 Chloe G. McKee
Chloe G. McKee Madison Hoffos
Madison Hoffos Haley A. Vecchiarelli
Haley A. Vecchiarelli Marie-Ève Tremblay
Marie-Ève Tremblay
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 09 February 2023
Sec. Neuropharmacology
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1125982
This article is part of the Research Topic Insights on Neuroinflammatory Response by Microglia-Targeted Pharmacology View all 7 articles
As individuals age, microglia, the resident immune cells of the central nervous system (CNS), become less effective at preserving brain circuits. Increases in microglial inflammatory activity are thought to contribute to age-related declines in cognitive functions and to transitions toward mild cognitive impairment (MCI) and Alzheimer’s disease (AD). As microglia possess receptors for communicating with the CNS environment, pharmacological therapies targeting these pathways hold potential for promoting homeostatic microglial functions within the aging CNS. Preclinical and early phase clinical trials investigating the therapeutic effects of pharmacological agents acting on microglia, including reactive oxygen species, TREM2, fractalkine signaling, the complement cascade, and the NLRP3 inflammasome, are currently underway; however, important questions remain unanswered. Current challenges include target selectivity, as many of the signaling pathways are expressed in other cell types. Furthermore, microglia are a heterogenous cell population with transcriptomic, proteomic, and microscopy studies revealing distinct microglial states, whose activities and abundance shift across the lifespan. For example, homeostatic microglia can transform into pathological states characterized by markers of oxidative stress. Selective pharmacological targeting aimed at limiting transitions to pathological states or promoting homeostatic or protective states, could help to avoid potentially harmful off-target effects on beneficial states or other cell types. In this mini-review we cover current microglial pathways of interest for the prevention and treatment of age-related cognitive decline and CNS disorders of aging focusing on MCI and AD. We also discuss the heterogeneity of microglia described in these conditions and how pharmacological agents could target specific microglial states.
Microglia, the resident immune cell of the central nervous system (CNS), play important roles across health and disease. They defend the CNS against infection and injury (Napoli and Neumann, 2009), contribute to neurogenesis (Sierra et al., 2014), refine synaptic connections (Paolicelli et al., 2011; Tremblay, 2011), and support neurons and other glial cells (Hansson and Rönnbäck, 2003). As microglia perform essential functions, their dysfunction is implicated in nearly all neurological pathologies, including the age-related neurodegenerative condition mild cognitive impairment (MCI), which can transition into Alzheimer’s disease (AD) (Tay et al., 2018). Genome-wide association studies (GWAS) found microglia to most strongly express 60% of single nucleotide polymorphisms (SNPs) associated with AD risk (Zhang et al., 2014; McQuade and Blurton-Jones, 2019). Microglial functions are also affected during normal aging, which describes a process of gradual decline in cognitive functions including reasoning, memory, and processing speed (Harada et al., 2013). Microglia are highly sensitive to their surrounding environment and respond by modulating their morphology and activity (Carvalho-Paulo et al., 2021). Microglia possess various receptors to sense their microenvironment that also regulate their functions (Song and Suk, 2017; Šimončičová et al., 2022), holding potential for the pharmacological treatment and prevention of MCI and AD.
During aging, the CNS environment becomes more inflammatory, triggering microglial reactivity (Norden et al., 2015), dystrophy (Streit et al., 2004), dysfunction (Mosher and Wyss-Coray, 2014), and senescence (Angelova and Brown, 2019). Microglial reactivity alters the balance between pro- and anti-inflammatory cytokine release, which can impair glial and neuronal functions leading to tissue damage. Notable age-related changes in microglia include their increased production of reactive oxygen species (ROS) and deficits in lysosomal digestion (Nakanishi and Wu, 2009). This compromises their maintenance of homeostasis, notably via reduced phagocytosis of apoptotic cells, aggregated proteins such as amyloid-beta (Aβ), and myelin debris (Harry, 2013; Rawji et al., 2016; Koellhoffer et al., 2017; Gabandé-Rodríguez et al., 2020). However, microglial phagocytosis is also involved in synaptic loss, a major correlate of cognitive decline hypothesized to be involved in the initiation and acceleration of AD (Terry et al., 1991; Scheff et al., 2006; Herrup, 2015). Thus, pharmacological approaches modulating microglial phagocytosis should consider cargo specificity. Classical pharmacological approaches, such as minocycline, focused on preventing microglial reactivity (Šimončičová et al., 2022) and have limited efficacy likely because they compromise beneficial microglial functions. New therapeutic approaches should promote beneficial microglial functions while preventing or reversing detrimental ones. Considering that microglia are highly heterogeneous, as notably identified by transcriptomic and microscopy studies (Table 1) (Paolicelli et al., 2022), such a goal could be accomplished by targeting specific microglial states which become more abundant with aging and AD pathology.
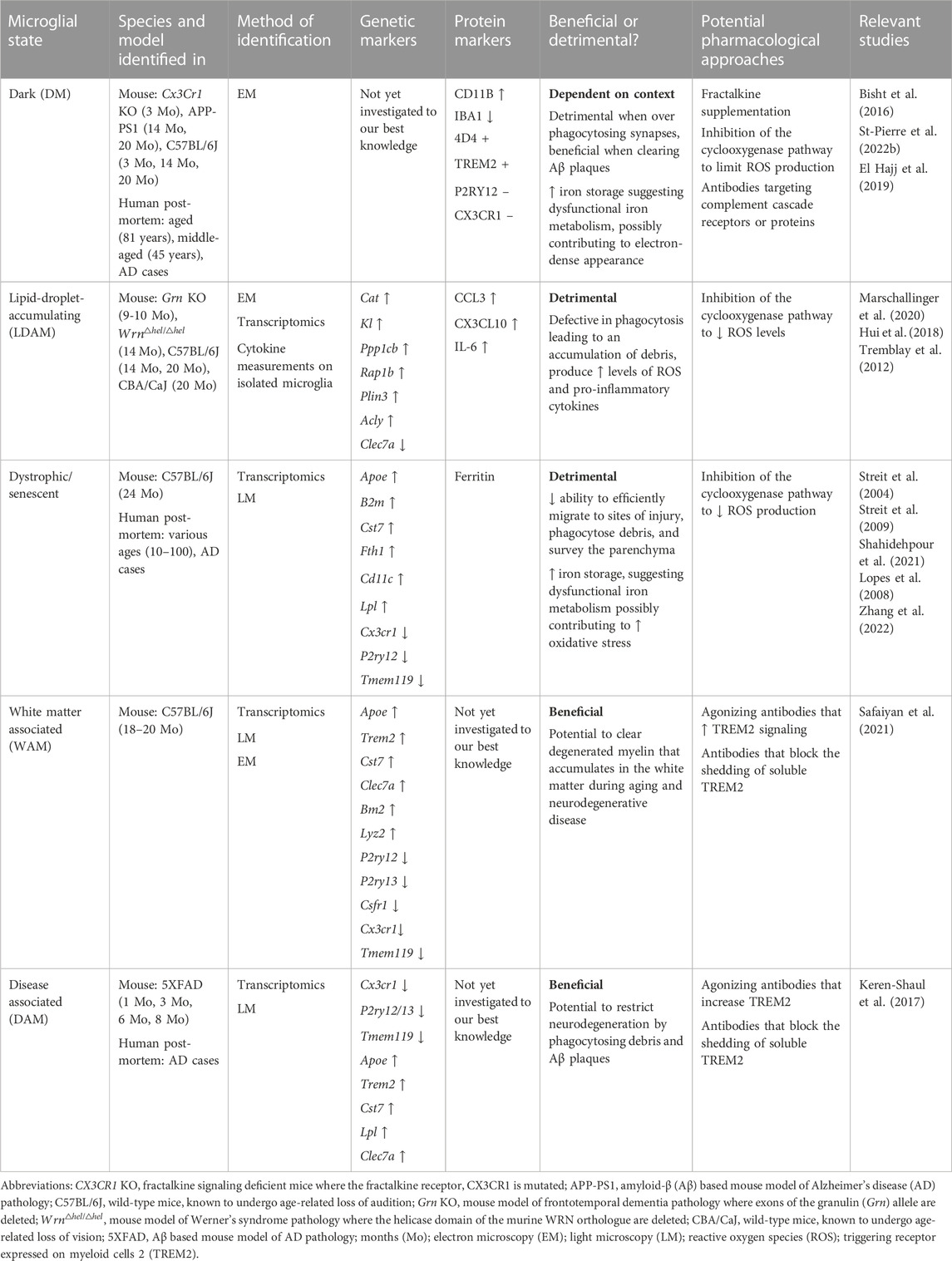
TABLE 1. Microglial states associated with normal cognitive aging and age-related neurodegenerative diseases which are discussed in our mini-review.
In this mini-review we cover current microglial pathways which have shown potential for the prevention or treatment of AD and slowing of cognitive aging. We also discuss how pharmacological agents could target specific microglial states to maintain protective functions or prevent detrimental ones during aging (Figure 1). Challenges surrounding microglial pharmacological targeting still remain, such as getting therapeutics across the blood-brain barrier (BBB) (Gabathuler, 2010) and avoiding off-target effects on other CNS cells that express many of the same receptors of interest as microglia.
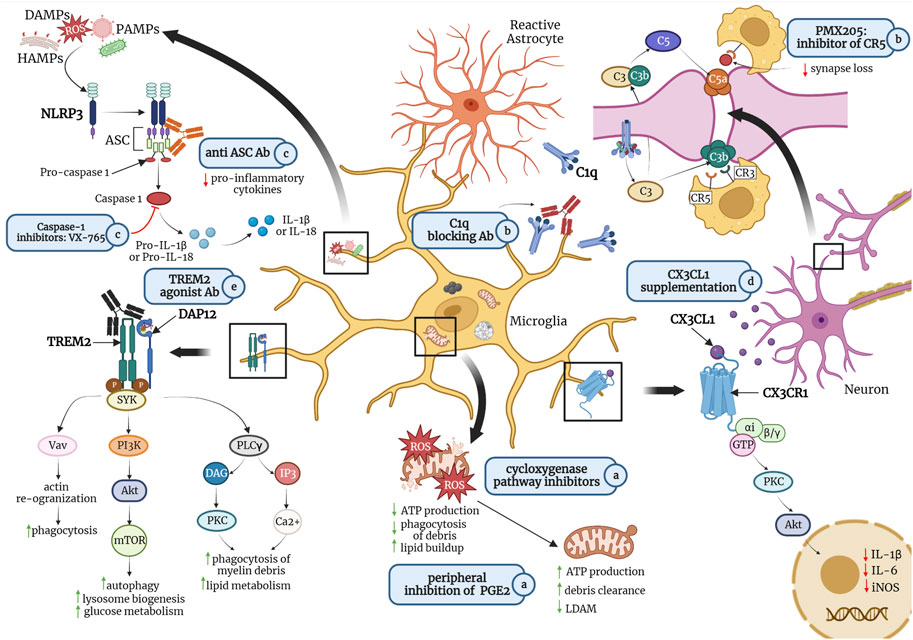
FIGURE 1. Potential pharmacological treatments for promoting homeostatic microglial states and preventing or reversing detrimental states include a) peripheral inhibition of lipid messenger prostaglandin E2 (PGE2) or cyclooxygenase inhibitors such as non-steroidal anti-inflammatory drugs (NSAIDs) to maintain mitochondrial morphology, thereby increasing adenosine triphosphate (ATP) production and debris clearance; b) antibodies against C1q, the initiating protein in the complement cascade, or small molecule inhibitors of complement receptor 5 (CR5), such as PMX205 to reduce microglia-mediated phagocytosis of synapses; c) caspase-1 inhibitors or antibodies against apoptosis-associated speck-like protein containing a CARD (ASC), a central adaptor protein of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, to inhibit the production of pro-inflammatory cytokines such as interleukin (IL)-1β and IL-18; d) fractalkine (CX3CL1) supplementation to reduce the production of pro-inflammatory cytokines; and e) agonist antibodies targeting triggering receptor expressed on myeloid cells (TREM)2 to promote microglial phagocytosis of myelin debris and amyloid-β (Aβ) plaques, as well as glucose metabolism. Abbreviations: ROS, reactive oxygen species; LDAM, lipid-droplet accumulating microglia; CX3CR1, fractalkine receptor; PKC, protein kinase C; Akt, protein kinase B; iNOS; inducible nitric oxide synthase; IL-6, interleukin 6; HAMPs, homeostasis-altering molecular patterns; DAMPs, danger-associated molecular patters; PAMPs, pathogen-associated molecular patterns; C3, C3 convertase; C3b, complement component 3b (an opsonin); CR3, complement receptor three (binds to C3b); C5, C5 convertase; C5a, complement component C5a (an anaphylatoxin); CR5, complement receptor five (binds to C5a); DAP12, DNAX-activating protein of 12 kDa; SYK, spleen tyrosine kinase; Vav, a guanine nucleotide exchange factor (GEF) for small G proteins of the Rho family; PI3K, phosphatidylinositol-3 kinase; mTOR, mammalian target of rapamycin; PLCγ, phospholipase C gamma; DAG, diacylglycerol, IP3; inositol trisphosphate. Figure is not drawn to scale. Created in Biorender.
ROS are molecules containing unpaired electrons formed by the partial reduction of oxygen within the mitochondrial electron transport chain (Guo et al., 2013). Under physiological conditions, ROS act as important signaling molecules (Ray et al., 2012); however, their levels increase during aging and in AD (Brieger et al., 2012; Manoharan et al., 2016). When ROS overwhelm antioxidant defense mechanisms, they can damage macromolecules and organelles, such as mitochondria (Guo et al., 2013), where disrupted cristae membranes are a marker of oxidative stress (Picca et al., 2020). Dark microglia (DM), a pathology associated microglial state (Table 1) found in mouse models of aging and AD pathology, and aging human post-mortem samples, often contain mitochondria with swollen and disrupted membranes (Bisht et al., 2016; St-Pierre et al., 2022a).
Lipid-droplet accumulating microglia (LDAM) which increase during aging in mice and humans, produce high levels of ROS and proinflammatory cytokines [e.g., interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)α] and have deficits in phagocytosis (Ye et al., 2017). Promoting microglial autophagy, responsible for transporting cytoplasmic organelles to lysosomes for degradation, through in vitro treatment with rapamycin, a Food and Drug Administration-approved immunosuppressant with anti-cognitive aging potential (Blagosklonny, 2019), decreased ROS and pro-inflammatory cytokine production (Ye et al., 2017). Thus, increasing microglial autophagy may limit lipid droplet buildup and transitions from homeostatic microglia to LDAM.
Increased signaling through the lipid messenger prostaglandin E2 (PGE2), a downstream component of the cyclooxygenase-2 pathway involved in inflammation was reported in aged mice (Minhas et al., 2021). This pathway converts glucose to glycogen, while reducing glucose flux through glycolysis and the Krebs cycle, thereby decreasing ATP production. Pharmacological inhibition of PGE2 in the periphery restored microglial mitochondrial morphology, ATP production, and long-term potentiation within the hippocampus to young adult levels, while resolving spatial memory deficits (Minhas et al., 2021). Furthermore, long-term treatment with drugs that inhibit cyclooxygenases, key enzymes involved in the production of PGE2, such as non-steroidal anti-inflammatory drugs (NSAIDs), delayed AD onset in pilot clinical trials and decreased microglial reactivity in rodent models of AD pathology (Ferencik et al., 2001; Krause and Müller, 2010).
Studies in models of Aβ pathology including 5XFAD (Baik et al., 2019) and APP/PS1 mice (McIntosh et al., 2019; Guillot-Sestier et al., 2021), have shown that microglia surrounding Aβ plaques shift from oxidative phosphorylation (OXPHOS) to the less efficient glycolytic metabolism. This reduces ATP production, decreases microglial phagocytosis, and leads to increased Aβ accumulation (McIntosh et al., 2019). Additionally, transitions to glycolytic metabolism caused microglia to become more iron retentive (McIntosh et al., 2019), a known trigger of ROS production (Picca et al., 2020). Increased ferritin immunoreactivity was reported in dystrophic and senescent microglia in human post-mortem samples from patients with and without dementia (Lopes et al., 2008). Although more research is needed, iron retention could contribute to the electron-dense appearance of DM. Overall, these findings suggest that reducing oxidative stress and maintaining microglial mitochondrial OXPHOS could help promote transitions from DM and LDAM to homeostatic states, while possibly limiting cognitive decline during aging.
The complement cascade is an innate immune system pathway that detects and removes pathogens and dying cells (Ricklin et al., 2010). Within the CNS, neurons, astrocytes, and primarily microglia produce the complement protein C1q (Kouser et al., 2015; Fonseca et al., 2017; Asano et al., 2020). When C1q is deposited onto synapses it acts as an “eat me signal” targeting them for phagocytosis (Hong et al., 2016b) or trogocytosis (Lim and Ruthazer, 2021).
C1q expression is consistently upregulated with age in the mouse and human CNS, especially in the hippocampus (Stephan et al., 2013), and may contribute to the synaptic loss observed during normal aging (Terry et al., 1991; Jackson et al., 2019). Furthermore, Aβ fibrils produce complement proteins in various mouse models of AD pathology (Rogers et al., 1992; Bradt et al., 1998). Knocking out the C3 convertase, located downstream of C1q, is protective against age-dependent synapse and neuron loss within the hippocampus, and results in improved memory and spatial learning in aged wild-type (Shi et al., 2015) and APP/PS1 (Shi et al., 2017) mice. Pharmacological approaches such as small molecule inhibitors and monoclonal antibodies have demonstrated some success in mouse models of AD pathology. Oral administration of PMX205, an antagonist of C5aR, a receptor which triggers microglial phagocytosis, significantly reduced Aβ load in Tg2576 mice (model of Aβ pathology), together with Aβ load and hyperphosphorylated Tau in 3xTg mice (model of Aβ/Tau pathology), compared to untreated controls (Fonseca et al., 2009). In Tau-P301S mice (model of Tau pathology), injection of a C1q-blocking antibody into the hippocampus reduced synaptic markers by 50% within microglial lysosomes, suggesting decreased synaptic phagocytosis (Dejanovic et al., 2018). The broad spectrum humanized monoclonal antibody against C1q, ANX005, is currently undergoing Phase II trials, while its murine precursor ANXM-1 inhibited the complement cascade in vivo in wild-type mice centrally receiving Aβ oligomers (Hong et al., 2016a).
In terms of targeting microglial states, DM highly express C3R in their processes surrounding synapses and Aβ (Bisht et al., 2016). Inhibition of the complement cascade could help lessen the pathological synapse loss associated with DM; however, challenges include a lack of specificity as neurons and astrocytes, among other cells, also express complement components (Barnum, 1995).
The NLRP3 inflammasome is a pattern recognition receptor expressed by many cell types (Holbrook et al., 2021), including microglia (Nayak et al., 2014), that recognizes homeostasis-altering molecular patterns (HAMPs), danger-associated molecular patterns (DAMPs), and pathogen-associated molecular patterns (PAMPs) (Kelley et al., 2019). HAMPs arise from cellular imbalances such as endoplasmic reticulum and Golgi stress (Liston and Masters, 2017), PAMPs are produced by microbial pathogens, and DAMPs by uric acid crystals (Liston and Masters, 2017) and ROS (Kelley et al., 2019), among other signals released during endogenous cell death.
When activated, NLRP3 induces caspase-1-mediated cleavage of IL-18 and IL-1β (Kelley et al., 2019). IL-1β which is elevated in the cerebrospinal fluid (CSF) and brain of patients with AD is considered toxic to microglia (Blevins et al., 2022) by triggering increased oxidative stress and apoptosis (Liu and Quan, 2018). Caspase-1 is also associated with inflammatory and cytoprotective responses that include cell death. Heneka et al. (2013) identified increased caspase-1 expression as an indication that NLRP3 may induce neurodegeneration, in patients with AD and APP/PS1 mice. APP/PS1 mice deficient in NLRP3 or caspase-1 displayed reduced spatial memory loss and IL-1β activation, suggesting beneficial outcomes of targeting caspase-1, or its upstream effectors, in AD.
In terms of targeting, VX-765, a small molecule caspase-1 inhibitor that is BBB permeable and approved for clinical trials, reversed impairment of episodic and spatial memory in J20 APPSw/Ind mice (model of Aβ pathology) (Sri et al., 2019), while preventing brain inflammation and Aβ build-up (Flores et al., 2018). Another small molecule capsase-1 inhibitor, MCC950, inhibits NLRP3 assembly, and IL-1β and Aβ build-up, while increasing microglial phagocytosis of Aβ (Dempsey et al., 2017). These changes were associated with improved working and recognition memory in APP/PS1 mice (Dempsey et al., 2017). MCC950 also prevented Tau pathology in Tau-P301S mice (Stancu et al., 2019).
Antagonizing antibodies that block the assembly of NLRP3 successfully modified disease outcomes in mouse models of AD pathology. Apoptosis-associated speck-like protein containing a CARD (ASC) is produced by microglia and is a central adaptor protein of NLRP3 that binds to Aβ and aids in the formation of Aβ oligomers (Venegas et al., 2017). Venegas et al. (2017) reported that an anti-ASC antibody prevented Aβ from accumulating in APP/PS1 mice. de Rivero Vaccari et al. (2022) also demonstrated that IC100, an anti-ASC antibody, which is BBB permeable and binds ASC filaments, blocks IL-1β production in human blood cell inflammasome assays. In vivo testing of IC100 is warranted to determine its therapeutic potential in treating AD.
In a rat model of AD pathology, systemic injection of mefenamic acid, a NSAID, was found to prevent memory deficits (Daniels et al., 2016). Similar behavioral results and decreased microglial reactivity and IL-1β expression, with IL-1β being a product of the NLRP3 signaling pathway, were observed in 3xTg mice centrally receiving mefenamic acid (Daniels et al., 2016). Thus far, animal models testing NLRP3 pathway inhibitors suggest that such treatments may have pharmacological relevance in reducing cognitive aging and AD pathology within patients.
Fractalkine signaling is one of the most important communication pathways between neurons and microglia (Mecca et al., 2018; Angelopoulou et al., 2020). Neurons primarily release the chemokine fractalkine (CX3CL1) which binds to the fractalkine receptor (CX3CR1), expressed primarily by microglia (Pawelec et al., 2020), although this is not universal in the CNS (Hatori et al., 2002). Fractalkine signaling is commonly viewed as an “OFF” switch that maintains microglia in a homeostatic state, by downregulating their production of IL-1β (Bachstetter et al., 2011). CX3CL1 expression decreases across the lifespan, being lower in middle-aged and further in aged versus young adult rats (Lyons et al., 2009; Bachstetter et al., 2011). Furthermore, human patients with AD compared to MCI and healthy controls have significantly less CX3CL1 in their CSF (Perea et al., 2018).
Targeting of the fractalkine pathway has demonstrated some success. Aged rats centrally treated with recombinant CX3CL1 exhibited increased hippocampal neurogenesis (Bachstetter et al., 2011), suggesting the potential of such treatments to decrease memory impairment in aging. Treatment with exogenous CX3CL1 attenuated microglial reactivity, by decreasing the expression of major histocompatibility complex class II and CD40 in hippocampal sections from aged rats (Lyons et al., 2009). Targeting the fractalkine pathway to treat AD has been more complex. CX3CR1 deficiency in APPS1 and R1.40 mice, models of Aβ pathology, resulted in reduced Aβ deposition potentially through enhanced microglial Aβ phagocytosis (Lee et al., 2010). mRNA expression of TNFα decreased while IL-1β increased in CX3CR1 deficient APP/PS1 mice. Contrastingly, CX3CR1 deficiency in the hAPP J20 mouse model of Aβ pathology did not significantly affect Aβ deposition and instead upregulated TNFα and IL-6 (Cho et al., 2011). Furthermore, CX3CR1 deficiency exacerbated Tau phosphorylation and worsened learning and memory in these mice, suggesting increased cognitive impairment. Similar results were observed in hTau mice, a model of tauopathy, where CX3CR1 deficiency increased tau phosphorylation and aggregation, and the expression of CD68, a marker of phagocytosis notably expressed by microglia (Bhaskar et al., 2010). Further work on simultaneously reducing Aβ deposition and tau pathology via targeting of the fractalkine pathway, combined with other targets, is warranted.
In terms of microglial diversity, CX3CR1 deficient young adult mice display a significantly greater density of DM in the hippocampus compared to age-matched WT controls (Bisht et al., 2016). This suggests that fractalkine supplementation may help to prevent increases in DM known to become abundant during aging and AD pathology. Although this supplementation holds potential as a clinical treatment, for cognitive aging especially, it will be necessary to limit its off-target effects on other cells and prevent potentially deleterious outcomes on tau pathology.
TREM2 is an immunoglobulin superfamily receptor, expressed most prominently in microglia within the CNS (Turnbull et al., 2006). The binding of TREM2 ligands including Aβ, apolipoproteins, phospholipids, and lipopolysaccharides (Kober and Brett, 2017) activates DNAX-activating protein of 12 kDa (DAP12), TREM2’s binding partner, increasing microglial migration, lipid metabolism, autophagy, and phagocytosis, notably of myelin (Mazaheri et al., 2017; McQuade et al., 2020; Qu and Li, 2021) and synapses (Filipello et al., 2018). GWAS revealed TREM2 SNPs to increase AD risk by 2–4 times (McQuade and Blurton-Jones, 2019), in addition to accelerating cognitive decline (Rajagopalan et al., 2013; Replogle et al., 2015). Knocking out TREM2 in PS2APP mice, a model of Aβ pathology, impairs microglia-mediated compaction of Aβ into dense plaques (Meilandt et al., 2020).
Activation of the program that leads to disease-associated microglia (DAM) and white-matter associated microglia (WAM), neuroprotective states found in AD and aging, respectively, requires TREM2 signaling. Pharmacological approaches aimed at increasing TREM2 signaling and promoting transitions to DAM hold potential for treating AD. Wang et al. (2020) found that the anti-human TREM2 agonistic antibody, AL002c, decreased filamentous plaques and neurite dystrophy in 5XFAD mice carrying the R47H TREM2 variant associated with AD risk. The clinical variant of AL002c, AL002, was found to be safe and well tolerated in a phase I clinical trial (NCT03635047) (Wang et al., 2020). Other antibody approaches include the monoclonal antibody, 4D9, which promotes TREM2 signaling by blocking shedding of soluble TREM2 (Schlepckow et al., 2020). 4D9 increased microglial phagocytosis of myelin debris and Aβ in vitro, while reducing amyloidogenesis in the APP-NL-G-F knock-in mouse model of Aβ pathology (Schlepckow et al., 2020). Increasing TREM2 signaling has the potential to limit the accumulation of lipid droplets and myelin debris which are thought to contribute to LDAM emergence (Marschallinger et al., 2020).
Although modulation of the TREM2 pathway could promote homeostatic microglial states, consideration of disease stage may be important. Work in Psen1 mice, a model of Aβ pathology, found that knockdown of TREM2 in the early to middle stages of AD was beneficial as it inhibited microglial phagocytosis of synapses, while this same approach in middle to late stages was detrimental by impairing microglial phagocytosis of Aβ (Sheng et al., 2019). Future work targeting TREM2 will need to balance promoting microglial phagocytosis of myelin debris and Aβ, while limiting synapse loss, perhaps in combination with other microglial targets.
Microglia are a heterogenous population whose morphology and activity shifts across the lifespan and between health and disease contexts (Grabert et al., 2016; Hammond et al., 2019). Although more research is needed to elucidate the functions of different microglial states, the diversity of this cell population holds selective pharmacotherapeutic potential for the prevention and treatment of cognitive aging, MCI, and AD. It will be important to identify differences in expressed receptors and signaling pathways, but also specific activities such as phagocytosis, metabolism, and inflammatory mediator production between microglial states (Šimončičová et al., 2022). This could allow for the design of drugs or antibody-mediated therapies that target specific microglial states, while preserving others and their critical functions.
However, several challenges remain. None of the receptors and associated pathways discussed herein or elsewhere (Rock and Peterson, 2006; Song and Suk, 2017; Šimončičová et al., 2022) are microglia-specific. Outstanding obstacles include getting therapeutics across the BBB and translation of the findings from animal models of AD pathology to human clinical trials (Shineman et al., 2011; Banik et al., 2015). Translation issues may be explained by genetic and epigenetic variations across patient profiles (Neuner et al., 2019), differences between human and murine microglia (Abels et al., 2021; Yvanka de Soysa et al., 2022), and the current inability of animal models to capture the complex pathology underlying AD including environmental risk factors (Bales, 2012; Onos et al., 2016). As the field evolves to tackle such issues, exploring microglial diversity will be an essential step to design more selective and effective therapeutics for treating and preventing AD and for slowing the overall cognitive aging process.
CGM wrote the introduction, the sections on reactive oxygen species, the complement cascade, fractalkine signaling, and TREM2, as well as the conclusion. MH wrote the section on the NLRP3 inflammasome. CGM conceived the figure and table. CGM, HAV, and M-ET edited the manuscript while HAV and MT provided supervisory guidance. All authors contributed to the article and approved the submitted version.
CGM is supported by a Jamie Cassels Undergraduate Research Award (JCURA) from the University of Victoria and a Boehm Family Award for Excellence in Science. HAV is supported by a fellowship from the Canadian Institutes of Health Research (CIHR) and is a Michael Smith Health Research BC Research Trainee. M-ET holds a Canada Research Chair (Tier 2) in Neurobiology of Aging and Cognition.
We acknowledge with respect the Lekwungen peoples on whose traditional territory the University of Victoria stands, and the Songhees, Esquimalt and WSÁNEĆ peoples whose historical relationships with the land continue to this day.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abels, E. R., Nieland, L., Hickman, S., Broekman, M. L. D., El Khoury, J., and Maas, S. L. N. (2021). Comparative analysis identifies similarities between the human and murine microglial sensomes. Int. J. Mol. Sci. 22, 1495. doi:10.3390/ijms22031495
Angelopoulou, E., Paudel, Y. N., Shaikh, M. F., and Piperi, C. (2020). Fractalkine (CX3CL1) signaling and neuroinflammation in Parkinson’s disease: Potential clinical and therapeutic implications. Pharmacol. Res. 158, 104930. doi:10.1016/j.phrs.2020.104930
Angelova, D. M., and Brown, D. R. (2019). Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 151, 676–688. doi:10.1111/jnc.14860
Asano, S., Hayashi, Y., Iwata, K., Okada-Ogawa, A., Hitomi, S., Shibuta, I., et al. (2020). Microglia–astrocyte communication via C1q contributes to orofacial neuropathic pain associated with infraorbital nerve injury. Int. J. Mol. Sci. 21, 6834. doi:10.3390/ijms21186834
Bachstetter, A. D., Morganti, J. M., Jernberg, J., Schlunk, A., Mitchell, S. H., Brewster, K. W., et al. (2011). Fractalkine and CX3CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 32, 2030–2044. doi:10.1016/j.neurobiolaging.2009.11.022
Baik, S. H., Kang, S., Lee, W., Choi, H., Chung, S., Kim, J.-I., et al. (2019). A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 30, 493–507. doi:10.1016/j.cmet.2019.06.005
Bales, K. R. (2012). The value and limitations of transgenic mouse models used in drug discovery for Alzheimer’s disease: An update. Expert Opin. Drug Discov. 7, 281–297. doi:10.1517/17460441.2012.666234
Banik, A., Brown, R. E., Bamburg, J., Lahiri, D. K., Khurana, D., Friedland, R. P., et al. (2015). Translation of pre-clinical studies into successful clinical trials for Alzheimer’s disease: What are the roadblocks and how can they Be overcome? J. Alzheimer’s Dis. 47, 815–843. doi:10.3233/JAD-150136
Barnum, S. R. (1995). Complement biosynthesis in the central nervous system. Crit. Rev. Oral Biol. Med. 6, 132–146. doi:10.1177/10454411950060020301
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi:10.1016/j.neuron.2010.08.023
Bisht, K., Sharma, K. P., Lecours, C., Gabriela Sánchez, M., El Hajj, H., Milior, G., et al. (2016). Dark microglia: A new phenotype predominantly associated with pathological states. Glia 64, 826–839. doi:10.1002/glia.22966
Blagosklonny, M. V. (2019). Rapamycin for longevity: Opinion article. Aging (Albany NY) 11, 8048–8067. doi:10.18632/aging.102355
Blevins, H. M., Xu, Y., Biby, S., and Zhang, S. (2022). The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front. Aging Neurosci. 14, 879021. doi:10.3389/fnagi.2022.879021
Bradt, B. M., Kolb, W. P., and Cooper, N. R. (1998). Complement-dependent proinflammatory properties of the Alzheimer’s disease. -Peptide. 8.
Brieger, K., Schiavone, S., Miller, , and Krause, K. (2012). Reactive oxygen species: From health to disease. Swiss Med. Wkly. 142, w13659. doi:10.4414/smw.2012.13659
Carvalho-Paulo, D., Bento Torres Neto, J., Filho, C. S., de Oliveira, T. C. G., de Sousa, A. A., dos Reis, R. R., et al. (2021). Microglial morphology across distantly related species: Phylogenetic, environmental and age influences on microglia reactivity and surveillance states. Frontiers in immunology 12. Available at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.683026 (Accessed November 7, 2022).
Cho, S.-H., Sun, B., Zhou, Y., Kauppinen, T. M., Halabisky, B., Wes, P., et al. (2011). CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of alzheimer disease. J. Biol. Chem. 286, 32713–32722. doi:10.1074/jbc.M111.254268
Daniels, M. J. D., Rivers-Auty, J., Schilling, T., Spencer, N. G., Watremez, W., Fasolino, V., et al. (2016). Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 7, 12504. doi:10.1038/ncomms12504
de Rivero Vaccari, J. P., Mim, C., Hadad, R., Cyr, B., Stefansdottir, T. A., and Keane, R. W. (2022). Mechanism of action of IC 100, a humanized IgG4 monoclonal antibody targeting apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). Transl. Res. 251, 27–40. doi:10.1016/j.trsl.2022.06.016
Dejanovic, B., Huntley, M. A., De Mazière, A., Meilandt, W. J., Wu, T., Srinivasan, K., et al. (2018). Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron 100, 1322–1336. doi:10.1016/j.neuron.2018.10.014
Dempsey, C., Rubio Araiz, A., Bryson, K. J., Finucane, O., Larkin, C., Mills, E. L., et al. (2017). Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain, Behav. Immun. 61, 306–316. doi:10.1016/j.bbi.2016.12.014
El Hajj, H., Savage, J. C., Bisht, K., Parent, M., Vallières, L., Rivest, S., et al. (2019). Ultrastructural evidence of microglial heterogeneity in Alzheimer’s disease amyloid pathology. J. Neuroinflammation 16, 87. doi:10.1186/s12974-019-1473-9
Ferencik, M., Novak, M., Rovensky, J., and Rybar, I. (2001). Alzheimer’s disease, inflammation and non-steroidal anti-inflammatory drugs. Bratisl. Lek. Listy 102, 123–132.
Filipello, F., Morini, R., Corradini, I., Zerbi, V., Canzi, A., Michalski, B., et al. (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48, 979–991. doi:10.1016/j.immuni.2018.04.016
Flores, J., Noël, A., Foveau, B., Lynham, J., Lecrux, C., and LeBlanc, A. C. (2018). Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 9, 3916. doi:10.1038/s41467-018-06449-x
Fonseca, M. I., Ager, R. R., Chu, S.-H., Yazan, O., Sanderson, S. D., LaFerla, F. M., et al. (2009). Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 183, 1375–1383. doi:10.4049/jimmunol.0901005
Fonseca, M. I., Chu, S.-H., Hernandez, M. X., Fang, M. J., Modarresi, L., Selvan, P., et al. (2017). Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflammation 14, 48. doi:10.1186/s12974-017-0814-9
Gabandé-Rodríguez, E., Keane, L., and Capasso, M. (2020). Microglial phagocytosis in aging and Alzheimer’s disease. J. Neurosci. Res. 98, 284–298. doi:10.1002/jnr.24419
Gabathuler, R. (2010). Approaches to transport therapeutic drugs across the blood–brain barrier to treat brain diseases. Neurobiol. Dis. 37, 48–57. doi:10.1016/j.nbd.2009.07.028
Grabert, K., Michoel, T., Karavolos, M. H., Clohisey, S., Baillie, J. K., Stevens, M. P., et al. (2016). Microglial brain region−dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516. doi:10.1038/nn.4222
Guillot-Sestier, M.-V., Araiz, A. R., Mela, V., Gaban, A. S., O’Neill, E., Joshi, L., et al. (2021). Microglial metabolism is a pivotal factor in sexual dimorphism in Alzheimer’s disease. Commun. Biol. 4, 711. doi:10.1038/s42003-021-02259-y
Guo, C., Sun, L., Chen, X., and Zhang, D. (2013). Oxidative stress, mitochondrial damage and neurodegenerative diseases****○. Neural Regeneration Research, 12.
Hammond, T. R., Dufort, C., Dissing-Olesen, L., Giera, S., Young, A., Wysoker, A., et al. (2019). Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253–271. doi:10.1016/j.immuni.2018.11.004
Hansson, E., and Rönnbäck, L. (2003). Glial neuronal signaling in the central nervous system. FASEB J. 17, 341–348. doi:10.1096/fj.02-0429rev
Harada, C. N., Natelson Love, M. C., and Triebel, K. L. (2013). Normal cognitive aging. Clin. Geriatric Med. 29, 737–752. doi:10.1016/j.cger.2013.07.002
Harry, G. J. (2013). Microglia during development and aging. Pharmacol. Ther. 139, 313–326. doi:10.1016/j.pharmthera.2013.04.013
Hatori, K., Nagai, A., Heisel, R., Ryu, J. K., and Kim, S. U. (2002). Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 69, 418–426. doi:10.1002/jnr.10304
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi:10.1038/nature11729
Herrup, K. (2015). The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18, 794–799. doi:10.1038/nn.4017
Holbrook, J. A., Jarosz-Griffiths, H. H., Caseley, E., Lara-Reyna, S., Poulter, J. A., Williams-Gray, C. H., et al. (2021). Neurodegenerative disease and the NLRP3 inflammasome. Front. Pharmacol. 12, 643254. doi:10.3389/fphar.2021.643254
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016a). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi:10.1126/science.aad8373
Hong, S., Dissing-Olesen, L., and Stevens, B. (2016b). New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 36, 128–134. doi:10.1016/j.conb.2015.12.004
Hui, C. W., St-Pierre, M.-K., Detuncq, J., Aumailley, L., Dubois, M.-J., Couture, V., et al. (2018). Nonfunctional mutant Wrn protein leads to neurological deficits, neuronal stress, microglial alteration, and immune imbalance in a mouse model of Werner syndrome. Brain, Behav. Immun. 73, 450–469. doi:10.1016/j.bbi.2018.06.007
Jackson, J., Jambrina, E., Li, J., Marston, H., Menzies, F., Phillips, K., et al. (2019). Targeting the synapse in Alzheimer’s disease. Front. Neurosci. 13, 735. doi:10.3389/fnins.2019.00735
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. IJMS 20, 3328. doi:10.3390/ijms20133328
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290. doi:10.1016/j.cell.2017.05.018
Kober, D. L., and Brett, T. J. (2017). TREM2-Ligand interactions in health and disease. J. Mol. Biol. 429, 1607–1629. doi:10.1016/j.jmb.2017.04.004
Koellhoffer, E., McCullough, L., and Ritzel, R. (2017). Old maids: Aging and its impact on microglia function. IJMS 18, 769. doi:10.3390/ijms18040769
Kouser, L., Madhukaran, S. P., Shastri, A., Saraon, A., Ferluga, J., Al-Mozaini, M., et al. (2015). Emerging and novel functions of complement protein C1q. Frontiers in immunology 6. Available at: https://www.frontiersin.org/articles/10.3389/fimmu.2015.00317 (Accessed November 11, 2022).
Krause, D. L., and Müller, N. (2010). Neuroinflammation, microglia and implications for anti-inflammatory treatment in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2010, e732806. doi:10.4061/2010/732806
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M., et al. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathology 177, 2549–2562. doi:10.2353/ajpath.2010.100265
Lim, T. K., and Ruthazer, E. S. (2021). Microglial trogocytosis and the complement system regulate axonal pruning in vivo. eLife 10, e62167. doi:10.7554/eLife.62167
Liston, A., and Masters, S. L. (2017). Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 17, 208–214. doi:10.1038/nri.2016.151
Liu, X., and Quan, N. (2018). Microglia and CNS interleukin-1: Beyond immunological concepts. Frontiers in neurology 9. Available at: https://www.frontiersin.org/articles/10.3389/fneur.2018.00008 (Accessed December 12, 2022).
Lopes, K. O., Sparks, D. L., and Streit, W. J. (2008). Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunoreactivity. Glia 56, 1048–1060. doi:10.1002/glia.20678
Lyons, A., Lynch, A. M., Downer, E. J., Hanley, R., O’Sullivan, J. B., Smith, A., et al. (2009). Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem. 110, 1547–1556. doi:10.1111/j.1471-4159.2009.06253.x
Manoharan, S., Guillemin, G. J., Abiramasundari, R. S., Essa, M. M., Akbar, M., and Akbar, M. D. (2016). The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and huntington’s disease: A mini review. Oxidative Med. Cell. Longev. 2016, 8590578–8590615. doi:10.1155/2016/8590578
Marschallinger, J., Iram, T., Zardeneta, M., Lee, S. E., Lehallier, B., Haney, M. S., et al. (2020). Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 23, 194–208. doi:10.1038/s41593-019-0566-1
Mazaheri, F., Snaidero, N., Kleinberger, G., Madore, C., Daria, A., Werner, G., et al. (2017). TREM 2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 18, 1186–1198. doi:10.15252/embr.201743922
McIntosh, A., Mela, V., Harty, C., Minogue, A. M., Costello, D. A., Kerskens, C., et al. (2019). Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 29, 606–621. doi:10.1111/bpa.12704
McQuade, A., and Blurton-Jones, M. (2019). Microglia in Alzheimer’s disease: Exploring how genetics and phenotype influence risk. J. Mol. Biol. 431, 1805–1817. doi:10.1016/j.jmb.2019.01.045
McQuade, A., Kang, Y. J., Hasselmann, J., Jairaman, A., Sotelo, A., Coburn, M., et al. (2020). Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 11, 5370. doi:10.1038/s41467-020-19227-5
Mecca, C., Giambanco, I., Donato, R., and Arcuri, C. (2018). Microglia and aging: The role of the TREM2–DAP12 and cx3cl1-cx3cr1 axes. IJMS 19, 318. doi:10.3390/ijms19010318
Meilandt, W. J., Ngu, H., Gogineni, A., Lalehzadeh, G., Lee, S.-H., Srinivasan, K., et al. (2020). Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the aβ42:aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J. Neurosci. 40, 1956–1974. doi:10.1523/JNEUROSCI.1871-19.2019
Minhas, P. S., Latif-Hernandez, A., McReynolds, M. R., Durairaj, A. S., Wang, Q., Rubin, A., et al. (2021). Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature 590, 122–128. doi:10.1038/s41586-020-03160-0
Mosher, K. I., and Wyss-Coray, T. (2014). Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 88, 594–604. doi:10.1016/j.bcp.2014.01.008
Nakanishi, H., and Wu, Z. (2009). Microglia-aging: Roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav. Brain Res. 201, 1–7. doi:10.1016/j.bbr.2009.02.001
Napoli, I., and Neumann, H. (2009). Microglial clearance function in health and disease. Neuroscience 158, 1030–1038. doi:10.1016/j.neuroscience.2008.06.046
Nayak, D., Roth, T. L., and McGavern, D. B. (2014). Microglia development and function. Annu. Rev. Immunol. 32, 367–402. doi:10.1146/annurev-immunol-032713-120240
Neuner, S. M., Heuer, S. E., Huentelman, M. J., O’Connell, K. M. S., and Kaczorowski, C. C. (2019). Harnessing genetic complexity to enhance translatability of Alzheimer’s disease mouse models: A path toward precision medicine. Neuron 101, 399–411. doi:10.1016/j.neuron.2018.11.040
Norden, D. M., Muccigrosso, M. M., and Godbout, J. P. (2015). Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 96, 29–41. doi:10.1016/j.neuropharm.2014.10.028
Onos, K. D., Sukoff Rizzo, S. J., Howell, G. R., and Sasner, M. (2016). Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res. Bull. 122, 1–11. doi:10.1016/j.brainresbull.2015.12.003
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi:10.1126/science.1202529
Paolicelli, R. C., Sierra, A., Stevens, B., Tremblay, M.-E., Aguzzi, A., Ajami, B., et al. (2022). Microglia states and nomenclature: A field at its crossroads. Neuron 110, 3458–3483. doi:10.1016/j.neuron.2022.10.020
Pawelec, P., Ziemka-Nalecz, M., Sypecka, J., and Zalewska, T. (2020). The impact of the cx3cl1/cx3cr1 Axis in neurological disorders. Cells 9, 2277. doi:10.3390/cells9102277
Perea, J. R., Lleó, A., Alcolea, D., Fortea, J., Ávila, J., and Bolós, M. (2018). Decreased CX3CL1 levels in the cerebrospinal fluid of patients with Alzheimer’s disease. Front. Neurosci. 12, 609. doi:10.3389/fnins.2018.00609
Picca, A., Calvani, R., Coelho-Junior, H. J., Landi, F., Bernabei, R., and Marzetti, E. (2020). Mitochondrial dysfunction, oxidative stress, and neuroinflammation: Intertwined roads to neurodegeneration. Antioxidants 9, 647. doi:10.3390/antiox9080647
Qu, W., and Li, L. (2021). Microglial TREM2 at the intersection of brain aging and Alzheimer’s disease. Neuroscientist, 107385842110407. doi:10.1177/10738584211040786
Rajagopalan, P., Hibar, D. P., and Thompson, P. M. (2013). TREM2 and neurodegenerative disease. N. Engl. J. Med. 369, 1565–1567. doi:10.1056/NEJMc1306509
Rawji, K. S., Mishra, M. K., Michaels, N. J., Rivest, S., Stys, P. K., and Yong, V. W. (2016). Immunosenescence of microglia and macrophages: Impact on the ageing central nervous system. Brain 139, 653–661. doi:10.1093/brain/awv395
Ray, P. D., Huang, B.-W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990. doi:10.1016/j.cellsig.2012.01.008
Replogle, J. M., Chan, G., White, C. C., Raj, T., Winn, P. A., Evans, D. A., et al. (2015). A TREM1 variant alters the accumulation of Alzheimer-related amyloid pathology. Ann. Neurology 77, 469–477. doi:10.1002/ana.24337
Ricklin, D., Hajishengallis, G., Yang, K., and Lambris, J. D. (2010). Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797. doi:10.1038/ni.1923
Rock, R. B., and Peterson, P. K. (2006). Microglia as a pharmacological target in infectious and inflammatory diseases of the brain. Jrnl NeuroImmune Pharm. 1, 117–126. doi:10.1007/s11481-006-9012-8
Rogers, J., Styren, S. D., Civin, W. H., Brachova, L., Wardi, P., and Lieberburg, I. (1992). Complement activation by f8-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 5.
Safaiyan, S., Besson-Girard, S., Kaya, T., Cantuti-Castelvetri, L., Liu, L., Ji, H., et al. (2021). White matter aging drives microglial diversity. Neuron 109, 1100–1117.e10. doi:10.1016/j.neuron.2021.01.027
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi:10.1016/j.neurobiolaging.2005.09.012
Schlepckow, K., Monroe, K. M., Kleinberger, G., Cantuti-Castelvetri, L., Parhizkar, S., Xia, D., et al. (2020). Enhancing protective microglial activities with a dual function TREM 2 antibody to the stalk region. EMBO Mol. Med. 12, e11227. doi:10.15252/emmm.201911227
Shahidehpour, R. K., Higdon, R. E., Crawford, N. G., Neltner, J. H., Ighodaro, E. T., Patel, E., et al. (2021). Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 99, 19–27. doi:10.1016/j.neurobiolaging.2020.12.003
Sheng, L., Chen, M., Cai, K., Song, Y., Yu, D., Zhang, H., et al. (2019). Microglial Trem2 induces synaptic impairment at early stage and prevents amyloidosis at late stage in APP/PS1 mice. FASEB J. 33, 10425–10442. doi:10.1096/fj.201900527R
Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., et al. (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9, eaaf6295. doi:10.1126/scitranslmed.aaf6295
Shi, Q., Colodner, K. J., Matousek, S. B., Merry, K., Hong, S., Kenison, J. E., et al. (2015). Complement C3-deficient mice fail to display age-related hippocampal decline. J. Neurosci. 35, 13029–13042. doi:10.1523/JNEUROSCI.1698-15.2015
Shineman, D. W., Basi, G. S., Bizon, J. L., Colton, C. A., Greenberg, B. D., Hollister, B. A., et al. (2011). Accelerating drug discovery for Alzheimer’s disease: Best practices for preclinical animal studies. Alzheimer’s Res. Ther. 3, 28. doi:10.1186/alzrt90
Sierra, A., Beccari, S., Diaz-Aparicio, I., Encinas, J. M., Comeau, S., and Tremblay, M.-È. (2014). Surveillance, phagocytosis, and inflammation: How never-resting microglia influence adult hippocampal neurogenesis. Neural Plast. 2014, 610343–610415. doi:10.1155/2014/610343
Šimončičová, E., Gonçalves de Andrade, E., Vecchiarelli, H. A., Awogbindin, I. O., Delage, C. I., and Tremblay, M.-È. (2022). Present and future of microglial pharmacology. Trends Pharmacol. Sci. 43, 669–685. doi:10.1016/j.tips.2021.11.006
Song, G. J., and Suk, K. (2017). Pharmacological modulation of functional phenotypes of microglia in neurodegenerative diseases. Front. Aging Neurosci. 9, 139. doi:10.3389/fnagi.2017.00139
Sri, S., Pegasiou, C.-M., Cave, C. A., Hough, K., Wood, N., Gomez-Nicola, D., et al. (2019). Emergence of synaptic and cognitive impairment in a mature-onset APP mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 7, 25. doi:10.1186/s40478-019-0670-1
St-Pierre, M.-K., Carrier, M., Gonzalez Ibanez, F., Simoncicova, E., Wallman, M.-J., Vallières, L., et al. (2022a). Dark microglia are present in human post-mortem brain samples and display ultrastructural and metabolic alterations during aging in a mouse model of amyloid deposition.
St-Pierre, M.-K., Carrier, M., González Ibáñez, F., Šimončičová, E., Wallman, M.-J., Vallières, L., et al. (2022b). Ultrastructural characterization of dark microglia during aging in a mouse model of Alzheimer’s disease pathology and in human post-mortem brain samples. J. Neuroinflammation 19, 235. doi:10.1186/s12974-022-02595-8
Stancu, I.-C., Cremers, N., Vanrusselt, H., Couturier, J., Vanoosthuyse, A., Kessels, S., et al. (2019). Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 137, 599–617. doi:10.1007/s00401-018-01957-y
Streit, W. J., Braak, H., Xue, Q.-S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485. doi:10.1007/s00401-009-0556-6
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212. doi:10.1002/glia.10319
Tay, T. L., Béchade, C., D’Andrea, I., St-Pierre, M.-K., Henry, M. S., Roumier, A., et al. (2018). Microglia gone rogue: Impacts on psychiatric disorders across the lifespan. Front. Mol. Neurosci. 10, 421. doi:10.3389/fnmol.2017.00421
Terry, R. D., Masliah, E., Salmon, D., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s Disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurology Official J. Am. Neurological Assoc. Child Neurology Soc. 30, 572–580. doi:10.1002/ana.410300410
Tremblay, M.-È. (2011). The role of microglia at synapses in the healthy CNS: Novel insights from recent imaging studies. Neuron Glia Biol. 7, 67–76. doi:10.1017/S1740925X12000038
Tremblay, M.-È., Zettel, M. L., Ison, J. R., Allen, P. D., and Majewska, A. K. (2012). Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 60, 541–558. doi:10.1002/glia.22287
Turnbull, I. R., Gilfillan, S., Cella, M., Aoshi, T., Miller, M., Piccio, L., et al. (2006). Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 177, 3520–3524. doi:10.4049/jimmunol.177.6.3520
Venegas, C., Kumar, S., Franklin, B. S., Dierkes, T., Brinkschulte, R., Tejera, D., et al. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361. doi:10.1038/nature25158
Wang, S., Mustafa, M., Yuede, C. M., Salazar, S. V., Kong, P., Long, H., et al. (2020). Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 217, e20200785. doi:10.1084/jem.20200785
Ye, J., Jiang, Z., Chen, X., Liu, M., Li, J., and Liu, N. (2017). The role of autophagy in pro-inflammatory responses of microglia activation via mitochondrial reactive oxygen species in vitro. J. Neurochem. 142, 215–230. doi:10.1111/jnc.14042
Yvanka de Soysa, T., Therrien, M., Walker, A. C., and Stevens, B. (2022). Redefining microglia states: Lessons and limits of human and mouse models to study microglia states in neurodegenerative diseases. Seminars Immunol. 60, 101651. doi:10.1016/j.smim.2022.101651
Zhang, X., Pearsall, V. M., Carver, C. M., Atkinson, E. J., Clarkson, B. D. S., Grund, E. M., et al. (2022). Rejuvenation of the aged brain immune cell landscape in mice through p16-positive senescent cell clearance. Nat. Commun. 13, 5671. doi:10.1038/s41467-022-33226-8
Keywords: microglia, microglial diversity, cognitive aging, mild cognitive impairment, Alzheimer’s disease, pharmacology
Citation: McKee CG, Hoffos M, Vecchiarelli HA and Tremblay M-È (2023) Microglia: A pharmacological target for the treatment of age-related cognitive decline and Alzheimer’s disease. Front. Pharmacol. 14:1125982. doi: 10.3389/fphar.2023.1125982
Received: 16 December 2022; Accepted: 03 February 2023;
Published: 09 February 2023.
Edited by:
Jacob Raber, Oregon Health and Science University, United StatesReviewed by:
Stergios Tsartsalis, Hôpitaux universitaires de Genève (HUG), SwitzerlandCopyright © 2023 McKee, Hoffos, Vecchiarelli and Tremblay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie-Ève Tremblay, ZXZldHJlbWJsYXlAdXZpYy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.