
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 22 November 2023
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1264216
This article is part of the Research Topic Intracellular ion channels in health and disease View all 5 articles
 Vera Paar1*†
Vera Paar1*† Michael Haslinger1†
Michael Haslinger1† Philipp Krombholz-Reindl2
Philipp Krombholz-Reindl2 Stefan Pittner3Matthias Neuner4Peter Jirak1,5Tobias Kolbitsch1
Stefan Pittner3Matthias Neuner4Peter Jirak1,5Tobias Kolbitsch1 Bernd Minnich6
Bernd Minnich6 Falk Schrödl7
Falk Schrödl7 Alexandra Kaser-Eichberger7Kristen Kopp1Andreas Koller8Clemens Steinwender9
Alexandra Kaser-Eichberger7Kristen Kopp1Andreas Koller8Clemens Steinwender9 Michael Lichtenauer1Fabio C. Monticelli3Rainald Seitelberger2Uta C. Hoppe1
Michael Lichtenauer1Fabio C. Monticelli3Rainald Seitelberger2Uta C. Hoppe1 Christian Dinges2‡Lukas J. Motloch1,9,10‡
Christian Dinges2‡Lukas J. Motloch1,9,10‡Introduction: Hypertrophies of the cardiac septum are caused either by aortic valve stenosis (AVS) or by congenital hypertrophic obstructive cardiomyopathy (HOCM). As they induce cardiac remodeling, these cardiac pathologies may promote an arrhythmogenic substrate with associated malignant ventricular arrhythmias and may lead to heart failure. While altered calcium (Ca2+) handling seems to be a key player in the pathogenesis, the role of mitochondrial calcium handling was not investigated in these patients to date.
Methods: To investigate this issue, cardiac septal samples were collected from patients undergoing myectomy during cardiac surgery for excessive septal hypertrophy and/or aortic valve replacement, caused by AVS and HOCM. Septal specimens were matched with cardiac tissue obtained from post-mortem controls without cardiac diseases (Ctrl).
Results and discussion: Patient characteristics and most of the echocardiographic parameters did not differ between AVS and HOCM. Most notably, the interventricular septum thickness, diastolic (IVSd), was the greatest in HOCM patients. Histological and molecular analyses showed a trend towards higher fibrotic burden in both pathologies, when compared to Ctrl. Most notably, the mitochondrial Ca2+ uniporter (MCU) complex associated proteins were altered in both pathologies of left ventricular hypertrophy (LVH). On the one hand, the expression pattern of the MCU complex subunits MCU and MICU1 were shown to be markedly increased, especially in AVS. On the other hand, PRMT-1, UCP-2, and UCP-3 declined with hypertrophy. These conditions were associated with an increase in the expression patterns of the Ca2+ uptaking ion channel SERCA2a in AVS (p = 0.0013), though not in HOCM, compared to healthy tissue. Our data obtained from human specimen from AVS or HOCM indicates major alterations in the expression of the mitochondrial calcium uniporter complex and associated proteins. Thus, in cardiac septal hypertrophies, besides modifications of cytosolic calcium handling, impaired mitochondrial uptake might be a key player in disease progression.
Cardiovascular diseases resulting in lethal arrhythmias and sudden cardiac death constitute one of the most common causes of death worldwide (Kumar et al., 2021). As young individuals without any preliminary symptoms are also frequently affected, they therefore represent a crucial health problem. Independent from symptoms and etiology, left ventricular hypertrophy (LVH) has been found to act as a strong independent risk factor for sudden cardiac death (Haider et al., 1998; Aro et al., 2017; Stewart et al., 2018). Generally, LVH may be caused by various factors, such as physiologic adaptions in athletes, or may be the result of genetic aberrations, as well as a compensatory mechanism in response to left ventricular outflow tract (LVOT) obstruction. Even concentric remodelling, the precursor of concentric and eccentric LVH, shows an increased risk for heart failure and sudden cardiac death (Aro et al., 2017), mainly through the generation of lethal (ventricular) arrhythmias.
Cardiac calcium (Ca2+) handling plays a fundamental role in the proper action potential (AP) generation and progression, contributing to the physiological contraction of the heart. Early studies have shown that hypertrophied myocytes present with a prolonged AP caused by changes in the ion channels’ expression and function, particularly Ca2+ related channels (McIntyre and Fry, 1997; Botchway et al., 2003). Hypertrophy also increases the predisposition for abnormal Ca2+ release events, the so-called spontaneous Ca2+ release from the sarcoplasmic reticulum (SR) (Brochet et al., 2012) by the ryanodine receptor (RyR) which provokes cytosolic Ca2+ overload. In LVH, this phenomenon is often counterbalanced by an enhanced expression of the sodium-Ca2+ exchanger (NCX). Additionally, the repolarization of hypertrophied myocytes is impaired due to a reduction in potassium currents. This triggers the generation of early or delayed afterdepolarizations (EADs or DADs) that in turn may lead to the re-opening of L-type Ca2+ channels (LTCCs), thus depolarizing the cell and inducing a premature ventricular beat (Tomita et al., 1994; Marionneau et al., 2008; Tse et al., 2016). Consequently, electrical waves may be initiated within the heart, predisposing cardiac tachyarrhythmias (Levy et al., 1987; Paar et al., 2019).
Besides disturbances in the cytosolic Ca2+ levels and fluxes, also mitochondrial Ca2+ handling has to be taken into consideration as it is a key regulator in diverse processes of cardiomyocyte physiology. First, mitochondria are the main providers of the energy needed for cardiac contraction via adenosine triphosphate (ATP). Increased mitochondrial Ca2+ cycling directly enhances the amount of ATP produced (Bers, 2002; Piquereau et al., 2013). Furthermore, mitochondria contribute in the modulation of the Ca2+ signalling within the cardiomyocyte, regulate the production of reactive oxygen species (Kohlhaas et al., 2010; Vakrou and Abraham, 2014), and may trigger cell death (Bernardi and Rasola, 2007; O'Rourke, 2007; Hoppe, 2010). The inner mitochondrial membrane is highly selective for Ca2+, which is tightly driven by the mitochondrial Ca2+ uniporter (MCU) complex, preventing the mitochondria from Ca2+ overload. The cardiac MCU complex is composed of the pore forming MCU protein (De Stefani et al., 2011) and multiple regulatory subunits, including the mitochondrial Ca2+ uptake 1 (MICU1) (Perocchi et al., 2010; Plovanich et al., 2013).
Additionally, besides the pore-forming subunits, several regulatory proteins determine the amount of Ca2+ that pass the inner mitochondrial membrane, such as protein arginine N-methyltransferase 1 (PRMT-1) and the mitochondrial uncoupling proteins (UCPs). PRMT-1 asymmetrically methylates MICU1 and reduces the sensitivity of MCU for Ca2+ (Madreiter-Sokolowski et al., 2016). In turn, the cardiac UCP-2 and UCP-3 isoforms normalize the inhibitory effects of PRMT-1 and re-activates the mitochondrial Ca2+ uptake (Trenker et al., 2007; Michels et al., 2009; Hoppe, 2010; Motloch et al., 2016a; Motloch et al., 2016b; Larbig et al., 2017; Paar et al., 2019).
Both pathologies, the acquired aortic valve stenosis (AVS) (Gunther and Grossman, 1979; Carabello and Paulus, 2009) and the congenital hypertrophic obstructive cardiomyopathy (HOCM) (Elliott et al., 2008; Elliott et al., 2014), may lead to a thickening of the left ventricle (LV) and concomitant cardiac remodelling on the tissue and cellular level. As both pathologies are known to promote cardiac arrhythmias, we hypothesized that cardiac remodelling might be the main cause for the development of an arrhythmogenic substrate. Of note, this accounts not only for the tissue level as in fibrosis, but also for changes on a molecular level. Regarding the crucial aspect of myocardial Ca2+ handling in cardiac arrhythmogenesis, we specially focussed on the expression of cytoplasmic and mitochondrial Ca2+ channels, as well as on the two most important SR-bound channels RyR2 and SR Ca2+-ATPase type 2a (SERCA2a). Due to the limited availability of human cardiac specimen, most of the previous studies were performed using animal models. To our knowledge, a molecular characterization of human myocardial cytoplasmic, mitochondrial, and SR Ca2+ channels and a direct comparison thereof in AVS and HOCM is still pending. Thus, we hypothesized, that both pathologies, AVS and HOCM, might be linked to major alterations in the expression of mitochondrial as well as cytosolic Ca2+ handling proteins. To investigate this issue, we analyzed septal specimen from cardiac surgery patients undergoing myectomy.
Patients suffering from severe, symptomatic AVS need to undergo aortic valve replacement to restore the functionality of the heart. As AVS may result in the asymmetric septal hypertrophy, the obstruction of the LVOT must be restored as well. Similarly, hypertrophic cardiomyopathy (HCM) may result in LVOT due do an excessive hypertrophy of the LV septum that obstructs the aortic blood flow, therefore termed HOCM. In order to restore physiological hemodynamics, part of this hypertrophic septal tissue is surgically removed in a process called myectomy (Kayalar et al., 2010; Hang et al., 2017). Thus, these specimens were further analysed in the present study.
The study was approved by the Austrian Ethics Committee of the State of Salzburg (415-E/2329/8-2018 and 415-E/2330/7-2018, respectively) and was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice. Post-mortem, subvalvular LV septal specimen from six forensic autopsy cases served as healthy control (Ctrl) tissue. Specifically, only cases were included without reported cardiac diseases and without any evidence of macroscopically observed cardiac pathologies, such as cardiomyopathy or significant coronary artery disease during forensic autopsy. Furthermore, no individuals with macroscopically observed cardiac or major thoracic injuries (including damage from eventual resuscitation), as well as reported or diagnosed drug abused were used in the present study. In total, 21 subjects were enrolled into the study, comprised of six Ctrl subjects, eight AVS patients and seven HOCM patients. All participants from the AVS and HOCM groups signed a written informed consent prior to study participation. For all three study groups, the inclusion criteria were defined as age ≥18 years and for AVS and HOCM, a significantly asymmetrical LVH which results in LVOT requiring surgical removal. All included patients had no history of previous thoracotomy.
A baseline echocardiographic examination was performed according to the ESC Guidelines for the management of cardiomyopathies (Ommen et al., 2020; Arbelo et al., 2023). These analyses included the functional assessment of the right- and left ventricle as well as evaluation of the valves prior to or at hospital admission and interpreted by a single cardiologist. The biplane Simpson method was used to calculate left ventricular ejection fraction (LVEF) and tricuspid annular plane systolic excursion (TAPSE) was used to delineate right ventricular function. Valvular heart disease was graded according to the 2021 ESC Valvular Heart Disease guidelines (Vahanian et al., 2022) using the baseline transthoracic and additional transesophageal echocardiographic data, if available. Important clinical data and the medical history of the patients was captured from in-hospital charts at the time of the surgical intervention.
The septal cardiac samples were gained during cardiac surgery or in course of a forensic autopsy and were processed or fixed immediately after harvesting, respectively. Ctrl cardiac septal tissue was retrieved from the subvalvular left ventricular part of the IVS, within 24 h after death. Until forensic autopsy, the corpses were kept under cooled conditions in order to keep degradation to a minimum. Before further processing, the endocardium was removed to solely analyse the septal myocardium excluding the endocardial composition to avoid false increase in fibrotic burden. For histological examinations, the samples were either fixed in 4% formaldehyde solution (buffered, pH 6.9, Merck Millipore, MA, United States) overnight or an appropriate portion of the septal tissue was shock-frozen in liquid nitrogen and stored at −80°C for later immunoblotting.
In order to determine potential tissue remodelling and the extent of fibrosis in hypertrophied septal specimen in comparison to healthy Ctrl tissue, the fixed tissue samples were dehydrated as described previously (Welsch et al., 2015). Then formalin-fixed paraffin-embedded (FFPE) tissue blocks were generated in Paraplast® (Leica, Austria). Thereafter, the FFPE samples were cut into 6 µm thick sections and dried overnight. The next day, the tissue slices of all three study cohorts were stained by haematoxylin-eosin (HE), or Masson’s trichrome stain (Riedelsheimer and Büchl-Zimmermann, 2015), respectively. They were then investigated by two independent observers using an inverse microscope (Axiovert 200, Carl Zeiss, Germany) at a magnification of ×10 and 20x. Inflammatory infiltrates were detected and distinguished by size and shape of the blue-coloured cells in the HE-stained sections. The infiltrative foci were then graded according to semi-quantitative scales, which were previously reported (Eriksson et al., 2003; Aly et al., 2007). The grades were determined as follows: 0: no inflammatory infiltrates, 1: small foci of inflammatory cells between myocytes, 2: larger foci of inflammatory cells, 3: 10%–30% of a cross-section, 4: >30% of a cross-section (Eriksson et al., 2003; Aly et al., 2007; Mirna et al., 2021). For the quantification of the extent of fibrosis and the assessment of the cell sizes, a minimum of 15 pictures were taken by Axiocam MRc3 (Carl Zeiss, Germany) and AxioVision SE64 Rel. 4.8 (Carl Zeiss, Germany) to generate a highly random collection of tissue sections. The measurement of the cell sizes was performed in Zen 2.6 (blue edition; Carl Zeiss, Germany); the quantification of the blue (fibrotic) versus the red (cardiac muscle) areas was performed using ImageJ (NIH, LOCI, United States). The relative fibrotic area (RFA) was calculated as the pixels measured in blue versus red color, given as percentages.
In addition, Congo red staining was performed according to Riedelsheimer and Büchl-Zimmermann (Riedelsheimer and Büchl-Zimmermann, 2015) to histologically exclude amyloidosis of all study participants. Thereafter, the stained tissue sections were analyzed by light microcopy with a ×10 and ×20 objective (Carl Zeiss, Germany) and exemplary pictures were taken by Axiocam MRc3 (Carl Zeiss, Germany) and AxioVision SE64 Rel. 4.8 software (Carl Zeiss, Germany). In case of amyloidosis, Congo red binds to amyloid and red color would have been detected within the sections (Yakupova et al., 2019).
The total protein content was extracted from the shock-frozen tissue sections by radio-immunoprecipitation assay (RIPA) lysis buffer (Santa Cruz Biotechnology, TX, United States) and the concentration was determined by a bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, NH, United States). Thereafter, 10–30 µg of total protein was deployed for Western blot analysis of the fibrosis-relevant proteins, such as collagen 1 (COL1; abcam, Cambridge, United Kingdom), collagen 3 (COL3; abcam, Cambridge, United Kingdom), transforming growth factor-beta 1 (TGF-β1; abcam, Cambridge, United Kingdom), and mothers against decapentaplegic homolog 3 (SMAD3; Cell Signaling Technology, MA, United States). Equal amounts of protein lysates per protein of interest were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on a 4%–15% Mini-PROTEAN TGX Gel (Bio-Rad Laboratories, Austria) with 4x Laemmli buffer (Bio-Rad Laboratories, Austria) serving as the loading buffer. Western blot bands were further transferred onto polyvinylidene fluoride (PVDF) Immun-Blot PVDF membranes (Bio-Rad Laboratories, Austria) by tank blotting. Membranes were blocked with an adequate blocking reagent, either EveryBlot Blocking Buffer (Bio-Rad Laboratories, Austria), ROTI®Block (Carl Roth, Germany), or 5% non-fat dry milk powder in TRIS-buffered saline with Tween-20 (TBS-T). Adequate blocking or diluting solutions were previously tested for each antibody. Membranes were incubated with the appropriate primary antibodies overnight at 4°C. The next day, after washing the membranes with TBS-T, the appropriate HRP-linked secondary antibodies (anti-rabbit or anti-mouse) were applied and incubated for 1 h at room temperature. The chemiluminescent signal was detected using Raytest Stella 8005 (Elysia-Raytest, Germany) and camera control software XStella Version 1.00.011 (Elysia-Raytest, Germany). In addition to the fibrosis-relevant proteins, Ca2+-associated ion channels and regulatory proteins were analyzed by Western blot. The analyses included: 1) the membrane-bound Ca2+channels: cardiac L-type Ca2+ channel (CaV1.2; Alomone Labs, Jerusalem, Israel) and the Ca2+ extruding channel NCX1 (Cell Signaling Technology, MA, United States); 2) the two most important Ca2+ SR proteins RyR2 (abcam, Cambridge, United Kingdom) and SERCA2a (Santa Cruz Biotechnology, TX, United States); 3) the mitochondrial proteins MCU (Cell Signaling Technology), MICU1 (Cell Signaling Technology), UCP-2 (ProteinTech Europe, Manchester, United Kingdom), UCP-3 (abcam), and PRMT-1 (Cell Signaling Technology). The secondary anti-mouse and anti-rabbit IgG antibodies were obtained from Cell Signaling Technology. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology) and cytochrome c oxidase 4 (COX 4; abcam) were used as housekeeping protein for the normalization of the proteins of interest, as previously described elsewhere (Dally et al., 2006; Holmström et al., 2015; Danielsen et al., 2020; Zheng et al., 2022; Dridi et al., 2023). Data analysis and normalization were performed using Image Lab 6.0.1 software (Bio-Rad, Vienna, Austria). The bands were detected manually and the normalization channel and reference lane (references sample) were selected. Additionally, the background subtraction was set to a disk size of 70.0 mm. The normalized result of each sample was gained by automatic calculation, named “Norm- Vol. (Int).” Since the inclusion of the study participants was sporadic over a three-year period, as stated in the limitations section, the samples were run on different gels. To ensure comparability of the bands on different gels, an interblot control (stated as reference sample) was applied onto each individual gel, as previously reported elsewhere (Pillai-Kastoori et al., 2020; Taylor et al., 2022). After normalization of all samples with the housekeeping protein, we set the intensity of the reference sample to the value 1 (100%). Finally, we mathematically calculated the relative protein volume (RPV) by dividing the normalized sample of interest by the normalized reference sample, equally to the “IBC ratio” or “fold change” as also described in the literature (Pillai-Kastoori et al., 2020; Taylor et al., 2022). To create graphically representative and descriptive figures, the bands were graphically connected within the Western blot band figures (Figure 3A, 4A, and 5A).
Statistical analysis was performed using GraphPad PRISM 9 software (GraphPad-Software, San Diego, CA, United States). The distribution of the data was assessed by Shapiro-Wilk test. Non-parametric data are expressed as median and interquartile range (IQR; 25% to 75%-quartile), normally distributed data are given as mean and standard deviation (SD). Comparisons of two datasets were either performed using Welch’s test (unpaired) or paired t-test for normally distributed datasets. Non-parametric datasets were compared by Mann-Whitney test (unpaired) or Wilcoxon matched-pairs rank test (paired). A Welch’s ANOVA test for normally distributed data or a Kruskal–Wallis test for non-parametric data were used for the comparison of more than two datasets. These were split by the post hoc test Dunnett’s T3 multiple comparisons test (normally distributed) or Dunn’s multiple comparisons test (non-parametric), respectively. As some figures included both, normally-distributed and non-parametric datasets, all graphs of these figures are presented with median and interquartile range. To investigate the relation of the proteins analyzed with the severity of LVH or to draw a connection between the concentration of the different Ca2+-related proteins, either a Pearson correlation coefficient analysis (normally distributed data), or a non-parametric Spearman correlation were performed, respectively. The alpha threshold and the confidence level were set to p ≤ 0.05, which was determined as statistically significant.
The analysis of all basic patient-related data showed a close similarity between the three study groups (see Table 1), with the exception of AVS: AVS patients displayed the highest age of all three study groups. Although ANOVA analysis of the ages of the three groups was significant (p = 0.0304), there was no significant difference between Ctrl vs. HOCM (p = 0.3036) or AVS vs. HOCM (p = 0.6063) groups, respectively.
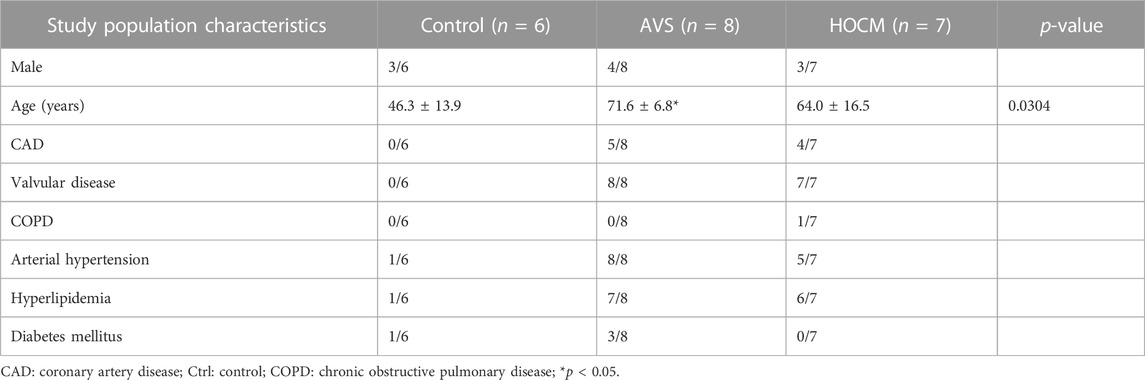
TABLE 1. Basic study population characteristics of the Ctrl group, as well as the AVS and HOCM cohorts. Due to the normal distribution of the data, the values are given as mean ± SD.
Furthermore, the major echocardiographic parameters as well as the clinical data, except from IVSd and AV Pmean, did not differ between the patients included in the AVS or HOCM study group, respectively (Table 2). Only a statistically significant elevation of IVSd in HOCM (p = 0.0204) in comparison to heathy Ctrl as well as a marked reduction in the mean atrial pressure in HOCM patients (p = 0.0302) was observed. No differences in right ventricular end-diastolic diameter were seen. In addition, none of the patients presented with typical echocardiography patterns suggesting cardiac amyloidosis, as previously described (Arbelo et al., 2023). This was also confirmed by Congo red stain of histological samples, shown in Supplementary Figure S1. Both AVS and HOCM presented as diffuse hypertrophy manifestations.
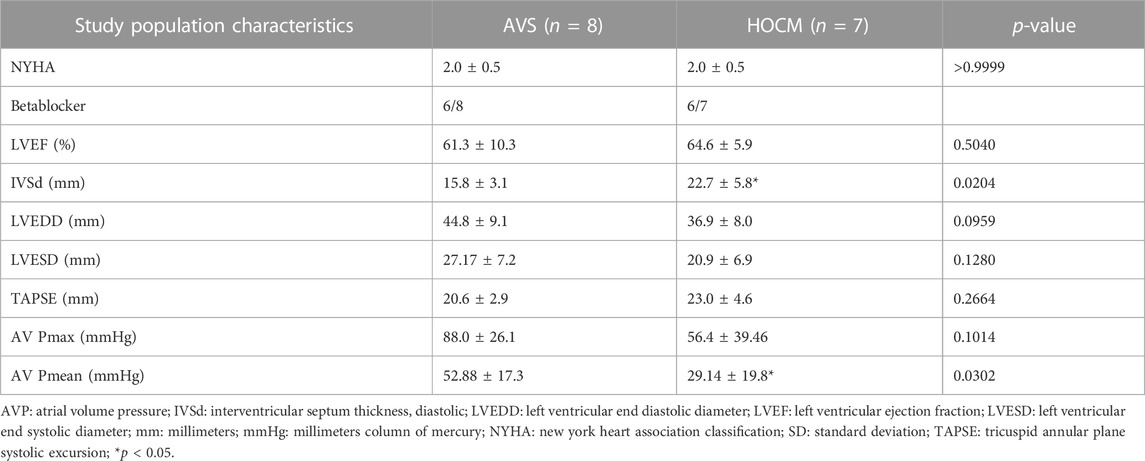
TABLE 2. Clinical data and echocardiographic parameters of the patients included in the AVS or HOCM study groups, respectively. Due to the normal distribution of the data, the values are given as mean ± SD.
Electrical signal transduction is highly dependent on the proper connection of two adjacent cardiomyocytes and an appropriate extent of extracellular matrix or connective tissue. In hypertrophy, several remodeling processes occur, with the increase in cardiomyocyte size as a primary sign. Furthermore, in acute cardiac diseases, inflammation is a major trigger for cardiac remodeling. Therefore, we investigated the extent of the cardiomyocyte growth in both diseases, as well as potential inflammatory infiltrates by standard histology (HE stain; Figure 1A). While we could not detect any inflammatory foci in any of the Ctrl samples (Score 0), the situation was slightly different in the pathological groups (depicted in Figure 1C): here 37.5% of the AVS (n = 8) and 57.1% of the HOCM sections analyzed had score 1 (n = 8). However, none of the samples analyzed had large foci of inflammatory infiltrates (score 3–4). When further analyzing cell sizes histologically (Figures 1A, B), the cardiomyocytes’ diameters reveal a substantial increase in the size of the hypertrophied cardiomyocytes in AVS as well as HOCM in comparison to the healthy Ctrl tissue (Figure 1D).
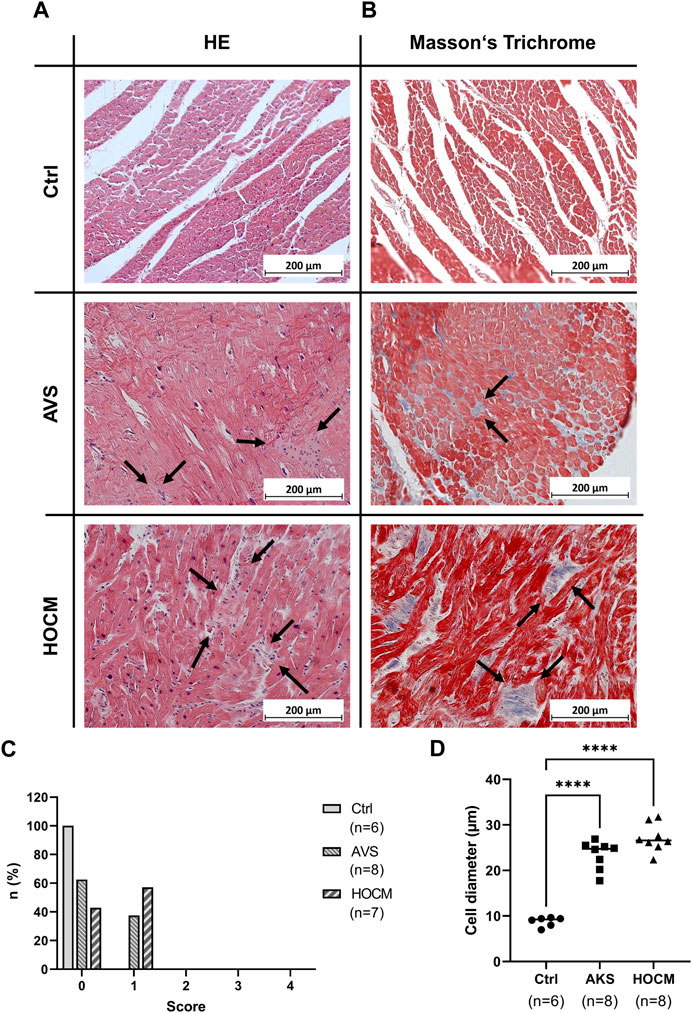
FIGURE 1. Light microscopy of cardiac tissue in Ctrl, AVS, and HOCM stained by HE (A) and Masson’s Trichrome (B). In HE, cardiomyocytes appear dark-purple with large and round nucleus, while inflammatory infiltrates (indicated by arrows) appear much lighter and contain small sized nuclei. In Masson’s Trichrome, cardiomyocytes are stained in red, while fibrous tissue is stained in blue (indicated by arrows). (C) Histogram of the inflammatory regions (0: no inflammatory infiltrates, 1: small foci of inflammatory cells between myocytes, 2: larger foci of inflammatory cells, 3: 10%–30% of a cross-section, 4: >30% of a cross-section). While we could not detect any inflammatory focus in Ctrl (score 0), approximately 40% of AVS, and approximately 60% of HOCM exhibited inflammatory infiltrate score 1, respectively. (D) Histogram of the cardiomyocyte diameters in Ctrl vs. AVS/HOCM tissue. In diseased tissue, there was a substantial and statistically significant (****p < 0.0001) increase in cell diameter compared to Ctrl tissue.
As cardiac hypertrophy is often accompanied by an extensive accumulation of fibrotic tissue (Vakrou and Abraham, 2014), the extent of fibrosis in the cardiac septum of healthy Ctrl individuals, AVS patients and HOCM patients was assessed histologically using Masson’s Trichrome. The qualitative examination of the microscopic pictures give rise to an increase of the fibrotic content (blue color) in AVS and the HOCM patients in comparison to the healthy Ctrl group (Figure 1B).
Quantitatively, the RFA was assessed as percentage between blue and red color in the whole section. Herein, the comparison of the RFA shows a slight trend towards an elevated fibrotic content in both pathologies, in comparison to the healthy Ctrl tissue (Figure 2A). Nevertheless, the RFA of all tissue analyzed strongly correlated with the echocardiographically measured IVSd. Confirming our assumptions, Figure 2B depicts a positive correlation of RFA with the IVSd (p = 0.0133) in both LVH pathologies, indicating that cardiac hypertrophy drives fibrotic accumulations.
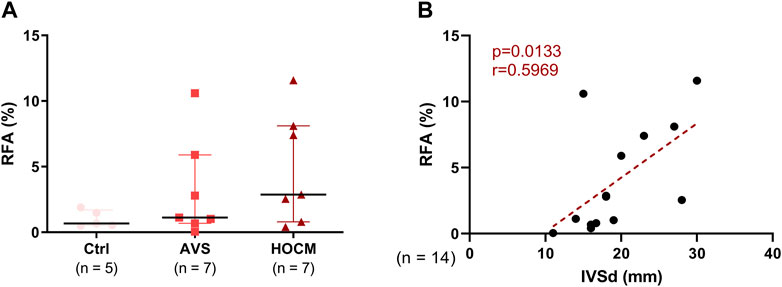
FIGURE 2. (A) Graphical comparison of the RFA of Ctrl, AVS, and HOCM tissue. The tissue sections were stained by Masson’s Trichrome stain and RFA was calculated by relating the resulting blue area (fibrous tissue) to the red area (cardiomyocytes). Due to non-Gaussian approximation, the data is depicted as median with IQR. (B) Relation of the RFA in both pathologies with the echocardiographically-measured diameter of the cardiac septum (IVSd) was obtained by Spearman correlation analysis. The dotted line depicts the regression curve of AVS and HOCM.
In a further analysis, the quantification of the contents of fibrosis-related proteins in all three study groups was performed in relation to a unique reference sample of human septal tissue. As depicted in Figures 3A, B, the structural proteins COL1 and COL3 revealed a trend for an elevation in both diseases, though without statistical significance (p > 0.05). Nevertheless, SMAD3 increased markedly in AVS (p = 0.0180), as well as in HOCM (p = 0.0277) in comparison to the healthy Ctrl group. Additionally, RPV of TGF-β1 was significantly elevated in AVS (7.111 ± 2.982) in comparison to the Ctrl tissue (0.369 ± 0.312; p = 0.0028).
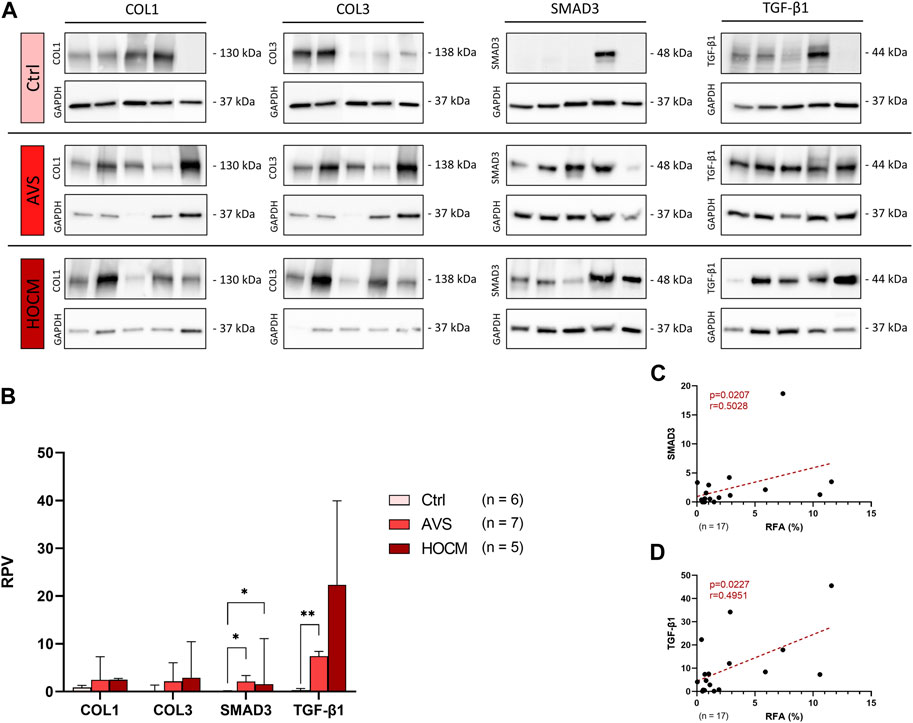
FIGURE 3. Western blot results of fibrosis-related proteins in Ctrl, AVS, and HOCM human cardiac tissue. RPV of each protein of interest is stated as a relative value to a unique reference sample. (A) Western blot bands of COL1, COL3, SMAD3, and TGF-β1 are normalized with GAPDH as housekeeping protein, respectively. Due to the study design, the depicted bands were graphically assembled from different blots. (B) Quantification of COL1, COL3, SMAD3, and TGF-β1 in human septal tissue, statistically analyzed by GraphPad PRISM. Due to non-Gaussian approximation of the figure’s data, RPVs are depicted as median and IQR with *p < 0.05 and **p < 0.01. (C) Spearman correlation of the RFA with the RPV of SMAD3 (n = 17). (D) Spearman correlation of the RFA with the RPV of TGF-β1 (n = 17). The dotted lines depict the regression curve of the Ctrl, AVS, and HOCM study groups.
Accordingly, SMAD3 and TGF-β1 RPV values positively correlated with the RFA in both pathologies, as well as in the Ctrl tissue (Figures 3C, D). Evaluated by Spearman correlation, these data indicate that SMAD3 and TGF-β1 are linked with the fibrotic remodeling in LVH in both diseases.
In order to clarify the role of Ca2+ AVS and HOCM, as well as unveil potential alterations in the hypertrophied tissue, we performed Western blot analyses of the most important Ca2+ trafficking ion channels of cardiomyocytes. Most importantly, under physiological conditions Ca2+ trafficking between the cytosol and the inner mitochondrial membrane is tightly regulated. Changes in mitochondrial Ca2+ levels are mainly associated with alterations in the expression of the MCU complex proteins or its regulatory proteins. Therefore, we first focused on the presence and potential alterations of MCU and MICU1 proteins as the most important proteins building the functional MCU complex (Figures 4A–C). As shown in Figure 4B, MCU is highly increased in AVS (p = 0.0111), as well as in HOCM (p = 0.0172) in comparison to the Ctrl group. Similarly, LVH also increased the levels of MICU1 in AVS (p = 0.0084). However, no statistically significant alteration was observed between the two diseases, neither for MCU, nor for MICU1. Most importantly, there is a markedly positive relation between MCU and MICU1 expression patterns, indicating an overall increase in the density of the whole MCU complex (Figure 4C).
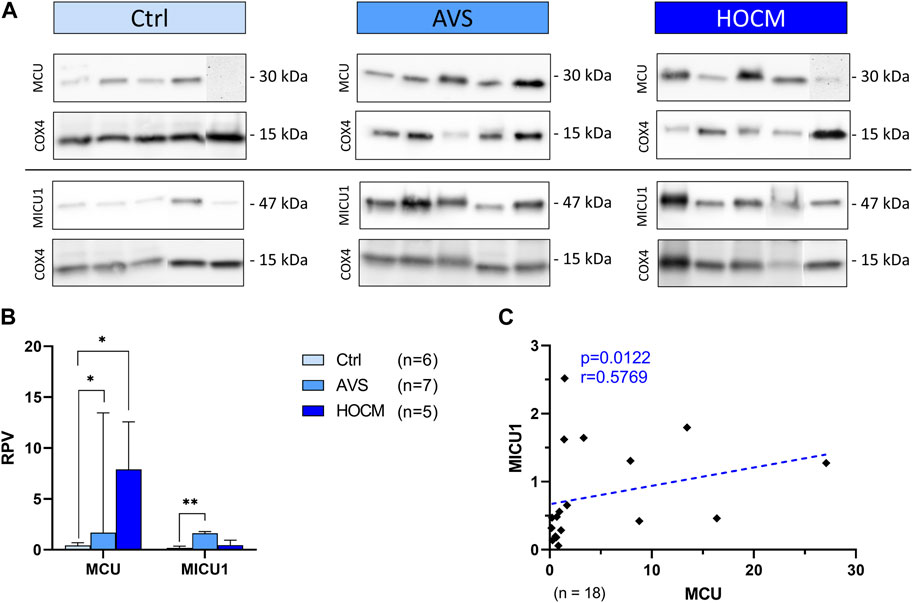
FIGURE 4. Western blot results of MCU complex-associated proteins in Ctrl, AVS, and HOCM human cardiac tissue. RPV of each protein of interest is stated as a relative value to a unique reference sample. (A) Western blot bands of MCU and MICU1 are normalized with COX4 as housekeeping protein. Due to the study design, the depicted bands were graphically assembled from different blots. (B) Quantification of MCU and MICU1 in human septal tissue. Due to non-normal distribution of MCU data, all data in the graph are given as median + IQR. The statistical significances were calculated using Kruskal–Wallis test for MCU and Welch ANOVA test for MICU1 with *p < 0.05 and **p < 0.01. (C) Spearman correlation of MCU with MICU1 of the whole study group. The dotted line depicts the regression curve.
As MICU1 prevents mitochondria from Ca2+ overload by reducing the sensitivity of MCU for Ca2+ uptake in its methylated form, it may counteract the effects of an increase of MCU in the diseased tissue. However, MCU activity and MICU1 methylation are highly affected by their regulatory proteins UCP-2 and UCP-3, as well as PRMT-1. Therefore, the protein concentrations of all three proteins were assessed and quantified (Figure 5). As shown in Figure 5A, UCP-2 resulted in two separate bands, one specific to the monomeric UCP-2 protein, and the other appropriate to the dimeric form of UCP-2. In order to ensure that the UCP-2 results gained are valid, both UCP-2 bands were statistically analyzed separately. Therefore, the results of both bands are given separately as dimeric, called “Dim,” or monomeric, called “Mono.”
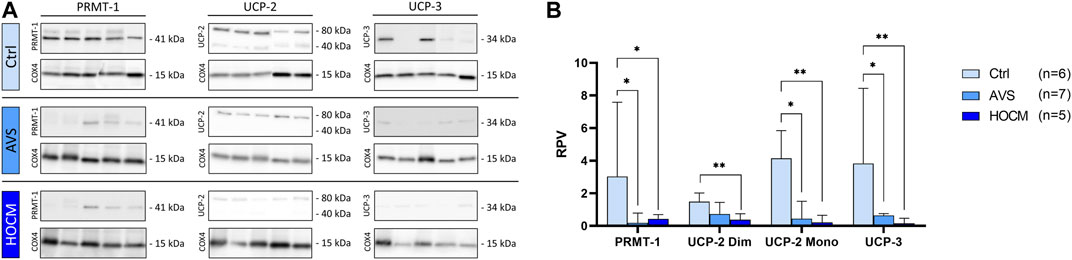
FIGURE 5. Western blot results of PRMT-1, UCP-2, and UCP-3 in Ctrl, AVS, and HOCM human cardiac tissue. RPV of each protein of interest is stated as a relative value to a unique reference sample. (A) Western blot bands of PRMT-1, UCP-2, and UCP-3 are normalized with COX4 as housekeeping protein. Due to the study design, the depicted bands were graphically assembled from different blots. (B) Quantification of PRMT-1, UCP-2, and UCP-3 in human septal tissue, statistically analyzed by GraphPad PRISM. Due to non-normal distribution, the statistical analysis was performed by Kruskal–Wallis test and the data is given as median + IQR with *p < 0.05 and **p < 0.01.
As seen in Figure 5B, PRMT-1 was greatly reduced in AVS (p = 0.0136), as well as in HOCM (p = 0.0400) in comparison to the Ctrl group. Functionally counteracting PRMT-1, the expression patterns of UCP-2 and UCP-3 were also significantly diminished in both diseases (AVS: p = 0.0485; HOCM: p = 0.0073) in comparison to the Ctrl group. Spearman correlation indicates the direct relation of the monomeric and dimeric UCP-2 bands (p < 0.0001, r = 0.8184; graph not shown).
To investigate the relationship of the MCU complex regulatory proteins with MCU and MICU1, a Spearman correlation analysis of PRMT-1 with MCU, or MICU1 was performed, respectively. Additionally, the counterparts UCP-2 and UCP-3 were also put into relation with both MCU complex proteins. As depicted in Figures 6A, B, PRMT-1 negatively correlates with MCU and MICU1 expression patterns (MCU: p = 0.0004; r = -0.7399; MICU1: p = 0.0032, r = −0.6553). Furthermore, these findings are supported by the negative correlation of the RPV of the uncoupling proteins with MCU and MICU1 (Figures 6C, D).
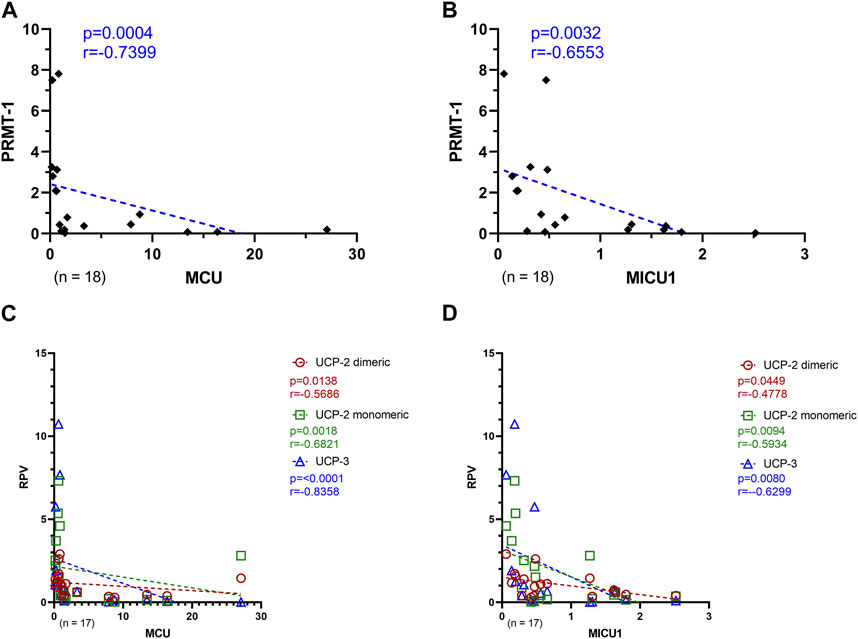
FIGURE 6. Relation of MCU complex regulatory proteins with MCU and MICU1 of Ctrl, AVS, and HOCM human cardiac tissue. (A) Spearman correlation of MCU with PRMT-1 including the whole study group. (B) Spearman correlation of MICU1 with PRMT-1. (C) Spearman correlation of MCU with the MCU or MICU1 regulatory proteins, UCP-2 Dim and Mono, as well as UCP-3, respectively. (D) Spearman correlation of MICU1 with the MCU or MICU1 regulatory proteins, UCP-2 Dim and Mono, as well as UCP-3, respectively. A p-value <0.05 is determined as statistically significant. The dotted lines depict the regression curve of all three study groups.
PRMT-1, as the opponent of UCP-2 and UCP-3, decreases the sensitivity of MCU complex for Ca2+ trafficking. Therefore, to investigate a potential compensatory expression pattern of UCP-2 and UCP-3 with PRMT-1, a Spearman correlation analysis was performed. Most notably, as seen in Figure 7, there is a strong positive relation of the PRMT-1 concentration with both UCP-2 (dimeric: p = 0.0058; monomeric: p = 0.0126) and UCP-3 (p = 0.0004).
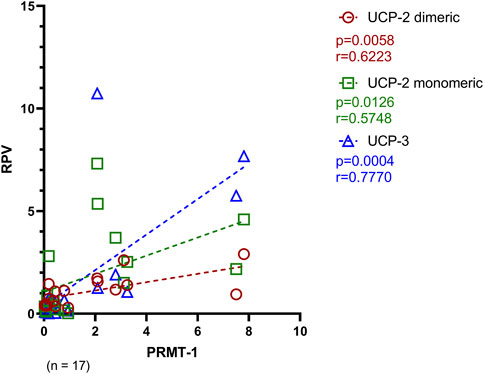
FIGURE 7. Spearman correlation of PRMT-1 with its counteracting proteins, UCP-2 and UCP-3 of Ctrl, AVS and HOCM. A p-value <0.05 is determined as statistically significant. The dotted lines depict the regression curve of all study groups.
As mitochondrial Ca2+ uptake is highly dependent on a distinct Ca2+ threshold, mainly mediated by the SR, we also addressed the SR Ca2+ channels RyR2 and SERCA2a in our analyses (Figure 8). Our Western blot results and statistical analyses showed no significant difference between Ctrl and both pathology groups. Nevertheless, when comparing RyR2 in AVS (3.315; IQR: 6.948–1.744) and HOCM (0.539; IQR: 1.782–0.424) there was a reduction of RyR2 in HOCM (p = 0.0140), as seen in Figure 8A. In contrast, SERCA2a protein levels showed a significant elevation in AVS (p = 0.0013), though just a trend for an increased concentration in HOCM (Figure 8B). Furthermore, we found a statistically positive correlation between MCU and SERCA2a (r = 0.8019, p < 0.0001), or MICU1 (r = 0.5542, p = 0.0170), as depicted in Figures 8D, E.
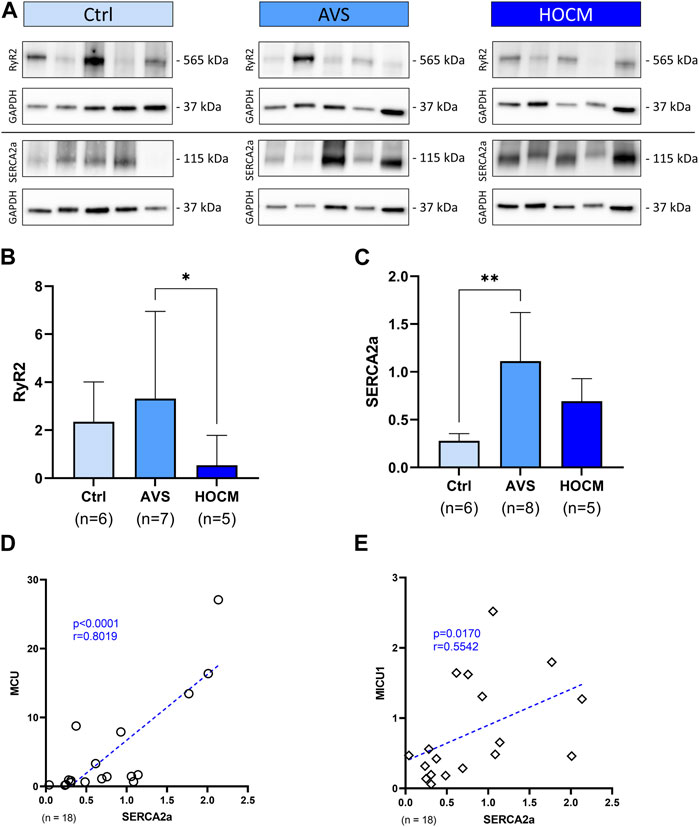
FIGURE 8. Western blot results of SR bound Ca2+-associated proteins in Ctrl, AVS, and HOCM human cardiac tissue. RPV of each protein of interest is stated as a relative value to a unique reference sample. (A) Western blot bands of RyR2 and SERCA2a are normalized with GAPDH as housekeeping protein. (B) Quantification of RyR2 in human septal tissue, statistically analyzed by GraphPad PRISM. Due to non-Gaussian approximation, the data was compared for their statistical differences using Kruskal–Wallis test and the bars are given as median + IQR with *p < 0.05 and **p < 0.01. (C) SERCA2a expression in human septal tissue. Due to normally distributed data, the statistical analysis was performed by Welch ANOVA test and the bars are given as mean + SD. (D,E) Spearman correlation of SERCA2a with MCU, or MICU1 Western blot results, respectively. The dotted line represents the regression line.
In order to bring the cardiac Ca2+ handling into a full circle of Ca2+ trafficking ion channels, we also quantified the presence of the membrane-bound Ca2+ uptaking CaV1.2 (the protein of the main cardiac LTCC) channel and vice versa the extruding ion pump NCX1. The results of the Western blot analyses of both proteins are depicted in Figure 9. Herein, we detected no significant alterations in CaV1.2 levels in the hypertrophied myocardium in comparison to the Ctrl tissue (p > 0.05). On the other hand, NCX1 was highly elevated in both pathologies in comparison to the healthy Ctrl tissue (AVS: p = 0.0295; HOCM: p = 0.0449).
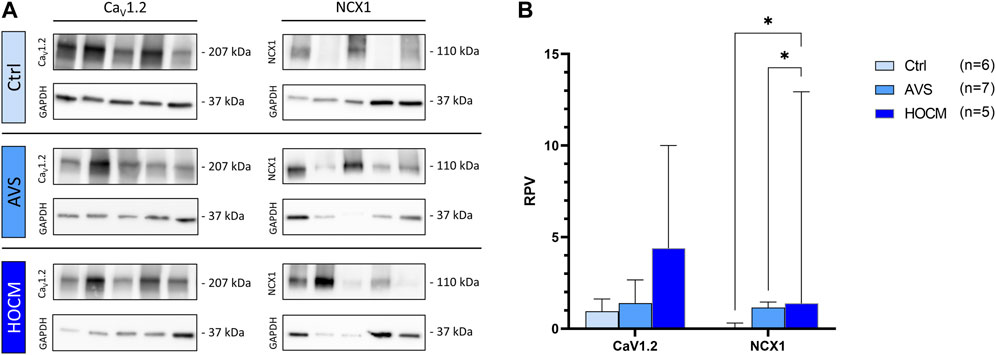
FIGURE 9. Western blot results of cell membrane-bound Ca2+-related channels in Ctrl, AVS, and HOCM human cardiac tissue. RPV of each protein of interest is stated as a relative value to a unique reference sample. (A) Western blot bands of CaV1.2 and NCX1 are normalized with GAPDH as housekeeping protein, respectively. Due to the study design, the depicted bands were graphically assembled from different blots. (B) Quantification of the cytoplasmic Ca2+ related ion channels CaV1.2 and NCX1, statistically analyzed by GraphPad PRISM. Comparison of the channels’ RPV in AVS, HOCM, and healthy Ctrl septal tissue. Due to non-Gaussian approximation, the values in the graph are depicted as median + IQR with *p < 0.05.
Our study reveals structural and molecular remodeling characteristics in the human myocardium affected by AVS and HOCM, respectively, compared to healthy Ctrl tissue. Cardiac specimen was extracted from patients undergoing AV replacement with concomitant myectomy, due to a massive asymmetrical hypertrophy that obstructs the LVOT. Healthy Ctrl tissue was collected from forensic autopsy cases and equally processed and analyzed as AVS and HOCM tissue, respectively. Furthermore, to potentially unveil structural or molecular differences between both etiologies, we compared the outcomes of the acquired disease AVS with the congenital HOCM data.
Our analyses included 1) Basic patient characteristics and echocardiographic data, 2) investigations of structural alterations, such as hypertrophy, inflammation, and hypertrophy; and most importantly, 3) the assessment of potential alterations in cardiac Ca2+ ion channels.
Most of the patient characteristics did not differ between the study groups included. Both pathologies underlie a pronounced and diffuse LVH, clinically characterized by an increased IVSd and an obstruction of the LVOT. Nevertheless, based on the underlying etiologies, HOCM patients had a greater manifestation of hypertrophy in the septum than the AVS group. Additionally, as AVS arises in advanced ages (Nkomo et al., 2006), this study group tendentially represented the oldest group. Furthermore, we could clearly exclude additional manifestations of amyloidosis in all study cohorts by histological assessments and echocardiographic analysis of the AVS and HOCM study patients (Yakupova et al., 2019; Arbelo et al., 2023). Considering the clinical data gathered, and the fact that both pathologies are characterized by LV hypertrophy, we expected structural and molecular alterations in both pathologies. Most importantly, to our knowledge, the present study is the first that addresses potential alterations in the expression patterns of the MCU complex. Detailed combinational analyses of cell membrane bound and SR Ca2+ channels, particularly in AVS, are still speculative. Furthermore, a comparative analysis of the given aspects in AVS versus HOCM was never conducted to date.
The hypertrophic alterations in AVS and HOCM cardiac specimen were confirmed by the increase of the cardiomyocyte diameter in both pathologies. Cell enlargement of cardiomyocytes did not differ between AVS and HOCM. As far as the extracellular matrix is concerned, inflammatory infiltrates and the fibrotic burden were tendentially increased in LVH in comparison to healthy Ctrl tissue, although without statistical significance. Nevertheless, the fibrosis-promoting proteins TGF-β1 and SMAD3 were significantly elevated in LVH in the present study. As both proteins play crucial roles in the TGF-β pathway and their signaling triggers the accumulation of collagens (Vaughan et al., 2000; Lijnen et al., 2003; Khan et al., 2014; Khalil et al., 2017; Ma et al., 2018), we speculate that their elevation is associated with fibrotic remodeling processes in both diseases. Concomitant with the molecular findings, we found a positive correlation of the RFA and the IVSd, indicating a direct relation of LVH remodeling and the septum thickness in both diseases (Lúdvíksson and Gunnlaugsdóttir, 2003). These alterations found may profoundly trigger the propensity for malignant ventricular arrhythmias, which may lead to sudden cardiac death in their most severe manifestation (Varnava et al., 2000; Nazarian, 2011; Aro et al., 2017; Minners et al., 2020).
Besides alterations in the extracellular space, also remodeling processes in the context of cellular ion handling may tremendously influence the cardiac rhythmogenesis. Disturbances in cellular and mitochondrial Ca2+ handling have major impacts on the physiology of the cardiac APs, triggering early or delayed afterdepolarizations (Minners et al., 2020). Previous studies in animal models have already unveiled the relation of a disturbed contractile function of hypertrophied cardiomyocytes with a prolonged AP. As Ca2+ is a key responsible ion for the electrophysiologic behavior, alterations in its transporting channel expression and function therein may fundamentally influence the heart’s contractile function. From animal models of aortic banding, it is known that Ca2+ trafficking is increased in the compensatory stage of hypertrophy, which is mainly achieved by alterations in Ca2+ channel density and their increased sensitivity for Ca2+. However, these observations stemming from data from animal models cannot necessarily be directly applied to humans. Furthermore, none of the previous studies focused on mitochondrial Ca2+ trafficking in LVH due to AVS or HOCM (Sumida et al., 1998; Gattoni et al., 2017; Lewalle et al., 2018).
Severe cardiac hypertrophy is associated with Ca2+ overload, generating an arrhythmogenic substrate (McIntyre and Fry, 1997; Sipido et al., 2000; Botchway et al., 2003; Coppini et al., 2018). In our study, we addressed the expression patterns of the MCU complex-associated proteins MCU and its mediator MICU1 as predictors for the mitochondrial Ca2+ uptake phenotype. Interestingly, our analyses unveiled significant increases in MCU and MICU1 protein levels in AVS and HOCM, in comparison to the Ctrl tissue. MICU1 and MICU2 mainly act as gatekeepers that tightly regulate the amount of Ca2+ taken up by MCU (Plovanich et al., 2013; Kamer and Mootha, 2014; Patron et al., 2014). Especially in its unmethylated state and during high concentrations of Ca2+, MICU1 may activate the MCU pore to transport Ca2+ into the mitochondria (Patron et al., 2014; Garg et al., 2021). Supporting our findings, Paillard et al. (Paillard et al., 2022) found an altered MCU complex composition in the failing human heart. Although they could not reveal an increase in MCU contents, MICU1 and MICU2 levels were highly elevated with an increase in MICU1/MCU ratio (Paillard et al., 2022). In the present study, MICU1/MCU ratio dif not significantly differ between the study groups (p = 0.2223; data not shown), though there was a positive correlation between MICU1 and MCU levels.
Triggering MCU-mediated Ca2+ uptake, we also addressed the expression patterns of the MICU1 regulatory proteins PRMT1, UCP-2, and UCP-3. PRMT-1 methylates MICU1 and leads the suppression of Ca2+ uptake via the MCU pore (Madreiter-Sokolowski et al., 2016). As PRMT-1 as well as its counter players UCP-2 and UCP-3 were decreased in both pathologies, we assume that Ca2+ uptake by the mitochondria are positively triggered in LVH in comparison to healthy cardiomyocytes. Our suspicions are supported by two animal studies addressing MCU expression patterns in hypertrophic adaptions in response to pressure-overloaded murine hearts. Herein, microRNA-1 selectively targets the MCU protein and negatively affects its expression. On the other hand, the repression of microRNA-1 was shown to trigger cardiac hypertrophy, and vice versa, microRNA-1 is inhibited by the activation of β-adrenoreceptors in hypertrophy, leading to the MCU translation (Zaglia et al., 2017). Finally, Yu et al. (Yu et al., 2018) showed a direct link of MCU density with ventricular enlargement together with a ventricular asynchrony in pressure-overloaded heart failure. In contrast, most recent studies indicate a positive effect of MCU upregulation in hypertrophy and heart failure. Since the heart samples in the present study represent rather a moderate, though not an end-stage, hypertrophy or heart failure, the increase in MCU expression found may indicate a compensatory stage of hypertrophy. Thus, the presence of elevated MCU may counteract SR Ca2+ leaks and potentially restore mitochondrial Ca2+ homeostasis (Hamilton et al., 2021; Liu et al., 2021). Nevertheless, since our study did not measure Ca2+-uptake this speculation need to be interpreted with caution. Consequently, MCU expression or function may indicate a potential target for future therapeutic interventions in AVS, or HOCM, respectively.
In addition to aforementioned marked alterations in MCU complex-associated proteins, hypertrophy leads also to a disarrangement of the transverse-tubules (t-tubules) and the SR, increasing the space between LTCC and the RyR2 of the SR. Consequently, the number of uncoupled RyR2 increase, resulting in a reduced or delayed CICR from the SR. This directly influences the synchrony of Ca2+ release, which alters the Ca2+ transient amplitude of the AP and has a direct effect on Ca2+-dependent arrhythmias (Hamilton and Terentyev, 2019). In our study, we found RyR2 to be reduced in HOCM in comparison to AVS. However, no statistically significant alteration was found in comparison to healthy Ctrl. We might cautiously interpret the slight (though without significant difference) increase of RyR2 in AVS by the previously reported need of RyR2 for compensated hypertrophy in response to pressure overload (Zou et al., 2011). More recently, an important study by Zheng et al. (Zheng et al., 2022) reported that RyR2 deficiency clearly affects Ca2+ sparks and amplitudes with mild desynchronization of the Ca2+ transients. Nevertheless, the functional performance and the global Ca2+ release is fully compensated (Zheng et al., 2022). Therefore, we suggest that the slight reduction in RyR2 in HOCM patients would not markedly affect the Ca2+ related excitation-contraction coupling.
As functional counter player of RyR2, we found SERCA2a to be significantly elevated in AVS in the present study in comparison to Ctrl. Supporting our present findings, an increase in SERCA2a expression is reported in the compensatory stage of hypertrophy during which the myocardial function and the contractile force is preserved (Ding et al., 2000; Ito et al., 2000; Silva et al., 2022). In this context, an improved Ca2+ trafficking in the SR, mediated by changes in expression levels or its regulating protein phospholamban, may be beneficial in hypertrophic or failing cardiomyocytes. However, the affinity of SERCA2a for Ca2+ has to be tightly regulated since it plays a prominent role in the progression of hypertrophy and cardiac dysfunction (Vangheluwe et al., 2006).
To further evaluate the interplay of cardiac Ca2+ channels, we finally addressed the expression levels of the membrane bound Ca2+ uptaking channel CaV1.2 (LTCC) and the extruding ion pump NCX1. Although without statistical significance, CaV1.2 levels were slightly increased in both pathologies. Most importantly, Coppini et al. (Coppini et al., 2018) found a cytosolic Ca2+ overload in HCM cardiomyocytes due to an increased trafficking of Ca2+ though LTCC. This was most recently confirmed by Medvedev et al. (2021) demonstrating a higher probability for open LTCCs in failing cardiomyocytes. In contrast, NXC1 is responsible for the extrusion of cytosolic Ca2+ into the extracellular space. Our data from AVS and HOCM patients confirm previous studies that found an upregulation of NCX1 in the hypertrophic or failing heart (Wang et al., 2001; Müller et al., 2002). This increase in expression may be associated with a decline in contractility and lead to abnormal Ca2+ transients, further promoting the propensity for arrhythmias (Wang et al., 2009; Ujihara et al., 2016). Nevertheless, since Wang et al. excluded the onset of heart failure 3–5 weeks after NCX1 transgene overexpression (Wang et al., 2009), we conclude that the present increase of NCX1 result from the LVH in AVS or HOCM, respectively, rather than being responsible for the progression of both pathologies (Menick et al., 2013).
Taken together, our findings confirm structural changes in the hypertrophied myocardium in AVS and HOCM. In line with earlier studies, these alterations may be associated with changes in the electrophysiological network of the heart. In the present study, there is a direct connection between the fibrotic burden and the grade of septal hypertrophy. Additionally, as shown by the analysis of the Ca2+ related ion channels of cardiomyocytes, several alterations of the cellular and mitochondrial Ca2+ take place. Most notably, we clearly demonstrated alterations in the levels of MCU complex associated proteins that indicate disturbances of the mitochondrial Ca2+ balance. Finally, our study shows that the alterations found in either AVS, or HOCM, were statistically even in both pathologies. To our knowledge, we unveiled significant structural changes concerning Ca2+ ion channels in LVH due to AVS, or HOCM in humans for the first time, respectively. Additionally, this is the first study that directly compares those structural changes in the acquired disease AVS with the congenial etiology of HOCM. Our study also provides first evidence for potential therapeutic targets in patients suffering from cardiomyopathies in LVH, triggered by AVS or HOCM, respectively.
The present study greatly supports and confirms previous studies performed in animal models. Of note, all individuals included into the Ctrl cohort were markedly younger than the patients enrolled in the AVS or HOCM groups. Furthermore, compared to the control group, patients in the AVS and HOCM group were affected by CADs, which might promote a higher fibrotic burden in both pathologies. As healthy cardiac tissue is only gained under special circumstances, such as death due to other (impaired health) reasons than cardiovascular diseases, we were restricted to specimens gained from post-mortem individuals. Therefore, we could not eliminate age-associated differences in the cardiac tissue analyzed. Although septal tissue was removed by myectomy in LVH, human cardiac tissue is not abundantly available. While immunohistochemistry was not performed, we were unable to discriminate between activated or non-activated endothelial cells in our HE-sections, while inflammatory cells were discernible by size and shape. Therefore, it seems unlikely that this issue would significantly alter our results. Additionally, an overall greater number of inclusions in each study cohort may have statistically confirmed the expected significant increase in the fibrotic burden in LVH in both diseases. As the study was designed and performed during the COVID-19 pandemic, cardiac surgery interventions were postponed or the patients were simply not included due to the additional efforts during that time. Consequently, inclusion numbers were reduced and patients not included in a timely or steady manner. Whether the alterations in the mitochondrial and SR Ca2+ related proteins have direct functional or cytotoxic effects on the contraction property or the electrophysiological properties of the diseased heart remains to be elucidated in future studies. As the isolation of human ventricular cardiomyocytes from small portions of cardiac tissue is rather difficult, we still failed to perform functional analyses, as well as electrophysiological examinations of the cellular and mitochondrial Ca2+ fluxes. Together with the clinical data, these analyses would deepen the understanding of the underlying pathophysiologies. This concern should be investigated in further trials.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Ethikkommission für das Bundesland Salzburg (415-E/2329/8-2018 and 415-E/2330/7-2018, respectively). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
VP: Conceptualization, Investigation, Methodology, Software, Visualization, Writing–original draft. MH: Formal Analysis, Investigation, Methodology, Writing–original draft. PK-R: Investigation, Resources, Writing–review and editing. SP: Resources, Writing–review and editing. MN: Resources, Writing–review and editing. PJ: Methodology, Writing–review and editing. TK: Methodology, Writing–review and editing. BM: Validation, Writing–review and editing. FS: Data curation, Validation, Writing–review and editing. AK-E: Formal Analysis, Writing–review and editing. KK: Validation, Writing–review and editing. AK: Methodology, Writing–review and editing. CS: Writing–review and editing. ML: Project administration, Resources, Software, Writing–review and editing. FM: Project administration, Resources, Writing–review and editing. RS: Resources, Writing–review and editing. UH: Data curation, Funding acquisition, Project administration, Resources, Software, Writing–review and editing. CD: Formal Analysis, Resources, Writing–review and editing. LM: Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Software, Supervision, Validation, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Paracelsus Medical University (R18/02/106-PAA) and by the Austrian Cardiology Society, as well as Anniversary Fund Oesterreichische Nationalbank (OeNB#18192).
We wish to thank the Department of Cardiac Surgery, Paracelsus Medical University, and the Department of Anesthesiology, Perioperative Medicine and Intensive Care Medicine, Paracelsus Medical University for their cooperation. In particular, they had the leading part in the inclusion of the study patients, the census of the clinical data, and the collection of the cardiac tissue. We also want to thank the members of the Research Program for Ophthalmology and Glaucoma Research, Paracelsus Medical University/SALK for their steady expertise. Furthermore, as parts of the presented data were used for a doctoral thesis, we also want to give special thanks to the Department of Environment and Biodiversity of Paris-Lodron University of Salzburg University of Salzburg, its head and research team.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1264216/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Histological samples of Ctrl, AVS, and HOCM patients, stained by Congo red in order to exclude amyloidosis. The cell nuclei were stained in blue. As amyloid was not detected, no red color is visible. Microscopic pictures were taken by a ×20 objective.
Aly, M., Wiltshire, S., Chahrour, G., Osti, J. C., and Vidal, S. M. (2007). Complex genetic control of host susceptibility to coxsackievirus B3-induced myocarditis. Genes Immun. 8 (3), 193–204. doi:10.1038/sj.gene.6364374
Arbelo, E., Protonotarios, A., Gimeno, J. R., Arbustini, E., Barriales-Villa, R., Basso, C., et al. (2023). 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 44 (37), 3503–3626. doi:10.1093/eurheartj/ehad194
Aro, A. L., Reinier, K., Phan, D., Teodorescu, C., Uy-Evanado, A., Nichols, G. A., et al. (2017). Left-ventricular geometry and risk of sudden cardiac arrest in patients with preserved or moderately reduced left-ventricular ejection fraction. Europace 19 (7), 1146–1152. doi:10.1093/europace/euw126
Bernardi, P., and Rasola, A. (2007). Calcium and cell death: the mitochondrial connection. Subcell. Biochem. 45, 481–506. doi:10.1007/978-1-4020-6191-2_18
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415 (6868), 198–205. doi:10.1038/415198a
Botchway, A. N., Turner, M. A., Sheridan, D. J., Flores, N. A., and Fry, C. H. (2003). Electrophysiological effects accompanying regression of left ventricular hypertrophy. Cardiovasc Res. 60 (3), 510–517. doi:10.1016/j.cardiores.2003.08.013
Brochet, D. X., Yang, D., Cheng, H., and Lederer, W. J. (2012). Elementary calcium release events from the sarcoplasmic reticulum in the heart. Adv. Exp. Med. Biol. 740, 499–509. doi:10.1007/978-94-007-2888-2_21
Carabello, B. A., and Paulus, W. J. (2009). Aortic stenosis. Lancet 373 (9667), 956–966. doi:10.1016/s0140-6736(09)60211-7
Coppini, R., Ferrantini, C., Mugelli, A., Poggesi, C., and Cerbai, E. (2018). Altered Ca(2+) and Na(+) homeostasis in human hypertrophic cardiomyopathy: implications for arrhythmogenesis. Front. Physiol. 9, 1391. doi:10.3389/fphys.2018.01391
Dally, S., Bredoux, R., Corvazier, E., Andersen, J. P., Clausen, J. D., Dode, L., et al. (2006). Ca2+-ATPases in non-failing and failing heart: evidence for a novel cardiac sarco/endoplasmic reticulum Ca2+-ATPase 2 isoform (SERCA2c). Biochem. J. 395 (2), 249–258. doi:10.1042/bj20051427
Danielsen, T. K., Sadredini, M., Manotheepan, R., Aronsen, J. M., Frisk, M., Hansen, M. H., et al. (2020). Exercise training stabilizes RyR2-dependent Ca(2+) release in post-infarction heart failure. Front. Cardiovasc Med. 7, 623922. doi:10.3389/fcvm.2020.623922
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476 (7360), 336–340. doi:10.1038/nature10230
Ding, B., Price, R. L., Goldsmith, E. C., Borg, T. K., Yan, X., Douglas, P. S., et al. (2000). Left ventricular hypertrophy in ascending aortic stenosis mice: anoikis and the progression to early failure. Circulation 101 (24), 2854–2862. doi:10.1161/01.cir.101.24.2854
Dridi, H., Liu, Y., Reiken, S., Liu, X., Argyrousi, E. K., Yuan, Q., et al. (2023). Heart failure-induced cognitive dysfunction is mediated by intracellular Ca(2+) leak through ryanodine receptor type 2. Nat. Neurosci. 26 (8), 1365–1378. doi:10.1038/s41593-023-01377-6
Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P., et al. (2008). Classification of the cardiomyopathies: a position statement from the European society of Cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 29 (2), 270–276. doi:10.1093/eurheartj/ehm342
Elliott, P. M., Anastasakis, A., Borger, M. A., Borggrefe, M., Cecchi, F., Charron, P., et al. (2014). 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European society of Cardiology (ESC). Eur. Heart J. 35 (39), 2733–2779. doi:10.1093/eurheartj/ehu284
Eriksson, U., Kurrer, M. O., Schmitz, N., Marsch, S. C., Fontana, A., Eugster, H. P., et al. (2003). Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation 107 (2), 320–325. doi:10.1161/01.cir.0000043802.38699.66
Garg, V., Suzuki, J., Paranjpe, I., Unsulangi, T., Boyman, L., Milescu, L. S., et al. (2021). The mechanism of MICU-dependent gating of the mitochondrial Ca(2+)uniporter. Elife 10, e69312. doi:10.7554/eLife.69312
Gattoni, S., Røe Å, T., Aronsen, J. M., Sjaastad, I., Louch, W. E., Smith, N. P., et al. (2017). Compensatory and decompensatory alterations in cardiomyocyte Ca(2+) dynamics in hearts with diastolic dysfunction following aortic banding. J. Physiol. 595 (12), 3867–3889. doi:10.1113/jp273879
Gunther, S., and Grossman, W. (1979). Determinants of ventricular function in pressure-overload hypertrophy in man. Circulation 59 (4), 679–688. doi:10.1161/01.cir.59.4.679
Haider, A. W., Larson, M. G., Benjamin, E. J., and Levy, D. (1998). Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 32 (5), 1454–1459. doi:10.1016/s0735-1097(98)00407-0
Hamilton, S., and Terentyev, D. (2019). Altered intracellular calcium homeostasis and arrhythmogenesis in the aged heart. Int. J. Mol. Sci. 20 (10), 2386. doi:10.3390/ijms20102386
Hamilton, S., Terentyeva, R., Perger, F., Hernández Orengo, B., Martin, B., Gorr, M. W., et al. (2021). MCU overexpression evokes disparate dose-dependent effects on mito-ROS and spontaneous Ca(2+) release in hypertrophic rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 321 (4), H615–h632. doi:10.1152/ajpheart.00126.2021
Hang, D., Nguyen, A., and Schaff, H. V. (2017). Surgical treatment for hypertrophic cardiomyopathy: a historical perspective. Ann. Cardiothorac. Surg. 6 (4), 318–328. doi:10.21037/acs.2017.04.03
Holmström, K. M., Pan, X., Liu, J. C., Menazza, S., Liu, J., Nguyen, T. T., et al. (2015). Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell Cardiol. 85, 178–182. doi:10.1016/j.yjmcc.2015.05.022
Hoppe, U. C. (2010). Mitochondrial calcium channels. FEBS Lett. 584 (10), 1975–1981. doi:10.1016/j.febslet.2010.04.017
Ito, K., Yan, X., Tajima, M., Su, Z., Barry, W. H., and Lorell, B. H. (2000). Contractile reserve and intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing hearts. Circ. Res. 87 (7), 588–595. doi:10.1161/01.res.87.7.588
Kamer, K. J., and Mootha, V. K. (2014). MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 15 (3), 299–307. doi:10.1002/embr.201337946
Kayalar, N., Schaff, H. V., Daly, R. C., Dearani, J. A., and Park, S. J. (2010). Concomitant septal myectomy at the time of aortic valve replacement for severe aortic stenosis. Ann. Thorac. Surg. 89 (2), 459–464. doi:10.1016/j.athoracsur.2009.10.065
Khalil, H., Kanisicak, O., Prasad, V., Correll, R. N., Fu, X., Schips, T., et al. (2017). Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest 127 (10), 3770–3783. doi:10.1172/jci94753
Khan, S., Joyce, J., Margulies, K. B., and Tsuda, T. (2014). Enhanced bioactive myocardial transforming growth factor-β in advanced human heart failure. Circ. J. 78 (11), 2711–2718. doi:10.1253/circj.cj-14-0511
Kohlhaas, M., Liu, T., Knopp, A., Zeller, T., Ong, M. F., Böhm, M., et al. (2010). Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121 (14), 1606–1613. doi:10.1161/circulationaha.109.914911
Kumar, A., Avishay, D. M., Jones, C. R., Shaikh, J. D., Kaur, R., Aljadah, M., et al. (2021). Sudden cardiac death: epidemiology, pathogenesis and management. Rev. Cardiovasc Med. 22 (1), 147–158. doi:10.31083/j.rcm.2021.01.207
Larbig, R., Reda, S., Paar, V., Trost, A., Leitner, J., Weichselbaumer, S., et al. (2017). Through modulation of cardiac Ca(2+) handling, UCP2 affects cardiac electrophysiology and influences the susceptibility for Ca(2+) -mediated arrhythmias. Exp. Physiol. 102 (6), 650–662. doi:10.1113/ep086209
Levy, D., Anderson, K. M., Savage, D. D., Balkus, S. A., Kannel, W. B., and Castelli, W. P. (1987). Risk of ventricular arrhythmias in left ventricular hypertrophy: the Framingham Heart Study. Am. J. Cardiol. 60 (7), 560–565. doi:10.1016/0002-9149(87)90305-5
Lewalle, A., Land, S., Carruth, E., Frank, L. R., Lamata, P., Omens, J. H., et al. (2018). Decreasing compensatory ability of concentric ventricular hypertrophy in aortic-banded rat hearts. Front. Physiol. 9, 37. doi:10.3389/fphys.2018.00037
Lijnen, P., Petrov, V., Rumilla, K., and Fagard, R. (2003). Transforming growth factor-beta 1 promotes contraction of collagen gel by cardiac fibroblasts through their differentiation into myofibroblasts. Methods Find. Exp. Clin. Pharmacol. 25 (2), 79–86. doi:10.1358/mf.2003.25.2.723680
Liu, T., Yang, N., Sidor, A., and O'Rourke, B. (2021). MCU overexpression rescues inotropy and reverses heart failure by reducing SR Ca(2+) leak. Circ. Res. 128 (8), 1191–1204. doi:10.1161/circresaha.120.318562
Lúdvíksson, B. R., and Gunnlaugsdóttir, B. (2003). Transforming growth factor-beta as a regulator of site-specific T-cell inflammatory response. Scand. J. Immunol. 58 (2), 129–138. doi:10.1046/j.1365-3083.2003.01297.x
Ma, Z. G., Yuan, Y. P., Wu, H. M., Zhang, X., and Tang, Q. Z. (2018). Cardiac fibrosis: new insights into the pathogenesis. Int. J. Biol. Sci. 14 (12), 1645–1657. doi:10.7150/ijbs.28103
Madreiter-Sokolowski, C. T., Klec, C., Parichatikanond, W., Stryeck, S., Gottschalk, B., Pulido, S., et al. (2016). PRMT1-mediated methylation of MICU1 determines the UCP2/3 dependency of mitochondrial Ca(2+) uptake in immortalized cells. Nat. Commun. 7, 12897. doi:10.1038/ncomms12897
Marionneau, C., Brunet, S., Flagg, T. P., Pilgram, T. K., Demolombe, S., and Nerbonne, J. M. (2008). Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ. Res. 102 (11), 1406–1415. doi:10.1161/circresaha.107.170050
McIntyre, H., and Fry, C. H. (1997). Abnormal action potential conduction in isolated human hypertrophied left ventricular myocardium. J. Cardiovasc Electrophysiol. 8 (8), 887–894. doi:10.1111/j.1540-8167.1997.tb00850.x
Medvedev, R. Y., Sanchez-Alonso, J. L., Mansfield, C. A., Judina, A., Francis, A. J., Pagiatakis, C., et al. (2021). Local hyperactivation of L-type Ca(2+) channels increases spontaneous Ca(2+) release activity and cellular hypertrophy in right ventricular myocytes from heart failure rats. Sci. Rep. 11 (1), 4840. doi:10.1038/s41598-021-84275-w
Menick, D. R., Li, M. S., Chernysh, O., Renaud, L., Kimbrough, D., Kasiganesan, H., et al. (2013). Transcriptional pathways and potential therapeutic targets in the regulation of Ncx1 expression in cardiac hypertrophy and failure. Adv. Exp. Med. Biol. 961, 125–135. doi:10.1007/978-1-4614-4756-6_11
Michels, G., Khan, I. F., Endres-Becker, J., Rottlaender, D., Herzig, S., Ruhparwar, A., et al. (2009). Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation 119 (18), 2435–2443. doi:10.1161/circulationaha.108.835389
Minners, J., Rossebo, A., Chambers, J. B., Gohlke-Baerwolf, C., Neumann, F. J., Wachtell, K., et al. (2020). Sudden cardiac death in asymptomatic patients with aortic stenosis. Heart 106 (21), 1646–1650. doi:10.1136/heartjnl-2019-316493
Mirna, M., Paar, V., Topf, A., Kraus, T., Sotlar, K., Aigner, A., et al. (2021). A new player in the game: treatment with antagomiR-21a-5p significantly attenuates histological and echocardiographic effects of experimental autoimmune myocarditis. Cardiovasc. Res. 118 (2), 556–572. doi:10.1093/cvr/cvab015
Motloch, L. J., Gebing, T., Reda, S., Schwaiger, A., Wolny, M., and Hoppe, U. C. (2016a). UCP3 regulates single-channel activity of the cardiac mCa1. J. Membr. Biol. 249 (4), 577–584. doi:10.1007/s00232-016-9913-2
Motloch, L. J., Larbig, R., Gebing, T., Reda, S., Schwaiger, A., Leitner, J., et al. (2016b). By regulating mitochondrial Ca2+-uptake UCP2 modulates intracellular Ca2+. PLoS One 11 (2), e0148359. doi:10.1371/journal.pone.0148359
Müller, J. G., Isomatsu, Y., Koushik, S. V., O'Quinn, M., Xu, L., Kappler, C. S., et al. (2002). Cardiac-specific expression and hypertrophic upregulation of the feline Na(+)-Ca(2+) exchanger gene H1-promoter in a transgenic mouse model. Circ. Res. 90 (2), 158–164. doi:10.1161/hh0202.103231
Nazarian, S. (2011). Is ventricular arrhythmia a possible mediator of the association between aortic stenosis-related midwall fibrosis and mortality? J. Am. Coll. Cardiol. 58 (12), 1280–1282. doi:10.1016/j.jacc.2011.04.045
Nkomo, V. T., Gardin, J. M., Skelton, T. N., Gottdiener, J. S., Scott, C. G., and Enriquez-Sarano, M. (2006). Burden of valvular heart diseases: a population-based study. Lancet 368 (9540), 1005–1011. doi:10.1016/s0140-6736(06)69208-8
Ommen, S. R., Mital, S., Burke, M. A., Day, S. M., Deswal, A., Elliott, P., et al. (2020). 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American college of Cardiology/American heart association joint committee on clinical Practice guidelines. Circulation 142 (25), e558–e631. doi:10.1161/cir.0000000000000937
O'Rourke, B. (2007). Mitochondrial ion channels. Annu. Rev. Physiol. 69, 19–49. doi:10.1146/annurev.physiol.69.031905.163804
Paar, V., Jirak, P., Larbig, R., Zagidullin, N. S., Brandt, M. C., Lichtenauer, M., et al. (2019). Pathophysiology of calcium mediated ventricular arrhythmias and novel therapeutic options with focus on gene therapy. Int. J. Mol. Sci. 20 (21), 5304. doi:10.3390/ijms20215304
Paillard, M., Huang, K. T., Weaver, D., Lambert, J. P., Elrod, J. W., and Hajnóczky, G. (2022). Altered composition of the mitochondrial Ca(2+)uniporter in the failing human heart. Cell Calcium 105, 102618. doi:10.1016/j.ceca.2022.102618
Patron, M., Checchetto, V., Raffaello, A., Teardo, E., Vecellio Reane, D., Mantoan, M., et al. (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53 (5), 726–737. doi:10.1016/j.molcel.2014.01.013
Perocchi, F., Gohil, V. M., Girgis, H. S., Bao, X. R., McCombs, J. E., Palmer, A. E., et al. (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467 (7313), 291–296. doi:10.1038/nature09358
Pillai-Kastoori, L., Schutz-Geschwender, A. R., and Harford, J. A. (2020). A systematic approach to quantitative Western blot analysis. Anal. Biochem. 593, 113608. doi:10.1016/j.ab.2020.113608
Piquereau, J., Caffin, F., Novotova, M., Lemaire, C., Veksler, V., Garnier, A., et al. (2013). Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front. Physiol. 4, 102. doi:10.3389/fphys.2013.00102
Plovanich, M., Bogorad, R. L., Sancak, Y., Kamer, K. J., Strittmatter, L., Li, A. A., et al. (2013). MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8 (2), e55785. doi:10.1371/journal.pone.0055785
Riedelsheimer, B., and Büchl-Zimmermann, S. (2015). “Färbungen,” in Romeis - mikroskopische technik. Editors M. Mulisch, and U. Welsch (Berlin, Heidelberg: Springer Berlin Heidelberg), 171–282.
Silva, V. L. D., Souza, S. L. B., Mota, G. A. F., Campos, D. H. S., Melo, A. B., Vileigas, D. F., et al. (2022). The dysfunctional scenario of the major components responsible for myocardial calcium balance in heart failure induced by aortic stenosis. Arq. Bras. Cardiol. 118 (2), 463–475. doi:10.36660/abc.20200618
Sipido, K. R., Volders, P. G., de Groot, S. H., Verdonck, F., Van de Werf, F., Wellens, H. J., et al. (2000). Enhanced Ca(2+) release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation 102 (17), 2137–2144. doi:10.1161/01.cir.102.17.2137
Stewart, M. H., Lavie, C. J., Shah, S., Englert, J., Gilliland, Y., Qamruddin, S., et al. (2018). Prognostic implications of left ventricular hypertrophy. Prog. Cardiovasc Dis. 61 (5-6), 446–455. doi:10.1016/j.pcad.2018.11.002
Sumida, E., Nohara, M., Muro, A., Sumida, E., Kaku, H., Koga, Y., et al. (1998). Altered calcium handling in compensated hypertrophied rat cardiomyocytes induced by pressure overload. Jpn. Circ. J. 62 (1), 36–46. doi:10.1253/jcj.62.36
Taylor, S. C., Rosselli-Murai, L. K., Crobeddu, B., and Plante, I. (2022). A critical path to producing high quality, reproducible data from quantitative western blot experiments. Sci. Rep. 12 (1), 17599. doi:10.1038/s41598-022-22294-x
Tomita, F., Bassett, A. L., Myerburg, R. J., and Kimura, S. (1994). Diminished transient outward currents in rat hypertrophied ventricular myocytes. Circ. Res. 75 (2), 296–303. doi:10.1161/01.res.75.2.296
Trenker, M., Malli, R., Fertschai, I., Levak-Frank, S., and Graier, W. F. (2007). Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 9 (4), 445–452. doi:10.1038/ncb1556
Tse, G., Wong, S. T., Tse, V., Lee, Y. T., Lin, H. Y., and Yeo, J. M. (2016). Cardiac dynamics: alternans and arrhythmogenesis. J. Arrhythm. 32 (5), 411–417. doi:10.1016/j.joa.2016.02.009
Ujihara, Y., Iwasaki, K., Takatsu, S., Hashimoto, K., Naruse, K., Mohri, S., et al. (2016). Induced NCX1 overexpression attenuates pressure overload-induced pathological cardiac remodelling. Cardiovasc Res. 111 (4), 348–361. doi:10.1093/cvr/cvw113
Vahanian, A., Beyersdorf, F., Praz, F., Milojevic, M., Baldus, S., Bauersachs, J., et al. (2022). 2021 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J. 43 (7), 561–632. doi:10.1093/eurheartj/ehab395
Vakrou, S., and Abraham, M. R. (2014). Hypertrophic cardiomyopathy: a heart in need of an energy bar? Front. Physiol. 5, 309. doi:10.3389/fphys.2014.00309
Vangheluwe, P., Tjwa, M., Van Den Bergh, A., Louch, W. E., Beullens, M., Dode, L., et al. (2006). A SERCA2 pump with an increased Ca2+ affinity can lead to severe cardiac hypertrophy, stress intolerance and reduced life span. J. Mol. Cell Cardiol. 41 (2), 308–317. doi:10.1016/j.yjmcc.2006.05.014
Varnava, A. M., Elliott, P. M., Sharma, S., McKenna, W. J., and Davies, M. J. (2000). Hypertrophic cardiomyopathy: the interrelation of disarray, fibrosis, and small vessel disease. Heart 84 (5), 476–482. doi:10.1136/heart.84.5.476
Vaughan, M. B., Howard, E. W., and Tomasek, J. J. (2000). Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 257 (1), 180–189. doi:10.1006/excr.2000.4869
Wang, J., Chan, T. O., Zhang, X. Q., Gao, E., Song, J., Koch, W. J., et al. (2009). Induced overexpression of Na+/Ca2+ exchanger transgene: altered myocyte contractility, [Ca2+]i transients, SR Ca2+ contents, and action potential duration. Am. J. Physiol. Heart Circ. Physiol. 297 (2), H590–H601. doi:10.1152/ajpheart.00190.2009
Wang, Z., Nolan, B., Kutschke, W., and Hill, J. A. (2001). Na+-Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. J. Biol. Chem. 276 (21), 17706–17711. doi:10.1074/jbc.M100544200
Welsch, U., Riedelsheimer, B., and Büchl-Zimmermann, S. (2015). “Schnittpräparation für die Lichtmikroskopie,” in Romeis - mikroskopische technik. Editors M. Mulisch, and U. Welsch (Berlin, Heidelberg: Springer Berlin Heidelberg), 99–120.
Yakupova, E. I., Bobyleva, L. G., Vikhlyantsev, I. M., and Bobylev, A. G. (2019). Congo Red and amyloids: history and relationship. Biosci. Rep. 39 (1). doi:10.1042/bsr20181415
Yu, Z., Chen, R., Li, M., Yu, Y., Liang, Y., Han, F., et al. (2018). Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement. Int. J. Cardiol. 271, 161–168. doi:10.1016/j.ijcard.2018.05.054
Zaglia, T., Ceriotti, P., Campo, A., Borile, G., Armani, A., Carullo, P., et al. (2017). Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 114 (43), E9006-E9015–e9015. doi:10.1073/pnas.1708772114
Zheng, J., Dooge, H. C., Pérez-Hernández, M., Zhao, Y. T., Chen, X., Hernandez, J. J., et al. (2022). Preserved cardiac performance and adrenergic response in a rabbit model with decreased ryanodine receptor 2 expression. J. Mol. Cell Cardiol. 167, 118–128. doi:10.1016/j.yjmcc.2022.04.004
Zou, Y., Liang, Y., Gong, H., Zhou, N., Ma, H., Guan, A., et al. (2011). Ryanodine receptor type 2 is required for the development of pressure overload-induced cardiac hypertrophy. Hypertension 58 (6), 1099–1110. doi:10.1161/hypertensionaha.111.173500
Keywords: left ventricular hypertrophy, aortic valve stenosis, hypertrophic obstructive cardiomyopathy, fibrosis, calcium, mitochondria
Citation: Paar V, Haslinger M, Krombholz-Reindl P, Pittner S, Neuner M, Jirak P, Kolbitsch T, Minnich B, Schrödl F, Kaser-Eichberger A, Kopp K, Koller A, Steinwender C, Lichtenauer M, Monticelli FC, Seitelberger R, Hoppe UC, Dinges C and Motloch LJ (2023) Hypertrophic cardiomyopathy is characterized by alterations of the mitochondrial calcium uniporter complex proteins: insights from patients with aortic valve stenosis versus hypertrophic obstructive cardiomyopathy. Front. Pharmacol. 14:1264216. doi: 10.3389/fphar.2023.1264216
Received: 20 July 2023; Accepted: 01 November 2023;
Published: 22 November 2023.
Edited by:
Giovanni Zifarelli, National Research Council (CNR), ItalyReviewed by:
Dmitry Terentyev, The Ohio State University, United StatesCopyright © 2023 Paar, Haslinger, Krombholz-Reindl, Pittner, Neuner, Jirak, Kolbitsch, Minnich, Schrödl, Kaser-Eichberger, Kopp, Koller, Steinwender, Lichtenauer, Monticelli, Seitelberger, Hoppe, Dinges and Motloch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vera Paar, di5wYWFyQHNhbGsuYXQ=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.