 Augustine Jing Jie Koh
Augustine Jing Jie Koh Varsha Thombare
Varsha Thombare Maytham Hussein
Maytham Hussein Gauri G. Rao
Gauri G. Rao Jian Li
Jian Li Tony Velkov
Tony Velkov
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 12 June 2023
Sec. Pharmacology of Infectious Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1158152
This article is part of the Research TopicGlobal Excellence in Pharmacology of Infectious Diseases: Australia and AsiaView all 6 articles
Antibiotic resistance is a top threat to human health and a priority across the globe. This problematic issue is accompanied by the decline of new antibiotics in the pipeline over the past 30 years. In this context, an urgent need to develop new strategies to combat antimicrobial resistance is in great demand. Lately, among the possible approaches used to deal with antimicrobial resistance is the covalent ligation of two antibiotic pharmacophores that target the bacterial cells through a dissimilar mode of action into a single hybrid molecule, namely hybrid antibiotics. This strategy exhibits several advantages, including better antibacterial activity, overcoming the existing resistance towards individual antibiotics, and may ultimately delay the onset of bacterial resistance. This review sheds light on the latest development of the dual antibiotic hybrids pipeline, their potential mechanisms of action, and challenges in their use.
The spread of antibiotic resistance has substantially diminished the efficacies of multiple clinical antibiotics in recent times owing to the emergence and proliferation of multidrug-resistant (MDR) bacterial pathogens (Aminov, 2010; Aslam et al., 2018; Annunziato, 2019). Six pathogenic species, collectively designated ESKAPE (Enterococcus faecium, Staphyloccocus aureus, Klebsiella pneumonia, Acinetobacter baumannii, Pseudomonas aeruginosa, and Escherichia coli) were ranked by the World Health Organization (WHO) as high-priority pathogens because of their ability to “escape” multiple antibiotics via resistance mechanisms including enzymatic inactivation, active efflux, and target modification, which can be genetically innate or acquired via horizontal gene transfer (Santajit and Indrawattana, 2016; Mulani et al., 2019; Zhen et al., 2019; de Oliveira et al., 2020; Cardoso et al., 2021). Hence, it is projected the annual mortality rate of antibiotic resistance (10 million) would supersede that of cancer (8.2 million) by 2050 (Dadgostar, 2019; Morris and Cerceo, 2020). This blight is exacerbated by the dearth of the antibiotic pipeline, in which the number of new antibiotics developed has steadily declined over the last 3 decades (Cardoso et al., 2021). With insufficient effective antibiotics, invasive and immunosuppressive treatments would be heavily restricted, and healthcare systems would find themselves extensively overwhelmed by the spread of MDR infections (Aslam et al., 2018; Piret and Boivin, 2021). While prudent stewardship of antibiotics may mitigate resistance, it remains paramount to expand the library of treatment options to compensate for various antibiotics rendered obsolete over the years. One of the means to revive antibiotic efficacy is through antibiotic combinations between synthetic antibiotics and/or last-resort antibiotics.
Co-administration of multiple antibiotics has gained increased popularity because the likelihood of pathogens developing resistance against two or more antibiotics concurrently is surmised to be extensively lower than against a single antibiotic administered (Mulani et al., 2019). In addition, any synergistic activity effectuated by the antibiotic combination far exceeds the antibacterial activity of either individual antibiotic in the combination (Figure 1) (Mulani et al., 2019; Theuretzbacher, 2020). Synergy occurs when antibiotics in the cocktail bind two or more targets, or inhibit different vital bacterial metabolic pathways simultaneously, inflicting substantial mortal damage on the bacterial population (Pokrovskaya and Baasov, 2010). However, antibiotic combination therapy incurs significant administrative and therapeutic monitoring costs as in-vitro synergy might not necessarily translate to the same effect in-vivo owing to a myriad of factors, including non-complementary pharmacodynamic/pharmacokinetic properties, which could have the unintended effect of exacerbating toxicity (Rybap and Mcgrath, 1996; Tamma et al., 2012). An example of antibiotic combinations augmenting toxic side effects is aminoglycoside/cephalosporin combinations, which aggravate nephrotoxicity and thereby, restrict their clinical use (Mannion et al., 1981).
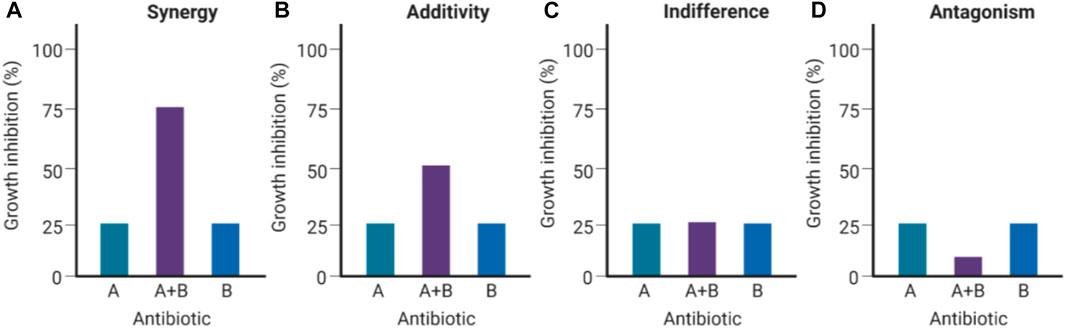
FIGURE 1. Types of antibacterial interactions for antibiotic combinations (Created with BioRender.com). If the Drug A/B combination is synergistic (A), the combined inhibition of growth (∼75%) would exceed the calculated cumulative antibacterial effect contributed by Drug A (∼25%) and Drug B (∼25%). If the Drug A/B combination is additive (B), the combined inhibition of growth would be equivalent to the calculated cumulative effect (∼50% growth inhibition). If Drugs A and B are indifferent (C), neither of them augments nor subtracts from each other’s efficacies when combined. If Drugs A and B are antagonistic (D), the resulting combined effect results in a considerable decrease in bacterial growth inhibition (Pokrovskaya and Baasov, 2010).
In a cocktail treatment of antibiotics, one of the antibiotics might be short-acting and readily inactivated metabolically or excreted, have different bacterial efflux susceptibilities, or have different tissue distributions compared to the other, where one antibiotic component is scarcely distributed, causing the other component to become vulnerable to resistance development (Domalaon et al., 2018). Differing antibiotic administration routes would also be a cause of inconvenience for patients (Fisher et al., 2020). Furthermore, different antibiotics may be chemically incompatible with one another when mixed. In the case of aminoglycoside/β-lactam co-administration, mixing some aminoglycosides (i.e., gentamicin, tobramycin) with certain β-lactams (i.e., carbenicillin, penicillin, and ticarcillin) in the same infusion fluids results in unwanted side reactions, producing inactive complexes (Kradjan and Burger, 1980; Tindula et al., 1983). The extent of inactivation is contingent on the β-lactam concentration, contact duration, and temperature (Aminoglycoside antibiotics, 2006).
Through scientific ingenuity, the idea of dual hybrid antibiotics materialized. The underlying objective behind ligating two antibiotics together via metabolically stable tethers is to construct a singular heterodimeric entity with a fixed pharmacokinetic profile while retaining the antibacterial mechanisms of the constituent pharmacophores. This could improve on-site targeting, impede bacterial efflux, sterically protect constituent pharmacophores from enzymatic degradation and reduce toxicity when administered in-vivo (Pokrovskaya and Baasov, 2010; Theuretzbacher, 2020). One pharmacophore could enhance the bioactivity of the neighbouring pharmacophore to which resistance is developed, potentially expanding its antibacterial spectrum and enabling rapid clearance of pathogens (Pokrovskaya et al., 2009). Moreover, different antibiotic targets (i.e., enzymes, precursor metabolites) can be involved in the same metabolic pathways and are in close physical proximity, which can be readily exploited with hybridized antibiotics through rapid simultaneous binding, readily impairing further development of resistance (Klahn and Brönstrup, 2017). To date, six dual hybrids were reported to have entered clinical trials (Table 1) (World Health Organization, 2021; fiercebiotech, 2018; Surur and Sun, 2021). Commercialization of any of these hybrids would significantly support research interest in developing hybrid antibiotics and provide humanity with a propitious advantage in overcoming the overlooked pandemic of antibiotic resistance. The following section summarizes the synthetic schemes, structural features, pharmacology, and antibacterial mechanisms of dual hybrid antibiotics presently in ongoing clinical evaluation.
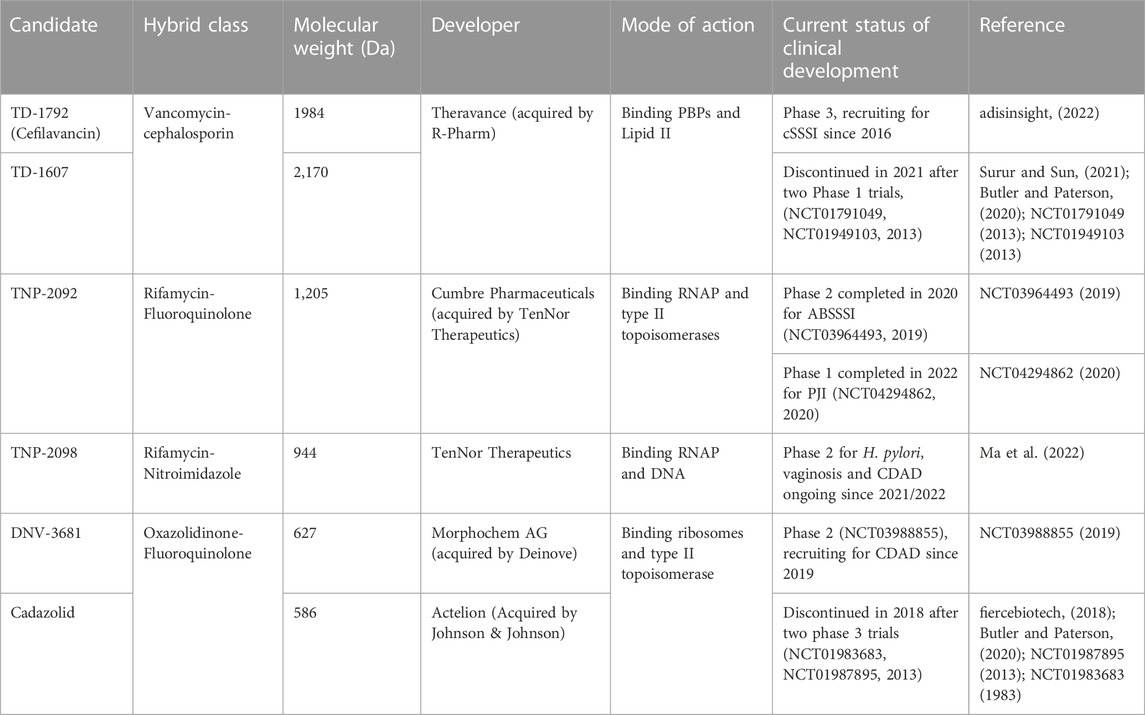
TABLE 1. Dual hybrid antibiotic clinical candidates.
The peptidoglycan cell wall is frequently exploited for antibiotic targeting due to its indispensability in protecting bacteria from osmotic lysis (Nikolaidis et al., 2014; Karas et al., 2020). During the late stage of peptidoglycan synthesis, membrane-bound precursors lipid II shuffle N-acetylglucosamine and N-acetylmuramic acid to the periplasmic space for peptidoglycan chain elongation. The elongated chains are cross-linked with the main chains, catalyzed by penicillin-binding proteins (PBPs) (Nikolaidis et al., 2014). β-Lactams (e.g., penicillin G, methicillin) and glycopeptides (e.g., vancomycin) target this critical stage by binding PBPs and lipid II, respectively (Sauvage and Terrak, 2016). Despite the fact that the discovery of additional β-lactam classes and the development of related β-lactam derivatives (i.e., 3rd/4th/5th generation cephalosporins and carbapenems), the expression of stronger classes of β-lactamases (i.e., extended-spectrum β-lactamases, and carbapenemases), low-affinity PBPs (e.g., PBP2a from methicillin-resistant S. aureus, MRSA) and overexpression of efflux pumps continue to substantially diminish β-lactam efficacy (Fisher et al., 2005; Worthington and Melander, 2013; Bush and Bradford, 2016; Fisher and Mobashery, 2016). Due to widespread β-lactam-resistance, glycopeptides have been administered as last resort alternatives (McCormick et al., 1955; Bush and Bradford, 2016; Cong et al., 2020).
However, even glycopeptide efficacy has been increasingly undermined by the emergence of vancomycin-intermediate S. aureus (VISA), and vancomycin-resistant Enterococcus (VRE) strains expressing seven resistance operons (VanA, VanB, VanC, VanD, VanE, VanF, and VanG) responsible for producing modified lipid II (Jubeh et al., 2020). VISA exhibits intermediate resistance to vancomycin via the excessive production of lipid II to sequester the glycopeptides (Sujatha and Praharaj, 2012; Butler et al., 2014). Moreover, the majority of VRE strains exhibit the VanA-phenotype, expressing lipid II with D-Ala-D-Lac epitopes of which vancomycin has overwhelmingly reduced binding affinity (Butler et al., 2014; Blaskovich et al., 2018). Expression of D-Ala-D-Lac is made possible primarily via vanA-operon expression and capable of being transferred interspecies via conjugation, which has led to the emergence of vancomycin-resistant S. aureus (VRSA) strains (Courvalin, 2006; Périchon and Courvalin, 2009; Sujatha and Praharaj, 2012). Therefore, it was surmised that to avert resistance to both β-lactams and glycopeptides, conjugating their pharmacophores to exploit the close molecular proximity of PBPs and lipid II could enable augmented bactericidal potency. Thus, Long and co-workers synthesized two β-lactam-glycopeptide hybrids, TD-1792 1 and TD-1607 4, from the pharmacophores of vancomycin 3 and the 3rd generation cephalosporin derivative THRX-169797 2 (Figure 2) (Long et al., 2008a; Long et al., 2008b).
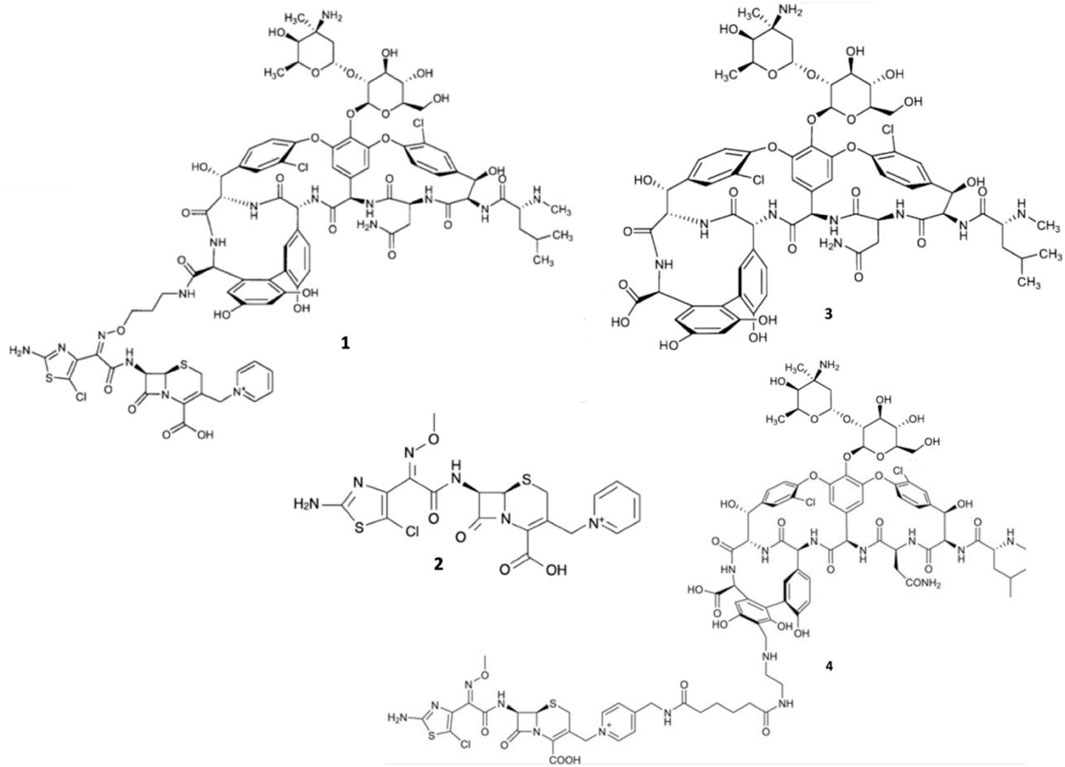
FIGURE 2. Structures of TD-1792 1, THRX-169797 2, vancomycin 3, and TD-1607 4.
Among the two vancomycin-cephalosporin hybrids, TD-1607 4, was discontinued after two phase 1 trials (NCT01791049, NCT01949103) possibly owing to poor tolerability, while TD-1792 1 cleared phase 2 trials for the treatment of Gram-positive complicated skin and soft-tissue infections (Butler and Paterson, 2020; NCT01791049, 2013; NCT01949103, 2013; Long et al., 2008b; Umscheid et al., 2011). Consistent with its in vitro potencies, TD-1792 exhibited strong antibacterial activity in vivo (≥1-log10CFU kill) against all Gram-positive strains from methicillin-susceptible S. aureus, methicillin-susceptible S. epidermidis, methicillin-resistant S. epidermidis, penicillin-susceptible S. pneumoniae, S. pyrogenes, MRSA and VISA present in the murine neutropenic thigh infection models investigated by Stryjewski and co-workers (Stryjewski et al., 2012a). TD-1792 is administered intravenously IV), possesses a half-life of 9–13-h, is moderately bound to plasma proteins (∼50%) in humans, and is primarily excreted via renal filtration. Compared to vancomycin, fewer adverse effects (pruritus, headaches, gastrointestinal disorders, etc) were reported in the TD-1792 treatment group in the phase two safety and efficacy study carried out by Stryjewski and co-workers (Stryjewski et al., 2012b). Ergo, TD-1792 is reported to have successfully progressed to phase three trials for Gram-positive complicated skin and soft tissue infections (adisinsight, 2022; Butler and Paterson, 2020).
Synthesis of TD-1792 1 (Figure 3) was commenced by coupling N-Boc–protected bromopropylamine 5 with the trityl-protected aminothiazolyl residue 6 to yield intermediate 7, which was then subjected to ester hydrolysis and chlorination with N-chlorosuccinimide (NCS), synthesizing Intermediate 8. Next, 8 was conjugated with 7-amino-3-chloromethyl-3-cephem-4-carboxylic acid p-methoxybenzyl ester 9. The resulting intermediate 10 was coupled with pyridine 11 and deprotected with trifluoroacetic acid (TFA) to yield THRX-169797 derivative 12. Finally, vancomycin 3 was conjugated directly to 12 through amide linkage to produce 1 (Long et al., 2008a). Cefilavancin effectively disrupts late-stage peptidoglycan synthesis via the inhibition of PBP and binding of lipid II D-Ala-D-Ala epitope, preventing peptidoglycan elongation and cross-linking concurrently (Figure 10A). Compared to penicillin G, cefilavancin exhibits substantial stability to staphylococcal β-lactamases congruent with that of THRX-169797 and is significantly unaffected by coexisting resistance mechanisms against multiple β-lactams (i.e., penicillin, methicillin, and oxacillin) and other antibiotic classes like fluoroquinolones (Blais et al., 2012; Stryjewski et al., 2012b). However, cefilavancin was primarily ineffective against Gram-negative pathogens just like vancomycin, owing to the impermeability of the outer membrane (Zhanel et al., 2008; Tyrrell et al., 2012).
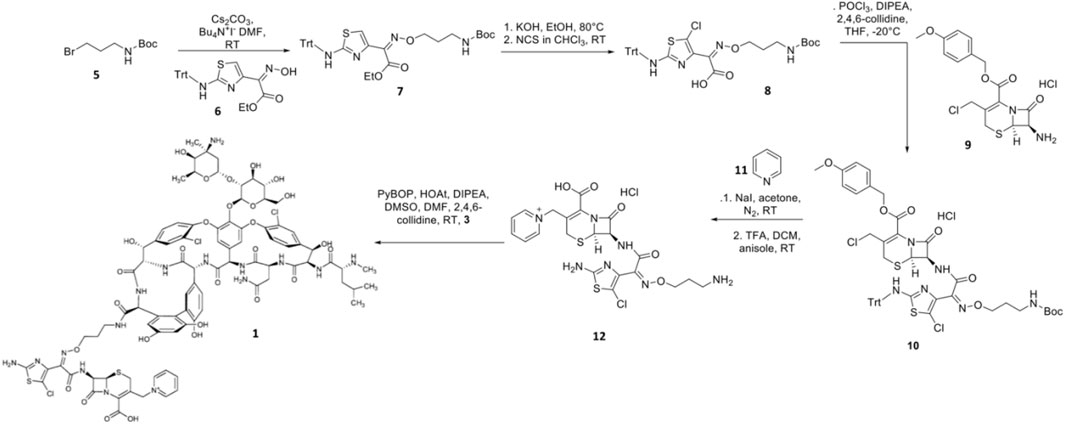
FIGURE 3. Synthetic scheme of TD-1792.
Crucial bacterial processes also exploited by antibiotics include chromosomal replication and gene expression. As bacteria have small-sized circular genomes, DNA replication and transcription occur in proximity (Proshkin et al., 1979; Merrikh et al., 2012). Hence, the neighbouring enzymes involved in DNA replication and expression can be readily exploited by the relevant antibiotic classes/families in combination or tethered together. The rifamycin family (Figure 4B) is a potent antibiotic class employed against biofilm pathogens, functioning as DNA-dependent RNA polymerase (RNAP) inhibitors (Ma et al., 2007). However, resistance against the rifamycin developed rapidly as point mutations resulted in the production of low-affinity bacterial RNA polymerases (Goldstein, 2014), requiring the use of rifamycin in combination therapy to potentiate antibacterial activity (Ma and Lynch, 2016; Fisher et al., 2020). Quinolones (and their fluorinated derivatives, fluoroquinolones, Figure 4A) are broad-spectrum bactericidal antibiotics that inhibit the enzymes topoisomerase IV and DNA gyrase, arresting DNA replication and transcription (Pham et al., 2019). Nevertheless, significant resistance against quinolones evolved via chromosomal mutations, and the acquisition of plasmids carrying relevant resistance genes. The overexpression of efflux pumps, mutations in DNA replication enzymes, and enzymatic drug modification substantially diminish quinolone efficacy, likewise prompting combination therapy to mitigate resistance (Aldred et al., 2014). However, as some rifamycin/quinolone cocktails exhibit a degree of antagonism in-vitro and in-vivo (Neu, 1991; Murillo et al., 2008; Balasubramanian et al., 2012), it was postulated that the synthesis of rifamycin-quinolone hybrids could effectively overcome both rifamycin and quinolone resistance by preventing undesired drug-drug interactions (Fisher et al., 2020).
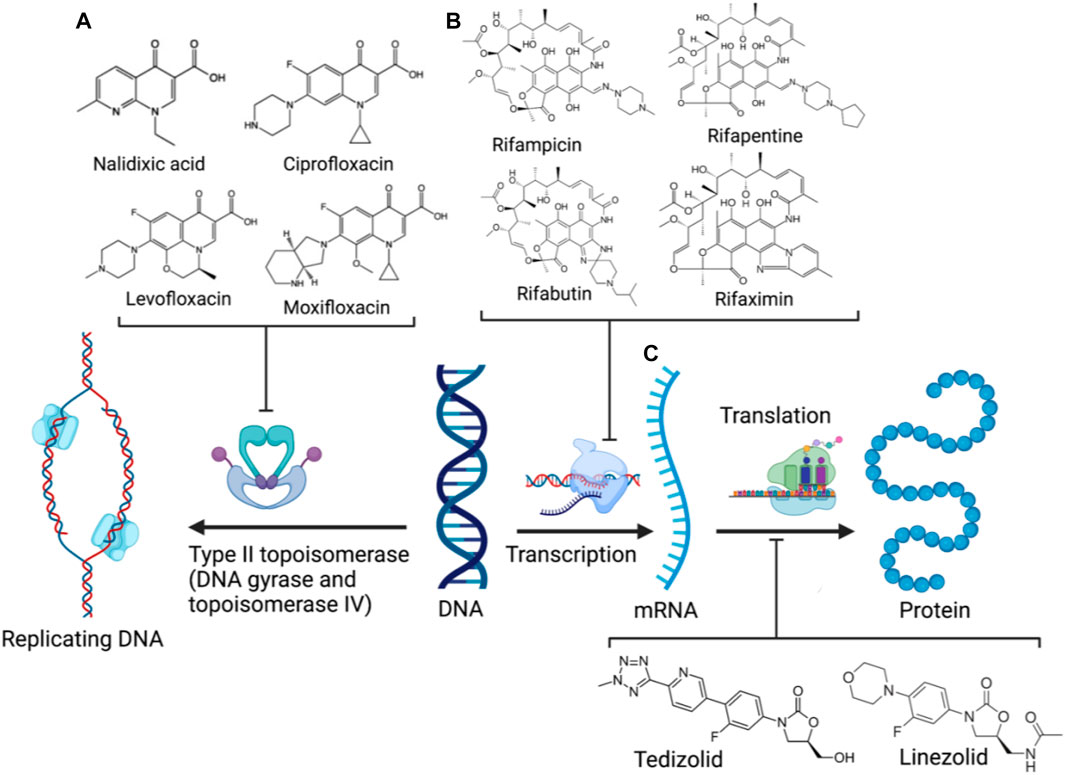
FIGURE 4. DNA replication and expression in bacteria and the corresponding clinical antibiotic classes, quinolones (A), rifamycins (B), and oxazolidinones (C) targeting the respective enzymes involved (Created with BioRender.com) (Bozdogan and Appelbaum, 2004; Goldstein, 2014; Pham et al., 2019).
One rifampicin-fluoroquinolone hybrid, TNP-2092, passed phase two trials (NCT03964493) for the treatment of acute bacterial skin and skin structure infections (ABSSSI) and was designated as an orphan drug for treating prosthetic joint infections (PJIs), which are often associated with substantial morbidity rates and healthcare expenses (NCT03964493, 2019; Fisher et al., 2020; Tande and Patel, 2014). TNP-2092 can be administered either intravenously or orally (World Health Organization, 2021). When administered orally (PO), TNP-2092 was observed to accumulate in the gastrointestinal tract due to low absorption, resulting in reduced systemic exposure and bioavailability compared to IV administration, and is excreted in feces. Ergo, TNP-2092 exhibits considerable potency in C. difficile murine infection models, capable of clearing infections at low doses (6.67 mg/kg). Moreover, TNP-2092 was observed to exert potent activity (MIC ≤ 0.12 μg/ml) against urease-producing bacteria, including Helicobacter pylori (H. pylori) and Salmonella, compared to rifaximin which is conventionally applied for these infections. The expedited clearance of urease-producing species considerably alleviates hepatic infections associated with liver cirrhosis. Hence, TNP-2092 is placed under clinical investigation for treating gastrointestinal and hepatic infections (Yuan et al., 2020).
TNP-2092 13 was designed via tethering the C-3 and C-8 side chains of rifampicin and the ciprofloxacin derivative ABT-719, respectively (Figure 5) (Ding et al., 2007; Ma and Lynch, 2016). Large-scale synthesis of 13 (Figure 6) commenced with the five-step synthesis of the linker component 19 from the precursor (S)-1-benzyl-3-hydroxypyrrolidine 14. Secondly, the linker 19 was conjugated with the ciprofloxacin derivative 20 under reflux in acetonitrile, producing 21, which was subjected to ester hydrolysis and deprotection with TFA, yielding 22. Finally, 22 is conjugated with the rifampin derivative 3-formalrifamycin 23 by converting the piperidine amino group of 22 into a hydrazine moiety, that is coupled with the C-3 aldehyde moiety of 23 to yield 13 (Ding et al., 2007). Biochemical characterizations of TNP-2092 indicated simultaneous inhibitory binding of RNAP and the type II topoisomerases, DNA gyrase, and DNA topoisomerase IV (Figure 10B), including common quinolone-resistant variants of the latter, accounting for potentiated bactericidal activity (Robertson et al., 2008). This enhanced activity of TNP-2092 was also confirmed by susceptibility tests involving a panel of S. aureus isolates carrying rifamycin or quinolone resistance alleles (or both), from which potency was observed (MIC ≤ 2 μg ml−1) against resistant isolates with either or both types of resistance alleles relative to rifampicin and ciprofloxacin, per se and in combination (MICs ≥8 μg ml−1) (Ma and Lynch, 2016; Fisher et al., 2020). Moreover, TNP-2092 is invulnerable to expulsion by fluoroquinolone efflux pumps, likely because of steric interference from the rifamycin pharmacophore, further verifying its low propensity for resistance development (Ma and Lynch, 2016). However, TNP-2092 shares a similar antibacterial spectrum of coverage as rifampicin, being less effective against most Gram-negative species owing to the limited permeability of the outer membrane to rifamycins (Yuan et al., 2020).
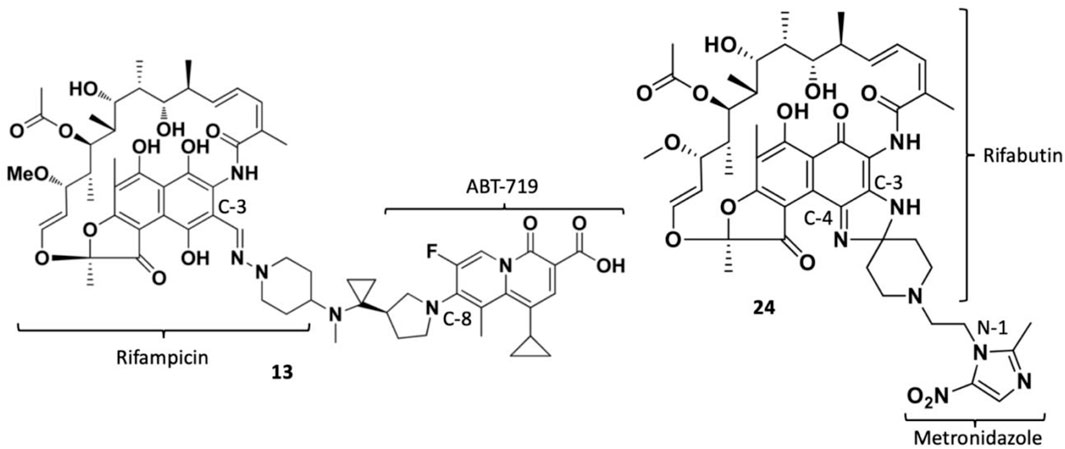
FIGURE 5. Structures of rifamycin hybrids TNP-2092 13 and TNP-2198 24.
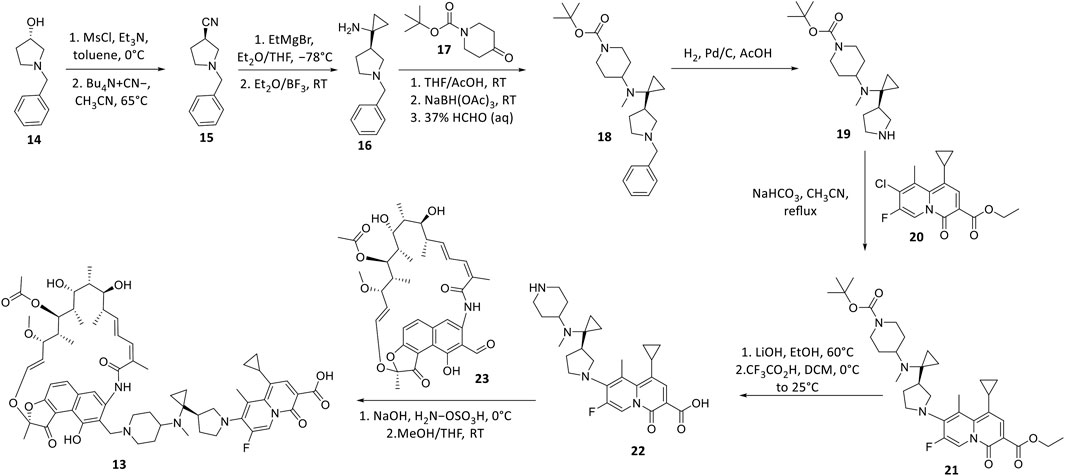
FIGURE 6. Synthetic scheme of TNP-2092.
Apart from fluoroquinolones, rifamycins were also hybridized with nitroimidazoles to overcome rifamycin resistance (Ma et al., 2022). Nitroimidazoles (E.g., metronidazole 25) are broad-spectrum antimicrobials utilized to treat anaerobic bacterial (Gram-positive and -negative), protozoal, and parasitic infections by yielding free radicals that inflict oxidative damage to bacterial DNA and other proteins (Mital, 2009; Ang et al., 2017). The rifamycin-nitroimidazole hybrid, TNP-2198, was produced by Ma and co-workers via tethering the C-3/C-4 and N-1 side chains of rifabutin and metronidazole, respectively, via an ethylene linker (Figure 5). Ma and co-workers synthesized TNP-2198 24 in milligram to kilogram quantities via a synthetic scheme (Figure 7) commencing from metronidazole 25. Firstly, 25 was subjected to Swern oxidation (using oxalyl chloride dissolved in DMSO, followed by quenching with triethylamine) to yield 26. Next, 26 was condensed with 4-hydroxypiperidine 27 and reduced using sodium triacetoxyborohydride in methanol to yield intermediate 28, which is used to produce intermediate 29 via Swern oxidation. Lastly, the ketone moiety of 29 is coupled with the C-3/C-4 amine and imine moieties of 3-amino-4-deoxy-4-imino-rifamycin S 30 to produce TNP-2198 24 (Omura and Swern, 1978; Ma et al., 2022).
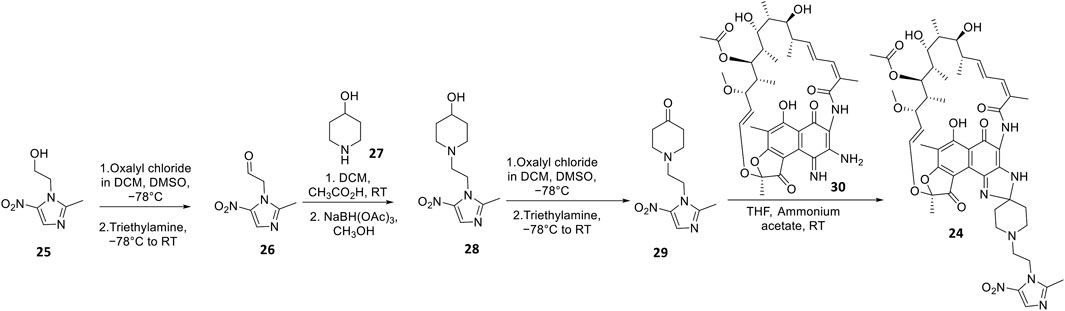
FIGURE 7. Synthetic scheme of TNP-2198.
TNP-2198 was observed to exert 4-fold greater potency than metronidazole against rifamycin-resistant H. pylori (MIC 0.5 μg ml−1 and 2 μg ml−1, respectively) and 64–500-fold potent activity against both lab-generated rifamycin- and ciprofloxacin-resistant C. difficile strains, suggesting the ability of the metronidazole scaffold to enhance the bioactivity of the adjacent rifabutin pharmacophore. The synergy between the rifabutin and metronidazole pharmacophores of TNP-2198 was verified via time-kill assays in which TNP-2198 displayed rapid bactericidal activity (≥ 3.0 log10 CFU) compared to either rifamycin, metronidazole monotherapy or 1:1 M rifamycin/metronidazole combination. As determined by X-ray crystallography, synergistic bactericidal mechanisms occur via the rifampicin pharmacophore occupying the RNAP RNA exit cleft and the metronidazole scaffold simultaneously binding to the DNA template strand in the RNAP active center cleft via hydrogen bonding. Furthermore, under microaerophilic/anaerobic conditions, nitroreductase-catalyzed reduction of the metronidazole nitro (-NO2) moiety yields additional toxic TNP-2198 free radicals forming fatal covalent cross-links with other bacterial nucleic acids and proteins (Figure 10C).
Similar to TNP-2092, TNP-2198 exhibits high tissue distribution in the stomach, large intestine, gums, and vaginal tissue. Consistent with its potent in vitro activity, TNP-2198 prolonged the survival rate of hamster C. difficile infection models (at 100% for 6 days) subjected to a low dosing regimen of 5 mg/kg compared to metronidazole monotherapy at 100 mg/kg from which 100% mortality was observed following 4 days. Moreover, TNP-2198 substantially reduced mean bacterial titers at low concentrations (5 mg/kg) to levels (∼2.8 log10 CFU) comparable to that of the potent anti-H. pylori macrolide clarithromycin (10 mg/kg) in H. pylori murine infection models (Ma et al., 2022). Nitroimidazoles are known for exhibiting cytotoxicity and genotoxicity, warranting their development as prodrugs for administration (Nepali et al., 2019). Promisingly, TNP-2198 exhibits lower distribution in the central nervous system, which eliminates neurotoxicity associated with the metronidazole pharmacophore, possibly due to the hybrid’s larger molecular weight preventing access through the blood-brain barrier and the rifabutin pharmacophore enabling better bacterial targeting (Hartmann et al., 1967; Koike et al., 2020; Ma et al., 2022). Hence, TNP-2198 has since entered phase two trials for treating vaginosis and gastrointestinal infections caused by H. pylori and C. difficile (World Health Organization, 2021).
Oxazolidinones (Figure 4C) are synthetic antibiotics derived from 2-oxazolidone, which occupy the P-site of the 50 S ribosomal subunit, inhibiting the assembly of the translation initiation complex and the translocation of peptidyl-tRNA from the A-site to the P-site, impeding mRNA translation (Bozdogan and Appelbaum, 2004). However, extensive resistance against oxazolidinones occurs via target modification (i.e., 23 S rRNA mutations in the 50 S subunit) and efflux (Brenciani et al., 2022). Several research groups have synthesized several oxazolidinone-fluoroquinolone hybrids, which were investigated for their potential to overcome oxazolidinone resistance and expand the spectrum to include Gram-negative pathogens, considering the ability of fluoroquinolones to permeate the outer membrane (Diver, 1989; Gordeev et al., 2003; Hubschwerlen et al., 2003). The oxazolidinone-fluoroquinolone hybrids with a 4-piperidinemethanol-derived linker were noted to display augmented potency and a reduced propensity for resistance development (Liu et al., 2019). Presently two such hybrids, cadazolid and DNV-3681 (Figure 8), cleared phase one trials (World Health Organization, 2021).
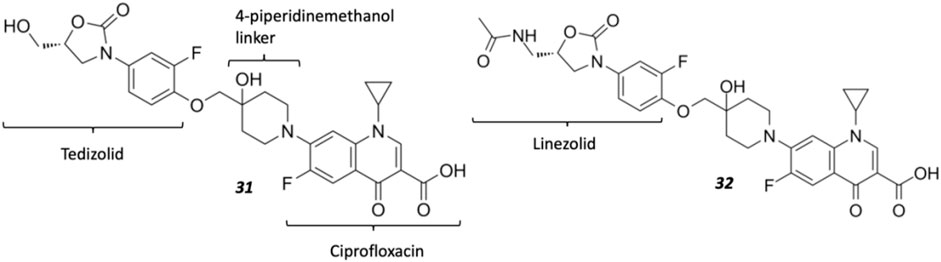
FIGURE 8. Structures of oxazolidinone hybrids Cadazolid 31 and DNV-3681 32.
Cadazolid, a tedizolid-ciprofloxacin hybrid, was investigated as an alternative antibiotic to vancomycin and metronidazole for treating C. difficile-associated diarrhea (CDAD) (Locher et al., 2014). Inhibition observed from DNA topoisomerase assays was minimal compared to fluoroquinolones moxifloxacin and ciprofloxacin, suggesting cadazolid is primarily a protein synthesis inhibitor with weak inhibition of DNA synthesis as a secondary effect. This was affirmed via cryo-electron microscopy analysis, indicating that cadazolid occupies the ribosomal P-site via its oxazolidinone pharmacophore, which consequently projects the fluoroquinolone pharmacophore into the A-site, sterically preventing the elongation of the peptide chain by blocking the next aminoacyl-tRNA from occupying the A-site of the ribosomal translation complex (Scaiola et al., 2019). Cadazolid is administered orally and accumulates in the colon due to its poor aqueous solubility, resulting in low plasma concentrations and its primary excretion in feces (Baldoni et al., 2014). However, after two identically designed phase three trials (NCT01983683, NCT01987895) involving the treatment of CDAD (Gerding et al., 2019), further clinical developments were reported to be discontinued owing to its failure to achieve non-inferiority to vancomycin in one of the two trials (Theuretzbacher, 2020; fiercebiotech, 2018; Butler and Paterson, 2020).
DNV-3681 32 is another oxazolidinone-fluoroquinolone hybrid, comprised of linezolid and ciprofloxacin. It can either be administered PO, or IV as a water-soluble prodrug DNV-3837 33 (Figure 9) [NO_PRINTED_FORM]Promisingly, DNV-3681 exhibited 4-8-fold greater potency (MIC50/90 0.032 μg ml−1/0.064 μg ml−1) against 114 C. difficile isolates compared to cadazolid (MIC50/90 0.125 μg ml−1/0.5 μg ml−1) in susceptibility tests conducted by Rashid and co-workers, suggesting a comparatively stronger inhibition of DNA replication and transcription than cadazolid (Figure 10D) (Rashid et al., 2014). Thus, DNV-3681 effectively overcomes dual quinolone-oxazolidinone-resistant C. difficile isolates and exhibits superior efficacy compared to standard antibiotics for treating CDAD, including vancomycin, metronidazole, and ciprofloxacin (DNV3837, 2022; Freeman et al., 2017). In a phase one investigation conducted by Dalhoff and co-workers involving twelve healthy male volunteers, DNV-3681 displayed minimal systemic absorption and was generally well tolerated following IV administration. DNV-3681 is distributed readily in the colon, where it exhibits potent activity against Gram-positive organisms (e.g., Clostridia, Enterococci, and S. aureus) whilst leaving normal Gram-negative microflora generally unscathed (Dalhoff et al., 2015). Currently, DNV-3681 progressed to phase two trials (NCT03988855) for the treatment of CDAD (Kullar et al., 2020).
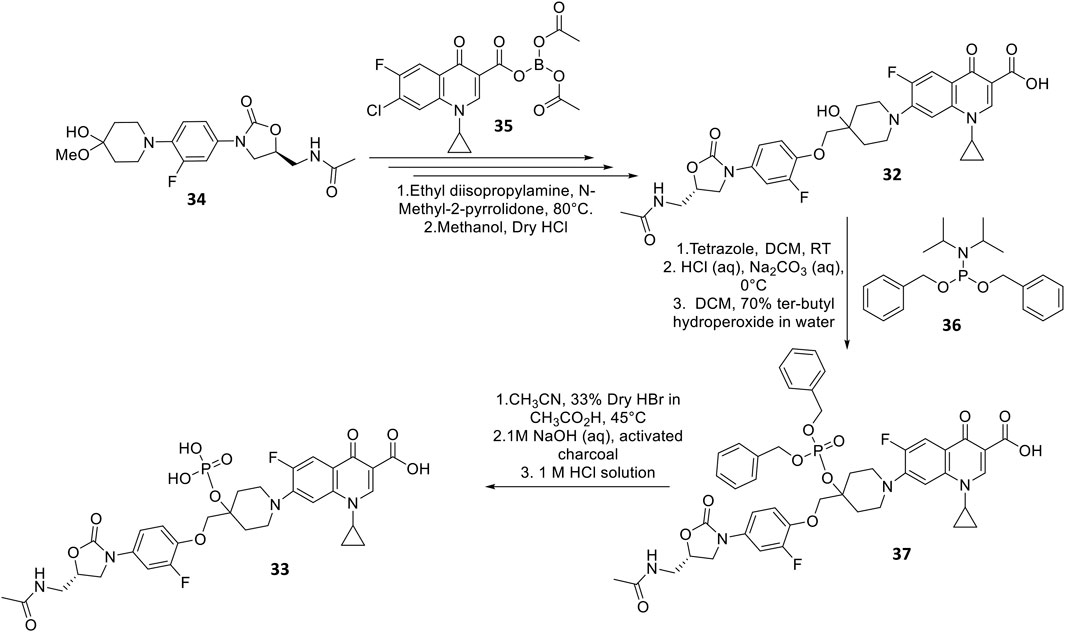
FIGURE 9. Synthetic scheme of DNV-3681 and DNV-3837 (Hubschwerlen et al., 2012).
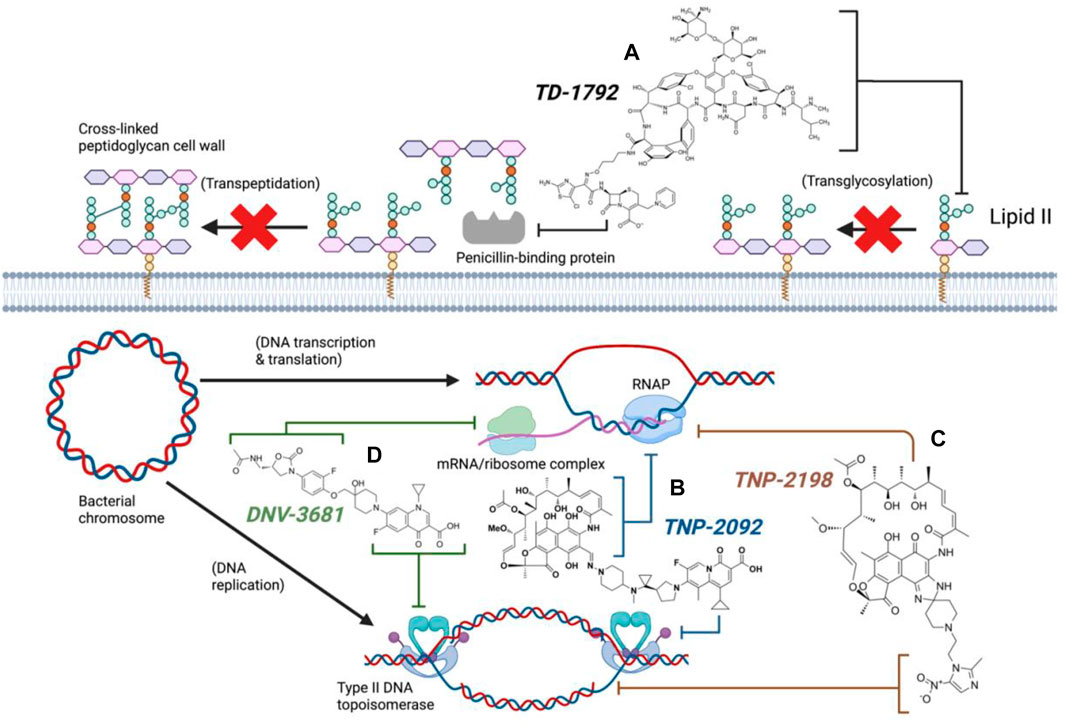
FIGURE 10. Antibacterial mechanisms of dual hybrid candidates (Created with BioRender.com). TD-1792 simultaneously binds lipid II and PBP (A, Black), TNP-2092 inhibits both RNA polymerase and type II DNA topoisomerases (B, Blue), TNP-2198 inhibits transcription and concurrently damages DNA (C, Brown), DNV-3681 inhibits both protein synthesis and DNA replication (D, Green).
The concept of dual-acting hybrid antibiotics currently holds significant promise in overcoming bacterial resistance since some compounds have progressed to phase three (E.g., cadazolid and cefilavancin). However, this novel approach is not without challenges. For instance, the complexity of designing chemical synthetic procedures may affect overall yields and potentially impair the intrinsic activity of the hybrids synthesized (Ma and Lynch, 2016; Domalaon et al., 2018; Lungu et al., 2022). Other obstacles involve the drug permeability impediments in Gram-negative bacteria due to increased molecular weight (> 600 Da), as well as the meticulous work needed to understand the mode of action and determine the benefits of the hybrid compounds over conventional antibiotics, such as the possibility of delaying the evolution of resistance (Pokrovskaya et al., 2009; Shavit et al., 2017; Koh Jing Jie et al., 2022). To remedy some of the challenges, the antibiotic hybridization strategy has expanded to include conjugation to adjuvants - small molecules or biologics without intrinsic antibacterial activity but potentiate antibiotic activity through anti-resistance mechanisms, bypassing membrane barriers or immune cell stimulation (Wright, 2016; Melander and Melander, 2017; Koh Jing Jie et al., 2022). To illustrate, siderophore adjuvants have been conjugated with antibiotics (E.g., cefiderocol, Fetroja®) to successfully bypass the outer membrane barrier of Gram-negative bacteria by exploiting the iron-uptake pathway (Fetroja FDA, 2022). With improvements in synthetic procedures, conjugation of adjuvants with the current dual hybrid candidates may assist in overcoming membrane permeability issues. Despite the challenges, the antibiotic hybrid strategy remains a viable approach to expanding the antimicrobial arsenal and minimizing the severe attrition experienced in the antibiotic discovery pipeline. As more data could be unraveled in the future, this field could be broadened and thereby, there will be a great optimism that human ingenuity will pave the way for the next generations of hybrids that can equip the antibiotic arsenal.
AK; conceptualization and investigation, AK and VT; resources, VT; molecular structure generation, AK; writing—original draft preparation, VT, MH, GR, and JL; writing—review and editing, TV; supervision. All authors contributed to the article and approved the submitted version.
GR, TV, and JL are supported by the National Institute of Allergy and Infectious Diseases, award number R01AI146241. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. TV is an MPT Connect REDI Fellow. JL is a National Australian Health and Medical Research Council Senior Principle Research Fellow.
JL is an Australian National Health and Medical Research Council (NHMRC) Principal Research Fellow, and TV is an Australian NHMRC Industry Career Development Level 2 Research Fellow.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
adisinsight Cefilavancin - theravance biopharma - AdisInsight. [Internet]. [cited 2022 Aug 30]. Available at: https://adisinsight.springer.com/drugs/800020586.
Aldred, K. J., Kerns, R. J., and Osheroff, N. (2014).Mechanism of quinolone action and resistance, Biochemistry, 53. 1565–1574.
Aminoglycoside antibiotics (2006). “Aminoglycoside antibiotics,” in Meyler’s side effects of drugs: The international encyclopedia of adverse drug reactions and interactions Netherlands, (Elsevier), 118–136.
Aminov, R. I. (2010). A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 1, 134. doi:10.3389/fmicb.2010.00134
Ang, C. W., Jarrad, A. M., Cooper, M. A., and Blaskovich, M. A. T. (2017). Nitroimidazoles: Molecular fireworks that combat a broad spectrum of infectious Diseases. J. Med. Chem. 60 (18), 7636–7657. doi:10.1021/acs.jmedchem.7b00143
Annunziato (2019). Strategies to overcome antimicrobial resistance (amr) making use of non-essential target inhibitors: A review. Int. J. Mol. Sci. 20 (23), 5844. doi:10.3390/ijms20235844
Aslam, B., Wang, W., Arshad, M. I., Khurshid, M., Muzammil, S., Rasool, M. H., et al. (2018). Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist 11, 1645–1658. doi:10.2147/IDR.S173867
Balasubramanian, V., Solapure, S., Gaonkar, S., Mahesh Kumar, K. N., Shandil, R. K., Deshpande, A., et al. (2012). Effect of coadministration of moxifloxacin and rifampin on Mycobacterium tuberculosis in a murine aerosol infection model. Antimicrob. Agents Chemother. 56 (6), 3054–3057. doi:10.1128/AAC.06383-11
Baldoni, D., Gutierrez, M., Timmer, W., and Dingemanse, J. (2014). Cadazolid, a novel antibiotic with potent activity against Clostridium difficile: Safety, tolerability and pharmacokinetics in healthy subjects following single and multiple oral doses. J. Antimicrob. Chemother. 69 (3), 706–714. doi:10.1093/jac/dkt401
Blais, J., Lewis, S. R., Krause, K. M., and Benton, B. M. (2012). Antistaphylococcal activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic. Antimicrob. Agents Chemother. 56 (3), 1584–1587. doi:10.1128/AAC.05532-11
Blaskovich, M. A. T., Hansford, K. A., Butler, M. S., Jia, Z., Mark, A. E., and Cooper, M. A. (2018). Developments in glycopeptide antibiotics. ACS Infect. Dis. 4 (5), 715–735. doi:10.1021/acsinfecdis.7b00258
Bozdogan, B., and Appelbaum, P. C. (2004). Oxazolidinones: Activity, mode of action, and mechanism of resistance. Int. J. Antimicrob. Agents 23, 113–119. doi:10.1016/j.ijantimicag.2003.11.003
Brenciani, A., Morroni, G., Schwarz, S., and Giovanetti, E. (2022). Oxazolidinones: Mechanisms of resistance and mobile genetic elements involved. J. Antimicrob. Chemother. 77, 2596–2621. doi:10.1093/jac/dkac263
Bush, K., and Bradford, P. A. (2016). β-Lactams and β-lactamase inhibitors: An overview. Cold Spring Harb. Perspect. Med. 6 (8), a025247. doi:10.1101/cshperspect.a025247
Butler, M. S., Hansford, K. A., Blaskovich, M. A. T., Halai, R., and Cooper, M. A. (2014).Glycopeptide antibiotics: Back to the future, J. Antibiotics, 67, 631–644. doi:10.1038/ja.2014.111
Butler, M. S., and Paterson, D. L. (2020).Antibiotics in the clinical pipeline in, J. Antibiotics, 73, 329–364. October 2019. doi:10.1038/s41429-020-0291-8
Cardoso, P., Glossop, H., Meikle, T. G., Aburto-Medina, A., Conn, C. E., Sarojini, V., et al. (2021). Molecular engineering of antimicrobial peptides: Microbial targets, peptide motifs and translation opportunities. Biophys. Rev. [Internet] 13, 35–69. Available from. doi:10.1007/s12551-021-00784-y
Cong, Y., Yang, S., and Rao, X. (2020). Vancomycin resistant Staphylococcus aureus infections: A review of case updating and clinical features. J. Adv. Res. 21, 169–176. doi:10.1016/j.jare.2019.10.005
Courvalin, P. Vancomycin resistance in gram-positive cocci clinical infectious Diseases [Internet]. Vol. 42,. 2006. Available at: https://academic.oup.com/cid/article-abstract/42/Supplement_1/S25/275393
Dadgostar, P. (2019). Antimicrobial resistance: Implications and costs. Infect. Drug Resist Vol. 12, 3903–3910. doi:10.2147/IDR.S234610
Dalhoff, A., Rashid, M. U., Kapsner, T., Panagiotidis, G., Weintraub, A., and Nord, C. E. (2015). Analysis of effects of MCB3681, the antibacterially active substance of prodrug MCB3837, on human resident microflora as proof of principle. Clin. Microbiol. Infect. 21 (8), 767.e1–e4. doi:10.1016/j.cmi.2015.05.025
de Oliveira, D. M. P., Forde, B. M., Kidd, T. J., Harris, P. N. A., Schembri, M. A., Beatson, S. A., et al. (2020). Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 33 (3), 001811. doi:10.1128/CMR.00181-19
Ding, C. Z., Ma, Z., Li, J., He, Y., Minor, K. P., Kim, I. H., et al. (2007). R/S) Rifamycin derivatives, their preparations and pharmaceutical compositions. Alexandria: U.S. Patent. No 7226931.
Diver, J. M. Quinolone uptake by bacteria and bacterial killing. Rev. Infect. Dis., Rev Infect Dis [Internet]. 1989;11:941–946. doi:10.1093/clinids/11.supplement_5.s941
DNV3837 DEINOVE [internet]. [cited 2022 Sep 15]. Available from: https://www.deinove.com/en/pipeline/dnv3837.
Domalaon, R., Idowu, T., Zhanel, G. G., and Schweizer, F. (2018). Antibiotic hybrids: The next generation of agents and adjuvants against gram-negative pathogens? Clin. Microbiol. Rev. 31 (2), e00077. doi:10.1128/CMR.00077-17
Fetroja FDA Fetroja FDA approval history [internet]. [cited 2022 Oct 15]. Available at: https://www.drugs.com/history/fetroja.html.
fiercebiotech Johnson & Johnson scraps phase 3 antibiotic program acquired in $30B Actelion takeover. [Internet]. 2018 [cited 2022 Sep 12]. Available at: https://www.fiercebiotech.com/biotech/j-j-scraps-phase-3-antibiotic-program-acquired-30b-actelion-takeover.
Fisher, C. R., Schmidt-Malan, S. M., Ma, Z., Yuan, Y., He, S., and Patel, R. (2020). In vitro activity of TNP-2092 against periprosthetic joint infection–associated staphylococci. Diagn Microbiol. Infect. Dis. 97 (3), 115040. doi:10.1016/j.diagmicrobio.2020.115040
Fisher, J. F., Meroueh, S. O., and Mobashery, S. Bacterial resistance to beta-lactam antibiotics: Compelling opportunism, compelling opportunity, compelling Oppor. Vol. 105, Chem. Rev. 2005. p. 395. doi:10.1021/cr030102i
Fisher, J. F., and Mobashery, S. (2016). β-Lactam resistance mechanisms: Gram-positive bacteria and Mycobacterium tuberculosis. Cold Spring Harb. Perspect. Med. 6 (5), a025221. doi:10.1101/cshperspect.a025221
Freeman, J., Pilling, S., Vernon, J., and Wilcox, M. H. (2017). In vitro activities of MCB3681 and eight comparators against Clostridium difficile isolates with known ribotypes and diverse geographical spread. Antimicrob. Agents Chemother. 61 (3), e02077. doi:10.1128/AAC.02077-16
Gerding, D. N., Cornely, O. A., Grill, S., Kracker, H., Marrast, A. C., Nord, C. E., et al. (2019). Cadazolid for the treatment of Clostridium difficile infection: Results of two double-blind, placebo-controlled, non-inferiority, randomised phase 3 trials. Lancet Infect. Dis. 19 (3), 265–274. doi:10.1016/S1473-3099(18)30614-5
Goldstein, B. P. (2014). Resistance to rifampicin: A review. J. Antibiot. (Tokyo) 67 (9), 625–630. doi:10.1038/ja.2014.107
Gordeev, M. F., Hackbarth, C., Barbachyn, M. R., Banitt, L. S., Gage, J. R., Luehr, G. W., et al. (2003). Novel oxazolidinone-quinolone hybrid antimicrobials. Bioorg Med. Chem. Lett. 13 (23), 4213–4216. doi:10.1016/j.bmcl.2003.07.021
Hartmann, G., Honikel, K. O., Knüsel, F., and Nüesch, J. (1967). The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochimica Biophysica Acta (BBA) - Nucleic Acids Protein Synthesis 145 (3), 843–844. doi:10.1016/0005-2787(67)90147-5
Hubschwerlen, C., Specklin, J. L., Baeschlin, D., Schmitt, C., Müller, S., and Cappi, M. W. (2012). Oxazolidinone-quinolone hybrid antibiotics. Alexandria: U.S. Patent. No 8158797.
Hubschwerlen, C., Specklin, J. L., Baeschlin, D. K., Borer, Y., Haefeli, S., Sigwalt, C., et al. (2003). Structure-activity relationship in the oxazolidinone-quinolone hybrid series: Influence of the central spacer on the antibacterial activity and the mode of action. Bioorg Med. Chem. Lett. 13 (23), 4229–4233. doi:10.1016/j.bmcl.2003.07.028
Jubeh, B., Breijyeh, Z., and Karaman, R. (2020). Resistance of gram-positive bacteria to current antibacterial agents and overcoming approaches, 25. Switzerland: Molecules MDPI AG.
Karas, J. A., Chen, F., Schneider-Futschik, E. K., Kang, Z., Hussein, M., Swarbrick, J., et al. (2020). Synthesis and structure−activity relationships of teixobactin. Ann. N. Y. Acad. Sci. 1459 (1), 86–105. doi:10.1111/nyas.14282
Klahn, P., and Brönstrup, M. (2017). Bifunctional antimicrobial conjugates and hybrid antimicrobials. Nat. Prod. Rep. R. Soc. Chem. 34, 832–885. doi:10.1039/c7np00006e
Koh Jing Jie, A., Hussein, M., Rao, G. G., Li, J., and Velkov, T. (2022). Drug repurposing approaches towards defeating multidrug-resistant gram-negative pathogens: Novel polymyxin/non-antibiotic combinations. Pathogens 11 (12), 1420. doi:10.3390/pathogens11121420
Koike, N., Kota, R., Naito, Y., Hayakawa, N., Matsuura, T., Hishiki, T., et al. (2020). 2-Nitroimidazoles induce mitochondrial stress and ferroptosis in glioma stem cells residing in a hypoxic niche. Commun. Biol. 3 (1), 450. doi:10.1038/s42003-020-01165-z
Kradjan, W. A., and Burger, R. (1980). In vivo inactivation of gentamicin by carbenicillin and ticarcillin. Arch. Intern Med. 140 (12), 1668–1670. doi:10.1001/archinte.1980.00330230114024
Kullar, R., Tran, M. C. N., and Goldstein, E. J. (2020). Investigational treatment agents for recurrent clostridioides difficile infection (rCDI). J. Exp. Pharmacol. 12, 371–384. doi:10.2147/JEP.S242959
Liu, L., Shao, L., Li, J., Cui, H., Li, B., Zhou, X., et al. (2019). Synthesis, antibacterial activities, mode of action and acute toxicity studies of new oxazolidinone-fluoroquinolone hybrids. Molecules 24 (8), 1641. doi:10.3390/molecules24081641
Locher, H. H., Caspers, P., Bruyère, T., Schroeder, S., Pfaff, P., Knezevic, A., et al. (2014). Investigations of the mode of action and resistance development of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob. Agents Chemother. 58 (2), 901–908. doi:10.1128/AAC.01831-13
Long, D. D., Aggen, J. B., Chinn, J., Choi, S. K., Christensen, B. G., Fatheree, P. R., et al. (2008a). Exploring the positional attachment of glycopeptide/beta-lactam heterodimers. J. Antibiot. (Tokyo) 61 (10), 603–614. doi:10.1038/ja.2008.80
Long, D. D., Aggen, J. B., Christensen, B. G., Judice, J. K., Hegde, S. S., Kaniga, K., et al. (2008b). A multivalent approach to drug discovery for novel antibiotics. J. Antibiot. (Tokyo) 61 (10), 595–602. doi:10.1038/ja.2008.79
Lungu, I. A., Moldovan, O. L., Biriș, V., and Rusu, A. (2022). Fluoroquinolones hybrid molecules as promising antibacterial agents in the fight against antibacterial resistance. Pharmaceutics 14 (8), 1749. doi:10.3390/pharmaceutics14081749
Ma, Z., Ginsberg, A. M., and Spigelman, M. (2007). “Antimycobacterium agents,” in Comprehensive medicinal chemistry II (Netherlands: Elsevier), 699–730.
Ma, Z., He, S., Yuan, Y., Zhuang, Z., Liu, Y., Wang, H., et al. (2022). Design, synthesis, and characterization of TNP-2198, a dual-targeted rifamycin-nitroimidazole conjugate with potent activity against microaerophilic and anaerobic bacterial pathogens. J. Med. Chem. 65 (6), 4481–4495. doi:10.1021/acs.jmedchem.1c02045
Ma, Z., and Lynch, A. S. (2016). Development of a dual-acting antibacterial agent (TNP-2092) for the treatment of persistent bacterial infections. J. Med. Chem. 59 (14), 6645–6657. doi:10.1021/acs.jmedchem.6b00485
Mannion, J. C., Bloch, R., and Popovich, N. G. (1981). Cephalosporin-aminoglycoside synergistic nephrotoxicity: Fact or fiction? Drug Intell. Clin. Pharm. 15 (4), 248–256. doi:10.1177/106002808101500401
McCormick, M. H., Mcguire, J. M., Pittenger, G. E., Pittenger, R. C., and Stark, W. M. (1955). Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot. Annu. 3, 606–611.
Melander, R. J., and Melander, C. (2017). The challenge of overcoming antibiotic resistance: An adjuvant approach? ACS Infect. Dis. 3 (8), 559–563. doi:10.1021/acsinfecdis.7b00071
Merrikh, H., Zhang, Y., Grossman, A. D., and Wang, J. D. (2012). Replication–transcription conflicts in bacteria. Nat. Rev. Microbiol. 10 (7), 449–458. doi:10.1038/nrmicro2800
Mital, A. (2009). Synthetic nitroimidazoles: Biological activities and mutagenicity relationships. Sci. Pharm. 77, 497–520. doi:10.3797/scipharm.0907-14
Morris, S., and Cerceo, E. (2020). Trends, epidemiology, and management of multi-drug resistant gram-negative bacterial infections in the hospitalized setting. Antibiotics 9 (4), 196. doi:10.3390/antibiotics9040196
Mulani, M. S., Kamble, E. E., Kumkar, S. N., Tawre, M. S., and Pardesi, K. R. (2019). Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: A review. Front. Microbiol. 10, 539. doi:10.3389/fmicb.2019.00539
Murillo, O., Pachón, M. E., Euba, G., Verdaguer, R., Tubau, F., Cabellos, C., et al. (2008). Antagonistic effect of rifampin on the efficacy of high-dose levofloxacin in staphylococcal experimental foreign-body infection. Antimicrob. Agents Chemother. 52 (10), 3681–3686. doi:10.1128/AAC.00458-08
NCT01791049 (2013). TD-1607 SAD study in healthy subjects. [Internet]. [cited 2022 Aug 30]. Available at: https://clinicaltrials.gov/ct2/show/NCT01791049.
NCT01949103 (2013). TD-1607 MAD study in healthy subjects. [Internet]. [cited 2022 Aug 30]. Available at: https://clinicaltrials.gov/ct2/show/NCT01949103.
NCT01983683 Efficacy and safety of cadazolid versus vancomycin in subjects with Clostridium difficile-associated diarrhea [internet]. [cited 2023 May 21]. Available at: https://clinicaltrials.gov/ct2/show/NCT01983683.
NCT01987895 (2013). Efficacy and safety of cadazolid versus vancomycin in subjects with Clostridium difficile - associated diarrhea. [Internet]. [cited 2023 May 22]. Available at: https://clinicaltrials.gov/ct2/show/NCT01987895.
NCT03964493 (2019). TNP-2092 to treat acute bacterial skin and skin structure infection (P2_ABSSSI). [Internet]. [cited 2022 Aug 30]. Available at: https://www.clinicaltrials.gov/ct2/show/NCT03964493.
NCT03988855 (2019). An Exploratory, Open-Label, Oligo-Center Study to Evaluate the Safety, Efficacy, and Pharmacokinetics of Intravenous DNV3837 in Subjects With Clostridium Difficile Infection. Available at: https://clinicaltrials.gov/ct2/show/NCT03988855 (Accessed June 6, 2023).
NCT04294862 (2020). Tissue distribution, pharmacokinetics, safety, and tolerability after a single dose of TNP-2092 in participants undergoing primary total hip or knee arthroplasty. [Internet]. [cited 2023 May 22]. Available at: https://clinicaltrials.gov/ct2/show/NCT04294862.
Nepali, K., Lee, H. Y., and Liou, J. P. (2019). Nitro-group-containing drugs. J. Med. Chem. 62 (6), 2851–2893. doi:10.1021/acs.jmedchem.8b00147
Neu, H. C. (1991). Synergy and antagonism of combinations with quinolones. Eur. J. Clin. Microbiol. Infect. Dis. 10 (4), 255–261. doi:10.1007/BF01966998
Nikolaidis, I., Favini-Stabile, S., and Dessen, A. (2014). Resistance to antibiotics targeted to the bacterial cell wall. Protein Sci. 23 (3), 243–259. doi:10.1002/pro.2414
Omura, K., and Swern, D. (1978). Oxidation of alcohols by “activated” dimethyl sulfoxide. a preparative, steric and mechanistic study. Tetrahedron 34 (11), 1651–1660. doi:10.1016/0040-4020(78)80197-5
Périchon, B., and Courvalin, P. (2009). VanA-type vancomycin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53, 4580–4587. doi:10.1128/AAC.00346-09
Pham, T. D. M., Ziora, Z. M., and Blaskovich, M. A. T. (2019). Quinolone antibiotics. Medchemcomm 10 (10), 1719–1739. doi:10.1039/c9md00120d
Piret, J., and Boivin, G. (2021). Pandemics throughout history. Front. Microbiol. 11, 11. doi:10.3389/fmicb.2020.631736
Pokrovskaya, V., and Baasov, T. (2010). Dual-acting hybrid antibiotics: A promising strategy to combat bacterial resistance. Expert Opin. Drug Discov. 5, 883–902. doi:10.1517/17460441.2010.508069
Pokrovskaya, V., Belakhov, V., Hainrichson, M., Yaron, S., and Baasov, T. (2009). Design, synthesis, and evaluation of novel Fluoroquinolone−Aminoglycoside hybrid antibiotics. J. Med. Chem. 52 (8), 2243–2254. doi:10.1021/jm900028n
Proshkin, S., Rahmouni, A. R., Mironov, A., and Nudler, E. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science (1979). 328(5977):504–508. doi:10.1126/science.1184939
Rashid, M. U., Dalhoff, A., Weintraub, A., and Nord, C. E. (2014). In vitro activity of MCB3681 against Clostridium difficile strains. Anaerobe 28, 216–219. doi:10.1016/j.anaerobe.2014.07.001
Robertson, G. T., Bonventre, E. J., Doyle, T. B., Du, Q., Duncan, L., Morris, T. W., et al. (2008). In vitro evaluation of CBR-2092, a novel rifamycin-quinolone hybrid antibiotic: Studies of the mode of action in Staphylococcus aureus. Antimicrob. Agents Chemother. 52 (7), 2313–2323. doi:10.1128/AAC.01649-07
Rybap, M. J., and Mcgrath, B. J. (1996). Combination antimicrobial therapy for bacterial infections. Guidelines for the clinician. Drugs 52, 390–405. doi:10.2165/00003495-199652030-00005
Santajit, S., and Indrawattana, N. (2016). Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed. Res. Int. 2016, 2475067. doi:10.1155/2016/2475067
Sauvage, E., and Terrak, M. (2016). Glycosyltransferases and transpeptidases/penicillin-binding proteins: Valuable targets for new antibacterials. Antibiotics 5 (1), 12. doi:10.3390/antibiotics5010012
Scaiola, A., Leibundgut, M., Boehringer, D., Caspers, P., Bur, D., Locher, H. H., et al. (2019). Structural basis of translation inhibition by cadazolid, a novel quinoxolidinone antibiotic. Sci. Rep. 9 (1), 5634. doi:10.1038/s41598-019-42155-4
Shavit, M., Pokrovskaya, V., Belakhov, V., and Baasov, T. (2017). Covalently linked kanamycin – ciprofloxacin hybrid antibiotics as a tool to fight bacterial resistance. Bioorg Med. Chem. 25 (11), 2917–2925. doi:10.1016/j.bmc.2017.02.068
Stryjewski, M. E., Potgieter, P. D., Li, Y. P., Barriere, S. L., Churukian, A., Kingsley, J., et al. (2012a). Pharmacodynamics of TD-1792, a novel glycopeptide-cephalosporin heterodimer antibiotic used against gram-positive bacteria, in a neutropenic murine thigh model. Antimicrob. Agents Chemother. 56 (3), 1578–1583. doi:10.1128/AAC.05382-11
Stryjewski, M. E., Potgieter, P. D., Li, Y. P., Barriere, S. L., Churukian, A., Kingsley, J., et al. (2012b). TD-1792 versus vancomycin for treatment of complicated skin and skin structure infections. Antimicrob. Agents Chemother. 56 (11), 5476–5483. doi:10.1128/AAC.00712-12
Sujatha, S., and Praharaj, I. (2012). Glycopeptide resistance in gram-positive cocci: A review. Interdiscip. Perspect. Infect. Dis. 2012, 781679. doi:10.1155/2012/781679
Surur, A. S., and Sun, D. (2021). Macrocycle-antibiotic hybrids: A path to clinical candidates. Front. Chem. 9, 659845. doi:10.3389/fchem.2021.659845
Tamma, P. D., Cosgrove, S. E., and Maragakis, L. L. (2012). Combination therapy for treatment of infections with gram-negative bacteria. Clin. Microbiol. Rev. 25, 450–470. doi:10.1128/CMR.05041-11
Tande, A. J., and Patel, R. (2014). Prosthetic joint infection. Clin. Microbiol. Rev. 27 (2), 302–345. doi:10.1128/CMR.00111-13
Theuretzbacher, U. (2020). Dual-mechanism antibiotics. Nat. Microbiol. 5 (8), 984–985. doi:10.1038/s41564-020-0767-0
Tindula, R. J., Ambrose, P. J., and Harralson, A. F. (1983). Aminoglycoside inactivation by penicillins and cephalosporins and its impact on drug-level monitoring. Drug Intell. Clin. Pharm. 17 (12), 906–908. doi:10.1177/106002808301701210
Tyrrell, K. L., Citron, D. M., Warren, Y. A., and Goldstein, E. J. C. (2012). In vitro activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic, against 377 strains of anaerobic bacteria and 34 strains of Corynebacterium species. Antimicrob. Agents Chemother. 56 (4), 2194–2197. doi:10.1128/AAC.06274-11
Umscheid, C. A., Margolis, D. J., and Grossman, C. E. (2011). Key concepts of clinical trials: A narrative review. Postgrad. Med. 123 (5), 194–204. doi:10.3810/pgm.2011.09.2475
World Health Organization. 2021 Antibacterial agents in clinical and preclinical development: an overview and analysis. Available at: https://www.who.int/publications/i/item/9789240047655 (Accessed June 6, 2023).
Worthington, R. J., and Melander, C. (2013). Overcoming resistance to β-Lactam antibiotics. J. Org. Chem. 78 (9), 4207–4213. doi:10.1021/jo400236f
Wright, G. D. (2016). Antibiotic adjuvants: Rescuing antibiotics from resistance. Trends Microbiol. 24 (11), 862–871. doi:10.1016/j.tim.2016.06.009
Yuan, Y., Wang, X., Xu, X., Liu, Y., Li, C., Yang, M., et al. (2020). Evaluation of a dual-acting antibacterial agent, TNP-2092, on gut microbiota and potential application in the treatment of gastrointestinal and liver disorders. ACS Infect. Dis. 6, 820–831. doi:10.1021/acsinfecdis.9b00374
Zhanel, G. G., Trapp, S., Gin, A. S., DeCorby, M., Lagacé-Wiens, P. R. S., Rubinstein, E., et al. (2008). Dalbavancin and telavancin: Novel lipoglycopeptides for the treatment of Gram-positive infections. Expert Rev. Anti-Infective Ther. 6, 67–81. doi:10.1586/14787210.6.1.67
Keywords: antibiotic resistance, drug combinations, hybrid antibiotics, antibiotic pipeline, drug synthesis
Citation: Koh AJJ, Thombare V, Hussein M, Rao GG, Li J and Velkov T (2023) Bifunctional antibiotic hybrids: A review of clinical candidates. Front. Pharmacol. 14:1158152. doi: 10.3389/fphar.2023.1158152
Received: 03 February 2023; Accepted: 02 June 2023;
Published: 12 June 2023.
Edited by:
Shan Cen, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Johannes Zuegg, The University of Queensland, AustraliaCopyright © 2023 Koh, Thombare, Hussein, Rao, Li and Velkov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Li, amlhbi5saUBtb25hc2guZWR1; Tony Velkov, dG9ueS52ZWxrb3ZAbW9uYXNoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.