 Xiaonan Li
Xiaonan Li Donghui Cao2
Donghui Cao2 Siming Sun
Siming Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 26 July 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1215020
Dysregulation of reactive oxygen species (ROS) production and ROS-regulated pathways in cancer cells leads to abnormal accumulation of reactive oxygen species, displaying a double-edged role in cancer progression, either supporting transformation/proliferation and stimulating tumorigenesis or inducing cell death. Cancer cells can accommodate reactive oxygen species by regulating them at levels that allow the activation of pro-cancer signaling pathways without inducing cell death via modulation of the antioxidant defense system. Therefore, targeting reactive oxygen species is a promising approach for cancer treatment. Ginsenosides, their derivatives, and related drug carriers are well-positioned to modulate multiple signaling pathways by regulating oxidative stress-mediated cellular and molecular targets to induce apoptosis; regulate cell cycle arrest and autophagy, invasion, and metastasis; and enhance the sensitivity of drug-resistant cells to chemotherapeutic agents of different cancers depending on the type, level, and source of reactive oxygen species, and the type and stage of the cancer. Our review focuses on the pro- and anticancer effects of reactive oxygen species, and summarizes the mechanisms and recent advances in different ginsenosides that bring about anticancer effects by targeting reactive oxygen species, providing new ideas for designing further anticancer studies or conducting more preclinical and clinical studies.
Reactive oxygen species (ROS), as by-products of cellular respiration and aerobic metabolism, are a group of highly active oxygenated molecules that play a pivotal role in body function (Zheng et al., 2022). Redox reactions maintain ROS balance in the organism and function as signaling molecules to trigger cellular regulatory pathways. Extensive evidence suggests that ROS and abnormal redox reactions damage DNA, proteins, and lipids; thus, ROS are thought to be genomically damaging and cancer-promoting (Ismail et al., 2019). It has been well-established that almost all cancer cells exhibit variable types of oxidative stress and higher levels of ROS (Hayes et al., 2020). To adapt to high ROS levels and optimize their pro-cancer effects, carcinoma cells intelligently strengthen their antioxidant capacity to maximize their profitability during different stages (Reczek and Chandel, 2017; Maslah et al., 2020; Attanasio et al., 2021). However, different opinions have been raised that ROS in cancers act as double-edged swords. ROS not only help in cancer cell transformation/proliferation but also exert cytotoxicity, activate anticancer signaling, and promote oxidative stress-induced cancer cell death (Schieber and Chandel, 2014; Cheung et al., 2020; Ding et al., 2020; Zhang et al., 2020; Cruz-Gregorio et al., 2021; Kuo et al., 2022). This property makes these cells more susceptible to redox manipulation or altered ROS levels than normal cells (Gorrini et al., 2013; Reczek and Chandel, 2017). Hence, ROS modulation is a prospective approach for cancer treatment.
Ginseng (Panax ginseng C.A. Meyer), a perennial herb of the genus Panax, has a medicinal history of thousands of years and is widely used as a health food worldwide because of its medicinal properties (Kim, 2018). Ginseng, with a variety of pharmacological effects, including anticancer, antioxidant, anti-inflammatory, and other biological effects (Fu et al., 2018; Metwaly et al., 2019; Wang et al., 2021a), contains many chemical components, of which the most dominant are ginsenosides. Numerous studies have suggested that ginsenosides protect cells by preventing oxidative damage, thereby preventing the occurrence and progression of diseases (Kang et al., 2013; Ong et al., 2015). Possible mechanisms mentioned in previous studies include their ability to inhibit oxidative damage by inhibiting malondialdehyde (MDA) formation, reducing lipid peroxidation, and regulating the activity of antioxidant enzymes, such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), and other antioxidant factors (Kim et al., 2004). In addition, it has also been suggested that modulation of oxidative stress-related oxidative signaling pathways, such as the Keap1/Nrf2/ARE, PI3K/Akt, and Wnt/β-catenin, and nuclear factor-k-gene binding (NF-κB) signaling pathways, is also an important way in which ginsenosides exert antioxidant damage (Li et al., 2016; Chen et al., 2019; Hagar et al., 2019).
Given the heightened sensitivity to changes in ROS levels, a strategy to modulate ROS levels is likely to be valid for cancer therapy. Coincidentally, ginsenosides have a superior antioxidant capacity, and studies have also suggested that ginsenosides hold a central position in cancer therapy by modulating this target to control apoptosis and autophagy, stall cancer invasion and metastasis, regulate cell cycle arrest, and enhance the sensitivity of drug-resistant cells to chemotherapeutic agents; however, the specific mechanisms studied have not been fully elucidated (Zheng et al., 2016; Hong and Fan, 2019b; Han et al., 2020; Ping et al., 2020; Lu et al., 2022). Therefore, in this article, we focus on the pro- and anticancer effects of ROS and investigate the particular mechanisms and latest advances of different ginsenosides, including their derivatives and drug delivery, exerting anticancer effects by targeting ROS to represent new insights for further design of anticancer studies or conduct more preclinical and clinical studies.
ROS, which are single unpaired electrons present in free radicals, ions, or molecules, are a group of highly reactive chemicals that contain oxygen, including superoxide anions (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH−) (Martínez-Cayuela, 1995). Of these, H2O2 performs a critical role in signaling by selectively modifying and regulating the function of numerous proteins, whereas other forms of ROS are more likely to cause damage and toxicity (Cheung and Vousden, 2022). The primary cellular source of ROS is the mitochondria, which produce ROS during respiration as a natural by-product of electron transport chain (ETC) activity (Bertram et al., 2006) (Figure 1). Superoxide molecules are produced in complexes I and III and subsequently released into the intermembrane space and mitochondrial matrix (Han et al., 2001; Chen et al., 2003). Next, superoxide molecules in the mitochondrial outer membrane leak into the cytoplasm through the mitochondrial permeability transition pore (MPTP), whereas superoxide molecules in the mitochondrial matrix or cytosol are disproportionated to H2O2 by MnSOD and Cu/ZnSOD, respectively (Liou and Storz, 2010). In addition to the mitochondria, active nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) are another major source of ROS (Brown and Griendling, 2009; Bae et al., 2011). Normally, the NOX family is activated by Racphox, p47phox, p22phox, p67phox, and 40phox to allow the catalytic subunit to remove an electron from the cell membrane of NADPH and transfer it to O2 to produce O2−(Moloney and Cotter, 2018) (Figure 1 and Figure 2). Moreover, there is striking evidence that ROS produced by NOXs as signaling molecules can regulate cell behaviors such as cell proliferation, differentiation, and apoptosis. The aberrant expression of NOXs can induce various diseases, including cancer (Bonner and Arbiser, 2012; Spencer and Engelhardt, 2014).
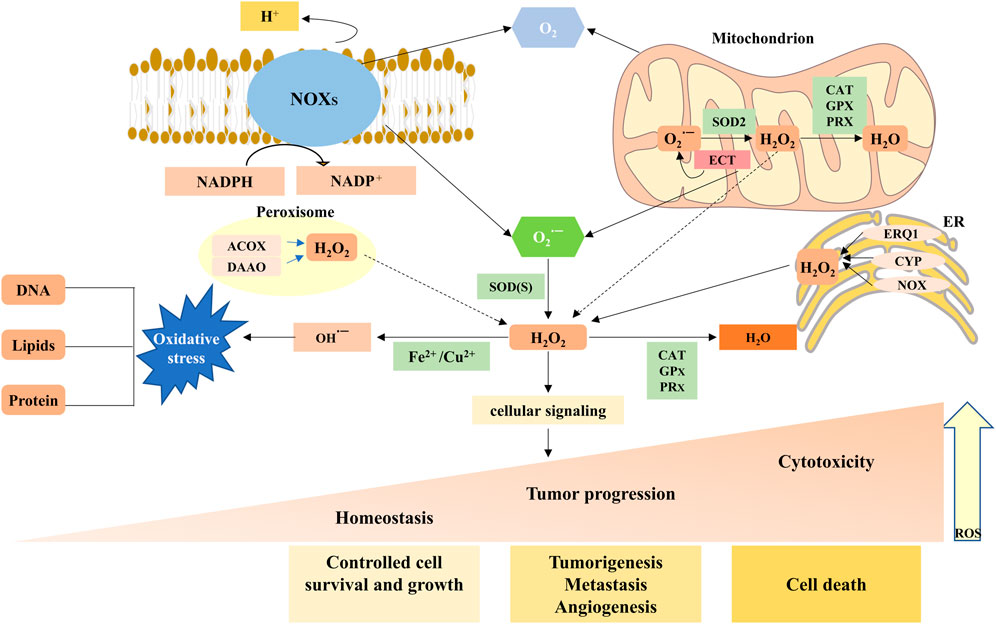
FIGURE 1. Main generation and modulation of ROS. Mitochondrial and membrane NADPH oxidases (NOXs) are the main sources of endogenous ROS production. The superoxide dismutase (SOD) enzymes transform superoxide radical anion (O2·−) into hydrogen peroxide (H2O2). H2O2 can undergo Fenton chemistry with Fe2+ to form a hydroxyl radical (OH·−), causing damage to DNA, proteins, and lipids. H2O2 can be reduced and converted to H2O by peroxiredoxins (PRXs), glutathione peroxidases (GPXs), and catalase (CAT). ROS, as a signaling molecule, impacts the development and progression of normal body and disease cells depending on its concentration.
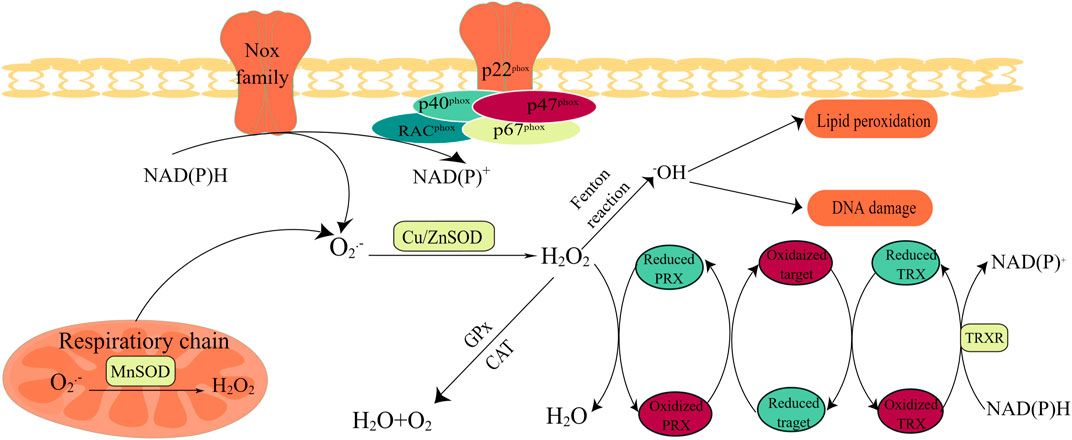
FIGURE 2. Oxidant and antioxidant enzymes. Activation of NOXs and the mitochondrial respiratory chain are the main sources of the superoxide radical anion (O2·−), and O2·− conversion to H2O2 can be performed by (SOD. The activation of the NOX family requires the involvement of subunits such as Racphox, p47phox, and p22phox; thus, the catalytic subunit can remove an electron from the cell membrane NADPH and transfer it to O2 to produce O2·−. CAT and/or GPX is modulated to maintain homeostasis in the body; when redox metals are present, increased concentrations of H2O2 produce hydroxyl radicals. Hydrogen peroxide oxidase (PRX) plays a critical role in hydrogen peroxide scavenging and redox regulation. Redox-regulated proteins can act as an extra redox relay base between PRX and thioredoxin (TRX).
Mitochondria and NOXs, the two main sources of ROS, mediate ROS-dependent signaling, an effect that can be achieved by spatial localization to oxidant receptors. H2O2 has strong diffusion properties and is brought into the cytoplasm exclusively by aquaporins (aquaporins 3 and 8) as secondary messengers that regulate multiple signaling pathways (Wang et al., 2021b). However, when accompanied by excessive ROS levels, H2O2 can move away from the site of production, inducing oxidative damage and cell death. Therefore, whether ROS are a poison or cure depends largely on the local concentration and type of ROS, as well as the abundance of antioxidants (Chandel and Tuveson, 2014).
Other sources of ROS come from misfolding of proteins through endoplasmic reticulum (ER) stress (Murphy, 2013); cytochrome P450 (CYP) (Kim et al., 2020); Fenton chemistry that involves transition metal ions (Halcrow et al., 2021) and external stimuli, such as epidermal growth factor (EGF), cancer necrosis factor-α (TNF-α), and interleukin-1β (IL-1β) irradiation; and hypoxia (Ilatovskaya et al., 2013; Clauzure et al., 2014; Roberge et al., 2014; Cheung and Vousden, 2022).
Unsurprisingly, healthy cells have evolved to overcome the damaging effects of ROS, including balanced ROS production, adequate antioxidant activity, and cellular repair, leading to the maintenance of ROS at an equilibrium concentration. At low-to-moderate levels, ROS can act as cell signaling messengers involved in regulating a variety of cellular functions; however, high levels of ROS can cause DNA damage, lipid peroxidation, and protein oxidation, causing cell damage (Bae et al., 2011; Chio and Tuveson, 2017; Reczek and Chandel, 2017). Under normal physiological conditions, contributing to a collectively powerful antioxidant system, intracellular ROS levels are steadily kept in balance to ensure that the ROS signaling process is maintained smoothly while avoiding oxidative damage. The generation and modulation of ROS, and the functions of oxidant and antioxidant enzymes are shown in Figure 1 and Figure 2, respectively.
SODs, categorized as cell membrane SOD1, mitochondrial SOD2, and extracellular SOD3, are vital enzymes in the antioxidant enzyme defense system, particularly superoxide anion radicals (which catalyze the conversion of O2− to H2O2 and O2) (Areti et al., 2014; Sheng et al., 2014).SODs may also attenuate NOX-dependent redox reactions by regulating diffuse H2O2 signaling and signaling-related activation of receptor tyrosine kinases and G-protein-coupled receptors (Parascandolo and Laukkanen, 2018). Despite being termed carcinogens, some SODs can also be upregulated during carcinogenesis (Hayes et al., 2020). To maximize the benefits of H2O2 levels for the body, several other antioxidants are involved in the intracellular conversion of H2O2 to H2O, including peroxidases (PRXs), glutathione peroxidases (GPXs), and CAT (Brigelius-Flohé and Maiorino, 2013). PRXs, which have a high-affinity binding site for H2O2 and are abundantly expressed in subcellular compartments, are considered to be ideal H2O2 scavengers (Wang et al., 2021b). GPXs convert H2O2 to H2O by oxidizing reduced glutathione (GSH) to glutathione disulfide (GSSG), and glutathione reductase (GR) uses NADPH as an electron donor to convert oxidized GSSG to GSH. Distinguishing from PRXs and GPXs, CAT participates in two different antioxidant reactions depending on the H2O2 concentration (Sepasi Tehrani and Moosavi-Movahedi, 2018). At high H2O2 levels, CAT exhibits catalytic activity, transforming H2O2 into H2O2 and O2. However, low H2O2 levels display peroxidative activity, decreasing one H2O2 molecule to two H2O molecules, by depleting two reducing equivalents from non-NADPH hydrogen donors, such as alcohols, phenols, hormones, and metals (Wang et al., 2021b).
The main members of the nonenzymatic antioxidant system include water-soluble small molecules, fat-soluble antioxidants, protein-based antioxidants, and trace elements, which form the body’s second line of defense (Birben et al., 2012; Gamcsik et al., 2012; Forman and Zhang, 2021; Jelic et al., 2021). Reduced GSH and NADPH levels have been the focus of many studies. GSH directly or indirectly reacts with oxidizing substances and is oxidized to GSSG (Gaucher et al., 2018). Radicals and ROS are directly quenched by GSH. Under enzymatic action, GSH functions as a cosubstrate for GPX, reducing H2O2 and lipid peroxide to H2O and lipid alcohols (lipid-OOH) (Niu et al., 2021). NADPH, an indispensable product of multiple intracellular metabolic pathways, is a highly desirable electron donor that restores oxidized GSH and thioredoxins (TXNs) produced by the reduction of GPXs and PRXs to a reduced state (Sies and Jones, 2020). As an electron carrier, NADPH is not only a producer (incompletely reduced) but also a scavenger (completely reduced) of ROS. Paradoxically, inadequate NADPH leads to ROS accumulation, and excessive NADPH leads to reductive stress, which is utilized by NOXs to produce ROS. Therefore, only if the NADP+/NADPH level remains in an equilibrium state can NADPH perform its antioxidant defensive roles (Wang et al., 2021b).
Many transcription factors, including nuclear factor E2-related factor (Nrf2), oncoprotein p53, activator protein 1 (AP-1), HIF-1a, nuclear factor κB (NF-κB), and forkhead box O class (FOXO), can be activated by ROS and regulate the redox state of cells (Marinho et al., 2014). (Marinho et al., 2014).
The function of ROS in cancer remains unknown and warrants in-depth exploration. Sustained exposure to high ROS levels can be detrimental to DNA, causing oncogene activation and cancer suppressor gene inactivation, mediating signaling events, and promoting carcinogenesis, progression, and metastasis (Chio and Tuveson, 2017; Rose Li et al., 2020; Aboelella et al., 2021). Unlike normal cells that die from prolonged exposure to these conditions, cancer cells strategically activate antioxidant systems and metabolic changes, including the elevated activity of antioxidants such as SOD2/MnSOD or inactivation of scavenging enzymes such as PRX1 stabilization of HIF, and activation of AMPK, to strengthen the generation of NADPH and GSH and the maintenance of redox homeostasis to thrive in harsh cancers (Jeon et al., 2012; Reczek and Chandel, 2017), thereby promoting carcinogenesis and progression (Chandel and Tuveson, 2014; Sies and Jones, 2020). However, when the accumulation of ROS crosses a certain threshold, their carcinogenic effects on proliferation and invasion are eliminated and transformed into antitumor effects via an induced regulated cell death (RCD) program that consists largely of apoptosis, autophagy, and ferroptosis (Wang et al., 2021b). Surprisingly, oxidative stress is a significant obstacle in the metastatic spread of cancer (Hayes et al., 2020). The role played by ROS in cancer, whether spear or shield, may depend on the genetic background of the cancer, the type of ROS involved, and the level and duration of ROS exposure. Figures 3–5 show the effects of ROS on normal cells, tumor progression, and cell death.
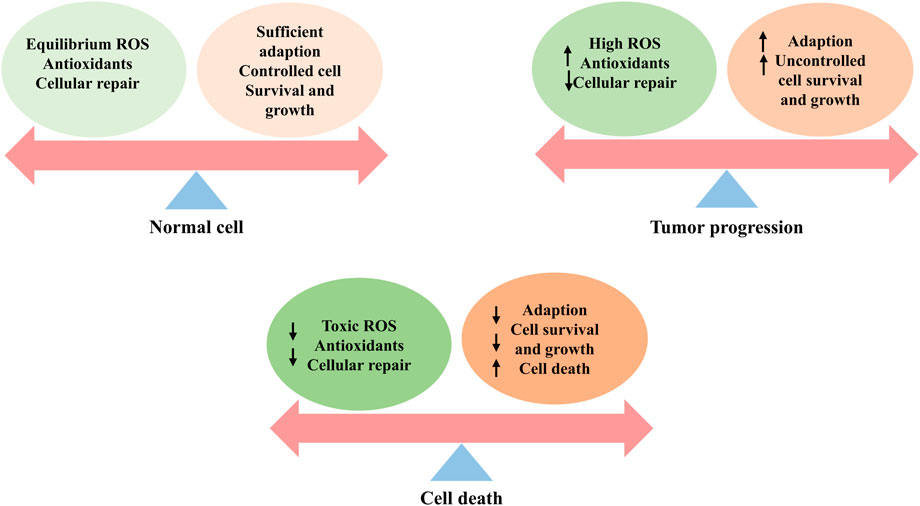
FIGURE 3. ROS effects on cells, including the normal cells, tumor progression, and cell death. Healthy cells have a well-balanced production of ROS, adequate antioxidant activity, and cellular repair, resulting in appropriate concentrations of ROS that limit cell survival and proliferation. Increased levels of ROS can cause cellular damage, yet tumor cells express the enhanced antioxidant activity and maintain pro-tumor signaling through adequate adaptation to conditions including hypoxia and through metabolic readjustment. However, ROS levels increase to toxic concentrations, and oxidative stress leads to irreparable damage to cells, inadequate adaptation, and, ultimately, tumor cell death.
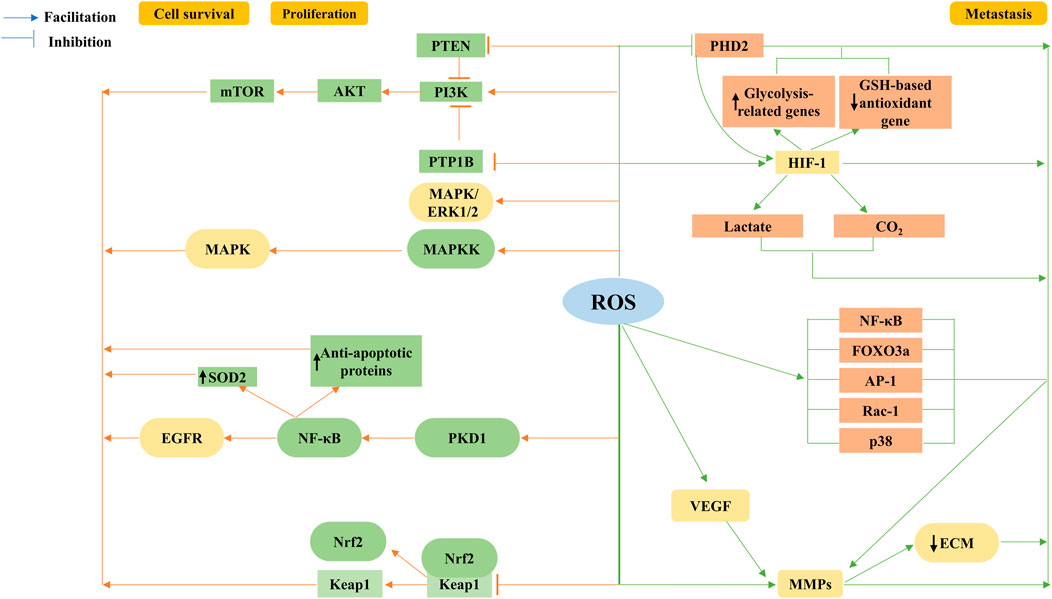
FIGURE 4. Tumorigenic role of ROS. ROS are a second messenger involved in growth factor activation via PI3K/Akt/mTOR, and mitogen-activated protein kinase (MAPK)/extracellular regulated protein kinase (ERK) proliferation survival-related signaling cascades, regulation of the nuclear factor κB (NF-κB) activation pathway, and mutations in nuclear factor E2-related factor (Nrf2) are involved. ROS enhance cancer cell migration and invasion by stabilizing hypoxia-inducible factor-1α (HIF-1α) subunits through inhibition of the prolyl hydroxylase domain protein (PHD2) in the hypoxia signaling pathway. HIF-1 upregulates lactate dehydrogenase and pyruvate dehydrogenase kinase 1, downregulates glutathione (GSH)-based antioxidant gene expression, and decreases mitochondrial ROS production, leading to increased lactate and carbon dioxide formation, all of which facilitate extracellular matrix degradation and cell invasion. The coordinated signaling cascade of HIF-1α completes growth factor-mediated angiogenesis, such as vascular endothelial growth factor (VEGF), while ROS also accelerate angiogenesis and promote metastasis by stimulating matrix metalloproteinase (MMP)-dependent extracellular matrix (ECM) protein degradation; in addition, ROS trigger transcription factors NF-κB and forkhead box O3 (FOXO3a) and Ras-related C3 botulinum toxin substrate 1 (Rac-1), activator protein-1 (AP-1), or p38 signaling pathways to alter MMP expression, promoting tumor progression.
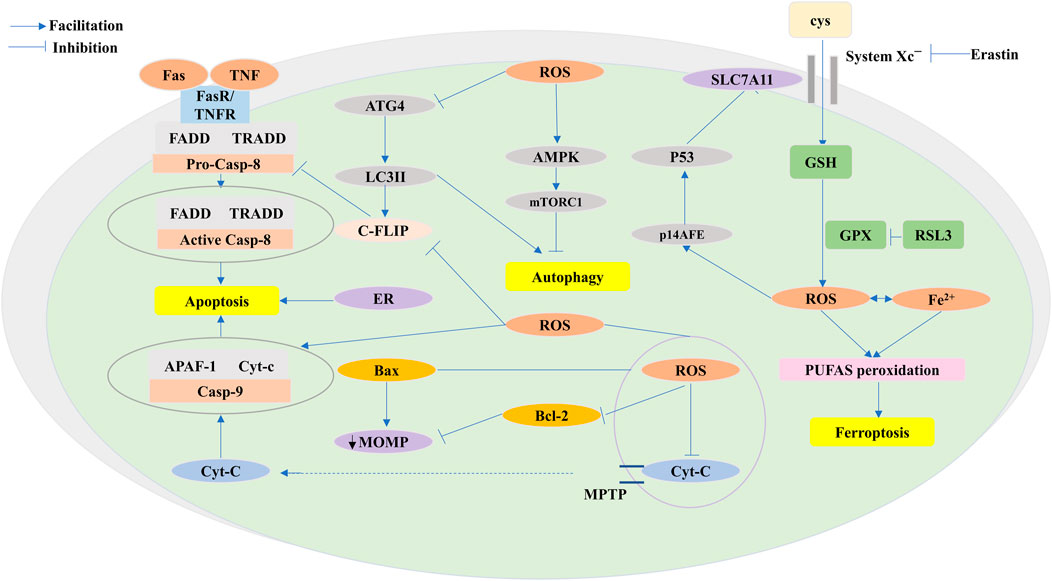
FIGURE 5. ROS functions of antitumor effects via programmed cell death (PCD) consisting largely of apoptosis, autophagy, and ferroptosis. ROS exert pressure on the open mitochondrial permeability transition pore (MPTP), leading to a decreased linear mitochondrial membrane potential (MMP), allowing cytochrome C (Cyt-c) to be released into the cytoplasm and form an apoptotic complex with apoptotic protease activating factor-1 (APAF-1) and procaspase-9 while triggering a caspase-9 signaling cascade that ultimately triggers apoptosis. ROS-dependent proteasomal degradation of cellular FADD-like IL-1β-converting enzyme (FLICE)-inhibitory protein (c-FLIP) enhances the exogenous apoptotic pathway, which is triggered by the splicing of death-inducing ligands with their cognate receptors (FasR). ROS-triggered endoplasmic reticulum (ER) stress is indispensable in apoptosis. ROS exert anticancer effects through autophagy. ROS-dependent inactivation of autophagy-related gene 4 (Atg4) leads to an increase in microtubule-associated protein 1 light chain 3 (LC3)-associated autophagosomes and induces autophagy. mTORC1, a negative regulator of autophagy, is activated by adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and inhibits autophagy. Ferroptosis is caused by ROS-induced iron-dependent lipid peroxidation of PCD. Fenton chemistry increases lipoxygenase activity/ROS production. Erastin impairs the GSH-dependent glutathione peroxidase (GPX) antioxidant system through the mediation of the cystine/glutamate reverse transporter protein (XC− system). Increasing ROS causes changes in the permeability of the outer mitochondrial membrane, and (1S,3R)-RSL3 (RSL3) causes changes in the outer mitochondrial membrane by blocking GPX that stimulates ferroptosis. The P14 alternate reading frame (p14ARF) is triggered by ROS, activates P53 to downregulate recombinant solute carrier family 7, member 11 (SLC7A11), and affects ferroptosis.
The ROS-dependent stimulation of cell survival and proliferation has been extensively studied. The most compelling accomplishment is the confirmation that ROS are secondary messengers involved in growth factor activation via the PI3K/Akt/mTOR and MAPK/extracellular regulated protein kinase (ERK) proliferation survival-related signaling cascades (Moloney and Cotter, 2018). Moreover, regulation of the NF-κB activation pathway and mutations in transcription factors and oncogenes, including Nrf2 and p53, are also involved.
The Akt pathway, through the phosphorylation and inactivation of its target proteins, consists of proapoptotic Bad, Bax, and Foxo transcription factors, which are responsible for their role in cell survival (Limaye et al., 2005; Moloney and Cotter, 2018). Numerous studies have shown that the ROS-induced PI3K/Akt survival pathway is activated in most cancer types. Phosphatases, such as PTEN and PTP1B, contain cysteine active sites that act as negative regulators of the PI3K signaling pathway and are oncogenic agents that are inactivated by increased H2O2 oxidation, thereby affecting cell survival. Considering the importance of PI3K/Akt in the mitogenic signaling cascade, excessive activation of this pathway by the destructive oxidation upstream of PTEN/PTP1B is a hallmark of malignancy (Pérez-Ramírez et al., 2015). Consistent with this finding, we found that PTEN was inactivated and the ROS-induced PI3K signaling pathway was activated in various cancers. NOXs in neuroblastoma lead to the inactivation of the oncogene PTEN and activation of the PI3K/Akt signaling pathway in an ROS-dependent manner (Calvo-Ochoa et al., 2017). Similarly, in breast cancer cells, the accumulation of transient H2O2 by CXCL12-activated NOX2 leads to the oxidation of PTEN and PTP1B, regulating the activation of the PI3K/Akt signaling pathway and leading to the maintenance of cell cycle protein D expression, which, in turn, stimulates cell proliferation (Deng et al., 2018).
Activation of the MAPK/ERK 1/2 pathway, as a function of increased cell proliferation, can be associated with the stimulation of growth factors and K-Ras (Khavari and Rinn, 2007). Mitochondrial ROS regulate the K-Ras-induced anchorage-independent growth of lung cancer cells through the MAPK/ERK pathway (Weinberg et al., 2010). The proliferative actions of ovarian cancer cells in the presence of high levels of endogenous ROS are facilitated by sustained ubiquitination and inactivation of endogenous mitogen-activated protein kinase phosphatase 3 (MKP3) and increased ERK1/2 activity (Chan et al., 2008). Identical results were obtained when breast cancer cells were treated with ROS scavengers/inhibitors targeting ERK1/2 or its upstream kinase MAPKK (Zhou et al., 2008). In addition to initiating its upstream signaling pathway, the MAPK phosphoenzyme is directly oxidized and inactivated, sustaining JNK activation via the reversible oxidation of cysteine to sulfenic acid at the catalytic site and the inhibition of JNK-inactivating phosphatase (Kamata et al., 2005; Son et al., 2011). As is well known, ERK1/2 not only plays a proliferative role but also functions in the survival, anchorage-dependent growth, and motility of a variety of cancer cells (Liou and Storz, 2010; Moloney and Cotter, 2018).
ROS contribute to cancer cell survival and proliferation by activating NF-κB and Nrf2. Studies identified that the formation of pancreatic precancerous lesions was attributed to K-Ras-derived mtROS activating NF-κB via PKD1 to upregulate proliferative EGFR signaling (Liou et al., 2016). Increased mitochondrial ROS levels would also induce the upregulation of antioxidant proteins, such as MnSOD, and antiapoptotic proteins through the aforementioned pathways (Storz et al., 2005), thus helping in cancer survival and proliferation. Studies have also shown that the occurrence of various cancers is strongly correlated with mutations in Nrf2 (Rojo de la Vega et al., 2018). These findings suggest that the upregulation of Nrf2, either through increased Nrf2 mRNA production by oncogenic gene transcription (DeNicola et al., 2011) or the inhibition of its blocker KEAP1 upregulation by ROS (Suzuki et al., 2019), promotes preneoplastic nodules in the liver (Zavattari et al., 2015). Furthermore, differential activation of Nrf2, caused by the previously mentioned factors, is linked to cancer recurrence and unfavorable prognosis (Wu et al., 2019).
Cancer metastasis is a complex multistep process, being one of the grounds for poor patient prognosis and treatment discouragement. The complex regulatory effects are reinforced by the fact that ROS function in cancer metastasis, allowing the regulation of key steps in cancer-increased migration, invasive potential, regulation of metabolism, epithelial–mesenchymal transition (EMT), and neovascularization (Liou and Storz, 2010).
The modulation of hypoxia contributes to the development of a malignant phenotype and aggressive cancer progression. PPP-dependent NADPH output is compromised by low glucose availability; subsequently, carcinoma cells accommodate glucose deprivation and protect against H2O2-induced apoptosis through the Warburg effect. Furthermore, the AMPK signaling pathway is activated to facilitate NADPH production and prevent anabolic processes that require NADPH depletion to optimize ROS yields, which significantly promotes the emergence of an aggressive phenotype (Aykin-Burns et al., 2009; Jeon et al., 2012; Ye et al., 2014; Vander Heiden and DeBerardinis, 2017).
The hypoxia-inducible factor-1 (HIF-1), composed of two subunits, HIF-1α and HIF-1β, potentially mediates cancer angiogenesis, metabolism, and metastasis, enhancing cancer survival and progression (Harris, 2002; Al Tameemi et al., 2019). ROS stabilize oxygen-sensitive HIF-α subunits by inhibiting PHD2 in the hypoxia signaling pathway (Bell et al., 2007), thereby enhancing cancer cell migration and invasion. Similarly, interest has been expressed in the increased expression of vascular endothelial growth factor (VEGF) genes and the activation of HIF-1α associated with metastatic disease (Semenza, 2012). The stability and activation of HIF-α through ROS-dependent mechanisms are strongly correlated with poor prognosis, increased cancer incidence (Horak et al., 2010), and aggressiveness in certain cancer cells. Furthermore, HIF-1 is responsible for the upregulation of glycolysis-related genes, such as lactate dehydrogenase and pyruvate dehydrogenase kinase 1, downregulation of GSH-based antioxidant gene expression (Lu et al., 2015; Stegen et al., 2016), and reduction of mitochondrial ROS production. It is equally intriguing to note that HIF-1 elicits increased lactate and carbon dioxide formation, and prevents intracellular acidification (Pouysségur et al., 2006), in both cases favoring extracellular matrix degradation and cell invasion (Rofstad et al., 2006).
EMT is the initial event in cancer cell metastasis. ROS promote metastasis by accelerating HIF-dependent angiogenesis through the stimulation of matrix metalloproteinase (MMP)-dependent ECM protein degradation (Chatterjee and Chatterjee, 2020; Aboelella et al., 2021). Another study also showed that ROS trigger the transcription factors NF-κB and FOXO3a and Rac-1, AP-1 (activator protein 1), or the p38 signaling pathway, to alter the expression of MMPs (Westermarck and Kähäri, 1999). It has also been reported that cancer cells induce the secretion of MMPs, such as MMP-1, via the upregulation of ROS to enhance vascular growth in the cancer microenvironment (Wartenberg et al., 2003). Angiogenesis, mediated by growth factors such as VEGF, can be accomplished through a coordinated signal transduction cascade by HIF-1α (Choudhry and Harris, 2018).
ROS have been proven to be excellent signaling molecules in the apoptotic process that directly induce cellular damage and activate caspase family protein-dependent activation of endogenous mitochondrial pathways and exogenous death receptor pathways (Redza-Dutordoir and Averill-Bates, 2016). ROS generate pressure against the open MPTP, causing a reduction in the mitochondrial transmembrane potential, thus contributing to the loss of membrane permeability and allowing the release of proapoptotic factors such as cytochrome c (Cyt-c) into the cytoplasm. Subsequently, once Cyt-c is released into the cytoplasm, its interaction with apoptosis protease-activating factor 1 (APAF-1) and pro-caspase-9 forms an apoptotic complex, simultaneously triggering a caspase-9 signaling cascade that breaks DNA and ultimately triggers apoptosis (Simon et al., 2000; Redza-Dutordoir and Averill-Bates, 2016; Moloney and Cotter, 2018). Furthermore, damaged mitochondria enable the production of more ROS, paradoxically accelerating apoptosis. In addition, ROS can modulate related signaling pathways and consequently cause apoptosis. Studies suggest that JNK, a subclass of the MAPK signaling pathway augmented by ROS, can respond by catalyzing the phosphorylation and downregulation of antiapoptotic proteins (Cadenas, 2004), increasing the expression of Bax, creating Bax homodimers, and ultimately destroying mitochondrial membrane integrity (Zhang et al., 2008). P38, another subclass of the MAPK signaling pathway, enthusiastically embraces apoptotic signaling enhanced by ROS production, and coincidentally, both of these can promote cell death through the involvement of apoptotic signal-regulated kinase 1 (Ask-1) (Saitoh et al., 1998; Liou and Storz, 2010). Moreover, it remains to be determined whether the ROS-mediated activity of the JNK and p38 signaling pathways is responsible for cell cycle arrest and inhibiting cancer cell growth and division (Wagner and Nebreda, 2009). Nevertheless, several other signaling proteins like P53 are also involved in ROS-induced apoptosis, and ROS above threshold levels initiate P53-induced genes, resulting in impaired mitochondrial function and triggering apoptotic effects (Safdar et al., 2016).
The extrinsic apoptotic pathway is triggered by the splicing of death-inducing ligands such as the Fas ligand (FasL) with their cognate receptors (FasR). After binding to the two receptors, the apoptotic message is transmitted to the cell interior, activating the linked death-inducing signaling complex (DISC), whose members include DD, DED, FADD (TRADD), and pro-caspase-8. Caspase-8 initiates a downstream caspase cascade that actively drives apoptosis. Cellular Fas-associated death domain-like interleukin 1β-converting enzyme inhibitor protein (c-FLIP) is a death receptor-mediated antiapoptotic factor. It has been shown that under conditions of threonine 166 phosphorylation and lysine 167 ubiquitination, the ROS-dependent degradation of the proteasome of c-FLIP reinforces the exogenous apoptotic pathway (Wilkie-Grantham et al., 2013). Additionally, in the case of N-acetylcysteine (NAC) pretreatment, the c-FLIP protein was efficiently stabilized and accelerated the initiation of apoptosis, confirming the role of ROS as an apoptosis-inducing factor (Moloney and Cotter, 2018). Moreover, ER stress and the regulation of energy metabolism triggered by ROS are indispensable for apoptosis.
Rather than acting as a mechanism for normal cell self-protection and survival, autophagy suppresses cancer and is referred to as type II programmed cell death. Ample evidence suggests that autophagy is directly mediated in malignancies and controlled by ROS levels (Poillet-Perez et al., 2015). The H2O2-dependent inactivation of autophagy-associated gene 4 (ATG4) leads to an increase in LC3-associated autophagosomes (Perillo et al., 2020), whereas oxidative stress in H2O2 and 2-ME-treated cancer cells also leads to autophagy-induced death (Chen et al., 2008). mTORC1, a negatively regulated regulator of autophagy (Moloney and Cotter, 2018), is suppressed by AMPK activation and contributes to autophagy induction. Autophagy in human bronchial epithelial cells is prevented by arsenic-induced ROS production. Notably, phosphorylation of AMPKK due to oxidative stress and the consequent alteration of the AMPK pathway further cause an increase in ROS and, eventually, apoptosis (Poillet-Perez et al., 2015). Last but not least, transcription factors, such as NF-κB, equally regulate autophagy by controlling the expression of genes that predominantly induce ROS-related autophagy in cancer, consequently regulating cell death (Boyer-Guittaut et al., 2014).
Ferroptosis is regulated by cell death (RCD), evoked by ROS-induced iron-dependent lipid peroxidation (Dixon et al., 2012). Peroxidized polyunsaturated fatty acids (PUFAs), triggered by oxidative stress involving excessive iron levels and insufficient GSH, are the underlying features of ferroptosis (Stockwell et al., 2017). There is ample evidence that intracellular iron, rendering a free state with redox activity, occurs through Fenton chemistry to increase lipoxygenase activity and ROS production (Stockwell et al., 2017). In contrast, membrane phospholipid-PUFAs are oxidized, which shows tremendous potential to shift membrane pores and integrity (Dixon and Stockwell, 2019). Studies have suggested that GPX4 utilizes GSH to reduce lipid hydroperoxides, thereby limiting ferroptosis (Kuang et al., 2020). In recent years, a mounting stream of data have emerged demonstrating that ferroptosis, specifically mediated by small molecules, exerts dramatic suppressive effects on cancer growth. Erastin, a synthetic small-molecule drug inhibiting cystine uptake, lends itself to an impairment of the GSH-dependent GPX antioxidant system via cystine/glutamate reverse transporter protein (system XC−) mediation, allowing for higher ROS levels and altered permeability of the outer mitochondrial membrane, causing cell death through ferroptosis in cancer cells, thus bearing mutant RAS (Yagoda et al., 2007; Dixon et al., 2012). The other agent, RAS-selective lethal compound 3 (RSL3), reportedly stimulates ferroptosis by blocking GPX, rather than the XC system (Imai et al., 2017). In addition, ROS trigger p14ARF (a cancer suppressor), which subsequently reactivates P53 and inhibits Nrf2 to activate ferroptosis and allow the downregulation of SLC7A11 and xCT activity (Chen et al., 2017). Numerous studies have confirmed that ferroptosis curtails cancer cell progression by eliminating these factors.
Cancer metastasis is inhibited by ROS overload (Piskounova et al., 2015). Remarkably, cancer cells can escape cell death during metastasis through metabolic alterations that are mediated by modulating the level of antioxidant capacity (Reczek and Chandel, 2017). Jiang et al. suggested that human melanoma cells promote metastasis by relying on NADPH-generating enzymes that rely on folate-mediated metabolism of single carbon units (Piskounova et al., 2015). Somewhat more interesting is the discovery that blood itself likely functioned as a prooxidant environment, provoking oxidative stress and hindering metastasis (Nieto et al., 2016).
Ginsenosides, the primary active components of ginseng, originate from the various pharmacological and biological effects of ginseng, affecting oxidative stress, metabolism, immunity, and the central nervous system, particularly when used in cancer therapy (Guo et al., 2019). Depending on the steroid backbone, hydroxyl groups attached, or number of sugar moieties, ginsenosides can be categorized into four types: protopanaxadiol-type (PPD), protopanaxatriol-type (PPT), oleanolic acid-type, and ocotillol-type. PPD-type ginsenosides include ginsenosides Rb1, Rb2, Rb3, Rc, Rd, Rg3, Ra1, and Ra2. PPT-type ginsenosides include Re, Rf, Rg1, Rg1, Rg2, and Rh1 (Shin et al., 2015). Ginsenosides with a pentacyclic triterpene backbone, such as Ro, are classified as oleanolic acids (Nag et al., 2012). The last ginsenoside is the ocotillol type, which has a five-membered epoxide ring at C-20, and the rare ginsenoside P-F11, which is classified in it (Nakamura et al., 2007). Figure 6 and Table 1 show the characteristic ginsenosides with different substituent groups that are commonly observed in the basic chemistry of the four ginsenosides. The number and position of sugar molecules, hydroxyl portion of the dammarane backbone, and stereoisomeric position of C-20 have profound effects on the biological activity of ginsenosides (Cheong et al., 2015). Some studies have indicated that 20(R)-Rg3 exhibits greater antioxidative stress activity than 20(S)-Rg3, depending on the stereoisomeric position of C-20 (Li et al., 2014). More reassuringly, additional studies have identified that modified versions of some secondary metabolites of ginsenoside conversion or relevant drug carriers possess enhanced biological activity and exhibit unique pharmacological activity relative to the parent compound (Zheng et al., 2017).
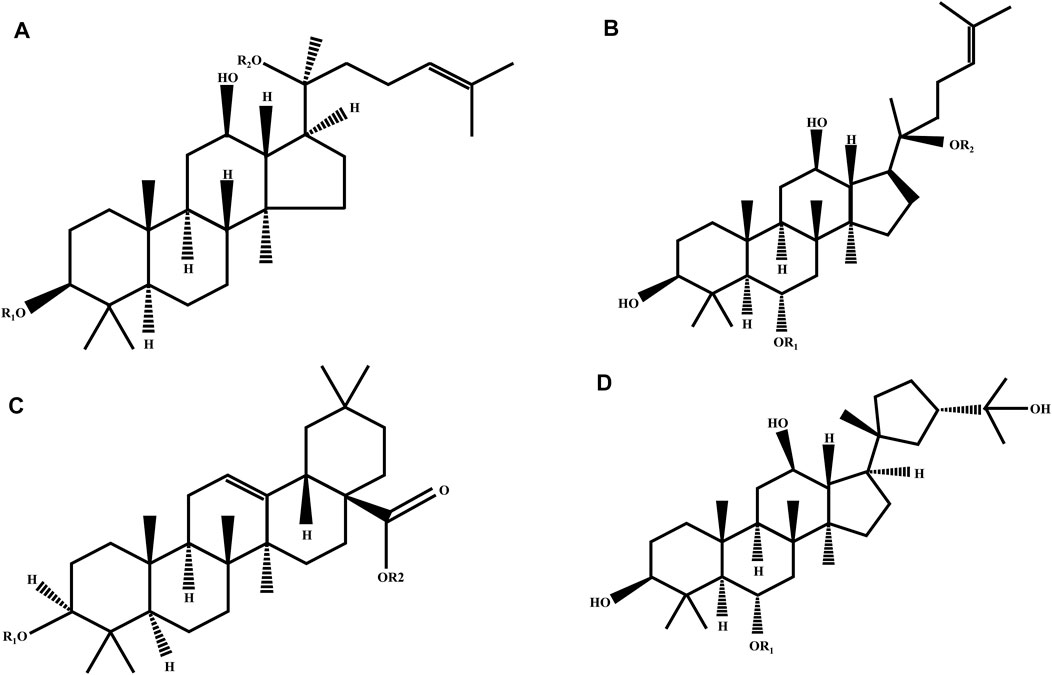
FIGURE 6. Chemical structure of four ginsenoside types. (A) Protopanaxadiol (PPD); (B) protopanaxatriol (PPD); (C) oleanolic acid; and (D) ocotillol ginsenosides.
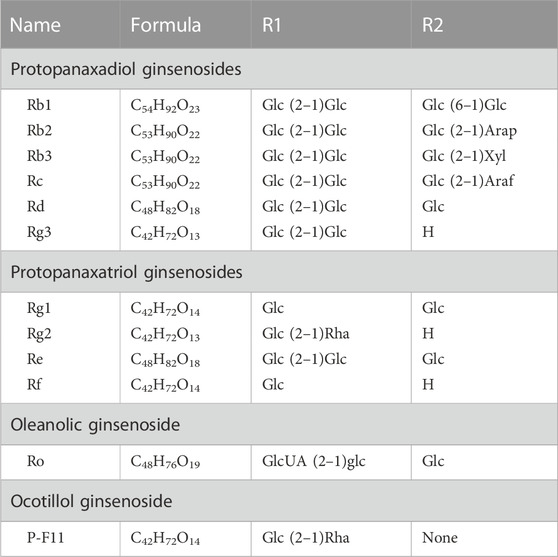
TABLE 1. Various types of ginsenosides with different substitute groups.
To date, a comprehensive description of the functions of ginsenosides is available, among which their anticancer effect has been in full swing (Zhang et al., 2021). Concerning different cancers, ginsenosides act as anticancer agents by modulating different mechanisms, namely, mediation of apoptosis, proliferation, cell cycle arrest, metastasis, angiogenic effects, autophagy, reversal of multidrug resistance, and enhancement of chemotherapeutic effects, all of which are possible via the modulation of ROS (Figures 7–10). The following section focuses on the exploration of the mechanisms underlying the anticancer activity of different classes of ginsenosides against this target.
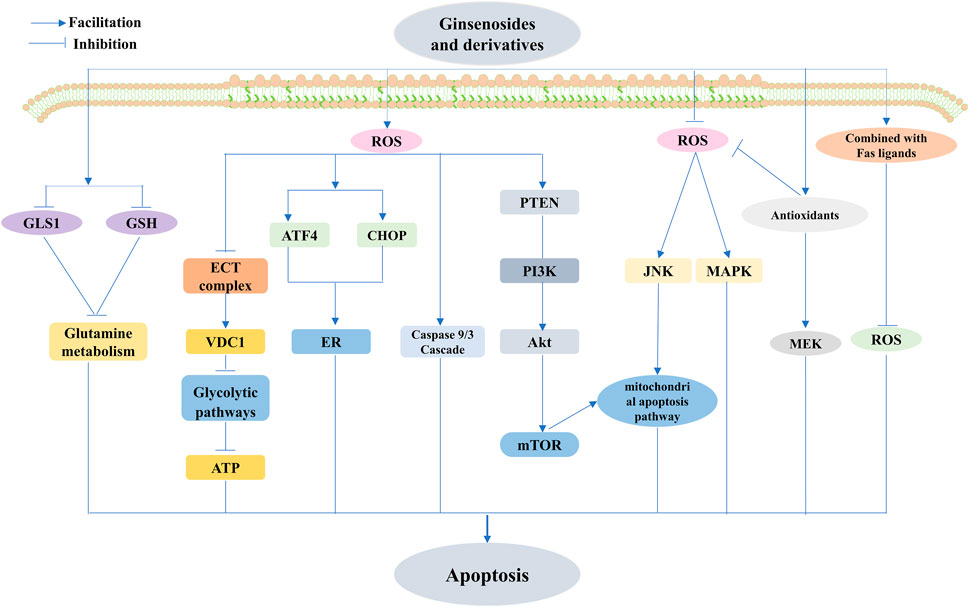
FIGURE 7. Ginsenosides modulate ROS-triggered apoptosis. Ginsenosides modulate ROS-triggered apoptosis involving mitochondria-associated, targeted endoplasmic reticulum (ER), and energy metabolism-mediated triggered apoptotic pathways. Ginsenosides promote apoptosis by increasing ROS production, and ginsenosides activate the caspase 9/3 cascade and mitochondria-mediated apoptotic pathways in cancer cells through ROS-dependent mechanisms and also induce mitochondria-dependent apoptosis by elevating the ROS-mediated PTEN/PI3K/Akt/mTOR signaling pathway. Increased ROS production also stimulates the upregulation of activating transcription factor-4 (ATF4) and C/Ebp-homologous protein (CHOP) The increased production of ROS also stimulates the upregulation of ATF4 and CHOP, as well as ER stress and inhibition of electron transport chain (ETC) complex formation, upregulation of voltage-dependent anion channel 1 (VDAC1), interference with the glycolytic pathway, reduction of glutaminase 1 (GLS1) and glutathione (GSH), and inhibition of glutamine metabolism to promote apoptosis; reduction of ROS induced by ginsenosides also mediates apoptosis, and reduced ROS-mediated upregulation of c-Jun N-terminal kinase (JNK) activation mediates the mitochondrial apoptosis pathway, and also, inhibition of antioxidant enzyme activity inhibits mitogen-activated extracellular signal-regulated kinase (MEK) and mitogen-activated protein kinase (MAPK). The combination of Rg3 and TNF-related apoptosis-inducing ligand (TRAIL) inhibited ROS to promote apoptosis.
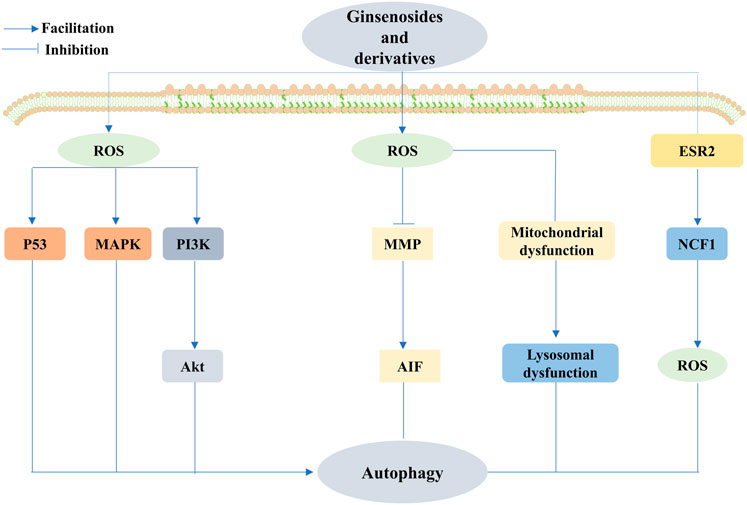
FIGURE 8. Ginsenosides promote or inhibit autophagy by regulating ROS to treat cancer. Ginsenosides can act as an autophagy inhibitor, mediating the production of ROS, inhibiting the mitochondrial membrane potential, and promoting the release of apoptosis-inducing factor (AIF) from mitochondria, thus causing nuclear translocation and promoting apoptosis; it can also increase ROS to impair mitochondrial function, leading to impaired lysosomal function and cell death. In addition, ginsenosides can also inhibit autophagy through the regulation of the estrogen receptor 2 (ESR2)-NCF1(neutrophil cytosolic factor 1)-ROS axis to exert anticancer effects. Conversely, ginsenosides exert anticancer effects through the regulation of ROS-mediated signaling pathways, including the upregulation of p53 signaling, and activation of mitogen-activated protein kinase (MAPK) and PI3K/Akt signaling pathways to promote autophagy to exert anticancer effects.
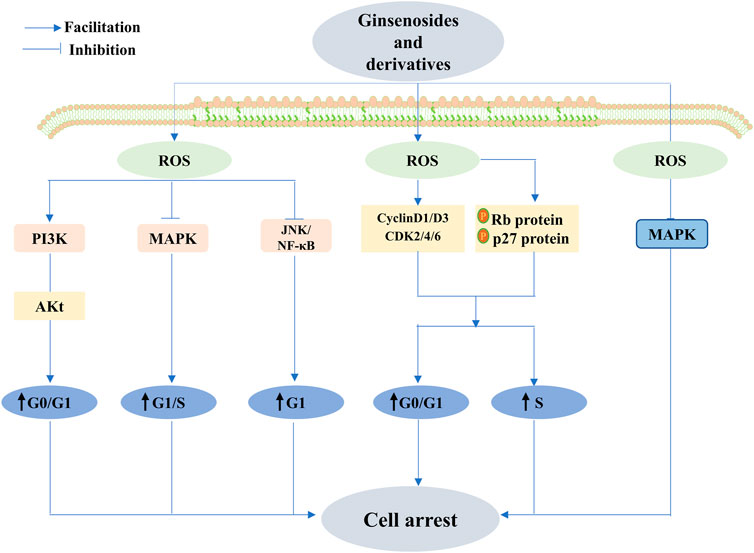
FIGURE 9. Ginsenoside exhibits anticancer activity through different stages of cell cycle arrest. Ginsenosides exert anticancer effects by reducing reactive oxygen species levels and activating MAPK. Conversely, ginsenosides activate the PI3K/Akt signaling pathway and c-Jun N-terminal kinase (JNK)/nuclear factor κB (NF-κB) and inhibit the MAPK signaling pathway by increasing ROS, causing cell cycle arrest in the G0/G1 phase, G1 phase, and G1/S phase, respectively, and promoting apoptosis. D1, cell cycle protein D3, cyclin-dependent kinase 2 (CDK2), cyclin-dependent kinase 4 (CDK4), and cyclin-dependent kinase 6 (CDK6), and a decrease in the expression of these proteins could promote the development of cell cycle arrest and anticancer effects.
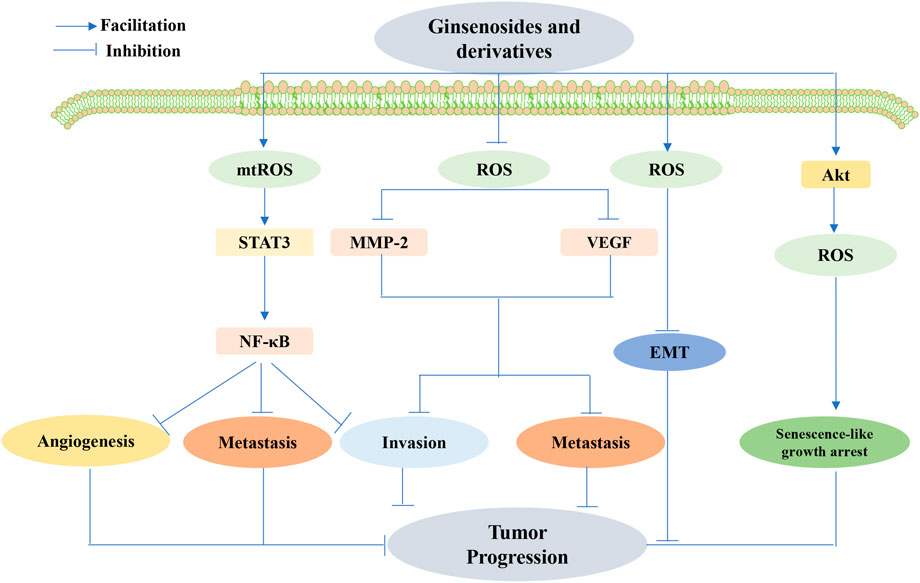
FIGURE 10. Ginsenoside-mediated interference with angiogenesis, metastasis, and invasion by ROS inhibited tumor development. Ginsenosides decreased ROS production, inhibited matrix metalloproteinases-2 (MMP-2) and VEGF expression, and suppressed tumor cell invasion and metastasis. Ginsenosides also promoted ROS produced in the mitochondria (MtROS)-mediated inhibition of the signal transducer and activator of the transcription 3(STAT3)/nuclear factor κB (NF-κB) signaling pathway, thereby inhibiting angiogenesis, metastasis, and invasion of cancer cells. Ginsenosides may also inhibit tumor progression by increasing ROS production and regulating the activation of epithelial–mesenchymal transition (EMT) conversion molecules. Targeted senescence may also be a promising approach for cancer treatment, where ginsenosides exert anticancer effects by inducing ROS production through the activation of Akt and senescence-like growth arrest.
Unsuitable ROS levels reinforce cell death, and inverse signals for cell survival are heightened. The survival signal is mediated by the mitochondrial ROS activation of protein kinases and NF-κB, leading to the upregulation of antioxidant proteins (Cao et al., 2020). Apoptosis triggered by natural ginsenosides via the modification of ROS, involving but not limited to endogenous and exogenous apoptotic pathways, tends to come into vogue in the treatment of cancer. In addition, many researchers have emphasized the superior role of ROS in targeting ER- or energy metabolism-mediated pathways that trigger apoptosis in various cancer cell lines. Natural ginsenoside extracts that modulate the role of ROS (lower or higher) in the aforementioned apoptotic pathways in cancer therapy should not be underestimated (Kim et al., 2017; Seervi et al., 2018; Cao et al., 2020; Zheng et al., 2020).
It was discovered that ultrasound-treated ginseng berry extract (UGBE), containing multiple ginsenosides, including Rh1, Rg2, and ginsenosides Rg3 and Rh2, could activate the mitochondria-mediated apoptotic pathway in cancer cells through an ROS-dependent mechanism. The same conclusion was obtained in ginsenoside Rh2- and Rg3-treated human leukemia Jurkat cells (Park et al., 2012; Chen et al., 2015; Jung et al., 2016; Xia et al., 2017). Consistent with previous studies, Rg3 enhanced the level of ROS and led to the caspase 9/3 cascade, yet its induction of apoptosis was not significantly disturbed by the application of NAC, suggesting that the increased ROS associated with Rg3 was not the culprit of apoptosis (Kim B.-M. et al., 2013). The underlying mechanism behind this process still requires further exploration. Several studies have uncovered a specific mechanism by which Rk1 induces mitochondria-dependent apoptosis by mediating the PTEN/PI3K/Akt/mTOR signaling pathway via elevated ROS levels in MCF-7 cells (Hong and Fan, 2019b). However, ginsenoside-induced reduction of ROS, leading to the activation of signaling pathways, is also an effective treatment for the induction of apoptosis (Huynh et al., 2021), which is consistent with the finding that diminished ROS-mediated upregulation of JNK activation plays an integral role in the ginsenoside-mediated mitochondrial apoptosis pathway (Ham et al., 2006; Mao et al., 2014). However, Chu et al. demonstrated that Rg1 exerts cytoprotective effects in MPP + -treated SHSY5 cells by reducing the ROS-mediated regulation of JNK (Chu et al., 2020).
Ginsenoside-mediated dysregulation of antioxidant enzyme activity is associated with endogenous apoptosis. Choi et al. identified that ginsenoside Rg3 regulates CAT activity and inhibits the MEK signaling pathway to mediate apoptosis (Choi et al., 2013). Rg5 and Rk1 mediate apoptosis in lung cancer cells through the regulation of GSH (Kwak and Pyo, 2016). Furthermore, Lu et al. found that Rk1 could promote apoptosis in cervical cancer cells by reducing GLS1 and GSH and inhibiting glutamine metabolism (Lu et al., 2022). However, one study pointed out that Rg1 could inhibit DMBA-mediated carcinogenesis by restoring antioxidant enzyme activity (Chu et al., 2020). FBG has cytoprotective functions by suppressing MAPK and lowering ROS levels caused by the induction of antioxidant enzyme activity (Bak et al., 2014). However, Rh2, by enhancing ROS, partially counteracts p53-induced apoptosis by activating AMPK and NF-κB signaling pathways to promote cancer survival and growth and can exert better anticancer effects when applied in combination with antioxidants (Li et al., 2011).
Rh2 and compound K evoke apoptosis in a cystathionine- and p38 MAPK-dependent manner in astrocytoma cells, and combined treatment with Fas ligands exerts synergistic cytotoxic effects by reducing intracellular ROS (Choi and Choi, 2009). Lee et al. found that the combination of Rg3 and TRAIL resulted in a noticeable improvement in ROS production to either one alone. Although the antioxidant NAC effectively inhibited ROS production, it failed to suppress apoptotic cell death induced by Rg3 combined with TRAIL, suggesting that ROS may not be required for Rg3 sensitivity to TRAIL (Lee et al., 2013).
ER stress-mediated apoptosis cannot be minimized by ginsenoside treatment. Ginsenoside Rh2 has been shown to facilitate apoptosis in lung cancer cells, especially through the induction of ROS production, stimulating the upregulation of ATF4 and CHOP, with ER stress further inhibiting cell proliferation (Ge et al., 2017). Similarly, Wu K. et al. found that ATF4 knockdown attenuated the proapoptotic effect of Rg3 in gallbladder cancer cell lines by inhibiting ROS (Wu K. et al., 2018). As such, it has been suggested that Rh2 enhances ROS, inhibits ETC complex formation, affects energy stress, upregulates VDAC1, and interferes with glycolytic pathways to promote apoptosis (Liu et al., 2020; Liu et al., 2021).
The dual role of autophagy in cancer progression and inhibition remains controversial. Natural ginsenosides can promote or inhibit autophagy by regulating ROS for cancer therapeutic effects.
It has become more unambiguous that Rg3, as an inhibitor of autophagy, can mediate the production of ROS, decrease the mitochondrial membrane potential, and consequently induce apoptosis, all of which induce the release of AIF from the mitochondria and enable nuclear translocation that leads to HeLa cancer cell death (Bian et al., 2019). G-Rh2 showed a significantly stronger inhibition of autophagy than Rg3. The specific mechanism is to impair mitochondrial function through increased ROS, leading to defective/impaired lysosomal acidification/function and, ultimately, HGC-27 cell death (Han et al., 2020). The conclusion that Rh2 inhibits autophagy in cervical cancer cells was also drawn by Wang et al. in cervical cancer cells (Wang J. et al., 2020). Furthermore, by the preapplication of N-acetyl-l-cysteine (NAC) to esophageal cancer cells, Bian et al. identified that ginsenoside Ro-induced elevated expression of autophagy-associated proteins, such as LC3B-II, was significantly reversed, precisely through the modulation of the ESR2-NCF1-ROS axis. Importantly, the study also observed that Ro enhanced the cytotoxicity of 5-fluorouracil (5-Fu) by delaying CHEK1 (checkpoint kinase 1) degradation and downregulating the DNA replication process, which strongly suggests that G-Ro can be used as an effective anticancer agent to overcome chemoresistance in combination therapy (Zheng et al., 2016). However, growing evidence suggests that the promotion of autophagy plays a critical role in cancer treatment. Hwang et al. demonstrated that the activation of mitochondrial autophagy is essential for combating lung cancer in Rg3-enriched red ginseng, which is initiated by the increase in ROS (Hwang et al., 2022). Likewise, several native ginsenosides exert anticancer effects through the ROS-mediated regulation of signaling pathways. Rh4 mediates ROS production to induce the upregulation of p53 signaling, thereby activating autophagy and positively regulating iron death to promote colorectal cancer cell death (Wu et al., 2022). However, Wu et al. revealed that Rh4 induces autophagy by facilitating the ROS/JNK/P53 pathway, and interestingly, the upregulation of autophagy-associated proteins ATG7 and Beclin-1 is involved in the regulation of autophagy (Wu Q. et al., 2018). Rg5 and Rh1 promote ROS production, and trigger MAPK activation and the PI3K/Akt signaling pathway, respectively, to promote autophagy and cell cycle arrest, inhibit cancer cell proliferation, and promote the onset of apoptosis (Liu and Fan, 2019).
Numerous studies have suggested that natural ginsenoside products display pronounced anticancer activity at different stages of cell cycle arrest. Lu et al. found that ginsenoside Rk1 effectively arrests the cell cycle in the S-phase and induces apoptosis in breast cancer MCF-7 cells. Moreover, increased p53 and p21 proteins and the downregulation of cyclin A and CDK2 might contribute to the anticancer effects of Rk1. Rk1 may regulate the proteins via the upregulation of the ROS-mediated PTEN/PI3K/Akt/mTOR signaling pathway, sequentially inhibiting proliferation and apoptosis (Lu et al., 2022). Hong et al. observed identical alterations in ROS and proteins in MDA-MB-231 triple-negative breast cancer cells, with the difference being that the cells blocked the cell cycle in the G0/G1 phase (Hong and Fan, 2019a). Huynh et al. revealed that the accumulation of the G0/G1 phase in cancer cells incubated with Rh1 was enhanced, along with marked improvement in phosphorylated Rb protein levels and p27 protein expression; meanwhile, the expression of cycle-related proteins—including cyclin D1, cyclin D3, CDK2, CDK4, and CDK6—diminished. Moreover, the mentioned reaction coincided with the ROS-mediated PI3K pathway (Huynh et al., 2021), and these results are similar to the findings of Jin et al. (2022). A decrease in ROS levels exerts an aggressive effect on cell cycle arrest. Shui et al. showed that Rg3 has promising anticancer activity against lung cancer by decreasing reactive oxygen levels, activating cell cycle-associated proteins, and controlling MAPK relevant to proliferation (Sun et al., 2016). Conversely, Jeon et al. applied NAC to counteract the G1-S phase arrest due to Rg2 via the activation of AMPK and the regulation of cell cycle regulators induced by increased ROS, implying that increased ROS can also exhibit excellent anticancer effects by promoting cell cycle arrest (Jeon et al., 2021). Comparably, Rg1 exerts apparent cancer-fighting actions on paclitaxel-resistant nasopharyngeal carcinoma cells by upregulating ROS to block the PI3k/Akt signaling pathway (Li et al., 2019). Rg18, in a similar manner, or at least in part, downregulated the JNK/NF-κB signaling pathway to lead to G1-phase cell arrest to curb cancer cell proliferation of A549 cells to combat cancer (Leem et al., 2018). It has, therefore, been abundantly demonstrated that ginsenosides can exhibit exceptional therapeutic anticancer abilities at different stages of the cell cycle via the modulation of ROS.
Targeting cancer metastasis by interfering with angiogenesis and reducing invasiveness is an important aspect of ginsenoside therapy. Previous research revealed that G-Rh2 shows both high antioxidant activity and low toxicity by decreasing the production of ROS and subsequently repressing MMP-2 and VEGF expression, thereby suppressing the invasion and metastasis of oral squamous cell carcinoma cells (Ping et al., 2020). Nonetheless, Jin et al. concluded that Rh1 repressed cancer cell angiogenesis, metastasis, and invasion by boosting MtROS-mediated inhibition of the STAT3/NF-KB signaling pathway (Jin et al., 2021). Additionally, the ginsenoside Rg1 may suppress the progression of DMBA-induced breast cancer via a concentration-dependent increase in ROS production and by modulating the activation of molecules involved in cell proliferation, apoptosis, invasion, angiogenesis, and EMT conversion (Chu et al., 2020). Targeting senescence is also a more promising approach to treat cancers, as suggested by Sin S et al., who delivered chronic treatment with sub-apoptotic concentrations of 20(S)-Rg3 to induce ROS production via Akt activation and p53/p21-dependent senescence-like growth arrest in glioma cells (Sin et al., 2012). This study provides valuable insights into the future development of 20(S)Rg3 as a novel anticancer agent.
This section highlights the different classes of ginsenoside derivatives, nanomaterial carriers, and other ginsenosides that mediate apoptosis, cell cycle arrest, and autophagy through direct or indirect modulation of ROS to exert anticancer effects. Studies have found that ginsenoside metabolite K (GCK) exhibits anticancer activity by boosting ROS-mediated alteration of the mitochondrial membrane potential in neuroblastoma or cervical cancer cells, functioning as an autophagy inhibitor or modulating caspase-3/9 and PARP intrinsic apoptotic pathways (Oh et al., 2019; Yin et al., 2021). Identically, Gao H. et al. and Wang X.D. et al. discovered that 2-deoxy-Rh2, a novel 20(S)-Rh2 derivative with enhanced anticancer activity, was designed and synthesized by the hybridization of protopanaxadiol and 2-deoxyglucose (2-DG), and 2-pyrazine-PPD, a derivative obtained by introducing a pyrazine ring into 25-OH-PPD, also mediated ROS-induced mitochondrial dysfunction to cause apoptosis in cancer cells. Dissimilarly, it is noted that the former also enhances the proapoptotic effect by affecting glycolysis and decreasing increased ATP (Gao et al., 2020), while the latter reinforces the antiapoptotic effect by affecting PERK/EIF2A/ATF4 and CHOP expression to affect ER stress, thus reinforcing the pro-cancer effect (Wang X. D. et al., 2020). An alternative derivative of PPD, 12-chloracetyl-PPD, a 25-OH-PPD analog synthesized by the addition of C-12-OH to chloroacetyl, can contribute to the counteracting effect by causing G2/M-phase cell cycle arrest via increased ROS levels, thus downregulating MDM2 and upregulating P53 (Wang et al., 2017). In addition, ROS-mediated signaling pathway activation positively contributes to anticancer effects. CK functions in colon and bladder cancers by mediating the activation of JNK and P38MAPK (Kim A. D. et al., 2013; Wang et al., 2013). AD-1, extracted from ginseng berries, can also promote apoptosis in lung cancer cells by mediating the activation of p38mapk, which has been validated using NAC (Zhang et al., 2013). 1c, a ginseng saponin derivative, functions as an anticancer agent through increased ROS-mediated inhibition of the Wnt/β-catenin signaling pathway (Wang et al., 2018).
The buildup of ginsenoside nanoparticles and related vectors has been shown in comparison to ginsenosides, which evoke stronger cytotoxicity by triggering a somewhat higher production of ROS, thereby inducing stronger cytotoxicity but lower toxicity to normal cells. Rh2HAZNO nanoparticles, a hyaluronic acid (HA)-functionalized zinc oxide (ZnO) nanocomposite (HA-ZnONcs) prepared using the coprecipitation method, were further functionalized with ginsenoside Rh2 through a cleavable ester bond of carbodiimide chemistry, generating ROS to induce apoptosis in cancer cells through the activation of the caspase-9/p38mapk signaling pathway (Kim et al., 2019). Xu et al. (2020) applied larulan polysaccharides grafted with allantoic acid and -lipoic acid (-LA) to obtain a pH and redox dual-responsive copolymer, LA-conjugated N-larulan allantoic acid (LA-URPA), enabling the copolymer LA-URPA-encapsulated ginsenoside Rh2 to form Rh2 nanoparticles (Rh2 NPs) that exert stronger proapoptotic effects by increasing ROS and downregulating antioxidant enzymes, such as SOD, CAT, and GSH (Xu et al., 2020). Moreover, the buildup of ginsenoside nanoparticles can reduce the side effects caused by ginsenosides. A previous study reported that DOX@Rg1 nanoparticles could attenuate free DOX-induced ROS generation and relevant apoptosis in H9C2 cells and mitigate cardiotoxicity; however, their toxicity was significantly increased in cancer cells (Li et al., 2021). Thus, constructing ginsenoside carriers can be a prospective therapeutic approach to enhance anticancer efficiency and reduce associated toxic side effects.
Despite studies supporting the therapeutic benefits of ginsenosides in combination with chemotherapeutic agents, their specific mechanisms have not been fully elucidated. A worthy case of back-citation is ginsenoside Ro, a novel autophagy inhibitor, that activates estrogen receptor 2 (ESR2), which consequentially activates a subunit of NADPH oxidase termed NCF1/p47PHOX (neutrophil lysyl factor 1); this cascade ultimately leads to 5-fluorouracil (5-Fu)-induced chemoresistant esophageal cancer cell death by ROS production and marked inhibition of autophagic fluxes (Zheng et al., 2016). Similarly, Chen et al. determined that Rh2, via regulatory autophagy, strengthened both NSCLC A549 and H1299 apoptosis caused by cisplatin, mostly through the induction of PD-L1 expression via the ROS-EGFR-PI3K-AKT-autophagy pathway. Interestingly, a significant increase in SOD activity was detected after Rh2 administration, whereas no significant change in GSH was noted, and the side effects of hearing loss due to ROS production were significantly attenuated (Chen et al., 2020). The upregulation of the Nrf2-driven antioxidative signaling pathway may also contribute to chemoresistance in cancer cells. Chian et al. studied this mechanism and verified that the ginsenoside Rd could, indeed, work against cisplatin-resistant lung cancer by downregulating this signaling pathway (Chian et al., 2019), which is consistent with the mechanism derived by Popov et al. in a study on the enhancement of the anticancer effect of DOX by RH2 (Popov et al., 2022). Moreover, both Rg1 and Rh2 may serve as chemosensitizers of doxorubicin, which suppresses the NF-κB signaling pathway to inhibit Dox-induced SASP (IL-8 and TNFα), thereby rescuing the viability of normal mammary epithelial cells and maintaining an inhibitory effect on cancer proliferation. Critically, such regulation is correlated with decreased ROS elicited by a significant increase in Rh2-mediated SOD1 and SOD2 and the regulators SIRT3 and SIRT5 at the protein level (Hou et al., 2020). However, when Rh2 was combined with DOX to treat breast cancer cells, the significantly higher ROS levels not only altered MMP, leading to cytochrome C release, but also acted as a synergistic cytotoxic agent by breaking single- and double-stranded DNA, thus activating apoptotic signaling (Liu et al., 2022). In addition, there is evidence of ginsenosides weakening the aggressiveness of antineoplastic drugs. Rg3 abrogated gemcitabine (GEM)-induced production of ROS-mediated activation of the Akt and extracellular signal-regulated kinase (ERK) pathways; moreover, it suppressed nuclear accumulation of NF-κB and HIF-1α to decrease PTX3, with significance in GEM-induced drug resistance, thereby dampening the GEM-induced aggressiveness of lung cancer cells (Ahmmed et al., 2019).
Taken together, there is an encouraging anticancer effect of ginsenosides in both cellular and animal models. However, preclinical studies on ginsenosides are scarce and the corresponding evidence is insufficient, possibly due to the following reasons: 1) a large molecular weight is composed of a large number of glycosides and needs to be metabolized into rare ginsenosides to have higher pharmacological activity in vivo. However, the low content of rare ginsenosides and the difficulty of isolation have limited the clinical application of ginsenosides (Fan et al., 2020; Hou et al., 2021); 2) due to poor absorption and bioavailability of ginsenosides, their anticancer effects are significantly reduced and species specificity render ginsenosides less dose-referenced for in vitro cellular and animal experiments. Using a monolayer model of human intestinal Caco-2 cells, Liu et al. demonstrated that G-Ra3 is poorly absorbed in the intestines (Liu et al., 2009) and that G-Rg1/Rb1 can achieve similar results, which may be related to the low permeability of the intestinal mucosa and first-pass action of the liver (Han and Fang, 2006; Kim, 2013). Encouragingly, given the aforementioned limitations of ginsenosides, studies on modified methods are emerging, investigating chemical modifications that alter the ginsenoside backbone structure (Ma et al., 2020) and the development of nano-delivery systems (Xu et al., 2020); ultimately, such modifications may effectively enhance pharmacological activity and bioavailability, as well as the targeting of ginsenosides to better treat cancer. It is reasonable to believe that with continuous technological development in pharmaceuticals and biochemistry fields, additional methods will be developed to improve the bioavailability of ginsenosides at a low economic cost, which is of great significance to fully utilize ginseng resources for the benefit of the public.
Given the growing body of literature, ginseng shows significant potential in cancer treatment. Ginsenosides exert their anticancer effects by modulating the majority of well-known carcinogenic modulators. The adjusted redox state of cancer cells can be used to design promising therapeutic strategies. ROS play a bidirectional role in cancer, which depends on the genetic background of the cancer, type of ROS involved, and level and duration of ROS exposure. Ginsenosides and their related derivatives exert superior anticancer effects by reacting with different cellular signaling cascades to directly or indirectly modulate ROS and impair redox homeostasis in cancer cells. Encouragingly, in the present cellular, animal, or preclinical studies involving ginsenosides, no cancer-promoting effects have been found, in the context of the possible mechanisms involved in the modulation of immunity and suppression of inflammatory responses besides those mentioned previously (Wong et al., 2015). In addition, since ginsenosides are known to lead to a variety of cell death processes, it is rare for cancer cells to be resistant to their induced cell death. Remarkably, ginsenosides can selectively kill tumor cells with relatively little toxicity to normal cells. Such selective toxicity and the optimization of this selection could be a valuable area of concern for future research and exploration.
YW came up with the idea, guided writing of the article, and provided financial support. XL wrote the manuscript. DC conducted drafting instructions and manuscript rewriting comments. SS provided and offered assistance in drawing technical support in drawing. All authors contributed to the article and approved the submitted version.
This paper was supported by the Jilin Science and Technology Development Program Project YDZJ202301ZYTS050.
The author acknowledges FiDraw for providing the drawing platform.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aboelella, N. S., Brandle, C., Kim, T., Ding, Z.-C., and Zhou, G. (2021). Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers (Basel) 13 (5), 986. doi:10.3390/cancers13050986
Ahmmed, B., Kampo, S., Khan, M., Faqeer, A., Kumar, S. P., Li, Y. L., et al. (2019). Rg3 inhibits gemcitabine-induced lung cancer cell invasiveness through ROS-dependent, NF-κB- and HIF-1α-mediated downregulation of PTX3. J. Cell. Physiology 234 (7), 10680–10697. doi:10.1002/jcp.27731
Al Tameemi, W., Dale, T. P., Al-Jumaily, R. M. K., and Forsyth, N. R. (2019). Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol. 7, 4. doi:10.3389/fcell.2019.00004
Areti, A., Yerra, V. G., Naidu, V., and Kumar, A. (2014). Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. 2, 289–295. doi:10.1016/j.redox.2014.01.006
Attanasio, U., Pirozzi, F., Poto, R., Cuomo, A., Carannante, A., Russo, M., et al. (2021). Oxidative stress in anticancer therapies-related cardiac dysfunction. Free Radic. Biol. Med. 169, 410–415. doi:10.1016/j.freeradbiomed.2021.04.021
Aykin-Burns, N., Ahmad, I. M., Zhu, Y., Oberley, L. W., and Spitz, D. R. (2009). Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 418 (1), 29–37. doi:10.1042/BJ20081258
Bae, Y. S., Oh, H., Rhee, S. G., and Yoo, Y. D. (2011). Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 32 (6), 491–509. doi:10.1007/s10059-011-0276-3
Bak, M. J., Jeong, W. S., and Kim, K. B. (2014). Detoxifying effect of fermented black ginseng on H2O2-induced oxidative stress in HepG2 cells. Int. J. Mol. Med. 34 (6), 1516–1522. doi:10.3892/ijmm.2014.1972
Bell, E. L., Klimova, T. A., Eisenbart, J., Moraes, C. T., Murphy, M. P., Budinger, G. R., et al. (2007). The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 177 (6), 1029–1036. doi:10.1083/jcb.200609074
Bertram, R., Gram Pedersen, M., Luciani, D. S., and Sherman, A. (2006). A simplified model for mitochondrial ATP production. J. Theor. Biol. 243 (4), 575–586. doi:10.1016/j.jtbi.2006.07.019
Bian, S., Zhao, Y., Li, F., Lu, S., Wang, S., Bai, X., et al. (2019). 20(S)-Ginsenoside Rg3 promotes HeLa cell apoptosis by regulating autophagy. Molecules 24 (20), 3655. doi:10.3390/molecules24203655
Birben, E., Sahiner, U. M., Sackesen, C., Erzurum, S., and Kalayci, O. (2012). Oxidative stress and antioxidant defense. World Allergy Organ J. 5 (1), 9–19. doi:10.1097/WOX.0b013e3182439613
Bonner, M. Y., and Arbiser, J. L. (2012). Targeting NADPH oxidases for the treatment of cancer and inflammation. Cell Mol. Life Sci. 69 (14), 2435–2442. doi:10.1007/s00018-012-1017-2
Boyer-Guittaut, M., Poillet, L., Liang, Q., Bôle-Richard, E., Ouyang, X., Benavides, G. A., et al. (2014). The role of GABARAPL1/GEC1 in autophagic flux and mitochondrial quality control in MDA-MB-436 breast cancer cells. Autophagy 10 (6), 986–1003. doi:10.4161/auto.28390
Brigelius-Flohé, R., and Maiorino, M. (2013). Glutathione peroxidases. Biochimica Biophysica Acta (BBA) - General Subj. 1830 (5), 3289–3303. doi:10.1016/j.bbagen.2012.11.020
Brown, D. I., and Griendling, K. K. (2009). Nox proteins in signal transduction. Free Radic. Biol. Med. 47 (9), 1239–1253. doi:10.1016/j.freeradbiomed.2009.07.023
Cadenas, E. (2004). Mitochondrial free radical production and cell signaling. Mol. Asp. Med. 25 (1-2), 17–26. doi:10.1016/j.mam.2004.02.005
Calvo-Ochoa, E., Sánchez-Alegría, K., Gómez-Inclán, C., Ferrera, P., and Arias, C. (2017). Palmitic acid stimulates energy metabolism and inhibits insulin/PI3K/AKT signaling in differentiated human neuroblastoma cells: The role of mTOR activation and mitochondrial ROS production. Neurochem. Int. 110, 75–83. doi:10.1016/j.neuint.2017.09.008
Cao, Y., Wang, J., Tian, H., and Fu, G. H. (2020). Mitochondrial ROS accumulation inhibiting JAK2/STAT3 pathway is a critical modulator of CYT997-induced autophagy and apoptosis in gastric cancer. J. Exp. Clin. Cancer Res. 39 (1), 119. doi:10.1186/s13046-020-01621-y
Chan, D. W., Liu, V. W., Tsao, G. S., Yao, K. M., Furukawa, T., Chan, K. K., et al. (2008). Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 29 (9), 1742–1750. doi:10.1093/carcin/bgn167
Chandel, N. S., and Tuveson, D. A. (2014). The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 371 (2), 177–178. doi:10.1056/NEJMcibr1405701
Chatterjee, R., and Chatterjee, J. (2020). ROS and oncogenesis with special reference to EMT and stemness. Eur. J. Cell Biol. 99 (2-3), 151073. doi:10.1016/j.ejcb.2020.151073
Chen, D., Tavana, O., Chu, B., Erber, L., Chen, Y., Baer, R., et al. (2017). NRF2 is a major target of ARF in p53-independent tumor suppression. Mol. Cell 68 (1), 224–232.e4. doi:10.1016/j.molcel.2017.09.009
Chen, F., Deng, Z., Xiong, Z., Zhang, B., Yang, J., and Hu, J. (2015). A ROS-mediated lysosomal-mitochondrial pathway is induced by ginsenoside Rh2 in hepatoma HepG2 cells. Food Funct. 6 (12), 3828–3837. doi:10.1039/c5fo00518c
Chen, Q., Vazquez, E. J., Moghaddas, S., Hoppel, C. L., and Lesnefsky, E. J. (2003). Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 278 (38), 36027–36031. doi:10.1074/jbc.M304854200
Chen, S., Li, X., Wang, Y., Mu, P., Chen, C., Huang, P., et al. (2019). Ginsenoside Rb1 attenuates intestinal ischemia/reperfusion-induced inflammation and oxidative stress via activation of the PI3K/Akt/Nrf2 signaling pathway. Mol. Med. Rep. 19 (5), 3633–3641. doi:10.3892/mmr.2019.10018
Chen, Y., McMillan-Ward, E., Kong, J., Israels, S. J., and Gibson, S. B. (2008). Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 15 (1), 171–182. doi:10.1038/sj.cdd.4402233
Chen, Y., Zhang, Y., Song, W., Zhang, Y., Dong, X., and Tan, M. (2020). Ginsenoside Rh2 improves the cisplatin anti-tumor effect in lung adenocarcinoma A549 cells via superoxide and PD-L1. Anticancer Agents Med. Chem. 20 (4), 495–503. doi:10.2174/1871520619666191209091230
Cheong, J. H., Kim, H., Hong, M. J., Yang, M. H., Kim, J. W., Yoo, H., et al. (2015). Stereoisomer-specific anticancer activities of ginsenoside Rg3 and Rh2 in HepG2 cells: Disparity in cytotoxicity and autophagy-inducing effects due to 20(S)-epimers. Biol. Pharm. Bull. 38 (1), 102–108. doi:10.1248/bpb.b14-00603
Cheung, E. C., DeNicola, G. M., Nixon, C., Blyth, K., Labuschagne, C. F., Tuveson, D. A., et al. (2020). Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 37 (2), 168–182.e4. doi:10.1016/j.ccell.2019.12.012
Cheung, E. C., and Vousden, K. H. (2022). The role of ROS in tumour development and progression. Nat. Rev. Cancer 22 (5), 280–297. doi:10.1038/s41568-021-00435-0
Chian, S., Zhao, Y., Xu, M., Yu, X., Ke, X., Gao, R., et al. (2019). Ginsenoside Rd reverses cisplatin resistance in non-small-cell lung cancer A549 cells by downregulating the nuclear factor erythroid 2-related factor 2 pathway. Anticancer Drugs 30 (8), 838–845. doi:10.1097/cad.0000000000000781
Chio, I. I. C., and Tuveson, D. A. (2017). ROS in cancer: The burning question. Trends Mol. Med. 23 (5), 411–429. doi:10.1016/j.molmed.2017.03.004
Choi, K., and Choi, C. (2009). Proapoptotic ginsenosides compound K and Rh enhance Fas-induced cell death of human astrocytoma cells through distinct apoptotic signaling pathways. Cancer Res. Treat. 41 (1), 36–44. doi:10.4143/crt.2009.41.1.36
Choi, Y. J., Lee, H. J., Kang, D. W., Han, I. H., Choi, B. K., and Cho, W. H. (2013). Ginsenoside Rg3 induces apoptosis in the U87MG human glioblastoma cell line through the MEK signaling pathway and reactive oxygen species. Oncol. Rep. 30 (3), 1362–1370. doi:10.3892/or.2013.2555
Choudhry, H., and Harris, A. L. (2018). Advances in hypoxia-inducible factor biology. Cell Metab. 27 (2), 281–298. doi:10.1016/j.cmet.2017.10.005
Chu, Y., Zhang, W., Kanimozhi, G., Brindha, G. R., and Tian, D. (2020). Ginsenoside Rg1 induces apoptotic cell death in triple-negative breast cancer cell lines and prevents carcinogen-induced breast tumorigenesis in sprague dawley rats. Evid. Based Complement. Altern. Med. 2020, 8886955. doi:10.1155/2020/8886955
Clauzure, M., Valdivieso, A. G., Massip Copiz, M. M., Schulman, G., Teiber, M. L., and Santa-Coloma, T. A. (2014). Disruption of interleukin-1β autocrine signaling rescues complex I activity and improves ROS levels in immortalized epithelial cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. PLoS One 9 (6), e99257. doi:10.1371/journal.pone.0099257
Cruz-Gregorio, A., Aranda-Rivera, A. K., Ortega-Lozano, A. J., Pedraza-Chaverri, J., and Mendoza-Hoffmann, F. (2021). Lipid metabolism and oxidative stress in HPV-related cancers. Free Radic. Biol. Med. 172, 226–236. doi:10.1016/j.freeradbiomed.2021.06.009
Deng, W., Wang, Y., Zhao, S., Zhang, Y., Chen, Y., Zhao, X., et al. (2018). MICAL1 facilitates breast cancer cell proliferation via ROS-sensitive ERK/cyclin D pathway. J. Cell Mol. Med. 22 (6), 3108–3118. doi:10.1111/jcmm.13588
DeNicola, G. M., Karreth, F. A., Humpton, T. J., Gopinathan, A., Wei, C., Frese, K., et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475 (7354), 106–109. doi:10.1038/nature10189
Ding, X., Yu, W., Wan, Y., Yang, M., Hua, C., Peng, N., et al. (2020). A pH/ROS-responsive, tumor-targeted drug delivery system based on carboxymethyl chitin gated hollow mesoporous silica nanoparticles for anti-tumor chemotherapy. Carbohydr. Polym. 245, 116493. doi:10.1016/j.carbpol.2020.116493
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dixon, S. J., and Stockwell, B. R. (2019). The hallmarks of ferroptosis. Annu. Rev. Cancer Biol. 3 (1), 35–54. doi:10.1146/annurev-cancerbio-030518-055844
Fan, W., Huang, Y., Zheng, H., Li, S., Li, Z., Yuan, L., et al. (2020). Ginsenosides for the treatment of metabolic syndrome and cardiovascular diseases: Pharmacology and mechanisms. Biomed. Pharmacother. 132, 110915. doi:10.1016/j.biopha.2020.110915
Forman, H. J., and Zhang, H. (2021). Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20 (9), 689–709. doi:10.1038/s41573-021-00233-1
Fu, W., Xu, H., Yu, X., Lyu, C., Tian, Y., Guo, M., et al. (2018). 20(S)-Ginsenoside Rg2 attenuates myocardial ischemia/reperfusion injury by reducing oxidative stress and inflammation: Role of SIRT1. RSC Adv. 8 (42), 23947–23962. doi:10.1039/c8ra02316f
Gamcsik, M. P., Kasibhatla, M. S., Teeter, S. D., and Colvin, O. M. (2012). Glutathione levels in human tumors. Biomarkers 17 (8), 671–691. doi:10.3109/1354750x.2012.715672
Gao, H., Liang, D., Li, C., Xu, G., Jiang, M., Li, H., et al. (2020). 2-Deoxy-Rh2: A novel ginsenoside derivative, as dual-targeting anti-cancer agent via regulating apoptosis and glycolysis. Biomed. Pharmacother. 124, 109891. doi:10.1016/j.biopha.2020.109891
Gaucher, C., Boudier, A., Bonetti, J., Clarot, I., Leroy, P., and Parent, M. (2018). Glutathione: Antioxidant properties dedicated to nanotechnologies. Antioxidants 7 (5), 62. doi:10.3390/antiox7050062
Ge, G. Q., Yan, Y., and Cai, H. (2017). Ginsenoside Rh2 inhibited proliferation by inducing ROS mediated ER stress dependent apoptosis in lung cancer cells. Biol. Pharm. Bull. 40 (12), 2117–2124. doi:10.1248/bpb.b17-00463
Gorrini, C., Harris, I. S., and Mak, T. W. (2013). Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12 (12), 931–947. doi:10.1038/nrd4002
Guo, Y. H., Kuruganti, R., and Gao, Y. (2019). Recent advances in ginsenosides as potential therapeutics against breast cancer. Curr. Top. Med. Chem. 19 (25), 2334–2347. doi:10.2174/1568026619666191018100848
Hagar, H., Husain, S., Fadda, L. M., Attia, N. M., Attia, M. M. A., and Ali, H. M. (2019). Inhibition of NF-κB and the oxidative stress -dependent caspase-3 apoptotic pathway by betaine supplementation attenuates hepatic injury mediated by cisplatin in rats. Pharmacol. Rep. 71 (6), 1025–1033. doi:10.1016/j.pharep.2019.06.003
Halcrow, P. W., Lynch, M. L., Geiger, J. D., and Ohm, J. E. (2021). Role of endolysosome function in iron metabolism and brain carcinogenesis. Semin. Cancer Biol. 76, 74–85. doi:10.1016/j.semcancer.2021.06.013
Ham, Y. M., Lim, J. H., Na, H. K., Choi, J. S., Park, B. D., Yim, H., et al. (2006). Ginsenoside-Rh2-induced mitochondrial depolarization and apoptosis are associated with reactive oxygen species- and Ca2+-mediated c-Jun NH2-terminal kinase 1 activation in HeLa cells. J. Pharmacol. Exp. Ther. 319 (3), 1276–1285. doi:10.1124/jpet.106.109926
Han, D., Williams, E., and Cadenas, E. (2001). Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 353 (2), 411–416. doi:10.1042/0264-6021:3530411
Han, M., and Fang, X. L. (2006). Difference in oral absorption of ginsenoside Rg1 between in vitro and in vivo models. Acta Pharmacol. Sin. 27 (4), 499–505. doi:10.1111/j.1745-7254.2006.00303.x
Han, Q., Han, L., Tie, F., Wang, Z., Ma, C., Li, J., et al. (2020). (20S)-Protopanaxadiol ginsenosides induced cytotoxicity via blockade of autophagic flux in HGC-27 cells. Chem. Biodivers. 17 (7), e2000187. doi:10.1002/cbdv.202000187
Harris, A. L. (2002). Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2 (1), 38–47. doi:10.1038/nrc704
Hayes, J. D., Dinkova-Kostova, A. T., and Tew, K. D. (2020). Oxidative stress in cancer. Cancer Cell 38 (2), 167–197. doi:10.1016/j.ccell.2020.06.001
Hong, Y., and Fan, D. (2019a). Ginsenoside Rk1 induces cell cycle arrest and apoptosis in MDA-MB-231 triple negative breast cancer cells. Toxicology 418, 22–31. doi:10.1016/j.tox.2019.02.010
Hong, Y., and Fan, D. (2019b). Ginsenoside Rk1 induces cell death through ROS-mediated PTEN/PI3K/Akt/mTOR signaling pathway in MCF-7 cells. J. Funct. Foods 57, 255–265. doi:10.1016/j.jff.2019.04.019
Horak, P., Crawford, A. R., Vadysirisack, D. D., Nash, Z. M., DeYoung, M. P., Sgroi, D., et al. (2010). Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc. Natl. Acad. Sci. U. S. A. 107 (10), 4675–4680. doi:10.1073/pnas.0907705107
Hou, J., Yun, Y., Xue, J., Jeon, B., and Kim, S. (2020). Doxorubicin-induced normal breast epithelial cellular aging and its related breast cancer growth through mitochondrial autophagy and oxidative stress mitigated by ginsenoside Rh2. Phytother. Res. 34 (7), 1659–1669. doi:10.1002/ptr.6636
Hou, M., Wang, R., Zhao, S., and Wang, Z. (2021). Ginsenosides in Panax genus and their biosynthesis. Acta Pharm. Sin. B 11 (7), 1813–1834. doi:10.1016/j.apsb.2020.12.017
Huynh, D. T. N., Jin, Y., Myung, C. S., and Heo, K. S. (2021). Ginsenoside Rh1 induces MCF-7 cell apoptosis and autophagic cell death through ROS-mediated Akt signaling. Cancers (Basel) 13 (8), 1892. doi:10.3390/cancers13081892
Hwang, S.-K., Jeong, Y.-J., Cho, H.-J., Park, Y.-Y., Song, K.-H., and Chang, Y.-C. (2022). Rg3-enriched red ginseng extract promotes lung cancer cell apoptosis and mitophagy by ROS production. J. Ginseng Res. 46 (1), 138–146. doi:10.1016/j.jgr.2021.05.005
Ilatovskaya, D. V., Pavlov, T. S., Levchenko, V., and Staruschenko, A. (2013). ROS production as a common mechanism of ENaC regulation by EGF, insulin, and IGF-1. Am. J. Physiol. Cell Physiol. 304 (1), C102–C111. doi:10.1152/ajpcell.00231.2012
Imai, H., Matsuoka, M., Kumagai, T., Sakamoto, T., and Koumura, T. (2017). Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr. Top. Microbiol. Immunol. 403, 143–170. doi:10.1007/82_2016_508
Ismail, T., Kim, Y., Lee, H., Lee, D. S., and Lee, H. S. (2019). Interplay between mitochondrial peroxiredoxins and ROS in cancer development and progression. Int. J. Mol. Sci. 20 (18), 4407. doi:10.3390/ijms20184407
Jelic, M. D., Mandic, A. D., Maricic, S. M., and Srdjenovic, B. U. (2021). Oxidative stress and its role in cancer. J. Cancer Res. Ther. 17 (1), 22–28. doi:10.4103/jcrt.JCRT_862_16
Jeon, H., Jin, Y., Myung, C. S., and Heo, K. S. (2021). Ginsenoside-Rg2 exerts anti-cancer effects through ROS-mediated AMPK activation associated mitochondrial damage and oxidation in MCF-7 cells. Arch. Pharm. Res. 44 (7), 702–712. doi:10.1007/s12272-021-01345-3
Jeon, S. M., Chandel, N. S., and Hay, N. (2012). AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485 (7400), 661–665. doi:10.1038/nature11066
Jin, Y., Huynh, D. T. N., and Heo, K. S. (2022). Ginsenoside Rh1 inhibits tumor growth in MDA-MB-231 breast cancer cells via mitochondrial ROS and ER stress-mediated signaling pathway. Arch. Pharm. Res. 45 (3), 174–184. doi:10.1007/s12272-022-01377-3
Jin, Y., Huynh, D. T. N., Myung, C. S., and Heo, K. S. (2021). Ginsenoside Rh1 prevents migration and invasion through mitochondrial ROS-mediated inhibition of STAT3/NF-κB signaling in MDA-MB-231 cells. Int. J. Mol. Sci. 22 (19), 10458. doi:10.3390/ijms221910458
Jung, H., Bae, J., Ko, S. K., and Sohn, U. D. (2016). Ultrasonication processed Panax ginseng berry extract induces apoptosis through an intrinsic apoptosis pathway in HepG2 cells. Arch. Pharm. Res. 39 (6), 855–862. doi:10.1007/s12272-016-0760-6
Kamata, H., Honda, S., Maeda, S., Chang, L., Hirata, H., and Karin, M. (2005). Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120 (5), 649–661. doi:10.1016/j.cell.2004.12.041
Kang, K. S., Ham, J., Kim, Y. J., Park, J. H., Cho, E. J., and Yamabe, N. (2013). Heat-processed Panax ginseng and diabetic renal damage: Active components and action mechanism. J. Ginseng Res. 37 (4), 379–388. doi:10.5142/jgr.2013.37.379
Khavari, T. A., and Rinn, J. L. (2007). Ras/erk MAPK signaling in epidermal homeostasis and neoplasia. Cell Cycle 6 (23), 2928–2931. doi:10.4161/cc.6.23.4998
Kim, A. D., Kang, K. A., Kim, H. S., Kim, D. H., Choi, Y. H., Lee, S. J., et al. (2013a). A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 4, e750. doi:10.1038/cddis.2013.273
Kim, B.-M., Kim, D.-H., Park, J.-H., Na, H.-K., and Surh, Y.-J. (2013b). Ginsenoside Rg3 induces apoptosis of human breast cancer (MDA-MB-231) cells. J. cancer Prev. 18 (2), 177–185. doi:10.15430/jcp.2013.18.2.177
Kim, D.-J., Seong, K.-S., Kim, D.-W., Kim, S.-R., and Chang, C.-C. (2004). Antioxidative effects of red ginseng saponins on paraquat-induced oxidative stress. J. Ginseng Res. 28, 5–10. doi:10.5142/JGR.2004.28.1.005
Kim, H. H., Choi, S. E., and Jeong, W. I. (2020). Oxidative stress and glutamate excretion in alcoholic steatosis: Metabolic synapse between hepatocyte and stellate cell. Clin. Mol. Hepatol. 26 (4), 697–704. doi:10.3350/cmh.2020.0152
Kim, H. K. (2013). Pharmacokinetics of ginsenoside Rb1 and its metabolite compound K after oral administration of Korean Red Ginseng extract. J. Ginseng Res. 37 (4), 451–456. doi:10.5142/jgr.2013.37.451
Kim, J. H. (2018). Pharmacological and medical applications of Panax ginseng and ginsenosides: A review for use in cardiovascular diseases. J. Ginseng Res. 42 (3), 264–269. doi:10.1016/j.jgr.2017.10.004
Kim, K. Y., Park, K. I., Kim, S. H., Yu, S. N., Lee, D., Kim, Y. W., et al. (2017). Salinomycin induces reactive oxygen species and apoptosis in aggressive breast cancer cells as mediated with regulation of autophagy. Anticancer Res. 37 (4), 1747–1758. doi:10.21873/anticanres.11507
Kim, Y. J., Perumalsamy, H., Castro-Aceituno, V., Kim, D., Markus, J., Lee, S., et al. (2019). Photoluminescent and self-assembled hyaluronic acid-zinc oxide-ginsenoside Rh2 nanoparticles and their potential caspase-9 apoptotic mechanism towards cancer cell lines. Int. J. Nanomedicine 14, 8195–8208. doi:10.2147/ijn.S221328
Kuang, F., Liu, J., Tang, D., and Kang, R. (2020). Oxidative damage and antioxidant defense in ferroptosis. Front. Cell Dev. Biol. 8, 586578. doi:10.3389/fcell.2020.586578
Kuo, C. L., Ponneri Babuharisankar, A., Lin, Y. C., Lien, H. W., Lo, Y. K., Chou, H. Y., et al. (2022). Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: Foe or friend? J. Biomed. Sci. 29 (1), 74. doi:10.1186/s12929-022-00859-2
Kwak, J. H., and Pyo, J. S. (2016). Characterization of apoptosis induced by ginsenosides in human lung cancer cells. Anal. Lett. 49 (6), 843–854. doi:10.1080/00032719.2015.1079208
Lee, J.-Y., Jung, K. H., Morgan, M. J., Kang, Y.-R., Lee, H.-S., Koo, G.-B., et al. (2013). Sensitization of TRAIL-induced cell death by 20(S)-Ginsenoside rg(3) via CHOP-mediated DR5 upregulation in human hepatocellular carcinoma cells. Mol. Cancer Ther. 12 (3), 274–285. doi:10.1158/1535-7163.Mct-12-0054
Leem, D. G., Shin, J. S., Kim, K. T., Choi, S. Y., Lee, M. H., and Lee, K. T. (2018). Dammarane-type triterpene ginsenoside-Rg18 inhibits human non-small cell lung cancer A549 cell proliferation via G(1) phase arrest. Oncol. Lett. 15 (4), 6043–6049. doi:10.3892/ol.2018.8057
Li, B., Zhao, J., Wang, C.-Z., Searle, J., He, T.-C., Yuan, C.-S., et al. (2011). Ginsenoside Rh2 induces apoptosis and paraptosis-like cell death in colorectal cancer cells through activation of p53. Cancer Lett. 301 (2), 185–192. doi:10.1016/j.canlet.2010.11.015
Li, C., Gou, X., and Gao, H. (2021). Doxorubicin nanomedicine based on ginsenoside Rg1 with alleviated cardiotoxicity and enhanced antitumor activity. Nanomedicine 16 (29), 2587–2604. doi:10.2217/nnm-2021-0329
Li, G., Zhang, X.-x., Lin, L., Liu, X.-n., Ma, C.-j., Li, J., et al. (2014). Preparation of ginsenoside Rg3 and protection against H2O2-induced oxidative stress in human neuroblastoma SK-N-sh cells. J. Chem. 2014, 848571. doi:10.1155/2014/848571
Li, J., Cai, D., Yao, X., Zhang, Y., Chen, L., Jing, P., et al. (2016). Protective effect of ginsenoside Rg1 on hematopoietic stem/progenitor cells through attenuating oxidative stress and the wnt/β-catenin signaling pathway in a mouse model of d-Galactose-induced aging. Int. J. Mol. Sci. 17 (6), 849. doi:10.3390/ijms17060849
Li, W., Li, G., She, W., Hu, X., and Wu, X. (2019). Targeted antitumor activity of Ginsenoside (Rg1) in paclitaxel-resistant human nasopharyngeal cancer cells are mediated through activation of autophagic cell death, cell apoptosis, endogenous ROS production, S phase cell cycle arrest and inhibition of m-TOR/PI3K/AKT signalling pathway. J. buon 24 (5), 2056–2061.
Limaye, V., Li, X., Hahn, C., Xia, P., Berndt, M. C., Vadas, M. A., et al. (2005). Sphingosine kinase-1 enhances endothelial cell survival through a PECAM-1-dependent activation of PI-3K/Akt and regulation of Bcl-2 family members. Blood 105 (8), 3169–3177. doi:10.1182/blood-2004-02-0452
Liou, G. Y., Döppler, H., DelGiorno, K. E., Zhang, L., Leitges, M., Crawford, H. C., et al. (2016). Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 14 (10), 2325–2336. doi:10.1016/j.celrep.2016.02.029
Liou, G. Y., and Storz, P. (2010). Reactive oxygen species in cancer. Free Radic. Res. 44 (5), 479–496. doi:10.3109/10715761003667554
Liu, H., Yang, J., Du, F., Gao, X., Ma, X., Huang, Y., et al. (2009). Absorption and disposition of ginsenosides after oral administration of Panax notoginseng extract to rats. Drug Metab. Dispos. 37 (12), 2290–2298. doi:10.1124/dmd.109.029819
Liu, S., Huang, J., Gao, F., Yin, Z., and Zhang, R. (2022). Ginsenoside RG1 augments doxorubicin-induced apoptotic cell death in MDA-MB-231 breast cancer cell lines. J. Biochem. Mol. Toxicol. 36 (1), e22945. doi:10.1002/jbt.22945
Liu, Y., and Fan, D. (2019). Ginsenoside Rg5 induces G2/M phase arrest, apoptosis and autophagy via regulating ROS-mediated MAPK pathways against human gastric cancer. Biochem. Pharmacol. 168, 285–304. doi:10.1016/j.bcp.2019.07.008
Liu, Y., Wang, J., Qiao, J., Liu, S., Wang, S., Zhao, D., et al. (2020). Ginsenoside Rh2 inhibits HeLa cell energy metabolism and induces apoptosis by upregulating voltage-dependent anion channel 1. Int. J. Mol. Med. 46 (5), 1695–1706. doi:10.3892/ijmm.2020.4725
Liu, Y., Yu, S., Xing, X., Qiao, J., Yin, Y., Wang, J., et al. (2021). Ginsenoside Rh2 stimulates the production of mitochondrial reactive oxygen species and induces apoptosis of cervical cancer cells by inhibiting mitochondrial electron transfer chain complex. Mol. Med. Rep. 24 (6), 873. doi:10.3892/mmr.2021.12513
Lu, H., Samanta, D., Xiang, L., Zhang, H., Hu, H., Chen, I., et al. (2015). Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. U. S. A. 112 (33), E4600–E4609. doi:10.1073/pnas.1513433112
Lu, H., Yin, H., Qu, L., Ma, X., Fu, R., and Fan, D. (2022). Ginsenoside Rk1 regulates glutamine metabolism in hepatocellular carcinoma through inhibition of the ERK/c-Myc pathway. Food Funct. 13 (7), 3793–3811. doi:10.1039/d1fo03728e
Ma, L., Wang, X., Li, W., Li, T., Xiao, S., Lu, J., et al. (2020). Rational design, synthesis and biological evaluation of triphenylphosphonium-ginsenoside conjugates as mitochondria-targeting anti-cancer agents. Bioorg. Chem. 103, 104150. doi:10.1016/j.bioorg.2020.104150
Mao, Q., Zhang, P.-H., Wang, Q., and Li, S.-L. (2014). Ginsenoside F-2 induces apoptosis in humor gastric carcinoma cells through reactive oxygen species-mitochondria pathway and modulation of ASK-1/JNK signaling cascade in vitro and in vivo. Phytomedicine 21 (4), 515–522. doi:10.1016/j.phymed.2013.10.013
Marinho, H. S., Real, C., Cyrne, L., Soares, H., and Antunes, F. (2014). Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2, 535–562. doi:10.1016/j.redox.2014.02.006
Martínez-Cayuela, M. (1995). Oxygen free radicals and human disease. Biochimie 77 (3), 147–161. doi:10.1016/0300-9084(96)88119-3
Maslah, H., Skarbek, C., Pethe, S., and Labruère, R. (2020). Anticancer boron-containing prodrugs responsive to oxidative stress from the tumor microenvironment. Eur. J. Med. Chem. 207, 112670. doi:10.1016/j.ejmech.2020.112670
Metwaly, A. M., Lianlian, Z., Luqi, H., and Deqiang, D. (2019). Black ginseng and its saponins: Preparation, phytochemistry and pharmacological effects. Molecules 24 (10), 1856. doi:10.3390/molecules24101856
Moloney, J. N., and Cotter, T. G. (2018). ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 80, 50–64. doi:10.1016/j.semcdb.2017.05.023
Murphy, M. P. (2013). Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. 18 (2), 145–146. doi:10.1016/j.cmet.2013.07.006
Nag, S. A., Qin, J. J., Wang, W., Wang, M. H., Wang, H., and Zhang, R. (2012). Ginsenosides as anticancer agents: In vitro and in vivo activities, structure-activity relationships, and molecular mechanisms of action. Front. Pharmacol. 3, 25. doi:10.3389/fphar.2012.00025
Nakamura, S., Sugimoto, S., Matsuda, H., and Yoshikawa, M. (2007). Medicinal flowers. XVII. New dammarane-type triterpene glycosides from flower buds of American ginseng, Panax quinquefolium L. Chem. Pharm. Bull. (Tokyo) 55 (9), 1342–1348. doi:10.1248/cpb.55.1342
Nieto, M. A., Huang, R. Y., Jackson, R. A., and Thiery, J. P. (2016). Emt: 2016. Cell 166 (1), 21–45. doi:10.1016/j.cell.2016.06.028
Niu, B., Liao, K., Zhou, Y., Wen, T., Quan, G., Pan, X., et al. (2021). Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials 277, 121110. doi:10.1016/j.biomaterials.2021.121110
Oh, J. M., Kim, E., and Chun, S. (2019). Ginsenoside compound K induces ros-mediated apoptosis and autophagic inhibition in human neuroblastoma cells in vitro and in vivo. Int. J. Mol. Sci. 20 (17), 4279. doi:10.3390/ijms20174279
Ong, W. Y., Farooqui, T., Koh, H. L., Farooqui, A. A., and Ling, E. A. (2015). Protective effects of ginseng on neurological disorders. Front. Aging Neurosci. 7, 129. doi:10.3389/fnagi.2015.00129
Parascandolo, A., and Laukkanen, M. O. (2018). Carcinogenesis and reactive oxygen species signaling: Interaction of the NADPH oxidase NOX1–5 and superoxide dismutase 1–3 signal transduction pathways. Antioxidants Redox Signal. 30 (3), 443–486. doi:10.1089/ars.2017.7268
Park, H.-M., Kim, S.-J., Kim, J.-S., and Kang, H.-S. (2012). Reactive oxygen species mediated ginsenoside Rg3-and Rh2-induced apoptosis in hepatoma cells through mitochondrial signaling pathways. Food Chem. Toxicol. 50 (8), 2736–2741. doi:10.1016/j.fct.2012.05.027
Pérez-Ramírez, C., Cañadas-Garre, M., Molina, M., Faus-Dáder, M. J., and Calleja-Hernández, M. (2015). PTEN and PI3K/AKT in non-small-cell lung cancer. Pharmacogenomics 16 (16), 1843–1862. doi:10.2217/pgs.15.122
Perillo, B., Di Donato, M., Pezone, A., Di Zazzo, E., Giovannelli, P., Galasso, G., et al. (2020). ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 52 (2), 192–203. doi:10.1038/s12276-020-0384-2
Ping, -.Z. B., Bo, -.L., Yang, -.C. J., Rui, -.C., Ting, -.G. S., Juan, -.H. C., et al. (2020). Anti-cancer effect of 20(S)-Ginsenoside-Rh2 on oral squamous cell carcinoma cells via the decrease in ROS and downregulation of MMP-2 and VEGF. Biomed. Environ. Sci. 33 (9)–713. doi:10.3967/bes2020.093
Piskounova, E., Agathocleous, M., Murphy, M. M., Hu, Z., Huddlestun, S. E., Zhao, Z., et al. (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527 (7577), 186–191. doi:10.1038/nature15726
Poillet-Perez, L., Despouy, G., Delage-Mourroux, R., and Boyer-Guittaut, M. (2015). Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 4, 184–192. doi:10.1016/j.redox.2014.12.003
Popov, A., Klimovich, A., Styshova, O., Tsybulsky, A., Hushpulian, D., Osipyants, A., et al. (2022). Probable mechanisms of doxorubicin antitumor activity enhancement by ginsenoside Rh2. Molecules 27 (3), 628. doi:10.3390/molecules27030628
Pouysségur, J., Dayan, F., and Mazure, N. M. (2006). Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441 (7092), 437–443. doi:10.1038/nature04871
Reczek, C. R., and Chandel, N. S. (2017). The two faces of reactive oxygen species in cancer. Annu. Rev. Cancer Biol. 1 (1), 79–98. doi:10.1146/annurev-cancerbio-041916-065808
Redza-Dutordoir, M., and Averill-Bates, D. A. (2016). Activation of apoptosis signalling pathways by reactive oxygen species. Biochimica Biophysica Acta 1863 (12), 2977–2992. doi:10.1016/j.bbamcr.2016.09.012
Roberge, S., Roussel, J., Andersson, D. C., Meli, A. C., Vidal, B., Blandel, F., et al. (2014). TNF-α-mediated caspase-8 activation induces ROS production and TRPM2 activation in adult ventricular myocytes. Cardiovasc Res. 103 (1), 90–99. doi:10.1093/cvr/cvu112
Rofstad, E. K., Mathiesen, B., Kindem, K., and Galappathi, K. (2006). Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 66 (13), 6699–6707. doi:10.1158/0008-5472.Can-06-0983
Rojo de la Vega, M., Chapman, E., and Zhang, D. D. (2018). NRF2 and the hallmarks of cancer. Cancer Cell 34 (1), 21–43. doi:10.1016/j.ccell.2018.03.022
Rose Li, Y., Halliwill, K. D., Adams, C. J., Iyer, V., Riva, L., Mamunur, R., et al. (2020). Mutational signatures in tumours induced by high and low energy radiation in Trp53 deficient mice. Nat. Commun. 11 (1), 394. doi:10.1038/s41467-019-14261-4
Safdar, A., Annis, S., Kraytsberg, Y., Laverack, C., Saleem, A., Popadin, K., et al. (2016). Amelioration of premature aging in mtDNA mutator mouse by exercise: The interplay of oxidative stress, PGC-1α, p53, and DNA damage. A hypothesis. Curr. Opin. Genet. Dev. 38, 127–132. doi:10.1016/j.gde.2016.06.011
Saitoh, M., Nishitoh, H., Fujii, M., Takeda, K., Tobiume, K., Sawada, Y., et al. (1998). Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. Embo J. 17 (9), 2596–2606. doi:10.1093/emboj/17.9.2596
Schieber, M., and Chandel, N. S. (2014). ROS function in redox signaling and oxidative stress. Curr. Biol. 24 (10), R453–R462. doi:10.1016/j.cub.2014.03.034
Seervi, M., Rani, A., Sharma, A. K., and Santhosh Kumar, T. R. (2018). ROS mediated ER stress induces Bax-Bak dependent and independent apoptosis in response to Thioridazine. Biomed. Pharmacother. 106, 200–209. doi:10.1016/j.biopha.2018.06.123
Semenza, G. L. (2012). Hypoxia-inducible factors in physiology and medicine. Cell 148 (3), 399–408. doi:10.1016/j.cell.2012.01.021
Sepasi Tehrani, H., and Moosavi-Movahedi, A. A. (2018). Catalase and its mysteries. Prog. Biophys. Mol. Biol. 140, 5–12. doi:10.1016/j.pbiomolbio.2018.03.001
Sheng, Y., Abreu, I. A., Cabelli, D. E., Maroney, M. J., Miller, A. F., Teixeira, M., et al. (2014). Superoxide dismutases and superoxide reductases. Chem. Rev. 114 (7), 3854–3918. doi:10.1021/cr4005296
Shin, B.-K., Kwon, S. W., and Park, J. H. (2015). Chemical diversity of ginseng saponins from Panax ginseng. J. Ginseng Res. 39 (4), 287–298. doi:10.1016/j.jgr.2014.12.005
Sies, H., and Jones, D. P. (2020). Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21 (7), 363–383. doi:10.1038/s41580-020-0230-3
Simon, H. U., Haj-Yehia, A., and Levi-Schaffer, F. (2000). Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 5 (5), 415–418. doi:10.1023/A:1009616228304
Sin, S., Kim, S. Y., and Kim, S. S. (2012). Chronic treatment with ginsenoside Rg3 induces Akt-dependent senescence in human glioma cells. Int. J. Oncol. 41 (5), 1669–1674. doi:10.3892/ijo.2012.1604
Son, Y., Cheong, Y.-K., Kim, N.-H., Chung, H.-T., Kang, D. G., and Pae, H.-O. (2011). Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J. Signal Transduct. 2011, 792639. doi:10.1155/2011/792639
Spencer, N. Y., and Engelhardt, J. F. (2014). The basic biology of redoxosomes in cytokine-mediated signal transduction and implications for disease-specific therapies. Biochemistry 53 (10), 1551–1564. doi:10.1021/bi401719r
Stegen, S., van Gastel, N., Eelen, G., Ghesquière, B., D’Anna, F., Thienpont, B., et al. (2016). HIF-1α promotes glutamine-mediated redox homeostasis and glycogen-dependent bioenergetics to support postimplantation bone cell survival. Cell Metab. 23 (2), 265–279. doi:10.1016/j.cmet.2016.01.002
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171 (2), 273–285. doi:10.1016/j.cell.2017.09.021
Storz, P., Döppler, H., Ferran, C., Grey, S. T., and Toker, A. (2005). Functional dichotomy of A20 in apoptotic and necrotic cell death. Biochem. J. 387 (1), 47–55. doi:10.1042/bj20041443
Sun, H. Y., Lee, J. H., Han, Y. S., Yoon, Y. M., Yun, C. W., Kim, J. H., et al. (2016). Pivotal roles of ginsenoside Rg3 in tumor apoptosis through regulation of reactive oxygen species. Anticancer Res. 36 (9), 4647–4654. doi:10.21873/anticanres.11015
Suzuki, H., Shibagaki, Y., Hattori, S., and Matsuoka, M. (2019). C9-ALS/FTD-linked proline–arginine dipeptide repeat protein associates with paraspeckle components and increases paraspeckle formation. Cell Death Dis. 10 (10), 746. doi:10.1038/s41419-019-1983-5
Vander Heiden, M. G., and DeBerardinis, R. J. (2017). Understanding the intersections between metabolism and cancer biology. Cell 168 (4), 657–669. doi:10.1016/j.cell.2016.12.039
Wang, X., Sun, Y. Y., Zhao, C., Qu, F. Z., and Zhao, Y. Q. (2017). 12-Chloracetyl-PPD, a novel dammarane derivative, shows anti-cancer activity via delay the progression of cell cycle G2/M phase and reactive oxygen species-mediate cell apoptosis. Eur. J. Pharmacol. 798, 49–56. doi:10.1016/j.ejphar.2016.12.027
Wagner, E. F., and Nebreda, A. R. (2009). Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9 (8), 537–549. doi:10.1038/nrc2694
Wang, H., Jiang, D., Liu, J., Ye, S., Xiao, S., Wang, W., et al. (2013). Compound K induces apoptosis of bladder cancer T24 cells via reactive oxygen species-mediated p38 MAPK pathway. Cancer Biotherapy Radiopharm. 28 (8), 607–614. doi:10.1089/cbr.2012.1468
Wang, J., Bian, S., Wang, S., Yang, S., Zhang, W., Zhao, D., et al. (2020a). Ginsenoside Rh2 represses autophagy to promote cervical cancer cell apoptosis during starvation. Chin. Med. 15 (1), 118. doi:10.1186/s13020-020-00396-w
Wang, X. D., Li, T., Li, Y., Yuan, W. H., and Zhao, Y. Q. (2020b). 2-Pyrazine-PPD, a novel dammarane derivative, showed anticancer activity by reactive oxygen species-mediate apoptosis and endoplasmic reticulum stress in gastric cancer cells. Eur. J. Pharmacol. 881, 173211. doi:10.1016/j.ejphar.2020.173211
Wang, X., Su, G. Y., Zhao, C., Qu, F. Z., Wang, P., and Zhao, Y. Q. (2018). Anticancer activity and potential mechanisms of 1C, a ginseng saponin derivative, on prostate cancer cells. J. Ginseng Res. 42 (2), 133–143. doi:10.1016/j.jgr.2016.12.014
Wang, Y., Fu, W., Xue, Y., Lu, Z., Li, Y., Yu, P., et al. (2021a). Ginsenoside Rc ameliorates endothelial insulin resistance via upregulation of angiotensin-converting enzyme 2. Front. Pharmacol. 12, 620524. doi:10.3389/fphar.2021.620524
Wang, Y., Qi, H., Liu, Y., Duan, C., Liu, X., Xia, T., et al. (2021b). The double-edged roles of ROS in cancer prevention and therapy. Theranostics 11 (10), 4839–4857. doi:10.7150/thno.56747
Wartenberg, M., Budde, P., De Mareés, M., Grünheck, F., Tsang, S. Y., Huang, Y., et al. (2003). Inhibition of tumor-induced angiogenesis and matrix-metalloproteinase expression in confrontation cultures of embryoid bodies and tumor spheroids by plant ingredients used in traditional Chinese medicine. Lab. Invest. 83 (1), 87–98. doi:10.1097/01.lab.0000049348.51663.2f
Weinberg, F., Hamanaka, R., Wheaton, W. W., Weinberg, S., Joseph, J., Lopez, M., et al. (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U. S. A. 107 (19), 8788–8793. doi:10.1073/pnas.1003428107
Westermarck, J., and Kähäri, V. M. (1999). Regulation of matrix metalloproteinase expression in tumor invasion. Faseb J. 13 (8), 781–792. doi:10.1096/fasebj.13.8.781
Wilkie-Grantham, R. P., Matsuzawa, S., and Reed, J. C. (2013). Novel phosphorylation and ubiquitination sites regulate reactive oxygen species-dependent degradation of anti-apoptotic c-FLIP protein. J. Biol. Chem. 288 (18), 12777–12790. doi:10.1074/jbc.M112.431320
Wong, A. S., Che, C. M., and Leung, K. W. (2015). Recent advances in ginseng as cancer therapeutics: A functional and mechanistic overview. Nat. Prod. Rep. 32 (2), 256–272. doi:10.1039/c4np00080c
Wu, K., Huang, J., Li, N., Xu, T., Cai, W., and Ye, Z. (2018a). Antitumor effect of ginsenoside Rg3 on gallbladder cancer by inducing endoplasmic reticulum stress-mediated apoptosis in vitro and in vivo. Oncol. Lett. 16 (5), 5687–5696. doi:10.3892/ol.2018.9331
Wu, Q., Deng, J., Fan, D., Duan, Z., Zhu, C., Fu, R., et al. (2018b). Ginsenoside Rh4 induces apoptosis and autophagic cell death through activation of the ROS/JNK/p53 pathway in colorectal cancer cells. Biochem. Pharmacol. 148, 64–74. doi:10.1016/j.bcp.2017.12.004
Wu, S., Lu, H., and Bai, Y. (2019). Nrf2 in cancers: A double-edged sword. Cancer Med. 8 (5), 2252–2267. doi:10.1002/cam4.2101
Wu, Y., Pi, D., Chen, Y., Zuo, Q., Zhou, S., and Ouyang, M. (2022). Ginsenoside Rh4 inhibits colorectal cancer cell proliferation by inducing ferroptosis via autophagy activation. Evidence-Based Complementary Altern. Med. 2022, 6177553. doi:10.1155/2022/6177553
Xia, T., Wang, Y.-N., Zhou, C.-X., Wu, L.-M., Liu, Y., Zeng, Q.-H., et al. (2017). Ginsenoside Rh2 and Rg3 inhibit cell proliferation and induce apoptosis by increasing mitochondrial reactive oxygen species in human leukemia Jurkat cells. Mol. Med. Rep. 15 (6), 3591–3598. doi:10.3892/mmr.2017.6459
Xu, Y., Li, X., Gong, W., Huang, H. B., Zhu, B. W., and Hu, J. N. (2020). Construction of ginsenoside nanoparticles with pH/reduction dual response for enhancement of their cytotoxicity toward HepG2 cells. J. Agric. Food Chem. 68 (32), 8545–8556. doi:10.1021/acs.jafc.0c03698
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447 (7146), 864–868. doi:10.1038/nature05859
Ye, J., Fan, J., Venneti, S., Wan, Y. W., Pawel, B. R., Zhang, J., et al. (2014). Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4 (12), 1406–1417. doi:10.1158/2159-8290.Cd-14-0250
Yin, Q., Chen, H., Ma, R. H., Zhang, Y. Y., Liu, M. M., Thakur, K., et al. (2021). Ginsenoside CK induces apoptosis of human cervical cancer HeLa cells by regulating autophagy and endoplasmic reticulum stress. Food Funct. 12 (12), 5301–5316. doi:10.1039/d1fo00348h
Zavattari, P., Perra, A., Menegon, S., Kowalik, M. A., Petrelli, A., Angioni, M. M., et al. (2015). Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 62 (3), 851–862. doi:10.1002/hep.27790
Zhang, H., Park, S., Huang, H., Kim, E., Yi, J., Choi, S. K., et al. (2021). Anticancer effects and potential mechanisms of ginsenoside Rh2 in various cancer types (Review). Oncol. Rep. 45 (4), 33. doi:10.3892/or.2021.7984
Zhang, L. H., Jia, Y. L., Lin, X. X., Zhang, H. Q., Dong, X. W., Zhao, J. M., et al. (2013). AD-1, a novel ginsenoside derivative, shows anti-lung cancer activity via activation of p38 MAPK pathway and generation of reactive oxygen species. Biochim. Biophys. Acta 1830 (8), 4148–4159. doi:10.1016/j.bbagen.2013.04.008
Zhang, R., Humphreys, I., Sahu, R. P., Shi, Y., and Srivastava, S. K. (2008). In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis 13 (12), 1465–1478. doi:10.1007/s10495-008-0278-6
Zhang, Y., Li, Q., Wei, S., Sun, J., Zhang, X., He, L., et al. (2020). ZNF143 suppresses cell apoptosis and promotes proliferation in gastric cancer via ROS/p53 Axis. Dis. Markers 2020, 5863178. doi:10.1155/2020/5863178
Zheng, D., Liu, J., Piao, H., Zhu, Z., Wei, R., and Liu, K. (2022). ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front. Immunol. 13, 1039241. doi:10.3389/fimmu.2022.1039241
Zheng, K., Li, Y., Wang, S., Wang, X., Liao, C., Hu, X., et al. (2016). Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy 12 (9), 1593–1613. doi:10.1080/15548627.2016.1192751
Zheng, M. M., Xu, F. X., Li, Y. J., Xi, X. Z., Cui, X. W., Han, C. C., et al. (2017). Study on transformation of ginsenosides in different methods. Biomed. Res. Int. 2017, 8601027. doi:10.1155/2017/8601027
Zheng, Q., Li, Q., Zhao, G., Zhang, J., Yuan, H., Gong, D., et al. (2020). Alkannin induces cytotoxic autophagy and apoptosis by promoting ROS-mediated mitochondrial dysfunction and activation of JNK pathway. Biochem. Pharmacol. 180, 114167. doi:10.1016/j.bcp.2020.114167
Keywords: ginsenoside, cancer, reactive oxygen species, treatment, signaling pathways
Citation: Li X, Cao D, Sun S and Wang Y (2023) Anticancer therapeutic effect of ginsenosides through mediating reactive oxygen species. Front. Pharmacol. 14:1215020. doi: 10.3389/fphar.2023.1215020
Received: 01 May 2023; Accepted: 10 July 2023;
Published: 26 July 2023.
Edited by:
Jianqiang Xu, Dalian University of Technology, ChinaReviewed by:
Shanshan Geng, Nanjing Medical University, ChinaCopyright © 2023 Li, Cao, Sun and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuehui Wang, eXVlaHVpd2FuZzMwMEBqbHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.