
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 20 April 2023
Sec. Experimental Pharmacology and Drug Discovery
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1151144
This article is part of the Research TopicThe Calcium-Sensing Receptor: from Physiology to PharmacologyView all 7 articles
 Valeriya Gushchina1
Valeriya Gushchina1 Nadja Kupper1
Nadja Kupper1 Michael Schwarzkopf1
Michael Schwarzkopf1 Gitta Frisch1Karina Piatek1Cornelia Aigner1Alexandra Michel1Hemma Schueffl2
Gitta Frisch1Karina Piatek1Cornelia Aigner1Alexandra Michel1Hemma Schueffl2 Luca Iamartino1,3
Luca Iamartino1,3 Taha Elajnaf1,4Teresa Manhardt1Andrea Vlasaty1
Taha Elajnaf1,4Teresa Manhardt1Andrea Vlasaty1 Petra Heffeter2
Petra Heffeter2 Marcella Bassetto5,6
Marcella Bassetto5,6 Enikö Kállay1
Enikö Kállay1 Martin Schepelmann1*
Martin Schepelmann1*Introduction: The prostaglandin E2 (PGE2) pathway is one of the main mediators of intestinal inflammation. As activation of the calcium-sensing receptor (CaSR) induces expression of inflammatory markers in the colon, we assessed the impact of the CaSR on the PGE2 pathway regulation in colon cancer cells and the colon in vitro and in vivo.
Methods and Results: We treated CaSR-transfected HT29 and Caco-2 colon cancer cell lines with different orthosteric ligands or modulators of the CaSR and measured gene expression and PGE2 levels. In CaSR-transfected HT29CaSR-GFP and Caco-2CaSR-GFP cells, the orthosteric CaSR ligand spermine and the positive allosteric CaSR modulator NPS R-568 both induced an inflammatory state as measured by IL-8 gene expression and significantly increased the expression of the PGE2 pathway key enzymes cyclooxygenase (COX)-2 and/or prostaglandin E2 synthase 1 (PGES-1). Inhibition of the CaSR with the calcilytic NPS 2143 abolished the spermine- and NPS R-568-induced pro-inflammatory response. Interestingly, we observed cell-line specific responses as e.g. PGES-1 expression was affected only in HT29CaSR-GFP but not in Caco-2CaSR-GFP cells. Other genes involved in the PGE2 pathway (COX-1, or the PGE2 receptors) were not responsive to the treatment. None of the studied genes were affected by any CaSR agonist in GFP-only transfected HT29GFP and Caco-2GFP cells, indicating that the observed gene-inducing effects of spermine and R-568 were indeed mediated by the CaSR.
In vivo, we had previously determined that treatment with the clinically approved calcimimetic cinacalcet worsened symptoms in a dextran sulfate sodium (DSS)-induced colitis mouse model. In the colons of these mice, cinacalcet significantly induced gene expression of PGES-2 and the EP3 receptor, but not COX-2; while NPS 2143 increased the expression of the PGE2-degrading enzyme 15-hydroxyprostaglandin dehydrogenase (15-PGDH). Importantly, neither treatment had any effect on the colons of non-DSS treated mice.
Discussion: Overall, we show that activation of the CaSR induces the PGE2 pathway, albeit with differing effects in vitro and in vivo. This may be due to the different microenvironment in vivo compared to in vitro, specifically the presence of a CaSR-responsive immune system. Since calcilytics inhibit ligand-mediated CaSR signaling, they may be considered for novel therapies against inflammatory bowel disease.
The calcium-sensing receptor (CaSR) is a versatile G protein-coupled receptor that is best known for its regulation of Ca2+ homeostasis in the parathyroid glands (Brown et al., 1993; Hannan et al., 2018).
Apart from Ca2+, the CaSR acts as a multimodal sensor for a plethora of other orthosteric agonists including divalent and trivalent cations, aromatic amino acids (e.g., Phe, Trp), polyamines (e.g., spermine), polypeptides, and even antibiotics (e.g., neomycin), (Conigrave and Brown, 2006; Díaz-Soto et al., 2016). Because of this diversity in ligands, and its tendency to couple to numerous G proteins (Gq/11, Gi/o and G12/13), the CaSR is involved in the initiation of multiple downstream signaling pathways by a process known as biased signaling (Leach et al., 2014). In addition to these natural ligands, small molecules, known as “calcimimetics” and “calcilytics,” have been developed that target the CaSR on its allosteric binding sites. NPS R-568 and cinacalcet are both calcimimetics, which bind to the receptor’s transmembrane domain and increase the sensitivity of the CaSR to its orthosteric ligands. Conversely, calcilytics, like NPS 2143, decrease the sensitivity of the CaSR to its ligands (Nemeth and Goodman, 2016).
The CaSR is expressed also in tissues outside of the Ca2+ homeostatic system, such as the vasculature, the lung and the intestine. In these tissues, the CaSR regulates a range of physiological processes which are responsible for vascular tone and mineral homeostasis (Schepelmann et al., 2016; Schepelmann et al., 2022), intestinal fluid secretion and water transport, epithelial layer integrity, inflammation, as well as proliferation, differentiation, and apoptosis, as reviewed in (Hannan et al., 2018; Iamartino et al., 2018).
The role of the CaSR in the intestines is not quite understood yet. While previous studies suggested an anti-inflammatory role for the CaSR in the colon (Cheng et al., 2014; Zhang et al., 2015a; Mine and Zhang, 2015), we have recently found that CaSR activation with calcimimetics (NPS R-568, and cinacalcet) promoted while calcilytics (NPS 2143) inhibited intestinal inflammation (Elajnaf et al., 2019; Iamartino et al., 2020; Schepelmann et al., 2021). We thus hypothesized that calcilytics may constitute a novel therapy for inflammatory bowel disease (IBD), just as in the lungs, where calcilytics are proposed as a safe and effective asthma treatment (Yarova et al., 2015; Yarova et al., 2021).
One of the central pathways involved in intestinal inflammation is the prostaglandin E2 (PGE2) pathway. Cyclooxygenases (COX) catalyze the rate-limiting steps of PGE2 synthesis, which is the conversion of arachidonic acid to PGH2. There are two primary isomers of COX. COX-1 is constitutively expressed and plays a role in tissue homeostasis. COX-2 is inducible and upregulated by various extracellular and intracellular stimuli, e.g., lipopolysaccharides, interleukin-1, tumor necrosis factor, epidermal growth factor, or oncogenes. PGH2 is then isomerized to PGE2 by one of the isoforms of prostaglandin E synthases (PGES). Secreted PGE2 can then bind to one of the four extracellular PGE2 receptors (EP1-4), (Greenhough et al., 2009). The accumulation of COX-2-derived PGE2 is subject to the upregulation of several signaling pathways and downregulation of apoptotic proteins (Gandhi et al., 2017). Thus, COX-2 over-expression contributes to inflammation, hence colitis and colitis-associated colorectal cancer. 15-Hydroxyprostaglandin dehydrogenase (15-PGDH) in turn inhibits prostaglandin signaling by degrading PGE2.
We have previously found that CaSR activation with a calcimimetic worsened clinical symptoms in a mouse model of colitis (Elajnaf et al., 2019), and upregulated various inflammatory pathways in CaSR-transfected human colorectal cancer cells (Iamartino et al., 2020). In the present study, we therefore aimed to determine the effect of the CaSR on the PGE2 pathway both in vitro and in vivo. We were able to show that activation of the CaSR induced gene expression of several PGE2 pathway genes as well as PGE2 secretion in a ligand-dependent manner, and that this effect could be prevented by calcilytics.
All solvents and reagents were obtained from Merck (United States), unless otherwise stated. CaSR allosteric modulators cinacalcet, NPS R-568 and NPS 2143 were obtained from Tocris Bioscience (United Kingdom). GSK3004774 was custom synthetized previously (Elajnaf et al., 2019).
We used two colon cancer cell lines which were lentivirally transduced to stably express the CaSR fused to GFP (HT29CaSR−GFP; Caco-2CaSR−GFP) or GFP alone as control (HT29GFP; Caco-2GFP), (Iamartino et al., 2020).
Both cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) medium, supplemented with 10% fetal calf serum (FCS), 10 mM HEPES buffer solution, 2 mM L-Glutamine, and 100 U/mL Pen-Strep (all Thermo Fischer Scientific, Gibco, United States). Cells were maintained at 37°C in a humid atmosphere with 5% CO2. Passaging occurred once a week, while selection for the stably transfected cells with puromycin occurred every 4 weeks. Once the cells reached 80% confluency, they were treated with vehicle (DMSO or H2O), different orthosteric ligands (5 mM spermine, 300 µM neomycin, 1 mM L-Phe, or 1 mM L-Trp) or allosteric modulators of the CaSR (1 µM NPS R-568, 10 µM GSK3004774, or NPS 2143), (for exact treatments and their concentrations, see individual figures). Concentrations of the orthosteric ligands were chosen based on the respective publications (Quinn et al., 1997; Ziegelstein et al., 2006; Mine and Zhang, 2015). The final concentration of DMSO was 0.1% (v/v) in all conditions.
For our in vivo experiments, we analyzed gene expression from colons of 8-week old female Balb/C mice with chemically induced colitis, treated with allosteric CaSR modulators (cinacalcet, GSK3004774, or NPS 2143; 10 mg/kg) or vehicle (20% cyclodextrin) by oral gavage for 2 weeks from a previous study (Elajnaf et al., 2019). In that experiment, colitis had been chemically induced by administration of 3.5% dextran sulphate sodium (DSS) in drinking water over 7 days (see Elajnaf et al., 2019 for experimental and treatment details). The non-colitis group received CaSR modulators (cinacalcet or NPS 2143; 30 mg/kg) or vehicle (20% cyclodextrin) for 2 weeks. Pieces of max. 20 mg of the proximal and distal colon were used for RNA isolation. All animal experiments were approved by the Ethics Committee of the Medical University of Vienna and the Austrian Federal Ministry of Education, Science and Research (BMWFW-66.009/0401-WF/V/3b/2017, BMWFW-66.009/0006-WF/V/3b/2017 and BMBWF-66.009/0394-V/3b/2018) and carried out in accordance with the European Union Regulations on Care and Use of Laboratory Animals.
Total RNA was isolated using TRIzol reagent (Invitrogen, United States) or EXTRAzol reagent (Blirt, PL) for cells and ReliaPrep RNA Miniprep Systems (Promega, United States) for homogenized colon tissue, following the respective manufacturer’s protocol. 1 μg of total RNA was used for cDNA synthesis using the High-Capacity RNA-to-cDNA Kit (Thermo Fischer Scientific, Applied Biosystems, United States). Obtained cDNA was used for reverse transcription quantitative PCR (RT-qPCR). RT-qPCR reactions were performed with Power SYBR Green PCR Master Mix (Thermo Fischer Scientific, Applied Biosystems). The primers were purchased from Merck and are listed in Supplementary Table S1. Results were normalized to the average of either hRPLP0 & hB2m (in vitro experiments), or mEef1β2 and mβ-actin (in vivo experiments) as housekeeping genes, using either commercially available human total RNA (Takara, Kusatsu, Japan) or mouse colon RNA (Takara, ClonTech, Japan) as calibrator. Relative gene expression was then determined using the ΔΔCT method.
Once cells reached 80% confluency, they were treated with either CaSR allosteric modulators (single treatment or double treatment) or vehicle control. After 4 h the supernatant was collected and PGE2 concentration was measured using the Prostaglandin E2 Parameter Assay Kit (R&D Systems, Catalog #KGE004B, United States), following the manufacturer’s protocol.
Statistical analysis and visualization were performed using GraphPad Prism 9.3.1 software (San Diego, CA, United States) and RStudio 2022.07.2 (Boston, MA, United States). Results are presented as mean ± standard deviation. Data were analyzed by one-way ANOVA or Mann-Whitney U test; p < 0.05 was considered statistically significant. The specific statistical tests for each dataset are indicated in each respective figure and table legends.
Because CaSR expression is lost in colon cancer cell lines and reliable “normal” colon epithelial cell lines are not available, we used CaSR-transfected (HT29CaSR−GFP and Caco-2CaSR−GFP) or empty vector-transfected (HT29GFP and Caco-2GFP) colorectal cancer cells as model systems for our in vitro experiments, as previously described (Iamartino et al., 2020). In previous studies, we have already shown that calcimimetics induced inflammatory gene expression via the CaSR (Iamartino et al., 2020; Schepelmann et al., 2021). The gastrointestinal tract is rich in nutrients of which some are known CaSR ligands. As cellular responses mediated by the CaSR are highly ligand-dependent, we assessed the effect of selected physiological orthosteric CaSR ligands on the PGE2 pathway. To confirm that the selected ligands indeed elicit inflammatory response in the cells, we first measured their effect on the expression of the inflammatory marker interleukin 8 (IL-8). The allosteric CaSR activators (calcimimetics) NPS R-568 and GSK3004774 (Sparks et al., 2017; Elajnaf et al., 2019) were used as positive controls for CaSR-specific inflammatory gene induction (Schepelmann et al., 2021).
Treatment with spermine and NPS R-568 significantly upregulated IL-8 expression (11.1-fold and 4.8-fold, respectively) in HT29CaSR−GFP cells, while treatment with neomycin, L-Phe, L-Trp or GSK3004774 did not affect the expression of IL-8 (Figure 1A). Higher concentrations of L-Phe and L-Trp (5 mM and 10 mM) were also unable to upregulate IL-8 (Supplementary Table S2). None of the treatments had an effect on IL-8 gene expression in HT29GFP cells (Figure 1B).
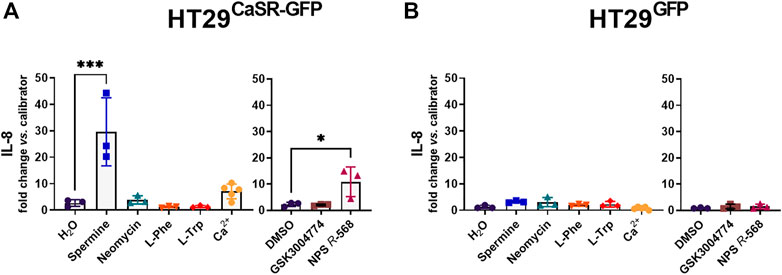
FIGURE 1. Induction of an inflammatory response in HT29 cells via the CaSR is ligand dependent. Relative gene expression of IL-8 in (A) HT29CaSR−GFP and (B) HT29GFP cells after 4 h treatment with CaSR ligands (left panel: H2O as vehicle control, 5 mM spermine, 300 µM neomycin, 1 mM L-Phe, 1 mM L-Trp, and 5 mM Ca2+) or CaSR modulators (right panel: 0.1% DMSO as vehicle control, 10 µM GSK3004774, and 1 µM NPS R-568). Mean ± SD, N = 3–5, one-way ANOVA with Dunnett’s post hoc test vs. vehicle control (H2O or 0.1% DMSO), *p < 0.05, ***p < 0.001.
We then assessed the effect of the ligands on the main genes constituting the PGE2 pathway (see introduction). As mentioned above, the PGE2 pathway plays a pivotal role in intestinal inflammation. Stimulating the CaSR with the orthosteric agonist spermine or the positive allosteric modulator NPS R-568 resulted in the same pattern for COX-2 and PGES-1 expression as for IL-8 expression. In HT29CaSR−GFP cells, both spermine and NPS R-568 significantly upregulated the expression of COX-2 (7.6-fold and 9.3-fold, respectively), (Figure 2B), and PGES-1 (2.9-fold and 3.3-fold, respectively), (Figure 2C). NPS R-568 upregulated the expression of EP1, while spermine upregulated EP4 expression (4.9-fold and 2.1-fold, respectively), (Figures 2G, H). Other genes involved in the PGE2 pathway were not affected (Figures 2A, D–F). In HT29GFP cells, the treatments had no significant effect on the expression of the PGE2 pathway genes (Supplementary Figure S1), indicating the presence of the CaSR to be necessary for both spermine- and NPS R-568-induced gene expression.
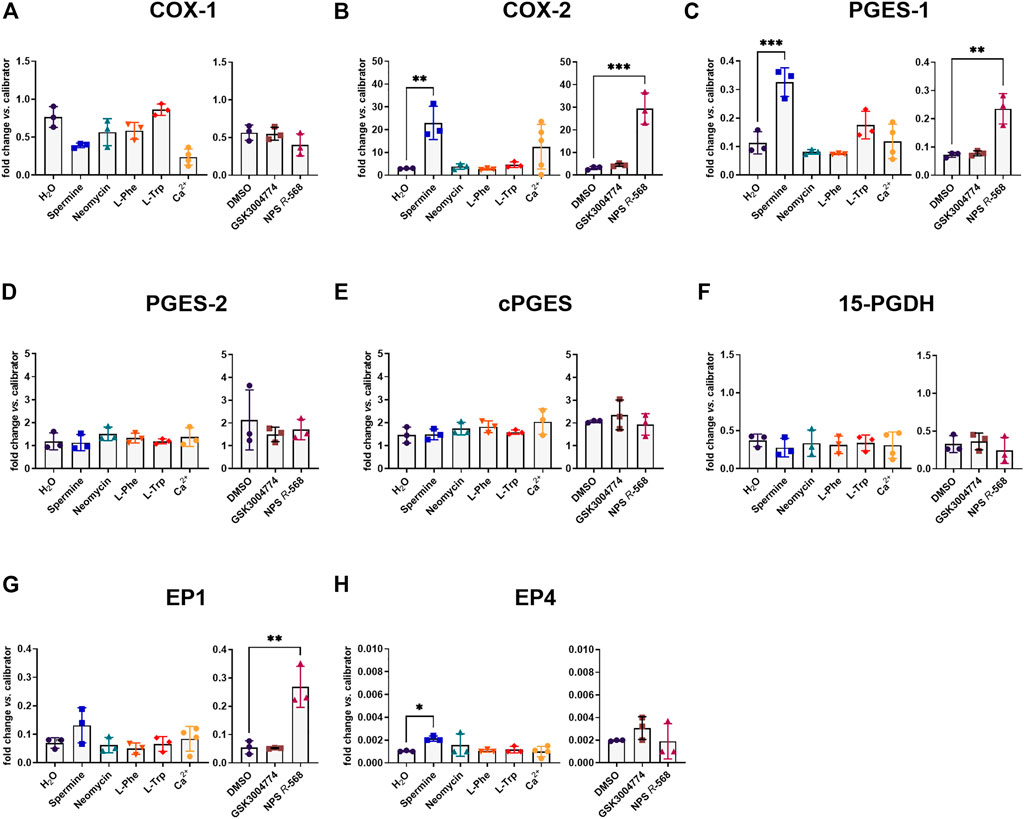
FIGURE 2. Induction of PGE2 pathway genes by CaSR ligands in HT29CaSR−GFP cells. Relative gene expression of (A) COX-1, (B) COX-2, (C) PGES-1, (D) PGES-2, (E) cPGES, (F) 15-PGDH, (G) EP1, and (H) EP4 in HT29CaSR−GFP cells after 4 h treatment with CaSR ligands (left panel: H2O as vehicle control, 5 mM spermine, 300 µM neomycin, 1 mM L-Phe, 1 mM L-Trp, and 5 mM Ca2+) or CaSR modulators (right panel: 0.1% DMSO as vehicle control, 10 µM GSK3004774, and 1 µM NPS R-568). Mean ± SD, N = 3–5, one-way ANOVA with Dunnett’s post hoc test vs. vehicle control (H2O or 0.1% DMSO), **p < 0.01, ***p < 0.001.
We also assessed the mRNA levels of the CaSR gene, as it has been suggested that the expression is increased upon activation of the CaSR due to a process called agonist-driven insertional signaling (ADIS), (Grant et al., 2011). Treatment of HT29CaSR−GFP cells with both spermine and NPS R-568 upregulated CaSR gene expression (4.6-fold and 5.2-fold, respectively). Elevated extracellular Ca2+ (5 mM) also led to a significant increase of CaSR gene expression (2.9-fold), (Supplementary Figure S2A). Again, no effect was observed in the HT29GFP cells, where CaSR expression was close to undetectable under all conditions (Supplementary Figure S2B).
All numerical results are provided in Supplementary Table S2. The normalized expression of all investigated genes in the HT29CaSR−GFP and HT29GFP cells are visualized in Supplementary Figure S3, demonstrating the dependence of gene expression on the individual ligand and presence of the CaSR.
Previous experiments have shown that pre-treatment with the calcilytic NPS 2143 prevented the induction of CaSR and IL-8 gene expression by the allosteric CaSR activator NPS R-568 (Schepelmann et al., 2021). We now found that pre-treatment of HT29CaSR−GFP cells with NPS 2143 also inhibited the upregulation of IL-8, CaSR, COX-2 and PGES-1 induced by the orthosteric CaSR-agonist spermine (Figures 3A–D).
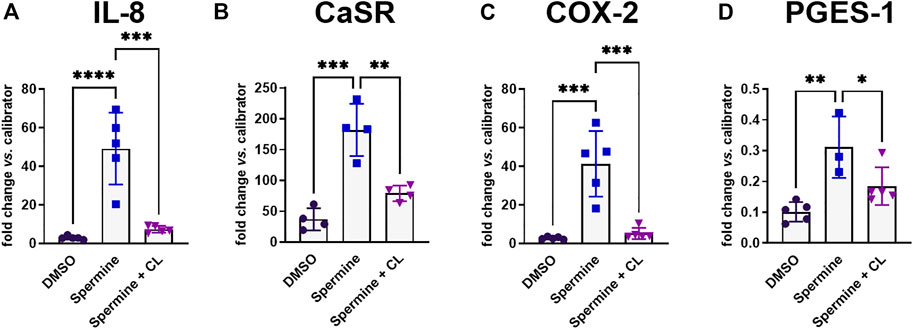
FIGURE 3. Gene induction by spermine can be suppressed by calcilytics. Relative gene expression of (A) IL-8, (B) CaSR, (C) COX-2, and (D) PGES-1 in HT29CaSR−GFP cells after treatment with 5 mM spermine alone or together with the calcilytic 1 µM NPS 2143 (CL) for 4 h (0.1% DMSO as vehicle control). Mean ± SD, N = 3–5, one-way ANOVA with Tukey’s post hoc test (0.1% DMSO), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
As we had observed a significant upregulation of COX-2 and mPGES-1 gene expression, we also assessed whether this translated into changes in secretion of PGE2 by the cells. Treatment with NPS R-568 for 4 h increased PGE2 secretion (17.1-fold) in HT29CaSR−GFP cells (Figure 4A), compared with vehicle, while pre-treatment with NPS 2143 inhibited the NPS R-568-induced PGE2 secretion. No effect of NPS R-568 treatment on PGE2 secretion was observed in HT29GFP cells (Figure 4B).
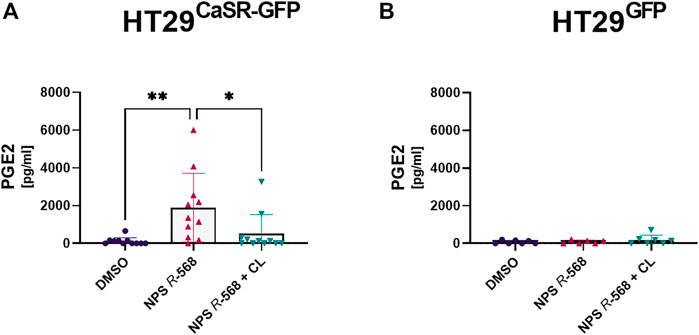
FIGURE 4. Induction and suppression of PGE2 secretion by CaSR activation and inhibition. PGE2 secretion in supernatant of (A) HT29CaSR−GFP and (B) HT29GFP after 4 h treatment with either vehicle control (0.1% DMSO), 1 µM NPS R-568 alone, or together with 1 µM of the calcilytic NPS 2143 (CL). Mean ± SD, N = 6–11, one-way ANOVA with Tukey’s post hoc test (0.1% DMSO), *p < 0.05, **p < 0.01.
As we had determined that both the orthosteric CaSR agonist spermine and the allosteric positive CaSR modulator NPS R-568 induced inflammatory responses in the HT29CaSR−GFP but not the HT29GFP cells, we were wondering whether the presence of the CaSR alone was also enough to increase susceptibility of the cells to inflammatory responses. Thus, we induced an inflammatory response in the cells using increasing concentrations (0.1 ng/μL–25 ng/μL) of the well-known inflammatory cytokine IL-1β, and measured IL-8 gene expression. IL-1β induced a strong dose-dependent increase of IL-8 only in HT29CaSR−GFP cells (Figure 5A), while a much weaker effect was observed in HT29GFP cells (Figure 5B).
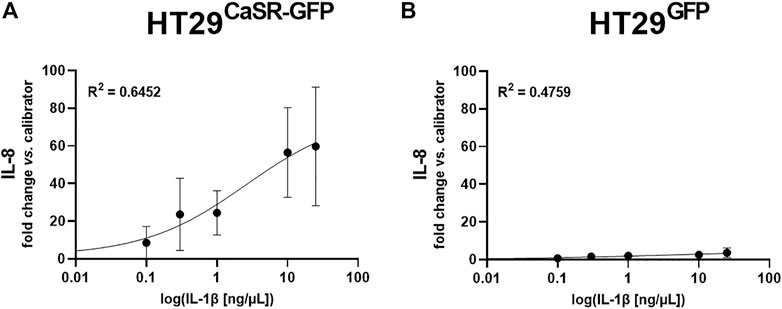
FIGURE 5. Induction of IL-8 gene expression via IL-1β is enhanced when the CaSR is present. Dose-response curve of relative gene expression of IL-8 in (A) HT29CaSR−GFP cells and (B) HT29GFP cells after 4 h treatment with IL-1β (0.1 ng/μL–25 ng/μL). Mean ± SD, N = 3–4. Sigmoidal 4-parameter fit, 0-concentration set to 10−20 ng/μL for logarithmic curve fitting. Calculated EC50 equals 2.49 ± 1.27 ng/μL.
To confirm our results, we repeated the experiments in CaSR and empty-vector transfected Caco-2 cells. Caco-2CaSR−GFP cells responded to the treatment with CaSR activators in a similar manner as HT29CaSR−GFP cells. Both NPS R-568 and spermine upregulated CaSR, and COX-2 expression, while IL-8 was affected significantly only by spermine but not NPS R-568 (Figures 6A–C). Treatment of Caco-2CaSR−GFP cells with Ca2+ upregulated only COX-2 gene expression, which in HT29CaSR−GFP cells affected only the expression of the CaSR itself (the observed upregulation of the CaSR in Caco-2CaSR−GFP following Ca2+ treatment Cells did not reach statistical significance with P ∼ 0.1). In Caco-2CaSR−GFP cells, pre-treatment with NPS 2143 was also successful in abolishing spermine-induced (inflammatory) gene expression. We also noticed that the expression levels of all affected genes were slightly lower in Caco-2CaSR−GFP cells than in the HT29CaSR−GFP cells. In contrast to the HT29CaSR−GFP cells, none of the treatments had any effect on PGES-1 gene expression in Caco-2CaSR−GFP cells (Figure 6D). As in the HT29GFP cells, the treatments had no effect on the gene expression of any of the selected targets in Caco-2GFP cells (Supplementary Figure S4). PGE2 secretion was nearly undetectable both in Caco-2CaSR−GFP and in Caco-2GFP cells. Compared with Caco-2GFP cells, treatment with NPS R-568 induced a slight elevation of PGE2 secretion by the Caco-2CaSR−GFP cells, but this did not reach statistical significance (Supplementary Figure S5). All numerical results for Caco-2CaSR−GFP and Caco-2GFP cells are presented in Supplementary Table S3.
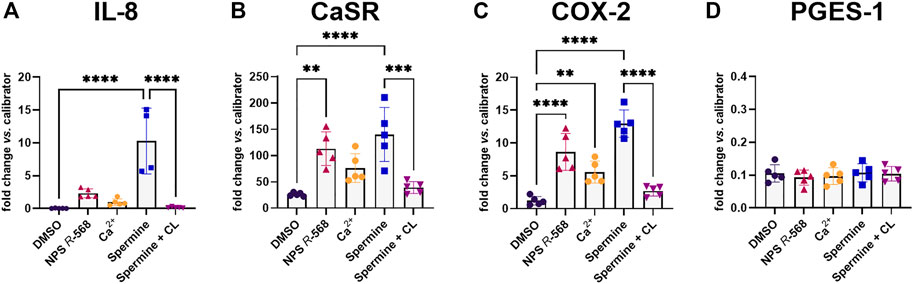
FIGURE 6. Gene induction and suppression by CaSR ligands in Caco-2CaSR−GFP cells. Relative gene expression of (A) IL-8, (B) CaSR, (C) COX-2, and (D) PGES-1 in Caco-2CaSR−GFP cells after 4 h treatment with either vehicle control (0.1% DMSO), 1 µM NPS R-568, CaSR ligands (5 mM Ca2+, and 5 mM spermine), or 5 mM spermine in combination with 1 µM NPS 2143 (CL). Mean ± SD, N = 3–5, one-way ANOVA with Tukey’s post hoc test (0.1% DMSO), **p < 0.01, ***p < 0.001, ****p < 0.0001.
We next tested whether the PGE2 pathway would also be affected by modulation of the CaSR in vivo. Thus, we measured gene expression levels of CaSR and several PGE2 pathway genes in non-inflamed mouse colons, and in the inflamed colons of mice from our previous study (Elajnaf et al., 2019), which had been treated with different CaSR modulators. Due to differences in gene expression, molecular patterns, and morphology between the proximal and distal colon (Goldstone et al., 2012; Missiaglia et al., 2014), we analyzed both parts of the colon separately.
CaSR gene expression was detected in all samples, but was unaffected by the treatment of the mice with CaSR modulators. In the inflamed colons the calcimimetics, cinacalcet and the non-gut-absorbable GSK3004774 (each 10 mg/kg), significantly increased expression of mPGES-2 in both the proximal and distal colon (Figure 7A). Cinacalcet also induced EP3 expression in the proximal colon (Figure 7B). We also observed a higher expression level of the PGE2 degrading enzyme 15-PGDH in the proximal colon after treatment with 10 mg/kg NPS 2143 (Figure 7C). Other genes of the PGE2 pathway were not affected (Supplementary Figure S6). In the non-inflamed colons, none of the CaSR modulators, even though administered at 3x higher concentration, affected gene expression of any of the investigated PGE2 pathway genes (Supplementary Figure S7). All numerical results are provided in Supplementary Table S4.
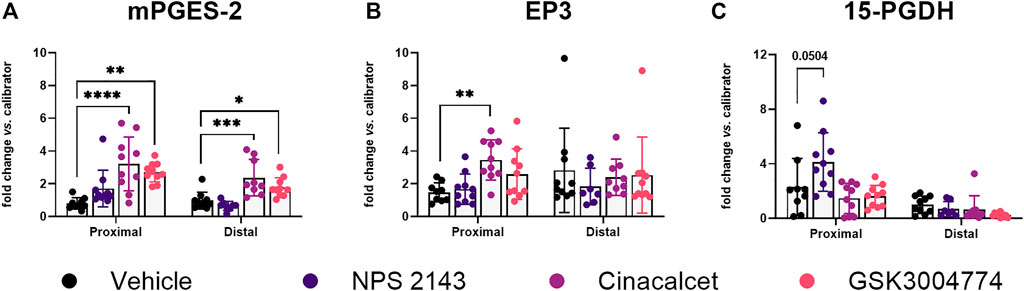
FIGURE 7. Altered PGE2 pathway genes in CaSR-ligand treated mice with colitis. Relative gene expression of (A) mPGES-2, (B) EP3, and (C) 15-PGDH in the colons of mice with DSS-induced colitis treated with CaSR allosteric modulators (each 10 mg/kg). Mean ± SD, N = 7–10. Proximal and distal colon were analyzed separately by one-way ANOVA with Dunnett’s post hoc test vs. vehicle control (20% cyclodextrin), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Recently, we have proposed a pro-inflammatory role of the CaSR in the intestine, as stimulation of the CaSR in transfected colon cancer cells with positive CaSR allosteric modulators induced the expression of genes involved in several inflammatory pathways (Iamartino et al., 2020). In the current study, we have now shown that the PGE2 pathway is indeed one of the involved pathways.
Both the orthosteric ligand spermine, and the allosteric modulator NPS R-568 increased COX-2 and PGES-1 expression in HT29CaSR−GFP cells, but not in HT29GFP cells, once again marking the CaSR as the mediator of these pro-inflammatory effects. It has been shown previously that COX-2 and PGES-1 are upregulated in the inflamed intestinal tissue and in colorectal tumors (Williams et al., 1999; Yoshimatsu et al., 2001; Wang and DuBois, 2010). Both COX and the PGES isoforms are enzymes involved in the synthesis of PGE2. COX catalyzes the first step of PGE2 synthesis, that is, the conversion of arachidonic acid to PGH2. PGES subsequently isomerizes PGH2 to PGE2. The combined increase in expression of both genes eventually leads to increased PGE2 production and secretion. Secreted PGE2 then binds to one of the 4 PGE2 receptors (EP1–EP4) which determines cellular response (Greenhough et al., 2009). The CaSR affects the PGE2 pathway in a cell-line specific manner, as we have observed some distinct differences in the effect of the CaSR modulators between HT29CaSR−GFP and Caco-2CaSR−GFP cells. In contrast to HT29CaSR−GFP cells, Caco-2CaSR−GFP cells responded to CaSR stimulation only with COX-2 upregulation, while neither PGES-1 expression nor PGE2 production was altered significantly. COX-2 and PGES are often expressed at different levels in individual colorectal tumors, possibly due to different expression control mechanisms (Yoshimatsu et al., 2001). Although both HT29 and Caco-2 cells are derived from colorectal adenocarcinomas, they differ in genetic background and their potential to differentiate. It is thus possible that Caco-2 cells lack certain downstream effectors of the CaSR which are essential for PGES-1 upregulation, or activate different signaling pathways that do not lead to PGES-1 upregulation. The lack of PGES-1 induction may also explain why in Caco-2CaSR−GFP cells PGE2 production was not increased. Even though both COX-2 and PGES-1 contribute to PGE2 production, it was suggested that activation of PGES-1 is more closely related to PGE2 induction in colon cancer (Kim and Kim, 2022). As shown before (Iamartino et al., 2020; Schepelmann et al., 2021), pre-incubation with the calcilytic NPS 2143 significantly reduced the NPS R-568-, and thus CaSR-induced gene expressions, as demonstrated before, indicating that the observed responses really were mediated via CaSR stimulation rather than unspecific actions of the tested ligands.
Spermine was identified as an orthosteric ligand of the CaSR already in 1997, shortly after the receptor’s first cloning (Quinn et al., 1997). Polyamines such as spermine, found in our daily diet, or produced by intestinal microbiota, are involved in the maintenance of proper gastrointestinal function (Pegg, 2014; Liu et al., 2022). The role of spermine seems to be multifold: spermine supports the barrier function of the gut, induces gut maturation and increases longevity (Bekebrede et al., 2020). It also suppresses cytokine synthesis in macrophages, preventing tissue damage-associated inflammation (Zhang et al., 2000). On the other hand, when acting as agonist of the CaSR, we have now demonstrated that spermine induces pro-inflammatory marker expression in the colon cells in vitro which can be inhibited by pre-incubation of the cells with the negative CaSR allosteric modulator NPS 2143. A similar situation has been observed in the lung. While some reports suggest an anti-inflammatory effect of spermine in the lungs (Wawrzyniak et al., 2021), spermine and other polyamines were shown to promote increased pulmonary fibrosis, airway hyperresponsiveness and asthma severity by activating the CaSR expressed in airway smooth muscle cells and epithelial cells. Calcilytics are able to prevent these CaSR-induced effects (Yarova et al., 2015; Wolffs et al., 2019). Here, we have demonstrated that calcilytics can exert the same effect also in the colon.
Interestingly, of the tested orthosteric CaSR ligands, only spermine affected gene expression, while i.a. the aromatic amino acids had no effect. Amino acids elicit slightly different intracellular signaling responses from the CaSR than spermine which may account for this difference. It is also possible that the colon cells simply would not be sensitive to amino acids as they would be constantly exposed to them in the form of the passing chyme. An important factor to take into consideration in these studies is the absence of an immune system within the highly artificial in vitro environment which consists only of a single type of cell.
The role of the CaSR in inflammation is very complex. Amongst the directly affected tissues, it is also present on a variety of immune cells (Iamartino and Brandi, 2022). We have demonstrated previously that the immune system in the colon is directly affected by CaSR modulators (Elajnaf et al., 2019). In that study we had used the DSS-induced colitis model of IBD, as it resembles human ulcerative colitis (Chassaing et al., 2014). While treatment with NPS 2143 reduced some of the colitis symptoms, the calcimimetic cinacalcet exhibited a rather pro-inflammatory effect. In the present study, we now tested whether activation of the PGE2 pathway is affected in the observed calcimimetic-mediated increase of disease severity, as suggested by the results from the transfected colon cancer cells. Interestingly, in the inflamed colons, treatment with the CaSR allosteric modulators did indeed affect the PGE2 pathway, but not via the expression of COX-2 (in contrast to the in vitro results where COX-2 was significantly upregulated in both HT29CaSR−GFP and Caco-2CaSR−GFP cells after stimulation of the CaSR).
Instead, stimulation of the CaSR in vivo affected the PGE2 pathway by upregulation of mPGES-2 and EP3. Treatment with both cinacalcet and GSK3004774 significantly increased the expression of mPGES-2 in the proximal and distal colon, while mPGES-1 was not affected at all. mPGES-1 is a highly inducible isoform of PGES, while the other two isoforms cPGES and mPGES-2 are constitutively expressed (Sampey et al., 2005). mPGES-1 is induced by inflammatory stimuli and the expression levels are increased in the inflamed mucosa of patients with IBD (Subbaramaiah et al., 2004). PGES isomers are functionally coupled to respective COX isomers. For instance, mPGES-1 is preferably coupled to COX-2 and thus produces PGE2 in a COX-2-dependent way (Murakami et al., 2000). Thus, the unaffected COX-2 gene expression levels might explain why in turn no upregulation of mPGES-1 was observed. Colorectal carcinoma is (as of yet) the only tissue in which an increased mPGES-2 expression was reported (Murakami et al., 2003).
We have also observed that in vivo, the effect of the CaSR modulators depend on an inflamed environment, as treatment with the CaSR modulators affected certain PGE2 pathway genes only in the colons of mice with DSS-induced colitis, but not in the colons of mice without colitis, indicating a selective action of the CaSR in an inflamed environment. This fits also well with our experiment of inducing exogenous inflammation in the HT29 cells using rising levels of IL-1β. The CaSR expressing HT29CaSR−GFP cells reacted with much stronger upregulation of IL-8 gene expression to IL-1β than the control HT29GFP cells, indicating that the presence of the CaSR alone already may increase the cells’ susceptibility, or “prime” them, towards inflammatory stimuli, e.g., as suggested by the observation that NPS R-568 upregulated IL-1R1 expression in HT29CaSR−GFP cells [RNASeq data from (Iamartino et al., 2020)].
In our previous study (Elajnaf et al., 2019) we found that the calcilytic NPS 2143 reduced the colitis symptoms in mice. In our current study, we could show that the anti-inflammatory effect of NPS 2143 was, at least partially, mediated via the upregulation of 15-PGDH. An antitumor potential of the PGE2-degrading enzyme 15-PGDH was already demonstrated as decreased 15-PGDH levels were shown to correlate with colitis-associated colon cancer development (Choi et al., 2014). Contrary to this, an inhibition of 15-PGDH was shown to promote tissue regeneration and reduced inflammatory cytokines in the colons of a DSS-induced colitis mouse model (Zhang et al., 2015b; Hu et al., 2022). Calcilytics, such as NPS 2143, have been shown to reduce inflammation, and disease-associated symptoms in asthma and pulmonary fibrosis, and are now in development for its therapy (Yarova et al., 2015; Yarova et al., 2018; Yarova et al., 2021). Moreover, NPS 2143 has been shown to ameliorate β-amyloid-induced cognitive deficits, which is partially mediated via the cytosolic phospholipase A2 and PGE2 pathway (Feng et al., 2020). In agreement with the before-mentioned findings, we suggest that calcilytics might also show potential as novel substances for the prevention of colitis and colitis-associated colon cancer, providing a safer alternative to non-steroidal anti-inflammatory drugs (NSAIDs) with less side-effects, as the use of NSAIDs to inhibit COX-2 was found to exacerbate IBD (Meyer et al., 2006; Long et al., 2016).
This study sheds further light on the pro-inflammatory actions of the CaSR. Despite the differences between the more artificial in vitro model system and the in vivo results, it is clear that activating the CaSR in the colon modulates the PGE2 pathway as one of the underlying mechanisms for the pro-inflammatory response following CaSR activation. Our results are in line with other studies that demonstrated increased PGE2 production after CaSR stimulation in neurons, dental pulp cells, and fibroblasts from jaw cysts and gingiva (Ogata et al., 2006; Feng et al., 2020; An et al., 2022). Apart from the PGE2 pathway, the CaSR also promotes inflammation via activation of the NLRP3 inflammasome in monocytes and adipocytes (Rossol et al., 2012; D'Espessailles et al., 2018; Jäger et al., 2020) as well as the NF-κB pathway in peripheral blood (Li et al., 2013). It is evident that the NLRP3 inflammasome (Bauer et al., 2010; Zhen and Zhang, 2019) and NF-κB pathway (Atreya et al., 2008) may contribute to intestinal inflammation, however the role of the CaSR in this context is yet to be determined.
Here, we demonstrate for the first time that activation of the CaSR induces the PGE2 pathway in CaSR-transfected colon cancer cells and in inflamed mouse colons. CaSR-mediated inflammatory responses in the colon may therefore be, at least in part, mediated through the PGE2 pathway.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the Ethics Committee of the Medical University of Vienna and the Austrian Federal Ministry of Education, Science and Research.
Conceptualization: MAS and EK; Methodology: LI, PH, and EK; Validation: VG and NK; Formal analysis: VG; Investigation: VG, NK, MIS, KP, CA, AM, GF, HS, LI, TE, TM, AV, EK, and MAS; Resources: PH, EK, MAS, and MB; Data curation: MAS, VG, and NK; Writing–original draft: VG; Writing–Review and Editing: MAS and EK; Visualization: VG; Supervision: MAS, PH, and EK; Project administration: MAS; Funding acquisition: MAS, PH, and EK.
This research was funded by the Austrian Science Fund (FWF) and the Herzfelder’sche Familienstiftung, Grant Number. P 32840-B (to MAS); FWF, Grant Number. P 29948-B28 (to EK) and P 32886 (to PH); the European Union’s Horizon 2020 research and innovation programme, Grant Agreement Number. 675228 (to EK). Open Access Funding by the FWF.
Author LI is employed by SiSaf Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1151144/full#supplementary-material
CaSR, Calcium-sensing receptor; IBD, Inflammatory bowel disease; PGE2, Prostaglandin E2; COX, Cyclooxygenase; PGES, Prostaglandin E synthase; 15-PGDH, 15-Hydroxyprostaglandin dehydrogenase; DSS, Dextran sulphate sodium.
An, S., Chen, Y., Yang, T., Huang, Y., and Liu, Y. (2022). A role for the calcium-sensing receptor in the expression of inflammatory mediators in LPS-treated human dental pulp cells. Mol. Cell Biochem. 477 (12), 2871–2881. doi:10.1007/s11010-022-04486-1
Atreya, I., Atreya, R., and Neurath, M. F. (2008). NF-kappaB in inflammatory bowel disease. J. Intern Med. 263 (6), 591–596. doi:10.1111/j.1365-2796.2008.01953.x
Bauer, C., Duewell, P., Mayer, C., Lehr, H. A., Fitzgerald, K. A., Dauer, M., et al. (2010). Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59 (9), 1192–1199. doi:10.1136/gut.2009.197822
Bekebrede, A. F., Keijer, J., Gerrits, W. J. J., and Boer, V. C. J. (2020). The molecular and physiological effects of protein-derived polyamines in the intestine. Nutrients 12 (1), 197. doi:10.3390/nu12010197
Brown, E. M., Gamba, G., Riccardi, D., Lombardi, M., Butters, R., Kifor, O., et al. (1993). Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366 (6455), 575–580. doi:10.1038/366575a0
Chassaing, B., Aitken, J. D., Malleshappa, M., and Vijay-Kumar, M. (2014). Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 104, 11–15. doi:10.1002/0471142735.im1525s104
Cheng, S. X., Lightfoot, Y. L., Yang, T., Zadeh, M., Tang, L., Sahay, B., et al. (2014). Epithelial CaSR deficiency alters intestinal integrity and promotes proinflammatory immune responses. FEBS Lett. 588 (22), 4158–4166. doi:10.1016/j.febslet.2014.05.007
Choi, S. H., Kim, B. G., Robinson, J., Fink, S., Yan, M., Sporn, M. B., et al. (2014). Synthetic triterpenoid induces 15-PGDH expression and suppresses inflammation-driven colon carcinogenesis. J. Clin. Invest. 124 (6), 2472–2482. doi:10.1172/jci69672
Conigrave, A. D., and Brown, E. M. (2006). Taste receptors in the gastrointestinal tract. II. L-Amino acid sensing by calcium-sensing receptors: Implications for GI physiology. Am. J. Physiol. Gastrointest. Liver Physiol. 291 (5), G753–G761. doi:10.1152/ajpgi.00189.2006
D'Espessailles, A., Mora, Y. A., Fuentes, C., and Cifuentes, M. (2018). Calcium-sensing receptor activates the NLRP3 inflammasome in LS14 preadipocytes mediated by ERK1/2 signaling. J. Cell Physiol. 233 (8), 6232–6240. doi:10.1002/jcp.26490
Díaz-Soto, G., Rocher, A., García-Rodríguez, C., Núñez, L., and Villalobos, C. (2016). The calcium-sensing receptor in Health and disease. Int. Rev. Cell Mol. Biol. 327, 321–369. doi:10.1016/bs.ircmb.2016.05.004
Elajnaf, T., Iamartino, L., Mesteri, I., Müller, C., Bassetto, M., Manhardt, T., et al. (2019). Nutritional and pharmacological targeting of the calcium-sensing receptor influences chemically induced colitis in mice. Nutrients 11 (12), 3072. doi:10.3390/nu11123072
Feng, C., Bao, X., Shan, L., Ling, Y., Ding, Y., Wang, J., et al. (2020). Calcium-sensing receptor mediates β-amyloid-induced synaptic formation impairment and cognitive deficits via regulation of cytosolic phospholipase A2/prostaglandin E2 metabolic pathway. Front. Aging Neurosci. 12, 144. doi:10.3389/fnagi.2020.00144
Gandhi, J., Khera, L., Gaur, N., Paul, C., and Kaul, R. (2017). Role of modulator of inflammation cyclooxygenase-2 in gammaherpesvirus mediated tumorigenesis. Front. Microbiol. 8, 538. doi:10.3389/fmicb.2017.00538
Goldstone, R., Itzkowitz, S., Harpaz, N., and Ullman, T. (2012). Dysplasia is more common in the distal than proximal colon in ulcerative colitis surveillance. Inflamm. Bowel Dis. 18 (5), 832–837. doi:10.1002/ibd.21809
Grant, M. P., Stepanchick, A., Cavanaugh, A., and Breitwieser, G. E. (2011). Agonist-driven maturation and plasma membrane insertion of calcium-sensing receptors dynamically control signal amplitude. Sci. Signal 4, ra78. doi:10.1126/scisignal.2002208
Greenhough, A., Smartt, H. J. M., Moore, A. E., Roberts, H. R., Williams, A. C., Paraskeva, C., et al. (2009). The COX-2/PGE 2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 30 (3), 377–386. doi:10.1093/carcin/bgp014
Hannan, F. M., Kallay, E., Chang, W., Brandi, M. L., and Thakker, R. V. (2018). The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 15 (1), 33–51. doi:10.1038/s41574-018-0115-0
Hu, B., Toda, K., Wang, X., Antczak, M. I., Smith, J., Geboers, S., et al. (2022). Orally bioavailable quinoxaline inhibitors of 15-prostaglandin dehydrogenase (15-PGDH) promote tissue repair and regeneration. J. Med. Chem. 65 (22), 15327–15343. doi:10.1021/acs.jmedchem.2c01299
Iamartino, L., and Brandi, M. L. (2022). The calcium-sensing receptor in inflammation: Recent updates. Front. Physiol. 13, 1059369. doi:10.3389/fphys.2022.1059369
Iamartino, L., Elajnaf, T., Gall, K., David, J., Manhardt, T., Heffeter, P., et al. (2020). Effects of pharmacological calcimimetics on colorectal cancer cells over-expressing the human calcium-sensing receptor. Biochim. Biophys. Acta Mol. Cell Res. 1867 (12), 118836. doi:10.1016/j.bbamcr.2020.118836
Iamartino, L., Elajnaf, T., Kallay, E., and Schepelmann, M. (2018). Calcium-sensing receptor in colorectal inflammation and cancer: Current insights and future perspectives. World J. Gastroenterol. 24 (36), 4119–4131. doi:10.3748/wjg.v24.i36.4119
Jäger, E., Murthy, S., Schmidt, C., Hahn, M., Strobel, S., Peters, A., et al. (2020). Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat. Commun. 11 (1), 4243. doi:10.1038/s41467-020-17749-6
Kim, Y. H., and Kim, K. J. (2022). Upregulation of prostaglandin E2 by inducible microsomal prostaglandin E synthase-1 in colon cancer. Ann. Coloproctol. 38 (2), 153–159. doi:10.3393/ac.2021.00374.0053
Leach, K., Sexton, P. M., Christopoulos, A., and Conigrave, A. D. (2014). Engendering biased signalling from the calcium-sensing receptor for the pharmacotherapy of diverse disorders. Br. J. Pharmacol. 171 (5), 1142–1155. doi:10.1111/bph.12420
Li, T., Sun, M., Yin, X., Wu, C., Wu, Q., Feng, S., et al. (2013). Expression of the calcium sensing receptor in human peripheral blood T lymphocyte and its contribution to cytokine secretion through MAPKs or NF-κB pathways. Mol. Immunol. 53 (4), 414–420. doi:10.1016/j.molimm.2012.09.010
Liu, G., Xu, X., Wu, C., Jia, G., Zhao, H., Chen, X., et al. (2022). Spermine protects intestinal barrier integrity through ras-related C3 botulinum toxin substrate 1/phospholipase C-γ1 signaling pathway in piglets. Anim. Nutr. 8 (1), 135–143. doi:10.1016/j.aninu.2021.06.016
Long, M. D., Kappelman, M. D., Martin, C. F., Chen, W., Anton, K., and Sandler, R. S. (2016). Role of nonsteroidal anti-inflammatory drugs in exacerbations of inflammatory bowel disease. J. Clin. Gastroenterology 50 (2), 152–156. doi:10.1097/MCG.0000000000000421
Meyer, A. M., Ramzan, N. N., Heigh, R. I., and Leighton, J. A. (2006). Relapse of inflammatory bowel disease associated with use of nonsteroidal anti-inflammatory drugs. Dig. Dis. Sci. 51 (1), 168–172. doi:10.1007/s10620-006-3103-5
Mine, Y., and Zhang, H. (2015). Anti-inflammatory effects of poly-L-lysine in intestinal mucosal system mediated by calcium-sensing receptor activation. J. Agric. Food Chem. 63 (48), 10437–10447. doi:10.1021/acs.jafc.5b03812
Missiaglia, E., Jacobs, B., D'Ario, G., Di Narzo, A. F., Soneson, C., Budinska, E., et al. (2014). Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 25 (10), 1995–2001. doi:10.1093/annonc/mdu275
Murakami, M., Nakashima, K., Kamei, D., Masuda, S., Ishikawa, Y., Ishii, T., et al. (2003). Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J. Biol. Chem. 278 (39), 37937–37947. doi:10.1074/jbc.M305108200
Murakami, M., Naraba, H., Tanioka, T., Semmyo, N., Nakatani, Y., Kojima, F., et al. (2000). Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 275 (42), 32783–32792. doi:10.1074/jbc.M003505200
Nemeth, E. F., and Goodman, W. G. (2016). Calcimimetic and calcilytic drugs: Feats, flops, and futures. Calcif. Tissue Int. 98 (4), 341–358. doi:10.1007/s00223-015-0052-z
Ogata, S., Kubota, Y., Satoh, S., Ito, S., Takeuchi, H., Ashizuka, M., et al. (2006). Ca2+ stimulates COX-2 expression through calcium-sensing receptor in fibroblasts. Biochem. Biophys. Res. Commun. 351 (4), 808–814. doi:10.1016/j.bbrc.2006.10.098
Quinn, S. J., Ye, C. P., Diaz, R., Kifor, O., Bai, M., Vassilev, P., et al. (1997). The Ca2+-sensing receptor: A target for polyamines. Am. J. Physiol. 273 (4), C1315–C1323. doi:10.1152/ajpcell.1997.273.4.C1315
Rossol, M., Pierer, M., Raulien, N., Quandt, D., Meusch, U., Rothe, K., et al. (2012). Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 3 (1), 1329. doi:10.1038/ncomms2339
Sampey, A. V., Monrad, S., and Crofford, L. J. (2005). Microsomal prostaglandin E synthase-1: The inducible synthase for prostaglandin E2. Arthritis Res. Ther. 7 (3), 114–117. doi:10.1186/ar1748
Schepelmann, M., Kupper, N., Sladczyk, M., Mansfield, B., Manhardt, T., Piatek, K., et al. (2021). Stereo-specific modulation of the extracellular calcium-sensing receptor in colon cancer cells. Int. J. Mol. Sci. 22 (18), 10124. doi:10.3390/ijms221810124
Schepelmann, M., Ranieri, M., Lopez-Fernandez, I., Webberley, T. S., Brennan, S. C., Yarova, P. L., et al. (2022). Impaired mineral ion metabolism in a mouse model of targeted calcium-sensing receptor (CaSR) deletion from vascular smooth muscle cells. J. Am. Soc. Nephrol. 33 (7), 1323–1340. doi:10.1681/asn.2021040585
Schepelmann, M., Yarova, P. L., Lopez-Fernandez, I., Davies, T. S., Brennan, S. C., Edwards, P. J., et al. (2016). The vascular Ca2+-sensing receptor regulates blood vessel tone and blood pressure. Am. J. Physiol. Cell Physiol. 310 (3), C193–C204. doi:10.1152/ajpcell.00248.2015
Sparks, S. M., Spearing, P. K., Diaz, C. J., Cowan, D. J., Jayawickreme, C., Chen, G., et al. (2017). Identification of potent, nonabsorbable agonists of the calcium-sensing receptor for GI-specific administration. Bioorg Med. Chem. Lett. 27 (20), 4673–4677. doi:10.1016/j.bmcl.2017.09.008
Subbaramaiah, K., Yoshimatsu, K., Scherl, E., Das, K. M., Glazier, K. D., Golijanin, D., et al. (2004). Microsomal prostaglandin E synthase-1 is overexpressed in inflammatory bowel disease: Evidence for involvement of the transcription factor egr-1. J. Biol. Chem. 279 (13), 12647–12658. doi:10.1074/jbc.M312972200
Wang, D., and DuBois, R. N. (2010). The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 29 (6), 781–788. doi:10.1038/onc.2009.421
Wawrzyniak, M., Groeger, D., Frei, R., Ferstl, R., Wawrzyniak, P., Krawczyk, K., et al. (2021). Spermidine and spermine exert protective effects within the lung. Pharmacol. Res. Perspect. 9 (4), e00837. doi:10.1002/prp2.837
Williams, C. S., Shattuck-Brandt, R. L., and DuBois, R. N. (1999). The role of COX-2 in intestinal cancer. Expert Opin. Investig. Drugs 8 (1), 1–12. doi:10.1517/13543784.8.1.1
Wolffs, K., Mansfield, B., Bruce, R., Verckist, L., Paes De Araújo, R., Attanoos, R., et al. (2019). S92 Calcium-sensing receptor as a therapeutic target for pulmonary fibrosis. Thorax 74 (2), A58. doi:10.1136/thorax-2019-BTSabstracts2019.98
Yarova, P. L., Huang, P., Schepelmann, M. W., Bruce, R., Ecker, R., Nica, R., et al. (2021). Characterization of negative allosteric modulators of the calcium-sensing receptor for repurposing as a treatment of asthma. J. Pharmacol. Exp. Ther. 376 (1), 51–63. doi:10.1124/jpet.120.000281
Yarova, P. L., Stewart, A. L., Sathish, V., Britt, R. D., Thompson, M. A., Ap, P. L., et al. (2015). Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci. Transl. Med. 7 (284), 284ra60. doi:10.1126/scitranslmed.aaa0282
Yarova, P., Schepelmann, M., Ferla, S., Huang, P., Telezhkin, V., Kidd, E., et al. (2018). Development of a new calcilytic for the treatment of inflammatory lung disease. Eur. Respir. J. 52. 1060. doi:10.1183/13993003.congress-2018.PA1060
Yoshimatsu, K., Golijanin, D., Paty, P. B., Soslow, R. A., Jakobsson, P. J., DeLellis, R. A., et al. (2001). Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 7 (12), 3971–3976.
Zhang, H., Desai, A., Yang, S. Y., Bae, K. B., Antczak, M. I., Fink, S. P., et al. (2015b). TISSUE REGENERATION. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 348 (6240), aaa2340. doi:10.1126/science.aaa2340
Zhang, H., Kovacs-Nolan, J., Kodera, T., Eto, Y., and Mine, Y. (2015a). γ-Glutamyl cysteine and γ-glutamyl valine inhibit TNF-α signaling in intestinal epithelial cells and reduce inflammation in a mouse model of colitis via allosteric activation of the calcium-sensing receptor. Biochim. Biophys. Acta 1852 (5), 792–804. doi:10.1016/j.bbadis.2014.12.023
Zhang, M., Wang, H., and Tracey, K. J. (2000). Regulation of macrophage activation and inflammation by spermine: A new chapter in an old story. Crit. Care Med. 28 (4), N60–N66. doi:10.1097/00003246-200004001-00007
Zhen, Y., and Zhang, H. (2019). NLRP3 inflammasome and inflammatory bowel disease. Front. Immunol. 10, 276. doi:10.3389/fimmu.2019.00276
Keywords: calcium-sensing receptor (CaSR), prostaglandins, inflammation, colitis, calcimimetic, calcilytic
Citation: Gushchina V, Kupper N, Schwarzkopf M, Frisch G, Piatek K, Aigner C, Michel A, Schueffl H, Iamartino L, Elajnaf T, Manhardt T, Vlasaty A, Heffeter P, Bassetto M, Kállay E and Schepelmann M (2023) The calcium-sensing receptor modulates the prostaglandin E2 pathway in intestinal inflammation. Front. Pharmacol. 14:1151144. doi: 10.3389/fphar.2023.1151144
Received: 25 January 2023; Accepted: 10 April 2023;
Published: 20 April 2023.
Edited by:
Leonel Pereira, University of Coimbra, PortugalReviewed by:
Katsuya Hirano, Kagawa University, JapanCopyright © 2023 Gushchina, Kupper, Schwarzkopf, Frisch, Piatek, Aigner, Michel, Schueffl, Iamartino, Elajnaf, Manhardt, Vlasaty, Heffeter, Bassetto, Kállay and Schepelmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Schepelmann, bWFydGluLnNjaGVwZWxtYW5uQG1lZHVuaXdpZW4uYWMuYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.