
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 13 October 2023
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1285754
This article is part of the Research Topic Reviews in Cardiovascular Pharmacology: 2023 View all 13 articles
 Karl Aramouni1†Roland K. Assaf1†
Karl Aramouni1†Roland K. Assaf1† Maria Azar1Karen Jabbour1Abdullah Shaito2Amirhossein Sahebkar3Assaad A. Eid1Manfredi Rizzo4
Maria Azar1Karen Jabbour1Abdullah Shaito2Amirhossein Sahebkar3Assaad A. Eid1Manfredi Rizzo4 Ali H. Eid5*
Ali H. Eid5*Atherosclerosis is a major instigator of cardiovascular disease (CVD) and a main cause of global morbidity and mortality. The high prevalence of CVD calls for urgent attention to possible preventive measures in order to curb its incidence. Traditional risk factors of atherosclerosis, like age, smoking, diabetes mellitus, dyslipidemia, hypertension and chronic inflammation, are under extensive investigation. However, these only account for around 50% of the etiology of atherosclerosis, mandating a search for different or overlooked risk factors. In this regard, chronic infections, by Helicobacter pylori for instance, are a primary candidate. H. pylori colonizes the gut and contributes to several gastrointestinal diseases, but, recently, the potential involvement of this bacterium in extra-gastric diseases including CVD has been under the spotlight. Indeed, H. pylori infection appears to stimulate foam cell formation as well as chronic immune responses that could upregulate key inflammatory mediators including cytokines, C-reactive protein, and lipoproteins. These factors are involved in the thickening of intima-media of carotid arteries (CIMT), a hallmark of atherosclerosis. Interestingly, H. pylori infection was found to increase (CIMT), which along with other evidence, could implicate H. pylori in the pathogenesis of atherosclerosis. Nevertheless, the involvement of H. pylori in CVD and atherosclerosis remains controversial as several studies report no connection between H. pylori and atherosclerosis. This review examines and critically discusses the evidence that argues for a potential role of this bacterium in atherogenesis. However, additional basic and clinical research studies are warranted to convincingly establish the association between H. pylori and atherosclerosis.
Atherosclerosis is precipitated by the pathological formation of plaques within blood vessels. These plaques are instigated by endothelium injury followed by infiltration of immune cells and proliferation of vascular smooth muscle cells. Thickening of the intima and hardening of the vessels then ensue, thus narrowing the vascular lumen which progresses into partial or complete obstruction of blood vessels. Atherosclerosis is a dominant cause and risk factor of cardiovascular disease (CVD), a major instigator of global mortality (Herrington et al., 2016; Libby et al., 2019). Some of the specific risk factors include age, smoking, diabetes mellitus, dyslipidemia, hypertension, and chronic inflammation (Siasos et al., 2014; Spence and Pilote, 2015; Aarabi et al., 2018; Libby et al., 2018; Yao et al., 2019; Lechner et al., 2020). These can account only for around 50% of the incidence of atherosclerosis (Herrington et al., 2016; Libby et al., 2019), making investigation into other risk factors of incidence a hot and rather attractive research area. In recent years, some attention has been given to newly identified potential atherosclerosis risk factors like chronic infections (Pothineni et al., 2017). Possible chronic infectious agents that have been reportedly linked to atherosclerosis include the infamous bacterium Helicobacter pylori (Karbasi-Afshar et al., 2015).
H. pylori is a gram-negative, microaerophilic, spiral-shaped bacterium. It is a key microorganism of the human microbiome and a colonizer of the gut of at least half of the world’s population (Eusebi et al., 2014; Mentis et al., 2015; Burucoa and Axon, 2017; Leja et al., 2019). In 1994, this bacterium was categorized as a human class I carcinogen by the World Health Organization (Wang, 2014). Not only that, but H. pylori infection is the major cause of several gastric diseases including acute and chronic active gastritis, chronic atrophic gastritis, peptic ulcer, gastric adenocarcinoma, and type B low-grade mucosa-associated lymphoid tissue lymphoma (Wang et al., 2014; Wroblewski and Peek, 2016; Chang et al., 2018; de Brito et al., 2019; Sugano, 2019; Ford et al., 2020). Importantly, H. pylori infection has been linked to more than 50 extra-gastrointestinal manifestations in a variety of medical specializations such as dermatology, endocrinology, hematology, and cardiology, among others (Campuzano-Maya, 2014; He et al., 2022). In particular, insulin resistance (Polyzos et al., 2011), liver disorders (Waluga et al., 2015), ventilator-associated pneumonia (Dadashi and Hosseinzadeh, 2018), osteoporosis (Mizuno et al., 2015), chronic kidney disease (Hata et al., 2021), hematological manifestations like iron deficiency and vitamin B12 (cobalamin) deficiency, hypertension, and atherosclerosis have been linked to H. pylori infection (Tsay and Hsu, 2018; Fang et al., 2019; Franceschi et al., 2019; Mansori et al., 2020a; Mansori et al., 2020b).
Involvement of H. pylori in this wide array of diseases may be partly related to the fact that this bacterial infection contributes not only to local inflammation but also to systemic inflammation (Tsay and Hsu, 2018; Fang et al., 2019; Franceschi et al., 2019; Mansori et al., 2020a; Mansori et al., 2020b). In turn, the latter can instigate several extra-gastrointestinal disorders such as metabolic syndrome, diabetes mellitus, insulin resistance, and CVD (Tsay and Hsu, 2018; Fang et al., 2019; Franceschi et al., 2019; Mansori et al., 2020a; Mansori et al., 2020b; Kountouras et al., 2021). Of particular interest, recent studies have found that H. pylori may be responsible for the initiation, progression, and complications of atherosclerotic plaque formation. Hence, this review was undertaken to critically examine the evidence concerning H. pylori chronic infection as a risk factor of atherosclerosis.
One early indication of atherosclerosis is thickening of the carotid artery. In atherosclerosis, this blood vessel narrows, and blood flow becomes compromised thereby increasing the risk of CVD. Indeed, CIMT, which measures the thickness of the inner two layers (the intima and media) of a carotid artery, is an early diagnostic tool of atherosclerosis as this thickening appears even in asymptomatic pre-atherosclerosis patients. Relatedly, several reports support a positive correlation between H. pylori infection and an increase of CIMT (Table 1). Indeed, CIMT values were significantly higher in H. pylori-infected versus non-infected subjects, and the levels of H. pylori-IgG were positively correlated with the increased CIMT measurements (Shan et al., 2018). Similar results were obtained in H. pylori-positive men, younger than 50 years of age, who showed a higher incidence of carotid atherosclerosis than non-H. pylori-infected counterparts (Zhang et al., 2019). In addition, subjects without carotid atherosclerosis, but with H. pylori infection, were found to have higher CIMT values than those free of the infection (Zhang et al., 2019). Importantly, a follow up of subjects with persistent H. pylori infection after 5 years revealed a significantly higher incidence of carotid atherosclerosis compared to subjects who were H. pylori negative (Zhang et al., 2019). This clearly shows a potentially causative effect of this bacterial infection with atherosclerosis. Indeed, a cross-sectional study shows a positive association between H. pylori infection, CIMT and carotid atherosclerosis, independent of classical risk factors (Zhang et al., 2021). In agreement with this, several meta-analyses show that H. pylori infection can significantly increase CIMT and lead to subclinical atherosclerosis (Wang et al., 2021b; Shi et al., 2022; Simon et al., 2022).
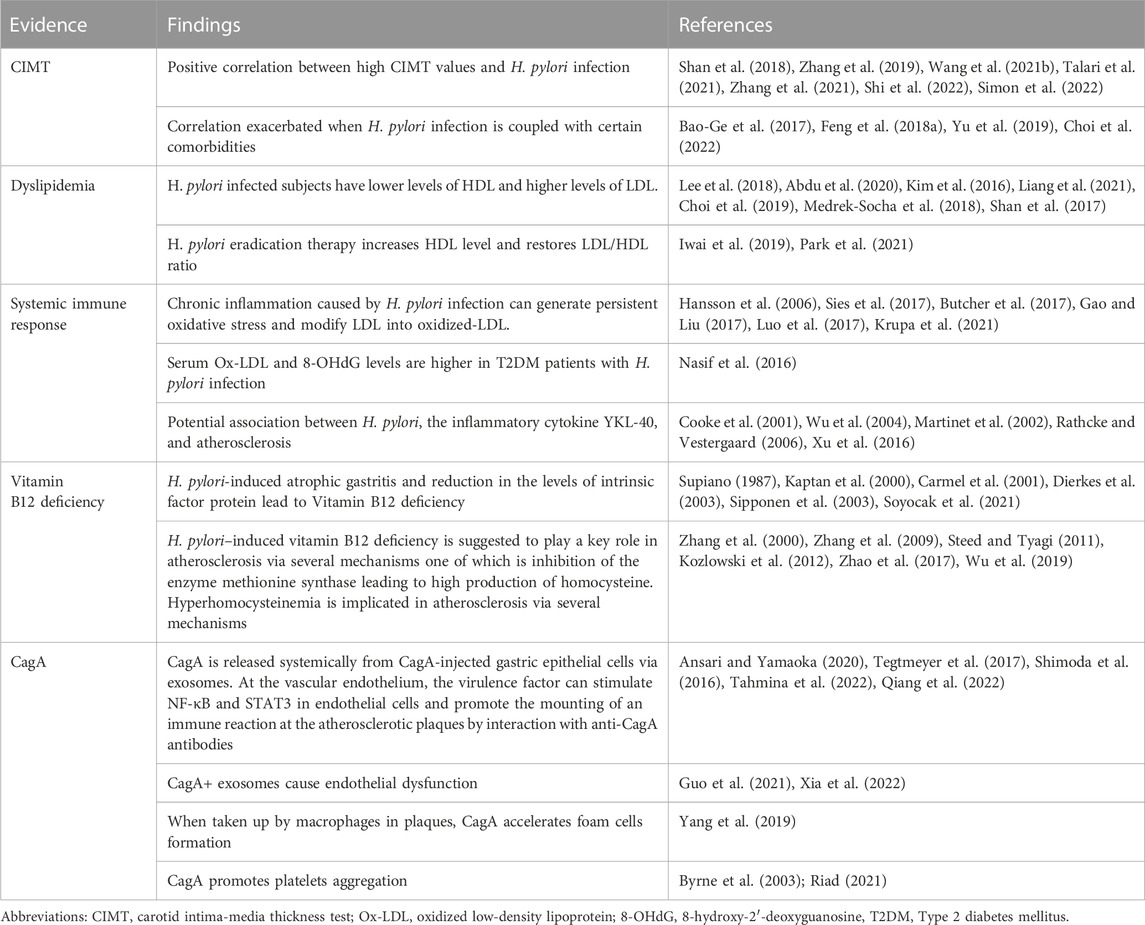
TABLE 1. Major evidence correlating H. pylori infection with atherosclerosis.
One of the key interplayers in the potential association between atherosclerosis and H. pylori infection is the cytotoxin-associated gene A (CagA). Indeed, studies involving both the right and left coronary artery of patients who underwent an upper GI endoscopy show higher CIMT values among patients infected with a H. pylori strain positive for CagA, compared to patients infected with CagA-negative H. pylori (Talari et al., 2021). The CagA-positive group also exhibited higher levels of high-sensitivity C-reactive protein (hsCRP), a marker of elevated inflammatory response (Talari et al., 2021). These studies led to the conclusion that CagA+ H. pylori strain can induce a systemic inflammatory response that may contribute to the development of atherosclerosis (Talari et al., 2021). Notably, changes in CIMT values were found to be even more prominent when H. pylori infection was coupled with certain comorbidities. For instance, CIMT values were highest among subjects with an H. pylori infection and alcoholic liver disease (ALD), compared to other groups with only one or neither of the conditions (Bao-Ge et al., 2017). Additionally, the coexistence of H. pylori infection and early-stage diabetic kidney disease (DKD) causes a significant increase in CIMT measurements, and potentiates the risk of developing atherosclerosis in type 2 diabetic patients (Feng et al., 2018a). In confirmation, two recent studies revealed that patients with both H. pylori infection and nonalcoholic fatty liver disease (NAFLD) exhibited the highest risk of carotid artery plaque formation and arterial stiffness (Yu et al., 2019; Choi et al., 2022). Taken together, these findings support the notion that H. pylori infection is associated with higher CIMT measurements, suggesting a correlation with the eventual incidence of atherosclerosis, in healthy individuals or ones with other co-morbidities.
Maintaining appropriate blood lipid profiles is critical for the prevention of atherosclerotic plaque buildup. Among serum lipids, cholesterol exhibits a high tendency to accumulate on vascular walls, potentially narrowing the lumen and obstructing blood flow. Interestingly, one of the major mechanisms by which H. pylori could precipitate the progression of atherosclerosis is by modifying serum lipid levels and profiles (Table 1) (Vijayvergiya and Vadivelu, 2015).
Current evidence suggests that H. pylori is responsible for an impairment in lipid metabolism (Adachi et al., 2018). A chronic infection with H. pylori can modify host body lipid distribution by activating pro-inflammatory factors, decreasing lipolysis, and enhancing de novo synthesis of fatty acids in the liver (Wang et al., 2022). H. pylori can also directly act on the liver to modify body lipid profiles by inducing liver dysfunction and elevating small intestinal mucosal permeability, facilitating the invasion of bacterial endotoxins to the liver through the portal vein, causing hepatic tissue damage (Wang et al., 2022). Importantly, H. pylori was reported as an independent risk factor for impaired lipid profiles, manifested as reduced high-density lipoprotein (HDL) and elevated low-density lipoprotein (LDL) levels (Buzás, 2014; Zhao et al., 2019; Hashim et al., 2022; Wang et al., 2022). It is not surprising then that dyslipidemia is prevalent in H. pylori-suspected patients where 87.2% of H. pylori positive subjects had at least one abnormality in lipid profile (Abdu et al., 2020). This argument is cemented by the finding that H. pylori-triggered deterioration in lipid metabolism or dyslipidemia is alleviated when this bacterium is eradicated (Park et al., 2021; Wang et al., 2022).
A recent study aiming to evaluate whether current H. pylori infection, detected using a rapid urease test [Campylobacter-like organism test (CLO)], is correlated with subclinical atherosclerosis showed that CLO-positive subjects are more likely to have significant coronary artery stenosis compared to CLO-negative subjects (Lee et al., 2018). The CLO-positive subjects also showed lower levels of HDL-cholesterol and higher mean levels of triglycerides compared to the CLO-negative subjects (Lee et al., 2018). The risk of having a coronary stenosis was even more prominent after adjusting for age, sex, and other factors that influence coronary artery stenosis, such as systolic blood pressure (BP), fasting glucose, HDL-cholesterol, anti-hypertension/diabetic medications, lipid-lowering agents, and antiplatelet agents (Lee et al., 2018). A recent cross-sectional study showed that H. pylori-infected patients exhibit higher LDL, triglycerides and cholesterol levels than control patients’ (Nigatie et al., 2022). This is further supported by other findings showing that H. pylori seropositivity is a significant risk factor for higher levels of LDL-cholesterol, triglycerides, BMI and lower levels of HDL-cholesterol, further establishing a role for H. pylori infection in dyslipidemia (Table 1) (Kim et al., 2016).
In asymptomatic healthy individuals, arterial stiffness, LDL-cholesterol levels, and the prevalence of dyslipidemia were significantly higher in the H. pylori-seropositive group compared to the H. pylori-seronegative group (Choi et al., 2019). In confirmation, findings from a recent study where participants were divided into three groups (healthy non-H. pylori infected, and symptomatic and asymptomatic H. pylori infected individuals), indicated that cholesterol levels were significantly higher in the symptomatic group than the asymptomatic and healthy groups (Medrek-Socha et al., 2018). Relevantly, both of the infected groups showed higher LDL and lower HDL concentrations than the healthy group (Medrek-Socha et al., 2018). Other supporting data include the finding that subjects infected by H. pylori, specifically males between 55 and 74 years of age, had significantly higher levels of LDL-cholesterol (Shan et al., 2017). A recent observational study in chronic gastritis patients showed that H. pylori eradication therapy causes a significant increase in HDL levels and a significant decrease of LDL/HDL ratio, a measure of the risk of atherosclerosis (Iwai et al., 2019). Taken together, it is becoming increasingly evident that infection with H. pylori is associated with dyslipidemia, a key contributing factor to atherosclerotic plaque formation.
Key parameters implicated in systemic inflammation are adhesion molecules and pro-inflammatory cytokines. In this context, it has been reported that H. pylori can induce upregulation of adhesion molecules on gastric epithelium as well as promote the release of several cytokines like IL-1, IL-6 and TNF -ɑ, which then activate leukocytes and precipitate systemic inflammation (Vijayvergiya and Vadivelu, 2015). This H. pylori-induced chronic inflammation can precipitate persistent oxidative stress, with various noxious effects such as DNA damage, mitochondrial membrane damage and pro-inflammatory immune responses (Butcher et al., 2017; Sies et al., 2017). This contributes to a wide range of extra-gastrointestinal tract abnormalities, including atherosclerosis.
Under normal oxidative conditions, LDL particles are taken up by macrophages via receptor-mediated endocytosis by the LDL-Receptors (LDL-Rs). LDL-Rs can be downregulated by elevated levels of cholesterol, limiting the uptake of fat. However, LDL particles are extremely sensitive to oxidative damage and can be modified into oxidized-LDL (Ox-LDL) during chronic H. pylori infection, for instance. Ox-LDL are atherogenic because they do not bind to LDL-Rs but have the capacity to bind and activate a group of receptors collectively known as scavenger receptors (SRs). These SRs are present on macrophages, but unlike LDL-Rs they are not downregulated by high levels of cholesterol, and can keep on accumulating cholesterol and fats, eventually causing the conversion of macrophages into foam cells, a hallmark of atherosclerosis (Figure 1) (Gao and Liu, 2017; Luo et al., 2017). When lipoproteins accumulate in the intima, the endothelium becomes poised to secrete a repertoire of chemokines and display several leukocyte adhesion molecules, favoring the recruitment of immune cells. An immune response is then mounted, leading to the accumulation of pro-inflammatory cytokines, proteases, and vasoactive molecules. This local inflammation can exacerbate plaque growth in blood vessels, furthering their obstruction (Hansson et al., 2006).
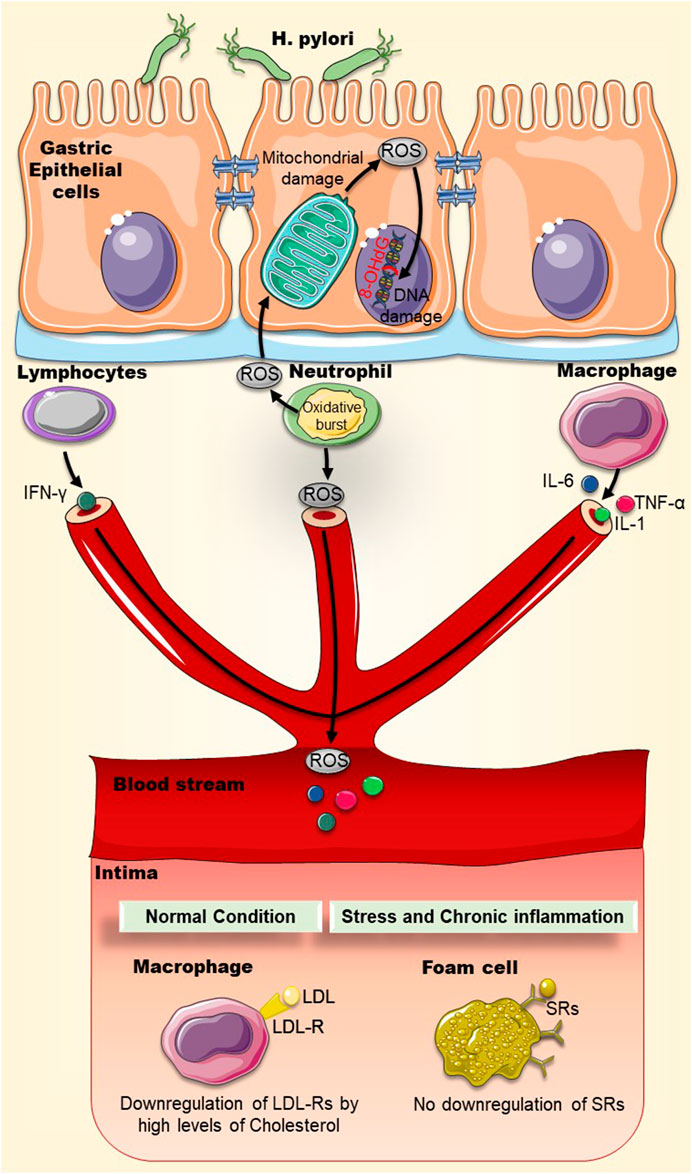
FIGURE 1. Chronic inflammation and oxidative stress may increase the uptake of oxidized-LDL particles by scavenger receptors on macrophages. Chronic H. pylori infection can cause local and systemic inflammation that may accelerate atherosclerotic plaque formation. Oxidative stress (ROS) and inflammatory markers (IL-1, IL-6, TNF-α, IFN-γ) generated by immune cells and mitochondrial membrane disruption can damage DNA where deoxyguanosine becomes oxidized into 8-OHdG. LDL particles are modified by oxidative stress into oxidized-LDL (Ox-LDL). Ox-LDL particles do not bind to LDL-Rs but rather bind to scavenger receptors (SRs), present on macrophages. Unlike LDL-Rs, SRs are not downregulated by high levels of cholesterol and accumulate more cholesterol and fats into macrophages, causing the eventual conversion of macrophages into foam cells. Abbreviations: LDL: Low-density lipoprotein; Ox-LDL: Oxidized low-density lipoprotein; LDL-R: low-density lipoprotein receptor; ROS: Reactive oxygen species; SRs: Scavenger receptors; 8-OHdG: 8-hydroxy-2′-deoxyguanosine.
Ox-LDL levels were also found to be higher in serum of H. pylori–infected type II diabetes mellitus (T2DM) patients than in serum of diabetic patients without H. pylori infection or non-diabetics (Table 1) (Nasif et al., 2016). More, the role of H. pylori infection in accelerating foam cell formation in animals with high-fat diet has also been reported (Krupa et al., 2021). In these animals, a dramatic increase in systemic inflammatory markers fueling a pro-atherogenic endothelial cell environment was noted. Infiltration of inflammatory cells, elevation of oxidative stress, formation of foam cells, and presence of pro-atherogenic molecules like 7-ketocholesterol and aldehydes were prominently evident (Krupa et al., 2021).
Oxidative damage also impacts both nuclear and mitochondrial DNA. For instance, deoxyguanosine becomes oxidized into 8-hydroxy-2′-deoxyguanosine (8-OHdG) (Cooke et al., 2001). Indeed, this oxidized DNA molecule is often used as a marker of oxidative damage. Moreover, it is reportedly found-in rather excessive amounts-in atherosclerotic plaques (Wu et al., 2004), inside macrophages, smooth muscle cells, and endothelial cells, demonstrating strong oxidative DNA damage and repair (Martinet et al., 2002). Likewise, levels of 8-OHdG and serum Ox-LDL levels in T2DM patients appear to be directly associated with H. pylori infection (Nasif et al., 2016). This lends support to the notion that H. pylori infection contributes to the pathogenesis of atherosclerosis by virtue of increasing serum Ox-LDL and 8-OHdG (Figure 1).
An interesting relation between vascular dementia, inflammation and atherosclerosis is emerging. Indeed, H. pylori-positive patients with vascular dementia (VD) had greater CIMT values and higher levels of YKL-40 cytokine [a biomarker of inflammation (Rathcke and Vestergaard, 2006)] than H. pylori-negative VD patients (Xu et al., 2016). Additionally, CIMT was positively correlated with serum levels of YKL-40 cytokine independent of traditional atherosclerotic risk factors (Xu et al., 2016). This finding suggests a potential association between H. pylori, YKL-40 cytokines and atherosclerosis, warranting further investigation (Xu et al., 2016). Overall, H. pylori infection-induced inflammation may be able to precipitate the development of atherosclerosis as summarized in Table 1.
Atrophic gastritis is chronic inflammation of the gastric mucosa that can result in stomach atrophy along with low or absent gastric acid secretion and inadequate production of intrinsic factor (IF). This factor is a protein that binds and facilitates the transit of vitamin B12 (cobalamin) through the small intestine to be absorbed into the bloodstream (Hall and Appelman, 2019). In the absence of gastric secretions or IF, the absorption of food-bound vitamin B12 is impaired (Neumann et al., 2013). As a result, people suffering from chronic atrophic gastritis have been documented to suffer from cobalamin deficiency (Allen, 2008). Of particular interest to this review, an untreated H. pylori infection can eventually cause atrophic gastritis, implicating H. pylori in vitamin B12 deficiency (Supiano, 1987). This causality was investigated in several studies, with the conclusion that there is a probable association between cobalamin deficiency and H. pylori infection (Kaptan et al., 2000; Carmel et al., 2001; Dierkes et al., 2003; Sipponen et al., 2003; Raut and Chandel, 2014; Civan, 2020; Soyocak et al., 2021). These findings have potential implications to atherosclerosis because vitamin B12 malabsorption can promote the progression of atherosclerosis (Zhao et al., 2017). In fact, cobalamin is a cofactor for the enzyme methionine synthase catalyzing the conversion of homocysteine to methionine (Kozlowski et al., 2012). In the case of vitamin B12 deficiency, methionine synthase is non-functional, escalating homocysteine levels (Sipponen et al., 2003). A recent study comparing homocysteine levels of H. pylori positive subjects to those of H. pylori negative individuals found that homocysteine levels were significantly higher in the infected subjects (Huang et al., 2022).
Hyperhomocysteinemia (HHcy) is suggested to play a key role in atherosclerosis via several mechanisms (Zhao et al., 2017). The auto-oxidation of homocysteine is a source of reactive oxygen species (ROS), which at high levels in the vasculature can cause lipid peroxidation, protein oxidation, and even cell death, eventually leading to vascular injuries (Steed and Tyagi, 2011). HHcy by itself as well as HHcy-generated ROS can induce peroxynitrite (ONOO−) formation, effectively reducing nitric oxide (NO) bioavailability in the vasculature. Generation of ONOO− and the decrease of NO levels can cause dysfunction of the vascular endothelium, instigating atherosclerosis and other diseases of the vasculature (Zhang et al., 2000). HHcy-induced oxidative stress can as well affect the endoplasmic reticulum of endothelial cells, thereby inducing apoptosis and inflammation, and dysregulation of lipid metabolism (Wu et al., 2019). Furthermore, elevated levels of homocysteine can induce the activation of nuclear factor kappa B (NF-κB) transcription factor which activates the expression of different cytokines, chemokines, and leukocyte adhesion molecules; important mediators of vascular inflammation, pro-thrombotic state, and atherogenesis (Figure 2) (Cattaneo, 1999; Zhang et al., 2009; Riad, 2021). Taken together, it is becoming more evident that H. pylori-induced vitamin B12 malabsorption can lead to elevated levels of homocysteine, which may then promote the development of atherosclerosis by various mechanisms (Figure 2).
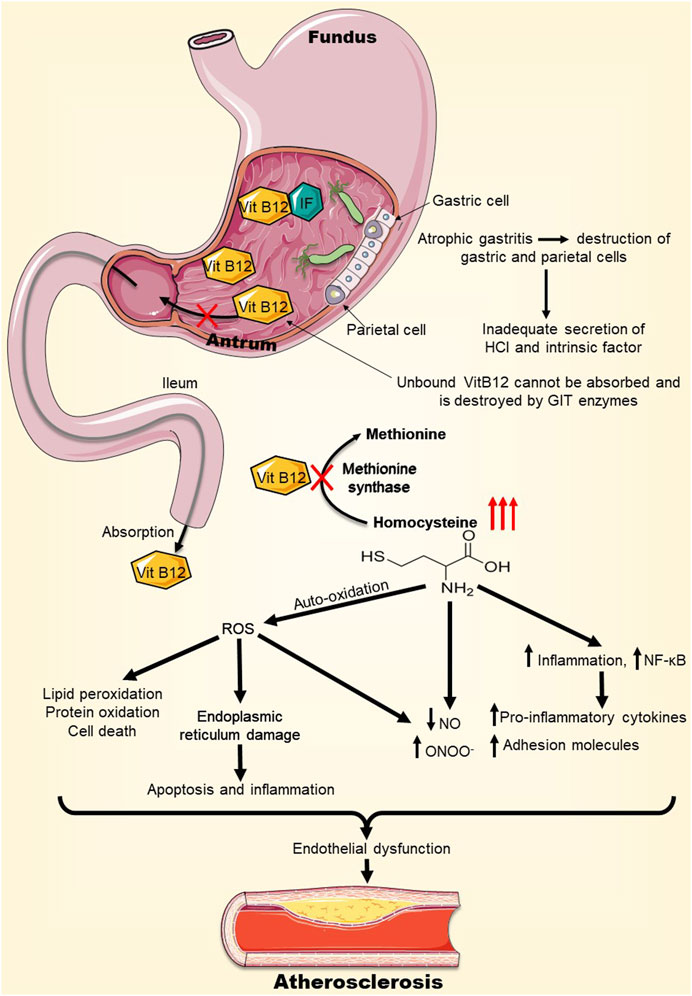
FIGURE 2. H. pylori-induced Vitamin B12 deficiency may precipitate atherosclerosis. H. pylori infection can lead to atrophic gastritis and destruction of parietal cells, which secrete intrinsic factor (IF), essential for vitamin B12 absorption. As a result, H. pylori infection can cause vitamin B12 deficiency. Vitamin B12 deficiency inhibits the enzyme methionine synthase, escalating homocysteine levels. High levels of homocysteine in the vasculature can lead to endoplasmic reticulum stress, oxidative stress, and inflammation, overall causing endothelial dysfunction which promotes atherosclerosis. Abbreviations: IF, Intrinsic Factor; ROS, Reactive oxygen species; NO, Nitric oxide; ONOO−, Peroxynitrite; NF-kB, Nuclear factor Kappa B.
CagA, a virulence factor protein expressed by certain strains of H. pylori, is responsible for the development of gastric diseases and is suspected to be involved in extra-gastric diseases (Ansari and Yamaoka, 2020). During infection, H. pylori injects this cytotoxin into host gastric cells, through a Type IV secretion system, where CagA can modify host cell signaling pathways (Tegtmeyer et al., 2017). H. pylori-infected gastric epithelial cells were found to release extracellular vesicles (exosomes) containing CagA. These exosomes can be delivered, through the bloodstream, to vascular endothelia distant from the primary site of infection (Table 1) (Shimoda et al., 2016). This finding may be one way to explain the systemic clinical effects thought to be mediated by H. pylori, including atherosclerosis. Indeed, these circulating CagA-containing exosomes may adhere to distant endothelial monolayers and deliver the CagA protein into endothelial cells, where it can stimulate NF-κB signaling. This signaling may lead to a chronic inflammatory condition, endothelial cell dysfunction, and consequently plaque formation. Similarly, Wang et al. reported that extracellular vesicle (outer membrane vesicles) derived from CagA+ H.pylori-infected cells accelerated atherosclerotic plaque formation in ApoE−/− mice via NF-κB signaling (Figure 3) (Wang et al., 2021a). Exosomes-delivered CagA to endothelial cells were also found to activate the pro-inflammatory transcription factor STAT3 (Tahmina et al., 2022). In another mechanism, CagA-containing exosomes may directly fuse with cells of the atherosclerotic plaques, exposing CagA on the plaque surface to be recognized by anti-CagA antibodies, leading to immune reactions at the atherosclerotic plaque (Figure 3) (Shimoda et al., 2016).
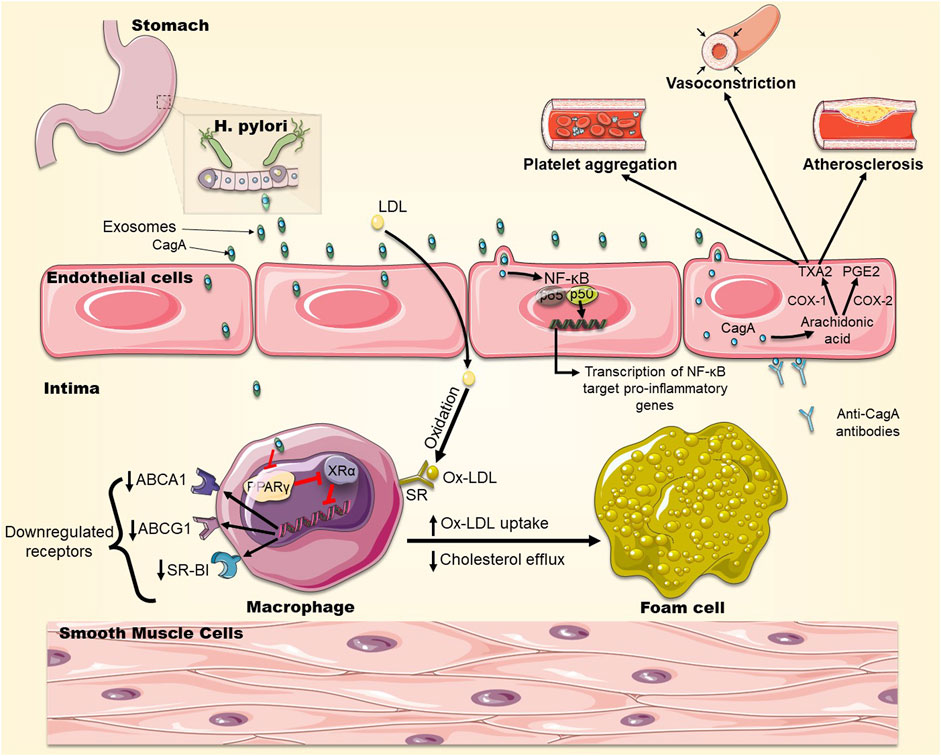
FIGURE 3. The role of CagA in foam cell formation. CagA is released in exosomes from H. pylori-infected gastric cells and becomes engulfed by macrophages in the intima of arteries. CagA downregulates the expression of transcription factors PPARγ and LXRα, which control the expression of the cholesterol efflux transporters ABCA1, ABCG1 and SR-BI on the surface of macrophages leading to cholesterol accumulation in macrophages and promoting foam cell formation. In addition, OxLDL infiltrates macrophages via SRs which are not downregulated by cholesterol, exacerbating foam cell formation. CagA also stimulates NF-kB signaling leading to the upregulation of expression of pro-inflammatory genes. CagA also elevates the levels of COX-1 and COX-2, leading to a higher production of TXA2 and PGE2, promoting platelets aggregation and vasoconstriction. CagA can be expressed on cells of the atherosclerotic plaque, to which anti-CagA antibodies will bind, exacerbating immune reactions.
PMSS1 is a CagA-positive H. pylori strain which can secrete the CagA virulence factor into host cells. PMSS1 H. pylori-infected mice have shown accelerated growth of atherosclerotic plaques that was dependent on dietary risk factors and genetic susceptibility (Yang et al., 2019). Immunohistochemical analysis of the vasculature of these mice revealed that the atherosclerotic plaques contained macrophage-derived foam cells. These cells significantly accumulate neutral lipid in the presence of PMSS1 infection, suggesting that the bacteria may help in the formation of foam cells (Yang et al., 2019).
This notion was reinforced by other in vitro and in vivo studies. Exosomes derived from H. pylori-infected gastric epithelial cells (Hp-GEC-EVs), were able to induce conversion of macrophages into foam cells when taken up by macrophages. Interestingly, CagA present in Hp-GEC-EVs would end up in the atherosclerotic plaques leading to exacerbation of the obstructive inflammatory process (Yang et al., 2019). It was also found that CagA mediated the formation of foam cells through downregulation of the expression of transcription factors PPARγ and LXRα, which control the expression of the cholesterol efflux transporters ABCA1, ABCG1 and SR-BI on the surface of macrophages, leading to cholesterol accumulation in macrophages and promoting foam cell formation (Figure 3) (Yang et al., 2019). A recent study comparing the virulence of CagA+ H. pylori to CagA− H. pylori in mice found that the former strain colonized gastric mucosa more effectively, causing endothelial dysfunction and enhancing atherosclerosis through ROS production, which was induced by CagA+ exosomes (Xia et al., 2022). Another study demonstrated that H. pylori infection was an independent risk factor for intracranial atherosclerosis, especially in women younger than 60 years of age, and that CagA+ exosomes significantly limited brain endothelial functioning in vitro (Guo et al., 2021). As well, a meta-analysis showed that H. pylori infection can significantly increase CIMT, especially in CagA+ individuals (Shi et al., 2022).
CagA-positive H. pylori strains can also promote platelet aggregation. Indeed, CagA is suspected to increase cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) production in the vascular endothelium resulting in higher thromboxane (TXA2) and prostaglandins levels, both of which are known to modify platelet aggregation and contribute to atherosclerosis (Figure 3) (Byrne et al., 2003; Riad, 2021).
Hence, CagA+ H. pylori infection may accelerate atherosclerotic plaque formation via several CagA-dependent mechanisms: CagA packaged in exosomes can modify macrophage intracellular cholesterol levels by increasing the import of cholesterol or decreasing cholesterol efflux which facilitates foam cell formation. Otherwise, CagA can promote the development of atherosclerotic plaques via the stimulation of NF-κB, platelet aggregation, and the interaction with anti-CagA antibodies (Figure 3).
Despite the multitude of studies demonstrating a positive association between H. pylori infection and atherosclerosis, some studies report otherwise (Ahmadnia et al., 2013; Jukic et al., 2017; Feng et al., 2018b; Wernly et al., 2022). A recent cross-sectional study aiming to examine the relationship between H. pylori infection and the severity of coronary atherosclerosis included patients with coronary artery disease (CAD) and who underwent coronary artery bypass grafting surgery. Coronary angiograms were scored by quantitative assessment based on three angiographic parameters: the vessel score, Gensini score, and angiographic severity score. The results showed that hypertension, systolic pressure, diastolic pressure, and total cholesterol values were higher in H. pylori-positive subjects compared to H. pylori-negative subjects. HDL-cholesterol values, on the other hand, were markedly lower in the H. pylori-positive than in the negative subjects. However, no significant differences were found in vessel score, Gensini score and angiographic severity score, or in the use of medications that affect atherogenesis, concluding that the pathogenesis of stable chronic CAD is mainly caused by traditional risk factors rather than the influence of H. pylori infection (Jukic et al., 2017). Furthermore, a cross-sectional investigating the correlation between the average of CIMT values and H. pylori infection did not report significant differences in CIMT measurements between H. pylori positive and negative subjects (Feng et al., 2018b). However, traditional atherosclerosis risk factors (like gender, BMI, waistline, lipid profiles) did correlate with significant differences in CIMT scores suggesting no correlation between H. pylori infection and early atherosclerosis (Feng et al., 2018b). Moreover, the presence of H. pylori in atherosclerotic plaques was investigated in the iliac arteries of 25 patients with end stage renal disease (ESRD) who were undergoing kidney transplantation. Although atherosclerotic plaques were present in 21 patients (84%), H. pylori was not detected by polymerase chain reaction and no significant relationship between atherosclerosis and gastric H. pylori infection could be established (Ahmadnia et al., 2013). Moreover, despite reporting an independent risk between H. pylori infection and CVD, the difference did not translate into CVD mortality between patients with and without the bacteria in a cohort (Wernly et al., 2022). Hence, conflicting data exist regarding the relationship between H. pylori and atherosclerosis and further investigations are needed in order to elucidate this relationship.
The extra-gastrointestinal manifestations of H. pylori infection have linked this pathogen to the progression of atherosclerosis. In addition, the bacteria may account for the development of atherosclerotic plaques through several mechanisms including dyslipidemia, systemic inflammation, oxidative stress and acceleration of foam cell formation. When different strains of the bacteria were compared, H. pylori strains positive for CagA showed a higher correlation with the incidence and pathogenesis of atherosclerosis, through various mechanisms involving exosomes, for example, (Figure 4). As such, interventions to eradicate H. pylori may help in the reduction of the prevalence of atherosclerosis. Paradoxically, some studies could not establish a role for H. pylori in atherosclerosis (Ahmadnia et al., 2013; Jukic et al., 2017; Feng et al., 2018b). The contradiction may be related to the presence of confounding variables, such as the methods used for diagnosis of H. pylori diagnostic (serology vs. rapid urease test), selection bias of geographic regions, or the study of different H. pylori strains, like CagA-positive or CagA-negative strains. In conclusion, H. pylori may exacerbate atherosclerosis, but further supporting evidence is warranted to place it alongside the traditional risk factors of atherosclerosis.
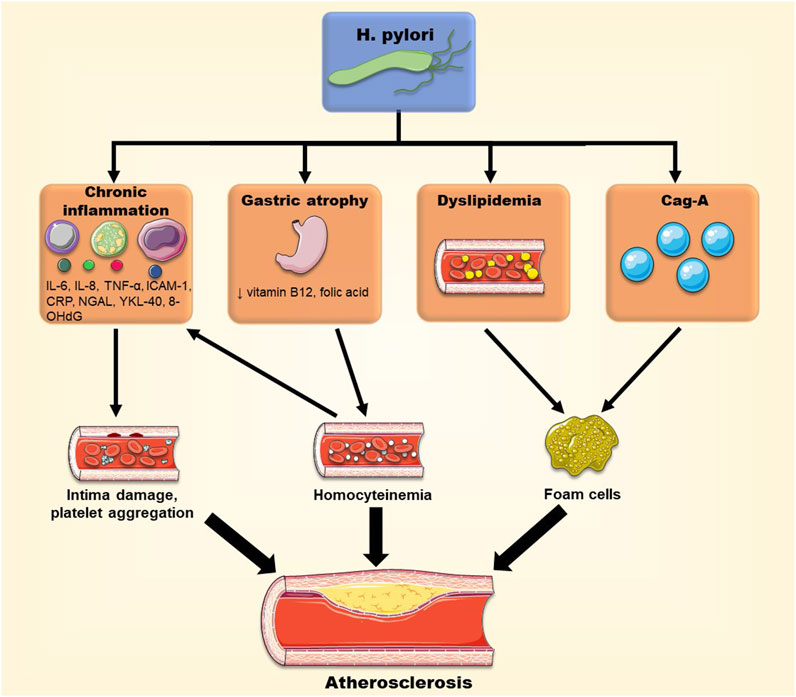
FIGURE 4. Different pathways through which H. pylori may promote atherosclerotic plaque formation. H. pylori infection can have systemic effects and cause chronic inflammation, dyslipidemia, Vitamin B12 deficiency and homocysteinemia, and promote foam cell formation, contributing to the progression of atherosclerosis.
KA: Writing–original draft. RA: Writing–original draft. MA: Writing–original draft. KJ: Writing–original draft. AbS: Writing: review and editing. AmS: Writing: review and editing. AAE: Writing: review and editing. MR: Writing: review and editing. AHE: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Open Access fees provided by the Qatar National Library.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Cag-A, Cytotoxin-Associated Gene; IL-6, Interleukin 6; IL-8, Interleukin 8; TNF-α, Tumor necrosis factor α; ICAM-1, Intercellular adhesion molecule 1; CRP, C-Reactive Protein; NGAL, neutrophil gelatinase-associated lipocalin; 8-OHdG, 8-hydroxy-2′-deoxyguanosine.
Aarabi, G., Heydecke, G., and Seedorf, U. (2018). Roles of oral infections in the pathomechanism of atherosclerosis. Int. J. Mol. Sci. 19 (7), 1978. doi:10.3390/ijms19071978
Abdu, A., Cheneke, W., Adem, M., Belete, R., and Getachew, A. (2020). Dyslipidemia and associated factors among patients suspected to have Helicobacter pylori infection at jimma university medical center, jimma, Ethiopia. Int. J. Gen. Med. 13, 311–321. doi:10.2147/IJGM.S243848
Adachi, K., Mishiro, T., Toda, T., Kano, N., Fujihara, H., Mishima, Y., et al. (2018). Effects of Helicobacter pylori eradication on serum lipid levels. J. Clin. Biochem. Nutr. 62 (3), 264–269. doi:10.3164/jcbn.17-88
Ahmadnia, H., Vossoughinia, H., Mansourian, E., and Gaffarzadegan, K. (2013). No detection of Helicobacter pylori in atherosclerotic plaques in end stage renal disease patients undergoing kidney transplantation. Indian J. Nephrol. 23 (4), 259–263. doi:10.4103/0971-4065.114483
Allen, L. H. (2008). Causes of vitamin B12 and folate deficiency. Food Nutr. Bull. 29 (2), S20–S34. doi:10.1177/15648265080292S105
Ansari, S., and Yamaoka, Y. (2020). Helicobacter pylori virulence factor cytotoxin-associated gene A (CagA)-Mediated gastric pathogenicity. Int. J. Mol. Sci. 21 (19), 7430. doi:10.3390/ijms21197430
Bao-Ge, Q., Hui, W., Yi-Guo, J., Ji-Liang, S., Zhong-Dong, W., Ya-Fei, W., et al. (2017). The correlation and risk factors between carotid intima-media thickening and alcoholic liver disease coupled with Helicobacter pylori infection. Sci. Rep. 7, 43059. doi:10.1038/srep43059
Burucoa, C., and Axon, A. (2017). Epidemiology of Helicobacter pylori infection. Helicobacter 22 (1). doi:10.1111/hel.12403
Butcher, L. D., den Hartog, G., Ernst, P. B., and Crowe, S. E. (2017). Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol. Gastroenterol. Hepatol. 3 (3), 316–322. doi:10.1016/j.jcmgh.2017.02.002
Buzás, G. M. (2014). Metabolic consequences of Helicobacter pylori infection and eradication. World J. Gastroenterol. 20 (18), 5226–5234. doi:10.3748/wjg.v20.i18.5226
Byrne, M. F., Murphy, J. F., Corcoran, P. A., Atherton, J. C., Sheehan, K. M., Cox, D., et al. (2003). Helicobacter pylori induces cyclooxygenase-1 and cyclooxygenase-2 expression in vascular endothelial cells. Scand. J. Gastroenterol. 38 (10), 1023–1030. doi:10.1080/00365520310005622
Campuzano-Maya, G. (2014). Hematologic manifestations of Helicobacter pylori infection. World J. Gastroenterol. 20 (36), 12818–12838. doi:10.3748/wjg.v20.i36.12818
Carmel, R., Aurangzeb, I., and Qian, D. (2001). Associations of food-cobalamin malabsorption with ethnic origin, age, Helicobacter pylori infection, and serum markers of gastritis. Am. J. Gastroenterol. 96 (1), 63–70. doi:10.1111/j.1572-0241.2001.03453.x
Cattaneo, M. (1999). Hyperhomocysteinemia, atherosclerosis and thrombosis. Thromb. Haemost. 81 (2), 165–176. doi:10.1055/s-0037-1614438
Chang, W. L., Yeh, Y. C., and Sheu, B. S. (2018). The impacts of H. pylori virulence factors on the development of gastroduodenal diseases. J. Biomed. Sci. 25 (1), 68. doi:10.1186/s12929-018-0466-9
Choi, J. M., Lim, S. H., Han, Y. M., Lee, H., Seo, J. Y., Park, H. E., et al. (2019). Association between Helicobacter pylori infection and arterial stiffness: Results from a large cross-sectional study. PLoS One 14 (8), e0221643. doi:10.1371/journal.pone.0221643
Choi, J. M., Park, H. E., Han, Y. M., Lee, J., Lee, H., Chung, S. J., et al. (2022). Non-alcoholic/Metabolic-Associated fatty liver disease and Helicobacter pylori additively increase the risk of arterial stiffness. Front. Med. (Lausanne) 9, 844954. doi:10.3389/fmed.2022.844954
Civan, H. A. (2020). Vitamin B12 status in children with Helicobacter pylori gastritis. Med. J. Bakirkoy 16 (2). doi:10.5222/BMJ.2020.03522
Cooke, M. S., Evans, M. D., Burd, R. M., Patel, K., Barnard, A., Lunec, J., et al. (2001). Induction and excretion of ultraviolet-induced 8-oxo-2'-deoxyguanosine and thymine dimers in vivo: Implications for PUVA. J. Invest. Dermatol 116 (2), 281–285. doi:10.1046/j.1523-1747.2001.01251.x
Dadashi, A., and Hosseinzadeh, N. (2018). High seroprevalence of anti-Helicobacter pylori antibodies in patients with ventilator-associated pneumonia. J. Res. Med. Sci. 23, 79. doi:10.4103/jrms.JRMS_117_18
de Brito, B. B., da Silva, F. A. F., Soares, A. S., Pereira, V. A., Santos, M. L. C., Sampaio, M. M., et al. (2019). Pathogenesis and clinical management of Helicobacter pylori gastric infection. World J. Gastroenterol. 25 (37), 5578–5589. doi:10.3748/wjg.v25.i37.5578
Dierkes, J., Ebert, M., Malfertheiner, P., and Luley, C. (2003). Helicobacter pylori infection, vitamin B12 and homocysteine. A review. Dig. Dis. 21 (3), 237–244. doi:10.1159/000073341
Eusebi, L. H., Zagari, R. M., and Bazzoli, F. (2014). Epidemiology of Helicobacter pylori infection. Helicobacter 19 (1), 1–5. doi:10.1111/hel.12165
Fang, Y., Fan, C., and Xie, H. (2019). Effect of Helicobacter pylori infection on the risk of acute coronary syndrome: A systematic review and meta-analysis. Med. Baltim. 98 (50), e18348. doi:10.1097/MD.0000000000018348
Feng, L., Deng, C., and Li, Y. (2018a). Assessment of the relationship between carotid intima-media thickening and early-stage diabetic kidney disease coupled with Helicobacter pylori infection. Dis. Markers 2018, 3793768. doi:10.1155/2018/3793768
Feng, Y., Zhou, W., Luo, L., and Xu, W. (2018b). Helicobacter pylori infection is not related to increased carotid intima-media thickness in general population. Sci. Rep. 8 (1), 14180. doi:10.1038/s41598-018-32465-4
Ford, A. C., Yuan, Y., Forman, D., Hunt, R., and Moayyedi, P. (2020). Helicobacter pylori eradication for the prevention of gastric neoplasia. Cochrane Database Syst. Rev. 7, CD005583. doi:10.1002/14651858.CD005583.pub3
Franceschi, F., Covino, M., and Roubaud Baudron, C. (2019). Review: Helicobacter pylori and extragastric diseases. Helicobacter 24 (1), e12636. doi:10.1111/hel.12636
Gao, S., and Liu, J. (2017). Association between circulating oxidized low-density lipoprotein and atherosclerotic cardiovascular disease. Chronic Dis. Transl. Med. 3 (2), 89–94. doi:10.1016/j.cdtm.2017.02.008
Guo, Y., Xu, C., Zhang, L., Chen, Z., and Xia, X. (2021). Helicobacter pylori infection acts as an independent risk factor for intracranial atherosclerosis in women less than 60 Years old. Front. Cardiovasc Med. 8, 819315. doi:10.3389/fcvm.2021.819315
Hall, S. N., and Appelman, H. D. (2019). Autoimmune gastritis. Arch. Pathol. Lab. Med. 143 (11), 1327–1331. doi:10.5858/arpa.2019-0345-RA
Hansson, G. K., Robertson, A. K., and Soderberg-Naucler, C. (2006). Inflammation and atherosclerosis. Annu. Rev. Pathol. 1, 297–329. doi:10.1146/annurev.pathol.1.110304.100100
Hashim, M., Mohammed, O., Egzeabeher, T., and Wolde, M. (2022). The association of Helicobacter Pylori infection with dyslipidaemia and other atherogenic factors in dyspeptic patients at St. Paul's Hospital Millennium Medical College. Heliyon 8 (5), e09430. doi:10.1016/j.heliyon.2022.e09430
Hata, K., Koyama, T., Ozaki, E., Kuriyama, N., Mizuno, S., Matsui, D., et al. (2021). Assessing the relationship between Helicobacter pylori and chronic kidney disease. Healthc. (Basel) 9 (2), 162. doi:10.3390/healthcare9020162
He, J., Liu, Y., Ouyang, Q., Li, R., Li, J., Chen, W., et al. (2022). Helicobacter pylori and unignorable extragastric diseases: Mechanism and implications. Front. Microbiol. 13, 972777. doi:10.3389/fmicb.2022.972777
Herrington, W., Lacey, B., Sherliker, P., Armitage, J., and Lewington, S. (2016). Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ. Res. 118 (4), 535–546. doi:10.1161/CIRCRESAHA.115.307611
Huang, X., Qiu, Y., Zheng, X., Luo, M., Wang, Y., Yi, J., et al. (2022). Correlation of Helicobacter pylori infection with atherosclerosis and its risk factors. doi:10.21203/rs.3.rs-1929881/v1
Iwai, N., Okuda, T., Oka, K., Hara, T., Inada, Y., Tsuji, T., et al. (2019). Helicobacter pylori eradication increases the serum high density lipoprotein cholesterol level in the infected patients with chronic gastritis: A single-center observational study. PLoS One 14 (8), e0221349. doi:10.1371/journal.pone.0221349
Jukic, A., Bozic, D., Kardum, D., Becic, T., Luksic, B., Vrsalovic, M., et al. (2017). Helicobacter pylori infection and severity of coronary atherosclerosis in patients with chronic coronary artery disease. Ther. Clin. Risk Manag. 13, 933–938. doi:10.2147/tcrm.S142193
Kaptan, K., Beyan, C., Ural, A. U., Cetin, T., Avcu, F., Gulsen, M., et al. (2000). Helicobacter pylori-is it a novel causative agent in Vitamin B12 deficiency? Arch. Intern Med. 160 (9), 1349–1353. doi:10.1001/archinte.160.9.1349
Karbasi-Afshar, R., Khedmat, H., and Izadi, M. (2015). Helicobacter pylori infection and atherosclerosis: a systematic review. Acta Med. Iran. 53 (2), 78–88.
Kim, T. J., Lee, H., Kang, M., Kim, J. E., Choi, Y. H., Min, Y. W., et al. (2016). Helicobacter pylori is associated with dyslipidemia but not with other risk factors of cardiovascular disease. Sci. Rep. 6, 38015. doi:10.1038/srep38015
Kountouras, J., Papaefthymiou, A., Polyzos, S. A., Deretzi, G., Vardaka, E., Soteriades, E. S., et al. (2021). Impact of Helicobacter pylori-related metabolic syndrome parameters on arterial hypertension. Microorganisms 9 (11), 2351. doi:10.3390/microorganisms9112351
Kozlowski, P. M., Kamachi, T., Kumar, M., and Yoshizawa, K. (2012). Reductive elimination pathway for homocysteine to methionine conversion in cobalamin-dependent methionine synthase. J. Biol. Inorg. Chem. 17 (4), 611–619. doi:10.1007/s00775-012-0881-4
Krupa, A., Gonciarz, W., Rusek-Wala, P., Rechciński, T., Gajewski, A., Samsel, Z., et al. (2021). Helicobacter pylori infection acts synergistically with a high-fat diet in the development of a proinflammatory and potentially proatherogenic endothelial cell environment in an experimental model. Int. J. Mol. Sci. 22 (7), 3394. doi:10.3390/ijms22073394
Lechner, K., von Schacky, C., McKenzie, A. L., Worm, N., Nixdorff, U., Lechner, B., et al. (2020). Lifestyle factors and high-risk atherosclerosis: Pathways and mechanisms beyond traditional risk factors. Eur. J. Prev. Cardiol. 27 (4), 394–406. doi:10.1177/2047487319869400
Lee, M., Baek, H., Park, J. S., Kim, S., Kyung, C., Baik, S. J., et al. (2018). Current Helicobacter pylori infection is significantly associated with subclinical coronary atherosclerosis in healthy subjects: A cross-sectional study. PLoS One 13 (3), e0193646. doi:10.1371/journal.pone.0193646
Leja, M., Grinberga-Derica, I., Bilgilier, C., and Steininger, C. (2019). Review: Epidemiology of Helicobacter pylori infection. Helicobacter 24 (1), e12635. doi:10.1111/hel.12635
Liang, H., Lin, S., Ji, Y., Xiao, Y., and Zheng, G. (2021). Helicobacter pylori increases the risk of carotid plaque formation: a clinical evidence. Ann. Med. 53 (1), 1448–1454. doi:10.1080/07853890.2021.1927169
Libby, P., Buring, J. E., Badimon, L., Hansson, G. K., Deanfield, J., Bittencourt, M. S., et al. (2019). Atherosclerosis. Nat. Rev. Dis. Prim. 5 (1), 56. doi:10.1038/s41572-019-0106-z
Libby, P., Loscalzo, J., Ridker, P. M., Farkouh, M. E., Hsue, P. Y., Fuster, V., et al. (2018). Inflammation, immunity, and infection in atherothrombosis: JACC review topic of the week. J. Am. Coll. Cardiol. 72 (17), 2071–2081. doi:10.1016/j.jacc.2018.08.1043
Luo, Y., Duan, H., Qian, Y., Feng, L., Wu, Z., Wang, F., et al. (2017). Macrophagic CD146 promotes foam cell formation and retention during atherosclerosis. Cell Res. 27 (3), 352–372. doi:10.1038/cr.2017.8
Mansori, K., Dehghanbanadaki, H., Naderpour, S., Rashti, R., Moghaddam, A. B., and Moradi, Y. (2020a). A systematic review and meta-analysis of the prevalence of Helicobacter pylori in patients with diabetes. Diabetes Metab. Syndr. 14 (4), 601–607. doi:10.1016/j.dsx.2020.05.009
Mansori, K., Moradi, Y., Naderpour, S., Rashti, R., Moghaddam, A. B., Saed, L., et al. (2020b). Helicobacter pylori infection as a risk factor for diabetes: a meta-analysis of case-control studies. BMC Gastroenterol. 20 (1), 77. doi:10.1186/s12876-020-01223-0
Martinet, W., Knaapen, M. W., De Meyer, G. R., Herman, A. G., and Kockx, M. M. (2002). Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 106 (8), 927–932. doi:10.1161/01.cir.0000026393.47805.21
Medrek-Socha, M., Chojnacki, J., Smigielski, J., Konrad, P., and Chojnacki, C. (2018). Changes in the lipid profile of patients with asymptomatic and symptomatic helicobacter pylori infection. Wiad. Lek. 71 (8), 1467–1473.
Mentis, A., Lehours, P., and Megraud, F. (2015). Epidemiology and diagnosis of Helicobacter pylori infection. Helicobacter 20 (1), 1–7. doi:10.1111/hel.12250
Mizuno, S., Matsui, D., Watanabe, I., Ozaki, E., Kuriyama, N., and Watanabe, Y. (2015). Serologically determined gastric mucosal condition is a predictive factor for osteoporosis in Japanese men. Dig. Dis. Sci. 60 (7), 2063–2069. doi:10.1007/s10620-015-3576-1
Nasif, W. A., Mukhtar, M. H., Nour Eldein, M. M., and Ashgar, S. S. (2016). Oxidative DNA damage and oxidized low density lipoprotein in Type II diabetes mellitus among patients with Helicobacter pylori infection. Diabetol. Metab. Syndr. 8, 34. doi:10.1186/s13098-016-0149-1
Neumann, W. L., Coss, E., Rugge, M., and Genta, R. M. (2013). Autoimmune atrophic gastritis-pathogenesis, pathology and management. Nat. Rev. Gastroenterol. Hepatol. 10 (9), 529–541. doi:10.1038/nrgastro.2013.101
Nigatie, M., Melak, T., Asmelash, D., and Worede, A. (2022). Dyslipidemia and its associated factors among Helicobacter pylori-infected patients attending at university of gondar comprehensive specialized hospital, gondar, north-west Ethiopia: A comparative cross-sectional study. J. Multidiscip. Healthc. 15, 1481–1491. doi:10.2147/jmdh.S368832
Park, Y., Kim, T. J., Lee, H., Yoo, H., Sohn, I., Min, Y. W., et al. (2021). Eradication of Helicobacter pylori infection decreases risk for dyslipidemia: A cohort study. Helicobacter 26 (2), e12783. doi:10.1111/hel.12783
Polyzos, S. A., Kountouras, J., Zavos, C., and Deretzi, G. (2011). The association between Helicobacter pylori infection and insulin resistance: a systematic review. Helicobacter 16 (2), 79–88. doi:10.1111/j.1523-5378.2011.00822.x
Pothineni, N. V. K., Subramany, S., Kuriakose, K., Shirazi, L. F., Romeo, F., Shah, P. K., et al. (2017). Infections, atherosclerosis, and coronary heart disease. Eur. Heart J. 38 (43), 3195–3201. doi:10.1093/eurheartj/ehx362
Qiang, L., Hu, J., Tian, M., Li, Y., Ren, C., Deng, Y., et al. (2022). Extracellular vesicles from helicobacter pylori-infected cells and helicobacter pylori outer membrane vesicles in atherosclerosis. Helicobacter 27 (2), e12877. doi:10.1111/hel.12877
Rathcke, C. N., and Vestergaard, H. (2006). YKL-40, a new inflammatory marker with relation to insulin resistance and with a role in endothelial dysfunction and atherosclerosis. Inflamm. Res. 55 (6), 221–227. doi:10.1007/s00011-006-0076-y
Raut, S. C., and Chandel, R. S. (2014). Comparison of vitamin B12 levels in gastritis with and without H. pylori. Walawalkar Int. Med. J. 1, 28–32.
Riad, M. (2021). Association of Helicobacter pylori infection with coronary artery disease: Is it an independent risk factor? Egypt Heart J. 73 (1), 61. doi:10.1186/s43044-021-00185-2
Shan, J., Bai, X., Han, L., Yuan, Y., Yang, J., and Sun, X. (2018). Association between atherosclerosis and gastric biomarkers concerning Helicobacter pylori infection in a Chinese healthy population. Exp. Gerontol. 112, 97–102. doi:10.1016/j.exger.2018.09.009
Shan, J. H., Bai, X. J., Han, L. L., Yuan, Y., and Sun, X. F. (2017). Changes with aging in gastric biomarkers levels and in biochemical factors associated with Helicobacter pylori infection in asymptomatic Chinese population. World J. Gastroenterol. 23 (32), 5945–5953. doi:10.3748/wjg.v23.i32.5945
Shi, H., Li, Y., Dong, C., Si, G., Xu, Y., Peng, M., et al. (2022). Helicobacter pylori infection and the progression of atherosclerosis: A systematic review and meta-analysis. Helicobacter 27 (1), e12865. doi:10.1111/hel.12865
Shimoda, A., Ueda, K., Nishiumi, S., Murata-Kamiya, N., Mukai, S. A., Sawada, S., et al. (2016). Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 6, 18346. doi:10.1038/srep18346
Siasos, G., Tsigkou, V., Kokkou, E., Oikonomou, E., Vavuranakis, M., Vlachopoulos, C., et al. (2014). Smoking and atherosclerosis: mechanisms of disease and new therapeutic approaches. Curr. Med. Chem. 21 (34), 3936–3948. doi:10.2174/092986732134141015161539
Sies, H., Berndt, C., and Jones, D. P. (2017). Oxidative stress. Annu. Rev. Biochem. 86, 715–748. doi:10.1146/annurev-biochem-061516-045037
Simon, O. A., Görbe, A., Hegyi, P., Szakó, L., Oštarijaš, E., Dembrovszky, F., et al. (2022). Helicobacter pylori infection is associated with carotid intima and media thickening: A systematic review and meta-analysis. J. Am. Heart Assoc. 11 (3), e022919. doi:10.1161/jaha.121.022919
Sipponen, P., Laxen, F., Huotari, K., and Harkonen, M. (2003). Prevalence of low vitamin B12 and high homocysteine in serum in an elderly male population: association with atrophic gastritis and Helicobacter pylori infection. Scand. J. Gastroenterol. 38 (12), 1209–1216. doi:10.1080/00365520310007224
Soyocak, A., Ergun, D. D., Koc, G., Ergun, S., and Ozsobaci, N. P. (2021). Investigation of aryl hydrocarbon receptor, zinc, and vitamin B12 levels in chronic gastritis with Helicobacter pylori infection. Biol. Trace Elem. Res. 199 (7), 2431–2437. doi:10.1007/s12011-021-02667-5
Spence, J. D., and Pilote, L. (2015). Importance of sex and gender in atherosclerosis and cardiovascular disease. Atherosclerosis 241 (1), 208–210. doi:10.1016/j.atherosclerosis.2015.04.806
Steed, M. M., and Tyagi, S. C. (2011). Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid. Redox Signal 15 (7), 1927–1943. doi:10.1089/ars.2010.3721
Sugano, K. (2019). Effect of Helicobacter pylori eradication on the incidence of gastric cancer: a systematic review and meta-analysis. Gastric Cancer 22 (3), 435–445. doi:10.1007/s10120-018-0876-0
Supiano, M. A. (1987). Atrophic gastritis and vitamin B12 deficiency. J. Am. Geriatr. Soc. 35 (5), 478. doi:10.1111/j.1532-5415.1987.tb04684.x
Tahmina, K., Hikawa, N., Takahashi-Kanemitsu, A., Knight, C. T., Sato, K., Itoh, F., et al. (2022). Transgenically expressed Helicobacter pylori CagA in vascular endothelial cells accelerates arteriosclerosis in mice. Biochem. Biophys. Res. Commun. 618, 79–85. doi:10.1016/j.bbrc.2022.06.010
Talari, H. R., Moniri, R., Mollaghanbari, M., Haddad Kashani, H., and Jalalian, M. N. (2021). Evaluating the relationship between Helicobacter pylori infection and carotid intima-media thickness a cross sectional study. Ann. Med. Surg. (Lond) 69, 102659. doi:10.1016/j.amsu.2021.102659
Tegtmeyer, N., Wessler, S., Necchi, V., Rohde, M., Harrer, A., Rau, T. T., et al. (2017). Helicobacter pylori employs a unique basolateral type IV secretion mechanism for CagA delivery. Cell Host Microbe 22 (4), 552–560. doi:10.1016/j.chom.2017.09.005
Tsay, F. W., and Hsu, P. I. (2018). H. pylori infection and extra-gastroduodenal diseases. J. Biomed. Sci. 25 (1), 65. doi:10.1186/s12929-018-0469-6
Vijayvergiya, R., and Vadivelu, R. (2015). Role of Helicobacter pylori infection in pathogenesis of atherosclerosis. World J. Cardiol. 7 (3), 134–143. doi:10.4330/wjc.v7.i3.134
Waluga, M., Kukla, M., Żorniak, M., Bacik, A., and Kotulski, R. (2015). From the stomach to other organs: Helicobacter pylori and the liver. World J. Hepatol. 7 (18), 2136–2146. doi:10.4254/wjh.v7.i18.2136
Wang, F., Meng, W., Wang, B., and Qiao, L. (2014). Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 345 (2), 196–202. doi:10.1016/j.canlet.2013.08.016
Wang, N., Zhou, F., Chen, C., Luo, H., Guo, J., Wang, W., et al. (2021a). Role of outer membrane vesicles from Helicobacter pylori in atherosclerosis. Front. Cell Dev. Biol. 9, 673993. doi:10.3389/fcell.2021.673993
Wang, X., He, Q., Jin, D., Ma, B., Yao, K., and Zou, X. (2021b). Association between helicobacter pylori infection and subclinical atherosclerosis: A systematic review and meta-analysis. Med. Baltim. 100 (46), e27840. doi:10.1097/md.0000000000027840
Wang, Y. C. (2014). Medicinal plant activity on Helicobacter pylori related diseases. World J. Gastroenterol. 20 (30), 10368–10382. doi:10.3748/wjg.v20.i30.10368
Wang, Z., Wang, W., Gong, R., Yao, H., Fan, M., Zeng, J., et al. (2022). Eradication of Helicobacter pylori alleviates lipid metabolism deterioration: a large-cohort propensity score-matched analysis. Lipids Health Dis. 21 (1), 34. doi:10.1186/s12944-022-01639-5
Wernly, S., Semmler, G., Völkerer, A., Flamm, M., Aigner, E., Niederseer, D., et al. (2022). Helicobacter pylori and cardiovascular risk: Only a dead Helicobacter is a good Helicobacter? Helicobacter 27 (6), e12928. doi:10.1111/hel.12928
Wroblewski, L. E., and Peek, R. M. (2016). Helicobacter pylori, cancer, and the gastric microbiota. Adv. Exp. Med. Biol. 908, 393–408. doi:10.1007/978-3-319-41388-4_19
Wu, L. L., Chiou, C. C., Chang, P. Y., and Wu, J. T. (2004). Urinary 8-OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin. Chim. Acta 339 (1-2), 1–9. doi:10.1016/j.cccn.2003.09.010
Wu, X., Zhang, L., Miao, Y., Yang, J., Wang, X., Wang, C. C., et al. (2019). Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 20, 46–59. doi:10.1016/j.redox.2018.09.021
Xia, X., Zhang, L., Wu, H., Chen, F., Liu, X., Xu, H., et al. (2022). CagA(+) Helicobacter pylori, not CagA(-) Helicobacter pylori, infection impairs endothelial function through exosomes-mediated ROS formation. Front. Cardiovasc Med. 9, 881372. doi:10.3389/fcvm.2022.881372
Xu, Y., Wang, Q., Liu, Y., Cui, R., Lu, K., and Zhao, Y. (2016). Association between Helicobacter pylori infection and carotid atherosclerosis in patients with vascular dementia. J. Neurol. Sci. 362, 73–77. doi:10.1016/j.jns.2016.01.025
Yang, S., Xia, Y. P., Luo, X. Y., Chen, S. L., Li, B. W., Ye, Z. M., et al. (2019). Exosomal CagA derived from Helicobacter pylori-infected gastric epithelial cells induces macrophage foam cell formation and promotes atherosclerosis. J. Mol. Cell Cardiol. 135, 40–51. doi:10.1016/j.yjmcc.2019.07.011
Yao, B. C., Meng, L. B., Hao, M. L., Zhang, Y. M., Gong, T., and Guo, Z. G. (2019). Chronic stress: a critical risk factor for atherosclerosis. J. Int. Med. Res. 47 (4), 1429–1440. doi:10.1177/0300060519826820
Yu, L. Y., Hu, K. C., Liu, C. J., Hung, C. L., Bair, M. J., Chen, M. J., et al. (2019). Helicobacter pylori infection combined with non-alcoholic fatty liver disease increase the risk of atherosclerosis: Focus in carotid artery plaque. Med. Baltim. 98 (9), e14672. doi:10.1097/MD.0000000000014672
Zhang, D., Jiang, X., Fang, P., Yan, Y., Song, J., Gupta, S., et al. (2009). Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation 120 (19), 1893–1902. doi:10.1161/CIRCULATIONAHA.109.866889
Zhang, L., Chen, Z., Xia, X., Chi, J., Li, H., Liu, X., et al. (2019). Helicobacter pylori infection selectively increases the risk for carotid atherosclerosis in young males. Atherosclerosis 291, 71–77. doi:10.1016/j.atherosclerosis.2019.10.005
Zhang, P., He, Q., Song, D., Wang, Y., Liu, X., Ding, G., et al. (2021). Association of Helicobacter pylori infection with carotid atherosclerosis in a northern Chinese population: A cross-sectional study. Front. Cardiovasc Med. 8, 795795. doi:10.3389/fcvm.2021.795795
Zhang, X., Li, H., Jin, H., Ebin, Z., Brodsky, S., and Goligorsky, M. S. (2000). Effects of homocysteine on endothelial nitric oxide production. Am. J. Physiol. Ren. Physiol. 279 (4), F671–F678. doi:10.1152/ajprenal.2000.279.4.F671
Zhao, J., Chen, H., Liu, N., Chen, J., Gu, Y., Chen, J., et al. (2017). Role of hyperhomocysteinemia and hyperuricemia in pathogenesis of atherosclerosis. J. Stroke Cerebrovasc. Dis. 26 (12), 2695–2699. doi:10.1016/j.jstrokecerebrovasdis.2016.10.012
Keywords: cardiovascular disease, oxidative stress, extra-gastric disease, CagA, vitamin B12 deficiency
Citation: Aramouni K, Assaf RK, Azar M, Jabbour K, Shaito A, Sahebkar A, Eid AA, Rizzo M and Eid AH (2023) Infection with Helicobacter pylori may predispose to atherosclerosis: role of inflammation and thickening of intima-media of carotid arteries. Front. Pharmacol. 14:1285754. doi: 10.3389/fphar.2023.1285754
Received: 30 August 2023; Accepted: 25 September 2023;
Published: 13 October 2023.
Edited by:
Ismail Laher, University of British Columbia, CanadaReviewed by:
Narasimham L. Parinandi, The Ohio State University, United StatesCopyright © 2023 Aramouni, Assaf, Azar, Jabbour, Shaito, Sahebkar, Eid, Rizzo and Eid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali H. Eid, YWxpLmVpZEBxdS5lZC51LnFh
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.