 Mingsong Shi1
Mingsong Shi1 Yan Zhou1
Yan Zhou1 Haoche Wei2Xinyu Zhang3Meng Du4
Haoche Wei2Xinyu Zhang3Meng Du4 Yanting Zhou5Yuan Yin1Xinghui Li3Xinyi Tang3Liang Sun6
Yanting Zhou5Yuan Yin1Xinghui Li3Xinyi Tang3Liang Sun6 Dingguo Xu4,7*Xiaoan Li1*
Dingguo Xu4,7*Xiaoan Li1*- 1NHC Key Laboratory of Nuclear Technology Medical Transformation, Mianyang Central Hospital, School of Medicine, University of Electronic Science and Technology of China, Mianyang, Sichuan, China
- 2State Key Laboratory of Biotherapy/Collaborative Innovation Center of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China
- 3West China School of Pharmacy, Sichuan University, Chengdu, Sichuan, China
- 4College of Chemistry, MOE Key Laboratory of Green Chemistry and Technology, Sichuan University, Chengdu, Sichuan, China
- 5Key Laboratory of Basic Pharmacology of Ministry of Education and Joint International Research Laboratory of Ethnocentric of Ministry of Education, Zunyi Medical University, Zunyi, Guizhou, China
- 6Shenzhen Shuli Tech Co., Ltd, Shenzhen, Guangdong, China
- 7Research Center for Material Genome Engineering, Sichuan University, Chengdu, Sichuan, China
Natural products are widely used for treating mitochondrial dysfunction-related diseases and cancers. Curcumin, a well-known natural product, can be potentially used to treat cancer. Human salt-induced kinase 3 (SIK3) is one of the target proteins for curcumin. However, the interactions between curcumin and human SIK3 have not yet been investigated in detail. In this study, we studied the binding models for the interactions between curcumin and human SIK3 using computational tools such as homology modeling, molecular docking, molecular dynamics simulations, and binding free energy calculations. The open activity loop conformation of SIK3 with the ketoenol form of curcumin was the optimal binding model. The I72, V80, A93, Y144, A145, and L195 residues played a key role for curcumin binding with human SIK3. The interactions between curcumin and human SIK3 were also investigated using the kinase assay. Moreover, curcumin exhibited an IC50 (half-maximal inhibitory concentration) value of 131 nM, and it showed significant antiproliferative activities of 9.62 ± 0.33 µM and 72.37 ± 0.37 µM against the MCF-7 and MDA-MB-23 cell lines, respectively. This study provides detailed information on the binding of curcumin with human SIK3 and may facilitate the design of novel salt-inducible kinases inhibitors.
1 Introduction
Mitochondrion plays indispensable role in programmed cell death, redox signaling, and energy metabolism, which suggests mitochondrion is a favorable target (Indran et al., 2011). Direct targeting mitochondria improves antiproliferative activity and decreases cancer invasion (Millard et al., 2013; Ye et al., 2017; Pan et al., 2018). In addition, mitochondria-targeted cytotoxic conjugates, which was combined one parent drug to mitochondria-directing compound with linker, are one the important approaches for targeting therapeutics (Zorova et al., 2018; Jeena et al., 2020). For example, the natural drug honokiol linked with the effective mitochondria-directing compound berberine were designed and synthesized a mitochondria-targeted conjugates (Shi et al., 2020). Those derivatives for honokiol shown effective antitumor activity than honokiol. Thus, based on natural mitochondria-targeted cytotoxic conjugates is one interesting field for cancer therapeutic.
Natural products have many notable characteristics, such as wide efficacy and low toxicity; they have recently attracted increasing research attention as lead compounds to treat mitochondrial dysfunction-related diseases, cancer, and antimicrobial-resistant infections (Du et al., 2021; An et al., 2022; Rahman M. A. et al., 2022; Bernal et al., 2022; Rahman M. M. et al., 2022; He et al., 2022; Liao et al., 2022; Nurumal et al., 2022; Su et al., 2022; Xu et al., 2022). Especially, the effects of many natural product on mitochondrial dysfunction-related diseases have been recently studied (An et al., 2022; Rahman M. A. et al., 2022; Rahman M. M. et al., 2022). Particularly, curcumin (bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione, CAS No. 458-37-7, Figure 1), a natural dietary polyphenol compound, has played close attention to its anti-inflammatory, antioxidant, neuroprotective, and antitumor activities (Golonko et al., 2019; Dev et al., 2021; Lei et al., 2022). Curcumin also induces paraptosis in breast cancer cell lines from swelling and fusion of mitochondria and endoplasmic reticulum (Yoon et al., 2010). Curcumin is a natural product which has been extracted from the turmeric, zedoary, and other ginger plants. Notably, it is the main bioactive compound in turmeric. Curcumin is mainly used for treating cancer. (Rajendran et al., 2022; Tang et al., 2022; Vadukoot et al., 2022; Wang et al., 2022; Yin et al., 2022). However, the anticancer activity of curcumin has not been fully elucidated in vivo and in vitro.
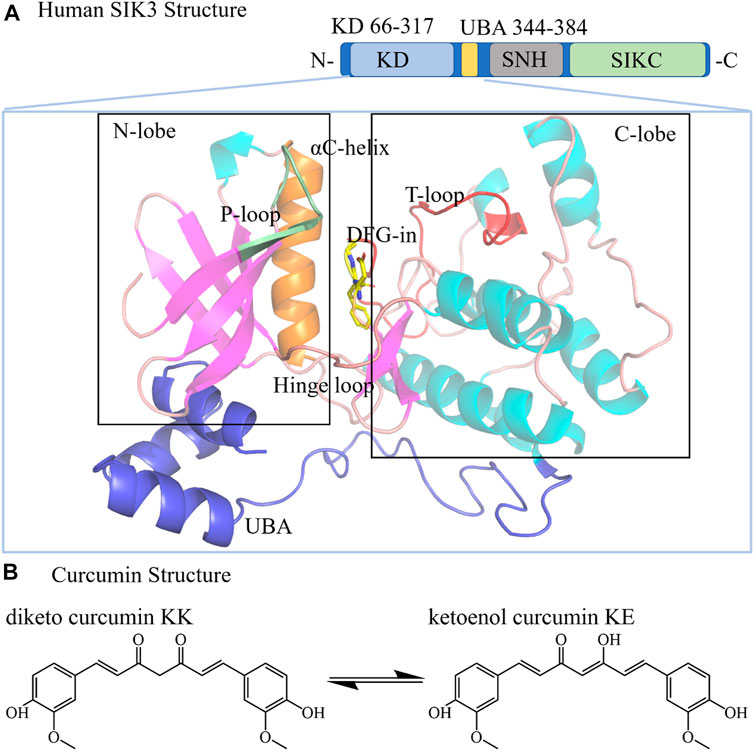
FIGURE 1. Structures of human SIK3 and curcumin. (A) Structure of human SIK3. Human SIK3 (UniProt ID: Q9Y2K2, residues: 1–1321) formed from a kinase domain (KD, residues: 66–317), central SNF protein kinase homology domain (SNH), and phosphorylation domain near the C-terminal (SIKC). The UBA domain (residues: 344–384) is a linker between KD and SNH. The kinase domain includes an N-terminal region (N-lobe), a C-terminal region (C-lobe), and a hinge loop between the N-lobe and C-lobe. The αC-helix, DFG motif, T-loop, and P-loop also play important roles in protein kinase activity. The ATP (adenosine triphosphate)-binding pocket (active pocket) was formed between the N-lobe and C-lobe. (B) Structure of the diketo (KK) and ketoenol (KE) forms of curcumin.
The mechanism for the antitumor activity of natural compound curcumin were related with multiple signaling pathways, such as the signal transducer, nuclear factor-κB, cyclooxygenase 2, mitogen-activated protein kinase, and activator of transcription 3, and TNF-α signaling pathways. Curcumin is a spectrum inhibitor that targets epidermal growth factor receptor (Gopal, 2022), protein kinase C (Hasmeda and Polya, 1996), tubulin (Chakraborti et al., 2011; Ota et al., 2018), cyclin-dependent kinase 2 (Sumirtanurdin et al., 2020), casein kinase 2 (Cozza et al., 2020), adenosine monophosphate-activated protein kinases (Soltani et al., 2019), and Abelson leukemia virus tyrosine kinase 1 (Rodrigues et al., 2021). A previous study reported that curcumin exhibits antitumor activity in triple-negative breast cancer patient-derived xenograft tumor mice by inhibiting the expression of salt-inducible kinase 3 (SIK3) (Cheng et al., 2021). However, the interactions between curcumin and SIK3 remain largely unclear, which hinders its use as a lead compound to design salt-inducible kinases inhibitors.
Salt-inducible kinases (SIKs) control the anti-inflammatory cytokine interleukin-10 (IL-10) in macrophages which is one of the cyclic adenosine monophosphate-dependent production (Okamoto et al., 2004; Du et al., 2016; Sakamoto et al., 2018; Wein et al., 2018; Chen F. Y. et al., 2019; Sun et al., 2020). Three isoforms exist for the SIKs family: SIK1, SIK2, and SIK3 (Katoh et al., 2004). SIKs are ubiquitously expressed and control the gene expression for increasing the intracellular levels of cyclic adenosine monophosphate. They are also found to regulate lipid and energy metabolism (Du et al., 2008; Sun et al., 2020). Moreover, SIKs inhibition can be considered as a therapeutic strategy for inflammatory disease (Antonio et al., 2022). In addition, SIKs are also dysregulated in prostate cancer, ovarian cancer, lung cancer, breast cancer, and other cancers (Du et al., 2016; Wein et al., 2018; Chen F. Y. et al., 2019), which indicates that SIKs are appealing pharmacological targets for treating cancer. Recently, numerous SIKs inhibitors have been developed and applied in the preclinical or clinical studies (Heap et al., 2017; Jin et al., 2020), such as HG-9-91-01 (Clark et al., 2012), KIN112 (Clark et al., 2012), YKL-06-062 (Mujahid et al., 2017), MRT67307, MRT199665, OMX-0407 (Hartl et al., 2021), and ARN-3236 (Supplementary Figure S1) (Zhou et al., 2017). However, no specifically SIKs inhibitors have been clinically approved. Thus, novel inhibitors specifically targeting SIKs is need to develop.
Curcumin inhibits the activity of SIK3 (Cheng et al., 2021), which suggests that it can be used as a lead compound for the optimization of SIKs inhibitors. However, reliable curcumin-SIKs structures have not yet been reported. The detailed structural information on drug–target binding plays a significant important role in computer–aided drug design. The ligand-receptor interaction is one of the incorporations to elucidate the drug–target binding. The target structure can be obtained from crystal structure or computational modeling. The homology modeling (HM) as one widely applied method can be used to predict target structures, which is based on amino acid sequences (Wieman et al., 2004; Xiang, 2006; Cavasotto and Phatak, 2009; Cavasotto, 2011; Tuccinardi and Martinelli, 2011; Gadhe et al., 2015; Munsamy and Soliman, 2017; Salam et al., 2018; Muhammed and Aki-Yalcin, 2019). The molecular docking method is employed to construct drug-target complexes for drug design (Hirayama, 2007; Kroemer, 2007; Bello et al., 2013; Wong, 2015; Scotti et al., 2017; Amaro et al., 2018; Pinzi and Rastelli, 2019; Saikia and Bordoloi, 2019; Jakhar et al., 2020). Molecular dynamics (MD) simulations have been applied to learn the interactions between inhibitors and proteins (Rognan, 1998; Durrant and McCammon, 2011; Aci-Seche et al., 2016; Hernandez-Rodriguez et al., 2016; Do et al., 2018; Liu et al., 2018; Yamashita, 2018; Bera and Payghan, 2019; Zou et al., 2019).
In this study, we obtained the curcumin–SIK3 binding models using homology modeling, molecular docking, and molecular dynamics simulation. We identified that the I72, V80, A93, Y144, A145, and L195 residues of curcumin showed antitumor activity. Moreover, curcumin inhibited human SIK3 protein kinase with an IC50 value (half maximal inhibitory concentration) of 131 nM. The antitumor activity of curcumin was also tested in human breast cancer cell line MDA-MB-231 and MCF-7. To the best of our knowledge, our study had identified the complexes for curcumin binding with human SIK3 based on computational analysis and experimental methods.
2 Materials and methods
2.1 Kinase assay
The IC50 values of curcumin against human SIK3 were determined by KinaseProfiler radiometric protein kinase assays obtained from Eurofins Pharma Discovery Services UK Limited (Eurofins). The adenosine triphosphate (ATP) concentrations used here represent the Km values of the corresponding kinases.
2.2 Cell assay
Human breast cancer cell line MDA-MB-231 and MCF-7 were obtained from the American Type Culture Collection (ATCC; Manassas, Virginia, United States). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, for MDA-MB-231) or minimum essential medium (MEM, for MCF-7), which was supplemented with 1% Penicillin-Streptomycin (PS) and 10% Fetal bovine serum (FBS). Approximate 4 × 103 cells, the cells were plated into each well in 96-well plate and were incubated in 5% CO2 at 37°C for 24 h. Curcumin (MDL No. MFCD00008365, CAS No. 458-37-7, purity 98%) was purchased from Shanghai Macklin Biochemical Co., Ltd. (Shanghai, China). HG-9-91-01 (TargeMol No. T4599, CAS No. 1456858-58-4, purity 99.64%) was purchased form TargetMol (TargetMol Chemicals Inc., Boston, United States). The tested compounds at the indicated final concentrations were added to the culture medium and incubated for 72 h (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) (MTT) was added in each cell. Subsequently, the cells were incubated for an additional 2 h in each well which were dissolved in 100 μL dimethyl sulfoxide (DMSO). The absorbance values (OD) of the 96-well tissue culture plates were read at 570 nm on a Spectra MAX M5 microplate spectrophotometer. The cell viability results were calculated using the GraphPad Prism 6.0 software. This assay was repeated three times.
2.3 SIK3 structure preparation
The protein sequence of human SIK3 (UniProt ID: Q9Y2K2, 1,321 residues) was downloaded from the UniProt database (Bateman et al., 2015; Bateman et al., 2017; Bateman et al., 2019). Experimentally, most SIKs inhibitors bind to the ATP-competitive inhibitors with bind into the ATP-binding pocket (Zhou et al., 2017; Liu et al., 2021; Tesch et al., 2021). Moreover, curcumin binds to the ATP-binding site of human DYRK2 [PDB ID: 6HDR (Elkins and Knapp, 2019)]. Thus, we speculated that curcumin may also bind to the ATP-binding site of human SIK3. The T-loop (also named activation loop or A-loop)) conformation of SIK2 had two class conformation with closed and open conformation, and so on the two class conformations of T-loop for SIK3 also presented. To construct the two conformations of SIK3 structure, two separate homology modeling experiments were run from the online service SWISS-MODEL (Waterhouse et al., 2018). Although the full sequence of human SIK3 contains three main domains, we only considered the protein kinase domain and ubiquitin-associated (UBA) domain (residues: 1–390) in this study. The crystal structure of catalytic and UBA domain of rat MARK2 [PDB ID: 2WZJ (Marx et al., 2010)] was employed to model the closed T-loop conformation for human SIK3 (labeled as SIK3-C). Meanwhile, kinase and UBA domain of human MARK2 [PDB ID: 3IEC (Nesic et al., 2010)] was used to construct the structure of the open conformation of T-loop for human SIK3 (defined as SIK3-O). Both SIK3 and MARK2 have similar structures and functions which results from belonging to the calcium/calmodulin-dependent protein kinase-like family (Gormand et al., 2011; Alexander et al., 2019). The VERIFY3D software (Bowie et al., 1991), ERRAT (Colovos and Yeates, 1993), the GMQE method (Benkert et al., 2011), and a Ramachandran plot obtained by the ProCheck software (Laskowski et al., 1993) were applied to validate the modeling SIK3 structure SIK3-C and SIK3-O.
2.4 Curcumin structure preparation
The two tautomers of curcumin, namely the diketo (KK) and ketoenol forms (KE), were constructed via the In Draw software (Integle chemical draw). The molecular structures of the two form curcumin KK and KE were optimized at the B3LYP/6-311G (d, p) level in the gas phase (Hohenberg and Kohn, 1964; Kohn and Sham, 1965; Becke, 1988; Lee et al., 1988; Miehlich et al., 1989; Blaudeau et al., 1997; Rassolov et al., 1998; Tran et al., 1999; Rassolov et al., 2001). After the optimization, frequency calculations were also performed at the B3LYP/6-311G (d, p) level. There are no imaginary frequencies for the optimization steps which indicate that those optimized structures for KK and KE curcumin are stable. All calculations were performed with the Gaussian 09 software package (Frisch et al., 2009).
2.5 Molecular docking
Currently, no crystalline structures have been reported for the binding of curcumin to human SIK3. Additionally, the T-loop conformation of SIK3 of curcumin during its binding is also unclear. Therefore, in this study, both the open and closed T-loop conformations (SIK3-C and SIK3-O) were considered. In addition, the two forms curcumin (KK and KE) were also applied to construct the curcumin-SIK3 complex structures. KK or KE form curcumins with an open or closed conformation of the T-loop (SIK3-C and SIK3-O) were employed to construct the curcumin/SIK3 complexes. PYMOL 2.1 (Schrödinger, 2010) was employed to prepare the receptor structure of human SIK3 based on the HM results of SIK3-C and SIK3-O. Subsequently, receptor protein SIK3 and ligand small molecule curcumin were pretreated by AutoDockTools 1.5.6, (Sanner, 1999), although three steps: 1) added hydrogen atoms; 2) adding Gasteiger charges (Gasteiger and Marsili, 1980); 3) adjusted the unreasonable atomic overlap. Subsequently, a grid box was estimated by AutoGrid v.4.2 (Morris et al., 2009) with a 0.375 Å grid spacing and a 40 × 40 × 40 grid size. The center of the ATP-binding pocket of human SIK3 was defined as the center of the grid box. Finally, 2000 conformations were constructed by the Lamarckian genetic algorithm (Fuhrmann et al., 2010) for the four systems (SIK3-C-KK, SIK3-C-KE, SIK3-O-KK, and SIK3-O-KE) in AutoDock v.4.2 (Morris et al., 2009). The conformation with the best rational orientation in the ATP-binding pocket, which was based on the curcumin with DYRK2 crystal structure (such as the key hydrogen bonds between curcumin and the hinge loop of DYRK2, PDB ID: 6HDR), was selected as the optimal conformation.
2.6 Molecular dynamics (MD) simulation
The four complex curcumin/SIK3 systems, SIK3-C-KK, SIK3-C-KE, SIK3-O-KK, and SIK3-O-KE, which were obtained from molecular docking, were run in molecular dynamics simulations to study the binding models. Thus far, no standard force field parameters have been generated for the KE and KK forms of curcumin. Therefore, the restrained electrostatic potential protocol (Bayly et al., 1993) and the general amber force field (GAFF, version 2) (Wang et al., 2004) were employed to generate the force field parameters for the KK and KE forms of curcumin with the Antechamber module in AMBERTools21 (Case et al., 2020). The partical aomic charges of KK and KE curcumin were obtained with HF/6-31G theory level. In addition, the standard protein force field [AMBER ff19SB force field (Tian et al., 2020)] was applied to generate the human SIK3 topology parameters. The protonation state of human SIK3 was determined with pH of 7.4 using the H++ online service (Bashford and Karplus, 1990; Gordon et al., 2005; Myers et al., 2006; Anandakrishnan et al., 2012). One Na+ ion was required to neutralize the curcumin/SIK3 complex systems. TIP3P water (Jorgensen et al., 1983) was used to solvate the complex systems in a 15 Å cuboid water box. Subsequently, 20,067, 20,065, 19,677, and 19,676 waters were added for SIK3-C-KK, SIK3-C-KE, SIK3-O-KK, and SIK3-O-KE, respectively. Lastly, the curcumin/SIK3 complex systems included 320 residues of human SIK3, one small molecule of curcumin, one Na+ ion, and approximately 19,670–20,100 water molecules. Firstly, the curcumin/SIK3 complex system was subjected to optimize with steepest descent method (9,000 steps) and the conjugate gradient method (1,000 steps) that all the solute molecules were fixed and not considered to minimize. Secondly, conjugate gradient method (10,000-step) was employed to minimize the entire system without restriction. After the two minimization steps, Langevin dynamics were employed to increase the temperature of the complex system from 0 to 300 K in 200 ps. The pressure was then maintained by isotropic position scaling at 1 bar for 200 ps. Subsequently, the system was pre-equilibrated within the NPT (isothermal-isobaric) ensemble at 300 K and 1 bar in 200 ps. Finally, the entire system underwent 500 ns MD simulations for data collection and analyses. In this work, all MD simulations were run with AMBER20 and AMBERTools21 (Case et al., 2020). The CPPTRAJ module (Roe and Cheatham, 2018; Cheatham et al., 2019) was then employed to analyze the data.
2.7 Binding free energy estimation
The MM/GBSA approach (Srinivasan et al., 1998; Lee et al., 2004) was used to calculate the binding free energies between curcumin and human SIK3. The MM/GBSA method has been widely employed to evaluate the binding affinities between ligands and enzyme (Tse and Verkhivker, 2015; Wang et al., 2017; Chen Q. Q. et al., 2019; Shi and Xu, 2019; Wei et al., 2019; Xu and Zheng, 2022). Previously, the MM/GBSA framework has been detailly introduced (Honig and Nicholls, 1995; Genheden and Ryde, 2015; Onufriev and Case, 2019). For the four curcumin/SIK3 complex systems, the MM/GBSA approach was employed to calculate the energy terms over 1,000 frames from the last 200 ns of the MD simulation. In addition, the entropy was obtained via the statistical average with 100 frames for 2 ns interval. In order to determine the contributions of each residue, the total binding energy between SIK3 and curcumin was decomposed with the MM/GBSA binding energy decomposition without considering the entropies (Gaillard and Simonson, 2014). The energy was then calculated using the MMPBA. py program in AMBERTools21 (Case et al., 2020). More information about the binding free energy calculation is provided in the Supplementary Material. A scheme of the methods employed is shown in Supplementary Figure S2.
3 Results and discussion
3.1 SIK3 structure modelling
Generally, the SIK3 protein comprises three domains (Figure 1): a serine-threonine kinase domain (KD) near the N-terminal, a central sucrose non-fermenting protein kinase homology domain (SNF), and a long phosphorylation domain (SIKC) near the C-terminal (Katoh et al., 2004; Antonio et al., 2022). A ubiquitin-associated (UBA) region followed by KD has also been defined. Specifically, the kinase domain phosphorylated threonine residues on the substrate proteins regulate downstream genes, such as CREB, class IIa HDAC, and PME-1 (Okamoto et al., 2004; Taub et al., 2010; Du et al., 2016; Wein et al., 2018; Chen F. Y. et al., 2019; Taub, 2019; Sun et al., 2020; Antonio et al., 2022). In this study, the kinase domain (residues: 66–317) and UBA region (344–384) were considered for following structural construction (residues: 1–400, labeled as SIK3 in the following text), and SNF and SIKC were excluded.
The three-dimensional structure of the human SIK3 kinase domain is composed of an N-terminal region (labeled as N-lobe) and a C-terminal region (defined as C-lobe). The N-lobe consists of five stranded β-sheets (named as β1–β5) and one α-helix (labeled as αC helix). The C-lobe is mostly α-helical (αD–αI) but contains important β-sheets (β6–β7). The hinge loop for protein kinase is referred for the linker between the C- and N-lobes (Supplementary Figure S3) (Hanks and Hunter, 1995; Cherry and Williams, 2004). The ATP-binding site is formed from the pocket between the N-lobe and C-lobe. In protein kinases, the DFG (Asp-Phe-Gly) motif in the C-lobe is highly conserved and follows T-loop. The T-loop serves as a key regulating region for protein kinase activities (Canagarajah et al., 1997; Stevenson et al., 2002; Cha et al., 2015; Mansueto et al., 2019). The activation of protein kinases depends considerably on the DFG conformation. For instance, when human FAK binds with N-Methyl-N-(3-((2-phenylamino-5-trifluoromethyl-pyrimidin-4-ylamino)-methyl)-pyridin-2-yl)-methanesulfonamide [PDB ID: 6YXV, (Berger et al., 2021), Supplementary Figure S4], the side chain of D toward into the active pocket, while the side chain of F orients outside of the pocket (defined as DFG-in conformation) (Modi and Dunbrack, 2019). Conversely, in the DFG-out conformation, the side chain of D orients out of the ATP-binding site, and that of F packs into the ATP-binding pocket (Roskoski, 2016). Based on the binding models of inhibitors and protein kinases, small molecule protein kinase inhibitors can be classified into six types (Supplementary Figure S5): type I (ATP-binding pocket, DFG-in), type II (ATP-binding pocket, DFG-out), type III (allosteric inhibitor binding next to ATP-binding pocket), type IV (allosteric inhibitor binding far away ATP-binding pocket), type V (bivalent inhibitor), and type VI (covalent inhibitor) (Roskoski, 2016; Modi and Dunbrack, 2019; Arter et al., 2022). The criterion for type-I inhibitors is that the protein kinases should have the DFG-in conformation. Previous studies have shown that when curcumin binds with DYRK2, it acts as a type I inhibitor. Thus, in this work, we assumed that curcumin behaves as type I inhibitor with SIK3 and exhibits the DFG-in and αC-in conformations.
However, some type-I inhibitors, such as dasatinib (Shi et al., 2021a), bosutinib (Shi et al., 2022b), MRT67307 (Shi et al., 2021b), MRT199665 (Shi et al., 2021b), KIN112 (Shi et al., 2021b), and HG-9-91-01 (Shi et al., 2021b), exhibit diverse T-loop conformations while binding with SIK2. Such diverse conformations of the of T-loop have also been reported for type-I inhibitors binding with human FAK (Shi et al., 2022a). Therefore, we speculated that curcumin may also bind with human SIK3 with diverse T-loop conformations, namely the open T-loop (labeled as SIK3-O that T-loop orients far away from the active site) and closed T-loop (defined as SIK3-C that T-loop orients toward the active site) conformations. Atomic information of the open and closed conformations is very important for exploring the binding models between curcumin and SIK3.
In this study, we constructed SIK3-C with closed T-loop and SIK3-O with open T-loop for SIK3 using HM, which were based on the crystal structures of MARK family. The published crystal structures for MARKs were obtained to verify the conformations, and four structures were observed in the overall T-loop residues. The 6C9D (PDB ID) (Emptage et al., 2018) 3FE3 (Nugoor et al., 2008) crystal structures were observed for MARK1 and MARK3, respectively. Meanwhile, the 3IEC and 2WZJ structures were published for MARK2. In addition, the T-loops in 3IEC exhibited the open conformation and those in 2WZJ exhibited the closed conformation. Thus, we selected two crystal structures as templates to construct the closed (known as SIK3-C, PDB ID: 2WZJ (Marx et al., 2010), Figure 2) and open conformation structures [labeled as SIK3-O, PDB ID: 3IEC (Nesic et al., 2010)]. The term root mean square deviation (RMSD) of the backbone was 0.05 Å between SIK3-C and 2WZJ and 0.08 Å between SIK3-O and 3IEC. The overall quality of the modeled structures was also evaluated from their non-bonded atomic interactions using the ERRAT analysis (86.49% and 89.61% for SIK3-O and SIK3-C, respectively, Supplementary Figure S6). The occupancy of residues with average 3D-1D score >0.2 from VERIFY-3D were 82.87% of SIK3-O and 72.27% of SIK3-C (Supplementary Figure S7). Additionally, one or more residues were located in the disallowed regions from the Ramachandran plot analysis. Meanwhile, 91.1% residues for SIK3-O and 89.3% residues for SIK3-C were found in the most favored regions (Supplementary Figure S8). These results indicate that the constructed human SIK3-C and SIK3-O structures were suitable and reliable for molecular docking. At the same time, the difference conformation of T-loop was the mainly diversity between SIK3-O and SIK3-C (Supplementary Figure S9) and the RMSD between SIK3-C and SIK3-O was 1.15 Å. Therefore, the constructed three-dimensional structures for human SIK3 (SIK3-C and SIK3-O) were well to be employed as receptor structures in the subsequent simulations.
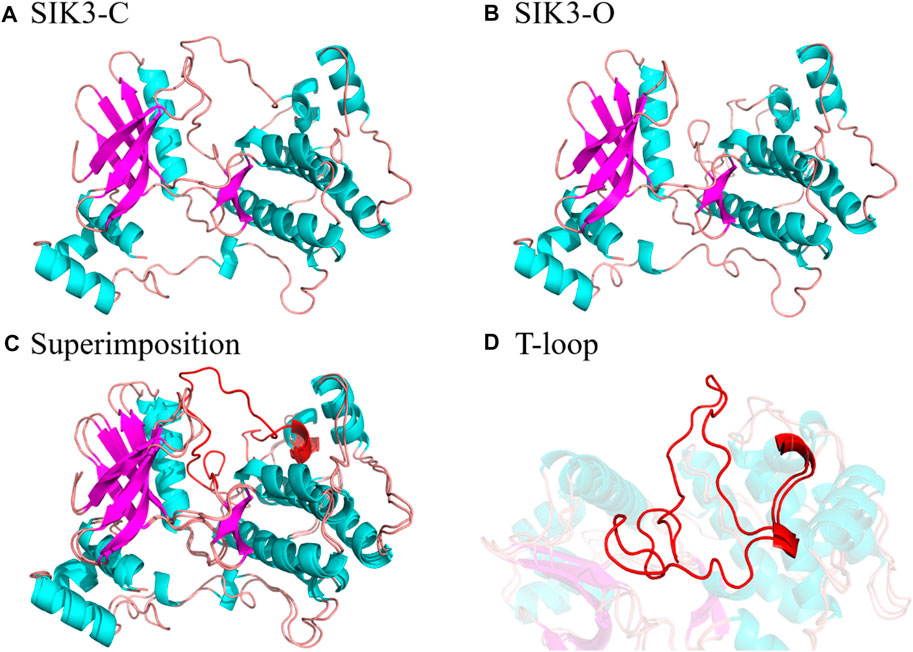
FIGURE 2. Structures of SIK3 with open and closed conformations of the T-loop. (A) Constructed structure of SIK3-C with a closed conformation of the T-loop generated by HM based on MARK2 (PDB ID: 2WZJ). (B) Constructed structure of SIK3-O with an open conformation of the T-loop generated by HM based on MARK2 (PDB ID: 3IEC). (C) Superimposition of SIK3-O to SIK3-C and (D) the enlarged T-loop conformation during the superimposition.
3.2 Curcumin structure
Similar to other β-diketo compounds, the diketo (KK) and ketoenol (KE) tautomers of curcumin exist in equilibrium. Both these forms have different potencies to bind with biomolecules (Rege et al., 2019; Osifova et al., 2022). Three polymorph crystal structures have been reported for curcumin (such as the monoclinic and orthorhombic forms) (Supplementary Figure S10) (Tonnesen et al., 1982; Parimita et al., 2007; Sanphui et al., 2011; Matlinska et al., 2018). The KE tautomer can be seen in those three polymorphs curcumin. Notably, the tautomeric forms of curcumin and curcumin derivatives are closely related with the biological activity. For example, the KK form of curcumin exerts antioxidant activity (Jovanovic et al., 1999), while the KE form curcumin is prone to degradation (Rege et al., 2019). In addition, the KE form can penetrate the blood–brain barrier and bind amyloid-beta aggregates. However, the curcumin tautomer that binds with SIK3 has not been identified. Thus, the KK and KE forms of curcumin were considered in this work.
When ligand molecule interacts with receptor protein, the geometry conformation of the ligand molecule must be complementary with the active site of receptor protein. The conformation of curcumin needs to be considered for it binding with human SIK3. Therefore, the density functional theory had been performed to regard the geometries of the KE and KK forms of curcumins. After optimized of the KK and KE forms of curcumin in the gaseous phase, the geometrical parameters are shown in Supplementary Figures S11, S12, respectively. In accordance with the optimized structures, the KE and KK forms adopt planar and non-planer conformations, respectively. Meanwhile, the charge distribution of the two tautomers is also different, especially between the diketo and ketoenol regions (Figure 3 and Supplementary Figure S13). The different protonation states between the KK and KE forms contribute to the differences in their binding models and antitumor activities. Therefore, these two forms of curcumin were docked into the ATP-pocket of human SIK3.
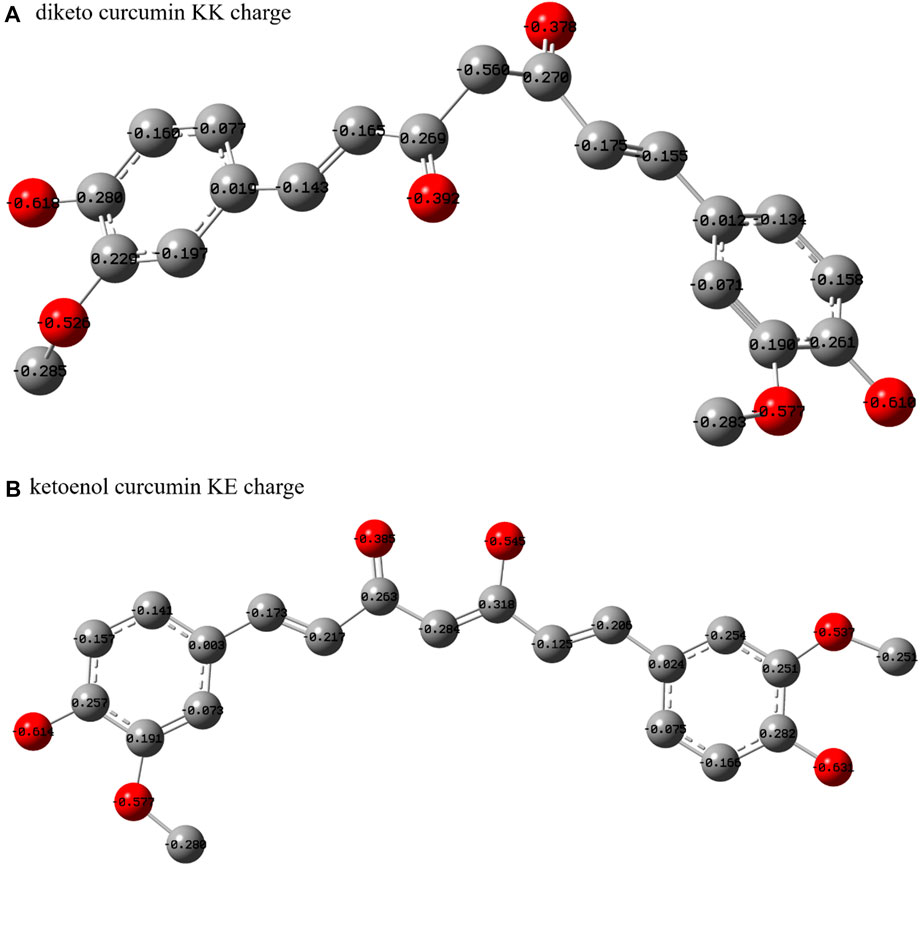
FIGURE 3. Heavy atom charges of curcumin. (A) For diketo curcumin and (B) for ketoenol curcumin.
3.3 Docking pose analysis
From the binding models of protein kinase inhibitors, we can assume that the binding model for curcumin with SIK3 is based on one key interaction, namely the hydrogen bonding between the hinge loop of SIK3 and the diketo or ketoenol moieties of curcumin. Two thousand conformations of the KK form of curcumin bound to SIK3-C were obtained from the docking experiments; these conformations were initially clustered into 33 clusters with a maximum RMS tolerance of 2.00 Å (Supplementary Figure S14). The occupancies of the top five clusters were >66.9%. Based on the speculated binding model and docking score, Cluster 1 was selected as the SIK3-C_KK conformation with a docking binding energy of −7.71 kcal/mol. Meanwhile, the occupancy of the top five clusters for the binding of KE with SIK3-C also exceeded 67.3% (Supplementary Figure S15). Based on the possible binding models, Cluster 2 was selected as the SIK3-C_KE conformation with a docking binding energy of −6.41 kcal/mol. In addition, Cluster 1 was selected for the possible binding models of KK or KE with SIK3-O, which had docking binding energies of −7.68 and −7.84 kcal/mol, respectively (Supplementary Figures S16, S17). Meanwhile, the occupancies of the top five clusters of SIK3-O_KK and SIK3-O_KE were 58.9% and 90.7%, respectively. As shown in Figure 2, we plotted four representative molecular docking models with different conformations, which were labeled as SIK3-C_KK, SIK3-C_KE, SIK3-O_KK, and SIK3-O_KE (Figure 4). Interestingly, the possible hydrogen bonding network remained between the diketo or ketoenol moieties of curcumin and the A145 residue of the hinge loop of the open or closed T-loop conformations of SIK3, which could potentially facilitate binding (Supplementary Figure S18). Additionally, unstable hydrogen bonds were found in all four complex systems; for instance, the I72 residue formed a hydrogen bond with the hydroxyphenyl group of the KK form of curcumin. However, this hydrogen bond was not observed for the KE form. Furthermore, some residues, such as I72, M117, L195, A205, and L212, formed van der Waals (vdW) interactions with the phenyl ring of curcumin. However, unfavorable donor-donor interactions were observed for SIK3-C_KE, SIK3-O_KK, and SIK3-O_KE systems. This indicates that the molecular dynamics simulations of the complex systems need to be optimized from the docking experiments.
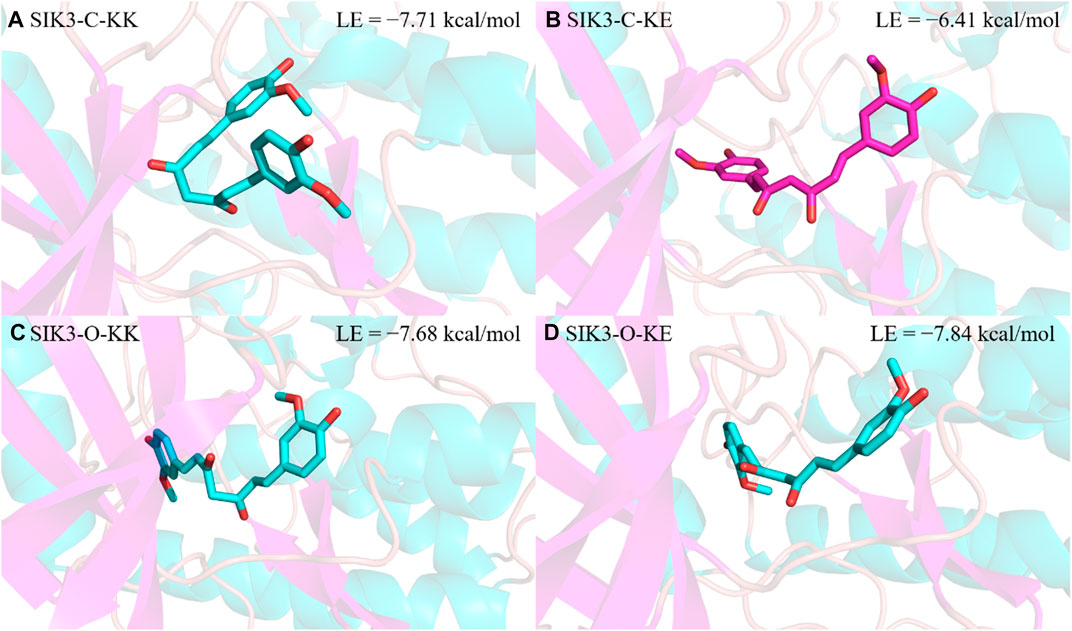
FIGURE 4. Selected results of curcumin docking with human SIK3. The lowest binding energy was obtained from the docking score. (A) For SIK3-C-KK, (B) for SIK3-C-KE, (C) for SIK3-O-KK, and (D) for SIK3-O-KE.
3.4 System stability
The RMSD value of the kinase domain (2.36 ± 0.25 Å for residues 66–317) was less than that of the SIK3 (3.17 ± 0.39 Å for residues 63–383) for SIK3-C-KK-2 system (Supplementary Table S1). This result indicates that the kinase domain is more stable than SIK3. This stable kinase domain also can be found in the other curcumin/SIK3 complex systems (Supplementary Figure S19). In addition, the RMSD value of the SIK3-C system was larger than that of the SIK3-O systems. For example, the lowest RMSD value of the kinase domain for the SIK3-C systems was 2.05 ± 0.31 Å, which was larger than the largest RMSD value of the kinase domain for the SIK3-O systems (1.73 ± 0.31 Å) (Figure 5). This phenomenon was also observed for the RMSD values of SIK3. This observation suggests that the SIK3-O complex systems are more stable than the SIK3-C complex systems. However, the RMSD values of both the KK and KE forms of curcumin were higher than those of SIK3 and the kinase domain, especially for the curcumin/SIK3-C systems. Nevertheless, the fluctuations in the RMSD values of curcumin were not related to the KK or KE forms as the RMSD values of curcumin for SIK3-O_KK-1 and SIK3-O_KE-1 were 1.98 ± 0.31 Å and 1.98 ± 0.25 Å, respectively. Simultaneously, we observed that the KE form of curcumin may cause more fluctuations for curcumin than the KK form. For instance, the RMSD values of curcumin for SIK3-C_KK-1 and SIK3-C_KE-1 were 3.64 ± 0.67 and 4.95 ± 0.74 Å, respectively. Thus, the conformation of curcumin fluctuated significantly in the simulation. The closed conformation of the T-loop induced more fluctuations than the open conformation. Supplementary Figure S20 shows the plots of RMSD values vs. time for the SIK3-C-Apo and SIK3-O-Apo systems, which excluded the curcumin molecule (Supplementary Figure S20). The receptor structure was also stable in the simulations after 100 ns, and the kinase domain was more stable than the overall SIK3 structure. The stability of these systems suggests that these simulations can be applied to following analyses.
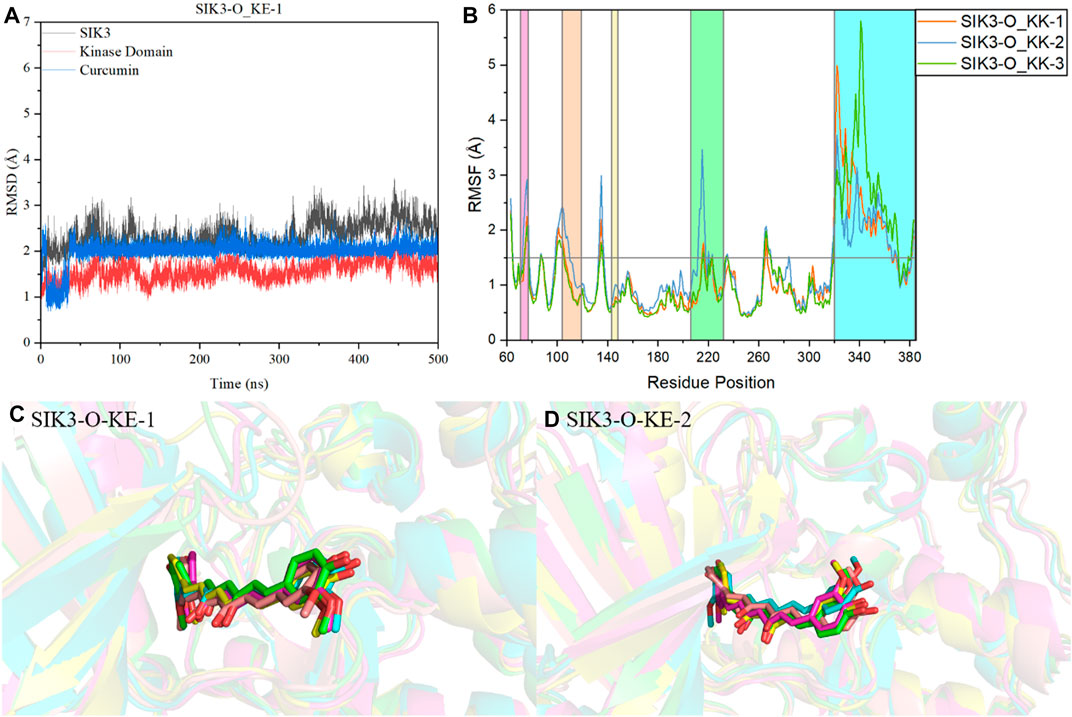
FIGURE 5. Analysis of molecular dynamics simulations. (A) Plot of RMSD of the Cα atom vs. time for the 500-ns MD simulation of SIK3-O-KE-1. (B) RMSF variations of the Cα atom of SIK3 from the 500-ns MD simulation of SIK3-O-KE. SIK3-O-KE-1 (C) and SIK3-O-KE-2 (D) snapshots along the dynamic simulation timeline at 100 ns, 200 ns, 300 ns, 400 ns, and 500 ns. For clarity, the water molecules have been removed. Curcumin is represented by a stick diagram, while SIK3 is depicted by a cartoon diagram.
The root-mean-square fluctuation (RMSF) of the residues of human SIK3 had been analyzed from the 500 ns simulations. Interestingly, there are two regions of human SIK3 with siginifacance fluctuations (Supplementary Figure S21), that is, the regions at positions 206–232 (T-loop) and 344–384 (UBA) (Figure 5). The conformation of the T-loop was different from the crystal structures of protein kinases; for instance, human FAK may exhibit open or closed T-loop conformation while binding with FAK-inhibitors (Supplementary Figure S22) (Guo et al., 2022). The T-loop was phosphorylated to activate the kinase domain and to regulate the downstream signal. In a previous study, protein-protein interactions were observed between SIK2 and 14-3-3 proteins when the T-loop was phosphorylated by LKB1 (Al-Hakim et al., 2005; Sonntag et al., 2018). Meanwhile, bosutinib binds with SIK2 while exhibiting multi-state conformations of the T-loop (Shi et al., 2022b). This flexibility of the T-loop can be observed in the SIK3-C and SIK3-O systems. These observations indicate that the different initial models of T-loops are very important for studying the interactions between inhibitors and SIKs. The UBA region was also found to be flexible; this region directly or indirectly regulates kinase activities (Jaleel et al., 2006; Marx et al., 2006; Wang et al., 2018). The UBA conformations were different for MELK, BRSK2, AMPK, and SNARK, which indicates UBA may form different conformations in SIKs (Wang et al., 2018). Moreover, the UBA region is an important domain for protein kinase autoinhibition (Marx et al., 2006; Cho et al., 2014; Wu et al., 2015; Emptage et al., 2018). In this work, we constructed the UBA region based on MARK2 structures. The RMSF value of the T-loop for the SIK3-O systems was less than that for the SIK3-C systems. In contrast, the RMSF value of the UBA region for the SIK3-O systems was larger than that for the SIK3-C systems. This shows that conformation of UBA and T-loop can be induced from curcumin binding into the ATP-binding site of human SIK3. Furthermore, the overall structure of the kinase domain was stable next to the T-loop region in the simulation.
For clearly demonstrating the conformation fluctuations of curcumin in the ATP-binding pocket of human SIK3, snapshots were obtained from the MD simulation at 100, 200, 300, 400, and 500 ns (Figures 5C, D, and Supplementary Figures S23–S34). However, both KK and KE forms produced binding complexes in the simulation, which indicates that curcumin can directly bind to the ATP-binding pocket of SIK3. In the SIK3-C-KK system, one 4-hydroxy-3-methoxyphenyl group of curcumin was approximately perpendicular to its other 4-hydroxy-3-methoxyphenyl group. This structure was obtained from the docking study. Meanwhile, this class formation was not observed in the other three systems. Curcumin exhibits line structure for the SIK3-C-KE, SIK3-O-KK, and SIK3-O-KE systems. Additionally, one 4-hydroxy-3-methoxyphenyl group of curcumin was exposed to the solvent environment, which may improve its bioavailability (Finlay et al., 2019; Wang et al., 2019). The 4-hydroxy-3-methoxyphenyl group was similar to the 4-methylpiperazine group of bosutinib and the piperazine moiety of the dasatinib (Shi et al., 2021a; Shi et al., 2022b). However, this region of the cyclin-dependent kinase 6 inhibitors can be applied to improve their selectivity among cyclin-dependent kinases (Lu and Schulze-Gahmen, 2006). This surface-exposed portion of the inhibitors can be considered as a modification. Thus, curcumin can form a stable binding model into the active site of the human SIK3 in simulation time.
3.5 Hydrogen bond analysis
The hydrogen bonds formed between small chemical scaffold molecules and the hinge loop of protein kinases are commonly required for potential inhibitors. Moreover, hydrogen bonding is an important feature of ATP-competitive inhibitors for protein kinases (Stavenger, 2008; Xing et al., 2015; Pang et al., 2021). Hydrogen bonds maybe significantly contribute to inhibitor binding with human SIK3 as one of protein kinase. Thus, the hydrogen bond numbers formed between curcumin and human SIK3 were first examined for the 12 curcumin/SIK3 complex systems from the simulation trajectory (Supplementary Figures S35). The wide range of hydrogen bond numbers (0–8) for the curcumin/SIK3 systems showed that some unstable hydrogen bonds were formed in the simulations. The SIK3-C-KK system had the lowest hydrogen bond number among the other class systems. The hydrogen bond numbers for SIK3-C-KK-1, SIK3-C-KK-2, and SIK3-C-KK-3 were 2.04 ± 0.80, 1.60 ± 0.70, and 1.76 ± 0.72, respectively (Supplementary Table S2). This indicates that disordered structure of the 4-hydroxy-3-methoxyphenyl group of curcumin forms hydrogen bonds with the back pocket of ATP-binding site of SIK3. Meanwhile, the SIK3-O-KE systems had the largest hydrogen bond numbers among the curcumin/SIK3 complexes (3.72 ± 0.93, 3.19 ± 0.99, and 3.17 ± 1.03 for SIK3-O_KE-1, SIK3-O_KE-2, and SIK3-O_KE-3, respectively). This shows that the KE form of curcumin forms higher number of hydrogen bonds with SIK3 than the KK form (Supplementary Figure S36).
The hydrogen bond occupancy analysis of the 12 complex systems was also performed. A hydrogen bond is formed when the distance between the acceptor and donor atoms is shorter than 3.5 Å, and the internal angle of the acceptor···H-donor is larger than 120°. The results of the hydrogen bond occupancy analysis are summarized in Figure 6. High occupancies were found for the diketo and ketoenol groups of curcumin with the hinge loop of SIK3. The occupancies of the KK form were 96.41%, 94.87%, and 97.85% for the SIK3-C-KK-1, SIK3-C-KK-2, and SIK3-C-KK-3 systems, respectively. Meanwhile, the occupancies of the KK form with the hinge loop for SIK3-O (87.34%–91.19%) were less than those of SIK3-C (>94.87%). In contrast, this hydrogen bond forming with A145 of hinge loop was more than 95% for SIK3-O systems with the KK and KE forms of curcumin except for the SIK3-O-KK-2 system (75.48%). However, this occupancy increased to 97.48% when the last 200 ns frames were applied to count the occupancy. Thus, the hydrogen bond between the A145 residue of the hinge loop and the diketo and ketoenol groups of curcumin play an important role in its binding with SIK3. These results agree with those of the experimental and theoretical studies (Shi et al., 2021a; Shi et al., 2021b; Tesch et al., 2021; Shi et al., 2022b). Moreover, the distance between the acceptor and donor atoms was calculated, and the angles between the acceptor atom, hydrogen atom, and donor atom were also checked (Supplementary Figures S37, S38). The distances for SIK3-C-KK-1, SIK3-C-KE-1, SIK3-O-KK-1, and SIK3-O-KE-1were 3.03 ± 0.21, 3.10 ± 0.36, 3.03 ± 0.30, and 3.15 ± 0.23 Å, respectively (Supplementary Table S3). This shows that the hydrogen bond between A145 and the diketo group is as strong as that between A145 and the ketoenol group. These results agreed with occupancy analysis. Especially, the length of the hydrogen bond in the SIK3-O-KK-2 system was greater than 3.5 Å in the first 100 ns, which indicates that no hydrogen bond was formed with the oxygen atom of the diketo group. This can be conformed from the occupancy analysis with 75.48% for 500 ns and 97.48% for the last 200 ns. The hydrogen bond formed between A145 and the hinge loop plays a significant role in the curcumin binding with human SIK3. Additionally, the two-dimensional interaction between curcumin and human SIK2 was drawn based on the representative frame of the maximum cluster for every system using the cluster analysis that had been performed with the average linkage cluster algorithm and epsilon = 2.5 Å from the last 200 ns of the simulation (Supplementary Figure S39). However, the diketo and ketoenol groups are related to the insatiability and low bioavailability of curcumin (Zhi et al., 2022). This limits the potential applications of curcumin. Some studies have overcome the structural disadvantages of curcumin, such as the methyl and methylene groups between 1,3-diketones (Anand et al., 2007; Tu et al., 2017). Thus, other scaffolds that form hydrogen bonds with the hinge loop can be replaced with the diketo and ketoenol groups. For example, adenine, 1H-pyrazole-3-amine, 2-aminopyridine, 1H-pyrrolo [2,3-b]pyridine, 1H-indazole, 7H-pyrrolo [2,3-d]pyrimidin-4-amine, pyrimidine-4,6-diamine, pyridine, 7H-pyrrolo [2,3-d]pyrimidine may be used for this purpose.
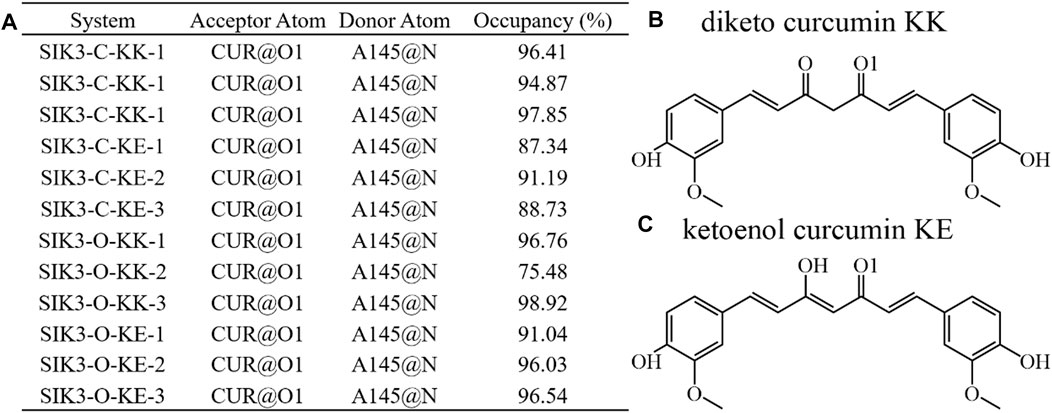
FIGURE 6. Hydrogen bonding network analysis of interactions between curcumin and human SIK3. (A) Occupancy is expressed as % of the period (500 ns, 50,000 frames) during which specific hydrogen bonds are formed. Hydrogen bond is defined as the distance between the acceptor and donor atoms (<3.5 Å), with an internal angle between the H-acceptor and H-donor (>120°). (B) and (C) labeled the O1 atom of curcumin for diketo and ketoenol curcumin.
3.6 Binding free energies
The simulations provide useful information regarding the binding of curcumin with human SIK3. However, the best binding model cannot be identified from these simulations. Thus, four different binding models of curcumin with SIK3 were applied to calculate the binding free energy using the MM/GBSA. The last 200 ns from the simulation trajectory were used herein. The binding free energies of the four complex systems are summarized in Table 1 and Supplementary Figures S4–S15. These calculations were repeated three times for every system. Both KK and KE forms of curcumin can be bound to the ATP-binding pocket of human SIK3 from the binding free energies of the 12 complex systems (−8.20 to −25.86 kcal/mol). The average binding free energies for SIK3-C-KK, SIK3-C-KE, SIK3-O-KK, and SIK3-O-KE were −9.38, −18.47, −18.65, and −22.75 kcal/mol, respectively. SIK3-C-KK showed the biggest binding free energy value, which indicates that the folding of curcumin was unfavorable for its binding with SIK3. In contrast, the line structures of curcumin exhibits better binding affinity for the other three complex systems. The dasatinib also bound with human SIK2 with line structure of dasatinib from our previous study (Shi et al., 2021a). Curcumin was also bound with protein kinase DYRK2 using a line structure (PDB ID: 5ZTN and 6HDR, Supplementary Figure S40) (Banerjee et al., 2018; Elkins and Knapp, 2019). In addition, curcumin was bound with human transthyretin (TTR) using a line structure (PDB ID: 4PME) (Ciccone et al., 2015). However, the non-line conformation of curcumin also binds with TTR (PDB ID: 4PMF) (Ciccone et al., 2015). Thus, the line conformation of curcumin is a better conformation than non-line conformation for binding with SIK3. Furthermore, the binding free energies of SIK3-O (−18.65 and −22.75 kcal/mol) were higher than those for SIK3-C (−9.38 and −18.47 kcal/mol). This suggests that SIK3-O is a more favorable conformation for curcumin binding than SIK3-C. Meanwhile, the KE form of curcumin showed binding affinities of −9.09 and −4.10 kcal/mol for SIK3-C and SIK3-O, respectively. Thus, the binding model for SIK3-O-KE (KE form curcumin binding with open T-loop conformation of human SIK3) is the optimal binding model when curcumin binding with human SIK3.
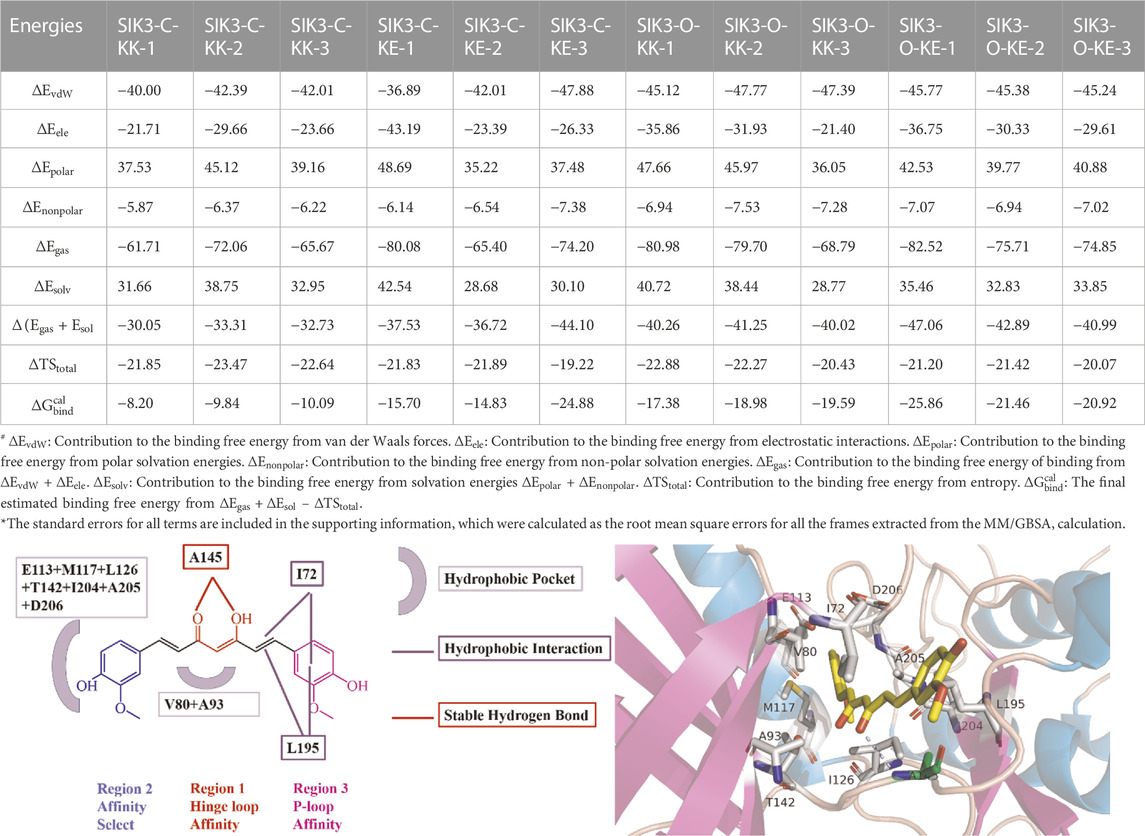
TABLE 1. Binding free energies, decomposition and electrostatic interactions (
The binding entropies and enthalpies for curcumin and SIK3 are in the range of −19.22 to −23.47 kcal/mol and −30.05–47.06 kcal/mol, respectively. The negative values of both entropy and enthalpy suggest that curcumin binding with human SIK3 is an enthalpy-driven process. The average entropies for curcumin binding with SIK3 are −22.65, −20.98, −21.86, and −20.90 kcal/mol for SIK3-C-KK, SIK3-C-KE, SIK3-O-KK, and SIK3-O-KE, respectively. The entropy values of the four binding systems did not differ significantly, which suggests that entropy does not affect the binding free energy of the systems. However, the differences in the enthalpies of the SIK3-C and SIK3-O conformations were 8.48 and 4.20 kcal/mol for the KK and KE forms of curcumin, respectively. The enthalpy difference between the KK and KE forms of curcumin for SIK3-C and SIK3-O were 7.42 and 3.14 kcal/mol, respectively. Therefore, enthalpy may affect the binding affinity of curcumin toward SIK3.
Meanwhile, the vdW interactions between curcumin and SIK3-C (−41.47 and −42.26 kcal/mol for SIK3-C-KK and SIK3-O-KE, respectively) are less than those between curcumin and SIK3-O (−46.76 and −45.46 kcal/mol for SIK3-C-KK and SIK3-C-KE, respectively). This suggests that the vdW interactions contribute more to the binding of curcumin with SIK3-O than that with SIK3-C. Electronic interactions are also favorable for the formation of the curcumin-SIK3 complex. The contribution of electrostatic interactions to the binding free energies of SIK3-C-KK, SIK3-C-KE, SIK3-O-KK, and SIK3-O-KE was −25.01, −30.97, 29.73, and −32.23 kcal/mol, respectively. Furthermore, the vdW interactions contribute more to the binding free energy than electronic interactions for the four binding models.
Usually, the binding interactions between inhibitors and targets depend on two major factors: the polar term (
In our previous study (Shi et al., 2022b), the binding free energies for bosutinib bound with ten different conformations of human SIK2 were between −12.81 and −24.72 kcal/mol. Meanwhile, the binding free energies for dasatinib with four different conformations of SIK2 were in the range of −20.09 to −29.37 kcal/mol (Shi et al., 2021a). In addition, the binding free energies for HG-9-91-01, KIN112, MRT67307, and MRT199665 were in the range of −18.81 to −28.40 kcal/mol for three different conformations (Shi et al., 2021b). In this work, the binding free energies of curcumin and human SIK3 were in the −18.47 to −22.75 kcal/mol range, excluding the unfavorable SIK3-C-KK system with the non-liner curcumin structure. Curcumin may bind with SIK3 with similarity binding affinities of other inhibitor binding with SIK2. Therefore, curcumin can be used as a lead compound to develop novel SIKs inhibitors.
3.7 Free energy decomposition
The binding of curcumin with human SIK3 can be explained using the four different binding models. However, the key residues that contribute to the complex formation are still unclear. Therefore, the per-residue free energy decomposition method was employed to find the key residues involved in the curcumin-SIK3 complex. The interaction energies for the 12 systems were estimated from the MM/GBSA method with per-residue decomposition (Supplementary Tables S16–S27).
In the SIK3-C-KK system, the I72, K74, V80, A93, L126, Y144, A145, G148, N193, L195, N211, and L212 residues contributed more than 0.5 kcal/mol (>0.5 kcal/mol in one of the three replicas) (Figure 7). The Y144, A145, and G148 residues are located on the hinge loop, which links the N- and C-lobes to form the ATP-binding pocket (Supplementary Figure S41). A145 forms hydrogen bonds with curcumin and contribute more than −1.35 kcal/mol. Meanwhile, the vdW interactions of Y144 in the SIK3-C-KK-1, SIK3-C-KK-2, and SIK3-C-KK3 systems were −2.24, −1.84, and −2.13 respectively. These interactions mainly arise from the side chain of Y144. The vdW interactions for the SIK3-C-KK-1, SIK3-C-KK-2, and SIK3-C-KK3 systems were −1.56, −1.39, and −1.55 kcal/mol, respectively. In addition, G148 forms vdW interactions with the phenyl ring of 4-hydroxy-3-methoxy phenyl group of curcumin and contributes more than 0.97 kcal/mol. In contrast, the 4-hydroxy-3-methoxy phenyl group of curcumin also interacts with I72 to form a clip between I72 and G148 for the phenyl ring (Supplementary Figure S40). Nevertheless, I72 was not perpendicular to the phenyl and was close to the diketo group of curcumin. Additionally, the L195 residue interacted with the phenyl ring and the diketo group. Thus, the diketo group can be replaced with aromatic groups to enhance the vdW interactions of I72.
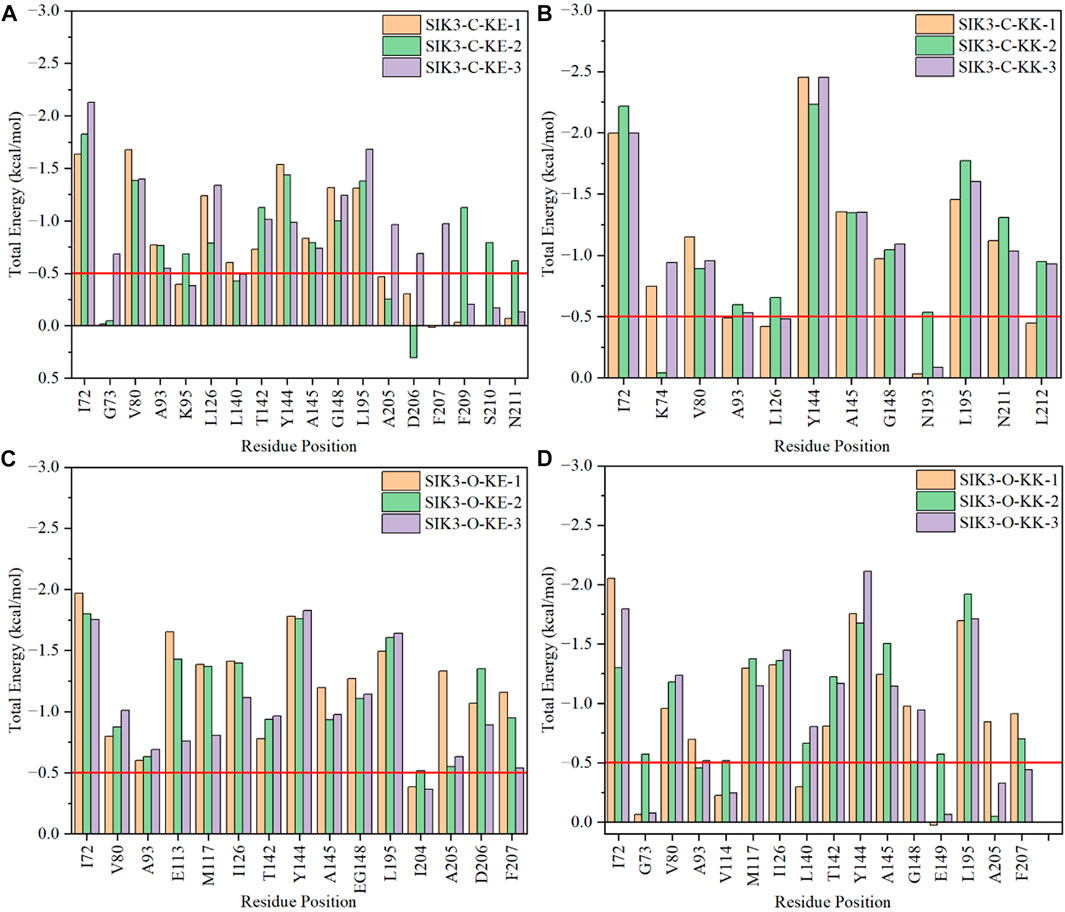
FIGURE 7. Per-residue binding free energies of selected amino acids in the interactions between curcumin and human SIK3. (A) For SIK3-C-KE, (B) for SIK3-C-KK, (C) for SIK3-O-KE, and (D) for SIK3-O-KK.
I72, G73, V80, A93, K95, L126, L140, T142, Y144, A145, G148, L195, A205, D206, F207, F209, S210, and N211 are the main residues of curcumin that bind with SIK3 in the SIK3-C-KE systems. The I72, Y144, A145, G148, and L195 residues interact similarly with the SIK3-C-KK systems (Supplementary Figure S42). One of the 4-hydroxy-3-methoxy phenyl groups of curcumin was located in the ATP-binding pocket along with V80, A93, K95, L126, L140, T142, A205, D206, and F209 (Supplementary Figure S43). This same pocket was occupied by the 2-chloro-6-methylphenyl ring of dasatinib and other six-membered rings (Shi et al., 2021a). G73 only interacted with the SIK3-C-KE-3 system and was not found in other replicated simulations, which suggest that G73 is unstable and does not interact with curcumin. Thus, if the aromaticity of the phenyl groups is modified to stabilize the interactions between G73 and curcumin, the binding affinity of curcumin toward SIK3 may improve.
The Y144 and A145 residues in the hinge loop of SIK3 play a similar role (hydrogen bond for A145 and vdW interaction for Y144) in the SIK3-O-KK, SIK3-C-KK, and SIK3-C-KE systems (Supplementary Figure S44). This contribution was more in the SIK3-O-KK system (less than −1.65 and −1.14 kcal/mol for Y144 and A145, respectively) than in the SIK3-C-KE systems (less than −1.54 and −0.83 kcal/mol for Y144 and A145, respectively). Meanwhile, the contribution was lower in the SIK3-O-KK system (average value of three replicas with −1.85 and −1.30 kcal/mol for Y144 and A145, respectively) than in the SIK3-C-KK system (average value of three replicas with −2.38 and −1.35 kcal/mol for Y144 and A145, respectively). Thus, Y144 and A145 are the key residues for curcumin binding with SIK3. I72 and L195 form a well flip for the phenyl group in the SIK3-O-KK-1 system but not in the SIK3-O-KK-2 and SIK3-O-KK-3 systems. Which results that I72 has −2.05, −1.30, and −1.80 kcal/mol for SIK3-O-KK-1, SIK3-O-KK-2, and SIK3-O-KK-3, respectively. Thus, if the flip for phenyl group between I72 and L195, the affinity will be increased for curcumin binding with SIK3.
The SIK3-O-KE system exhibited the lowest binding free energy (−22.75 kcal/mol), which suggests that the KE form of curcumin had the high binding affinity with the T-loop open conformation of human SIK3. The hinge loop also formed similar interactions with the other three models (Supplementary Figure S45). Moreover, the I72 residue played a similar role in SIK3-O-KE (−1.84 kcal/mol), SIK3-O-KK (−1.72 kcal/mol), and SIK3-C-KE (−1.86 kcal/mol); however, its contribution was less in the SIK3-C-KK system (−2.07 kcal/mol). This may have occurred because I72 interacts with both the phenyl and diketo groups in SIK3-C-KK. In addition, the back pocket related to 204IADFGFSNLF213 was important for the line structure of curcumin binding with human SIK3. The DFG motif can be applied to control the kinase activity of protein kinases; DFG-in can be used as the active conformation and DFG-out as the inactive conformation (Modi and Dunbrack, 2019). However, in this work, the DFG-in conformation of SIK3 was considered for type-I kinase inhibitors (Roskoski, 2016; Arter et al., 2022). The conformation of DFG motif may affect the binding models for curcumin and SIK3. Moreover, the DFG motif of human FAK is an important region, which improves the affinity of the FAK inhibitors by forming a hydrogen bond between the DFG motif and inhibitors (Shi et al., 2022a). This indicates that the DFG motif of SIK3 can also be employed to increase the affinity between curcumin and SIK3 by replacing the 4-hydroxy-3-methoxy phenyl group of curcumin.
Particularly, some hydrophobic residues are shown to have significant subtotal binding free energies for curcumin binding with SIK3. For instance, the I72, V80, A93, Y144, A145, and L195 residues contributed more than −1.30, −0.80, −0.46, −0.98, −0.74, and −1.31 kcal/mol, respectively (Figure 8). Especially, the I72, A145, and L195 residues were the key resides in the four binding models. The A145 residue forms hydrogen bonds with the diketo and ketoenol groups of curcumin. I72 is located in the P-loop, which interacts with the 4-hydroxy-3-methoxy phenyl group. Meanwhile, L195 also forms van der Walls interactions with the 4-hydroxy-3-methoxy phenyl group and the diketo and ketoenol groups of curcumin. Thus, the diketo and ketoenol groups can be replaced with aromatic rings, which remain hydrogen bonds with hinge loop to improve the affinity of curcumin.
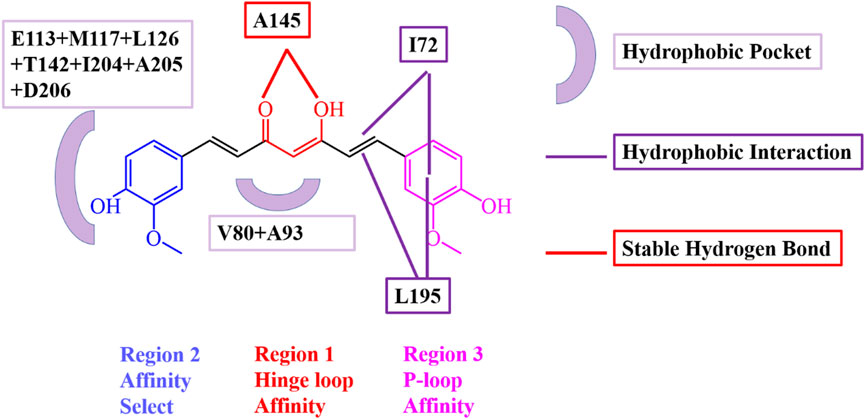
FIGURE 8. Interaction model of curcumin and human SIK3 obtained from the MD simulations for SIK3-O-KE system. Key residues involved in hydrogen bonding and hydrophobic interactions were highlighted.
3.8 Kinase assay
The commercial KinaseProfiler Service (Eurofins Scientific, Inc.) was used to evaluate the IC50 of curcumin against human SIK3. KinaseProfiler assay protocols measure the percentage of inhibition of the phosphorylation of a peptide substrate in the presence of fixed concentrations of ATP, which are close to the Km values of human SIK3. Curcumin can inhibit the kinase activity of SIK3 with IC50 = 131 nM, with reference to the PKR inhibitor (CAS NO. 608512-97-6). Curcumin remarkably inhibits the kinase activity of human SIK3, which suggests that it can be used as a lead compound for designing and optimizing curcumin derivatives that target human SIK3.
Additionally, the antiproliferative effect of curcumin and HG-9-91-01 was tested for human breast cancer cell lines MDA-MB-231 and MCF-7. The antiproliferative activity of curcumin was significantly higher than that of HG-9-91-01. The IC50 values for the antiproliferative activity of curcumin and HG-9-91-01 against MCF-7 were 9.62 ± 0.33 and 15.92 ± 0.90 µM, respectively (Supplementary Figure S46). Meanwhile, the IC50 values for curcumin and HG-9-91-01 against MDA-MB-23 were 72.37 ± 0.37 and 55.41 ± 5.11 µM, respectively. These results suggest that curcumin can directly target human SIK3 to inhibit the proliferative activity of MDA-MB-231 and MCF-7.
4 Conclusion
In this study, the binding models of curcumin and human SIK3 were studied using computational methods. HM was used to construct the SIK3 kinase domain and UBA region with open/closed conformations of the T-loop (SIK3-C and SIK3-O). Quantum chemistry was employed to calculate the optimized structures and atomic charges of the KK and KE forms of curcumin. Molecular docking was used to obtain the initial complex structures for curcumin and human SIK3 (SIK3-C-KK, SIK3-C-KE, SIK3-O-KK, and SIK3-O-KE). Molecular dynamics simulation was used to obtain the qualitative model for the binding of curcumin with SIK3. Additionally, the binding free energies were calculated for four binding models, and SIK3-O-KE was found to be the optimal model. The three regions of curcumin were identified to increase its binding affinity and to improve its selectivity among protein kinases. Furthermore, in the kinase assay, curcumin showed an IC50 value of 131 nM and antiproliferative activities of 9.62 ± 0.33 µM and 72.37 ± 0.37 µM against MCF-7 and MDA-MB-23 cell lines, respectively. This study provides detailed information on the binding of curcumin with human SIK3, which may facilitate the design of novel SIKs inhibitors. The computed and modeled results obtained in this study can be validated by designing curcumin derivatives based on the binding models reported herein. Meanwhile, novel SIKs inhibitors may be developed in the future to treat breast cancer.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
Conceptualization, MS, DX, and XaL; methodology, MS, YanZ, and HW; software, MS and DX; validation, MS, XZ, MD, XhL, and XT; formal analysis, MS, XhL, YtZ, and YY; investigation, MS, YanZ, and YtZ; resources, MS, DX, and XaL; data curation, MS, YY, and LS; writing—original draft preparation, MS; writing—review and editing, MS, DX, and XaL; visualization, MS and DX; supervision, MS, DX, and XaL; project administration, MS and XaL; funding acquisition, MS, HW, and DX. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 22203056 and 21973064), Natural Science Foundation of Sichuan, China (2022NSFSC1467), NHC Key Laboratory of Nuclear Technology Medical Transformation (Mianyang Central Hospital) (No. 2022HYX001), GHfund B (ghfund202302022385), and the Science and Technology Planning Project of Sichuan Province (2021YJ0483).
Acknowledgments
Certain data were obtained from the National Supercomputing Center of Guangzhou, the Supercomputing Center of Sichuan University, and the Chengdu Supercomputing Center. The authors thank Editage (www.editage.cn) for English language editing.
Conflict of interest
LS was employed by Shenzhen Shuli Tech Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1116098/full#supplementary-material
References
Aci-Seche, S., Ziada, S., Braka, A., Arora, R., and Bonnet, P. (2016). Advanced molecular dynamics simulation methods for kinase drug discovery. Future Med. Chem. 8 (5), 545–566. doi:10.4155/fmc.16.9
Al-Hakim, A. K., Goransson, O., Deak, M., Toth, R., Campbell, D. G., Morrice, N. A., et al. (2005). 14-3-3 cooperates with lkb1 to regulate the activity and localization of qsk and sik. J. Cell Sci. 118 (23), 5661–5673. doi:10.1242/jcs.02670
Alexander, S. P. H., Fabbro, D., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., et al. (2019). The concise guide to pharmacology 2019/20: Enzymes. Br. J. Pharmacol. 176, S297–S396. doi:10.1111/bph.14752
Amaro, R. E., Baudry, J., Chodera, J., Demir, O., McCammon, J. A., Miao, Y. L., et al. (2018). Ensemble docking in drug discovery. Biophys. J. 114 (10), 2271–2278. doi:10.1016/j.bpj.2018.02.038
An, J., An, S., Choi, M., Jung, J. H., and Kim, B. (2022). Natural products for esophageal cancer therapy: From traditional medicine to modern drug discovery. Int. J. Mol. Sci. 23 (21), 13558. doi:10.3390/ijms232113558
Anand, P., Kunnumakkara, A. B., Newman, R. A., and Aggarwal, B. B. (2007). Bioavailability of curcumin: Problems and promises. Mol. Pharm. 4 (6), 807–818. doi:10.1021/mp700113r
Anandakrishnan, R., Aguilar, B., and Onufriev, A. V. (2012). H++3.0: Automating pk prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40 (W1), W537–W541. doi:10.1093/nar/gks375
Antonio, T., Soares-da-Silva, P., Pires, N. M., and Gomes, P. (2022). Salt-inducible kinases: New players in pulmonary arterial hypertension? Trends Pharmacol. Sci. 43 (10), 806–819. doi:10.1016/j.tips.2022.06.008
Arter, C., Trask, L., Ward, S., Yeoh, S., and Bayliss, R. (2022). Structural features of the protein kinase domain and targeted binding by small-molecule inhibitors. J. Biol. Chem. 298 (8), 102247. doi:10.1016/j.jbc.2022.102247
Banerjee, S., Ji, C. G., Mayfield, J. E., Goel, A., Xiao, J. Y., Dixon, J. E., et al. (2018). Ancient drug curcumin impedes 26s proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. U. S. A. 115 (32), 8155–8160. doi:10.1073/pnas.1806797115
Bashford, D., and Karplus, M. (1990). pKa's of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry 29 (44), 10219–10225. doi:10.1021/bi00496a010
Bateman, A., Martin, M. J., O'Donovan, C., Magrane, M., Alpi, E., Antunes, R., et al. (2017). Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 45 (D1), D158–D169. doi:10.1093/nar/gkw1099
Bateman, A., Martin, M. J., O'Donovan, C., Magrane, M., Apweiler, R., Alpi, E., et al. (2015). Uniprot: A hub for protein information. Nucleic Acids Res. 43 (D1), D204–D212. doi:10.1093/nar/gku989
Bateman, A., Martin, M. J., Orchard, S., Magrane, M., Alpi, E., Bely, B., et al. (2019). Uniprot: A worldwide hub of protein knowledge. Nucleic Acids Res. 47 (D1), D506–D515. doi:10.1093/nar/gky1049
Bayly, C. I., Cieplak, P., Cornell, W. D., and Kollman, P. A. (1993). A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The resp model. J. Phys. Chem. 97 (40), 10269–10280. doi:10.1021/j100142a004
Becke, A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 38 (6), 3098–3100. doi:10.1103/PhysRevA.38.3098
Bello, M., Martinez-Archundia, M., and Correa-Basurto, J. (2013). Automated docking for novel drug discovery. Expert. Opin. Drug Discov. 8 (7), 821–834. doi:10.1517/17460441.2013.794780
Benkert, P., Biasini, M., and Schwede, T. (2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27 (3), 343–350. doi:10.1093/bioinformatics/btq662
Bera, I., and Payghan, P. V. (2019). Use of molecular dynamics simulations in structure-based drug discovery. Curr. Pharm. Des. 25 (31), 3339–3349. doi:10.2174/1381612825666190903153043
Berger, B. T., Amaral, M., Kokh, D. B., Nunes-Alves, A., Musil, D., Heinrich, T., et al. (2021). Structure-kinetic relationship reveals the mechanism of selectivity of fak inhibitors over pyk2. Cell Chem. Biol. 28 (5), 686–698.e7. doi:10.1016/j.chembiol.2021.01.003
Bernal, F. A., Hammann, P., and Kloss, F. (2022). Natural products in antibiotic development: Is the success story over? Curr. Opin. Biotechnol. 78, 102783. doi:10.1016/j.copbio.2022.102783
Blaudeau, J. P., McGrath, M. P., Curtiss, L. A., and Radom, L. (1997). Extension of Gaussian-2 (g2) theory to molecules containing third-row atoms k and ca. J. Chem. Phys. 107 (13), 5016–5021. doi:10.1063/1.474865
Bowie, J. U., Luthy, R., and Eisenberg, D. (1991). A method to identify protein sequences that fold into a known three-dimensional structure. Science 253 (5016), 164–170. doi:10.1126/science.1853201
Canagarajah, B. J., Khokhlatchev, A., Cobb, M. H., and Goldsmith, E. J. (1997). Activation mechanism of the map kinase erk2 by dual phosphorylation. Cell 90 (5), 859–869. doi:10.1016/s0092-8674(00)80351-7
Case, D. A., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., Cheatham, T. E., Cruzeiro, V. W. D., et al. (2020). Amber 2020. san francisco: University of California.
Cavasotto, C. N. (2011). Homology models in docking and high-throughput docking. Curr. Top. Med. Chem. 11 (12), 1528–1534. doi:10.2174/156802611795860951
Cavasotto, C. N., and Phatak, S. S. (2009). Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 14 (13-14), 676–683. doi:10.1016/j.drudis.2009.04.006
Cha, Y., Cho, Y. S., Kang, Y., Kim, K., Cha, Y., and Cho, H. S. (2015). The structures of the kinase domain and UBA domain of MPK38 suggest the activation mechanism for kinase activity. Acta Crystallogr. Sect. A 71, S183. doi:10.1107/s2053273315097326
Chakraborti, S., Das, L., Kapoor, N., Das, A., Dwivedi, V., Poddar, A., et al. (2011). Curcumin recognizes a unique binding site of tubulin. J. Med. Chem. 54 (18), 6183–6196. doi:10.1021/jm2004046
Cheatham, T., Galindo, R., and Roe, D. (2019). Parallel analysis of large ensembles of molecular dynamics simulation derived trajectories with the open-source cpptraj tools. Abstr. Pap. Am. Chem. Soc. 257, 1.
Chen, F. Y., Chen, L. W., Qin, Q., and Sun, X. C. (2019a). Salt-inducible kinase 2: An oncogenic signal transmitter and potential target for cancer therapy. Front. Oncol. 9, 18. doi:10.3389/fonc.2019.00018
Chen, Q. Q., Wang, Y., Shi, S. S., Li, K. H., Zhang, L., and Gao, J. (2019b). Insights into the interaction mechanisms of the proviral integration site of moloney murine leukemia virus (pim) kinases with pan-pim inhibitors pim447 and azd1208: A molecular dynamics simulation and mm/gbsa calculation study. Int. J. Mol. Sci. 20 (21), 5410. doi:10.3390/ijms20215410
Cheng, T. C., Sayseng, J. O., Tu, S. H., Juan, T. C., Fang, C. L., Liao, Y. C., et al. (2021). Curcumin-induced antitumor effects on triple-negative breast cancer patient-derived xenograft tumor mice through inhibiting salt-induced kinase-3 protein. J. Food Drug Anal. 29 (4), 622–637. doi:10.38212/2224-6614.3387
Cherry, M., and Williams, D. H. (2004). Recent kinase and kinase inhibitor x-ray structures: Mechanisms of inhibition and selectivity insights. Curr. Med. Chem. 11 (6), 663–673. doi:10.2174/0929867043455792
Cho, Y. S., Yoo, J., Park, S., and Cho, H. S. (2014). The structures of the kinase domain and uba domain of mpk38 suggest the activation mechanism for kinase activity. Acta Crystallogr. Sect. d-struct. Biol. 70, 514–521. doi:10.1107/s1399004713027806
Ciccone, L., Tepshi, L., Nencetti, S., and Stura, E. A. (2015). Transthyretin complexes with curcumin and bromo-estradiol: Evaluation of solubilizing multicomponent mixtures. New Biotech. 32 (1), 54–64. doi:10.1016/j.nbt.2014.09.002
Clark, K., MacKenzie, K. F., Petkevicius, K., Kristariyanto, Y., Zhang, J. Z., Choi, H. G., et al. (2012). Phosphorylation of crtc3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. U. S. A. 109 (42), 16986–16991. doi:10.1073/pnas.1215450109
Colovos, C., and Yeates, T. O. (1993). Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2 (9), 1511–1519. doi:10.1002/pro.5560020916
Cozza, G., Zonta, F., Dalle Vedove, A., Venerando, A., Dall'Acqua, S., Battistutta, R., et al. (2020). Biochemical and cellular mechanism of protein kinase ck2 inhibition by deceptive curcumin. Febs J. 287 (9), 1850–1864. doi:10.1111/febs.15111
Dev, S. S., Abidin, S. A. Z., Farghadani, R., Othman, I., and Naidu, R. (2021). Receptor tyrosine kinases and their signaling pathways as therapeutic targets of curcumin in cancer. Front. Pharmacol. 12, 26. doi:10.3389/fphar.2021.772510
Do, P. C., Lee, E. H., and Le, L. (2018). Steered molecular dynamics simulation in rational drug design. J. Chem. Inf. Model. 58 (8), 1473–1482. doi:10.1021/acs.jcim.8b00261
Du, J., Chen, Q., Takemori, H., and Xu, H. Y. (2008). Sik2 can be activated by deprivation of nutrition and it inhibits expression of lipogenic genes in adipocytes. Obesity 16 (3), 531–538. doi:10.1038/oby.2007.98
Du, W. Q., Zheng, J. N., and Pei, D. S. (2016). The diverse oncogenic and tumor suppressor roles of salt-inducible kinase (sik) in cancer. Expert Opin. Ther. Targets 20 (4), 477–485. doi:10.1517/14728222.2016.1101452
Du, Y. P., Ye, Z., Zheng, Z. J., Li, Y. D., Chen, J., Zaaboul, F., et al. (2021). Brucea javanica oil inhibits proliferation of hepatocellular carcinoma cells and induces apoptosis via the pi3k/akt pathway. Tradit. Med. Res. 6 (2), 14. doi:10.53388/tmr20200421174
Durrant, J. D., and McCammon, J. A. (2011). Molecular dynamics simulations and drug discovery. BMC Biol. 9, 71. doi:10.1186/1741-7007-9-71
Elkins, J. M., and Knapp, S. (2019). Dyrk2 bound to curcumin (PDB ID: 6HDR). Available at: https://www.rcsb.org/structure/6hDR (Accessed April 6, 2023).
Emptage, R. P., Lemmon, M. A., Ferguson, K. M., and Marmorstein, R. (2018). Structural basis for mark1 kinase autoinhibition by its ka1 domain. Structure 26(8), 1137–1143.e3. doi:10.1016/j.str.2018.05.008
Finlay, M. R. V., Anderton, M., Bailey, A., Boyd, S., Brookfield, J., Cairnduff, C., et al. (2019). Discovery of a thiadiazole-pyridazine-based allosteric glutaminase 1 inhibitor series that demonstrates oral bioavailability and activity in tumor xenograft models. J. Med. Chem. 62 (14), 6540–6560. doi:10.1021/acs.jmedchem.9b00260
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. Wallingford, CT, USA: Gaussian, Inc.
Fuhrmann, J., Rurainski, A., Lenhof, H. P., and Neumann, D. (2010). A new lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 31 (9), 1911–1918. doi:10.1002/jcc.21478
Gadhe, C. G., Balupuri, A., and Cho, S. J. (2015). In silico characterization of binding mode of ccr8 inhibitor: Homology modeling, docking and membrane based md simulation study. J. Biomol. Struct. Dyn. 33 (11), 2491–2510. doi:10.1080/07391102.2014.1002006
Gaillard, T., and Simonson, T. (2014). Pairwise decomposition of an mmgbsa energy function for computational protein design. J. Comput. Chem. 35 (18), 1371–1387. doi:10.1002/jcc.23637
Gasteiger, J., and Marsili, M. (1980). Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 36 (22), 3219–3228. doi:10.1016/0040-4020(80)80168-2
Genheden, S., and Ryde, U. (2015). The mm/pbsa and mm/gbsa methods to estimate ligand-binding affinities. Expert. Opin. Drug Discov. 10 (5), 449–461. doi:10.1517/17460441.2015.1032936
Golonko, A., Lewandowska, H., Swislocka, R., Jasinska, U. T., Priebe, W., and Lewandowski, W. (2019). Curcumin as tyrosine kinase inhibitor in cancer treatment. Eur. J. Med. Chem. 181, 111512. doi:10.1016/j.ejmech.2019.07.015
Gopal, S. (2022). Virtual screening and molecular docking analysis of degradation products of curcumin as inhibitors of egfr. J. Comput. Biophys. Chem. 21 (06), 629–646. doi:10.1142/s2737416522500247
Gordon, J. C., Myers, J. B., Folta, T., Shoja, V., Heath, L. S., and Onufriev, A. (2005). H++: A server for estimating pk(a)s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33, W368–W371. doi:10.1093/nar/gki464
Gormand, A., Henriksson, E., Strom, K., Jensen, T. E., Sakamoto, K., and Goransson, O. (2011). Regulation of amp-activated protein kinase by lkb1 and camkk in adipocytes. J. Cell. Biochem. 112 (5), 1364–1375. doi:10.1002/jcb.23053
Guo, C., Li, Q. X., Xiao, J. J., Ma, F., Xia, X., and Shi, M. S. (2022). Identification of defactinib derivatives targeting focal adhesion kinase using ensemble docking, molecular dynamics simulations and binding free energy calculations. J. Biomol. Struct. Dyn. 17, 1–17. doi:10.1080/07391102.2022.2135601
Hanks, S. K., and Hunter, T. (1995). The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification 1. Faseb J. 9 (8), 576–596. doi:10.1096/fasebj.9.8.7768349
Hartl, C., Michels, T., Milde, R., Klein, V., Beckhove, P., Loferer, H., et al. (2021). 301 OMX-0407, a highly potent SIK3 inhibitor, sensitizes tumor cells to apoptosis and eradicates tumors in combination with PD-1 inhibition. J. Immunother. Cancer 9, A324. doi:10.1136/jitc-2021-SITC2021.301
Hasmeda, M., and Polya, G. M. (1996). Inhibition of cyclic amp-dependent protein kinase by curcumin. Phytochemistry 42 (3), 599–605. doi:10.1016/0031-9422(96)00091-x
He, X., Wang, N., Zhang, Y., Huang, X. B., and Wang, Y. (2022). The therapeutic potential of natural products for treating pancreatic cancer. Front. Pharmacol. 13, 1051952. doi:10.3389/fphar.2022.1051952
Heap, R. E., Hope, A. G., Pearson, L. A., Reyskens, K., McElroy, S. P., Hastie, C. J., et al. (2017). Identifying inhibitors of inflammation: A novel high-throughput MALDI-TOF screening assay for salt-inducible kinases (SIKs). Slas Discov. 22 (10), 1193–1202. doi:10.1177/2472555217717473
Hernandez-Rodriguez, M., Rosales-Hernandez, M. C., Mendieta-Wejebe, J. E., Martinez-Archundia, M., and Basurto, J. C. (2016). Current tools and methods in molecular dynamics (md) simulations for drug design. Curr. Med. Chem. 23 (34), 3909–3924. doi:10.2174/0929867323666160530144742
Hirayama, N. (2007). Docking method for drug discovery. Yakugaku Zasshi-J. Pharm. Soc. Jpn. 127 (1), 113–122. doi:10.1248/yakushi.127.113
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous electron gas. Phys. Rev. B 136 (3B), B864–B871. doi:10.1103/PhysRev.136.B864
Honig, B., and Nicholls, A. (1995). Classical electrostatics in biology and chemistry. Science 268 (5214), 1144–1149. doi:10.1126/science.7761829
Indran, I. R., Tufo, G., Pervaiz, S., and Brenner, C. (2011). Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochimica Biophysica Acta-Bioenergetics 1807 (6), 735–745. doi:10.1016/j.bbabio.2011.03.010
Jakhar, R., Dangi, M., Khichi, A., and Chhillar, A. K. (2020). Relevance of molecular docking studies in drug designing. Curr. Bioinform. 15 (4), 270–278. doi:10.2174/1574893615666191219094216
Jaleel, M., Villa, F., Deak, M., Toth, R., Prescott, A. R., Van Aalten, D. M. F., et al. (2006). The ubiquitin-associated domain of ampk-related kinases regulates conformation and lkb1-mediated phosphorylation and activation. Biochem. J. 394, 545–555. doi:10.1042/bj20051844
Jeena, M. T., Kim, S., Jin, S., and Ryu, J.-H. (2020). Recent progress in mitochondria-targeted drug and drug-free agents for cancer therapy. Cancers 12 (1), 4. doi:10.3390/cancers12010004
Jin, H. Y., Tudor, Y., Choi, K., Shao, Z. F., Sparling, B. A., McGivern, J. G., et al. (2020). High-throughput implementation of the nanobret target engagement intracellular kinase assay to reveal differential compound engagement by sik2/3 isoforms. SLAS Discov. 25 (2), 215–222. doi:10.1177/2472555219893277
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 (2), 926–935. doi:10.1063/1.445869
Jovanovic, S. V., Steenken, S., Boone, C. W., and Simic, M. G. (1999). H-atom transfer is a preferred antioxidant mechanism of curcumin. J. Am. Chem. Soc. 121 (41), 9677–9681. doi:10.1021/ja991446m
Katoh, Y., Takemori, H., Horike, N., Doi, J., Muraoka, M., Min, L., et al. (2004). Salt-inducible kinase (sik) isoforms: Their involvement in steroidogenesis and adipogenesis. Mol. Cell. Endocrinol. 217 (1-2), 109–112. doi:10.1016/j.mce.2003.10.016
Kohn, W., and Sham, L. J. (1965). Self-consistent equations including exchange and correlation effects. Phys. Rev. 140 (4A), 1133–A1138. doi:10.1103/PhysRev.140.A1133
Kroemer, R. T. (2007). Structure-based drug design: Docking and scoring. Curr. Protein Pept. Sci. 8 (4), 312–328. doi:10.2174/138920307781369382
Laskowski, R. A., Macarthur, M. W., Moss, D. S., and Thornton, J. M. (1993). Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. doi:10.1107/s0021889892009944
Lee, C. T., Yang, W. T., and Parr, R. G. (1988). Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 37 (2), 785–789. doi:10.1103/PhysRevB.37.785
Lee, M. S., Salsbury, F. R., and Olson, M. A. (2004). An efficient hybrid explicit/implicit solvent method for biomolecular simulations. J. Comput. Chem. 25 (16), 1967–1978. doi:10.1002/jcc.20119
Lei, Z., Hong, H., Han, W., Jun-lan, Z., Xiong, C., and Fu-yuan, F. (2022). Research progress on molecular mechanism of curcumin against pulmonary fibrosis. Chin. Traditional Herb. Drugs 53 (21), 6909–6918. doi:10.7501/j.issn.0253-2670.2022.21.030
Liao, K., Gong, L. Y., Yang, Y., He, Y. M., Wang, F. L., Huang, Y. Q., et al. (2022). A comprehensive review of research progress in Chinese medicines for primary liver cancer treatment. Tradit. Med. Res. 7 (2), 10. doi:10.53388/tmr20220207263
Liu, X. W., Shi, D. F., Zhou, S. Y., Liu, H. L., Liu, H. X., and Yao, X. J. (2018). Molecular dynamics simulations and novel drug discovery. Expert. Opin. Drug Discov. 13 (1), 23–37. doi:10.1080/17460441.2018.1403419
Liu, Y., Tang, W. Q., Ji, C. H., Gu, J. H., Chen, Y. M., Huang, J., et al. (2021). The selective sik2 inhibitor arn-3236 produces strong antidepressant-like efficacy in mice via the hippocampal crtc1-creb-bdnf pathway. Front. Pharmacol. 11, 624429. doi:10.3389/fphar.2020.624429
Lu, H., and Schulze-Gahmen, U. (2006). Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. J. Med. Chem. 49 (13), 3826–3831. doi:10.1021/jm0600388
Mansueto, M. S., Reens, A., Rakhilina, L., Chi, A., Pan, B. S., and Miller, J. R. (2019). A reevaluation of the spleen tyrosine kinase (syk) activation mechanism. J. Biol. Chem. 294 (19), 7658–7668. doi:10.1074/jbc.RA119.008045
Marx, A., Nugoor, C., Muller, J., Panneerselvam, S., Timm, T., Bilang, M., et al. (2006). Structural variations in the catalytic and ubiquitin-associated domains of microtubule-associated protein/microtubule affinity regulating kinase (mark) 1 and mark2. J. Biol. Chem. 281 (37), 27586–27599. doi:10.1074/jbc.M604865200
Marx, A., Nugoor, C., Panneerselvam, S., and Mandelkow, E. (2010). Structure and function of polarity-inducing kinase family mark/par-1 within the branch of ampk/snf1-related kinases. Faseb J. 24 (6), 1637–1648. doi:10.1096/fj.09-148064
Matlinska, M. A., Wasylishen, R. E., Bernard, G. M., Terskikh, V. V., Brinkmann, A., and Michaelis, V. K. (2018). Capturing elusive polymorphs of curcumin: A structural characterization and computational study. Cryst. Growth Des. 18 (9), 5556–5563. doi:10.1021/acs.cgd.8b00859
Miehlich, B., Savin, A., Stoll, H., and Preuss, H. (1989). Results obtained with the correlation-energy density functionals of becke and lee, yang and parr. Chem. Phys. Lett. 157 (3), 200–206. doi:10.1016/0009-2614(89)87234-3
Millard, M., Gallagher, J. D., Olenyuk, B. Z., and Neamati, N. (2013). A selective mitochondrial-targeted chlorambucil with remarkable cytotoxicity in breast and pancreatic cancers. J. Med. Chem. 56 (22), 9170–9179. doi:10.1021/jm4012438
Modi, V., and Dunbrack, R. L. (2019). Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. U. S. A. 116 (14), 6818–6827. doi:10.1073/pnas.1814279116
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30 (16), 2785–2791. doi:10.1002/jcc.21256
Muhammed, M. T., and Aki-Yalcin, E. (2019). Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 93 (1), 12–20. doi:10.1111/cbdd.13388
Mujahid, N., Liang, Y. K., Murakami, R., Choi, H. G., Dobry, A. S., Wang, J. H., et al. (2017). A uv-independent topical small-molecule approach for melanin production in human skin. Cell Rep. 19 (11), 2177–2184. doi:10.1016/j.celrep.2017.05.042
Munsamy, G., and Soliman, M. E. S. (2017). Homology modeling in drug discovery-an update on the last decade. Lett. Drug Des. Discov. 14 (9), 1099–1111. doi:10.2174/1570180814666170110122027
Myers, J., Grothaus, G., Narayanan, S., and Onufriev, A. (2006). A simple clustering algorithm can be accurate enough for use in calculations of pks in macromolecules. Proteins 63 (4), 928–938. doi:10.1002/prot.20922
Nesic, D., Miller, M. C., Quinkert, Z. T., Stein, M., Chait, B. T., and Stebbins, C. E. (2010). Helicobacter pylori caga inhibits par1-mark family kinases by mimicking host substrates. Nat. Struct. Mol. Biol. 17 (1), 130–132. doi:10.1038/nsmb.1705
Nugoor, C., Marx, A., Panneerselvam, S., Mandelkow, E.-M., and Mandelkow, E. (2008). Crystal structure of the kinase mark3/par-1: T211a-s215a double mutant. PDB Data Protein Bank. 2008. doi:10.2210/pdb3FE3/pdb
Nurumal, S. R., Ramli, N. S., Mohammad, Z., and Shah, S. A. (2022). Traditional herbal medicine as adjunctive therapy for colorectal cancer: A scoping review. Tradit. Med. Res. 7 (2), 10. doi:10.53388/tmr20220127260
Okamoto, M., Takemori, H., and Katoh, Y. (2004). Salt-inducible kinase in steroidogenesis and adipogenesis. Trends Endocrinol. Metab. 15 (1), 21–26. doi:10.1016/j.tem.2003.11.002
Onufriev, A. V., and Case, D. A. (2019). “Generalized born implicit solvent models for biomolecules,” in Annual review of biophysics. Editor K. A. Dill (Palo Alto: Annual Reviews), 275–296.
Osifova, Z., Reiberger, R., Cisarova, I., Machara, A., and Dracinsky, M. (2022). Diketo–ketoenol tautomers in curcuminoids: Synthesis, separation of tautomers, and kinetic and structural studies. J. Org. Chem. 87 (15), 10309–10318. doi:10.1021/acs.joc.2c01357
Ota, S., Tomioka, S., Sogawa, H., Satou, R., Fujimori, M., Karpov, P., et al. (2018). Binding properties between curcumin and malarial tubulin: Molecular-docking and ab initio fragment molecular orbital calculations. Chem-Bio Inf. J. 18, 44–57. doi:10.1273/cbij.18.44
Pan, J., Lee, Y., Cheng, G., Zielonka, J., Zhang, Q., Bajzikova, M., et al. (2018). Mitochondria-targeted honokiol confers a striking inhibitory effect on lung cancer via inhibiting complex i activity. iScience 3, 192–207. doi:10.1016/j.isci.2018.04.013
Pang, X. J., Liu, X. J., Liu, Y., Liu, W. B., Li, Y. R., Yu, G. X., et al. (2021). Drug discovery targeting focal adhesion kinase (fak) as a promising cancer therapy. Molecules 26 (14), 4250. doi:10.3390/molecules26144250
Parimita, S. P., Ramshankar, Y. V., Suresh, S., and Row, T. N. G. (2007). Redetermination of curcumin: (1e,4z,6e)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,4,6-trien -3-one. Acta Crystallogr. Sect. e.-crystallogr. Commun. 63, O860–O862. doi:10.1107/s160053680700222x
Pinzi, L., and Rastelli, G. (2019). Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 20 (18), 4331. doi:10.3390/ijms20184331
Rahman, M. A., Akter, S., Dorotea, D., Mazumder, A., Uddin, M. N., Hannan, M. A., et al. (2022a). Renoprotective potentials of small molecule natural products targeting mitochondrial dysfunction. Front. Pharmacol. 13, 925993. doi:10.3389/fphar.2022.925993
Rahman, M. M., Wang, X. Y., Islam, M. R., Akash, S., Supti, F. A., Mitu, M. I., et al. (2022b). Multifunctional role of natural products for the treatment of Parkinson's disease: At a glance. Front. Pharmacol. 13, 976385. doi:10.3389/fphar.2022.976385
Rajendran, J. V., Thomas, S., Jafari, Z., Fariborzi, N., Khorasani, S., Mofrad, Z. K., et al. (2022). Recent advances on large-scale manufacture of curcumin and its nanoformulation for cancer therapeutic application. Biointerface Res. Appl. Chem. 12 (6), 7863–7885. doi:10.33263/briac126.78637885
Rassolov, V. A., Pople, J. A., Ratner, M. A., and Windus, T. L. (1998). 6-31g* basis set for atoms k through zn. J. Chem. Phys. 109 (4), 1223–1229. doi:10.1063/1.476673
Rassolov, V. A., Ratner, M. A., Pople, J. A., Redfern, P. C., and Curtiss, L. A. (2001). 6-31g*basis set for third-row atoms. J. Comput. Chem. 22 (9), 976–984. doi:10.1002/jcc.1058
Rege, S. A., Arya, M., and Momin, S. A. (2019). Structure activity relationship of tautomers of curcumin: A review. Ukr. Food J. 8 (1), 45–60. doi:10.24263/2304-974x-2019-8-1-6
Rodrigues, F. C., Hari, G., Pai, K. S. R., Suresh, A., Nayak, U. Y., Anilkumar, N. V., et al. (2021). Molecular modeling piloted analysis for semicarbazone derivative of curcumin as a potent abl-kinase inhibitor targeting colon cancer. 3 Biotech. 11 (12), 506. doi:10.1007/s13205-021-03051-9
Roe, D. R., and Cheatham, T. E. (2018). Parallelization of cpptraj enables large scale analysis of molecular dynamics trajectory data. J. Comput. Chem. 39 (25), 2110–2117. doi:10.1002/jcc.25382
Rognan, D. (1998). Molecular dynamics simulations: A tool for drug design. Perspect. Drug Discov. Des. 9-11, 181–209. doi:10.1023/a:1027268223451
Roskoski, R. (2016). Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 103, 26–48. doi:10.1016/j.phrs.2015.10.021
Saikia, S., and Bordoloi, M. (2019). Molecular docking: Challenges, advances and its use in drug discovery perspective. Curr. Drug Targets 20 (5), 501–521. doi:10.2174/1389450119666181022153016
Sakamoto, K., Bultot, L., and Goransson, O. (2018). The salt-inducible kinases: Emerging metabolic regulators. Trends Endocrinol. Metab. 29 (12), 827–840. doi:10.1016/j.tem.2018.09.007
Salam, A. A. A., Nayek, U., and Sunil, D. (2018). Homology modeling and docking studies of bcl-2 and bcl-xl with small molecule inhibitors: Identification and functional studies. Curr. Top. Med. Chem. 18 (31), 2633–2663. doi:10.2174/1568026619666190119144819
Sanner, M. F. (1999). Python: A programming language for software integration and development. J. Mol. Graph. 17 (1), 57–61.
Sanphui, P., Goud, N. R., Khandavilli, U. B. R., Bhanoth, S., and Nangia, A. (2011). New polymorphs of curcumin. Chem. Commun. 47 (17), 5013–5015. doi:10.1039/c1cc10204d
Scotti, L., Mendona, F. J. B., Ishiki, H. M., Ribeiro, F. F., Singla, R. K., Barbosa, J. M., et al. (2017). Docking studies for multi-target drugs. Curr. Drug Targets 18 (5), 592–604. doi:10.2174/1389450116666150825111818
Shi, M., Chen, T., Wei, S., Zhao, C., Zhang, X., Li, X., et al. (2022a). Molecular docking, molecular dynamics simulations, and free energy calculation insights into the binding mechanism between vs-4718 and focal adhesion kinase. ACS Omega 7 (36), 32442–32456. doi:10.1021/acsomega.2c03951
Shi, M., Wang, L., Li, P., Liu, J., Chen, L., and Xu, D. (2021a). Dasatinib-sik2 binding elucidated by homology modeling, molecular docking, and dynamics simulations. ACS Omega 6 (16), 11025–11038. doi:10.1021/acsomega.1c00947
Shi, M., Wang, L., Liu, K., Chen, Y., Hu, M., Yang, L., et al. (2022b). Molecular dynamics simulations of the conformational plasticity in the active pocket of salt-inducible kinase 2 (sik2) multi-state binding with bosutinib. Comput. Struct. Biotechnol. J. 20, 2574–2586. doi:10.1016/j.csbj.2022.05.039
Shi, M., and Xu, D. (2019). Molecular dynamics investigations suggest a non-specific recognition strategy of 14-3-3σ protein by tweezer: Implication for the inhibition mechanism. Front. Chem. 7 (237), 237. doi:10.3389/fchem.2019.00237
Shi, M., Zhao, M., Wang, L., Liu, K., Li, P., Liu, J., et al. (2021b). Exploring the stability of inhibitor binding to sik2 using molecular dynamics simulation and binding free energy calculation. Phys. Chem. Chem. Phys. 23 (23), 13216–13227. doi:10.1039/d1cp00717c
Shi, X. J., Zhang, T., Lou, H. X., Song, H. N., Li, C. H., and Fan, P. H. (2020). Anticancer effects of honokiol via mitochondrial dysfunction are strongly enhanced by the mitochondria-targeting carrier berberine. J. Med. Chem. 63 (20), 11786–11800. doi:10.1021/acs.jmedchem.0c00881
Soltani, A., Salmaninejad, A., Jalili-Nik, M., Soleimani, A., Javid, H., Hashemy, S. I., et al. (2019). 5 '-adenosine monophosphate-activated protein kinase: A potential target for disease prevention by curcumin. J. Cell. Physiol. 234 (3), 2241–2251. doi:10.1002/jcp.27192
Sonntag, T., Vaughan, J. M., and Montminy, M. (2018). 14-3-3 proteins mediate inhibitory effects of camp on salt-inducible kinases (siks). Febs J. 285 (3), 467–480. doi:10.1111/febs.14351
Srinivasan, J., Cheatham, T. E., Cieplak, P., Kollman, P. A., and Case, D. A. (1998). Continuum solvent studies of the stability of DNA, rna, and phosphoramidate - DNA helices. J. Am. Chem. Soc. 120 (37), 9401–9409. doi:10.1021/ja981844+
Stavenger, R. A. (2008). “Rho-kinase inhibitors for cardiovascular disease,” in Annual reports in medicinal chemistry. Editor J. E. Macor (San Diego: Elsevier Academic Press Inc), 87–102.
Stevenson, L. M., Deal, M. S., Hagopian, J. C., and Lew, J. (2002). Activation mechanism of cdk2: Role of cyclin binding versus phosphorylation. Biochemistry 41 (26), 8528–8534. doi:10.1021/bi025812h
Su, X., Zhou, M. Y., Li, Y. J., Zhang, J. Z., An, N., Yang, F., et al. (2022). Protective effects of natural products against myocardial ischemia/reperfusion: Mitochondria-targeted therapeutics. Biomed. Pharmacother. 149, 112893. doi:10.1016/j.biopha.2022.112893
Sumirtanurdin, R., Sungkar, S., Hisprastin, Y., Sidharta, K. D., and Nurhikmah, D. D. (2020). Molecular docking simulation studies of curcumin and its derivatives as cyclin-dependent kinase 2 inhibitors. Turk. J. Pharm. Sci. 17 (4), 417–423. doi:10.4274/tjps.galenos.2019.55822
Sun, Z. C., Jiang, Q. W., Li, J., and Guo, J. P. (2020). The potent roles of salt-inducible kinases (siks) in metabolic homeostasis and tumorigenesis. Signal Transduct. Target. Ther. 5 (1), 150. doi:10.1038/s41392-020-00265-w
Tang, C. Y., Liu, J. T., Yang, C. S., Ma, J., Chen, X. J., Liu, D. W., et al. (2022). Curcumin and its analogs in non-small cell lung cancer treatment: Challenges and expectations. Biomolecules 12 (11), 1636. doi:10.3390/biom12111636
Taub, M. (2019). Salt inducible kinase signaling networks: Implications for acute kidney injury and therapeutic potential. Int. J. Mol. Sci. 20 (13), 3219. doi:10.3390/ijms20133219
Taub, M., Springate, J. E., and Cutuli, F. (2010). Targeting of renal proximal tubule na,k-atpase by salt-inducible kinase. Biochem. Biophys. Res. Commun. 393 (3), 339–344. doi:10.1016/j.bbrc.2010.02.037
Tesch, R., Rak, M., Raab, M., Berger, L. M., Kronenberger, T., Joerger, A. C., et al. (2021). Structure-based design of selective salt-inducible kinase inhibitors. J. Med. Chem. 64 (12), 8142–8160. doi:10.1021/acs.jmedchem.0c02144
Tian, C., Kasavajhala, K., Belfon, K. A. A., Raguette, L., Huang, H., Migues, A. N., et al. (2020). Ff19sb: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16 (1), 528–552. doi:10.1021/acs.jctc.9b00591
Tonnesen, H. H., Karlsen, J., Mostad, A., Samuelsson, B., Enzell, C. R., and Berg, J. E. (1982). Structural studies of curcuminoids. I. The crystal structure of curcumin. Acta Chem. Scand. Ser. B-Organic Chem. Biochem. 36 (7), 475–479. doi:10.3891/acta.chem.scand.36b-0475
Tran, J. M., Rassolov, V. A., Ratner, M. A., and Pople, J. A. (1999). Diffuse functions in the 6-31g basis set for atoms k through zn. Abstr. Pap. Am. Chem. Soc. 217, U443.
Tse, A., and Verkhivker, G. M. (2015). Molecular dynamics simulations and structural network analysis of c-abl and c-src kinase core proteins: Capturing allosteric mechanisms and communication pathways from residue centrality. J. Chem. Inf. Model. 55 (8), 1645–1662. doi:10.1021/acs.jcim.5b00240
Tu, Z. S., Wang, Q., Sun, D. D., Dai, F., and Zhou, B. (2017). Design, synthesis, and evaluation of curcumin derivatives as nrf2 activators and cytoprotectors against oxidative death. Eur. J. Med. Chem. 134, 72–85. doi:10.1016/j.ejmech.2017.04.008
Tuccinardi, T., and Martinelli, A. (2011). Protein kinase homology models: Recent developments and results. Curr. Med. Chem. 18 (19), 2848–2853. doi:10.2174/092986711796150441
Vadukoot, A. K., Mottemmal, S., and Vekaria, P. H. (2022). Curcumin as a potential therapeutic agent in certain cancer types. Cureus J. Med. Sci. 14 (3), e22825. doi:10.7759/cureus.22825
Wang, J. M., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case, D. A. (2004). Development and testing of a general amber force field. J. Comput. Chem. 25 (9), 1157–1174. doi:10.1002/jcc.20035
Wang, J. Y., Chen, Q., Wang, M., and Zhong, C. (2017). The opening/closure of the p-loop and hinge of bcr-abl1 decodes the low/high bioactivities of dasatinib and axitinib. Phys. Chem. Chem. Phys. 19 (33), 22444–22453. doi:10.1039/c7cp03443a
Wang, S. M., Gao, X. L., Li, J. L., Wei, S. X., Shao, Y. F., Yin, Y. P., et al. (2022). The anticancer effects of curcumin and clinical research progress on its effects on esophageal cancer. Front. Pharmacol. 13, 1058070. doi:10.3389/fphar.2022.1058070
Wang, X. J., Blackaby, W., Allen, V., Chang, G. K. Y., Chang, J. H., Chiang, P. C., et al. (2019). Optimization of pan-pim kinase activity and oral bioavailability leading to diaminopyrazole (gdc-0339) for the treatment of multiple myeloma. J. Med. Chem. 62 (4), 2140–2153. doi:10.1021/acs.jmedchem.8b01857
Wang, Y. L., Wang, J., Chen, X., Wang, Z. X., and Wu, J. W. (2018). Crystal structure of the kinase and uba domains of snrk reveals a distinct uba binding mode in the ampk family. Biochem. Biophys. Res. Commun. 495 (1), 1–6. doi:10.1016/j.bbrc.2017.10.105
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). Swiss-model: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (1), W296–W303. doi:10.1093/nar/gky427
Wei, W. Q., Chen, Y. N., Xie, D. Q., and Zhou, Y. Z. (2019). Molecular insight into chymotrypsin inhibitor 2 resisting proteolytic degradation. Phys. Chem. Chem. Phys. 21 (9), 5049–5058. doi:10.1039/c8cp07784c
Wein, M. N., Foretz, M., Fisher, D. E., Xavier, R. J., and Kronenberg, H. M. (2018). Salt-inducible kinases: Physiology, regulation by camp, and therapeutic potential. Trends Endocrinol. Metab. 29 (10), 723–735. doi:10.1016/j.tem.2018.08.004
Wieman, H., Tondel, K., Anderssen, E., and Drablos, F. (2004). Homology-based modelling of targets for rational drug design. Mini-Rev. Med. Chem. 4 (7), 793–804. doi:10.2174/1389557043403639
Wong, C. F. (2015). Flexible receptor docking for drug discovery. Expert. Opin. Drug Discov. 10 (11), 1189–1200. doi:10.1517/17460441.2015.1078308
Wu, J. X., Cheng, Y. S., Wang, J., Chen, L., Ding, M., and Wu, J. W. (2015). Structural insight into the mechanism of synergistic autoinhibition of sad kinases. Nat. Commun. 6, 8953. doi:10.1038/ncomms9953
Xiang, Z. X. (2006). Advances in homology protein structure modeling. Curr. Protein Pept. Sci. 7 (3), 217–227. doi:10.2174/138920306777452312
Xing, L., Klug-Mcleod, J., Rai, B., and Lunney, E. A. (2015). Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 23 (19), 6520–6527. doi:10.1016/j.bmc.2015.08.006
Xu, D., and Zheng, Q. C. (2022). Theoretical investigations on the effects of mutations in important residues of ns1b on its rna-binding using molecular dynamics simulations. Comput. Biol. Med. 145, 105412. doi:10.1016/j.compbiomed.2022.105412
Xu, M. W., Peng, R., Min, Q., Hui, S. W., Chen, X., Yang, G., et al. (2022). Bisindole natural products: A vital source for the development of new anticancer drugs. Eur. J. Med. Chem. 243, 114748. doi:10.1016/j.ejmech.2022.114748
Yamashita, F. (2018). Dynamic simulation of drug drug interactions by using multi-level physiological modeling & simulation platforms. Yakugaku Zasshi-J. Pharm. Soc. Jpn. 138 (3), 347–351. doi:10.1248/yakushi.17-00191-2
Ye, Y., Zhang, T., Yuan, H., Li, D., Lou, H., and Fan, P. (2017). Mitochondria-targeted lupane triterpenoid derivatives and their selective apoptosis-inducing anticancer mechanisms. J. Med. Chem. 60 (14), 6353–6363. doi:10.1021/acs.jmedchem.7b00679
Yin, Y., Tan, Y., Wei, X. N., Li, X. S., Chen, H. F., Yang, Z. H., et al. (2022). Recent advances of curcumin derivatives in breast cancer. Chem. Biodivers. 19 (10), e202200485. doi:10.1002/cbdv.202200485
Yoon, M. J., Kim, E. H., Lim, J. H., Kwo, T. K., and Choi, K. S. (2010). Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Radic. Biol. Med. 48 (5), 713–726. doi:10.1016/j.freeradbiomed.2009.12.016
Zhi, T. X., Liu, K. Q., Cai, K. Y., Zhao, Y. C., Li, Z. W., Wang, X., et al. (2022). Anti-lung cancer activities of 1,2,3-triazole curcumin derivatives via regulation of the mapk/nf-kappa b/stat3 signaling pathways. ChemMedChem 17 (3), e202100676. doi:10.1002/cmdc.202100676
Zhou, J. H., Alfraidi, A., Zhang, S., Santiago-O'Farrill, J. M., Reddy, V. K. Y., Alsaadi, A., et al. (2017). A novel compound arn-3236 inhibits salt-inducible kinase 2 and sensitizes ovarian cancer cell lines and xenografts to paclitaxel. Clin. Cancer Res. 23 (8), 1945–1954. doi:10.1158/1078-0432.Ccr-16-1562
Zorova, L. D., Popkov, V. A., Plotnikov, E. Y., Silachev, D. N., Pevzner, I. B., Jankauskas, S. S., et al. (2018). Mitochondrial membrane potential. Anal. Biochem. 552, 50–59. doi:10.1016/j.ab.2017.07.009
Keywords: curcumin, salt-induced kinase, molecular dynamics simulation, molecular docking, binding free energy, binding model
Citation: Shi M, Zhou Y, Wei H, Zhang X, Du M, Zhou Y, Yin Y, Li X, Tang X, Sun L, Xu D and Li X (2023) Interactions between curcumin and human salt-induced kinase 3 elucidated from computational tools and experimental methods. Front. Pharmacol. 14:1116098. doi: 10.3389/fphar.2023.1116098
Received: 12 December 2022; Accepted: 03 April 2023;
Published: 13 April 2023.
Edited by:
Sajjad Gharaghani, University of Tehran, IranReviewed by:
Mohammad Ghattas, Al Ain University, United Arab EmiratesLarance Ronsard, Ragon Institute, United States
Copyright © 2023 Shi, Zhou, Wei, Zhang, Du, Zhou, Yin, Li, Tang, Sun, Xu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dingguo Xu, ZGd4dUBzY3UuZWR1LmNu; Xiaoan Li, bGl4aWFvYW5Ac2MtbWNoLmNu