 Junzhe Zhao
Junzhe Zhao Keene Lee
Keene Lee Han Chong Toh
Han Chong Toh Kong Peng Lam3,4,5
Kong Peng Lam3,4,5 Shi Yong Neo
Shi Yong Neo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 30 March 2023
Sec. Experimental Pharmacology and Drug Discovery
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1163160
This article is part of the Research Topic Cancer Metabolism: Targets, Biomarkers, Mechanism and Drugs View all 6 articles
The dysregulation of the biochemical pathways in cancer promotes oncogenic transformations and metastatic potential. Recent studies have shed light on how obesity and altered lipid metabolism could be the driving force for tumor progression. Here, in this review, we focus on liver cancer and discuss how obesity and lipid-driven metabolic reprogramming affect tumor, immune, and stroma cells in the tumor microenvironment and, in turn, how alterations in these cells synergize to influence and contribute to tumor growth and dissemination. With increasing evidence on how obesity exacerbates inflammation and immune tolerance, we also touch upon the impact of obesity and altered lipid metabolism on tumor immune escape.
There is accumulating evidence of a link between environmental factors and cancer incidence rates. Environmental factors related to lifestyle choices, which include high-carbohydrate and high-lipid diets, have been strongly correlated with metabolic disruptions leading to obesity, insulin resistance, and even metabolic syndrome. Obesity, a result of excessive systemic adipose tissue accumulation leading to increased body weight, is strongly associated with the development of a host of chronic diseases such as type 2 diabetes mellitus, hypertension, cardiovascular disease, and hepatic steatosis. Furthermore, it has been implicated as a risk factor in the initiation and progression of a wide variety of cancers (Dobbins et al., 2013) which include breast, ovary (Liu et al., 2015), prostate, liver, colon, and pancreatic cancers.
Cancer can be regarded as a metabolic disease. Despite obesity being a risk factor for cancer development, the scientific link between obesity and tumor progression is not apparent. In a meta-analysis of 33 studies across different cancer types, obesity is revealed as a prognostic but not an independent predictor for the survival outcomes of cancer patients (Parekh et al., 2012). Several obesity-associated molecular factors and signaling pathways such as adipokine production, alterations in the insulin-like growth factor 1 (IGF-1) signaling pathway, excessive steroidal sex hormone production, chronic low-grade inflammation, oxidative stress, and gut dysbiosis were identified to influence tumor cell biology, but the underlying mechanisms have not been fully elucidated (Parekh et al., 2012; Park et al., 2014a; Avgerinos et al., 2019).
In this review, we cover how dysregulated lipid metabolism could fuel cancer progression. Using hepatic steatosis and liver cancer as a classic example, we discuss how obesity could lead to metabolic disease and also oncogenesis. Finally, we will also highlight the role of lipids as a metabolic immune checkpoint in the stroma and immune compartments of the tumor microenvironment (TME) (Figure 1).
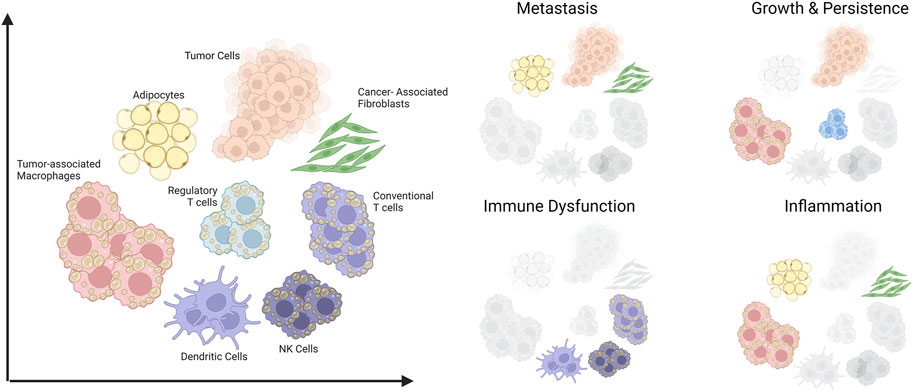
FIGURE 1. Schematic summary of cells in the tumor microenvironment that are amenable to lipid-induced metabolic programming and their contributions to aspects of cancer progression. The scope of this review covers various cell types within the tumor microenvironment (Left) and their involvement in four key aspects of tumor progression (Right). Created with BioRender.com.
Given that lipids play an integral role in cellular metabolism (Carracedo et al., 2013), signaling, and membrane structure, altered lipid metabolism has been regarded as one of the more important metabolic alterations associated with tumor cell growth and cancer progression (Rohrig and Schulze, 2016). A systematic analysis of omics data from TCGA has revealed that fatty acids (FAs), arachidonic acid (AA), and cholesterol metabolism, together with peroxisome proliferator-activated receptor (PPAR) signaling came up as the top hits of altered lipid metabolism pathways (Hao et al., 2019). Furthermore, the pathways involving altered lipid uptake, storage, recycling, and de novo synthesis have been associated with cancer progression and metastasis (reviewed in detail in Mehla and Singh, 2019; Fu et al., 2020; Vasseur and Guillaumond, 2022), making lipid metabolism enzymes potentially attractive targets for cancer treatments (Figure 2).
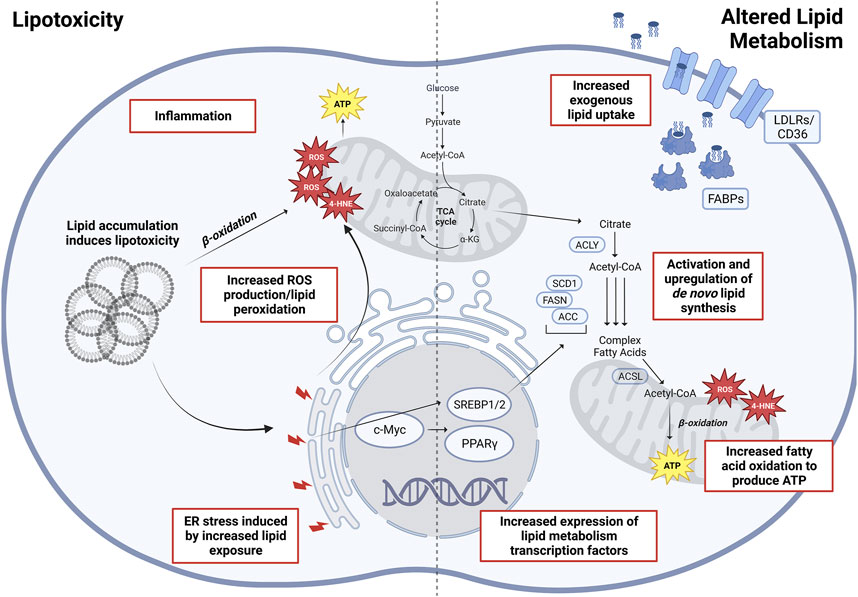
FIGURE 2. Altered lipid metabolism and lipotoxicity in oncogenesis. Depicted (Left) are various factors associated with lipid accumulation and are lipotoxicity induced, eventually increasing overall cellular stress leading to oncogenesis and malignant transformations. (Right) Main altered pathways in lipid metabolism associated with cancers. Abbreviations: ROS, reactive oxygen species; 4-HNE, 4-hydroxynonenal; SREBP1/2, sterol regulatory element–binding proteins 1/2; PPARγ, peroxisome proliferator-activated receptor gamma; ACLY, ATP-citrate lyase; SCD1, stearoyl-CoA desaturase 1; ACSL, long-chain fatty acid–CoA ligase 1; FASN, fatty acid synthase; ACC, acetyl-CoA carboxylase; FABP, fatty acid–binding proteins; LDLR, low-density lipoprotein receptor. Created with BioRender.com.
The oncogenic activation of de novo synthesis of cholesterol and FAs would allow tumor cells to rely less on exogenous uptake of lipids (Pizer et al., 1996; Roongta et al., 2011; Menendez et al., 2016). Multiple enzymes within the FA synthesis pathway which include ATP-citrate lyase (ACLY) (Bauer et al., 2005; Hatzivassiliou et al., 2005), acetyl-CoA carboxylases (ACC1/2) (Jones et al., 2017; Ye et al., 2019), FA synthase (FASN) (Flavin et al., 2010; Fhu and Ali, 2020), and stearoyl-CoA desaturase (SCD1) (Roongta et al., 2011; Mason et al., 2012; Chen et al., 2016; Luis et al., 2021) have all been found to correlate strongly with tumor growth and have been proposed as biomarkers for specific types of cancers. Modulating expression levels of these enzymes are transcription factors from the SREBP family (SREBP1/2), interacting with or modulated by key oncogenic signaling nodes which include the PI3K/AKT signaling axis (Ricoult et al., 2016; Yi et al., 2020) and c-Myc (Wu et al., 2016). Importantly, SREBP1/2 upregulations have been observed in several cancers (Bao et al., 2016; Ricoult et al., 2016), making these potential therapeutic targets that globally downregulate de novo lipid synthesis. Another transcription factor PPARγ controls the expression levels of the mediators of lipid metabolism, playing important roles in regulating FA oxidation (FAO), FA storage, and cholesterogenesis (Grygiel-Gorniak, 2014). Its downregulation has been implicated in a worse prognosis in non–small-cell lung cancer (Sasaki et al., 2002), while its higher expression is correlated with increased survival in colorectal cancer (CRC) patients (Ogino et al., 2009) and reported as a favorable prognosis marker in breast cancer (Abduljabbar et al., 2015). Conversely, tumors could at the same time often upregulate the expression of lipid uptake receptors (Kuemmerle et al., 2011). In particular, the upregulation of fatty acid transporter CD36 has been associated with many different cancer types such as oral, breast, ovarian, cervical, and gastric cancers (Pascual et al., 2017; Luo et al., 2021). Furthermore, CD36 overexpression has been associated with faster tumor growth, cancer progression, and metastasis initiation (Pascual et al., 2017; Luo et al., 2021; Guerrero-Rodriguez et al., 2022; Ruan et al., 2022).
Increases in exogenous uptake and de novo synthesis of lipids have to be complemented by concomitant increases in lipolysis, particularly to capitalize on the increased availability of lipids as a bioenergetic source. Consequently, FAO has been observed as another metabolic adaptation in tumor cells that is necessary for continued tumor growth and metastasis (aside from aerobic glycolysis and increased reliance on glutaminolysis), especially under unfavorable and possibly poorly vascularized and hypoxic tumor microenvironments (Carracedo et al., 2013; Ma et al., 2018a). Shifts in FAO reliance for the production of ATP and reducing metabolites such as NADPH, classified as a “lipolytic phenotype” (Ma et al., 2018a), can be attributed to the dysregulation of expression levels of key FAO genes such as CD36 (discussed above), carnitine palmitoyltransferase I/II (CPT1/2) (Xiong et al., 2020), carnitine transporter 2 (CT2) (Wang et al., 2021), and various classes of acyl-CoA synthetase long-chain family members (ACSLs) (Padanad et al., 2016). This lipolytic phenotype provides the metabolic flexibility of cancer cells to increase lipid availability, whether it be for energy production, membrane synthesis, or cell signaling. The reliance on FAO in cancer cells has been shown to be necessary for tumor proliferation, survival, maintenance of stemness, drug resistance, and even metastatic progression, as reviewed by Ma et al. (2018a). Considering that tumor cells could rely more on fatty acid oxidation to fuel their metabolic needs, an in vitro study found that dampening lipid metabolism with etomoxir treatment results in reduced metastatic potential and growth arrest of bladder cancer cells (Cheng et al., 2019). Notably, metastatic cells within lymph nodes are protected by oleic acid within the tumor microenvironment, conferring resistance to oxidative stress and ferroptosis that enable them to subsequently form secondary tumors (Ubellacker et al., 2020). Taken together, the various alterations to lipid metabolism in cancers is clearly integral to multiple cellular and signaling processes involved in continued tumor survival and cancer progression.
Altered lipid metabolism has a significant influence in the development of non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), and even hepatocellular carcinoma (HCC). Fatty liver disease (hepatic steatosis) is the hallmark of NAFLD/NASH and commonly the first step in the tumorigenesis cascade. Epidemiology shows that one in four in the world’s population has NAFLD at some point in their life and, alarmingly, the prevalence of this global epidemic has risen by almost 8% in the past 5 years (Younossi et al., 2019; Riazi et al., 2022). NASH is a more severe form of NAFLD, where steatosis is accompanied by inflammation and hepatocellular ballooning, with or without fibrosis (Chalasani et al., 2018). NAFLD/NASH represents a significant risk factor for HCC where 10%–20% of NAFLD/NASH patients may eventually develop HCC, overtaking viral hepatitis as the leading HCC etiology globally (Huang et al., 2021). Cancers like HCC are not simply driven by oncogenic mutations or malignancy emerged from chronic inflammation, HCC entails considerable metabolic reprogramming such as altered lipid metabolism (Anstee et al., 2019). Gross histopathology examination of resected HCC tumors often shows irregular nodules that are more yellow and soft than the adjacent normal (Schlageter et al., 2014), indicating steatosis and altered lipid metabolism. A recent characterization of the NASH-HCC transcriptome shows apparent upregulation of the pathways involved in FA metabolism (Pinyol et al., 2021). The complex metabolic and inflammatory aberrations lead to increased proliferation of hepatobiliary progenitors (Gadd et al., 2014), deranged genomic and epigenomic stability (Nishida et al., 2016), and escape from immune surveillance (Pfister et al., 2021), which ultimately drive tumorigenesis.
The development of NAFLD/NASH follows the natural trajectory of disease progression, and hyperinsulinemia (an indication of insulin resistance) correlates with HCC incidence (Chettouh et al., 2015). A high fat diet or high intake of carbohydrates and calories, in general, can lead to insulin resistance and subsequently to NAFLD/NASH or even metabolic syndrome. Insulin resistance sits at the center of metabolic syndrome, comprising abdominal obesity, hyperglycemia, dyslipidemia, and hypertension—the co-occurrence of which significantly predisposes to 2 diabetes (T2DM) and atherosclerotic cardiovascular diseases (ASCVD). While the exact causal relationships between insulin resistance and increased HCC risk is not fully understood, insulin resistance leads to increased circulating free FAs and glucose, as well as de novo lipogenesis in the hepatocytes (Smith et al., 2020). It comes as no surprise that drugs that increase insulin sensitivity, such as metformin, can improve NAFLD/NASH histology and subsequently reduce the risk of HCC (Plaz Torres et al., 2022). Synergistically, deranged free cholesterols could potentially lead to malignant progression. The accumulation of free cholesterols may be due to increased de novo synthesis (Simonen et al., 2011; Min et al., 2012), as there is a high substrate load and upregulation of HMG-CoA reductase. There seems to be a decreased absorption of plasma cholesterol (Simonen et al., 2011) and decreased LDL receptor expression (Min et al., 2012) by the hepatocytes, which may explain the dyslipidemia and increased ASCVD risk. To add on, a high fructose diet is also notorious for its effect in driving NAFLD/NASH. Fructose drives de novo lipogenesis, suppresses fatty acid oxidation, and damages mitochondrial integrity in hepatocytes, hence increasing lipid accumulation and hepatocellular damage (Yu et al., 2021a). Interestingly, genetic predisposition has been indicated in the development of lipotoxicity and hepatic steatosis. PNPLA3 polymorphism or mutation is probably the best-known genetic predisposition to NAFLD/NASH, fibrosis, and HCC (Liu et al., 2014; Dong, 2019). PNPLA3 encodes a triglyceride lipase that often binds to intracellular lipid droplets. The altered forms of PNPLA3 protein lead to impaired lipid metabolism and hence, increased lipid droplet accumulation, resulting in lipotoxicity and downstream cellular stresses (Dong, 2019). Experiments have also shown a seemingly genetic predisposition of NAFLD/NASH when abnormal hepatic differentiation is observed from patient-derived iPSCs when compared with those from healthy individuals (Kimura et al., 2022).
Further understanding of the molecular basis of NAFLD/NASH has proven to be critical to better design potential therapeutic interventions. Several transcription factors, enzymes, and transporters have been implicated in the accumulation of lipid via dysregulation of processes such as de novo lipogenesis, circulating free FAs (e.g., from diet), and adipolysis. FXR agonists (e.g., obeticholic acid) (Neuschwander-Tetri et al., 2015) and PPARγ agonists (e.g., pioglitazone) (Chalasani et al., 2018) have been shown to improve NAFLD/NASH histology, with both targets regulating lipid uptake and metabolism. Sterol response element binding protein 1c (SREBP-1c) is also a central transcription factor controlling cellular FA metabolism (Wang et al., 2015). Induced by insulin, it increases the expression of acetyl-CoA carboxylase (ACC) (Lally et al., 2019) and fatty acid synthase (FASN) (Jones and Infante, 2015), hence upregulating the production of malonyl-CoA and FAs. SREBP-1c is upregulated in the hepatocytes during insulin resistance, NAFLD/NASH, and HCC (Williams et al., 2013), indicating that de novo lipogenesis represents an indispensable source of lipid accumulation. In addition, interstitial FAs are transported into hepatocytes by fatty acid binding protein 1 (FABP1), lipid scavenger receptors, and fatty acid transport proteins (FATPs), depending on the length of the FAs. These carrier proteins may directly influence lipid accumulation and hence accelerate disease progression. While silencing of FABP1 directly ameliorates hepatic steatosis (Mukai et al., 2017), its effect on HCC development seems to depend more on increased secretion of vascular endothelial growth factor-A (VEGF-A) (Ku et al., 2016), therefore promoting angiogenesis. Of note, one of the scavenger receptor, CD36, is significantly expressed in higher levels in HCC than in adjacent normal tissues (Nath et al., 2015), which leads to increased FA uptake (Nath et al., 2015) and activates the Warburg effect via the mTOR/PI3K/AKT pathways (Luo et al., 2021).
Apart from the bioenergetics fueled by alterations in lipid metabolism, lipotoxicity could lead to intrinsic cellular stress that may also drive the oncogenic progression of NAFLD/NASH to HCC (Figure 2). Increased substrate loading, mitochondrial metabolic activity, and inflammation inevitably led to increased production of reactive oxygen species (ROS) and products of lipid peroxidation (Begriche et al., 2013; Anstee et al., 2019). The exhaustion of cellular antioxidant mechanisms in turn causes increased lipid accumulation, forming a vicious cycle. It is also well known that increased oxidative stress can lead to DNA damage and genomic/epigenomic instability, which is more prominent in NAFLD/NASH patients and those who have progressed with HCC (Tanaka et al., 2013; Nishida et al., 2016). Likewise, studies have elucidated the cross talk between endoplasmic reticulum (ER) stress and lipid metabolism. Lipid exposure leads to increased ER stress in hepatocytes and HCC cells (Cao et al., 2012), which in turn causes increased ROS production and hepatocellular damage. The MUP-uPA mouse model of NASH-HCC relies on both a high fat diet and the overexpression and ER accumulation of urokinase plasminogen activator (uPA) in the hepatocytes for tumorigenesis (Febbraio et al., 2019). ER stress also modulates lipid metabolism by activating SREBP-1c and causing oxidative stress (Egnatchik et al., 2014; Han and Kaufman, 2016). Such cross talk complements the tumorigenic role of TNF upregulation and inflammation induced by ER stress (Nakagawa et al., 2014). In addition, the accumulation of cholesterols also contributes to ER stress and mitochondrial dysfunction, resulting in hepatocellular damage and inflammation (Malhotra et al., 2020; Zhou and Sun, 2021). While the above discussion of hepatic steatosis focuses primarily on hepatocyte metabolism, the complexity of NAFLD/NASH and HCC involve an extensive remodeling of the liver microenvironment with lipid-driven fibrosis and chronic inflammation. Within the stroma, cholesterol can trigger the metabolic reprogramming of Kupffer cells (KCs) and hepatic stellate cells (HSCs) which in turn activate these cells into a pro-inflammatory state (Arguello et al., 2015). Subsequent sections of the review will further discuss the effects of lipid dysregulation on adipocytes, cancer-associated fibroblasts, immune myeloid cells, and lymphocytes.
Within the lipid-laden tumor microenvironment, the involvement of adipocytes could in part explain the association between obesity and cancer progression. Of note, adipocytes produce pro-inflammatory cytokines such as IL-6, IL-8, and TNFα (Bruun et al., 2003; Ritchie and Connell, 2007), and angiogenic factors such as VEGF, prostaglandins and leukotrienes, which not only have pro-tumorigenic correlations but have been associated with cancer progression, metastasis, and even immune evasion (Wang and DuBois, 2016; Tian et al., 2020). With obesity, the accumulation of adipocytes within the breast tumor microenvironment could also result in high collagen IV production implicating chronic inflammation and tissue fibrosis (Park and Scherer, 2012). Inhibition of hypoxia-inducible factor 1α (HIF1α) was demonstrated to prevent the activation of adipocytes leading to reduced fibrosis and inflammation during obesity (Sun et al., 2013). It was also shown that excessive adipocyte accumulation, particularly in visceral areas around tumor initiation sites, can induce a hypoxic microenvironment (Park et al., 2014b), promoting angiogenesis and contributing to cancer metastasis.
Cancer cells can secrete factors that modulate adipocyte FA metabolism, forming a reciprocal interaction which enhances the availability of FA, fueling cancer growth (Lengyel et al., 2018). For example, breast cancer cells can secrete exosomes containing pro-lipolytic factors such as miR-144 and miR-126, which stimulate lipolysis within neighboring adipocytes through the activation of AMPK and autophagy. As a result, enhanced lipolysis and the release of FAs shift the metabolic dependencies of migrating cancer cells toward increasing exogenous FA uptake and their reliance on β-oxidation for energy supply (Balaban et al., 2017). Likewise, inflammatory cytokines secreted by adipocytes were found to enhance the metastatic potential of breast cancer cells (Dirat et al., 2011). Interestingly, multiple cancers such as renal, gastric, breast and colon cancers have been shown to preferentially grow in adipocyte-rich environments. Furthermore, metastatic prostate,breast, and ovarian cancer cells preferentially home toward adipocyte tissues in the vicinity, including peri-glandular regions and the visceral omentum.
Conversely, the activation of brown adipose tissue (BAT) may have opposing roles that are beneficial to the tumor-bearing host. A recent study has demonstrated that the UCP1-dependent activation of brown adipose tissue dampens the glycolytic pathways within cancer cells to influence the growth of several cancer types as demonstrated in mouse models and a human study (Seki et al., 2022). Indeed, our current understanding is that there is an interesting functional plasticity of adipocytes in their involvement during cancer progression. Yet, the role of adipocytes remains underappreciated in the field and should be further studied in specific cancers.
An emerging cell type that has been revealed to heavily influence tumor progression and metastasis within the TME is cancer-associated fibroblasts (CAFs). These cells are known to modulate the TME through various mechanisms (reviewed by Sahai et al., 2020; Ping et al., 2021) which include, but are not limited to 1) extracellular matrix remodeling, 2) immune cross talk through chemokine and cytokine production, 3) soluble factor secretions which include angiogenic and growth factors (VEGF and HGF), microRNAs (in exosomes), and lastly, the most relevant to this discussion, 4) metabolic modulation through lactate, alanine, and aspartate shuttling and amino acid depletion (Wu et al., 2017). Apart from carbohydrate and amino acid modulations within the TME, CAFs are also known to modulate the bioavailability of lipids both directly and indirectly to fuel the metabolism of cancer cells within the often nutrient-starved TME. Of note, Santi et al. (2015) have revealed the direct transfer of lipids from CAFs to cancer cells via ectosomes, fueling cancer growth. Gong et al. (2020) also confirmed lipidomic reprogramming of CAFs in CRC having enhanced de novo FA synthesis and reduced lipid catabolism phenotype. The study further reveals FASN upregulation in CAFs, which together with CD36 upregulation in CRC cells, promotes this intimate metabolic cross talk and facilitates both CRC cell migration and growth. A complementary study done in lung cancer revealed that CAF-derived lipids, particularly oleic acid, can activate lipid metabolism in cancer cells via the upregulation of SCD1 under glucose-starved conditions. Furthermore, oleic acid transfer from CAFs could enhance cancer cell stemness (Hwang et al., 2022). Further examples of metabolic reprogramming occurring in CAFs to facilitate increased lipid availability and transfer to cancer cells include the role of pancreatic stellate cells, which upon activation, releases a wide range of lipids, such as lysophosphatidylcholines (LPCs). These LPCs go on to enhance phosphatidylcholine synthesis in pancreatic ductal adenocarcinoma (PDAC) cells, activating wound healing mediators which enhance PDAC cell proliferation and migration (Auciello et al., 2019). The emerging roles of CAFs as a lipid source fueling cancer cell growth, proliferation, and metastasis place emphasis on the requirement for further studies to dissect lipid cross talk between CAFs and cancer cells and present an additional interface of cancer therapeutic design.
Different macrophage populations possess distinct influences on homeostasis and disease beyond phagocytosis, as discovered by Elie Metchnikoff (PMID: 27477126). In 2004, the M1-M2 spectrum of macrophage polarization was proposed (Mantovani et al., 2004; Mantovani et al., 2013), and the concept of tumor-associated macrophages (TAMs) was subsequently pioneered (Mantovani et al., 2017). Indeed, macrophages can be a “friend” or “foe” in almost every step of pathogenesis and disease progression for a vast number of diseases (Mantovani et al., 2022; Wculek et al., 2022). A recent study has demonstrated that CAF reprogramming of blood monocytes converts them into lipid-associated macrophages with an immune suppressive phenotype. These macrophages are STAB1+ and TREM2high and reported to be expanded in non-responders of immune checkpoint therapy (Timperi et al., 2022).
A reciprocal relationship exists between macrophage phenotype and metabolism. On one hand, the macrophage activation pattern leads to its metabolic reprogramming. M1 macrophages adopt a glycolytic metabolic profile downstream of LPS/IFN-γ activation of toll-like receptors (TLRs) and interferon gamma receptors, along with increased induced nitric oxide synthase (iNOS) activity (Viola et al., 2019; Liu et al., 2021; Wculek et al., 2022). This is to facilitate the rapid turnover of metabolites during phagocytosis and production of pro-inflammatory cytokines and NO. FAs are rapidly synthesized and deployed for eicosanoid synthesis, while fatty acid oxidation (FAO) is blocked (Batista-Gonzalez et al., 2019; Yan and Horng, 2020). Together with lipid overload and triglyceride synthesis, there is increased intracellular lipid accumulation in the M1 state (Xiang et al., 2018; Rosas-Ballina et al., 2020; Morgan et al., 2021). By contrast, M2 macrophages activated by IL-4 primarily utilize oxidative phosphorylation (OXPHOS) and FAO (Viola et al., 2019; Wu et al., 2020; Liu et al., 2021; Wculek et al., 2022). IL-4 stimulation activates STAT6, which in turn increases the expression of lysosomal acid lipase (LAL) and scavenger receptors like CD36 (Huang et al., 2014) and MARCO (Georgoudaki et al., 2016), upregulating lipoprotein/FA uptake and lipolysis. However, FAO is not required for M2 polarization (Nomura et al., 2016), suggesting compensatory metabolic pathways that may be induced by AMPK or PPARγ (Namgaladze and Brune, 2016).
On the other hand, metabolic derangements can alter macrophage phenotype, which depends on the local environment which includes its biochemical (metabolic) environment (Mehla and Singh, 2019). The overload of saturated fatty acids (SFAs) can directly bind to TLRs and cause M1 polarization (Namgaladze and Brune, 2016). The increased accumulation of lipids (Heymann et al., 2015), ROS (Yan and Horng, 2020), and lactic acid (Colegio et al., 2014) will in turn cause cellular damage and death, causing further M1 activation. However, a lipid-/lactate-rich environment can cause both a transient “burst” of M1 polarization as well as the selection of M2 for survival (Camell and Smith, 2013; Colegio et al., 2014), which may explain the discrepancy between a pro-inflammatory secretome and an anti-inflammatory cellular profile. High glucose (Pavlou et al., 2018) and fructose (Yu et al., 2021a) exposure may also shift macrophage phenotypes. While it causes an increase in both pro-inflammatory cytokines in M1 and anti-inflammatory cytokines in M2, there is also an aberrant macrophage metabolism with reduced glycolytic capacity, implying a decrease in phagocytosis and NO production (Pavlou et al., 2018). Abnormal cholesterol metabolism has also been indicated in different disease settings but generally leads to pro-inflammatory macrophage phenotypes (Malhotra et al., 2020; Morgan et al., 2021). Finally, hypoxia may alter macrophage functioning as well, yet the precise action remains elusive (Sadiku and Walmsley, 2019).
It has been shown that macrophages in vivo adopt a spectrum of pro- and anti-inflammatory phenotypes, where a dichotomy of M1 versus M2 is an oversimplification (Liu et al., 2021). Indeed, much evidence has suggested that macrophages can possess surface markers classically defined for both M1 and M2, and their phenotypes contribute to disease progression or improvement in a spatiotemporally dependent manner. In cancer, it is recognized that these global biochemical and metabolic aberrations generally lead to a pro-inflammatory polarization at the pro-tumor necroinflammation stage, and an anti-inflammatory polarization in the full-blown tumor (Lee-Rueckert et al., 2022). The same also applies to ASCVD development (Lee-Rueckert et al., 2022), where chronic inflammation leads to M1-like cholesterol ester accumulation and foam cell formation, followed by M2-driven remodeling before acute plaque rupture and thrombosis via inflammation again (Sukhorukov et al., 2020; Wculek et al., 2022). TAMs are subject to extreme metabolic conditions such as hypoxia, nutrient deprivation, environmental acidity, and a myriad of cytokines both pro-inflammatory (TNFα, IL-1, IL-6, and IFN-γ) and anti-inflammatory (adenosine, IL-10, and TGF-β) (Mantovani et al., 2022). Hence, it is important therapeutically that we target specific metabolic pathways at different stages of tumor development. For instance, systemic glycolytic inhibitors may reduce lactic acid–driven M2 polarization yet compromising M1 activation at the same time. Although blocking lipid uptake or FAO may result in abrogation of anti-inflammatory TAM functions (Georgoudaki et al., 2016), therapeutics targeting TAM metabolism have yet a long way to go before clinical impact (Mantovani et al., 2022). Interestingly, a recent study has shown glycolytic TAMs are the major PD-L1 expressors in HCC, and PD-L1 blockade can unleash glycolysis-dependent tumoricidal activity (Lu et al., 2022). Future studies may investigate the relationship between metabolism and immune checkpoints (Yu et al., 2021b) and design novel immuno-oncology interventions powered by metabolomics (Bleve et al., 2020).
The metabolic profile of dendritic cells (DCs) in cancer bears resemblance to that of macrophages. DC activation causes a metabolic switch from OXPHOS to glycolysis (Krawczyk et al., 2010; Williford et al., 2019), with increasing lipid accumulation that may facilitate antigen presentation (Bougneres et al., 2009). However, the influence of metabolic dysregulation on DC phenotype is more prominent in tumors. The antigen processing and presentation in lipid-laden DCs are compromised, resulting in defective T-cell stimulation (Herber et al., 2010). Metabolic reprogramming of DCs from glycolysis to FAO depends on PPARγ, conferring an immunosuppressive effect similar to M2 activation (Zhao et al., 2018). Lipid peroxidation and accumulation can also lead to ER stress (Cubillos-Ruiz et al., 2015) and decreased antigen processing at the ER (Veglia et al., 2017), abrogating antitumor immunity. While neoantigen-based DC vaccine is a promising immuno-oncology strategy, it may be necessary to interrogate the extracellular or intracellular biochemical environment to avoid the negative impact by metabolic derangements on DC functions (Wculek et al., 2019; Moller et al., 2022). The use of a carnitine palmitoyltransferase-1 (CPT1) mitochondrial fatty acid transporter inhibitor—etomoxir—could be a promising candidate for combinatory therapy with conventional immune checkpoint inhibitors where the metabolic reprogramming of DCs can be abrogated to prevent polarization into a tolerogenic phenotyping that drives the generation of regulatory T cells (Zhao et al., 2018).
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous group of immunosuppressive myeloid cells that are differentiated in chronic inflammatory conditions such as cancer (Hegde et al., 2021). These cells may not possess typical granulocyte or monocyte/macrophage phenotypes and hence a monolithic view of MDSCs can see a dichotomy of polymorphonuclear (PMN) and monocyte (M) MDSCs. The current understanding has uncovered much heterogeneity of MDSCs and an alternative framework has been proposed (Hegde et al., 2021). MDSCs reprogram their metabolism in ways resembling immunosuppressive macrophages and dysfunctional DCs. MDSCs upregulate FA uptake and oxidation (Al-Khami et al., 2017), which is abrogated by FAO inhibitors that delay tumor growth and synergize with other therapies (Hossain et al., 2015). Evidence suggests that this process is mediated by inflammation, hypoxia, and environmental acidity, which in turn upregulate HIF-1α (Corzo et al., 2010), PPARγ (Xin et al., 2021), AMPK (Yan et al., 2019), and STAT3 (Ma et al., 2018b). Increased eicosanoid metabolism (Gabrilovich, 2017), characterized by increased COX-2 activity and concentrations of AA and prostaglandin E2 (Prima et al., 2017; Wang et al., 2022), reciprocally increases lipid accumulation and confers immunosuppressive activity. The nitrogen metabolism is more complex, as increase in both arginase-1 (Raber et al., 2012), a potent T-cell suppressor, and iNOS (Enioutina et al., 2011), the pro-inflammatory NO synthase, has been reported. The immunosuppressive effect is a delicate balance of mTOR/PI3K/AKT regulation as well as stimulation by Th1 or Th2 cytokines (Yan et al., 2019). Finally, ER stress may also confer MDSC immunosuppression via the PERK-NRF2 pathway (Mohamed et al., 2020). Ablating PERK, a kinase on the ER, upregulates type I interferon response and reprograms the MDSCs toward tumor-limiting myeloid cells (Mohamed et al., 2020). Taken together, metabolic reprogramming of MDSCs lead to an increased expression of myeloid checkpoints [e.g., PD-L1 (Prima et al., 2017; Xin et al., 2021) and arginase-1 (Grzywa et al., 2020)] and immunosuppressive cytokines [e.g., TGF-β (Gabrilovich, 2017), IL-10 (Grzywa et al., 2020), and PGE2 (Wang et al., 2022)]. Targeting MDSC metabolism, such as lipid transport (Al-Khami et al., 2016)/FAO inhibitors (Hossain et al., 2015; Xin et al., 2021), LXR agonists (Tavazoie et al., 2018), and COX-2 inhibitors (Prima et al., 2017), may synergize with immune checkpoint blockade and confer clinical benefit.
Studies in recent years have started to shed light on the link between obesity and antitumor immune responses. With an increasing focus on immunotherapy in cancer, there is a greater need to understand how altered lipid metabolism would influence the performance of cytotoxic lymphocytes within the tumor microenvironment.
A systematic analysis has revealed that an abundance of CD8 T cells was the most predictive parameter of a beneficial response to anti-PD-1/PD-L1 therapy across multiple cancer types (Lee and Ruppin, 2019). As exemplified in mouse models, a high-fat diet resulted in poor CD8 T-cell infiltration into several types of solid tumors. Colorectal tumors of patients with higher BMI of more than 35 were also associated with lower CD8 immune scores (Ringel et al., 2020). Our current understanding is that the shift toward lipid metabolism drives cellular stress and T-cell dysfunction. In fact, it has been elucidated that PD-1 ligation elevated FAO in T cells during immune suppression which can be modulated by conventional immune checkpoint blockade (Patsoukis et al., 2015). In addition, accumulation of LCFAs dampens mitochondrial respiratory capacity and triggers extensive transcriptional reprogramming of CD8 T cells (Manzo et al., 2020). Considering that the bioenergetics of intra-tumoral T-cells have shifted toward FAO rather than glycolysis, the activation of PPARγ enables beneficial metabolic adaptation that boosts mitochondrial functions and oxidative phosphorylation despite the altered lipid metabolism (Chowdhury et al., 2018).
As a scavenger receptor for LCFAs and oxidized LDL, CD36 is highlighted in recent studies as an emerging target for cancer therapeutics. It has been demonstrated that a high-fat diet enhances the potential of CD36+ tumor cells to initiate the formation of metastases (Pascual et al., 2017). Being both a signal transducer and lipid transporter, the expression of CD36 influences the activation of both conventional and regulatory T cells, implying that CD36 could be a potential metabolic immune checkpoint to target. The deletion of CD36 would reduce the uptake of oxidized lipids that is associated with ferroptosis and impaired CD8 T-effector functions. At the same time, CD36-triggered AMPK and PPARγ signaling could also be suppressive pathways that are distinct from metabolic stress (Chen et al., 2022). Unlike conventional T cells, regulatory T cells are programmed to express lower GLUT-1 with a higher reliance on lipid oxidation (Michalek et al., 2011). It has been demonstrated that CD36 mediates altered lipid metabolism that enables PPARβ-mediated adaptation to high lactate environment for the survival of regulatory T cells (Wang et al., 2020).
Similar to T cells, obesity has also been recently reported to influence natural killer (NK) cell–mediated antitumor immunity. In a comparison of normal weight and obese human subjects, it was found that the frequency of NKG2D+ and CD56dim cytotoxic NK cells in the peripheral blood was reduced (Bahr et al., 2018). Reduced frequencies of NK cells in the spleen and liver were also reported in obese mice which would potentially increase the risk of cancer development (Bahr et al., 2017). Lipotoxicity impairs mTOR activation in NK cells and glycolytic activity which are both essential for its effector functions. Scavenger receptors such as CD36, CD68, and MSR1 were found upregulated in dysfunctional NK cells which were reported to be induced by granulocytic MDSCs (Niavarani et al., 2019). Mechanistically, lipid-treated NK cells acquire defects in lytic granule polarization despite successfully forming an immune synapse with its tumor target (Michelet et al., 2018). Likewise, etomoxir was also reported to restore NK cell functionality under lipid stress (Michelet et al., 2018). While we previously reported that NK cells could experience cumulative oxidative stress in lung cancer patients with smoking history, it was also found that obesity increases the susceptibility of NK cells to the harmful effects of cigarette smoke (Yang et al., 2020; O’Shea et al., 2010). Obesity not only has suppressive effects on NK cell cytotoxicity but also influences the immune-regulatory functions of NK cells. An increased proportion of a NK cell subset expressing IL6Ra was observed in obese mice and humans in which transcriptomics analysis revealed an enrichment of inflammatory genes that is suggestive that these regulatory NK cells could contribute to obesity-associated inflammation (Theurich et al., 2017).
So far, the oncogenic role of obesity on the tumor-bearing host has been mostly studied in HCC where hepatic steatosis is a prominent driver of malignancy. Despite the existing vast literature on altered lipid metabolism, most studies largely focus on characterizing the metabolic changes that reprograms the bioenergetics of tumor cells. Within the gut–liver axis, the obesity-augmented microbiome and metabolites may have significant influences on the host’s immune responses during tumor progression. A recent study has reported differential bile acids and gut microbiome profiles comparing HCC responders versus non-responders to immune checkpoint blockade (Lee et al., 2022). Additionally, the gut microbiome influences levels of short-chain fatty acids that influence the functional plasticity of hepatic immune cells (Hu et al., 2023). Given that obesity is often implicated in chronic inflammation, future studies should seek to understand how obesity influences the systemic immune responses of the tumor-bearing host. Ultimately, these novel insights on metabolism could change existing treatment and lifestyle modification paradigms to provide the next breakthroughs in oncology.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work is supported by the National Research Foundation of Singapore (NRF-CRP26-2021RS-0001) and the National Medical Research Council of Singapore (NMRC/OFLCG/003/2018).
The authors thank all funding agencies for supporting their research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, editors, and reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abduljabbar, R., Al-Kaabi, M. M., Negm, O. H., Jerjees, D., Muftah, A. A., Mukherjee, A., et al. (2015). Prognostic and biological significance of peroxisome proliferator-activated receptor-gamma in luminal breast cancer. Breast Cancer Res. Treat. 150 (3), 511–522. doi:10.1007/s10549-015-3348-9
Al-Khami, A. A., Rodriguez, P. C., and Ochoa, A. C. (2016). Metabolic reprogramming of myeloid-derived suppressor cells (MDSC) in cancer. Oncoimmunology 5 (8), e1200771. doi:10.1080/2162402X.2016.1200771
Al-Khami, A. A., Zheng, L., Del Valle, L., Hossain, F., Wyczechowska, D., Zabaleta, J., et al. (2017). Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 6 (10), e1344804. doi:10.1080/2162402X.2017.1344804
Anstee, Q. M., Reeves, H. L., Kotsiliti, E., Govaere, O., and Heikenwalder, M. (2019). From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 16 (7), 411–428. doi:10.1038/s41575-019-0145-7
Arguello, G., Balboa, E., Arrese, M., and Zanlungo, S. (2015). Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta 1852 (9), 1765–1778. doi:10.1016/j.bbadis.2015.05.015
Auciello, F. R., Bulusu, V., Oon, C., Tait-Mulder, J., Berry, M., Bhattacharyya, S., et al. (2019). A stromal lysolipid-autotaxin signaling Axis promotes pancreatic tumor progression. Cancer Discov. 9 (5), 617–627. doi:10.1158/2159-8290.CD-18-1212
Avgerinos, K. I., Spyrou, N., Mantzoros, C. S., and Dalamaga, M. (2019). Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 92, 121–135. doi:10.1016/j.metabol.2018.11.001
Bahr, I., Goritz, V., Doberstein, H., Hiller, G. G., Rosenstock, P., Jahn, J., et al. (2017). Diet-induced obesity is associated with an impaired NK cell function and an increased colon cancer incidence. J. Nutr. Metab. 2017, 4297025. doi:10.1155/2017/4297025
Bahr, I., Jahn, J., Zipprich, A., Pahlow, I., Spielmann, J., and Kielstein, H. (2018). Impaired natural killer cell subset phenotypes in human obesity. Immunol. Res. 66 (2), 234–244. doi:10.1007/s12026-018-8989-4
Balaban, S., Shearer, R. F., Lee, L. S., van Geldermalsen, M., Schreuder, M., Shtein, H. C., et al. (2017). Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 5, 1. doi:10.1186/s40170-016-0163-7
Bao, J., Zhu, L., Zhu, Q., Su, J., Liu, M., and Huang, W. (2016). SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 12 (4), 2409–2416. doi:10.3892/ol.2016.4988
Batista-Gonzalez, A., Vidal, R., Criollo, A., and Carreno, L. J. (2019). New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front. Immunol. 10, 2993. doi:10.3389/fimmu.2019.02993
Bauer, D. E., Hatzivassiliou, G., Zhao, F., Andreadis, C., and Thompson, C. B. (2005). ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24 (41), 6314–6322. doi:10.1038/sj.onc.1208773
Begriche, K., Massart, J., Robin, M. A., Bonnet, F., and Fromenty, B. (2013). Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58 (4), 1497–1507. doi:10.1002/hep.26226
Bleve, A., Durante, B., Sica, A., and Consonni, F. M. (2020). Lipid metabolism and cancer immunotherapy: Immunosuppressive myeloid cells at the crossroad. Int. J. Mol. Sci. 21 (16), 5845. doi:10.3390/ijms21165845
Bougneres, L., Helft, J., Tiwari, S., Vargas, P., Chang, B. H., Chan, L., et al. (2009). A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity 31 (2), 232–244. doi:10.1016/j.immuni.2009.06.022
Bruun, J. M., Lihn, A. S., Verdich, C., Pedersen, S. B., Toubro, S., Astrup, A., et al. (2003). Regulation of adiponectin by adipose tissue-derived cytokines: In vivo and in vitro investigations in humans. Am. J. Physiol. Endocrinol. Metab. 285 (3), E527–E533. doi:10.1152/ajpendo.00110.2003
Camell, C., and Smith, C. W. (2013). Dietary oleic acid increases M2 macrophages in the mesenteric adipose tissue. PLoS One 8 (9), e75147. doi:10.1371/journal.pone.0075147
Cao, J., Dai, D. L., Yao, L., Yu, H. H., Ning, B., Zhang, Q., et al. (2012). Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell Biochem. 364 (1-2), 115–129. doi:10.1007/s11010-011-1211-9
Carracedo, A., Cantley, L. C., and Pandolfi, P. P. (2013). Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 13 (4), 227–232. doi:10.1038/nrc3483
Chalasani, N., Younossi, Z., Lavine, J. E., Charlton, M., Cusi, K., Rinella, M., et al. (2018). The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67 (1), 328–357. doi:10.1002/hep.29367
Chen, L., Ren, J., Yang, L., Li, Y., Fu, J., Li, Y., et al. (2016). Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci. Rep. 6, 19665. doi:10.1038/srep19665
Chen, Y., Zhang, J., Cui, W., and Silverstein, R. L. (2022). CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 219 (6), e20211314. doi:10.1084/jem.20211314
Cheng, S., Wang, G., Wang, Y., Cai, L., Qian, K., Ju, L., et al. (2019). Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARγ-mediated pathway in bladder cancer. Clin. Sci. (Lond). 133 (15), 1745–1758. doi:10.1042/CS20190587
Chettouh, H., Lequoy, M., Fartoux, L., Vigouroux, C., and Desbois-Mouthon, C. (2015). Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int. 35 (10), 2203–2217. doi:10.1111/liv.12903
Chowdhury, P. S., Chamoto, K., Kumar, A., and Honjo, T. (2018). PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8(+) T cells and facilitates anti-PD-1 therapy. Cancer Immunol. Res. 6 (11), 1375–1387. doi:10.1158/2326-6066.CIR-18-0095
Colegio, O. R., Chu, N. Q., Szabo, A. L., Chu, T., Rhebergen, A. M., Jairam, V., et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513 (7519), 559–563. doi:10.1038/nature13490
Corzo, C. A., Condamine, T., Lu, L., Cotter, M. J., Youn, J. I., Cheng, P., et al. (2010). HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 207 (11), 2439–2453. doi:10.1084/jem.20100587
Cubillos-Ruiz, J. R., Silberman, P. C., Rutkowski, M. R., Chopra, S., Perales-Puchalt, A., Song, M., et al. (2015). ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161 (7), 1527–1538. doi:10.1016/j.cell.2015.05.025
Dirat, B., Bochet, L., Dabek, M., Daviaud, D., Dauvillier, S., Majed, B., et al. (2011). Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 71 (7), 2455–2465. doi:10.1158/0008-5472.CAN-10-3323
Dobbins, M., Decorby, K., and Choi, B. C. (2013). The association between obesity and cancer risk: A meta-analysis of observational studies from 1985 to 2011. ISRN Prev. Med. 2013, 680536. doi:10.5402/2013/680536
Dong, X. C. (2019). PNPLA3-A potential therapeutic target for personalized treatment of chronic liver disease. Front. Med. (Lausanne). 6, 304. doi:10.3389/fmed.2019.00304
Egnatchik, R. A., Leamy, A. K., Jacobson, D. A., Shiota, M., and Young, J. D. (2014). ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol. Metab. 3 (5), 544–553. doi:10.1016/j.molmet.2014.05.004
Enioutina, E. Y., Bareyan, D., and Daynes, R. A. (2011). A role for immature myeloid cells in immune senescence. J. Immunol. 186 (2), 697–707. doi:10.4049/jimmunol.1002987
Febbraio, M. A., Reibe, S., Shalapour, S., Ooi, G. J., Watt, M. J., and Karin, M. (2019). Preclinical models for studying NASH-driven HCC: How useful are they? Cell Metab. 29 (1), 18–26. doi:10.1016/j.cmet.2018.10.012
Fhu, C. W., and Ali, A. (2020). Fatty acid synthase: An emerging target in cancer. Molecules 25 (17), 3935. doi:10.3390/molecules25173935
Flavin, R., Peluso, S., Nguyen, P. L., and Loda, M. (2010). Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 6 (4), 551–562. doi:10.2217/fon.10.11
Fu, Y., Zou, T., Shen, X., Nelson, P. J., Li, J., Wu, C., et al. (2020). Lipid metabolism in cancer progression and therapeutic strategies. MedComm 2 (1), 27–59. doi:10.1002/mco2.27
Gabrilovich, D. I. (2017). Myeloid-derived suppressor cells. Cancer Immunol. Res. 5 (1), 3–8. doi:10.1158/2326-6066.CIR-16-0297
Gadd, V. L., Skoien, R., Powell, E. E., Fagan, K. J., Winterford, C., Horsfall, L., et al. (2014). The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 59 (4), 1393–1405. doi:10.1002/hep.26937
Georgoudaki, A. M., Prokopec, K. E., Boura, V. F., Hellqvist, E., Sohn, S., Ostling, J., et al. (2016). Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 15 (9), 2000–2011. doi:10.1016/j.celrep.2016.04.084
Gong, J., Lin, Y., Zhang, H., Liu, C., Cheng, Z., Yang, X., et al. (2020). Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 11 (4), 267. doi:10.1038/s41419-020-2434-z
Grygiel-Gorniak, B. (2014). Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications--a review. Nutr. J. 13, 17. doi:10.1186/1475-2891-13-17
Grzywa, T. M., Sosnowska, A., Matryba, P., Rydzynska, Z., Jasinski, M., Nowis, D., et al. (2020). Myeloid cell-derived arginase in cancer immune response. Front. Immunol. 11, 938. doi:10.3389/fimmu.2020.00938
Guerrero-Rodriguez, S. L., Mata-Cruz, C., Perez-Tapia, S. M., and Velasco-Velazquez, M. A. (2022). Role of CD36 in cancer progression, stemness, and targeting. Front. Cell Dev. Biol. 10, 1079076. doi:10.3389/fcell.2022.1079076
Han, J., and Kaufman, R. J. (2016). The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 57 (8), 1329–1338. doi:10.1194/jlr.R067595
Hao, Y., Li, D., Xu, Y., Ouyang, J., Wang, Y., Zhang, Y., et al. (2019). Investigation of lipid metabolism dysregulation and the effects on immune microenvironments in pan-cancer using multiple omics data. BMC Bioinforma. 20 (7), 195. doi:10.1186/s12859-019-2734-4
Hatzivassiliou, G., Zhao, F., Bauer, D. E., Andreadis, C., Shaw, A. N., Dhanak, D., et al. (2005). ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8 (4), 311–321. doi:10.1016/j.ccr.2005.09.008
Hegde, S., Leader, A. M., and Merad, M. (2021). Mdsc: Markers, development, states, and unaddressed complexity. Immunity 54 (5), 875–884. doi:10.1016/j.immuni.2021.04.004
Herber, D. L., Cao, W., Nefedova, Y., Novitskiy, S. V., Nagaraj, S., Tyurin, V. A., et al. (2010). Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16 (8), 880–886. doi:10.1038/nm.2172
Heymann, F., Peusquens, J., Ludwig-Portugall, I., Kohlhepp, M., Ergen, C., Niemietz, P., et al. (2015). Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 62 (1), 279–291. doi:10.1002/hep.27793
Hossain, F., Al-Khami, A. A., Wyczechowska, D., Hernandez, C., Zheng, L., Reiss, K., et al. (2015). Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 3 (11), 1236–1247. doi:10.1158/2326-6066.CIR-15-0036
Hu, C., Xu, B., Wang, X., Wan, W. H., Lu, J., Kong, D., et al. (2023). Gut microbiota-derived short-chain fatty acids regulate group 3 innate lymphoid cells in HCC. Hepatology 77 (1), 48–64. doi:10.1002/hep.32449
Huang, D. Q., El-Serag, H. B., and Loomba, R. (2021). Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 18 (4), 223–238. doi:10.1038/s41575-020-00381-6
Huang, S. C., Everts, B., Ivanova, Y., O'Sullivan, D., Nascimento, M., Smith, A. M., et al. (2014). Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 15 (9), 846–855. doi:10.1038/ni.2956
Hwang, S. H., Yang, Y., Jung, J. H., and Kim, Y. (2022). Oleic acid from cancer-associated fibroblast promotes cancer cell stemness by stearoyl-CoA desaturase under glucose-deficient condition. Cancer Cell Int. 22 (1), 404. doi:10.1186/s12935-022-02824-3
Jones, J. E., Esler, W. P., Patel, R., Lanba, A., Vera, N. B., Pfefferkorn, J. A., et al. (2017). Inhibition of acetyl-CoA carboxylase 1 (ACC1) and 2 (ACC2) reduces proliferation and de novo lipogenesis of EGFRvIII human glioblastoma cells. PLoS One 12 (1), e0169566. doi:10.1371/journal.pone.0169566
Jones, S. F., and Infante, J. R. (2015). Molecular pathways: Fatty acid synthase. Clin. Cancer Res. 21 (24), 5434–5438. doi:10.1158/1078-0432.CCR-15-0126
Kimura, M., Iguchi, T., Iwasawa, K., Dunn, A., Thompson, W. L., Yoneyama, Y., et al. (2022). En masse organoid phenotyping informs metabolic-associated genetic susceptibility to NASH. Cell 185 (22), 4216–4232.e16. doi:10.1016/j.cell.2022.09.031
Krawczyk, C. M., Holowka, T., Sun, J., Blagih, J., Amiel, E., DeBerardinis, R. J., et al. (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115 (23), 4742–4749. doi:10.1182/blood-2009-10-249540
Ku, C. Y., Liu, Y. H., Lin, H. Y., Lu, S. C., and Lin, J. Y. (2016). Liver fatty acid-binding protein (L-FABP) promotes cellular angiogenesis and migration in hepatocellular carcinoma. Oncotarget 7 (14), 18229–18246. doi:10.18632/oncotarget.7571
Kuemmerle, N. B., Rysman, E., Lombardo, P. S., Flanagan, A. J., Lipe, B. C., Wells, W. A., et al. (2011). Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 10 (3), 427–436. doi:10.1158/1535-7163.MCT-10-0802
Lally, J. S. V., Ghoshal, S., DePeralta, D. K., Moaven, O., Wei, L., Masia, R., et al. (2019). Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 29 (1), 174–182.e5. doi:10.1016/j.cmet.2018.08.020
Lee, J. S., and Ruppin, E. (2019). Multiomics prediction of response rates to therapies to inhibit programmed cell death 1 and programmed cell death 1 ligand 1. JAMA Oncol. 5, 1614–1618. doi:10.1001/jamaoncol.2019.2311
Lee, P. C., Wu, C. J., Hung, Y. W., Lee, C. J., Chi, C. T., Lee, I. C., et al. (2022). Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor-treated unresectable hepatocellular carcinoma. J. Immunother. Cancer 10 (6), e004779. doi:10.1136/jitc-2022-004779
Lee-Rueckert, M., Lappalainen, J., Kovanen, P. T., and Escola-Gil, J. C. (2022). Lipid-laden macrophages and inflammation in atherosclerosis and cancer: An integrative view. Front. Cardiovasc Med. 9, 777822. doi:10.3389/fcvm.2022.777822
Lengyel, E., Makowski, L., DiGiovanni, J., and Kolonin, M. G. (2018). Cancer as a matter of fat: The crosstalk between adipose tissue and tumors. Trends Cancer 4 (5), 374–384. doi:10.1016/j.trecan.2018.03.004
Liu, Y., Xu, R., Gu, H., Zhang, E., Qu, J., Cao, W., et al. (2021). Metabolic reprogramming in macrophage responses. Biomark. Res. 9 (1), 1. doi:10.1186/s40364-020-00251-y
Liu, Y. L., Patman, G. L., Leathart, J. B., Piguet, A. C., Burt, A. D., Dufour, J. F., et al. (2014). Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 61 (1), 75–81. doi:10.1016/j.jhep.2014.02.030
Liu, Z., Zhang, T. T., Zhao, J. J., Qi, S. F., Du, P., Liu, D. W., et al. (2015). The association between overweight, obesity and ovarian cancer: A meta-analysis. Jpn. J. Clin. Oncol. 45 (12), 1107–1115. doi:10.1093/jjco/hyv150
Lu, L. G., Zhou, Z. L., Wang, X. Y., Liu, B. Y., Lu, J. Y., Liu, S., et al. (2022). PD-L1 blockade liberates intrinsic antitumourigenic properties of glycolytic macrophages in hepatocellular carcinoma. Gut 71 (12), 2551–2560. doi:10.1136/gutjnl-2021-326350
Luis, G., Godfroid, A., Nishiumi, S., Cimino, J., Blacher, S., Maquoi, E., et al. (2021). Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 43, 102006. doi:10.1016/j.redox.2021.102006
Luo, X., Zheng, E., Wei, L., Zeng, H., Qin, H., Zhang, X., et al. (2021). The fatty acid receptor CD36 promotes HCC progression through activating Src/PI3K/AKT axis-dependent aerobic glycolysis. Cell Death Dis. 12 (4), 328. doi:10.1038/s41419-021-03596-w
Ma, J., Xu, H., and Wang, S. (2018). Immunosuppressive role of myeloid-derived suppressor cells and therapeutic targeting in lung cancer. J. Immunol. Res. 2018, 6319649. doi:10.1155/2018/6319649
Ma, Y., Temkin, S. M., Hawkridge, A. M., Guo, C., Wang, W., Wang, X. Y., et al. (2018). Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 435, 92–100. doi:10.1016/j.canlet.2018.08.006
Malhotra, P., Gill, R. K., Saksena, S., and Alrefai, W. A. (2020). Disturbances in cholesterol homeostasis and non-alcoholic fatty liver diseases. Front. Med. (Lausanne). 7, 467. doi:10.3389/fmed.2020.00467
Mantovani, A., Allavena, P., Marchesi, F., and Garlanda, C. (2022). Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 21 (11), 799–820. doi:10.1038/s41573-022-00520-5
Mantovani, A., Biswas, S. K., Galdiero, M. R., Sica, A., and Locati, M. (2013). Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 229 (2), 176–185. doi:10.1002/path.4133
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L., and Allavena, P. (2017). Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14 (7), 399–416. doi:10.1038/nrclinonc.2016.217
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25 (12), 677–686. doi:10.1016/j.it.2004.09.015
Manzo, T., Prentice, B. M., Anderson, K. G., Raman, A., Schalck, A., Codreanu, G. S., et al. (2020). Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J. Exp. Med. 217 (8), e20191920. doi:10.1084/jem.20191920
Mason, P., Liang, B., Li, L., Fremgen, T., Murphy, E., Quinn, A., et al. (2012). SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS One 7 (3), e33823. doi:10.1371/journal.pone.0033823
Mehla, K., and Singh, P. K. (2019). Metabolic regulation of macrophage polarization in cancer. Trends Cancer 5 (12), 822–834. doi:10.1016/j.trecan.2019.10.007
Menendez, J. A., Vellon, L., Espinoza, I., and Lupu, R. (2016). The metastasis inducer CCN1 (CYR61) activates the fatty acid synthase (FASN)-driven lipogenic phenotype in breast cancer cells. Oncoscience 3 (7-8), 242–257. doi:10.18632/oncoscience.314
Michalek, R. D., Gerriets, V. A., Jacobs, S. R., Macintyre, A. N., MacIver, N. J., Mason, E. F., et al. (2011). Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186 (6), 3299–3303. doi:10.4049/jimmunol.1003613
Michelet, X., Dyck, L., Hogan, A., Loftus, R. M., Duquette, D., Wei, K., et al. (2018). Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 19 (12), 1330–1340. doi:10.1038/s41590-018-0251-7
Min, H. K., Kapoor, A., Fuchs, M., Mirshahi, F., Zhou, H., Maher, J., et al. (2012). Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 15 (5), 665–674. doi:10.1016/j.cmet.2012.04.004
Mohamed, E., Sierra, R. A., Trillo-Tinoco, J., Cao, Y., Innamarato, P., Payne, K. K., et al. (2020). The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity 52 (4), 668–682.e7. doi:10.1016/j.immuni.2020.03.004
Moller, S. H., Wang, L., and Ho, P. C. (2022). Metabolic programming in dendritic cells tailors immune responses and homeostasis. Cell Mol. Immunol. 19 (3), 370–383. doi:10.1038/s41423-021-00753-1
Morgan, P. K., Huynh, K., Pernes, G., Miotto, P. M., Mellett, N. A., Giles, C., et al. (2021). Macrophage polarization state affects lipid composition and the channeling of exogenous fatty acids into endogenous lipid pools. J. Biol. Chem. 297 (6), 101341. doi:10.1016/j.jbc.2021.101341
Mukai, T., Egawa, M., Takeuchi, T., Yamashita, H., and Kusudo, T. (2017). Silencing of FABP1 ameliorates hepatic steatosis, inflammation, and oxidative stress in mice with nonalcoholic fatty liver disease. FEBS Open Bio 7 (7), 1009–1016. doi:10.1002/2211-5463.12240
Nakagawa, H., Umemura, A., Taniguchi, K., Font-Burgada, J., Dhar, D., Ogata, H., et al. (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26 (3), 331–343. doi:10.1016/j.ccr.2014.07.001
Namgaladze, D., and Brune, B. (2016). Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim. Biophys. Acta 1861 (11), 1796–1807. doi:10.1016/j.bbalip.2016.09.002
Nath, A., Li, I., Roberts, L. R., and Chan, C. (2015). Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 5, 14752. doi:10.1038/srep14752
Neuschwander-Tetri, B. A., Loomba, R., Sanyal, A. J., Lavine, J. E., Van Natta, M. L., Abdelmalek, M. F., et al. (2015). Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 385 (9972), 956–965. doi:10.1016/S0140-6736(14)61933-4
Niavarani, S. R., Lawson, C., Bakos, O., Boudaud, M., Batenchuk, C., Rouleau, S., et al. (2019). Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer 19 (1), 823. doi:10.1186/s12885-019-6045-y
Nieman, K. M., Romero, I. L., Van Houten, B., and Lengyel, E. (2013). Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 1831 (10), 1533–1541. doi:10.1016/j.bbalip.2013.02.010
Nishida, N., Yada, N., Hagiwara, S., Sakurai, T., Kitano, M., and Kudo, M. (2016). Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 31 (9), 1646–1653. doi:10.1111/jgh.13318
Nomura, M., Liu, J., Rovira, , Gonzalez-Hurtado, E., Lee, J., Wolfgang, M. J., et al. (2016). Fatty acid oxidation in macrophage polarization. Nat. Immunol. 17 (3), 216–217. doi:10.1038/ni.3366
O'Shea, D., Cawood, T. J., O'Farrelly, C., and Lynch, L. (2010). Natural killer cells in obesity: Impaired function and increased susceptibility to the effects of cigarette smoke. PLoS One 5 (1), e8660. doi:10.1371/journal.pone.0008660
Ogino, S., Shima, K., Baba, Y., Nosho, K., Irahara, N., Kure, S., et al. (2009). Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology 136 (4), 1242–1250. doi:10.1053/j.gastro.2008.12.048
Padanad, M. S., Konstantinidou, G., Venkateswaran, N., Melegari, M., Rindhe, S., Mitsche, M., et al. (2016). Fatty acid oxidation mediated by acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 16 (6), 1614–1628. doi:10.1016/j.celrep.2016.07.009
Parekh, N., Chandran, U., and Bandera, E. V. (2012). Obesity in cancer survival. Annu. Rev. Nutr. 32, 311–342. doi:10.1146/annurev-nutr-071811-150713
Park, J., Cho, S. Y., Lee, S. B., Son, H., and Jeong, H. (2014). Obesity is associated with higher risk of prostate cancer detection in a biopsy population in Korea. BJU Int. 114 (6), 891–895. doi:10.1111/bju.12600
Park, J., Morley, T. S., Kim, M., Clegg, D. J., and Scherer, P. E. (2014). Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 10 (8), 455–465. doi:10.1038/nrendo.2014.94
Park, J., and Scherer, P. E. (2012). Adipocyte-derived endotrophin promotes malignant tumor progression. J. Clin. Invest. 122 (11), 4243–4256. doi:10.1172/JCI63930
Pascual, G., Avgustinova, A., Mejetta, S., Martin, M., Castellanos, A., Attolini, C. S., et al. (2017). Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541 (7635), 41–45. doi:10.1038/nature20791
Patsoukis, N., Bardhan, K., Chatterjee, P., Sari, D., Liu, B., Bell, L. N., et al. (2015). PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692. doi:10.1038/ncomms7692
Pavlou, S., Lindsay, J., Ingram, R., Xu, H., and Chen, M. (2018). Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol. 19 (1), 24. doi:10.1186/s12865-018-0261-0
Pfister, D., Nunez, N. G., Pinyol, R., Govaere, O., Pinter, M., Szydlowska, M., et al. (2021). NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 592 (7854), 450–456. doi:10.1038/s41586-021-03362-0
Ping, Q., Yan, R., Cheng, X., Wang, W., Zhong, Y., Hou, Z., et al. (2021). Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 28 (9), 984–999. doi:10.1038/s41417-021-00318-4
Pinyol, R., Torrecilla, S., Wang, H., Montironi, C., Pique-Gili, M., Torres-Martin, M., et al. (2021). Corrigendum to 'Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis'. J. Hepatol. 75, 1515. doi:10.1016/j.jhep.2021.09.014
Pizer, E. S., Jackisch, C., Wood, F. D., Pasternack, G. R., Davidson, N. E., and Kuhajda, F. P. (1996). Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res. 56 (12), 2745–2747.
Plaz Torres, M. C., Jaffe, A., Perry, R., Marabotto, E., Strazzabosco, M., and Giannini, E. G. (2022). Diabetes medications and risk of HCC. Hepatology 76 (6), 1880–1897. doi:10.1002/hep.32439
Prima, V., Kaliberova, L. N., Kaliberov, S., Curiel, D. T., and Kusmartsev, S. (2017). COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. U. S. A. 114 (5), 1117–1122. doi:10.1073/pnas.1612920114
Raber, P., Ochoa, A. C., and Rodriguez, P. C. (2012). Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol. Invest. 41 (6-7), 614–634. doi:10.3109/08820139.2012.680634
Riazi, K., Azhari, H., Charette, J. H., Underwood, F. E., King, J. A., Afshar, E. E., et al. (2022). The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 7 (9), 851–861. doi:10.1016/S2468-1253(22)00165-0
Ricoult, S. J., Yecies, J. L., Ben-Sahra, I., and Manning, B. D. (2016). Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 35 (10), 1250–1260. doi:10.1038/onc.2015.179
Ringel, A. E., Drijvers, J. M., Baker, G. J., Catozzi, A., Garcia-Canaveras, J. C., Gassaway, B. M., et al. (2020). Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell 183 (7), 1848–1866.e26. doi:10.1016/j.cell.2020.11.009
Ritchie, S. A., and Connell, J. M. (2007). The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr. Metab. Cardiovasc Dis. 17 (4), 319–326. doi:10.1016/j.numecd.2006.07.005
Rohrig, F., and Schulze, A. (2016). The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 16 (11), 732–749. doi:10.1038/nrc.2016.89
Roongta, U. V., Pabalan, J. G., Wang, X., Ryseck, R. P., Fargnoli, J., Henley, B. J., et al. (2011). Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res. 9 (11), 1551–1561. doi:10.1158/1541-7786.MCR-11-0126
Rosas-Ballina, M., Guan, X. L., Schmidt, A., and Bumann, D. (2020). Classical Activation of Macrophages Leads to Lipid Droplet Formation Without de novo Fatty Acid Synthesis. Front. Immunol. 11, 131. doi:10.3389/fimmu.2020.00131
Ruan, C., Meng, Y., and Song, H. (2022). CD36: An emerging therapeutic target for cancer and its molecular mechanisms. J. Cancer Res. Clin. Oncol. 148 (7), 1551–1558. doi:10.1007/s00432-022-03957-8
Sadiku, P., and Walmsley, S. R. (2019). Hypoxia and the regulation of myeloid cell metabolic imprinting: Consequences for the inflammatory response. EMBO Rep. 20 (5), e47388. doi:10.15252/embr.201847388
Sahai, E., Astsaturov, I., Cukierman, E., DeNardo, D. G., Egeblad, M., Evans, R. M., et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20 (3), 174–186. doi:10.1038/s41568-019-0238-1
Santi, A., Caselli, A., Ranaldi, F., Paoli, P., Mugnaioni, C., Michelucci, E., et al. (2015). Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim. Biophys. Acta 1853 (12), 3211–3223. doi:10.1016/j.bbamcr.2015.09.013
Sasaki, H., Tanahashi, M., Yukiue, H., Moiriyama, S., Kobayashi, Y., Nakashima, Y., et al. (2002). Decreased perioxisome proliferator-activated receptor gamma gene expression was correlated with poor prognosis in patients with lung cancer. Lung Cancer 36 (1), 71–76. doi:10.1016/s0169-5002(01)00449-4
Schlageter, M., Terracciano, L. M., D'Angelo, S., and Sorrentino, P. (2014). Histopathology of hepatocellular carcinoma. World J. Gastroenterol. 20 (43), 15955–15964. doi:10.3748/wjg.v20.i43.15955
Seki, T., Yang, Y., Sun, X., Lim, S., Xie, S., Guo, Z., et al. (2022). Brown-fat-mediated tumour suppression by cold-altered global metabolism. Nature 608 (7922), 421–428. doi:10.1038/s41586-022-05030-3
Simonen, P., Kotronen, A., Hallikainen, M., Sevastianova, K., Makkonen, J., Hakkarainen, A., et al. (2011). Cholesterol synthesis is increased and absorption decreased in non-alcoholic fatty liver disease independent of obesity. J. Hepatol. 54 (1), 153–159. doi:10.1016/j.jhep.2010.05.037
Smith, G. I., Shankaran, M., Yoshino, M., Schweitzer, G. G., Chondronikola, M., Beals, J. W., et al. (2020). Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Invest. 130 (3), 1453–1460. doi:10.1172/JCI134165
Sukhorukov, V. N., Khotina, V. A., Chegodaev, Y. S., Ivanova, E., Sobenin, I. A., and Orekhov, A. N. (2020). Lipid metabolism in macrophages: Focus on atherosclerosis. Biomedicines 8 (8), 262. doi:10.3390/biomedicines8080262
Sun, K., Halberg, N., Khan, M., Magalang, U. J., and Scherer, P. E. (2013). Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol. Cell Biol. 33 (5), 904–917. doi:10.1128/MCB.00951-12
Tanaka, S., Miyanishi, K., Kobune, M., Kawano, Y., Hoki, T., Kubo, T., et al. (2013). Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J. Gastroenterol. 48 (11), 1249–1258. doi:10.1007/s00535-012-0739-0
Tavazoie, M. F., Pollack, I., Tanqueco, R., Ostendorf, B. N., Reis, B. S., Gonsalves, F. C., et al. (2018). LXR/ApoE activation restricts innate immune suppression in cancer. Cell 172 (4), 825–840.e18. doi:10.1016/j.cell.2017.12.026
Theurich, S., Tsaousidou, E., Hanssen, R., Lempradl, A. M., Mauer, J., Timper, K., et al. (2017). IL-6/Stat3-Dependent induction of a distinct, obesity-associated NK cell subpopulation deteriorates energy and glucose homeostasis. Cell Metab. 26 (1), 171–184.e6. doi:10.1016/j.cmet.2017.05.018
Tian, W., Jiang, X., Kim, D., Guan, T., Nicolls, M. R., and Rockson, S. G. (2020). Leukotrienes in tumor-associated inflammation. Front. Pharmacol. 11, 1289. doi:10.3389/fphar.2020.01289
Timperi, E., Gueguen, P., Molgora, M., Magagna, I., Kieffer, Y., Lopez-Lastra, S., et al. (2022). Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 82 (18), 3291–3306. doi:10.1158/0008-5472.CAN-22-1427
Ubellacker, J. M., Tasdogan, A., Ramesh, V., Shen, B., Mitchell, E. C., Martin-Sandoval, M. S., et al. (2020). Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585 (7823), 113–118. doi:10.1038/s41586-020-2623-z
Vasseur, S., and Guillaumond, F. (2022). Lipids in cancer: A global view of the contribution of lipid pathways to metastatic formation and treatment resistance. Oncogenesis 11 (1), 46. doi:10.1038/s41389-022-00420-8
Veglia, F., Tyurin, V. A., Mohammadyani, D., Blasi, M., Duperret, E. K., Donthireddy, L., et al. (2017). Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 8 (1), 2122. doi:10.1038/s41467-017-02186-9
Viola, A., Munari, F., Sanchez-Rodriguez, R., Scolaro, T., and Castegna, A. (2019). The metabolic signature of macrophage responses. Front. Immunol. 10, 1462. doi:10.3389/fimmu.2019.01462
Wang, D., and DuBois, R. N. (2016). The role of prostaglandin E(2) in tumor-associated immunosuppression. Trends Mol. Med. 22 (1), 1–3. doi:10.1016/j.molmed.2015.11.003
Wang, H., Franco, F., Tsui, Y. C., Xie, X., Trefny, M. P., Zappasodi, R., et al. (2020). CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 21 (3), 298–308. doi:10.1038/s41590-019-0589-5
Wang, M., Wang, K., Liao, X., Hu, H., Chen, L., Meng, L., et al. (2021). Carnitine palmitoyltransferase system: A new target for anti-inflammatory and anticancer therapy? Front. Pharmacol. 12, 760581. doi:10.3389/fphar.2021.760581
Wang, Q., Zhang, X., Li, C., Xiong, M., Bai, W., Sun, S., et al. (2022). Intracellular lipid accumulation drives the differentiation of decidual polymorphonuclear myeloid-derived suppressor cells via arachidonic acid metabolism. Front. Immunol. 13, 868669. doi:10.3389/fimmu.2022.868669
Wang, Y., Viscarra, J., Kim, S. J., and Sul, H. S. (2015). Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 16 (11), 678–689. doi:10.1038/nrm4074
Wculek, S. K., Dunphy, G., Heras-Murillo, I., Mastrangelo, A., and Sancho, D. (2022). Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol. Immunol. 19 (3), 384–408. doi:10.1038/s41423-021-00791-9
Wculek, S. K., Khouili, S. C., Priego, E., Heras-Murillo, I., and Sancho, D. (2019). Metabolic control of dendritic cell functions: Digesting information. Front. Immunol. 10, 775. doi:10.3389/fimmu.2019.00775
Williams, K. J., Argus, J. P., Zhu, Y., Wilks, M. Q., Marbois, B. N., York, A. G., et al. (2013). An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 73 (9), 2850–2862. doi:10.1158/0008-5472.CAN-13-0382-T
Williford, J. M., Ishihara, J., Ishihara, A., Mansurov, A., Hosseinchi, P., Marchell, T. M., et al. (2019). Recruitment of CD103(+) dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 5 (12), eaay1357. doi:10.1126/sciadv.aay1357
Wu, D., Zhuo, L., and Wang, X. (2017). Metabolic reprogramming of carcinoma-associated fibroblasts and its impact on metabolic heterogeneity of tumors. Semin. Cell Dev. Biol. 64, 125–131. doi:10.1016/j.semcdb.2016.11.003
Wu, L., Zhang, X., Zheng, L., Zhao, H., Yan, G., Zhang, Q., et al. (2020). RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol. Res. 8 (5), 710–721. doi:10.1158/2326-6066.CIR-19-0261
Wu, Y., Chen, K., Liu, X., Huang, L., Zhao, D., Li, L., et al. (2016). Srebp-1 interacts with c-myc to enhance somatic cell reprogramming. Stem Cells 34 (1), 83–92. doi:10.1002/stem.2209
Xiang, W., Shi, R., Kang, X., Zhang, X., Chen, P., Zhang, L., et al. (2018). Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 9 (1), 2574. doi:10.1038/s41467-018-04999-8
Xin, G., Chen, Y., Topchyan, P., Kasmani, M. Y., Burns, R., Volberding, P. J., et al. (2021). Targeting PIM1-mediated metabolism in myeloid suppressor cells to treat cancer. Cancer Immunol. Res. 9 (4), 454–469. doi:10.1158/2326-6066.CIR-20-0433
Xiong, X., Wen, Y. A., Fairchild, R., Zaytseva, Y. Y., Weiss, H. L., Evers, B. M., et al. (2020). Upregulation of CPT1A is essential for the tumor-promoting effect of adipocytes in colon cancer. Cell Death Dis. 11 (9), 736. doi:10.1038/s41419-020-02936-6
Yan, D., Adeshakin, A. O., Xu, M., Afolabi, L. O., Zhang, G., Chen, Y. H., et al. (2019). Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front. Immunol. 10, 1399. doi:10.3389/fimmu.2019.01399
Yan, J., and Horng, T. (2020). Lipid metabolism in regulation of macrophage functions. Trends Cell Biol. 30 (12), 979–989. doi:10.1016/j.tcb.2020.09.006
Yang, Y., Neo, S. Y., Chen, Z., Cui, W., Chen, Y., Guo, M., et al. (2020). Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. J. Clin. Invest. 130 (10), 5508–5522. doi:10.1172/JCI137585
Ye, B., Yin, L., Wang, Q., and Xu, C. (2019). ACC1 is overexpressed in liver cancers and contributes to the proliferation of human hepatoma Hep G2 cells and the rat liver cell line BRL 3A. Mol. Med. Rep. 19 (5), 3431–3440. doi:10.3892/mmr.2019.9994
Yi, J., Zhu, J., Wu, J., Thompson, C. B., and Jiang, X. (2020). Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. U. S. A. 117 (49), 31189–31197. doi:10.1073/pnas.2017152117
Younossi, Z., Tacke, F., Arrese, M., Chander Sharma, B., Mostafa, I., Bugianesi, E., et al. (2019). Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 69 (6), 2672–2682. doi:10.1002/hep.30251
Yu, S., Li, C., Ji, G., and Zhang, L. (2021). The contribution of dietary fructose to non-alcoholic fatty liver disease. Front. Pharmacol. 12, 783393. doi:10.3389/fphar.2021.783393
Yu, Y., Liang, Y., Li, D., Wang, L., Liang, Z., Chen, Y., et al. (2021). Glucose metabolism involved in PD-L1-mediated immune escape in the malignant kidney tumour microenvironment. Cell Death Discov. 7 (1), 15. doi:10.1038/s41420-021-00401-7
Zhao, F., Xiao, C., Evans, K. S., Theivanthiran, T., DeVito, N., Holtzhausen, A., et al. (2018). Paracrine wnt5a-beta-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity 48 (1), 147–160.e7. doi:10.1016/j.immuni.2017.12.004
Keywords: hepatocellular carcinoma, NASH, lipid, adipocytes, obesity
Citation: Zhao J, Lee K, Toh HC, Lam KP and Neo SY (2023) Unravelling the role of obesity and lipids during tumor progression. Front. Pharmacol. 14:1163160. doi: 10.3389/fphar.2023.1163160
Received: 10 February 2023; Accepted: 16 March 2023;
Published: 30 March 2023.
Edited by:
Shuijie Li, Harbin Medical University, ChinaReviewed by:
Chao Sun, The Second Hospital of Shandong University, ChinaCopyright © 2023 Zhao, Lee, Toh, Lam and Neo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shi Yong Neo, bmVvX3NoaV95b25nQGltbXVub2wuYS1zdGFyLmVkdS5zZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.