. J. Inorg. Biochem. 151, 107–114. doi:10.1016/j.jinorgbio.2015.05.004
Pollesello, P., Papp, Z., and Papp, J. G. (2016). Calcium sensitizers: what have we learned over the last 25 years? Int. J. Cardiol. 203, 543–548. doi:10.1016/j.ijcard.2015.10.240
Purchase, R. (2013). The link between copper and Wilson's disease. Sci. Prog. 96, 213–223. doi:10.3184/003685013X13712193905878
Quagliariello, V., De Laurentiis, M., Rea, D., Barbieri, A., Monti, M. G., Carbone, A., et al. (2021). The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. 20, 150. doi:10.1186/s12933-021-01346-y
Qu, Y., Gao, R., Wei, X., Sun, X., Yang, K., Shi, H., et al. (2022). Gasdermin D mediates endoplasmic reticulum stress via FAM134B to regulate cardiomyocyte autophagy and apoptosis in doxorubicin-induced cardiotoxicity. Cell Death Dis. 13, 901. doi:10.1038/s41419-022-05333-3
Refaie, M. M. M., Shehata, S., Ibrahim, R. A., Bayoumi, A. M. A., and Abdel-Gaber, S. A. (2021). Dose-dependent cardioprotective effect of hemin in doxorubicin-induced cardiotoxicity via nrf-2/HO-1 and TLR-5/NF-κB/TNF-α signaling pathways. Cardiovasc Toxicol. 21, 1033–1044. doi:10.1007/s12012-021-09694-7
Ren, J., Xu, X., Wang, Q., Ren, S. Y., Dong, M., and Zhang, Y. (2016). Permissive role of AMPK and autophagy in adiponectin deficiency-accentuated myocardial injury and inflammation in endotoxemia. J. Mol. Cell Cardiol. 93, 18–31. doi:10.1016/j.yjmcc.2016.02.002
Renu, K., Abilash, V. G., Tirupathi Pichiah, P. B., and Arunachalam, S. (2018). Molecular mechanism of doxorubicin-induced cardiomyopathy - an update. Eur. J. Pharmacol. 818, 241–253. doi:10.1016/j.ejphar.2017.10.043
Schimmer, A. D., Jitkova, Y., Gronda, M., Wang, Z., Brandwein, J., Chen, C., et al. (2012). A phase I study of the metal ionophore clioquinol in patients with advanced hematologic malignancies. Clin. Lymphoma Myeloma Leuk. 12, 330–336. doi:10.1016/j.clml.2012.05.005
Sheibani, M., Azizi, Y., Shayan, M., Nezamoleslami, S., Eslami, F., Farjoo, M. H., et al. (2022). Doxorubicin-induced cardiotoxicity: an overview on pre-clinical therapeutic approaches. Cardiovasc Toxicol. 22, 292–310. doi:10.1007/s12012-022-09721-1
Sun, L., Wang, H., Yu, S., Zhang, L., Jiang, J., and Zhou, Q. (2022). Herceptin induces ferroptosis and mitochondrial dysfunction in H9c2 cells. Int. J. Mol. Med. 12.
Sun, X., Zhou, Z., and Kang, Y. J. (2001). Attenuation of doxorubicin chronic toxicity in metallothionein-overexpressing transgenic mouse heart. Cancer Res. 61, 3382–3387.
Swain, S. M., Whaley, F. S., and Ewer, M. S. (2003). Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 97, 2869–2879. doi:10.1002/cncr.11407
Syamprasad, N. P., Jain, S., Rajdev, B., Panda, S. R., Gangasani, J. K., Challa, V. S., et al. (2023). AKR1B1 inhibition using NARI-29-an Epalrestat analogue-alleviates Doxorubicin-induced cardiotoxicity via modulating Calcium/CaMKII/MuRF-1 axis. Chem. Biol. Interact. 381, 110566. doi:10.1016/j.cbi.2023.110566
Tadokoro, T., Ikeda, M., Ide, T., Deguchi, H., Ikeda, S., Okabe, K., et al. (2020). Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5, e132747. doi:10.1172/jci.insight.132747
Tainer, J. A., Getzoff, E. D., Richardson, J. S., and Richardson, D. C. (1983). Structure and mechanism of copper, zinc superoxide dismutase. Nature 306, 284–287. doi:10.1038/306284a0
Thomas, J. P., Bachowski, G. J., and Girotti, A. W. (1986). Inhibition of cell membrane lipid peroxidation by cadmium- and zinc-metallothioneins. Biochim. Biophys. Acta 884, 448–461. doi:10.1016/0304-4165(86)90195-9
Townsend, D. M., Tew, K. D., and Tapiero, H. (2003). The importance of glutathione in human disease. Biomed. Pharmacother. 57, 145–155. doi:10.1016/s0753-3322(03)00043-x
Tuncay, E., Bitirim, C. V., Olgar, Y., Durak, A., Rutter, G. A., and Turan, B. (2019). Zn(2+)-transporters ZIP7 and ZnT7 play important role in progression of cardiac dysfunction via affecting sarco(endo)plasmic reticulum-mitochondria coupling in hyperglycemic cardiomyocytes. Mitochondrion 44, 41–52. doi:10.1016/j.mito.2017.12.011
Tuncay, E., Bitirim, V. C., Durak, A., Carrat, G. R. J., Taylor, K. M., Rutter, G. A., et al. (2017). Hyperglycemia-induced changes in ZIP7 and ZnT7 expression cause Zn(2+) release from the sarco(endo)plasmic reticulum and mediate ER stress in the heart. Diabetes 66, 1346–1358. doi:10.2337/db16-1099
Turan, B. (2019). A brief overview from the physiological and detrimental roles of zinc homeostasis via zinc transporters in the heart. Biol. Trace Elem. Res. 188, 160–176. doi:10.1007/s12011-018-1464-1
Wang, A. J., Tang, Y., Zhang, J., Wang, B. J., Xiao, M., Lu, G., et al. (2022a). Cardiac SIRT1 ameliorates doxorubicin-induced cardiotoxicity by targeting sestrin 2. Redox Biol. 52, 102310. doi:10.1016/j.redox.2022.102310
Wang, G. W., Klein, J. B., and Kang, Y. J. (2001). Metallothionein inhibits doxorubicin-induced mitochondrial cytochrome c release and caspase-3 activation in cardiomyocytes. J. Pharmacol. Exp. Ther. 298, 461–468.
Wang, L., Liu, Y., Du, T., Yang, H., Lei, L., Guo, M., et al. (2020a). ATF3 promotes erastin-induced ferroptosis by suppressing system Xc-. Cell Death Differ. 27, 662–675. doi:10.1038/s41418-019-0380-z
Wang, L., Yin, Y. L., Liu, X. Z., Shen, P., Zheng, Y. G., Lan, X. R., et al. (2020b). Current understanding of metal ions in the pathogenesis of Alzheimer's disease. Transl. Neurodegener. 9, 10. doi:10.1186/s40035-020-00189-z
Wang, X., Chen, X., Zhou, W., Men, H., Bao, T., Sun, Y., et al. (2022b). Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm. Sin. B 12, 708–722. doi:10.1016/j.apsb.2021.10.005
Wang, Y., Liao, J., Luo, Y., Li, M., Su, X., Yu, B., et al. (2023). Berberine alleviates doxorubicin-induced myocardial injury and fibrosis by eliminating oxidative stress and mitochondrial damage via promoting nrf-2 pathway activation. Int. J. Mol. Sci. 24, 3257. doi:10.3390/ijms24043257
Wang, Y., Mei, X., Yuan, J., Lu, W., Li, B., and Xu, D. (2015). Taurine zinc solid dispersions attenuate doxorubicin-induced hepatotoxicity and cardiotoxicity in rats. Toxicol. Appl. Pharmacol. 289, 1–11. doi:10.1016/j.taap.2015.08.017
Wang, Y., Yan, S., Liu, X., Deng, F., Wang, P., Yang, L., et al. (2022c). PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 29, 1982–1995. doi:10.1038/s41418-022-00990-5
Wang, Z., Yao, M., Jiang, L., Wang, L., Yang, Y., Wang, Q., et al. (2022d). Dexmedetomidine attenuates myocardial ischemia/reperfusion-induced ferroptosis via AMPK/GSK-3β/Nrf2 axis. Biomed. Pharmacother. 154, 113572. doi:10.1016/j.biopha.2022.113572
Weiss, G., Loyevsky, M., and Gordeuk, V. R. (1999). Dexrazoxane (ICRF-187). Gen. Pharmacol. 32, 155–158. doi:10.1016/s0306-3623(98)00100-1
Wu, L., Fan, Z., Gu, L., Liu, J., Cui, Z., Yu, B., et al. (2023). QiShenYiQi dripping pill alleviates myocardial ischemia-induced ferroptosis via improving mitochondrial dynamical homeostasis and biogenesis. J. Ethnopharmacol. 308, 116282. doi:10.1016/j.jep.2023.116282
Wu, R., Mei, X., Wang, J., Sun, W., Xue, T., Lin, C., et al. (2019). Zn(ii)-Curcumin supplementation alleviates gut dysbiosis and zinc dyshomeostasis during doxorubicin-induced cardiotoxicity in rats. Food Funct. 10, 5587–5604. doi:10.1039/c9fo01034c
Wu, X., Wang, L., Wang, K., Li, J., Chen, R., Wu, X., et al. (2022). ADAR2 increases in exercised heart and protects against myocardial infarction and doxorubicin-induced cardiotoxicity. Mol. Ther. 30, 400–414. doi:10.1016/j.ymthe.2021.07.004
Ye, Y., Chen, A., Li, L., Liang, Q., Wang, S., Dong, Q., et al. (2022). Repression of the antiporter SLC7A11/glutathione/glutathione peroxidase 4 axis drives ferroptosis of vascular smooth muscle cells to facilitate vascular calcification. Kidney Int. 102, 1259–1275. doi:10.1016/j.kint.2022.07.034
Yoshii, J., Yoshiji, H., Kuriyama, S., Ikenaka, Y., Noguchi, R., Okuda, H., et al. (2001). The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int. J. Cancer 94, 768–773. doi:10.1002/ijc.1537
Zaidi, Y., Arjmand, F., Zaidi, N., Usmani, J. A., Zubair, H., Akhtar, K., et al. (2014). A comprehensive biological insight of trinuclear copper(II)-tin(IV) chemotherapeutic anticancer drug entity: in vitro cytotoxicity and in vivo systemic toxicity studies. Metallomics 6, 1469–1479. doi:10.1039/c4mt00035h
Zhang, W. B., Lai, X., and Guo, X. F. (2021). Activation of Nrf2 by miR-152 inhibits doxorubicin-induced cardiotoxicity via attenuation of oxidative stress, inflammation, and apoptosis. Oxid. Med. Cell Longev. 2021, 8860883. doi:10.1155/2021/8860883
Zhang, J., Wang, X., Guan, B., Wang, X., An, X., Wang, T., et al. (2023a). Qing-Xin-Jie-Yu Granule inhibits ferroptosis and stabilizes atherosclerotic plaques by regulating the GPX4/xCT signaling pathway. J. Ethnopharmacol. 301, 115852. doi:10.1016/j.jep.2022.115852
Zhang, Y., Xu, J., Liu, C., Long, X., Zheng, M., He, J., et al. (2023b). Curative effect of zinc-selenium tea on rat's cardiotoxicity induced by long-term exposure to nonylphenol. Environ. Toxicol. 38, 101–114. doi:10.1002/tox.23667
Zhao, L., Zhou, X., Xie, F., Zhang, L., Yan, H., Huang, J., et al. (2022). Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. (Lond) 42, 88–116. doi:10.1002/cac2.12250
Zhao, X., Jin, Y., Li, L., Xu, L., Tang, Z., Qi, Y., et al. (2019). MicroRNA-128-3p aggravates doxorubicin-induced liver injury by promoting oxidative stress via targeting Sirtuin-1. Pharmacol. Res. 146, 104276. doi:10.1016/j.phrs.2019.104276
Zhu, J., Zhang, X., Xie, H., Wang, Y., Zhang, X., and Lin, Z. (2021). Cardiomyocyte Stim1 deficiency exacerbates doxorubicin cardiotoxicity by magnification of endoplasmic reticulum stress. J. Inflamm. Res. 14, 3945–3958. doi:10.2147/JIR.S304520
Keywords: doxorubicin, cardiotoxicity, iron, copper, zinc, calcium
Citation: Zhou N, Wei S, Sun T, Xie S, Liu J, Li W and Zhang B (2023) Recent progress in the role of endogenous metal ions in doxorubicin-induced cardiotoxicity. Front. Pharmacol. 14:1292088. doi: 10.3389/fphar.2023.1292088
Received: 11 September 2023; Accepted: 28 November 2023;
Published: 08 December 2023.
Edited by:
Irena Kostova, Medical University Sofia, BulgariaReviewed by:
Amany Mohammed Gad, Sinai University, EgyptCopyright © 2023 Zhou, Wei, Sun, Xie, Liu, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenqun Li, bGl3cTEyMDRAY3N1LmVkdS5jbg==; Bikui Zhang, NTA1OTk1QGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.