 Muhammad Shafique1
Muhammad Shafique1 Maqsood Ur Rehman2,3Zul Kamal4Rami M. Alzhrani5Sameer Alshehri5Ali H. Alamri6Mohammed Ali Bakkari7
Maqsood Ur Rehman2,3Zul Kamal4Rami M. Alzhrani5Sameer Alshehri5Ali H. Alamri6Mohammed Ali Bakkari7 Fahad Y. Sabei7
Fahad Y. Sabei7 Awaji Y. Safhi7
Awaji Y. Safhi7 Ahmed M. Mohammed8Mohamed A. El Hamd1,9
Ahmed M. Mohammed8Mohamed A. El Hamd1,9 Saud Almawash1*
Saud Almawash1*- 1Department of Pharmaceutical Sciences, College of Pharmacy, Shaqra University, Shaqra, Saudi Arabia
- 2Department of Pharmaceutics, School of Pharmacy, University College London, London, United Kingdom
- 3Department of Pharmacy, University of Malakand, Chakdara, (Dir Lower), Pakistan
- 4Department of Pharmacy, Shaheed Benazir Bhutto University, Sheringal, (Dir Upper), Pakistan
- 5Department of Pharmaceutics and Industrial Pharmacy, College of Pharmacy, Taif University, Taif, Saudi Arabia
- 6Department of Pharmaceutics, College of Pharmacy, King Khalid University, Abha, Saudi Arabia
- 7Department of Pharmaceutics, College of Pharmacy, Jazan University, Jazan, Saudi Arabia
- 8Department of pharmaceutics and pharmaceutical technology Faculty of Pharmacy Al-azhar University, Assiut, Egypt
- 9Department of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, South Valley University, Qena, Egypt
The purpose of this study was to assess the parameters of doxorubicin (DOX) loaded lipid polymer hybrid nanoparticles (LPHNs) formulation development, and then the bioavailability of DOX were determined in the rabbit model, in order to evaluate the intrinsic outcome of dosage form improvement after the oral administration. LPHNs were prepared by combine approach, using both magnetic stirring and probe sonication followed by its characterization in terms of size-distribution (Zeta Size), entrapment efficiency (EE), loading capacity, and the kinetics of DOX. LPHNPs were further characterized by using scanning electron microscopy (SEM), powder X-Ray diffractometry (P-XRD), Fourier transform infrared spectroscopy (FT-IR), differential scanning calorimetry (DSC), in vitro and in vivo studies. The molecular modeling was determined through the density functional theory (DFT) simulations and interactions. DOX loaded and unloaded LPHNs were administered orally to the rabbits for bioavailability and pharmacokinetic parameters determinations. The plasma concentration of DOX was determined through high performance liquid chromatography (HPLC). The average size of DOX-loaded LPHNs was 121.90 ± 3.0 nm. The drug loading of DOX was 0.391% ± 0.01 of aqueous dispersion, where its encapsulation efficiency was 95.5% ± 1.39. After oral administration of the DOX-LPHNs, the area under the plasma drug concentration-time curve (AUC) improved about 2-folds comparatively (p < 0.05). DFT simulations were used to understand the interactions of polymers with different sites of DOX molecule. The larger negative binding energies (−9.33 to −18.53 kcal/mol) of the different complexes evince that the polymers have stronger affinity to bind with the DOX molecule while the negative values shows that the process is spontaneous, and the synthesis of DOX-LPHNs is energetically favorable. It was concluded that DOX-LPHNs provides a promising new formulation that can enhance the oral bioavailability, which have optimized compatibilities and improve the pharmacokinetic of DOX after oral administration.
1 Introduction
Globally, cancer causes a major burden of diseases, affecting millions of people out of which half the patients die. A number of chemotherapeutic agents have been modified into various formulations, in order to enhance their therapeutic performances. Doxorubicin (DOX) was the first drug which received clinical approval from the Food and Drug Administration (FDA) as an anticancer drug encapsulated in the form of liposomes DOX belongs to the anthracycline group and is the most commonly used member of this group (Abraham et al., 2005). DOX has been used with common applications in different kinds of cancer including breast cancer, ovarian cancer, lung cancer, and malignant lymphoma (Chan et al., 1999). However, the major side effect of DOX is cardiotoxicity which prevents its long-term use (Lage, 2003; Duggan and Keating, 2011). Besides cardiotoxicity, DOX has some other major shortcomings like poor water solubility, short half-life, gastric instability, and first pass hepatic effects (Peltier et al., 2006; Gou et al., 2011).
Various approaches have been used to improve the oral efficacy of drugs including polymer-prodrugs (PD), polymer-conjugates (PG), polymeric-nanoparticles (PNPs), liposomes, solid lipid nanoparticles (SLNs), etc. (Zhou et al., 2011; Du et al., 2013; Zhang et al., 2015; Ahmad et al., 2018 such as using SLN, layersomes, and dendrimer for DOX (Yang et al., 1999; Ke et al., 2008; Jain et al., 2012), polymeric-micelles for paclitaxel (Yao et al., 2011), and polymeric-nanoparticles for etoposide (Fatma et al., 2016). Poly (lactic-co-glycolic acid) based nanoparticles (PLGA-NPs) were also studied whereby enhancement in gemcitabine bioavailability was observed, (Joshi et al., 2014) in addition, a boosted pharmacodynamics profiles were observed for DOX and paclitaxel (Bhardwaj et al., 2009). Additionally, polyethylene glycol (PEG) containing NPs have been shown to have a more diffusion property with greater penetration properties across the thick layer of mucosa. This adhesive feature leads to enhanced oral bioavailability (Huang et al., 2000; Yoncheva et al., 2007).
DOX loaded to LPHNs can enhanced the oral bioavailability and therapeutic efficacy. Inadequate oral bioavailability is due to the hydrophobic nature of DOX and its poor absorption from duodenal sites (Gold and Moellering, 1996; Walsh, 2000; Huh and Kwon, 2011) and that’s why, it has been categorized as Class-IV based on the Biopharmaceutical Classification System (BCS-IV).
LPHNs were developed as a drug delivery system with characteristic features of both liposomes and polymeric-NPs (Zhang et al., 2008). This hybrid system is a smart drug delivery system with high stability, enhanced entrapment efficiency, attired release kinetics, and fine targeting properties. This study focused at developing physically stable DOX-LPHNs formulation for augmenting its aqueous solubility and enhancing its oral bioavailability, and compatibility. Eudragit RS-100 (polymer), stearic Acid (solid lipid), oleic acid (liquid lipid), and ethyl cellulose (Hepler polymer) were used as excipients in the nano-formulation of LPHNs. The DOX-LPHNs were synthesized and characterized for their surface physico-chemical properties, drug loading, and entrapment efficiencies. The molecular modelling, interaction, and simulations were determined through DFTs. The in vitro and in vivo release kinetics (plasma concentrations) were determined by various pharmacokinetics models using HPLC, its nano-formulations stabilities and acute toxicities were also determined.
2 Materials and methods
2.1 Materials
Doxorubicin (Atco Labs Pakistan), stearic acid (SA) (Acros-Organics TFS (United States), Eudragit (Acros-Organics TFS (United State), ethyl cellulose (Acros-Organics TFS (United States), sodium lauryl sulphate (Sigma), and oleic acid (Sigma). All of the solvents used in all the experiments were of analytical grade.
2.2 Preparation and optimization of LPHNs (unloaded)
LPHNs were fabricated by a combined process, using both probe sonication and magnetic stirring processes. The unloaded LPHNs were prepared by melting the stearic acid (lipid) at 80°C. The eudragit and sodium lauryl sulphate (SLS) were dissolved in ethanol and were added to the melted stearic acid. The organic phase (ethanol) was removed by stirring while the final volume was adjusted with deionized water. This resultant mixture was passed through probe sonication at 30% amplitude to get LPHNs dispersion. Various approaches regarding the material ratios were adopted for nano-formulations’ optimization, which were compared against the obtained particles size, from LPHNs-1 to LPHNs-6.
2.3 Preparation of loaded LPHNs (DOX-LPHNs)
DOX-LPHNs were fabricated by the addition of DOX (40 mg) to the solution of polymer and surfactant in organic phase. Co-encapsulation was carried out by dissolving ethyl cellulose and oleic acid in the same organic solution and the same procedure as were used for LPHNs was followed. The freeze drying or lyophilization was conducted to give stability to LPHNs and DOX-LPHNs and their further conversion to dry powder. Before lyophilization, the samples were subjected to addition of cryoprotectant (glucose solution, 10%) followed by cooling at −20°C overnight. LPHNs/DOX-LPHNs were then transferred to the freeze dryer for lyophilization at temperature of −75°C for about 2 days (48 h), while the increasing temperature was kept around 5°C/h (Abdelwahed et al., 2006).
2.4 Entrapment efficiency and drug loading capacity
The optimized formulations of DOX-LPHNs, fabricated by the mentioned technique were centrifuged. The supernatant was separated and further analyzed for un-entrapped drug by UV Visible Spectrophotometer. DOX-LPHNs entrapment efficiency (EE) and drug loading capacity (DLC) for all the prepared samples were calculated by using the following formulae (Ghanshyam et al., 2011; Sadiq and Abdul Rassol, 2014),
2.5 Surface characterization
2.5.1 Size, zeta-potential and polydispersity index
Size (Z), zeta-potential (ζ), and polydispersity index (PDI) were determined by dynamic light scattering (DLS) technique using the Macrotac Zeta Instruments. To obtain suitable scattering, the LPHNs formulations (both loaded and unloaded) were diluted with deionized water. Measurements were then taken at scattering angle of 90 at room temperature. The particles size, PDI and zeta potential of nanoformulations were calculated by taking the average of three results.
2.5.2 Infrared spectroscopy
IR Prestige 21 Shimadzu (Japan) was used to study the IR Spectra of LPHNs and DOX-LPHNs (Tiţa et al., 2011). During FT-IR studies, scanning was performed at a frequency range of 4,000 cm−1 to 450 cm−1. For the compatibility of the formulation components, the peaks and patterns shaped by the unprocessed drug were compared with their processed formulations of LPHNs.
2.5.3 Scanning electron microscopy (SEM)
The surface morphology of DOX-LPHNs was studied by using SEM (JEOL, Japan) (Dubes et al., 2003; Uprit et al., 2013). Prior to conducting SEM analysis, deionized water was used to dilute all nanoformulations to form clear and visible samples. Double ended adhesive carbon tape was employed to fixed sample drops of on metallic stub of microscope followed by drying under vacuum for further analysis. Magnification power in the range of 15,000–60000X has been used with varied voltage.
2.5.4 Powder X-Ray diffraction
Powder X-ray diffractometer (JEOL, Japan) was used for unprocessed DOX and processed DOX (DOX-4) to determine changes in the physical state of the drug (Racault et al., 1994). Thus, P-XRD analysis was conducted to study the variations in the crystalline nature and physical state of different samples. P-XRD study was performed using plain plastic holder for sample in the scan range of 2θ = 5–80° with Cu Kα radiation. Tube was operated at 40 kV, 30 mA, step size 0.05°, step time 1.0 s, receiving slit 0.2 mm, scattering slit 1.0° and divergence slit 1.0°.
2.5.5 Differential scanning calorimetry
Thermal analysis of pure DOX, DOX-LPHNs, stearic acid and physical mixture were carried out by DSC (Perkin Elmer-USA). Samples were investigated in aluminum pans at a rate of 10°C/min and DSC thermogram was determined from 40ºC to 400°C (Hou et al., 2003).
2.5.6 Formulation stability studies
Physical stability study was conducted for optimized formulations of LPHNs. The freshly fabricated sample was divided into two parts. Each was then put in two different vials and stored at two different temperatures, i.e., 4°C and 25°C (del Pozo-Rodríguez et al., 2009). After specific intervals of time (1st day, 2nd week, 4thweek, 8th week, and 12th week), both the particle size and Polydispersity index (PDI) were determined through DLS. Data was analyzed statistically by two-tailed t-test. Probability ˂0.05 was considered significant.
2.5.7 In-vitro release of DOX-LPHNs
Dialysis membrane method was used to study the release of DOX from the DOX-LPHNs polymeric nanoparticles (Bhardwaj and Burgess, 2010). The dialysis membrane soaked in water at least 12 h prior to its use. One ml of DOX-LPHNs (each formulation) was decanted into the dialysis membrane which was then kept at pH 7.4 (50 rpm) using 250 mL of phosphate buffer solution (PBS). We took sample from each formulation after specific time (1–12 h) and analyzed it by means of an UV-spectrophotometer (λmax = 278 nm) (Moffat et al., 2011). The release data was tailored into diverse kinetic models to learn both the drug release rate and mechanism of drug release [ (Roohullah et al., 2013), (Taninaka et al., 2000)].
2.5.8 Comparative in vivo study
For conducting in vivo pharmacokinetic study, healthy rabbits (2 ± 0.3 kg) were used. All experimental animals (rabbits/rats) were screened and accepted for experimental purpose by the Ethical Committee, Department of Pharmacy, University of Malakand (Ref no: UOM/Pharm-IRB-2022/07). The in-vivo pharmacokinetic studies were completed in line with the ethical committee of the University of Malakand (Pakistan) and pertinent byelaws, 2008 (Scientific procedure issue 1). All the experimental animals (rabbits) were kept in fasted state (12 h) before dosing but access to water was given. Any experimental animal having dis-comfort was expelled from studies. Prior to oral drug administration, six groups of animals were made, each having n = 6 rabbits/group. The optimized LPHNs nanosuspension was administered to Group-I, prepared capsules to Group-II while marketed product to Group-III. At various time interval (0_24 h), sample of blood (0.5 mL) was taken from marginal ear vein of rabbits. Blood samples were kept in 3 mL tubes (heparinized), plasma was separated through centrifugation and stored (−20°C) for further analysis.
Different pharmacokinetic parameters were determined for non−compartmental model. From concentration−time curve, Area Under Curve (AUC0→t) was determined via trapezoidal rule. From the individual plasma concentration−time curve, peak plasma concentration (Cmax) and peak plasma concentration time (tmax) were calculated. Total area under the curve (AUC0→24) was determined by Eq. 3:
Ct is drug concentration at 24th hour and Ke is apparent elimination rate constant.
Relative bioavailability (Fr) after 24 h for equal dose was determined by Eq. 4:
One−way analysis of variance and t−test (p < 0.05) were used for statistical analysis of data.
2.5.9 Computational details
The simulations were performed by Gaussian 09 code (Abraham et al., 2005). The Grimme’s dispersion corrected DFT-D3 (Chan et al., 1999; Duggan and Keating, 2011) B3LYP functional was used for all the calculations. The 6-311G (d,p) basis set was applied for geometries optimization. The binding energy (Eb) was calculated using the following equation:
Where, Ecomplex, EM1 and EM2 are the total electronic energies of complex system (polymer bind with doxorubicin drug), monomer-1 (polymer), and monomer-2 (doxorubicin drug), respectively.
2.5.10 Acute toxicity test
Acute toxicity test was based on the chemical testing guidelines of the OECD (Organization for Economic Cooperation for Development) (Jonsson et al., 2013). Mice were used as subject animals which were divided into groups (each n = 6). DOX-LPHNs was administered at doses of 50 mg, 100 mg, 200 mg, 400 mg, 800 mg, and 1,600 mg per kg body weight to each group. Morbidity was studied for the first 2 h while mortality was observed post 24 h of dosing. The experimental animals were checked for any behavioral changes as well. The 50% mortality among the rabbits was premeditated by means of the Probit analysis method.
2.5.11 Statistical analysis
All data are presented as the mean ± standard error means (SEM). Statistically significant differences were assessed by one and two-way ANOVA, t-test using the graph pad prism software, and the differences were considered significant statistically when p < 0.05. Statistical values were indicated in the figure by the following symbols: * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001. Probit analysis was used for calculating acute toxicities in the experimental animals for dose-response relationships.
3 Results and discussion
3.1 Preparation and optimization of LPHNs (unloaded) and DOX-LPHNs (loaded)
The detailed schematic illustration of the step-wise procedure for LPHNs and DOX-LPHNs fabrication is shown in Figure 1. The diagrammatical representation of LPHNs is actually combinative approach employing magnetic stirring and probe sonication. For fabrication of LPHNs, stearic acid was used as solid lipid, oleic acid as liquid lipid, sodium lauryl sulphate as surfactant, PEG as co-surfactant, eudragit RS-100 as polymer and ethyl cellulose as helper polymer. Optimization was carried out using different variable factors like concentration of excipients, magnetic stirring and sonication time (Table 1).
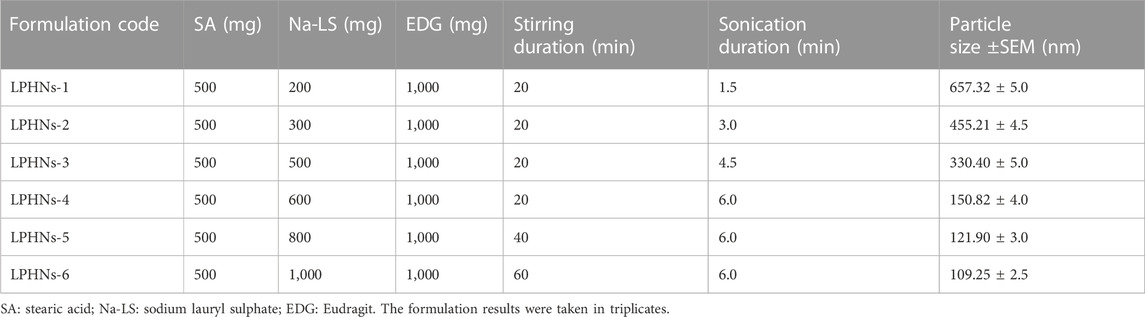
TABLE 1. Preparation and optimization of unloaded lipid polymer hybrid nanoparticles (LPHNs) nano-formulations.
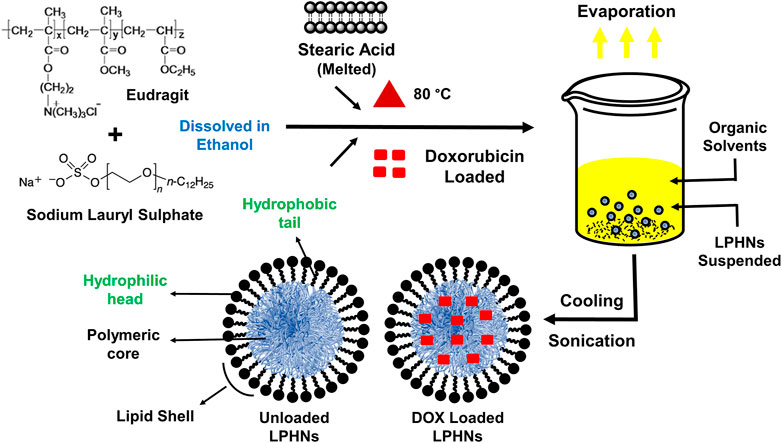
FIGURE 1. Schematic illustrations showing the preparation of LPHNs (unloaded) and DOX-LPHNs (loaded) formulations.
3.1.1 Concentration of surfactant
During optimization of LPHNs sodium laural sulphate is used as surfactant. When the concentration of surfactant was increased, it caused abrupt reduction in particles size. As, further increase in concentration of surfactant showed almost no effect on particle size. It has been reported in literature that higher concentration of surfactant showed lower particle size and also offer better stability to small lipid droplets as it prevent them from coalescence (Kovacevic et al., 2011).
3.1.2 Concentration of co-surfactant
Further decrease in particle size was achieved with addition of co-surfactant. PEG being employed as co-surfactant, further reduced particle size. As, LPHNs fabricated with surfactant/co-surfactant mixture have lower particle size and better stability as compared to LPHNs of unadded co-surfactants.
3.1.3 Stirring time
During variation in magnetic stirring time, reduction in particle size and PDI reduced to the desired acceptable range. During increase in the magnetic stirring time, it has been noticed that particle size also reduced to some extent but it mainly controlled the PDI. Thus, PDI was controlled and reduced by increasing stirring time which has shown almost little bit effect on particle size reduction (Baharifar et al., 2015).
3.1.4 Sonication
During variation in sonication parameters, reduction in particle size was observed. By increasing the sonication time/Hz, particle size reduced to the desired acceptable range. Finally, size was controlled and reduced by sonication which has shown excellent effect on particle size reduction. Important variations in terms of particle size and PDI were seen by changing the mentioned four variable parameters.
During optimization process of blank LPHNs, desired particles size and acceptable PDI were produced with stearic acid, sodium laurel sulphate, PVP and magnetic stirring time (15 min). After optimization of different formulation parameters (concentration of surfactant, magnetic stirring and sonication time) LPHNs and DOX-LPHNs showed optimized average particle size of 150.82 ± 4.37 nm and 185.43 ± 4.43 nm, average PDI of 0.238 ± 0.009 and 0.256 ± 0.003 and zeta potential (ζ) of −31.1 ± 3.0 and −33.95 ± 3.53 respectively (Figure 2). The PDI <0.5 and ZP in the range of ±30 revealed that the fabricated LPHNs would be stable in nature (Ali et al., 2011). For our prepared LPHNs both PDI and ZP were within the acceptable range, which exhibit electrostatic stabilization to avoid aggregation thus preventing particles growth and Ostwald ripening (Liu et al., 2007). As the fabricated loaded LPHNs are for oral administration, so, the produced average particles size is less than 400 nm having the ability of easily crossing the linings of gastro-intestinal cells to achieve the desired boosted oral bioavailability (Suh et al., 2009). Moreover, the fabricated LPHNs comprised of 100–200 nm size range, since particles having size less than 200 nm are undetectable to the Reticulo endothelial system (RES) and remain in circulatory system for a prolonged time period (Bhandari and Kaur, 2013).
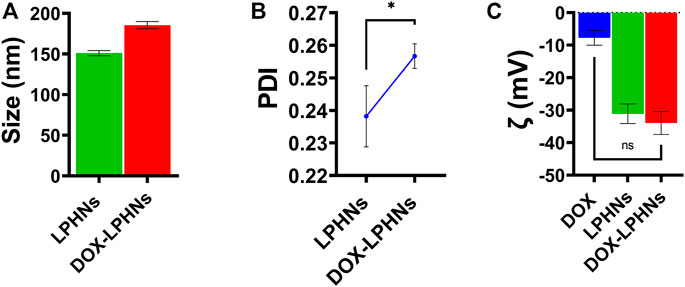
FIGURE 2. Surface characterization of LPHNs and DOX-LPHNs. (A) Average particle size of LPHNs and DOX-LPHNs (B) Polydispersity Index of LPHNs and DOX-LPHNs (C) Zeta potential (ζ) of LPHNs and DOX-LPHNs. One sample t-test (two tailed), p value = 0.0999.
3.2 Loading capacity and entrapment efficiency
The fabricated DOX-LPHNs were optimized based on the concentrations of oleic acid and ethyl-cellulose to determine EE (%) and DLC (%). All optimized formulations (DOX-LPHNs-1 to DOX-LPHNs-5), along with the concentrations of oleic acid and ethyl-cellulose and EE and DLC are mentioned in Table 2; Figure 3. These results show that addition and then increasing the concentration of oleic acid and ethyl-cellulose increases the EE and DLC significantly.
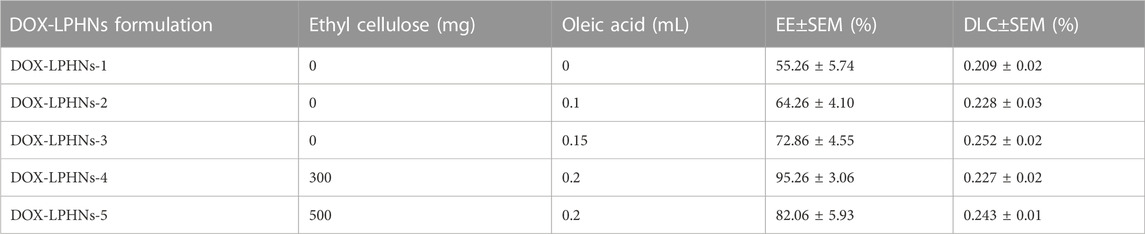
TABLE 2. EE and DLC of different DOX-LPHNs (1–6) formulations with different concentrations.
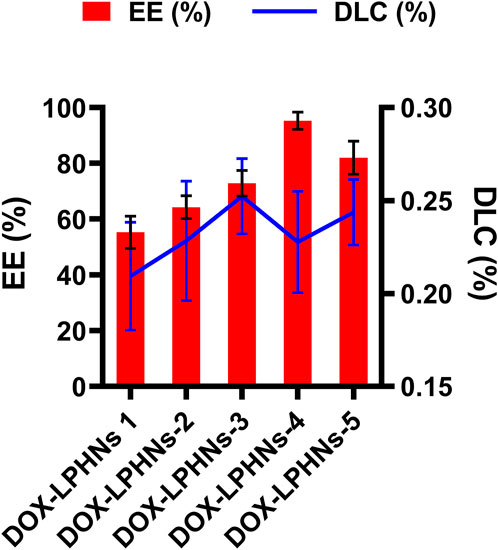
FIGURE 3. Encapsulation efficiency % (EE) and Drug Loading Contents % (DLC) of doxorubicin loaded lipid polymeric hybrid nanoparticle formulations.
The optimized formulation of DOX-LPHNs-4 showed 95.26% ± 3.06% for EE and 0.227% ± 0.02% for DLC. Figure 4 shows the 3D model of EE and DLC of DOX-LPHNs.
FIGURE 4. 3D Model surface graph for entrapment efficiency (EE).
The combination and specified concentrations of DOX, stearic acid, and polymer were found effective to demonstrate maximum encapsulation of the drug. It has been reported in literature that in polymer and lipid based nano-particulate drug delivery systems, high binding energy of the drugs with the polymers and lipids is required for the successful encapsulation of drugs in polymers as well as lipid layers (Liu et al., 2010). In the reported work, maximum entrapment efficacy and drug loading capacity can be credited to the higher binding energy of the drugs with stearic acid (Liu et al., 2010).
3.3 Infrared spectroscopy (drug-excipient’s interaction)
Figure 5 shows the compatibility of DOX with the formulation components. The peak of –OH in the spectra of DOX-LPHNs has a minor shift to the lower-band and spread to a value of 3,310 cm−1. The distinctive peaks at 1,077 cm−1, 1,448 cm−1, 1723 cm−1, and 2,918 cm−1 are allocated to carbonyl groups, ketone, and quinone, respectively. The stretching bands of the C–H groups are indicated in peak at 2,918 cm−1. The stretching bands of the C=O group(s) are indicated in peak at 1723 cm−1. C–C groups stretching bands are at peak of 1,410 cm−1. The peak at 1,071 cm−1 indicates the stretching bands of the C=O group(s). The minor peak at different places, i.e., 707 cm−1, 854 cm−1, and 980 cm−1 are the stretching bands of the C–O–C groups.

FIGURE 5. Fourier transform infrared (FT-IR) spectra of (A) DOX (Pure) (B) DOX-LPHNs.
This clearly indicated that the unprocessed samples and their respective prepared loaded LPHNs have similar chemical structure. Thus, no interaction of DOX and excipients was proved by FTIR spectra of unprocessed drugs and processed nanoformulations. This analysis exposed that the formation of a new complex has not been observed among the formulation components, which confirm the compatibility of the drugs with the formulation components. Thus, on the basis of FT-IR analysis, representing no chemical interactions, the prepared loaded nanoparticles can be further processed to achieve the desired boosted oral bioavailability results.
3.4 Surface morphology
The surface morphology of DOX-LPHNs was determined by scanning electron microscopy. White patches in micrograph showed solid, identical and fairly spherical shaped nanoparticles with a well-defined periphery (Figure 6). Most of the LPHNs were present in dispersed form with homogeneous distribution which exhibit amorphous nature of the produced nanoparticles. There were some masses of particles which were due to agglomeration. SEM representing nanometric size particles confirmed the results of zeta sizer analysis. Furthermore, the blunt and non-spiky white patches in the micrographs revealed amorphous nature nanoparticles, which plays a vital role in the solubility enhancement of the drugs being a successful outcome of pharmaceutical nanoengineering.

FIGURE 6. SEM micrograph of DOX-LPHNs.
3.5 X-ray diffractometry
The crystallinity of DOX-LPHNs formulations were determined by powder X-ray diffraction. As shown in Figure 7, the pure DOX had sharp peaks which indicate its crystalline nature while DOX- LPHNs-4 had some diffused peaks which suggests change or decrease in crystallinity of DOX in.
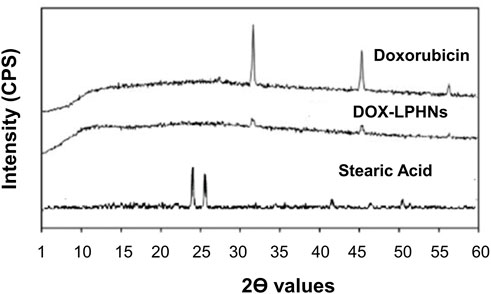
FIGURE 7. Powder X-RAY Diffraction of DOX, DOX-LPHNs-4, and Stearic acid.
LPHNs formation. Disappearance and reduction in intensities of the peaks in the diffractograms of DOX-LPHNs-4 nanoformulations is indicative for reduction in the crystalline nature (Ali et al., 2011; Khan et al., 2013). Reduction in the crystalline nature to semi-crystalline form or conversion to amorphous form favors increased solubility which in-turn boosted the oral bioavailability (Dang et al., 2009). Semi-crystalline and amorphous drugs have greater free energy compared to crystalline form, so, easily solubilized favoring enhanced oral bioavailability (Müller and Junghanns, 2006; Murdande et al., 2010a; Murdande et al., 2010b; Kakran et al., 2010). Thus, modification in the crystalline nature via nano-sizing approach being confirmed by P-XRD studies is highly appreciated and reported in literature (Kakran et al., 2010).
3.6 Differential scanning calorimetry
Differential scanning calorimetry was carried out to determine the melting points which further indicates the changes in crystallinity of DOX. DSC study was accomplished for pure DOX, DOX- LPHNs-4, oleic acid, stearic acid and ethyl-cellulose (Figure 8). The mentioned results indicating reduction in particles size, increased surface area as well as closed contact of solid lipid (stearic acid) and polymer with the drug. This change could be considered as a proof for the reduction in the crystallinity of nanoformulations. The mentioned results also showed the dispersion of the drugs in lipid and polymer layers as the level of melting point lowered along with fading of the peaks along with other formulation components. In the literature of LPHNs, the shifting of the melting point peak of drugs to the decreased level has been previously reported (Fang et al., 2008; Farboud et al., 2011).
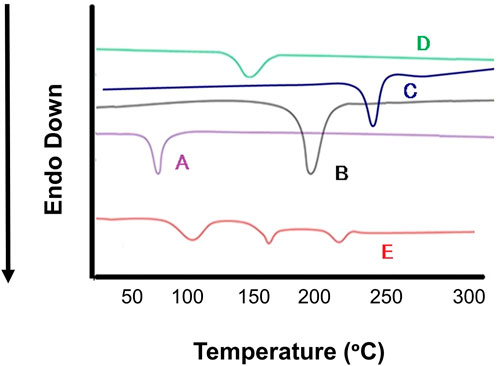
FIGURE 8. Differential scanning calorimetry (DSC) thermogram of (A) Stearic Acid (B) Doxorubicin, (C) Ethyl Cellulosecellulose, (D) Oleic Acid (E) DOX-LPHNS-4.
3.7 Stability study
The physical stability of the prepared DOX-LPHNs formulations were assessed both at refrigerated and room temperatures. The Figure 9 shows change in size and PDI of the DOX-LPHNs formulations that were kept for three months, which proposes a long lasting stability of the DOX- LPHNS formulations. At 25°C ± 3.00°C, some rapid growth might be observed for the initial 30 days which may be because of the amorphous nature of the drug loaded LPHNs followed by stabilization for rest of the period. This might be attributed to the dissolution of the small particles while depositing onto the surface of the large particles which is common in amorphous particles (Ali et al., 2011; Khan et al., 2013). Additionally, at room temperature, amorphous solids have increased free energy due to which chemical and physical stability is decreased (Hancock and Zografi, 1997; Khawam and Flanagan, 2006).
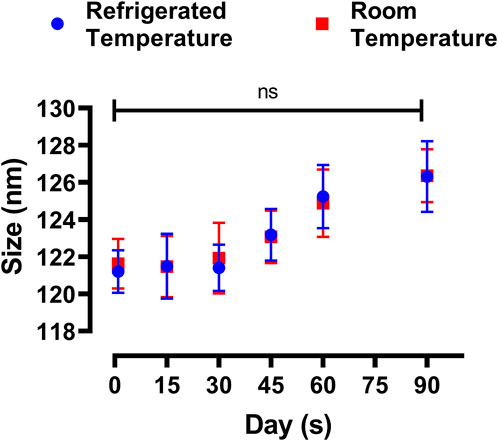
FIGURE 9. Change in particle size of DOX-LPHNs-4 formulation (DOX-4).
3.8 In-vitro release of the drug
The release of DOX from the DOX-LPHNs formulations (DOX-1TO DOX-5) was studied (Zur Mühlen and Mehnert, 1998). It was observed that all nano-formulations (DOX-LPHNs) showed good in vitro drug release profile. Initially, burst drug release was observed but later a gradual drug release was observed as shown in Figures 10, 11. This clearly indicated that when drug pay-load increased, cumulative percent drug release decreased and vice versa. Thus, it is concluded that increased payload of drugs resulted in prolonged drug release time (Rehman et al., 2015).
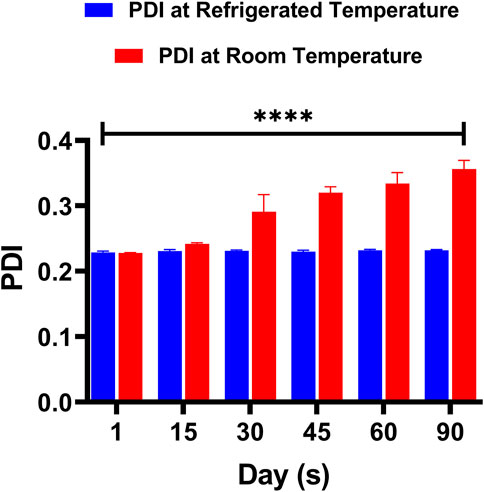
FIGURE 10. Change in the PDI of DOX-LPHNS-4 formulation (DOX-4).
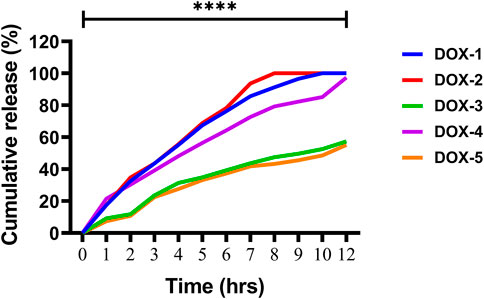
FIGURE 11. In-vitro drug release of the DOX nano-formulations.
3.9 Pharmacokinetic modeling and in vivo evaluation
The rate and mechanism of DOX release from DOX-LPHNs was studied by putting the release data in different kinetic models. It was observed that the Korsmeyar-Peppas model offered DOX release in the best way. This model showed that the release exponent (n) was more 0.5 which confirmed anomalous transport (Non-Fickian diffusion kinetics) (Barzegar-Jalali et al., 2008; Sadiq and Abdul Rassol, 2014).The in vitro drug release rate from LPHNs can be modified on the choice of appropriate surfactant, fabrication variables, polymer concentration and lipid form. A vigorous sustained drug release rate from the polymer hybrid drug delivery system can be provided by the helper polymer and lipid with the optimized concentrations. A stable drug polymer complex lead by compacted interactions between the polymer and drug molecules. Moreover, it leads to a higher sustained drug release profile in contrast with the looser interactions (Ullah et al., 2018). To probe the mechanism of drug release from the hybrid system, various kinetic models were used. It was elucidated that the drug release mechanism from LPHNs has been transformed to anomalous transport (Non-Fickian diffusion kinetics) from diffusion controlled. Dissolution erosion and diffusion is controlling the release of drugs from LPHNs in non-Fickian diffusion kinetics.
The in vivo pharmacokinetic parameters of DOX-LPHNs and marketed DOX, i.e., Cmax, Tmax AUC, and t1/2 are present in Tables 3, 4, while the Figure 12 shows comparative in-vivo release of drug from DOX-LPHNs and marketed DOX. The plot shows the plasma concentration vs. time curve. The data obtained from this study as compared to DOX-treated rabbits at the respective time-period are shown here as mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.00). The data was statistically significant after two-way analysis and post-hoc Bonferroni’s analysis. DOX loaded LPHNs at a dose of 20 mg/kg body weight showed higher Cmax (3.333 μg/mL) as compared to the marketed drug (1.65 μg/mL). Similarly, the tmax for DOX loaded LPHNs was observed as 0.31 h while for marketed DOX as 0.634 h. Similarly, the t1/2 for marketed drug was 9.14 h and for DOX loaded LPHNs was 26.07 h. The area under concentration-time curve from time zero to 24 h for DOX loaded LPHNs was 33.23 μg h/mL while for marketed DOX was 17.20 μg h/mL. Optimized DOX loaded LPHNs showed considerable variations in the pharmacokinetics of DOX. A notable rise in the peak plasma concentration (Cmax) and elimination half-life (t1/2) with a significant drop-in time required for peak plasma concentration in comparison with marketed DOX. Correspondingly, the DOX loaded LPHNs (DOX-4) also showed enhancement bioavailability of DOX. Area under concentration time curve (AUC) for the marketed doxorubicin decreased by 50% as compared to DOX LPHNs (DOX-4) in the bloodstream after oral administration (Zhang et al., 2012).
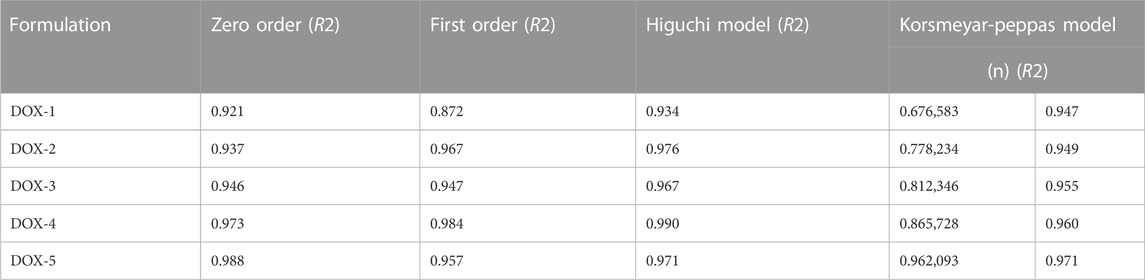
TABLE 3. Pharmacokinetic modelling of the DOX-LPHNS formulations.
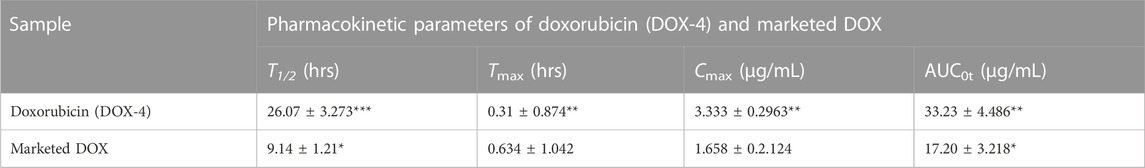
TABLE 4. Pharmacokinetic parameters of Doxorubicin (DOX-4) and marketed DOX.
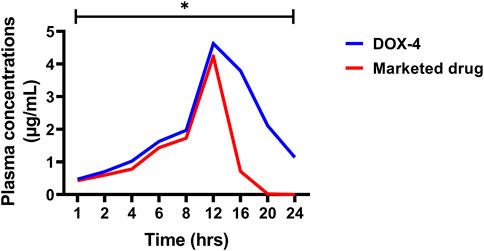
FIGURE 12. In-vivo pharmacokinetic profile of doxorubicin (DOX-4) and marketed DOX.
3.10 Computational analysis
We performed density functional theory (DFT) simulations to gain deeper insights in the interaction mechanism of the drug molecule with the polymers. Before simulating the drug interaction mechanism, initially, we optimized the geometries of the monomers (drug and polymers) system, to understand the reactive sites in the monomers system, which will interact during the reaction process. The optimized geometries of the monomers are illustrated in Figure 13. The structure of doxorubicin drug, Eudragit RS-100 (polymer), stearic Acid (solid lipid), oleic acid (liquid lipid), and ethyl cellulose (Hepler polymer) are represented by DD, ERS-100, SA, OA, and EC, respectively. The white, grey, blue, and red balls in the structures show the hydrogen, carbon, nitrogen, and oxygen atoms, respectively. The values of Mullikan atomic charges extracted from the geometries of monomers are listed in Table 5. The Mullikan atomic charges for nitrogen (−0.61 e) and oxygen atoms (−0.46 to −0.51 e) of both DD and polymers are highly negative while the values of hydrogen atoms as strongly positive. This shows that the O and N atoms are strong nucleophilic sites, and the hydrogen atoms are the electrophilic sites. Thus, we made different complexes, where the nucleophilic and electrophilic sites of DD and polymers are directed to each other and full geometry relaxation were performed. Thus, on the basis of the atomic charges, we interacted the DD with the EC unit of polymer via C=O and OH sites with OH site of EC represented in Figure 14A. The values of intermolecular bond distances, adsorption energies and charge transfer are given in Table 5. The geometry relaxation evinces that the polymer (EC) makes between two intermolecular hydrogen bonds with the DD via its OH group, i.e., OH---OH with bond distance of 1.89 Å and OH---OC with bond distance of 1.85 Å, respectively. The binding energy (Eb) value obtained for this complex was −12.67 kcal/mol. The charge transfer analysis showed that −0.021 e is transferred to the O atom from H in OH---OH bond and −0.044 e in OH---OC bond. While in complex-2 (Figure 14B, the ERS-100 polymer interacted with the DD through the OH site. In this complex, only one intermolecular hydrogen-bond (H-bond) with the OH group of the DD formed. The bond length value calculated for OH---OH H-bond was 1.92 Å and the Eb value obtained was −9.83 kcal/mol, while −0.043 e is transferred from the O atom to the H atom after complexation. Moreover, in complex-3 (Figure 14C, the COOH group OA is directed towards the OH and CO sites of DD. The complex-3 shows two H-bonds with the COOH group of the OA, i.e., OH---OC having bond length of 1.80 Å, on the other hand the bond length of CO----HO is 1.81 Å which is almost similar in strength with the OH---OC bond. The shorted H-bond formation depicts the stronger electrostatic interaction between the OA polymer and DD. This leads to the larger Eb value of −18.53 kcal/mol, which is larger compared to the other complexes. Due to stronger electrostatic interaction, the charge transfer observed between the H and O atom was very large (−0.059 and −0.057 e). Similarly, in complex-4, the COOH group interacted with the C=O and O—H sites of DD. The geometry relaxation evinces the formation of two stronger H-bond formation with binding distance of 1.88 Å and 1.93 Å, which results in Eb value of −17.32 kcal/mol while the charge transferred value noticed was −0.052 and −0.049 e. Thus, the DFT simulations showed that the polymers molecules have greater affinity to bind with the DD molecule and the binding distances, binding energies and charge transferred results demonstrated that the synthesis of DOX-LPHNs is strongly favorable.
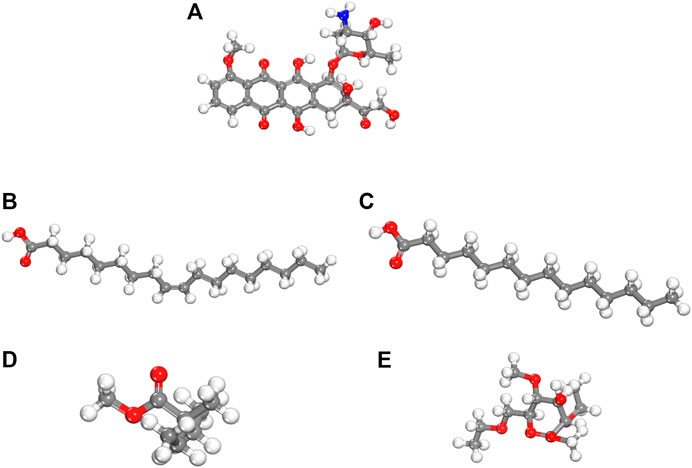
FIGURE 13. dft optimized geometry of (A) dd molecule (B) oa (C) sa (D) ec and (E) ers-100. White, grey, and red balls represent hydrogen, carbon, and oxygen atoms, respectively.
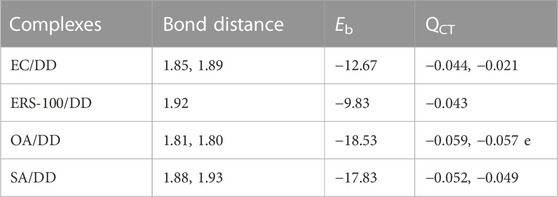
TABLE 5. Bond distances (Å), binding energy (Eb kcal/mol) and charge transferred (QCT e) for the different complexes of DD with polymers obtained though DFT simulations.
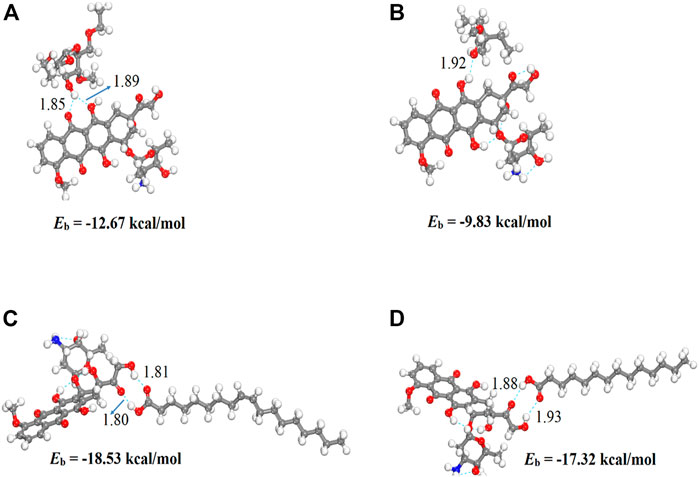
FIGURE 14. Optimized structures of DD complexes with polymers unit (A) EC/DD (B) ERS-100/DD (C) OA/DD AND (D) SA/DD. Bond distances are in Å.
3.11 Toxicity study
No behavioral change was observed in the first 2 h after administration of DOX-loaded LPHNs. Similarly, post 24 h of DOX-loaded LPHNs administration (50 mg/kg to 400 mg/kg), no death was noted. When the dose of the drug was increase from 400 mg/kg to 800 mg/kg, 16% death (mortality) rate was observed. In the same pattern, when experimental dose was increased to 1,600 mg/kg, the mortality rate almost doubled to 32.6%. This study revealed that acute toxicity or LD50 for DOX loaded LPHNs is more than 1,600 mg/kg (Table 6).
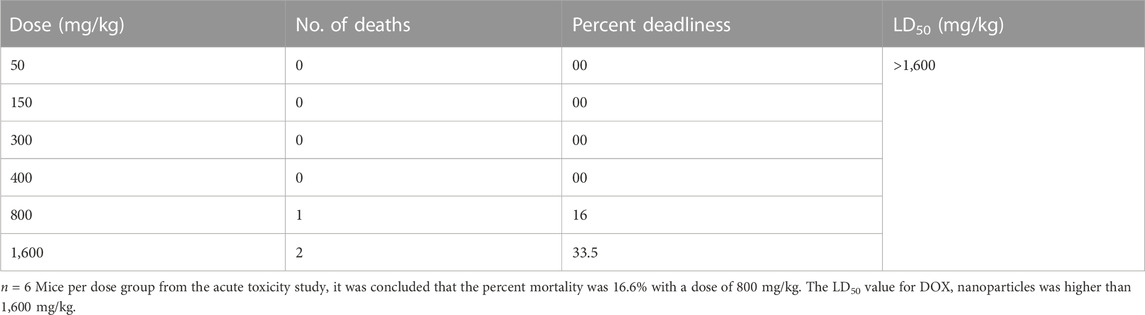
TABLE 6. Acute toxicity test of DOX nanoparticles.
4 Conclusion
It is proved that DOX loaded in lipid polymer hybrid nanoparticles (LPHNs) is a good nanomedicine having the desired value−added characteristics. Similarly, it has been shown that DOX and excipients have an excellent interaction as well affinity. LPHNs were fabricated via combinative approach of magnetic stirring and sonication. No sophisticated apparatus was used during the fabrication procedure. Surfactant and co-surfactant were used during LPHNs fabrication to stabilize the developed formulation. They were more stable at cold temperature (5°C ± 3.00°C). The developed formulation was easy, simple, and reproducible with the potential to easily scale up for large scale production. In vitro and in vivo studies confirmed sustained drug release behavior and improved bioavailability. The DFT calculations demonstrated that the polymers have stronger affinity towards the DD molecule. The polymers interacted with different sites of the DD through stronger electrostatic intermolecular interactions with shorter bond distances. In addition, the larger negative binding energies (−9.83 to −18.53 kcal/mol) showed that the interaction mechanism is spontaneous, and the polymers have greater affinity to stably deliver the DD molecule. Thus, it is concluded that we successfully prepared LPHNs loaded with DOX showing sustained release.Thus, it can be concluded that an attempt can be made to produce DOX loaded in lipid polymer hybrid nanoparticles (LPHNs) which could potentially be converted into a suitable solid dosage form followed by its comparative in vitro and in vivo assessments.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material further inquiries can be directed to the corresponding author.
Ethics statement
The Animals used in current study were under the Institutional Animal Care and Use Committee. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
MS: manuscript compilation and experimental designing. MR, ZK, RA, and SA: graphical designing and statistical analysis. AA and MB: in vivo model studies. FS, AS, and AM: DFTs and PK studies. MH: references and manuscript formatting. SA: manuscript revision.
Acknowledgments
The authors would like to thank the Deanship of Scientific Research at Shaqra University for supporting this work. We would like to thank Editage (www.editage.com) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1025013/full#supplementary-material
References
Abdelwahed, W., Degobert, G., Stainmesse, S., and Fessi, H. (2006). Freeze-drying of nanoparticles: Formulation, process and storage considerations. Adv. drug Deliv. Rev. 58 (15), 1688–1713. doi:10.1016/j.addr.2006.09.017
Abraham, S. A., Waterhouse, D. N., Mayer, L. D., Cullis, P. R., Madden, T. D., and Bally, M. B. (2005). The liposomal formulation of doxorubicin. Methods Enzym. 391, 71–97. doi:10.1016/S0076-6879(05)91004-5
Ahmad, N., Ahmad, R., Alam, M. A., and Ahmad, F. J. (2018). Enhancement of oral bioavailability of doxorubicin through surface modified biodegradable polymeric nanoparticles. Chem. Central J. 12 (1), 65–14. doi:10.1186/s13065-018-0434-1
Ali, H. S., York, P., Ali, A. M. A., and Blagden, N. (2011). Hydrocortisone nanosuspensions for ophthalmic delivery: A comparative study between microfluidic nanoprecipitation and wet milling. J. Control. release 149 (2), 175–181. doi:10.1016/j.jconrel.2010.10.007
Baharifar, H., Tavoosidana, G., Karimi, R., Bidgoli, S. A., Ghanbari, H., Faramarzi, M. A., et al. (2015). Optimization of self-assembled chitosan/streptokinase nanoparticles and evaluation of their cytotoxicity and thrombolytic activity. J. Nanosci. Nanotechnol. 15 (12), 10127–10133. doi:10.1166/jnn.2015.11696
Barzegar-Jalali, M., Adibkia, K., Valizadeh, H., Shadbad, M. R. S., Nokhodchi, A., Omidi, Y., et al. (2008). Kinetic analysis of drug release from nanoparticles. J. Pharm. Pharm. Sci. 11 (1), 167–177. doi:10.18433/j3d59t
Bhandari, R., and Kaur, I. P. (2013). Pharmacokinetics, tissue distribution and relative bioavailability of isoniazid-solid lipid nanoparticles. Int. J. Pharm. 441 (1), 202–212. doi:10.1016/j.ijpharm.2012.11.042
Bhardwaj, U., and Burgess, D. J. (2010). A novel USP apparatus 4 based release testing method for dispersed systems. Int. J. Pharm. 388 (1-2), 287–294. doi:10.1016/j.ijpharm.2010.01.009
Bhardwaj, V., Ankola, D. D., Gupta, S. C., Schneider, M., Lehr, C. M., and Kumar, M. N. V. R. (2009). PLGA nanoparticles stabilized with cationic surfactant: Safety studies and application in oral delivery of paclitaxel to treat chemical-induced breast cancer in rat. Pharm. Res. 26 (11), 2495–2503. doi:10.1007/s11095-009-9965-4
Chan, S., Friedrichs, K., Noel, D., Pinter, T., Van Belle, S., Vorobiof, D., et al. (1999). Prospective randomized trial of docetaxel versus doxorubicin in patients with metastatic breast cancer. J. Clin. Oncol. 17 (8), 2341–2354. doi:10.1200/JCO.1999.17.8.2341
Dang, L., Yang, H., Black, S., and Wei, H. (2009). The effect of temperature and solvent composition on transformation of β-to α-Glycine as monitored in situ by FBRM and PVM. Org. Process Res. Dev. 13 (6), 1301–1306. doi:10.1021/op900134w
del Pozo-Rodríguez, A., del Pozo-Rodriguez, A., Gascon, A. R., and Pedraz, J. L. (2009). Short-and long-term stability study of lyophilized solid lipid nanoparticles for gene therapy. Eur. J. Pharm. Biopharm. 71 (2), 181–189. doi:10.1016/j.ejpb.2008.09.015
Du, C., Deng, D., Shan, L., Wan, S., Cao, J., Tian, J., et al. (2013). A pH-sensitive doxorubicin prodrug based on folate-conjugated BSA for tumor-targeted drug delivery. Biomaterials 34 (12), 3087–3097. doi:10.1016/j.biomaterials.2013.01.041
Dubes, A., Parrot-Lopez, H., Abdelwahed, W., Degobert, G., Fessi, H., Shahgaldian, P., et al. (2003). Scanning electron microscopy and atomic force microscopy imaging of solid lipid nanoparticles derived from amphiphilic cyclodextrins. Eur. J. Pharm. Biopharm. 55 (3), 279–282. doi:10.1016/s0939-6411(03)00020-1
Duggan, S. T., and Keating, G. M. (2011). Pegylated liposomal doxorubicin: A review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related kaposi's sarcoma. Drugs 71 (18), 2531–2558. doi:10.2165/11207510-000000000-00000
Fang, J.-Y., Fang, C. L., Liu, C. H., and Su, Y. H. (2008). Lipid nanoparticles as vehicles for topical psoralen delivery: Solid lipid nanoparticles (SLN) versus nanostructured lipid carriers (NLC). Eur. J. Pharm. Biopharm. 70 (2), 633–640. doi:10.1016/j.ejpb.2008.05.008
Farboud, E. S., Nasrollahi, S. A., and Tabbakhi, Z. (2011). Novel formulation and evaluation of a Q10-loaded solid lipid nanoparticle cream: In vitro and in vivo studies. Int. J. nanomedicine 6, 611–617. doi:10.2147/IJN.S16815
Fatma, S., Talegaonkar, S., Iqbal, Z., Panda, A. K., Negi, L. M., Goswami, D. G., et al. (2016). Novel flavonoid-based biodegradable nanoparticles for effective oral delivery of etoposide by P-glycoprotein modulation: An in vitro, ex vivo and in vivo investigations. Drug Deliv. 23 (2), 500–511. doi:10.3109/10717544.2014.923956
Ghanshyam, U., Patel, P., and Patel, J. (2011). Formulation and characterization of solid lipid nanoparticles dry powder inhaler containing triamcinolone acetonide. Int. J. Res. Pharm. Chem. 1 (3), 662–673.
Gold, H. S., and Moellering, R. C. (1996). Antimicrobial-drug resistance. N. Engl. J. Med. 335 (19), 1445–1453. doi:10.1056/NEJM199611073351907
Gou, M., Shi, H., Guo, G., Men, K., Zhang, J., Zheng, L., et al. (2011). Improving anticancer activity and reducing systemic toxicity of doxorubicin by self-assembled polymeric micelles. Nanotechnology 22 (9), 095102. doi:10.1088/0957-4484/22/9/095102
Hancock, B. C., and Zografi, G. (1997). Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 86 (1), 1–12. doi:10.1021/js9601896
Hou, D., Xie, C., Huang, K., and Zhu, C. (2003). The production and characteristics of solid lipid nanoparticles (SLNs). Biomaterials 24 (10), 1781–1785. doi:10.1016/s0142-9612(02)00578-1
Huang, Y., Leobandung, W., Foss, A., and Peppas, N. A. (2000). Molecular aspects of muco-and bioadhesion:: Tethered structures and site-specific surfaces. J. Control. release 65 (1-2), 63–71. doi:10.1016/s0168-3659(99)00233-3
Huh, A. J., and Kwon, Y. J. (2011). Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control. Release 156 (2), 128–145. doi:10.1016/j.jconrel.2011.07.002
Jain, S., Patil, S. R., Swarnakar, N. K., and Agrawal, A. K. (2012). Oral delivery of doxorubicin using novel polyelectrolyte-stabilized liposomes (layersomes). Mol. Pharm. 9 (9), 2626–2635. doi:10.1021/mp300202c
Joshi, G., Kumar, A., and Sawant, K. (2014). Enhanced bioavailability and intestinal uptake of Gemcitabine HCl loaded PLGA nanoparticles after oral delivery. Eur. J. Pharm. Sci. 60, 80–89. doi:10.1016/j.ejps.2014.04.014
Kakran, M., Sahoo, N. G., Li, L., Judeh, Z., Wang, Y., Chong, K., et al. (2010). Fabrication of drug nanoparticles by evaporative precipitation of nanosuspension. Int. J. Pharm. 383 (1), 285–292. doi:10.1016/j.ijpharm.2009.09.030
Ke, W., Zhao, Y., Huang, R., Jiang, C., and Pei, Y. (2008). Enhanced oral bioavailability of doxorubicin in a dendrimer drug delivery system. J. Pharm. Sci. 97 (6), 2208–2216. doi:10.1002/jps.21155
Khan, S., Matas, M. d., Zhang, J., and Anwar, J. (2013). Nanocrystal preparation: Low-energy precipitation method revisited. Cryst. Growth and Des. 13 (7), 2766–2777. doi:10.1021/cg4000473
Khawam, A., and Flanagan, D. R. (2006). Basics and applications of solid-state kinetics: A pharmaceutical perspective. J. Pharm. Sci. 95 (3), 472–498. doi:10.1002/jps.20559
Kovacevic, A., Vuleta, G., Muller, R. H., and Keck, C. M. (2011). Polyhydroxy surfactants for the formulation of lipid nanoparticles (SLN and NLC): Effects on size, physical stability and particle matrix structure. Int. J. Pharm. 406 (1), 163–172. doi:10.1016/j.ijpharm.2010.12.036
Lage, H. (2003). ABC-Transporters: Implications on drug resistance from microorganisms to human cancers. Int. J. Antimicrob. agents 22 (3), 188–199. doi:10.1016/s0924-8579(03)00203-6
Liu, J., Hu, W., Chen, H., Ni, Q., Xu, H., and Yang, X. (2007). Isotretinoin-loaded solid lipid nanoparticles with skin targeting for topical delivery. Int. J. Pharm. 328 (2), 191–195. doi:10.1016/j.ijpharm.2006.08.007
Liu, Y., Pan, J., and Feng, S.-S. (2010). Nanoparticles of lipid monolayer shell and biodegradable polymer core for controlled release of paclitaxel: Effects of surfactants on particles size, characteristics and in vitro performance. Int. J. Pharm. 395 (1), 243–250. doi:10.1016/j.ijpharm.2010.05.008
Moffat, A. C., Osselton, M. D., Widdop, B., and Watts, J. (2011). Clarke's analysis of drugs and poisons: in pharmaceuticals, body fluids and postmortem material s, Vol. 3. London: Pharmaceutical Press.
Müller, R. H., and Junghanns, J. (2006). Drug nanocrystals/nanosuspensions for the delivery of poorly soluble drugs. Nanoparticulates as drug carriers 1, 307–328.
Murdande, S. B., Pikal, M. J., Shanker, R. M., and Bogner, R. H. (2010). Solubility advantage of amorphous pharmaceuticals: I. A thermodynamic analysis. J. Pharm. Sci. 99 (3), 1254–1264. doi:10.1002/jps.21903
Murdande, S. B., Pikal, M. J., Shanker, R. M., and Bogner, R. H. (2010). Solubility advantage of amorphous pharmaceuticals: II. Application of quantitative thermodynamic relationships for prediction of solubility enhancement in structurally diverse insoluble pharmaceuticals. Pharm. Res. 27 (12), 2704–2714. doi:10.1007/s11095-010-0269-5
Jonsson, M., Jestoi, M., Nathanail, A. V., Kokkonen, U. M., Anttila, M., and Koivisto, P. (2013). Application of OECD Guideline 423 in assessing the acute oral toxicity of moniliformin. Food Chem. Toxicol. 53, 27–32.
Peltier, S., Oger, J. M., Lagarce, F., Couet, W., and Benoit, J. P. (2006). Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharm. Res. 23 (6), 1243–1250. doi:10.1007/s11095-006-0022-2
Racault, C., Langlais, F., and Naslain, R. (1994). Solid-state synthesis and characterization of the ternary phase Ti3SiC2. J. Mater. Sci. 29 (13), 3384–3392. doi:10.1007/bf00352037
Rehman, M., Madni, A., Ihsan, A., Khan, W. S., Khan, M. I., Mahmood, M. A., et al. (2015). Solid and liquid lipid-based binary solid lipid nanoparticles of diacerein: In vitro evaluation of sustained release, simultaneous loading of gold nanoparticles, and potential thermoresponsive behavior. Int. J. nanomedicine 10, 2805–2814. doi:10.2147/IJN.S67147
Roohullah, Z. I., Iqbal, F., Nasir, M., Akhlaq, A., Sadozai, S K., Zada, A., et al. (2013). Sustained release carbamezapine matrix tablets prepared by solvent-evaporation technique using different polymers. Middle-East J. Sci. Res. 15 (10), 1368–1374. doi:10.5829/idosi.mejsr.2013.15.10.7553
Sadiq, A. A., and Abdul Rassol, A. (2014). Formulation and evaluation of silibinin loaded solid lipid nanoparticles for peroral use targeting lower part of gastrointestinal tract. Int. J. Pharm. Pharm. Sci. 6 (1), 55–67.
Suh, W. H., Suslick, K. S., Stucky, G. D., and Suh, Y. H. (2009). Nanotechnology, nanotoxicology, and neuroscience. Prog. Neurobiol. 87 (3), 133–170. doi:10.1016/j.pneurobio.2008.09.009
Taninaka, C., Ohtani, H., Hanada, E., Kotaki, H., Sato, H., and Iga, T. (2000). Determination of erythromycin, clarithromycin, roxithromycin, and azithromycin in plasma by high-performance liquid chromatography with amperometric detection. J. Chromatogr. B Biomed. Sci. Appl. 738 (2), 405–411. doi:10.1016/s0378-4347(99)00512-5
Tiţa, B., Fulias, A., Bandur, G., Marian, E., and Tita, D. (2011). Compatibility study between ketoprofen and pharmaceutical excipients used in solid dosage forms. J. Pharm. Biomed. analysis 56 (2), 221–227. doi:10.1016/j.jpba.2011.05.017
Ullah, N., Khan, S., Ahmed, S., Govender, T., Faidah, H. S., de Matas, M., et al. (2018). Dexibuprofen nanocrystals with improved therapeutic performance: Fabrication, characterization, in silico modeling, and in vivo evaluation. Int. J. nanomedicine 13, 1677–1692. doi:10.2147/IJN.S151597
Uprit, S., Kumar Sahu, R., Roy, A., and Pare, A. (2013). Preparation and characterization of minoxidil loaded nanostructured lipid carrier gel for effective treatment of alopecia. Saudi Pharm. J. 21 (4), 379–385. doi:10.1016/j.jsps.2012.11.005
Walsh, C. (2000). Molecular mechanisms that confer antibacterial drug resistance. Nature 406 (6797), 775–781. doi:10.1038/35021219
Yang, S., Zhu, J., Lu, Y., Liang, B., and Yang, C. (1999). Body distribution of camptothecin solid lipid nanoparticles after oral administration. Pharm. Res. 16 (5), 751–757. doi:10.1023/a:1018888927852
Yao, H.-J., Ju, R. J., Wang, X. X., Zhang, Y., Li, R. J., Yu, Y., et al. (2011). The antitumor efficacy of functional paclitaxel nanomicelles in treating resistant breast cancers by oral delivery. Biomaterials 32 (12), 3285–3302. doi:10.1016/j.biomaterials.2011.01.038
Yoncheva, K., Guembe, L., Campanero, M. A., and Irache, J. M. (2007). Evaluation of bioadhesive potential and intestinal transport of pegylated poly (anhydride) nanoparticles. Int. J. Pharm. 334 (1-2), 156–165. doi:10.1016/j.ijpharm.2006.10.016
Zhang, J., Tao, W., Chen, Y., Chang, D., Wang, T., Zhang, X., et al. (2015). Doxorubicin-loaded star-shaped copolymer PLGA-vitamin E TPGS nanoparticles for lung cancer therapy. J. Mater. Sci. Mater. Med. 26 (4), 165. doi:10.1007/s10856-015-5498-z
Zhang, L., Chan, J. M., Gu, F. X., Rhee, J. W., Wang, A. Z., Radovic-Moreno, A. F., et al. (2008). Self-assembled lipid− polymer hybrid nanoparticles: A robust drug delivery platform. ACS nano 2 (8), 1696–1702. doi:10.1021/nn800275r
Zhang, Z., Gao, F., Bu, H., Xiao, J., and Li, Y. (2012). Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: In vitro characteristics and absorption mechanism in rats. Nanomedicine Nanotechnol. Biol. Med. 8 (5), 740–747. doi:10.1016/j.nano.2011.08.016
Zhou, L., Cheng, R., Tao, H., Ma, S., Guo, W., Meng, F., et al. (2011). Endosomal pH-activatable poly (ethylene oxide)-graft-doxorubicin prodrugs: Synthesis, drug release, and biodistribution in tumor-bearing mice. Biomacromolecules 12 (5), 1460–1467.
Keywords: bioavailability, doxorubicin, lipid polymer hybrid nanoparticles, in vitro, in vivo
Citation: Shafique M, Ur Rehman M, Kamal Z, Alzhrani RM, Alshehri S, Alamri AH, Bakkari MA, Sabei FY, Safhi AY, Mohammed AM, Hamd MAE and Almawash S (2023) Formulation development of lipid polymer hybrid nanoparticles of doxorubicin and its in-vitro, in-vivo and computational evaluation. Front. Pharmacol. 14:1025013. doi: 10.3389/fphar.2023.1025013
Received: 22 August 2022; Accepted: 23 January 2023;
Published: 07 February 2023.
Edited by:
Hussain Ali, Quaid-i-Azam University, PakistanReviewed by:
Dilpreet Singh, Indo-Soviet Friendship College of Pharmacy, IndiaJianmin Chen, Putian University, China
Muhammad Usman Munir, Jouf University, Saudi Arabia
Abid Hussain, Beijing Institute of Technology, China
Copyright © 2023 Shafique, Ur Rehman, Kamal, Alzhrani, Alshehri, Alamri, Bakkari, Sabei, Safhi, Mohammed, Hamd and Almawash. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saud Almawash, c2FsbWF3YXNoQHN1LmVkdS5zYQ==