 Yujin Seo†
Yujin Seo† Jiyeon Kim
Jiyeon Kim- Department of Biomedical Laboratory Science, School of Health Science, Dankook University, Cheonan, Republic of Korea
Long-term administration of tyrosine kinase inhibitors (TKIs) used for the treatment of non-small cell lung cancer (NSCLC) induces TKI resistance in cells. The appearance of resistant cells requires the combined administration of another therapeutic agent and may cause side effects in the gastrointestinal and central nervous system. In previous studies, we found that derivatives of cilengitide, a cyclic Arg-Gly-Asp (RGD) peptide, exert NSCLC apoptotic and anti–epithelial-mesenchymal transition (EMT) effects. In particular, cRGDwV and cRGDyV, which are cyclic peptides containing aromatic amino acids, were found to inhibit NSCLC cell growth, TGF-β1-induced EMT, and invasion. In this study, we confirmed the effects of cRGDwV and cRGDyV on proliferation, TGF-β1-induced EMT marker expression, migration, and invasion in gefitinib-resistant NSCLC A549 (A549GR) cells. In A549GR cells, cRGDwV and cRGDyV showed inhibitory effects on the expression of mesenchymal marker expression, migration, and invasion. These results indicate that cyclic RGD peptides containing aromatic amino acids can be used to inhibit mesenchymal marker expression as well as migration and invasion in gefitinib-resistant cells.
1 Introduction
The most common mutation seen in non-small cell lung cancer (NSCLC), accounting for approximately 85% of all lung cancer types, is in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) (Testa et al., 2018). A subset of NSCLC patients have active EGFR mutations, most of which are the deletion of exon 19 (Ex19del) and the L858R point mutation in exon 21 (Westover et al., 2018). In lung cancer cells, EGFR mutations are associated with anti-apoptotic signaling (e.g., through AKT and STAT), which promotes survival. In addition, there is a possibility of metastasis to the liver or central nervous system (CNS), so tyrosine kinase inhibitors (TKIs) targeting EGFR mutations are preferentially used as therapeutic agents (Sordella et al., 2004; Doebele et al., 2012; Shin et al., 2014; Iuchi et al., 2015). EGFR-TKIs were developed for the treatment of NSCLC and have been used through their first, second, and third generations to date (Westover et al., 2018; He et al., 2021).
Gefitinib, together with erlotinib and icotinib, is a first-generation TKI that reversibly binds to the ATP-binding domain of tyrosine kinase and inhibits EGFR downstream signaling, thereby exhibiting anticancer effects (Huang and Fu, 2015). Approved by the U.S. Food and Drug Administration (FDA) in 2015, gefitinib has been used for the first-line treatment of NSCLC patients with EGFR mutations, and many clinical studies have demonstrated that it prolongs progression-free survival when compared with existing treatment methods (Mok et al., 2009; Kazandjian et al., 2016). However, continuous use of TKIs for more than 9 months results in the disease not reaching full cure and spreading through the body due to resistance. Through many studies over the past few years, the mechanism of resistance has been investigated, and there has been continuous research on methods to increase treatment efficacy by suppressing the resistance mechanism (Mitsudomi et al., 2010; Sequist et al., 2011; Yu et al., 2013; Maemondo et al., 2020).
Peptides containing the Arg-Gly-Asp (RGD) sequence show high binding affinity for integrin αvβ3 expressed in cancer cells, thereby reducing side effects and increasing the delivery rate of anticancer drugs to the body (Temming et al., 2005; Bianchini et al., 2019; Sartori et al., 2019). In particular, cilengitide [c (RGDf (NMe)V], a cyclic RGD peptide showing nanomolar activity and affinity for integrin receptors, has been used as a single or combined treatment for various diseases (Mas-Moruno et al., 2010). According to recent studies, the combined administration of these RGD sequence-containing peptides and existing anticancer agents is effective in inducing apoptosis and suppressing EMT and invasion by lung cancer cells (Park and Kim, 2020; Jeong and Kim, 2021; Jeong and Kim, 2022). In addition, we have demonstrated that combined treatment with the first-generation TKI gefitinib (or erlotinib) and the cyclic RGD peptide cilengitide and its derivatives have synergistic inhibitory effects on the growth of NSCLC cells and TGF-β1-induced EMT (Jeong and Kim, 2021; Jeong and Kim, 2022; Park et al., 2022; Seo and Kim, 2022).
EMT is a phenomenon caused by changes in the morphology of epithelial cells during processes such as embryogenesis, wound healing, and organ fibrosis, and is one of the main ways in which cancer cells acquire migratory and invasive properties, especially in the tumor microenvironment (Thiery et al., 2009; Siegel et al., 2011). EMT-triggered changes in cancer cells during the progression of lung cancer cause poor prognosis, such as increased mortality and metastasis to other organs, and thus can be an important target for lung cancer treatment. Transforming growth factor (TGF)-β is one of the major cytokines that induce EMT and is involved in cancer cell invasion and metastasis (Behrens et al., 1989; Buck and Knabbe, 2006; Barcellos-Hoff and Akhurst, 2009; Drabsch and ten Dijke, 2012). EMT induction by TGF-β occurs through the activation of signaling pathways such as Smad or non-Smad pathways including the Wnt/β-catenin and MAPK/ERK pathways (Heldin et al., 1997; Massagué, 1998; Massagué et al., 2005; Galliher et al., 2006; Medici et al., 2008; Chen et al., 2012). Therefore, inhibiting the signaling pathways associated with EMT and inducing the apoptosis of NSCLC cells can prevent changes in cell morphology that can affect metastasis and invasion.
In this study, we evaluated the anticancer and EMT inhibitory effects of the cilengitide derivatives cRGDwV and cRGDyV in gefitinib-resistant NSCLC cells. In gefitinib-resistant NSCLC A549 (A549GR) cells, cRGDwV and cRGDyV suppressed cell growth and inhibited TGF-β1-induced mesenchymal marker expression, migration, and invasion. In addition, we found that the inhibitory effects of cRGDwV and cRGDyV were increased when it was combined with gefitinib. These results suggest the use of cyclic RGD peptides can be an alternative means of reducing side effects that may occur when other types of TKIs are co-administered to patients with TKI-resistant NSCLC.
2 Materials and methods
2.1 Materials
Gefitinib (#SML1657) was purchased from Merck (United States), and cilengitide derivatives (cRGDwV and cRGDyV) were synthesized by Dr. Park (CHA Meditech, Korea) (Supplementary Figures S1–S17) (Park et al., 2022; Seo and Kim, 2022). Fetal bovine serum (FBS) was purchased from Thermo Fisher Scientific (United States). RPMI 1640 medium and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) were purchased from Corning (United States). Recombinant human TGF-β1 was purchased from R&D Systems (United States). Antibodies against E-cadherin (#3195), α-smooth muscle actin (α-SMA) (#19245), p-ERK1/2 (#4370), ERK1/2 (#4695), and p-Smad2/3 (#8828) were purchased from Cell Signaling Technology (United States). Antibodies against Smad2/3 (#sc-133098), vimentin (#sc-6260), N-cadherin (#sc-59987), β-catenin (#sc-7199), and horseradish peroxidase (HRP)-conjugated secondary antibodies (#sc-516102 and #sc-2357), HRP-conjugated β-actin (#sc-47778), and HRP-conjugated GAPDH (#47724) were purchased from Santa Cruz Biotechnology (United States).
2.2 Cell line and determination of cell proliferation
Bovine pulmonary epithelium CPAE (KCLB No. 10209) and NSCLC A549 (KCLB No. 10185) cell lines were obtained from the Korean Cell Line Bank (Korea). CPAE and all NSCLC cells were maintained in RPMI 1640 medium containing 10% heat-inactivated FBS and 1% antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) and maintained at 37°C in a humidified atmosphere of 5% CO2. The gefitinib-resistant A549 (A549GR) cell line was established by treatment with a high concentration (2 μM) of gefitinib for at least 6 months (Supplementary Figure S18) (Shien et al., 2013).
Cell viability was detected using an EZ-Cytox Enhanced Cell Viability Assay Kit (DoGen, Korea). CPAE or A549GR cells were treated with gefitinib or peptides for 24–72 h in culture medium containing 10% FBS. The absorbance was measured by a Multiscan FC microplate photometer (Thermo Fisher Scientific, United States), and cell viability is presented as a percentage of control (untreated) cell viability.
2.3 Western blot analysis
A549GR cells were treated with TGF-β1 (5 ng/mL) or in combination with cRGDwV or cRGDyV. After a 48–72 h incubation, lysates of pooled cells were prepared using Nuclear and Cytoplasmic Extraction Reagent (Thermo Fisher Scientific, United States). The extracted protein was separated by SDS-PAGE and transferred to a nitrocellulose membrane, and the expression of each specific protein was detected with a primary antibody and an HRP-conjugated secondary antibody. The amount of each protein expressed was visualized using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, United States) and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, United States) and quantified using ImageJ software, and the relative signal ratio of each sample to the GAPDH or β-actin loading control is shown in the figures.
2.4 Analysis of combined drug effects
The effect of the combination of gefitinib with cRGDwV or cRGDyV was analyzed using the CalcuSyn software program (Biosoft, UK). To determine whether the outcome of combined treatment with two compounds was additive or synergistic, a combination index (CI) method derived from the median effect principle of Chou and Talalay was applied, and the CI was calculated with the formula published by Zhao et al. (Chou and Talalay, 1984; Zhao et al., 2004). In Supplementary Table S1, a CI of 1 indicates an additive effect between the two compounds, CI > 1 indicates antagonism, and CI < 1 indicates a synergistic effect.
2.5 Wound healing assay
A549GR cells were grown in 24-well plates. After 24-h serum starvation, cells were scratched using a SPLScar scratcher (SPL Life Sciences, Co., Ltd.), rinsed with PBS to remove debris, and incubated with TGF-β1 (5 ng/mL) and cRGDwV or cRGDyV in serum-free RPMI 1640 medium for 48 h. Images of migrated A549GR cells were captured at 0, 24, and 48 h. The wound healing index was expressed as a percentage and was calculated based on the length of three randomly selected zones at 0 h.
2.6 Invasion assay
A549GR cells were cultured in serum-free medium for 24 h and then treated with TGF-β1 (5 ng/mL) and cRGDwV or cRGDyV for 48 h. After harvesting, the cells were resuspended in serum-free medium for counting. Culture medium (30 μL) containing 5% FBS was added to the bottom of a Boyden chamber, and a gelatin-coated membrane filter, silicone gasket, and upper chamber were placed in this order, followed by cell suspension (5 × 103 cells/50 μL) in the upper chamber and incubation at 37°C in 5% CO2. After 4 h of incubation, cells that had penetrated the membrane were stained using the Differential Quick III stain kit (Polysciences, United States).
2.7 Statistical analysis
All experiments were conducted in triplicate (n = 3), and representative figures of the results of at least three independent experiments are shown. Data are presented as the mean ± SD. Significant differences were evaluated by one-way ANOVA with post-hoc Tukey HSD test using Microsoft Excel. p < 0.05 was considered significant (* or #).
3 Results
3.1 Effect of cRGDwV and cRGDyV on cell viability in A549GR cells
Among the seven cilengitide derivatives synthesized in previous studies, cRGDwV (R-1) and cRGDyV (R-7) showed strong inhibitory effects on NSCLC cell growth without significant cytotoxicity to normal lung endothelial cells (Park et al., 2022; Seo and Kim, 2022). In addition, it was confirmed that these peptides exhibit a synergistic effect in inducing NSCLC cell death when co-administered with gefitinib (Park et al., 2022; Seo and Kim, 2022). These two derivatives did not show significant cytotoxicity as measured by the growth of normal bovine lung epithelium CPAE cells (Park et al., 2022; Seo and Kim, 2022). On the basis of these results, we confirmed the effect of co-treatment with gefitinib and cRGDwV or cRGDyV on gefitinib-resistant A549 (A549GR) cells. As shown in Figure 1, A549GR cells were not significantly affected by gefitinib treatment alone, but A549GR cell growth was suppressed when gefitinib was used in combination with cRGDwV (Figures 1A–D) or cRGDyV (Figures 1E–H). The combination index (CI) was calculated using the raw data in Figure 1 (Supplementary Table S1). At a concentration of 0.3 μM or higher, all CI values were less than 1. This result means that cell death can be induced more effectively in gefitinib-resistant cells when treated with either cRGDwV or cRGDyV together with gefitinib. The combination treatment of gefitinib and cRGDwV or cRGDyV did not significantly affect the growth of CPAE cells (Supplementary Figures S19, S20). This result indicates that cRGDwV and cRGDyV can increase the growth inhibitory effect of gefitinib on gefitinib-resistant cells without significant cytotoxicity to normal lung endothelial cells.
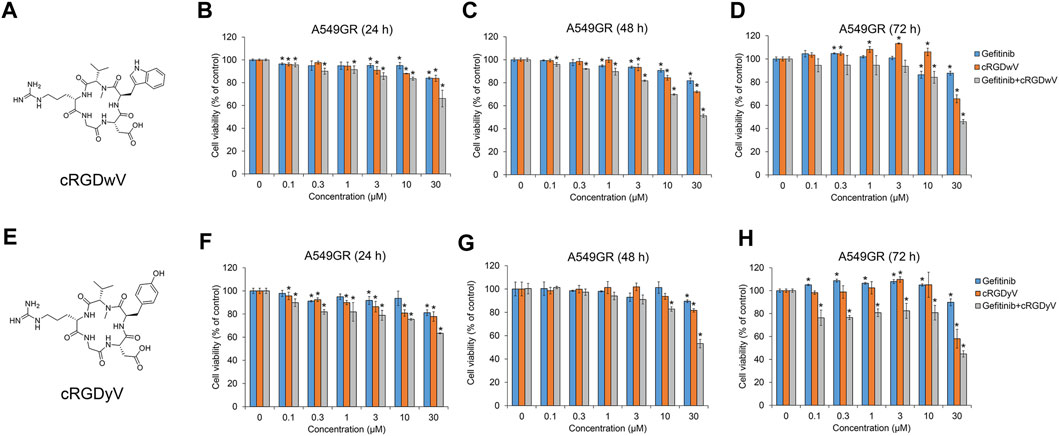
FIGURE 1. Combination effect of gefitinib with cilengitide derivatives on gefitinib-resistant A549 (A549GR) cell proliferation. (A,E) Chemical structures of cRGDwV (A) and cRGDyV (E). (B–D) A549GR cells were treated with gefitinib or cRGDwV or gefitinib with cRGDwV for 24–72 h (F–H) A549GR cells were treated with gefitinib or cRGDyV or gefitinib with cRGDyV for 24–72 h. After incubation, cell viability was measured by the CCK-8 assay. Experiments were performed in triplicate. Data are the mean ± SD.
3.2 Effects of cRGDwV and cRGDyV on the expression of EMT markers and related signaling in A549GR cells
TGF-β1 treatment decreases the expression of the epithelial marker E-cadherin, but increases the expression of mesenchymal markers such as N-cadherin, vimentin, and α-SMA (Kalluri and Weinberg, 2009; Jakobsen et al., 2016). We have confirmed in previous studies that cRGDwV (R-1) and cRGDyV (R-7) inhibit the expression of N-cadherin, vimentin, and α-smooth muscle actin (SMA), which are TGF-β1-induced mesenchymal markers (Park et al., 2022; Seo and Kim, 2022). Those studies also confirmed that mesenchymal marker expression and related signaling processes are suppressed when wild-type A549 cells are treated with gefitinib.
To verify the effects of cRGDwV and cRGDyV on gefitinib-resistant A549 cells, the expression of TGF-β1-induced EMT markers was confirmed in A549GR cells. As shown in Figures 2A, B, the expression of E-cadherin suppressed by TGF-β1 was restored by cRGDwV and cRGDyV, and the increases in N-cadherin and α-SMA expression were suppressed. Although not as strongly inhibited as N-cadherin expression, at 30 μM peptide vimentin expression was also inhibited.
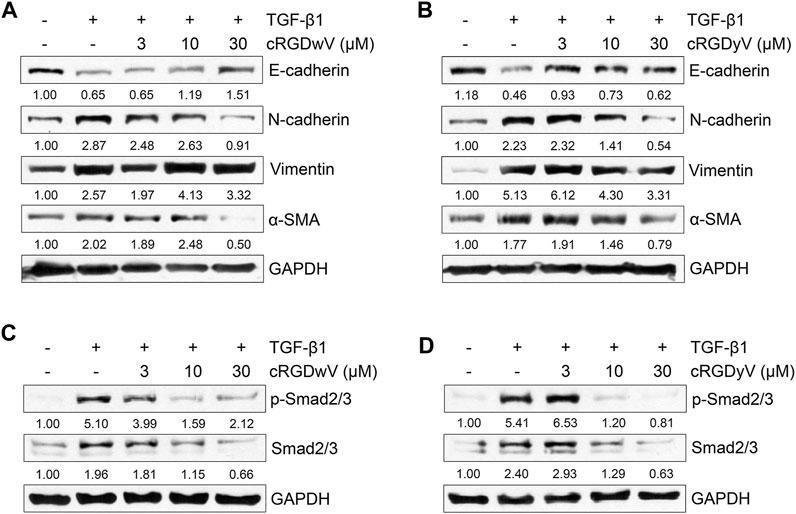
FIGURE 2. Effect of cilengitide derivatives on TGF-β1-induced EMT marker expression and related signaling pathways in A549GR cells. Serum-deprived A549GR cells were treated with TGF-β1 (5 ng/mL) and cRGDwV (A,C) or cRGDyV (B,D) for 72 h. Expression of EMT markers and phosphorylation of nuclear Smad2/3 was determined by western blot analysis. GAPDH was used as a loading control.
To verify the inhibitory effects of cRGDwV and cRGDyV on TGF-β1-induced mesenchymal marker expression, we also confirmed the phosphorylation of Smad2/3, a key mediator in the EMT-related signaling pathway, and the expression of non-Smad signaling-related proteins. Recent studies have reported that cRGDwV or cRGDyV alone inhibits Smad-dependent or Smad-independent signaling processes in gefitinib-sensitive A549 cells, thereby affecting EMT phenotype change (Park et al., 2022; Seo and Kim, 2022). As shown in Figures 2C, D, treatment with cRGDwV or cRGDyV inhibits TGF-β1-induced phosphorylation of nuclear Smad2/3 in a dose-dependent manner.
In addition, we checked the phosphorylation of ERK1/2 and the expression of β-catenin to confirm the effects of cRGDwV and cRGDyV on non-Smad signaling. In Smad-independent signaling, cRGDwV or cRGDyV did not significantly inhibit phosphorylation of ERK1/2 and nuclear translocation of β-catenin in A549GR cells (Supplementary Figure S21). These results indicate that Smad-dependent signaling is maintained more strongly in gefitinib-resistant cells than in cells that are not resistant, and that the combined administration of gefitinib with either one of two cilengitide derivatives may inhibit EMT by inhibiting Smad-dependent signaling.
3.3 Effect of cRGDwV and cRGDyV on migration and invasion in A549GR cells
EMT in cancer cells plays a very important role in metastasis by promoting migration and invasion (Mittal, 2018). In recent studies on A549 cells, we confirmed the inhibitory effect of combined treatment with gefitinib and either cilengitide derivative cRGDwV or cRGDyV on TGF-β1-induced invasion across a gelatin-coated membrane (Seo and Kim, 2022). The combined treatment of gefitinib and cRGDwV or cRGDyV inhibited invasion more effectively than gefitinib treatment alone. Based on these previous studies, we also investigated inhibitory effects of cRGDwV and cRGDyV on A549GR cell migration and invasion. As shown in Figures 3A, B, TGF-β1-treated A549GR cells in the scratched monolayer showed a pattern of gathering together over time, and all scratches were filled after 72 h. However, in the cRGDwV- or cRGDyV-treated group, scratches were not filled, and they persisted. This result was similar to those of the invasion assay. Treatment with TGF-β1 induced A549GR cells to cross the gelatin-coated membrane, but the number of infiltrating cells was reduced (Figures 3C, D). These results show that cRGDwV and cRGDyV alone are excellent inhibitors of migration and invasion, and demonstrate the possibility that they can act synergistically when used together with drugs showing anticancer effects in NSCLC.
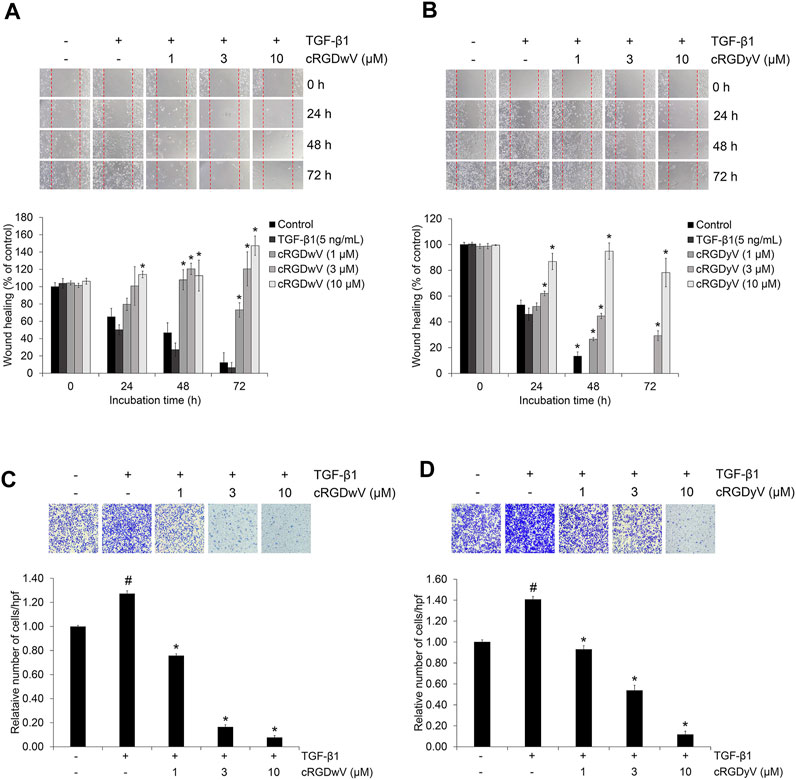
FIGURE 3. Effect of cilengitide derivatives on TGF-β1-induced migration and invasion in A549GR cells. Serum-deprived A549GR cells were scratched and treated with TGF-β1 (5 ng/mL) and cRGDwV (A,C) or cRGDyV (B,D) for 72 h. Images of A549GR cells that had migrated were captured at 0, 24, and 48 h. The wound healing index is expressed as a percentage and was calculated based on the length of three randomly selected zones at 0 h #p < 0.05 versus control; *p < 0.05 versus the group treated with TGF-β1 only.
3.4 Combination effect of gefitinib with cRGDwV and cRGDyV on the expression of EMT markers, related signaling and invasion in A549GR cells
We confirmed the combined effects of cRGDwV and cRGDyV with gefitinib on the expression of EMT markers and invasion in A549GR cells. As shown in Figures 4A, B, treatment with gefitinib (1 μM) or cRGDwV (3 μM) or cRGDyV (3 μM) alone did not significantly inhibit mesenchymal marker expression, whereas treatment with cRGDwV or cRGDyV inhibited the expression of mesenchymal makers such as N-cadherin, vimentin, and α-SMA. In particular, cRGDwV was effective in suppressing the expression of N-cadherin and vimentin, and cRGDyV seemed to help suppress the expression of α-SMA. This inhibitory effect of mesenchymal marker expression following the combined administration is thought to originate from inhibition of phosphorylation of Smad2/3 (Figures 4C, D), and the non-Smad signaling pathway was not suppressed (Supplementary Figure S22).
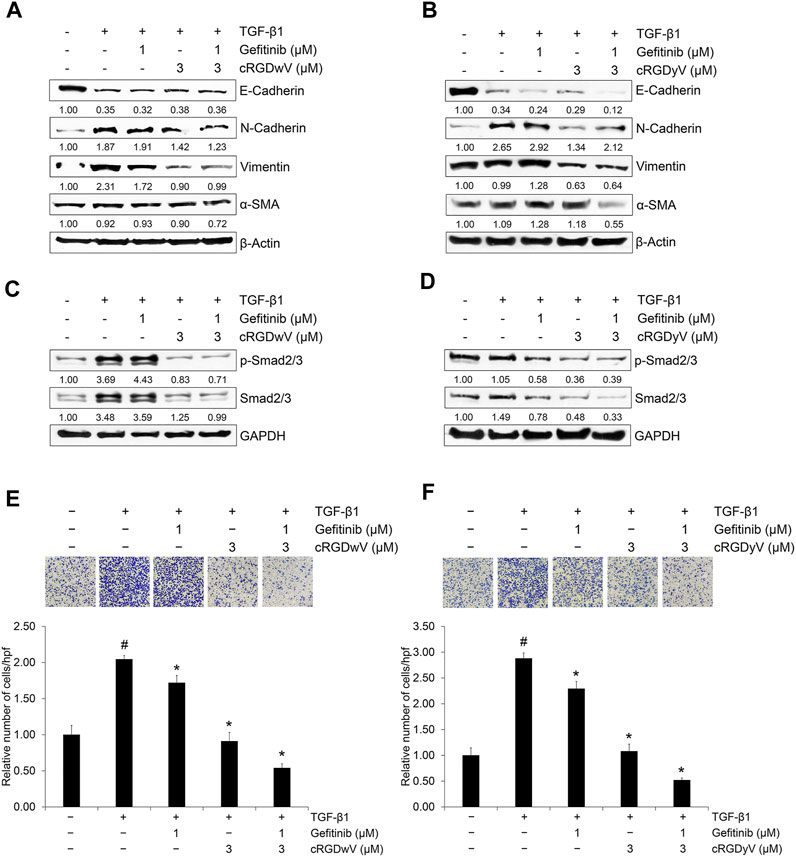
FIGURE 4. Combination effect of gefitinib with cilengitide derivatives on TGF-β1-induced expression of EMT markers, related signaling and invasion in A549GR cells. (A–F) Serum-deprived A549GR cells were treated with TGF-β1 (5 ng/mL) or gefitinib (1 μM) or in combination with cRGDwV (3 μM) (A,C,E) or cRGDyV (3 μM) (B,D,F) for 72 h (A–D) Expression of EMT markers and nuclear expression of phosphorylated Smad2/3 and endogenous Smad2/3 was determined by western blot analysis. β-actin was used as a loading control. (E,F) The number of invading cells was evaluated using a Boyden chamber assay. The number of cells that invaded the gelatin-coated membrane is represented by an average number of cells per three randomly selected high-power fields (HPFs). The detailed experimental procedure is described in the Materials and Methods section. #p < 0.05 versus control; *p < 0.05 versus the group treated with TGF-β1 only.
In addition, the invasion-inhibitory effect of low-dose gefitinib (1 μM) was not significant in A549GR cells, whereas cRGDwV and cRGDyV alone or in combination with gefitinib inhibited invasion significantly (Figures 4E, F). These results suggest that the effect of combined administration of gefitinib with cyclic peptides such as cRGDwV and cRGDyV may be more effective than treatment with high concentrations of gefitinib to inhibit the migration and invasion of NSCLC cells resistant to gefitinib.
4 Discussion
We have already confirmed in previous studies that first-generation TKIs such as gefitinib or erlotinib inhibit the growth of NSCLC and are effective in inhibiting TGF-β1-induced EMT and invasion. In addition, a synergistic effect was confirmed when cyclic peptides such as a cilengitide were used in combination treatment (Jeong and Kim, 2021; Jeong and Kim, 2022; Park et al., 2022; Seo and Kim, 2022). RTK inhibitors such as erlotinib and gefitinib are used to treat NSCLC but have limitations because the long-term use of them alone is the main cause of the emergence of resistant cells due to compensatory activation of intracellular signaling mechanisms (Sequist et al., 2011; Yu et al., 2013; Huang and Fu, 2015). In the process of acquiring EGFR-TKI resistance, cells are known to undergo changes such as loss of EGFR mutation, secondary EGFR T790M and minor mutations, MET amplification, and the activation of the MET/HGF axis and EMT (Huang and Fu, 2015; He et al., 2021). Third-generation EGFR-TKIs such as abivertinib, rociletinib, nazartinib, and olmutinib, which target the EGFR T790M mutation, can be used to overcome resistance caused by first- and second-generation EGFR-TKIs used as first-line treatment for NSCLC. However, in the case of long-term use, resistance to third-generation EGFR-TKIs cannot be avoided, and the mechanism of resistance is very complex and incompletely understood (He et al., 2021).
In order to overcome the resistance caused by the use of TKIs, a new chemotherapy that can suppress the proliferation and metastasis of NSCLC is needed, rather than one co-administered with TKIs that can cause resistance again. Thus, in recent studies, we confirmed the inhibitory effects on NSCLC cell growth and EMT of the first-generation TKIs gefitinib and erlotinib and the RGD-based cyclic peptide cilengitide (Jeong and Kim, 2021; Jeong and Kim, 2022). Cilengitide induced cell death in NSCLC by itself, but showed a synergistic effect on cell growth inhibition when combined with gefitinib or erlotinib. It was also found to be effective in suppressing mesenchymal marker expression and related signaling processes induced by TGF-β1 (Jeong and Kim, 2021; Jeong and Kim, 2022). Subsequent studies confirmed the effects of cilengitide derivatives: the peptides cRGDwV and cRGDyV containing aromatic amino acids inhibited NSCLC cell growth and EMT (Park et al., 2022; Seo and Kim, 2022). However, in NSCLC chemotherapy using TKIs, it was considered necessary to evaluate the effects of the peptides on TKI-resistant cells because it is expected that TKI resistance will eventually appear in NSCLC cells even with long-term peptide co-administration.
To confirm whether the cilengitide derivatives cRGDwV and cRGDyV have anticancer effects in TKI-resistant NSCLC, we established A549 cells resistant to gefitinib, a first-generation TKI, and used them to confirm the effects of the cilengitide derivatives. The experiments demonstrated that gefitinib-resistant A549 (A549GR) cells showed less gefitinib-induced cell death than did wild-type A549 cells, and high phosphorylation of Smad2/3, which is related to EMT. Since gefitinib alone does not show a sufficient anticancer effect in gefitinib-resistant cells, the result was confirmed by co-administration of cilengitide derivatives, which were shown to induce cell death and inhibit EMT in previous studies (Park et al., 2022; Seo and Kim, 2022). In A549GR cells, the cilengitide derivatives cRGDwV and cRGDyV had cell death-inducing effects similar to those of gefitinib, but a synergistic effect was observed when either derivative and gefitinib were administered together. In addition, regarding the expression of EMT markers induced by TGF-β1, when administered at the concentration that inhibits mesenchymal marker expression in gefitinib-sensitive A549 cells, gefitinib did not inhibit mesenchymal marker expression significantly in A549GR cells, and its inhibitory effect in combination with cRGDwV or cRGDyV was confirmed (Jeong and Kim, 2021). This inhibition was judged to affect migration and invasion processes associated with EMT. Mesenchymal marker expression was not significantly inhibited in A549GR cells when treated with cRGDwV or cRGDyV alone, but migration and invasion were effectively inhibited as the concentration increased, and a synergistic effect was observed when cells were co-treated with gefitinib. As in the previous study, we expected that cilengitide derivatives would suppress the expression of TGF-β1-induced mesenchymal makers in A549GR cells, and that this effect would also affect migration and invasion. However, genetic or other changes in A549GR cells are presumed to interfere with the EMT inhibitory effect of cRGDwV and cRGDyV. Although the anti-cancer effect of these cilengitide derivatives is somewhat insufficient in inhibiting EMT, it has been confirmed that the effect of inhibiting migration and invasion is good, so a greater anti-cancer effect can be expected when they are administered in combination with second- or third-generation lung cancer drugs.
We plan to continue to study the establishment of NSCLC cells resistant to second- and third-generation TKIs and the anti-cancer effects of cilengitide derivatives by extending the results of this work on A549GR cells. In addition, we will aim to develop peptides that act as antagonists of integrin receptors and that help therapeutic agents such as TKIs enter cancer cells more easily by designing and synthesizing new cyclic peptides containing aromatic amino acids. Peptide therapeutics are continuously being studied as an alternative approach to reducing the side effects of small molecule compound-based drugs, but they take a lot of time and money to develop due to in vivo degradation and low stability. For this reason, many pharmaceutical companies are applying existing safety-tested drugs to new diseases before developing new drugs, in what is called “drug repositioning” (Chong and Sullivan, 2007; Hanusova et al., 2015). Therefore, in NSCLC, the combined administration of conventional therapeutic agents such as TKIs and RGD-based cyclic peptides showing anti-cancer effects is expected to provide a new treatment method that can reduce side effects and overcome resistance. It is still necessary to verify their effects in other TKI-resistant cells and the effect of co-administering these peptides with second- and third-generation TKIs, and to conduct additional studies such as animal experiments, but the results of this study provide a new clue to the development of NSCLC treatments using peptides.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
YS: Data curation, Formal Analysis, Investigation, Methodology, Visualization, Writing–review and editing. MS: Data curation, Formal Analysis, Investigation, Methodology, Visualization, Writing–review and editing. JK: Conceptualization, Data curation, Funding acquisition, Project administration, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of Science and NCT; 2023R1A2C1002467).
Acknowledgments
We thank Dr. Kyeong-Yong Park from CHA Meditech for providing synthetic and analytical data on the cilengitide derivatives cRGDwV and cRGDyV.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1277199/full#supplementary-material
References
Barcellos-Hoff, M. H., and Akhurst, R. J. (2009). Transforming growth factor-beta in breast cancer: Too much, too late. Breast Cancer Res. 11, 202. doi:10.1186/bcr2224
Behrens, J., Mareel, M. M., Van Roy, F. M., and Birchmeier, W. (1989). Dissecting tumor cell invasion: Epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J. Cell Biol. 108, 2435–2447. doi:10.1083/jcb.108.6.2435
Bianchini, F., Portioli, E., Ferlenghi, F., Vacondio, F., Andreucci, E., Biagioni, A., et al. (2019). Cell-targeted c(AmpRGD)-sunitinib molecular conjugates impair tumor growth of melanoma. Cancer Lett. 446, 25–37. doi:10.1016/j.canlet.2018.12.021
Buck, M. B., and Knabbe, C. (2006). TGF-beta signaling in breast cancer. Ann. N. Y. Acad. Sci. 1089, 119–126. doi:10.1196/annals.1386.024
Chen, X. F., Zhang, H. J., Wang, H. B., Zhu, J., Zhou, W. Y., Zhang, H., et al. (2012). Transforming growth factor-β1 induces epithelial-to-mesenchymal transition in human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling pathways. Mol. Biol. Rep. 39, 3549–3556. doi:10.1007/s11033-011-1128-0
Chong, C. R., and Sullivan, D. J. (2007). New uses for old drugs. Nature 448, 645–646. doi:10.1038/448645a
Chou, T. C., and Talalay, P. (1984). Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22, 27–55. doi:10.1016/0065-2571(84)90007-4
Doebele, R. C., Lu, X., Sumey, C., Maxson, D. A., Weickhardt, A. J., Oton, A. B., et al. (2012). Oncogene status predicts patterns of metastatic spread in treatment-naive nonsmall cell lung cancer. Cancer 118, 4502–4511. doi:10.1002/cncr.27409
Drabsch, Y., and ten Dijke, P. (2012). TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 31, 553–568. doi:10.1007/s10555-012-9375-7
Galliher, A. J., Neil, J. R., and Schiemann, W. P. (2006). Role of transforming growth factor-beta in cancer progression. Future Oncol. 2, 743–763. doi:10.2217/14796694.2.6.743
Hanusova, V., Skalova, L., Kralova, V., and Matouskova, P. (2015). Potential anti-cancer drugs commonly used for other indications. Curr. Cancer Drug Targets 15, 35–52. doi:10.2174/1568009615666141229152812
He, J., Huang, Z., Han, L., Gong, Y., and Xie, C. (2021). Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review). Int. J. Oncol. 59, 90. doi:10.3892/ijo.2021.5270
Heldin, C. H., Miyazono, K., and ten Dijke, P. (1997). TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471. doi:10.1038/37284
Huang, L., and Fu, L. (2015). Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta. Pharm. Sin. B 5, 390–401. doi:10.1016/j.apsb.2015.07.001
Iuchi, T., Shingyoji, M., Itakura, M., Yokoi, S., Moriya, Y., Tamura, H., et al. (2015). Frequency of brain metastases in non-small-cell lung cancer, and their association with epidermal growth factor receptor mutations. Int. J. Clin. Oncol. 20, 674–679. doi:10.1007/s10147-014-0760-9
Jakobsen, K. R., Demuth, C., Sorensen, B. S., and Nielsen, A. L. (2016). The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 5, 172–182. doi:10.21037/tlcr.2016.04.07
Jeong, J., and Kim, J. (2022). Combination effect of cilengitide with erlotinib on TGF-β1-induced epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. Int. J. Mol. Sci. 23, 3423. doi:10.3390/ijms23073423
Jeong, J., and Kim, J. (2021). Cyclic RGD pentapeptide cilengitide enhances efficacy of gefitinib on TGF-β1-induced epithelial-to-mesenchymal transition and invasion in human non-small cell lung cancer cells. Front. Pharmacol. 12, 639095. doi:10.3389/fphar.2021.639095
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi:10.1172/JCI39104
Kazandjian, D., Blumenthal, G. M., Yuan, W., He, K., Keegan, P., and Pazdur, R. (2016). FDA approval of gefitinib for the treatment of patients with metastatic EGFR mutation-positive non-small cell lung cancer. Clin. Cancer Res. 22, 1307–1312. doi:10.1158/1078-0432.CCR-15-2266
Maemondo, M., Inoue, A., Kobayashi, K., Sugawara, S., Oizumi, S., Isobe, H., et al. (2020). Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388. doi:10.1056/NEJMoa0909530
Mas-Moruno, C., Rechenmacher, F., and Kessler, H. (2010). Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 10, 753–768. doi:10.2174/187152010794728639
Massagué, J., Seoane, J., and Wotton, D. (2005). Smad transcription factors. Genes Dev. 19, 2783–2810. doi:10.1101/gad.1350705
Massagué, J. (1998). TGF-beta signal transduction. Annu. Rev. Biochem. 67, 753–791. doi:10.1146/annurev.biochem.67.1.753
Medici, D., Hay, E. D., and Olsen, B. R. (2008). Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol. Biol. Cell 19, 4875–4887. doi:10.1091/mbc.e08-05-0506
Mitsudomi, T., Morita, S., Yatabe, Y., Negoro, S., Okamoto, I., Tsurutani, J., et al. (2010). Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 11, 121–128. doi:10.1016/S1470-2045(09)70364-X
Mittal, V. (2018). Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 13, 395–412. doi:10.1146/annurev-pathol-020117-043854
Mok, T. S., Wu, Y. L., Thongprasert, S., Yang, C. H., Chu, D. T., Saijo, N., et al. (2009). Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957. doi:10.1056/NEJMoa0810699
Park, K. Y., Jeong, J., and Kim, J. (2022). Synthesis and biological evaluation of cilengitide derivatives on TGF-β1-induced epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. Pep. Sci. 114, e24285. doi:10.1002/pep2.24285
Park, K. Y., and Kim, J. (2020). Cyclic pentapeptide cRGDfK enhances the inhibitory effect of sunitinib on TGF-β1-induced epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. PLoS One 15, e0232917. doi:10.1371/journal.pone.0232917
Sartori, A., Corno, C., De Cesare, M., Scanziani, E., Minoli, L., Battistini, L., et al. (2019). Efficacy of a selective binder of αVβ3 integrin linked to the tyrosine kinase inhibitor sunitinib in ovarian carcinoma preclinical models. Cancers (Basel) 11, 531. doi:10.3390/cancers11040531
Seo, M., and Kim, J. (2022). Combination effect of cilengitide derivatives with gefitinib on TGF-β1-induced epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. Bull. Korean Chem. Soc. 43, 1212–1216. doi:10.1002/bkcs.12601
Sequist, L. V., Waltman, B. A., Dias-Santagata, D., Digumarthy, S., Turke, A. B., Fidias, P., et al. (2011). Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3, 75ra26. doi:10.1126/scitranslmed.3002003
Shien, K., Toyooka, S., Yamamoto, H., Soh, J., Jida, M., Thu, K. L., et al. (2013). Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 73, 3051–3061. doi:10.1158/0008-5472.CAN-12-4136
Shin, D. Y., Na, I. I., Kim, C. H., Park, S., Baek, H., and Yang, S. H. (2014). EGFR mutation and brain metastasis in pulmonary adenocarcinomas. J. Thorac. Oncol. 9, 195–199. doi:10.1097/JTO.0000000000000069
Siegel, R., Ward, E., Brawley, O., and Jemal, A. (2011). Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. Ca. Cancer J. Clin. 61, 212–236. doi:10.3322/caac.20121
Sordella, R., Bell, D. W., Haber, D. A., and Settleman, J. (2004). Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 305, 1163–1167. doi:10.1126/science.1101637
Temming, K., Schiffelers, R. M., Molema, G., and Kok, R. J. (2005). RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug resist. updat. 8, 381–402. doi:10.1016/j.drup.2005.10.002
Testa, U., Castelli, G., and Pelosi, E. (2018). Lung cancers: Molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers (Basel) 10, 248. doi:10.3390/cancers10080248
Thiery, J. P., Acloque, H., Huang, R. Y., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. doi:10.1016/j.cell.2009.11.007
Westover, D., Zugazagoitia, J., Cho, B. C., Lovly, C. M., and Paz-Ares, L. (2018). Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 29, i10–i19. doi:10.1093/annonc/mdx703
Yu, H. A., Arcila, M. E., Rekhtman, N., Sima, C. S., Zakowski, M. F., Pao, W., et al. (2013). Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 19, 2240–2247. doi:10.1158/1078-0432.CCR-12-2246
Keywords: Cilengitide, cRGDwV, cRGDyV, EMT, NSCLC, A549GR
Citation: Seo Y, Seo M and Kim J (2023) Effects of cilengitide derivatives on TGF-β1-induced epithelial-to-mesenchymal transition and invasion in gefitinib-resistant non-small cell lung cancer cells. Front. Pharmacol. 14:1277199. doi: 10.3389/fphar.2023.1277199
Received: 14 August 2023; Accepted: 26 September 2023;
Published: 20 October 2023.
Edited by:
Selim Cellek, Anglia Ruskin University, United KingdomReviewed by:
Alice Lapthorn, Anglia Ruskin University, United KingdomKun Zhou, Hong Kong University of Science and Technology, Hong Kong SAR, China
Copyright © 2023 Seo, Seo and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiyeon Kim, eWVvbkBkYW5rb29rLmFjLmty
†These authors have contributed equally to this work