 Wenye Xu1,2†
Wenye Xu1,2† Ting Wu
Ting Wu Zhihong Zuo
Zhihong Zuo- 1Department of Critical Care Medicine, Xiangya Hospital, Central South University, Changsha, Hunan, China
- 2Hunan Key Laboratory of Molecular Precision Medicine, Department of Critical Care Medicine, Xiangya Hospital, Central South University, Changsha, Hunan, China
- 3Department of Cardiovascular Medicine, Xiangya Hospital, Central South University, Changsha, Hunan, China
- 4Department of Cardiovascular Medicine, The Third Xiangya Hospital, Central South University, Changsha, Hunan, China
Background: Elexacaftor-Tezacaftor-Ivacaftor (ELE/TEZ/IVA) is believed to be an effective and well-tolerated treatment for cystic fibrosis (CF), but the exact efficacy and safety profile are still unknown.
Objective: This study aimed to clarify the extent of functional restoration when patients are given with triple combination treatment and demonstrate the prevalence of adverse events, to evaluate the overall profile of ELE/TEZ/IVA on CF.
Methods: A literature search was conducted in PubMed, Web of Science and Cochrane Library. Random effects single-arm meta-analysis was performed to decipher the basal characteristics of CF, the improvement and safety profile after ELE/TEZ/IVA treatment.
Results: A total 53 studies were included in this analysis. For all the patients in included studies. 4 weeks after ELE/TEZ/IVA treatment, the increasement of percentage of predicted Forced Expiratory Volume in the first second (ppFEV1) was 9.23% (95%CI, 7.77%–10.70%), the change of percentage of predicted Forced Vital Capacity (ppFVC) was 7.67% (95%CI, 2.15%–13.20%), and the absolute change of Cystic Fibrosis Questionnaire–Revised (CFQ-R) score was 21.46 points (95%CI, 18.26–24.67 points). The Sweat chloride (SwCl) was significantly decreased with the absolute change of −41.82 mmol/L (95%CI, −44.38 to −39.25 mmol/L). 24 weeks after treatment, the increasement of ppFEV1 was 12.57% (95%CI, 11.24%–13.90%), the increasement of ppFVC was 10.44% (95%CI, 7.26%–13.63%), and the absolute change of CFQ-R score was 19.29 points (95%CI, 17.19–21.39 points). The SwCl was significantly decreased with the absolute change of −51.53 mmol/L (95%CI, −56.12 to −46.94 mmol/L). The lung clearance index2.5 (LCI2.5) was also decreased by 1.74 units (95%CI, −2.42 to −1.07 units). The body mass index increased by 1.23 kg/m2 (95%CI, 0.89–1.57 kg/m2). As for adverse events, 0.824 (95%CI, 0.769–0.879) occurred during ELE/TEZ/IVA period, while the incidence of severe adverse events was 0.066 (95%CI, 0.028–0.104).
Conclusion: ELE/TEZ/IVA is a highly effective strategy and relatively safe for CF patients and needs to be sustained to achieve better efficacy.
Systematic Review Registration: Identifier: CRD42023441840.
Introduction
Cystic fibrosis (CF), an autosomal recessive disease most commonly in Caucasian populations, limits and shortens an individual’s life, in which the main cause of mortality is respiratory failure secondary to end-stage lung disease. It has an impact on multi-organs, with clinical manifestations including liver dysfunction, pancreatic insufficiency, malnutrition, increase of sweat chloride and the end-stage respiratory failure. Currently, early diagnosis through newborn screening, multi-divisional professional medical care, the strategy of medication, and availability of therapies are necessary for improving the survival rate of patients.
The pathogenesis of cystic fibrosis is now clearly proposed as the mutation of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, which was first described in 1989 (Berger et al., 1991). A low conductance cAMP-dependent chloride channel encoded by CFTR, is located at apical membrane of epithelial cells in several tissues, including respiratory tract, the gastro-intestinal tract, sweat glands duct, and the male reproductive tract (Crawford et al., 1991). CFTR mutations are divided into 6 groups, in which the p.Phe508del is class II and the most prevalent. p.Phe508del contributes to protein incorrectly folded and rapidly degraded, impairing protein trafficking to the cell surface and results in severe reduction of CFTR activity. In class I mutations, the production of CFTR protein is decreased; in class III mutations, the CFTR protein is not functional (“gating mutations”–for example G551D mutation); in class IV mutations, ions transport is diminished; class V mutations produce inadequate quantities of the CFTR protein and class VI mutations produce a less stable CFTR protein (Crawford et al., 1991; Ren et al., 2018). The phenotype of CF caused by class I-III mutations compared to class IV-VI mutations is much severer. Frequently, the mutations lead to more than one mechanism of protein failure, thus belong to more than one class.
Traditional therapies were the management of symptoms including airway clearance, antibiotics, and nutritional support. Until recent years, researchers pay attention to the recovery of CFTR function (Burgener and Moss, 2018; Bell et al., 2020). CFTR function might be partially rescued by small molecules known as modulators, which contains potentiators and correctors, that increase conductance of the CFTR channel and improve CFTR trafficking to the cell surface, respectively (Bardin et al., 2021; Shteinberg et al., 2021). Ivacaftor, one of the potentiators, was proven to be effective in a III clinical trial, leading to significant pulmonary and nutritional improvements, which also gave clues for further precise medicine in CF (Sermet-Gaudelus, 2013). Correctors, such as tezacaftor (TEZ), lumacaftor, and the next-generation elexacaftor (ELE), correct p.Phe508del folding and trafficking defect (Haggie et al., 2017; Donaldson et al., 2018; Southern et al., 2018). The combination of Ivacaftor (IVA) and Lumacaftor was firstly approved by the North American and European regulatory agencies, resulting in a modest improvement of lung function and other clinical outcomes (Wainwright et al., 2015). Hence, combined molecule therapy is getting more and more attentions.
Afterwards, it was found that treatment with TEZ/IVA was effective in terms of lung function and resulted in a significantly lower rate of pulmonary exacerbations than placebo (Southern et al., 2020). Additionally, compared to TEZ/IVA treatment, triple combination of elexacaftor-tezacaftor-ivacaftor (ELX/TEZ/IVA) has been shown to be the most effective strategy to improve the expression of corrected p.Phe508del CFTR protein at the cell surface according to in vitro experiments and clinical results (Furstova et al., 2022; Shakir et al., 2022; Pallenberg et al., 2023). Two CFTR correctors, ELX and TEZ, increase cell surface expression by improving folding and trafficking of F508del, and the CFTR potentiator IVA augments gating of CFTR channels inserted into the apical cell membrane (Wainwright et al., 2015; Taylor-Cousar et al., 2017). Triple combination showed unprecedented improvements in clinical outcomes including spirometry, nutritional outcomes, and patient-reported respiratory symptoms in patients with one or two F508del alleles (Zemanick et al., 2021). Significant reduction of sweat chloride concentration, a direct indicator of systemic CFTR function, was also demonstrated after triple therapy (Fidler et al., 2017). This triple combination was first approved in the United States of America in October 2019 for patients ≥12 years old, but has become available for children ≥6 years old since June 2021 (Goetz and Savant, 2021). However, the extent of functional restoration achieved by ELX/TEZ/IVA and the duration of this treatment strategy in these CF target organs in patients with F508del mutations has not been studied. The safety profile of this triple treatment remains to be elucidated as well.
In the systemic review and meta-analysis, we included 53 studies and analyzed the characteristics of patients, the endpoints, and adverse events after using ELE/TEZ/IVA, to evaluate the overall efficacy and safety.
Materials and methods
This study was registered in PROSPERO, with registration No. CRD42023441840.
Search strategy
We searched PubMed, Web of Science, and the Cochrane Library from 1998 to 4 August 2023. The following items were used: “elexacaftor-tezacaftor-ivacaftor”, “Trikafta” “VX445” “ELE/TEZ/IVA” and “cystic fibrosis” alone or in combination. The references of literature reviews and original articles were also scanned to avoid missing any qualified studies.
Inclusion and exclusion criteria
The inclusion criteria were as follows: 1) prospective clinical studies (including randomized control trials and single-arm studies); 2) observational studies involving elexacaftor-tezacaftor-ivacaftor on cystic fibrosis; and 3) studies reporting the efficacy and safety of elexacaftor-tezacaftor-ivacaftor on cystic fibrosis. The exclusion criteria were as follows: 1) article type: letters, editorials, expert opinions, case reports and reviews; 2) studies without useable data; and 3) duplicate publications.
Study Selection
All studies from the electronic search were uploaded into Endnote X9 and duplicates were removed. Two independent investigators reviewed remaining identified trials to confirm that they fulfilled the inclusion criteria. Finally, reference lists of included studies were screened to assess other potentially relevant studies. All disagreements were discussed and solved after rechecking the source data with a third investigator; in all cases one person recognized an error.
Data extraction
Two investigators extracted data from the eligible studies independently, and any disagreements were resolved by discussion with a third investigator. For each study, the following characteristic information was recorded: the first author, year of publication, number of patients, CF related diabetes, pancreatic insufficiency, p.Phe508del mutation, percentage of predicted Forced Expiratory Volume in the first second (ppFEV1), percentage of predicted Forced Vital Capacity (ppFVC), lung clearance index2.5 (LCI2.5), Cystic Fibrosis Questionnaire–Revised (CFQ-R) respiratory domain score, sweat chloride concentration, body mass index (BMI), Hemoglobin A1C(HbA1c); absolute change in sweat chloride concentration from baseline at week 4 and week 24, absolute change in percentage of predicted FEV1 from baseline at week 4 and week 24, absolute change in percentage of predicted FVC from baseline through week 4 and week 24, absolute change in CFQ-R respiratory domain score from baseline at week 4 and week 24, absolute change in BMI from baseline at week 24, absolute change in LCI2.5 from baseline through week 24; the incidence of adverse events during elexacaftor-tezacaftor-ivacaftor treatment. Severe adverse events were defined by clinicians or investigators according to the severity of clinical manifestations in the included studies.
Quality assessment
The quality assessment forms recommended by the Agency for Healthcare Research and Quality were used for quality assessment of the included trials. We resolved all disagreements through a discussion.
Statistical analysis
The analysis software used in our study was Stata 14.0(Stata Corporation, College Station, Texas, United States of America). Meta-analysis was performed using the “metaprop” command. Since most of the included studies were single-arm studies, there may be high heterogeneity. Therefore, the combined results were expressed as the benefit value plus 95% confidence interval (CI) under the random effects model, and the incidence of each variable was analyzed. For all analysis results, p < 0.05 was considered statistically significant.
Results
Selection of included studies and study characteristics
A total of 453 relevant articles were identified by searching online databases. Figure 1 showed the screening and selection process of the eligible trials. This meta-analysis included 53 studies, of which 11 articles were clinical trials and 42 articles were observational studies. All these studies were conducted in Caucasian countries. The age of included subjects ranged from 2 years old to 72 years old. The characteristics of the included trials were listed in Table 1. The results of the quality assessments were presented in Table 1.
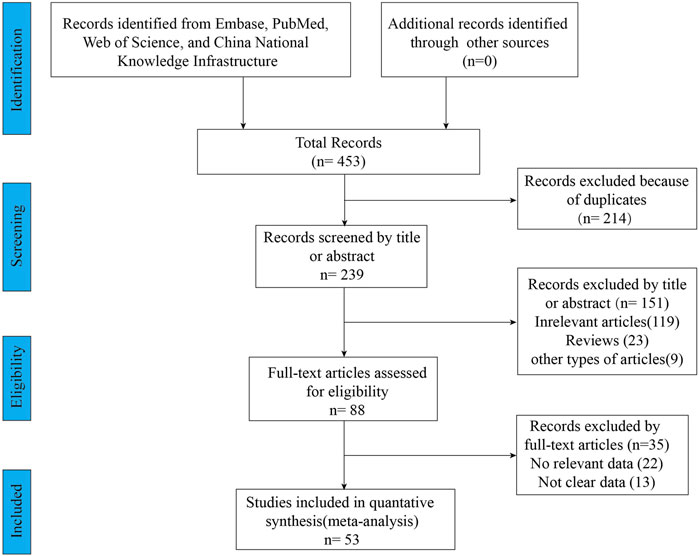
FIGURE 1. PRISMA diagram of study selection.
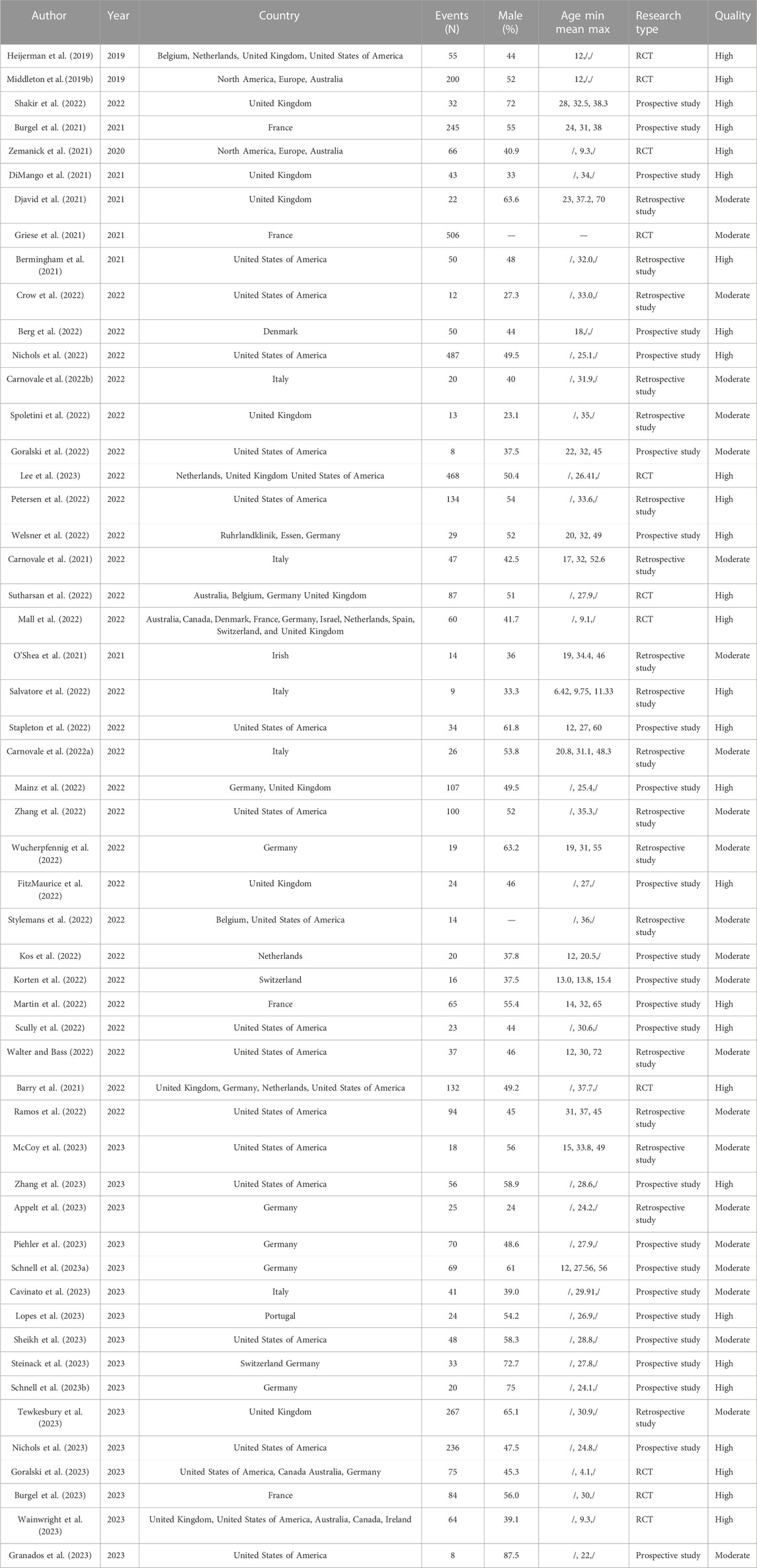
TABLE 1. Study characteristics of included studies.
The basic characteristics of pulmonary function, nutritional status, secretion of sweat glands in CF patients
The pooled meta-analysis results indicated that ppFEV1 was 57.53% (95%CI, 50.99%–64.06%), ppFVC was 70.20% (95%CI, 62.95%–77.45%), CFQ-R score was 65.59 points (95%CI, 60.44–70.75 points), and LCI2.5 was 10.90 units (95%CI, 9.35–12.44 units), which revealed the overall pulmonary function of included patients. Patients with cystic fibrosis were inclined to suffer from diabetes, with the incidence of 0.406 (95%CI, 0.357–0.455). The HbA1c was 5.98% (95%CI, 5.64%–6.31%), close to the threshold limit value, which suggesting patients were at high risk of diabetes. Pancreatic insufficiency commonly occurred in 89.5% of CF patients, with 95%CI (0.849,0.941) in this meta-analysis. Among these patients, over a half were F508del homozygous, the incidence was 0.552 (95%CI, 0.445–0.659). As for the nutritional evaluation, the BMI of included subjects was 21.29 kg/m2 (95%CI, 20.55–21.83 kg/m2). SwCl, directly associated with the severity of CF, was 94.55 mmol/L (95%CI, 91.48–97.42 mmol/L). All these data show the basal characteristics of CF patients, which were seen in Figures 2–5 and Table2.
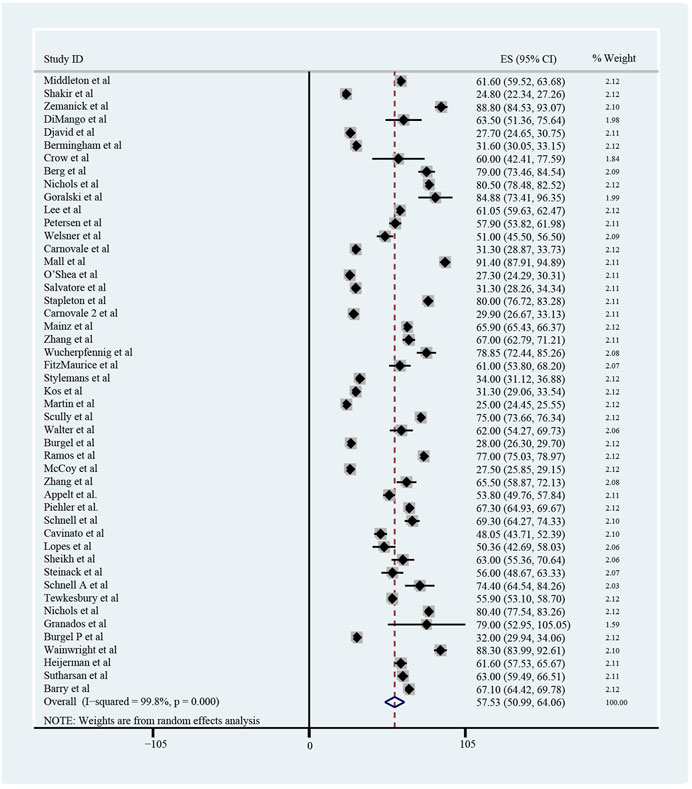
FIGURE 2. Percentage of Predicted Forced Expiratory Volume in the first second (ppFEV1) of included patients before ELX/TEZ/IVA treatment.
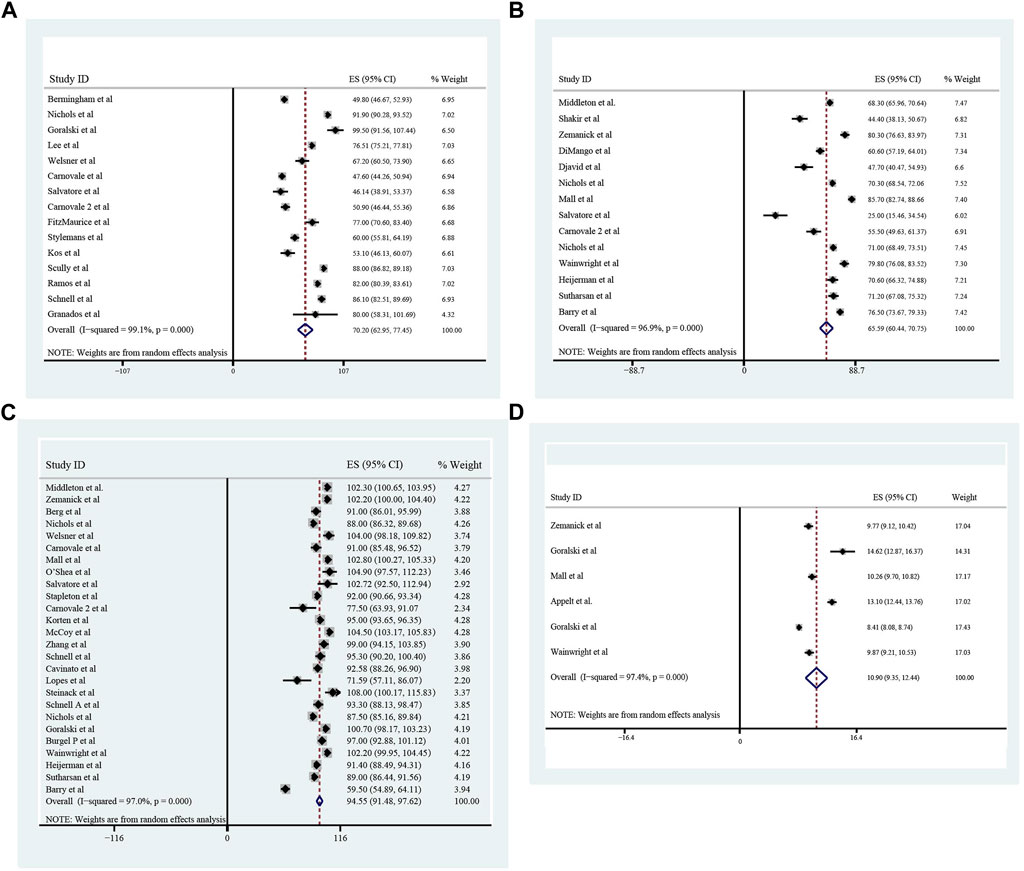
FIGURE 3. The Percentage of predicted Forced Vital Capacity (A), Cystic Fibrosis Questionnaire-Revised Score (B), Sweat Chloride (C), and Lung Clearance Index2.5 (D) of included patients before ELX/TEZ/IVA treatment.
FIGURE 4. Body Mass Index of included patients before ELX/TEZ/IVA treatment.
FIGURE 5. Homozygous F508 mutation (A), pancreatic insufficiency (B), cystic fibrosis related diabetes (C) and HbA1c (D) of included patients before ELX/TEZ/IVA treatment.
TABLE 2. The general characteristics of included subjects.
Improvement of pulmonary function, nutrition, sweat chloride after triple treatment with ETX/TEZ/IVA for 4 weeks and 24 weeks
All these included patients were eligible for ETX/TEZ/IVA triple treatment. After being treated with triple combination for 4 weeks and 24 weeks respectively (Figures 6, 7), the pulmonary function, nutrition status and concentration of sweat chloride were improved at certain extent (Table 3). After 4 weeks, the increasement of ppFEV1 was 9.23% (95%CI, 7.77%–10.70%), the increasement of ppFVC was 7.67% (95%CI, 2.15%–13.20%), and the absolute change of CFQ-R was 21.46points (95%CI, 18.26–24.67points). The SwCl was significantly decreased with the absolute change at −41.82 mmol/L (95%CI, −44.38 to −39.25 mmol/L). At 24 weeks after treatment, the increasement of ppFEV1 was 12.57% (95%CI, 11.24%–13.90%), the increasement of ppFVC was 10.44% (95%CI, 7.26%–13.63%), and the absolute change of CFQ-R was 19.29points (95%CI, 17.19–21.39points). The SwCl was significantly decreased with the absolute change at −51.53 mmol/L (95%CI, −56.12 to −46.94 mmol/L). The LCI2.5 was also decreased with the value at −1.74units (95%CI, −2.42 to −1.07units). The BMI increased by 1.23 kg/m2 (95%CI, 0.89–1.57 kg/m2). All the data were shown in Table 3. Prolonged period of triple combination treatment contributes to better improvement.
FIGURE 6. The absolute changes of Percentage of Predicted Forced Expiratory Volume in the first second (A), Sweat Chloride (B), Cystic Fibrosis Questionnaire-Revised Score (C), and Percentage of predicted Forced Vital Capacity (D) among included patients after 4 weeks of ELX/TEZ/IVA treatment.
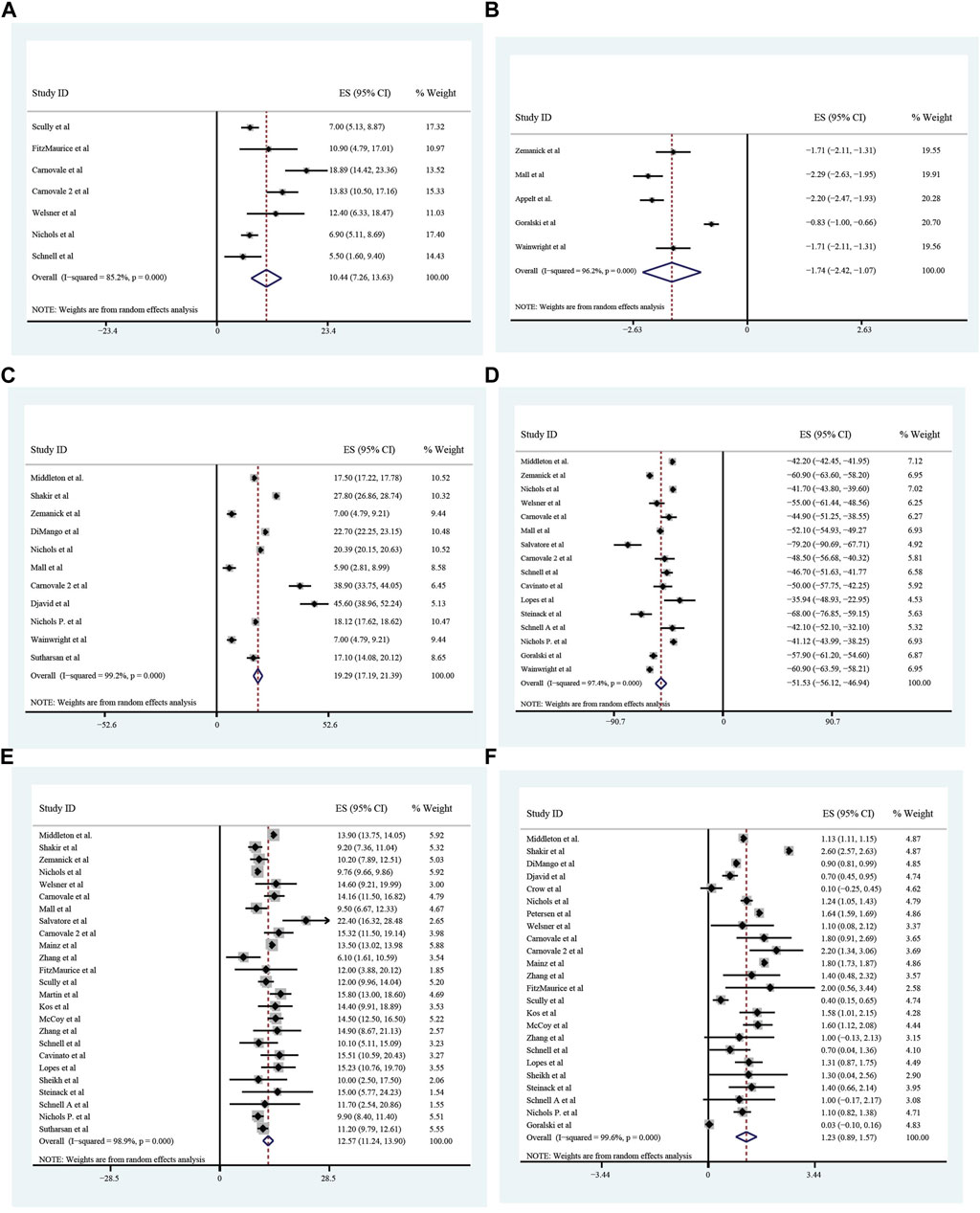
FIGURE 7. The absolute changes of Percentage of predicted Forced Vital Capacity (A), Lung Clearance Index2.5 (B), Cystic Fibrosis Questionnaire-Revised Score (C), Sweat Chloride (D), and Percentage of Predicted Forced Expiratory Volume in the first second (E) and Body Mass Index (F) among included patients after 24 weeks of ELX/TEZ/IVA treatment.
TABLE 3. Improvement of lung function, nutrition and sweat chloride after triple treatment.
The adverse events occurred during ETX/TEZ/IVA treatment
The overall incidence of adverse events associated with ETX/TEZ/IVA treatment was 0.824 (95%CI, 0.769–0.879), among which the incidence of severe adverse events was 0.066 (95%CI, 0.028–0.104). (Figures 8, 9).The specific symptoms of adverse events included cough 0.238 (95%CI, 0.141–0.335), rhinorrhea 0.212 (95%CI, 0.119–0.305), headache 0.161 (95%CI, 0.111–0.211), rash 0.101 (95%CI, 0.065–0.138), oropharyngeal pain 0.117 (95%CI, 0.082–0.152), abdominal pain 0.096 (95%CI, 0.051–0.142), nasopharyngitis 0.125 (95%CI, 0.100–0.149), upper respiratory tract infection 0.119 (95%CI, 0.097–0.140), transaminase increase 0.075 (95%CI, 0.057–0.093), infective pulmonary exacerbation of CF 0.148 (95%CI, 0.019–0.277).
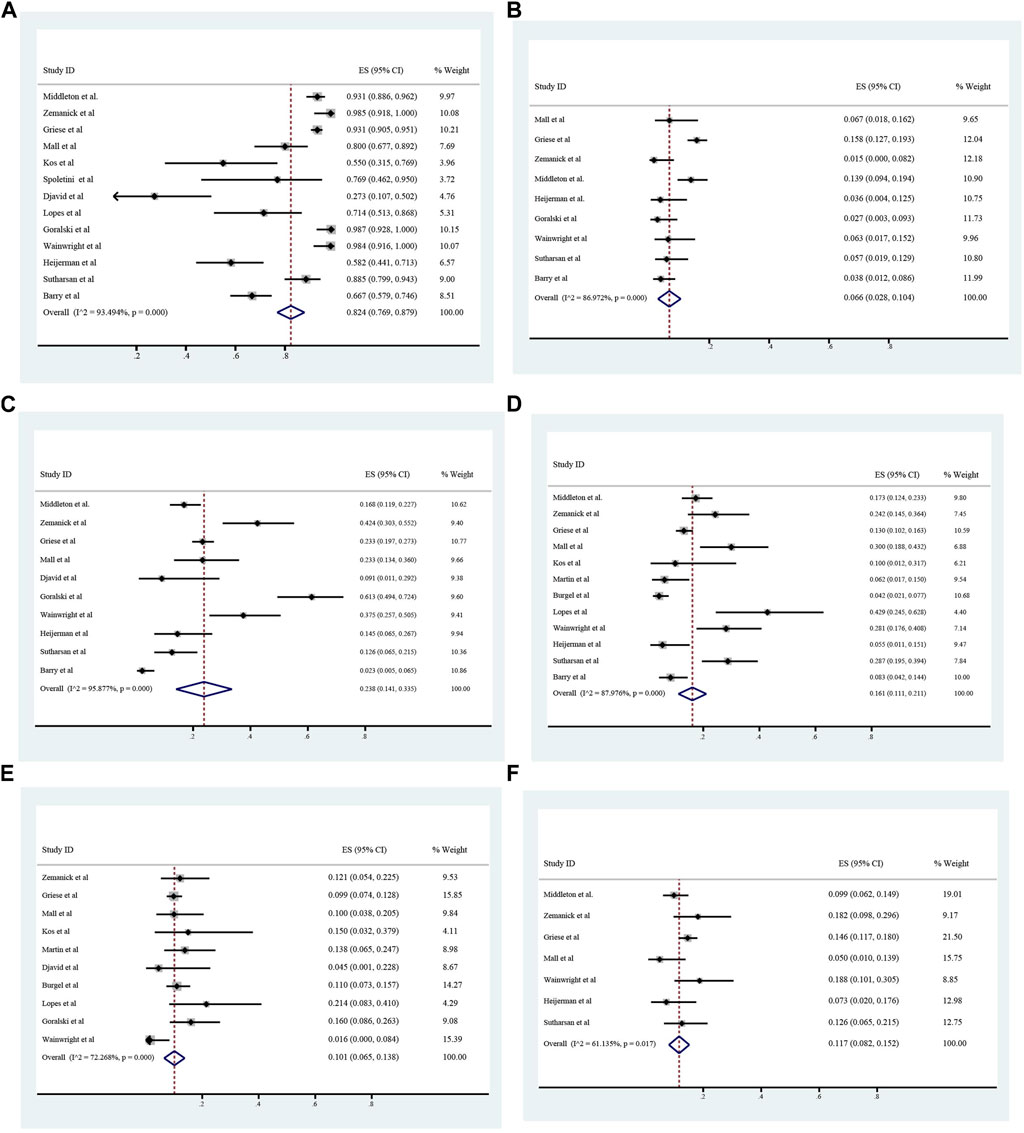
FIGURE 8. Incidence of total and severe adverse events (A and B), cough (C), headache (D), rash (E), and oropharyngeal pain (F) during ELX/TEZ/IVA therapy.
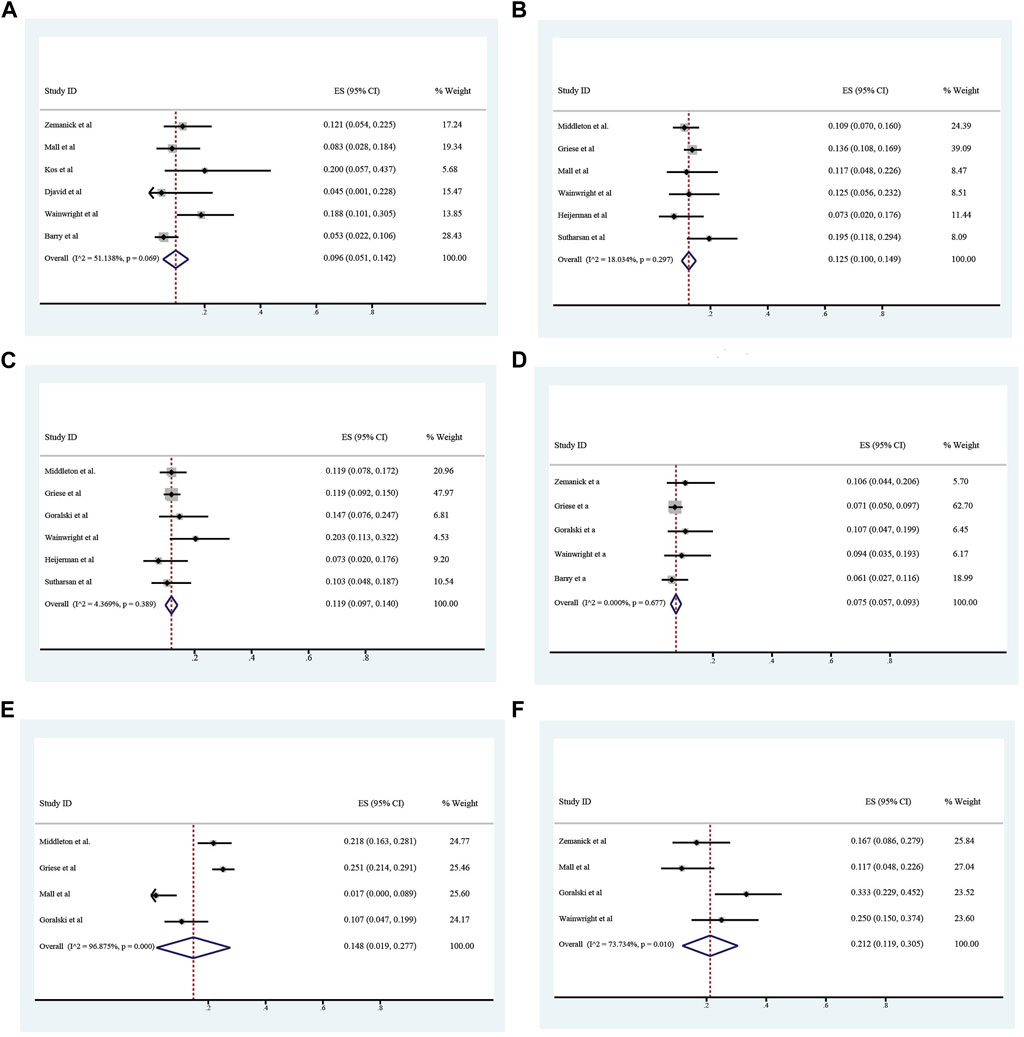
FIGURE 9. Incidence of abdominal pain (A), nasopharyngeal pain (B), upper respiratory tract infection (C), alamine transferase increase (D), infective pulmonary exacerbation of cystic fibrosis (E) and rhinorrhea (F) during ELX/TEZ/IVA therapy.
Discussion
In this single-arm meta-analysis, we included 53 studies in almost Caucasian countries, with the subjects ranging from children aged 2 years old to adults. For all the patients in included studies, 4 weeks after ELE/TEZ/IVA treatment, the increasement of percentage of ppFEV1 was 9.23% (95%CI, 7.77%–10.70%), the change of ppFVC was 7.67% (95%CI, 2.15%–13.20%), and the absolute change of CFQ-R score was 21.46 points (95%CI, 18.26–24.67 points). The SwCl was significantly decreased with the absolute change of −41.82 mmol/L (95%CI, −44.38 to −39.25 mmol/L). 24 weeks after treatment, the increasement of ppFEV1 was 12.57% (95%CI, 11.24%–13.90%), the increasement of ppFVC was 10.44% (95%CI, 7.26%–13.63%), and the absolute change of CFQ-R score was 19.29 points (95%CI, 17.19–21.39 points). The SwCl was significantly decreased with the absolute change of −51.53 mmol/L (95%CI, −56.12 to −46.94 mmol/L). The lung clearance index2.5 (LCI2.5) was also decreased by 1.74 units (95%CI, −2.42 to −1.07 units). The body mass index increased by 1.23 kg/m2 (95%CI, 0.89–1.57 kg/m2). Unsurprisingly, the prolonged course of treatment brings more improvement. Additionally, these triple combination led to an overall 82.4% incidence of adverse events, while the incidence of severe ones was only 6.6%. Thes adverse events were common symptoms, such as cough, headache, rash, and oropharyngeal pain, which were unlikely to cause medication discontinuation or disease exacerbation.
SwCl is believed to be directly associated with CFTR function. Different functional classes of CFTR mutations would have differences in epithelial chloride conductance, thus leading to variations of SwCl in children and adults (Wilschanski et al., 1995; Lebecque et al., 2002). Multicenter trials suggested SwCl as a biomarker of CFTR activity and to test the effect of CFTR potentiators (Accurso et al., 2014). Therefore, the test of SwCl is used as the gold standard for CF diagnosis among symptomatic patients, also within the infant screening and in the follow-up of CF patients during molecular therapies (Sontag, 2016). Although the sweat test is considered a robust measure, SwCl measurements in patients with CF and a G551D mutation had an inherent biological variability that was higher than commonly considered (Vermeulen et al., 2017). Notably, the normal SwCl could not exclude CF (Stewart et al., 1995). During infant screening, SwCl concentrations in CF patients did not change in a meaningful way during the first year of life (LeGrys et al., 2019). Additionally, compared to males, SwCl response was larger in females, which also explained the negative correlation of weight with the response in sweat chloride concentration after start of lumacaftor/ivacaftor (Aalbers et al., 2021). Furthermore, the value of SwCl also varied among different age (Traeger et al., 2014). Nevertheless, SwCl concentration did not necessarily predict a milder pulmonary course in patients with cystic fibrosis (Davis et al., 2004). Heltshe et al. found it was difficult to identify a minimum threshold for change in SwCl that was associated with FEV1 improvement (Heltshe et al., 2013). There was significant variability in sweat chloride distribution across CFTR class 2–5 genotypes. The relationship between sweat chloride and mortality varied by genotype with a relatively strong relationship in R117H/F508del patients (Espel et al., 2018). SwCl concentration might be a useful predictor of mortality and clinical phenotype when CFTR genotype functional class was unclassified (McKone et al., 2015). A clinical trial proposed SwCl was as a clinical endpoint to measure efficacy based on ivacaftor as well (Durmowicz et al., 2013). Changes in SwCl at early stage of treatment had the sufficient predictive potential to identify the individuals that showed large improvement at later stage (Seliger et al., 2013). ELX/TEZ/IVA treatment of children with CF led to greater improvements in sweat chloride than those previously seen in adults and adolescents (Middleton et al., 2019a; Shakir et al., 2022). Even though the concentration of SwCl is influenced by gender, age, and types of mutations and might be associated with lung function, both observational studies and clinical trials consider it as one of endpoints in evaluating therapy. As shown in this meta-analysis, regardless of age distribution, male percentage and previous medicine take, SwCl decreased by 41.82 mmol/L and 51.53 mmol/L after 4 weeks and 24 weeks respectively, which indicated triple treatment improved CFTR function efficiently and longer treatment duration brought about more reduction. Moreover, the decrease of SwCl was related to improved survival in patients with CF (McKone et al., 2015). In conclusion, substantial improvements in sweat chloride in response to ELX/TEZ/IVA therapy may presage improvements in long-term clinical outcomes.
Improvement of pulmonary function is another manifestation of molecular therapy. Lung is one of the major target organs of CF. In the worst scenario, patients would develop pulmonary fibrosis and require lung transplant or even die. Spirometric variables, FEV1 and FVC, are employed to measure the lung disease progression of CF. FEV1 has served as the critical primary endpoint of most clinical trials (Fuchs et al., 1994; Saiman et al., 2003). The rate of lung function decline was shown to be related to survival of CF (Corey et al., 1997; Schluchter et al., 2002), while decline in lung function did not predict future decline in CF patients (Rosenfeld et al., 2015). In a retrospective study, even with attenuated lung function decline, there were still reduced survival in adult patients (Keating et al., 2017). Hence, studies evaluated efficacy based on improvement of FEV1 and FVC rather than rate of decline. A decrease in ppFEV1 predicted a decrease in health-related quality of life over time, which demonstrated ppFEV1 should be increased to improve patients’ life quality (Abbott et al., 2013). The value of FEV1 at age 6 was proved to be an independent predictor for CF-related mortality by a retrospective study (Vandenbroucke et al., 2020). Also, a marked improvement of ppEFV1 occurred in survival patients with an FEV1 below 30% (George et al., 2011). Basically, children had a higher baseline of ppFEV1, ppFVC and CFQ-R score than adolescents and adults (Middleton et al., 2019a; Shakir et al., 2022). Despite different baselines, ELX/TEZ/IVA therapy led to a 9.23% improvement in mean ppFEV1, a 7.67% increase in mean ppFVC, and 21.67 point in CFQ-R score (a questionnaire based on patients’ self-evaluation) at week 4 in present study. LCI2.5, a measure of ventilation inhomogeneity that might be more sensitive than spirometry in detecting lung function changes during childhood, had a mean decrease of 1.74 units after 24-week triple therapy, which further corroborated the improvement of ppFEV1. LCI2.5 was proved to be able to discover early stage of lung injury in CF based on a large longitudinal study (Kraemer et al., 2005) and it was also indictive for the presence of lung structure changes after 3 years in 87% CF cases (Fuchs et al., 2014). Early initiation of ELX/TEZ/IVA in children with CF is helpful for improving lung function and minimizing the decline of ppFEV1 and LCI2.5 tightly associated with disease progression (VanDevanter et al., 2016). In addition, most studies focused on the efficacy on patients older than 6 years old, while a recent clinical trial found even in children 2 through 5 years of age, ELX/TEZ/IVA led to clinically meaningful reductions in SwCl concentration and lung clearance index (Goralski et al., 2023). Regardless of age, gender or mutation type, ELX/TEZ/IVA is an effective therapy, rapidly ameliorating the lung function in CF patients.
Benefits of ELX/TEZ/IVA combination were observed on other important endpoints, including surrogates for nutritional health. On one hand, CFRD, a complex and multifactorial disease, attributing to insulin secretion insufficiency and peripheral insulin resistance, represents capacity of dealing with carbohydrate. The mean prevalence of CRFD in this meta-analysis was 40.6% and HbA1c was 5.98%, close the upper range of reference. CFTR modulators were shown to be beneficial for controlling blood glucose level (Crow et al., 2022; Korten et al., 2022). However, relevant studies were not adequately conducted to quantify its efficacy. On the other hand, weight and BMI are accompanied with nutritional status; however, BMI is more commonly used to measure an individual’s nutrition. BMI in CF was closely correlated with improvements in lung function and was independent predictors of survival (Liou et al., 2001; Yen et al., 2013). Underweight status was independently associated with adverse clinical outcomes in CF, including worsening lung function, morbidity, and mortality (Culhane et al., 2013). Even for severe cases requiring lung transplantation, low BMI was suggested to portend a poor outcome after transplantation (Snell et al., 1998). CF management guidelines have focused primarily on attenuation of nutritional failure with recommendations to maintain a BMI above 22 kg/m2 in women and 23 kg/m2 in men (Castellani et al., 2018). Nevertheless, overweight and obesity were risk factors in adult CF patients with severe scenario (Harindhanavudhi et al., 2020), which was further validated in an Italy multicenter study (Gramegna et al., 2022). In addition, a meta-analysis also concluded that the currently recommended target BMI in patients with CF should be reconsidered, since higher BMI was not beneficial for outcomes of CF (Nagy et al., 2022). In present study, the mean BMI of included subjects is 21.29 kg/m2, and the increase of BMI after 24-week triple treatment is 1.23 kg/m2, indicating that ELX/TEZ/IVA is promising for improving patients’ nutrition status.
Adverse events are prevalent in ELX/TEZ/IVA triple therapy with an incidence at 0.824, which were almost associated with CF development and mild to moderate, while rate of severe adverse events was only 6.6%, rarely leading to the discontinuation of treatment. All clinical trials in present study concluded consistent results that ELX/TEZ/IVA was generally safe and well-tolerated. Cough, rhinorrhea, and headache ranked top 3 among these adverse events. The notable adverse events, infective pulmonary exacerbation of CF, occurred in 14.8% patients. Pulmonary exacerbations were associated with a higher risk of lung function decline and decreased survival (Waters et al., 2012). Compared to placebo treatment, the incidence of pulmonary exacerbations in children aged 6 to 11 receiving ELX/TEZ/IVA treatment was only 1.7% in a randomized clinical trial (Mall et al., 2022). However, in another clinical trial conducted in patients 12 years of age or older, pulmonary exacerbations in ELX/TEZ/IVA group was 21.8%, while in placebo group it was 47.3% (Middleton et al., 2019b). The differences between these clinical trials resulted from age and dosage of medicine. Even relative to the TEZ/IVA group, there was still a reduction in reported adverse events of infective pulmonary exacerbation of CF in the ELX/TEZ/IVA group (Shakir et al., 2022). Although commonly seen during the treatment, these adverse events were still under control and unlikely to cause deterioration of cystic fibrosis. Nevertheless, small size of samples in most included studies limited the ability to detect uncommon and rare adverse events, which were not categorized and shown in the analysis, for example, influenza. In summary, the overall safety profile of present study was consistent with clinical trials and real-world findings.
Among these studies included in this meta-analysis, most followed up the efficacy and safety after 4 and 24 weeks of ELX/TEZ/IVA treatment and patients achieved the initial improvement as early as 4 weeks after receiving medications. 48-week and 2-year period were investigated by several studies, and it was found no loss of pulmonary function or any new safety concern, suggesting ELX/TEZ/IVA is persistently effective and well-tolerated (Carnovale et al., 2022a; Lee et al., 2023). However, it is urgent to determine the exact follow-up periods to validate the observed improvements with respect to clinical stability, slowing down or stopping the decline in pulmonary function, reducing the incidence of complications, improving the survival and enhancing the compliance of patients. Further prospective research and clinical trials are needed to determine the optimal duration of therapy. Additionally, patients at different age would be prescribed for different dosage, bringing in the variance of improvement and safety events. Therefore, the best dosage for different age groups requires further exploration.
Limitations
Due to ethical issues, the majority of clinical trials did not include a placebo group. The incidence of safety events attributable to the underlying disease process was not ascertained and the efficacy could not be compared quantitively and directly. Secondly, CF is prevalent in children to adult patients and has several mutations, so it is difficult to do sub-analysis for age and mutation, respectively. The mean incidence of homozygous F508del mutation in present meta-analysis is 55.2%, which predominated among all mutations. Moreover, most study follow-ups were conducted during the pandemic of COVID−19, and the safety results reported were based on both in-clinic and in-home safety assessments, while the in-home safety assessments were not presented in study.
Conclusion
In conclusion, this single-arm meta-analysis demonstrated that ELX/TEZ/IVA therapy is effective and well-tolerated among CF patients in different age groups. Prolonged treatment duration would bring more improvement of pulmonary function, CFTR function and nutritional status without extra risk of adverse events. Clinicians are supposed to encourage the application of ELX/TEZ/IVA and adjust the dosage and period according to the individual’s variance.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
WX: Investigation, Methodology, Writing–original draft, Writing–review and editing. TW: Data curation, Formal Analysis, Methodology, Software, Writing–review and editing. ZiZ: Conceptualization, Writing–review and editing. ZhZ: Conceptualization, Investigation, Methodology, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aalbers, B. L., Hofland, R. W., Bronsveld, I., de Winter-de Groot, K. M., Arets, H. G. M., de Kiviet, A. C., et al. (2021). Females with cystic fibrosis have a larger decrease in sweat chloride in response to lumacaftor/ivacaftor compared to males. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 20 (1), e7–e11. doi:10.1016/j.jcf.2020.05.004
Abbott, J., Hurley, M. A., Morton, A. M., and Conway, S. P. (2013). Longitudinal association between lung function and health-related quality of life in cystic fibrosis. Thorax 68 (2), 149–154. doi:10.1136/thoraxjnl-2012-202552
Accurso, F. J., Van Goor, F., Zha, J., Stone, A. J., Dong, Q., Ordonez, C. L., et al. (2014). Sweat chloride as a biomarker of CFTR activity: proof of concept and ivacaftor clinical trial data. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 13 (2), 139–147. doi:10.1016/j.jcf.2013.09.007
Appelt, D., Steinkamp, G., Sieber, S., and Ellemunter, H. (2023). Early and sustained improvements of lung clearance index from two to sixteen weeks of elexacaftor/tezacaftor/ivacaftor therapy in patients with cystic fibrosis-a real world study. Front. Pharmacol. 14, 1125853. doi:10.3389/fphar.2023.1125853
Bardin, E., Pastor, A., Semeraro, M., Golec, A., Hayes, K., Chevalier, B., et al. (2021). Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 213, 113195. doi:10.1016/j.ejmech.2021.113195
Barry, P. J., Mall, M. A., Álvarez, A., Colombo, C., de Winter-de Groot, K. M., Fajac, I., et al. (2021). Triple therapy for cystic fibrosis phe508del-gating and -residual function genotypes. N. Engl. J. Med. 385 (9), 815–825. doi:10.1056/NEJMoa2100665
Bell, S. C., Mall, M. A., Gutierrez, H., Macek, M., Madge, S., Davies, J. C., et al. (2020). The future of cystic fibrosis care: a global perspective. Lancet Respir. Med. 8 (1), 65–124. doi:10.1016/S2213-2600(19)30337-6
Berg, P., Sorensen, M. V., Rousing, A. Q., Vebert Olesen, H., Jensen-Fangel, S., Jeppesen, M., et al. (2022). Challenged urine bicarbonate excretion as a measure of cystic fibrosis transmembrane conductance regulator function in cystic fibrosis. Ann. Intern. Med. 175 (11), 1543–1551. doi:10.7326/M22-1741
Berger, H. A., Anderson, M. P., Gregory, R. J., Thompson, S., Howard, P. W., Maurer, R. A., et al. (1991). Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J. Clin. investigation 88 (4), 1422–1431. doi:10.1172/JCI115450
Bermingham, B., Rueschhoff, A., Ratti, G., Nesmith, A., Goodwin, D., et al. (2021). Short-term effect of elexacaftor-tezacaftor-ivacaftor on lung function and transplant planning in cystic fibrosis patients with advanced lung disease. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 20 (5), 768–771. doi:10.1016/j.jcf.2021.05.009
Burgel, P. R., Durieu, I., Chiron, R., Ramel, S., Danner-Boucher, I., Prevotat, A., et al. (2021). Rapid improvement after starting elexacaftor-tezacaftor-ivacaftor in patients with cystic fibrosis and advanced pulmonary disease. Am. J. Respir. Crit. care Med. 204 (1), 64–73. doi:10.1164/rccm.202011-4153OC
Burgel, P. R., Sermet-Gaudelus, I., Durieu, I., Kanaan, R., Macey, J., Grenet, D., et al. (2023). The French Compassionate Program of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis with advanced lung disease and no F508del CFTR variant. Eur. Respir. J., 2202437. doi:10.1183/13993003.02437-2022
Burgener, E. B., and Moss, R. B. (2018). Cystic fibrosis transmembrane conductance regulator modulators: precision medicine in cystic fibrosis. Curr. Opin. Pediatr. 30 (3), 372–377. doi:10.1097/MOP.0000000000000627
Carnovale, V., Iacotucci, P., Terlizzi, V., Colangelo, C., Ferrillo, L., Pepe, A., et al. (2022a). Elexacaftor/Tezacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation and Advanced Lung Disease: a 48-Week Observational Study. J. Clin. Med. 11 (4), 1021. doi:10.3390/jcm11041021
Carnovale, V., Iacotucci, P., Terlizzi, V., Colangelo, C., Medio, P., Ferrillo, L., et al. (2021). Effectiveness and safety of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease with the Phe508del/minimal function genotype. Respir. Med. 189, 106646. doi:10.1016/j.rmed.2021.106646
Carnovale, V., Scialò, F., Gelzo, M., Iacotucci, P., Amato, F., Zarrilli, F., et al. (2022b). Cystic Fibrosis Patients with F508del/Minimal Function Genotype: laboratory and Nutritional Evaluations after One Year of Elexacaftor/Tezacaftor/Ivacaftor Treatment. J. Clin. Med. 11 (23), 6900. doi:10.3390/jcm11236900
Castellani, C., Duff, A. J. A., Bell, S. C., Heijerman, H. G. M., Munck, A., Ratjen, F., et al. (2018). ECFS best practice guidelines: the 2018 revision. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 17 (2), 153–178. doi:10.1016/j.jcf.2018.02.006
Cavinato, L., Luly, F. R., Pastore, V., Chiappetta, D., Sangiorgi, G., Ferrara, E., et al. (2023). Elexacaftor/tezacaftor/ivacaftor corrects monocyte microbicidal deficiency in cystic fibrosis. Eur. Respir. J. 61 (4), 2200725. doi:10.1183/13993003.00725-2022
Corey, M., Edwards, L., Levison, H., and Knowles, M. (1997). Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J. Pediatr. 131 (6), 809–814. doi:10.1016/s0022-3476(97)70025-8
Crawford, I., Maloney, P. C., Zeitlin, P. L., Guggino, W. B., Hyde, S. C., Turley, H., et al. (1991). Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc. Natl. Acad. Sci. U. S. A. 88 (20), 9262–9266. doi:10.1073/pnas.88.20.9262
Crow, H., Bengtson, C., Shi, X., Graves, L., and Anabtawi, A. (2022). CGM patterns in adults with cystic fibrosis-related diabetes before and after elexacaftor-tezacaftor-ivacaftor therapy. J. Clin. Transl. Endocrinol. 30, 100307. doi:10.1016/j.jcte.2022.100307
Culhane, S., George, C., Pearo, B., and Spoede, E. (2013). Malnutrition in cystic fibrosis: a review. Nutr. Clin. Pract. official Publ. Am. Soc. Parenter. Enter. Nutr. 28 (6), 676–683. doi:10.1177/0884533613507086
Davis, P. B., Schluchter, M. D., and Konstan, M. W. (2004). Relation of sweat chloride concentration to severity of lung disease in cystic fibrosis. Pediatr. Pulmonol. 38 (3), 204–209. doi:10.1002/ppul.20054
DiMango, E., Overdevest, J., Keating, C., Francis, S. F., Dansky, D., and Gudis, D. (2021). Effect of highly effective modulator treatment on sinonasal symptoms in cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 20 (3), 460–463. doi:10.1016/j.jcf.2020.07.002
Djavid, A. R., Thompson, A. E., Irace, A. L., Gusman, E., Altman, K., DiMango, E. A., et al. (2021). Efficacy of elexacaftor/tezacaftor/ivacaftor in advanced cystic fibrosis lung disease. Ann. Am. Thorac. Soc. 18 (11), 1924–1927. doi:10.1513/AnnalsATS.202102-220RL
Donaldson, S. H., Pilewski, J. M., Griese, M., Cooke, J., Viswanathan, L., Tullis, E., et al. (2018). Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. care Med. 197 (2), 214–224. doi:10.1164/rccm.201704-0717OC
Durmowicz, A. G., Witzmann, K. A., Rosebraugh, C. J., and Chowdhury, B. A. (2013). Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience. Chest 143 (1), 14–18. doi:10.1378/chest.12-1430
Espel, J. C., Palac, H. L., Bharat, A., Cullina, J., Prickett, M., Sala, M., et al. (2018). The relationship between sweat chloride levels and mortality in cystic fibrosis varies by individual genotype. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 17 (1), 34–42. doi:10.1016/j.jcf.2017.11.002
Fidler, M. C., Beusmans, J., Panorchan, P., and Van Goor, F. (2017). Correlation of sweat chloride and percent predicted FEV(1) in cystic fibrosis patients treated with ivacaftor. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 16 (1), 41–44. doi:10.1016/j.jcf.2016.10.002
FitzMaurice, T. S., McCann, C., Nazareth, D., Shaw, M., McNamara, P. S., and Walshaw, M. J. (2022). Measuring the effect of elexacaftor/tezacaftor/ivacaftor combination therapy on the respiratory pump in people with CF using dynamic chest radiography. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (6), 1036–1041. doi:10.1016/j.jcf.2022.01.007
Fuchs, H. J., Borowitz, D. S., Christiansen, D. H., Morris, E. M., Nash, M. L., Ramsey, B. W., et al. (1994). Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N. Engl. J. Med. 331 (10), 637–642. doi:10.1056/NEJM199409083311003
Fuchs, S. I., Gappa, M., Eder, J., Unsinn, K. M., Steinkamp, G., and Ellemunter, H. (2014). Tracking Lung Clearance Index and chest CT in mild cystic fibrosis lung disease over a period of three years. Respir. Med. 108 (6), 865–874. doi:10.1016/j.rmed.2014.03.011
Furstova, E., Dousova, T., Beranek, J., Libik, M., Fila, L., Modrak, M., et al. (2022). Response to elexacaftor/tezacaftor/ivacaftor in intestinal organoids derived from people with cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (2), 243–245. doi:10.1016/j.jcf.2021.07.006
George, P. M., Banya, W., Pareek, N., Bilton, D., Cullinan, P., Hodson, M. E., et al. (2011). Improved survival at low lung function in cystic fibrosis: cohort study from 1990 to 2007. BMJ Clin. Res. ed) 342, d1008. doi:10.1136/bmj.d1008
Goetz, D. M., and Savant, A. P. (2021). Review of CFTR modulators 2020. Pediatr. Pulmonol. 56 (12), 3595–3606. doi:10.1002/ppul.25627
Goralski, J. L., Chung, S. H., Ceppe, A. S., Powell, M. Z., Sakthivel, M., Handly, B. D., et al. (2022). Dynamic perfluorinated gas MRI shows improved lung ventilation in people with cystic fibrosis after elexacaftor/tezacaftor/ivacaftor: an observational study. J. Clin. Med. 11 (20), 6160. doi:10.3390/jcm11206160
Goralski, J. L., Hoppe, J. E., Mall, M. A., McColley, S. A., McKone, E., Ramsey, B., et al. (2023). Phase 3 Open-Label Clinical Trial of Elexacaftor/Tezacaftor/Ivacaftor in Children Aged 2-5 Years with Cystic Fibrosis and at Least One F508del Allele. Am. J. Respir. Crit. care Med. 208 (1), 59–67. doi:10.1164/rccm.202301-0084OC
Gramegna, A., Aliberti, S., Contarini, M., Savi, D., Sotgiu, G., Majo, F., et al. (2022). Overweight and obesity in adults with cystic fibrosis: an Italian multicenter cohort study. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (1), 111–114. doi:10.1016/j.jcf.2021.05.002
Granados, A., Chan, C. L., Moheet, A., Vigers, T., Arbeláez, A. M., and Larson Ode, K. (2023). The impact of elexacaftor/tezacaftor/ivacaftor on body composition in a small cohort of youth with cystic fibrosis. Pediatr. Pulmonol. 58 (6), 1805–1811. doi:10.1002/ppul.26388
Griese, M., Costa, S., Linnemann, R. W., Mall, M. A., McKone, E. F., Polineni, D., et al. (2021). Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. care Med. 203 (3), 381–385. doi:10.1164/rccm.202008-3176LE
Haggie, P. M., Phuan, P. W., Tan, J. A., Xu, H., Avramescu, R. G., Perdomo, D., et al. (2017). Correctors and potentiators rescue function of the truncated W1282x-cystic fibrosis transmembrane regulator (CFTR) translation product. J. Biol. Chem. 292 (3), 771–785. doi:10.1074/jbc.M116.764720
Harindhanavudhi, T., Wang, Q., Dunitz, J., Moran, A., and Moheet, A. (2020). Prevalence and factors associated with overweight and obesity in adults with cystic fibrosis: a single-center analysis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 19 (1), 139–145. doi:10.1016/j.jcf.2019.10.004
Heijerman, H. G. M., McKone, E. F., Downey, D. G., Van Braeckel, E., Rowe, S. M., Tullis, E., et al. (2019). Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet (London, Engl. 394 (10212), 1940–1948. doi:10.1016/S0140-6736(19)32597-8
Heltshe, S. L., Mayer-Hamblett, N., and Rowe, S. M. (2013). Understanding the relationship between sweat chloride and lung function in cystic fibrosis. Chest 144 (4), 1418. doi:10.1378/chest.13-1320
Keating, C., Poor, A. D., Liu, X., Chiuzan, C., Backenroth, D., Zhang, Y., et al. (2017). Reduced survival in adult cystic fibrosis despite attenuated lung function decline. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 16 (1), 78–84. doi:10.1016/j.jcf.2016.07.012
Korten, I., Kieninger, E., Krueger, L., Bullo, M., Flück, C. E., Latzin, P., et al. (2022). Short-term effects of elexacaftor/tezacaftor/ivacaftor combination on glucose tolerance in young people with cystic fibrosis-an observational pilot study. Front. Pediatr. 10, 852551. doi:10.3389/fped.2022.852551
Kos, R., Neerincx, A. H., Fenn, D. W., Brinkman, P., Lub, R., Vonk, S. E. M., et al. (2022). Real-life efficacy and safety of elexacaftor/tezacaftor/ivacaftor on severe cystic fibrosis lung disease patients. Pharmacol. Res. Perspect. 10 (6), e01015. doi:10.1002/prp2.1015
Kraemer, R., Blum, A., Schibler, A., Ammann, R. A., and Gallati, S. (2005). Ventilation inhomogeneities in relation to standard lung function in patients with cystic fibrosis. Am. J. Respir. Crit. care Med. 171 (4), 371–378. doi:10.1164/rccm.200407-948OC
Lebecque, P., Leal, T., De Boeck, C., Jaspers, M., Cuppens, H., and Cassiman, J. J. (2002). Mutations of the cystic fibrosis gene and intermediate sweat chloride levels in children. Am. J. Respir. Crit. care Med. 165 (6), 757–761. doi:10.1164/ajrccm.165.6.2104073
Lee, T., Sawicki, G. S., Altenburg, J., Millar, S. J., Geiger, J. M., Jennings, M. T., et al. (2023). EFFECT OF ELEXACAFTOR/TEZACAFTOR/IVACAFTOR ON ANNUAL RATE OF LUNG FUNCTION DECLINE IN PEOPLE WITH CYSTIC FIBROSIS. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 22 (3), 402–406. doi:10.1016/j.jcf.2022.12.009
LeGrys, V. A., Moon, T. C., Laux, J., Accurso, F., and Martiniano, S. A. (2019). A multicenter evaluation of sweat chloride concentration and variation in infants with cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 18 (2), 190–193. doi:10.1016/j.jcf.2018.12.006
Liou, T. G., Adler, F. R., Fitzsimmons, S. C., Cahill, B. C., Hibbs, J. R., and Marshall, B. C. (2001). Predictive 5-year survivorship model of cystic fibrosis. Am. J. Epidemiol. 153 (4), 345–352. doi:10.1093/aje/153.4.345
Lopes, K., Custódio, C., Lopes, C., Bolas, R., and Azevedo, P. (2023). Elexacaftor/tezacaftor/ivacaftor-real-world clinical effectiveness and safety. A single-center Portuguese study. J. Bras. Pneumol. publicacao Of. Soc. Bras. Pneumol. Tisilogia 49 (2), e20220312. doi:10.36416/1806-3756/e20220312
Mainz, J. G., Zagoya, C., Polte, L., Naehrlich, L., Sasse, L., Eickmeier, O., et al. (2022). Elexacaftor-tezacaftor-ivacaftor treatment reduces abdominal symptoms in cystic fibrosis-early results obtained with the CF-specific CFabd-score. Front. Pharmacol. 13, 877118. doi:10.3389/fphar.2022.877118
Mall, M. A., Brugha, R., Gartner, S., Legg, J., Moeller, A., Mondejar-Lopez, P., et al. (2022). Efficacy and Safety of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 Through 11 Years of Age with Cystic Fibrosis Heterozygous for F508del and a Minimal Function Mutation: a Phase 3b, Randomized, Placebo-controlled Study. Am. J. Respir. Crit. care Med. 206 (11), 1361–1369. doi:10.1164/rccm.202202-0392OC
Martin, C., Reynaud-Gaubert, M., Hamidfar, R., Durieu, I., Murris-Espin, M., Danner-Boucher, I., et al. (2022). Sustained effectiveness of elexacaftor-tezacaftor-ivacaftor in lung transplant candidates with cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (3), 489–496. doi:10.1016/j.jcf.2022.01.012
McCoy, K. S., Blind, J., Johnson, T., Olson, P., Raterman, L., Bai, S., et al. (2023). Clinical change 2 years from start of elexacaftor-tezacaftor-ivacaftor in severe cystic fibrosis. Pediatr. Pulmonol. 58 (4), 1178–1184. doi:10.1002/ppul.26318
McKone, E. F., Velentgas, P., Swenson, A. J., and Goss, C. H. (2015). Association of sweat chloride concentration at time of diagnosis and CFTR genotype with mortality and cystic fibrosis phenotype. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 14 (5), 580–586. doi:10.1016/j.jcf.2015.01.005
Middleton, P. G., Mall, M. A., Drevinek, P., Lands, L. C., McKone, E. F., Polineni, D., et al. (2019a). Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. New Engl. J. Med. 381 (19), 1809–1819. doi:10.1056/NEJMoa1908639
Middleton, P. G., Mall, M. A., Dřevínek, P., Lands, L. C., McKone, E. F., Polineni, D., et al. (2019b). Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 381 (19), 1809–1819. doi:10.1056/NEJMoa1908639
Nagy, R., Gede, N., Ocskay, K., Dobai, B. M., Abada, A., Vereczkei, Z., et al. (2022). Association of body mass index with clinical outcomes in patients with cystic fibrosis: a systematic review and meta-analysis. JAMA Netw. open 5 (3), e220740. doi:10.1001/jamanetworkopen.2022.0740
Nichols, D. P., Morgan, S. J., Skalland, M., Vo, A. T., Van Dalfsen, J. M., Singh, S. B., et al. (2023). Pharmacologic improvement of CFTR function rapidly decreases sputum pathogen density, but lung infections generally persist. J. Clin. investigation 133 (10), e167957. doi:10.1172/JCI167957
Nichols, D. P., Paynter, A. C., Heltshe, S. L., Donaldson, S. H., Frederick, C. A., Freedman, S. D., et al. (2022). Clinical effectiveness of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis: a clinical trial. Am. J. Respir. Crit. care Med. 205 (5), 529–539. doi:10.1164/rccm.202108-1986OC
O’Shea, K. M., O'Carroll, O. M., Carroll, C., Grogan, B., Connolly, A., O'Shaughnessy, L., et al. (2021). Efficacy of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease. Eur. Respir. J. 57 (2), 2003079. doi:10.1183/13993003.03079-2020
Pallenberg, S. T., Held, I., Dopfer, C., Minso, R., Nietert, M. M., Hansen, G., et al. (2023). Differential effects of ELX/TEZ/IVA on organ-specific CFTR function in two patients with the rare CFTR splice mutations c.273+1G>A and c.165-2A>G. Front. Pharmacol. 14, 1153656. doi:10.3389/fphar.2023.1153656
Petersen, M. C., Begnel, L., Wallendorf, M., and Litvin, M. (2022). Effect of elexacaftor-tezacaftor-ivacaftor on body weight and metabolic parameters in adults with cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (2), 265–271. doi:10.1016/j.jcf.2021.11.012
Piehler, L., Thalemann, R., Lehmann, C., Thee, S., Röhmel, J., Syunyaeva, Z., et al. (2023). Effects of elexacaftor/tezacaftor/ivacaftor therapy on mental health of patients with cystic fibrosis. Front. Pharmacol. 14, 1179208. doi:10.3389/fphar.2023.1179208
Ramos, K. J., Guimbellot, J. S., Valapour, M., Bartlett, L. E., Wai, T. H., Goss, C. H., et al. (2022). Use of elexacaftor/tezacaftor/ivacaftor among cystic fibrosis lung transplant recipients. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (5), 745–752. doi:10.1016/j.jcf.2022.04.009
Ren, C. L., Morgan, R. L., Oermann, C., Resnick, H. E., Brady, C., Campbell, A., et al. (2018). Cystic fibrosis foundation pulmonary guidelines. Use of cystic fibrosis transmembrane conductance regulator modulator therapy in patients with cystic fibrosis. Ann. Am. Thorac. Soc. 15 (3), 271–280. doi:10.1513/AnnalsATS.201707-539OT
Rosenfeld, M., VanDevanter, D. R., Ren, C. L., Elkin, E. P., Pasta, D. J., Konstan, M. W., et al. (2015). Decline in lung function does not predict future decline in lung function in cystic fibrosis patients. Pediatr. Pulmonol. 50 (9), 856–862. doi:10.1002/ppul.23227
Saiman, L., Marshall, B. C., Mayer-Hamblett, N., Burns, J. L., Quittner, A. L., Cibene, D. A., et al. (2003). Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. Jama 290 (13), 1749–1756. doi:10.1001/jama.290.13.1749
Salvatore, D., Cimino, G., Troiani, P., Bignamini, E., Esposito, I., Leonetti, G., et al. (2022). Elexacaftor/tezacaftor/ivacaftor in children aged 6-11 years with cystic fibrosis, at least one F508DEL allele, and advanced lung disease: a 24-week observational study. Pediatr. Pulmonol. 57 (9), 2253–2256. doi:10.1002/ppul.25980
Schluchter, M. D., Konstan, M. W., and Davis, P. B. (2002). Jointly modelling the relationship between survival and pulmonary function in cystic fibrosis patients. Statistics Med. 21 (9), 1271–1287. doi:10.1002/sim.1104
Schnell, A., Hober, H., Kaiser, N., Ruppel, R., Geppert, A., Tremel, C., et al. (2023a). Elexacaftor - tezacaftor - Ivacaftor treatment improves systemic infection parameters and Pseudomonas aeruginosa colonization rate in patients with cystic fibrosis a monocentric observational study. Heliyon 9 (5), e15756. doi:10.1016/j.heliyon.2023.e15756
Schnell, A., Jüngert, J., Klett, D., Hober, H., Kaiser, N., Ruppel, R., et al. (2023b). Increase of liver stiffness and altered bile acid metabolism after triple CFTR modulator initiation in children and young adults with cystic fibrosis. Liver Int. official J. Int. Assoc. Study Liver 43 (4), 878–887. doi:10.1111/liv.15544
Scully, K. J., Marchetti, P., Sawicki, G. S., Uluer, A., Cernadas, M., Cagnina, R. E., et al. (2022). The effect of elexacaftor/tezacaftor/ivacaftor (ETI) on glycemia in adults with cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (2), 258–263. doi:10.1016/j.jcf.2021.09.001
Seliger, V. I., Rodman, D., Van Goor, F., Schmelz, A., and Mueller, P. (2013). The predictive potential of the sweat chloride test in cystic fibrosis patients with the G551D mutation. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 12 (6), 706–713. doi:10.1016/j.jcf.2013.03.004
Sermet-Gaudelus, I. (2013). Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation. Eur. Respir. Rev. official J. Eur. Respir. Soc. 22 (127), 66–71. doi:10.1183/09059180.00008512
Shakir, S., Echevarria, C., Doe, S., Brodlie, M., Ward, C., and Bourke, S. J. (2022). Elexacaftor-Tezacaftor-Ivacaftor improve Gastro-Oesophageal reflux and Sinonasal symptoms in advanced cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (5), 807–810. doi:10.1016/j.jcf.2022.06.003
Sheikh, S., Britt, R. D., Ryan-Wenger, N. A., Khan, A. Q., Lewis, B. W., Gushue, C., et al. (2023). Impact of elexacaftor-tezacaftor-ivacaftor on bacterial colonization and inflammatory responses in cystic fibrosis. Pediatr. Pulmonol. 58 (3), 825–833. doi:10.1002/ppul.26261
Shteinberg, M., Haq, I. J., Polineni, D., and Davies, J. C. (2021). Cystic fibrosis. Lancet (London, Engl. 397 (10290), 2195–2211. doi:10.1016/S0140-6736(20)32542-3
Snell, G. I., Bennetts, K., Bartolo, J., Levvey, B., Griffiths, A., Williams, T., et al. (1998). Body mass index as a predictor of survival in adults with cystic fibrosis referred for lung transplantation. J. heart lung Transplant. official Publ. Int. Soc. Heart Transplant. 17 (11), 1097–1103.
Sontag, M. K. (2016). Sweat chloride: the critical biomarker for cystic fibrosis trials. Am. J. Respir. Crit. care Med. 194 (11), 1311–1313. doi:10.1164/rccm.201606-1286ED
Southern, K. W., Murphy, J., Sinha, I. P., and Nevitt, S. J. (2020). Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del). Cochrane database Syst. Rev. 12 (12), Cd010966. doi:10.1002/14651858.CD010966.pub3
Southern, K. W., Patel, S., Sinha, I. P., and Nevitt, S. J. (2018). Correctors (specific therapies for class II CFTR mutations) for cystic fibrosis. Cochrane database Syst. Rev. 8 (8), Cd010966. doi:10.1002/14651858.CD010966.pub2
Spoletini, G., Gillgrass, L., Pollard, K., Shaw, N., Williams, E., Etherington, C., et al. (2022). Dose adjustments of Elexacaftor/Tezacaftor/Ivacaftor in response to mental health side effects in adults with cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (6), 1061–1065. doi:10.1016/j.jcf.2022.05.001
Stapleton, A. L., Kimple, A. J., Goralski, J. L., Nouraie, S. M., Branstetter, B. F., Shaffer, A. D., et al. (2022). Elexacaftor-Tezacaftor- Ivacaftor improves sinonasal outcomes in cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (5), 792–799. doi:10.1016/j.jcf.2022.03.002
Steinack, C., Ernst, M., Beuschlein, F., Hage, R., Roeder, M., Schuurmans, M. M., et al. (2023). Improved glucose tolerance after initiation of Elexacaftor/Tezacaftor/Ivacaftor in adults with cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 22, 722–729. doi:10.1016/j.jcf.2023.01.004
Stewart, B., Zabner, J., Shuber, A. P., Welsh, M. J., and McCray, P. B. (1995). Normal sweat chloride values do not exclude the diagnosis of cystic fibrosis. Am. J. Respir. Crit. care Med. 151 (3 Pt 1), 899–903. doi:10.1164/ajrccm/151.3_Pt_1.899
Stylemans, D., Darquenne, C., Schuermans, D., Verbanck, S., and Vanderhelst, E. (2022). Peripheral lung effect of elexacaftor/tezacaftor/ivacaftor in adult cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (1), 160–163. doi:10.1016/j.jcf.2021.03.016
Sutharsan, S., McKone, E. F., Downey, D. G., Duckers, J., MacGregor, G., Tullis, E., et al. (2022). Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: a 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir. Med. 10 (3), 267–277. doi:10.1016/S2213-2600(21)00454-9
Taylor-Cousar, J. L., Munck, A., McKone, E. F., van der Ent, C. K., Moeller, A., Simard, C., et al. (2017). Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 377 (21), 2013–2023. doi:10.1056/NEJMoa1709846
Tewkesbury, D. H., Athwal, V., Bright-Thomas, R. J., Jones, A. M., and Barry, P. J. (2023). Longitudinal effects of elexacaftor/tezacaftor/ivacaftor on liver tests at a large single adult cystic fibrosis centre. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 22 (2), 256–262. doi:10.1016/j.jcf.2023.01.007
Traeger, N., Shi, Q., and Dozor, A. J. (2014). Relationship between sweat chloride, sodium, and age in clinically obtained samples. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 13 (1), 10–14. doi:10.1016/j.jcf.2013.07.003
Vandenbroucke, N. J., Zampoli, M., and Morrow, B. (2020). Lung function determinants and mortality of children and adolescents with cystic fibrosis in South Africa 2007-2016. Pediatr. Pulmonol. 55 (6), 1381–1387. doi:10.1002/ppul.24726
VanDevanter, D. R., Kahle, J. S., O'Sullivan, A. K., Sikirica, S., and Hodgkins, P. S. (2016). Cystic fibrosis in young children: a review of disease manifestation, progression, and response to early treatment. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 15 (2), 147–157. doi:10.1016/j.jcf.2015.09.008
Vermeulen, F., Le Camus, C., Davies, J. C., Bilton, D., Milenković, D., and De Boeck, K. (2017). Variability of sweat chloride concentration in subjects with cystic fibrosis and G551D mutations. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 16 (1), 36–40. doi:10.1016/j.jcf.2016.02.015
Wainwright, C., McColley, S. A., McNally, P., Powers, M., Ratjen, F., Rayment, J. H., et al. (2023). Long-Term Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor in Children Aged ⩾6 Years with Cystic Fibrosis and at Least One F508del Allele: a Phase 3, Open-Label Clinical Trial. Am. J. Respir. Crit. care Med. 208 (1), 68–78. doi:10.1164/rccm.202301-0021OC
Wainwright, C. E., Elborn, J. S., Ramsey, B. W., Marigowda, G., Huang, X., Cipolli, M., et al. (2015). Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 373 (3), 1783–1784. doi:10.1056/NEJMc1510466
Walter, E., and Bass, J. L. (2022). The effect of elexacaftor/tezacaftor/ivacaftor on hospitalizations and intravenous antibiotic use. Perm. J. 26 (1), 73–79. doi:10.7812/TPP/21.089
Waters, V., Stanojevic, S., Atenafu, E. G., Lu, A., Yau, Y., Tullis, E., et al. (2012). Effect of pulmonary exacerbations on long-term lung function decline in cystic fibrosis. Eur. Respir. J. 40 (1), 61–66. doi:10.1183/09031936.00159111
Welsner, M., Schulte, T., Dietz-Terjung, S., Weinreich, G., Stehling, F., Taube, C., et al. (2022). Effect of triple combination CFTR modulator therapy on sleep in adult patients with cystic fibrosis. Respir. Int. Rev. Thorac. Dis. 101 (8), 766–774. doi:10.1159/000524773
Wilschanski, M., Zielenski, J., Markiewicz, D., Tsui, L. C., Corey, M., Levison, H., et al. (1995). Correlation of sweat chloride concentration with classes of the cystic fibrosis transmembrane conductance regulator gene mutations. J. Pediatr. 127 (5), 705–710. doi:10.1016/s0022-3476(95)70157-5
Wucherpfennig, L., Triphan, S. M. F., Wege, S., Kauczor, H. U., Heussel, C. P., Schmitt, N., et al. (2022). Magnetic resonance imaging detects improvements of pulmonary and paranasal sinus abnormalities in response to elexacaftor/tezacaftor/ivacaftor therapy in adults with cystic fibrosis. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 21 (6), 1053–1060. doi:10.1016/j.jcf.2022.03.011
Yen, E. H., Quinton, H., and Borowitz, D. (2013). Better nutritional status in early childhood is associated with improved clinical outcomes and survival in patients with cystic fibrosis. J. Pediatr. 162 (3), 530–535. doi:10.1016/j.jpeds.2012.08.040
Zemanick, E. T., Taylor-Cousar, J. L., Davies, J., Gibson, R. L., Mall, M. A., McKone, E. F., et al. (2021). A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am. J. Respir. Crit. care Med. 203 (12), 1522–1532. doi:10.1164/rccm.202102-0509OC
Zhang, L., Albon, D., Jones, M., and Bruschwein, H. (2022). Impact of elexacaftor/tezacaftor/ivacaftor on depression and anxiety in cystic fibrosis. Ther. Adv. Respir. Dis. 16, 17534666221144211. doi:10.1177/17534666221144211
Keywords: elexacaftor-tezacaftor-ivacaftor, cystic fibrosis, pulmonary function, sweat chloride, adverse events
Citation: Xu W, Wu T, Zhou Z and Zuo Z (2023) Efficacy and safety profile of elexacaftor-tezacaftor-ivacaftor triple therapy on cystic fibrosis: a systematic review and single arm meta-analysis. Front. Pharmacol. 14:1275470. doi: 10.3389/fphar.2023.1275470
Received: 28 August 2023; Accepted: 20 November 2023;
Published: 14 December 2023.
Edited by:
Onofrio Laselva, University of Foggia, ItalyReviewed by:
Arturo Solis-Moya, Dr. Carlos Sáenz Herrera National Children’s Hospital, Costa RicaRoberto Plebani, G. D’Annunzio University of Chieti-Pescara, Italy
Copyright © 2023 Xu, Wu, Zhou and Zuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihong Zuo, emhpaG9uZy56dW8xOTk1QGdtYWlsLmNvbQ==
†These authors have contributed equally to this work