 S. Herrera-Pérez
S. Herrera-Pérez L. Rueda-Ruzafa
L. Rueda-Ruzafa A. Campos-Ríos
A. Campos-Ríos D. Fernández-Fernández
D. Fernández-Fernández J.A. Lamas
J.A. Lamas
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 15 September 2022
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.997188
Background and Purpose: Verapamil, a drug widely used in certain cardiac pathologies, exert its therapeutic effect mainly through the blockade of cardiac L-type calcium channels. However, we also know that both voltage-dependent and certain potassium channels are blocked by verapamil. Because sympathetic neurons of the superior cervical ganglion (SCG) are known to express a good variety of potassium currents, and to finely tune cardiac activity, we speculated that the effect of verapamil on these SCG potassium channels could explain part of the therapeutic action of this drug. To address this question, we decided to study, the effects of verapamil on three different potassium currents observed in SCG neurons: delayed rectifier, A-type and TREK (a subfamily of K2P channels) currents. We also investigated the effect of verapamil on the electrical behavior of sympathetic SCG neurons.
Experimental Approach: We employed the Patch-Clamp technique to mouse SCG neurons in culture.
Key Results: We found that verapamil depolarizes of the resting membrane potential of SCG neurons. Moreover, we demonstrated that this drug also inhibits A-type potassium currents. Finally, and most importantly, we revealed that the current driven through TREK channels is also inhibited in the presence of verapamil.
Conclusion and Implications: We have shown that verapamil causes a clear alteration of excitability in sympathetic nerve cells. This fact undoubtedly leads to an alteration of the sympathetic-parasympathetic balance which may affect cardiac function. Therefore, we propose that these possible peripheral alterations in the autonomic system should be taken into consideration in the prescription of this drug.
Some drugs used in the treatment of heart diseases, like verapamil or nifedipine, have been developed and are used based on their interaction with the L-type voltage-gated calcium channel, expressed by cardiac and vascular smooth muscle cells. This is the case for class IV antiarrhythmic drugs used in pathological conditions such as chronic angina pectoris, cardiac arrhythmias or hypertension (Kato et al., 2004). In fact, the phenylalkylamine verapamil exerts its therapeutic action predominantly on cardiac cells, reducing heart contractility and rate (Cohen et al., 1987), by blocking L-type calcium channels (Keith et al., 1994; Bergson et al., 2011). Notwithstanding, verapamil has also been shown to exert a significant inhibition of T-type calcium channels expressed in both native and heterologous systems (Freeze et al., 2006; Bergson et al., 2011). Although attracting less attention, those calcium antagonists also inhibit several potassium channels. Concentrations in the low micromolar range inhibit delayed rectifier (IKDR; KV1.3, KV1.5) and ether a go-go (IKeag; KV11.1) potassium currents in both native and heterologous systems (Chouabe et al., 1998; Rauer and Grissmer, 1999; Baba et al., 2015; Diesch and Grissmer, 2017).
The effect of verapamil and analog drugs on potassium channels has also been verified in native heart cells. In fact, verapamil inhibits the acetylcholine-induced potassium current (IKAch) in atrial myocytes (Ito et al., 1989). However, and despite the importance that the voltage sensitive A-type potassium current (IA) has both in the heart (Liu et al., 2011; Devenyi et al., 2017; Grandi et al., 2017) and in the autonomic nervous system (ANS) (Lamas et al., 1997; Doan and Kunze, 1999), the potential effect of verapamil modulating these channels has not been investigated. TREK-1, TREK-2 and TRAAK constitute the TREK subfamily of K2P family. These channels (mainly TREK-2) are abundantly expressed in the superior cervical ganglion (SCG) neurons (Cadaveira-Mosquera et al., 2011) and also in atrial and ventricular cardiomyocytes (Limberg et al., 2011; Bond et al., 2014; Bodnar et al., 2015), where they function as major regulators of the resting membrane potential (RMP) (Cadaveira-Mosquera et al., 2011; Unudurthi et al., 2016; Grandi et al., 2017). Indeed, TREK channels have been related to various heart diseases (Schmidt et al., 2012; Wiedmann et al., 2016; Decher et al., 2017) and although it has been previously shown that verapamil blocks some members of the K2P family, including TRESK (Park et al., 2018) and TASK-4 (Staudacher et al., 2018), its effect on TREK channels remains unknown.
It is common to check the effect of substances that affect the functioning of the heart on non-neuronal cardiac cells, mainly cardiomyocytes and comparatively, less effort has been devoted to test the interaction of drugs with the neurons regulating heart working. However, we know that the neurons of the parasympathetic intracardiac ganglion (ICG) and sympathetic SCG innervate both muscle fibers and pacemakers. The SCG hosts a critical population of sympathetic neurons projecting to the cardiac tissue (Pather et al., 2003; De Gama et al., 2012), and several studies have suggested that the SCG might be directly implicated in the pathophysiology of different cardiovascular diseases (Hernandez-Ochoa et al., 2009; Kong et al., 2013; Na et al., 2014; Cheng et al., 2018).
In the present study, we have used cultured sympathetic neurons, isolated from the mouse SCG (mSCG), to study the effects that verapamil produces on their RMP and excitability. Using the Patch-Clamp technique we found that verapamil evokes a dose-dependent depolarization of the RMP, without affecting the number of action potentials (AP) fired upon application of depolarizing current pulses. We were also able to confirm the blockade that verapamil exerts on voltage-dependent potassium (IKv) currents IKDR and IA. Probably more important, we show for the first time that verapamil induces a robust inhibition of TREK channels activated by riluzole.
Swiss CD1 mice were obtained from the Biomedical Research Center (CINBIO) of the University of Vigo. Mice were housed under 12 h light/dark cycle in a pathogen-free area, with food and water freely available. All experiments were approved by the Spanish Research Council and the University of Vigo Scientific Committee, under Spanish and European directives for the protection of experimental animals (RD 05/03/2013; EU 06/03/2010).
The culture of mSCG neurons was performed as described previously (Romero et al., 2004). Mice (30–60 days of age) were terminally anesthetized with CO2 and immediately decapitated. The SCG were removed in cold Leibowitz medium (L-15) under a binocular microscope and, once cleaned, the ganglia were incubated in collagenase (2.5 mg/ml in HBSS) for 15 min at 37°C. The ganglia were then incubated for 30 min in trypsin (1 mg/ml). Finally, the neurons were mechanically isolated, centrifuged, and plated in 35 mm Petri dishes previously treated with laminin (10 μg/ml in EBSS). Neurons were cultured for 1 day at 37°C and 5% CO2 in L-15 medium containing the following: 24 mM NaHCO3, 10% fetal calf serum, 2 mM l-glutamine, 38 mM D-glucose, 100 UI/ml penicillin, 100 μg/ml streptomycin and 50 ng/ml nerve growth factor.
On the day of the experiment, cultured mSCG neurons were placed on an inverted microscope and continuously perfused by gravity (10 ml/min) with a standard solution at room temperature. Recordings were obtained using a HEKA (EPC 800) amplifier. Sampling frequency was 2 kHz (filtered at 0.5 kHz) for voltage-clamp and 10 kHz (filtered at 5 kHz) for current-clamp (Bridge mode) experiments. Patch pipette resistance varied from 4 to 6 MΩ. Data were digitized using a Digidata 1440A and analyzed offline using the software pClamp10 (Molecular Devices). Plotting and statistical analysis were performed with Origin (Pro) 8.5 (OriginLab Corporation, Northampton, MA, United States). Membrane conductance (G) was estimated in voltage-clamp experiments (holding potential = −30 mV) by the application of negative 15 mV brief voltage steps (50 ms) at 0.5 Hz. After measuring the current (I) obtained during these steps, G was calculated using the Ohm’s Law, where G = 1/R and R = 15 mV/I. Assuming that the Perforated whole-cell Patch-Clamp limits washout and drift problems to the maximum. We took 20 random points before and after treatment and compared the average conductances of each cell in the two conditions (control and drug-treated).
The intracellular pipette solution contained (in mM) 90 K-acetate, 20 KCl, 3 MgCl2, 1 CaCl2, 3 EGTA, and 40 HEPES (pH adjusted to 7.2 with NaOH), and the standard extracellular solution (standard solution) contained (in mM) 140 NaCl, 3 KCl, 1 MgCl2, 2 CaCl2, 10 D-glucose and 10 HEPES (pH adjusted to 7,2 with Tris (Tris (hydroxymethyl)-amino methane)). All solutions were kept between 290 and 300 mOsm.When specified, tetraethylammonium chloride (TEA, 15 mM), 4-aminopyridine (4-AP, 2 mM), cesium chloride (CsCl, 1 mM), cadmium chloride (CdCl2, 100 μM) and TTX (0.5 μM), were added to the extracellular solution in order to block voltage-dependent potassium, cationic, calcium and sodium currents. All these chemicals were purchased from Sigma-Aldrich. This combination of drugs constituted the standard cocktail solution (referred to as Cocktail A). In some experiments, this cocktail was supplemented with apamin (200 nM), paxilline (1 μM), 4-aminopyridine (4-AP, 2 mM) and clemizole (10 μM) (referred to as Cocktail B) in order to block, respectively, calcium dependent potassium channels (SK and BK), TRP channels and A-Type potassium channels. All these chemicals were purchased from Tocris Bioscience. Verapamil (Sigma-Aldrich) was dissolved in distilled water (10 nM stock solution), and maintained in darkness and cold. Riluzole (Tocris Bioscience) was made up in DMSO at a stock concentration of 10 mM. Final DMSO concentration ranged 0.1–0.2%, which has been tested not to affect cell physiology.
Data are represented as the mean ± SEM and statistical differences were assessed using a Paired Sample t-Test or a Two Sample t-Test. For the comparison of more than two groups the One-way ANOVA test has been applied. The differences among groups were considered significant when *p < 0.05, **p < 0.02 or ***p < 0.01. For the calculation of the TAU (63% of decay) value of the A-current decay, a mono-exponential curve fit based on the following equation for one term was applied using Clampfit 10.7 (Molecular Devices, United Kingdom):
Dose-response curves were fitted using the Hill equation:
where K corresponds to the EC50 and N is the Hill coefficient.
We studied the effect of verapamil on the RMP of mSCG neurons, these neurons showed a mean membrane potential of -64 ± 3 mV (n = 39) at rest. The acute application of different concentrations of verapamil (from 3 to 300 µM) induced a dose-dependent depolarization of the membrane potential (Figures 1A,B) a maximal depolarization of 13 mV was obtained at 300 µM. Data points were fitted using the Hill equation, with an extrapolated half-maximal (EC50) concentration of 50.19 µM and a Hill coefficient of 0.82. This Hill coefficient is consistent with a single binding site (Monod et al., 1965; Weiss, 1997). Regardless of the clear depolarization induced by verapamil on the RMP, the addition of 50 µM verapamil did not affect the number of action potentials of mSCG neurons (n = 14) in response to depolarizing current pulses (Figure 1C). Because verapamil induces a depolarization of the RPM, after verapamil was added we set the RMP at −60 mV (VHold = −60 mV) to confirm that the lack of effect of verapamil on the firing pattern was independent on the membrane potential before the current injections (Table 1; Figure 1D).
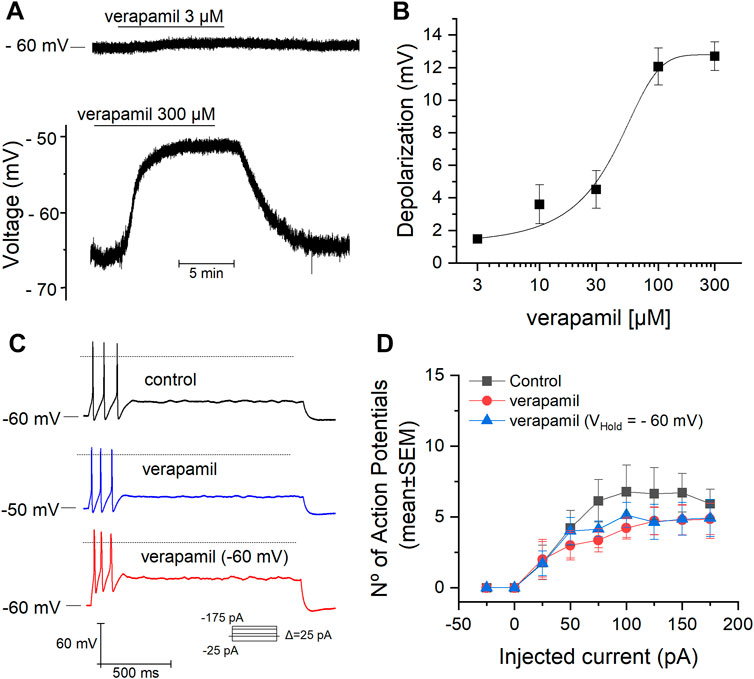
FIGURE 1. Effect of verapamil on the RMP and firing properties. (A) Membrane potential measured in CC conditions (gap-free mode). The addition of verapamil causes a clear depolarization when applied at 300 µM (bottom). (B) Dose-response curve for the verapamil-induced depolarization of the RMP. The depolarization induced by verapamil 3, 10, 30, 100 and 300 μM was of 1.5 ± 0.2 mV (n = 10), 3.5 ± 1 mV (n = 6), 4.5 ± 1 mV (n = 7), 12 ± 1 mV (n = 8) and 13 ± 1 mV (n = 8) respectively. (C,D) Number of action potentials (mean ± SEM) elicited in response to a protocol of increasing depolarizing currents pulses in the absence (black) and presence of verapamil (blue) and verapamil with the RMP hold at -60 mV (red). The dotted line represents the level at 0 mv.

TABLE 1. Number of action potentials elicited in response to depolarizing current steps in the presence and absence of verapamil. The values are expressed as mean ± SEM. Average number of AP in control condition, after the addition of verapamil, and in the presence of verapamil plus fixation of the RMP at -60 mV are indicated in columns. The p values were obtained with a One-way repeated measures ANOVA test followed by a Bonferroni post-hoc test, the average AP obtained in the three conditions was compared within each intensity.
To evaluate the effect of verapamil on the IKv current, a (100 ms) voltage-clamp protocol was applied using a voltage step from -50 to + 10 mV, in the presence of TTX 0.5 μM and CdCl2 100 μM, and IKv was measured at the end of this step (Figure 2A). The application of verapamil (50 μM) induced a significant decrease of IKv from 1001.14 ± 65.05 pA in control conditions to 860.63 ± 50.27 pA in the presence of the drug, reducing the IKv current by 13.5 ± 2.92 % (Figure 2B, n = 7).
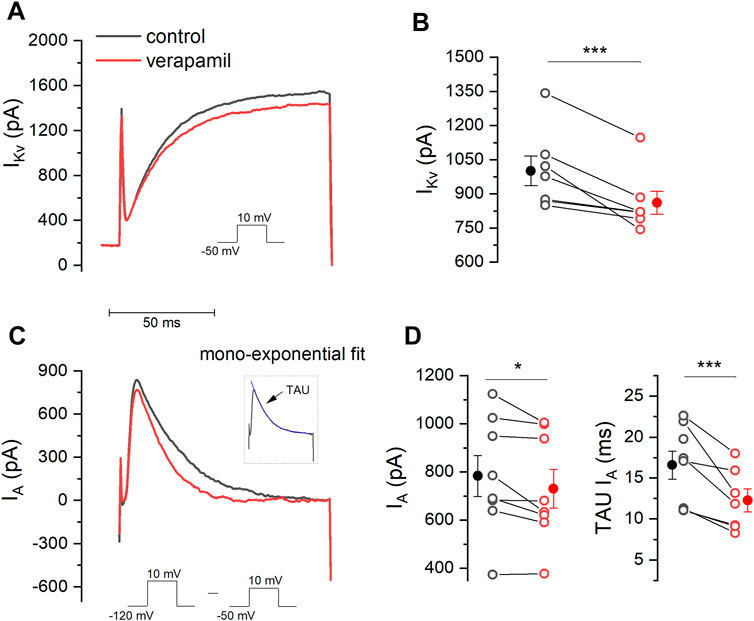
FIGURE 2. Effect of verapamil on IKv and IA. (A) A step protocol (−50 mV–10 mV) was elicited in order to activate IKv in the absence and presence of verapamil. IKv was measure at the end of this step. (B) Verapamil causes a significant reduction in the amplitude of the IKv current (n = 7, p = 0.005, paired sample t-Test). (C) To measure IA we subtracted the current elicited by a step protocol from −120 mV to 10 mV from that evoked by the protocol used in. (A). Inset illustrates how TAU (blue) was estimated. (D) 50 μM verapamil reduces peak current (n = 7, p = 0.034, paired sample t-Test) and accelerates the rate of decay, represented by the TAU (n = 7, p = 0.009, paired sample t-Test). Filled dots and error bars represent the mean ± SEM.
The experimental protocol for the recording of the A-type current is shown in Figure 2C. Two different currents are activated (IKv + IA) simultaneously when a voltage step from -120 mV to +10 mV is given, however a step from -50 mV to 10 mV will only activate IKv (Lamas 1997). It is therefore possible to obtain the A current in isolation by subtracting the second recording from the first one, thus allowing to fit the A current inactivation and to measure the inactivation time constant. IA was measured at the maximum of the peak (Figure 2C and inset). Using this strategy, we found that verapamil induced a decrease of A current, showing 783.46 ± 85.49 pA in control conditions versus 730.60 ± 80.0 pA in the presence of verapamil. Also, we found that verapamil decrease of the IA inactivation time constant. In control conditions, IA TAU was 16.06 ± 1.9 ms, which was significantly reduced to 12.21 ± 1.4 ms after the application of 50 µM verapamil, reducing the inactivating TAU value by 22.9 ± 4.1% (Figure 2D).
Besides IKv and IA, we also wanted to evaluate whether verapamil is an inhibitor of the M-current. The M-current, known to be blocked by TEA (Hadley et al., 2003), was activated using voltage-ramps ranging from -30 to -100 mV (7 s) in the presence of TTX 0.5 μM, CdCl2 100 μM and CsCl 1 mM. As seen in Figure 3A, the application of verapamil did not significantly change the magnitude of the current at any level of the ramp. In control conditions, the current at -30 mV was 118.39 ± 20.29 pA (black trace), and when verapamil was added, the current was 107.23 ± 19.74 pA (red trace). On the contrary, when TEA 15 mM was applied (green trace) a clear inhibition of the outward current was observed, and the current elicited at -30 mV (56.91 ± 10.26 pA) was significantly smaller than that observed in control and in verapamil conditions (Figure 3B). Next, we evaluated the kinetics of the currents sensitive to both drugs TEA and verapamil. For this, we subtracted from the control the current in the presence of TEA and verapamil (ITEA and IVerapamil respectively). Both currents show a different kinetics, while the ITEA (blue) is clearly open at values above -60 mV, the IVerapamil (grey) has a linear kinetic, much like a leakage (Miranda et al., 2013) current that is slightly open at all voltages from -100 to -30 mV (Figure 3C).
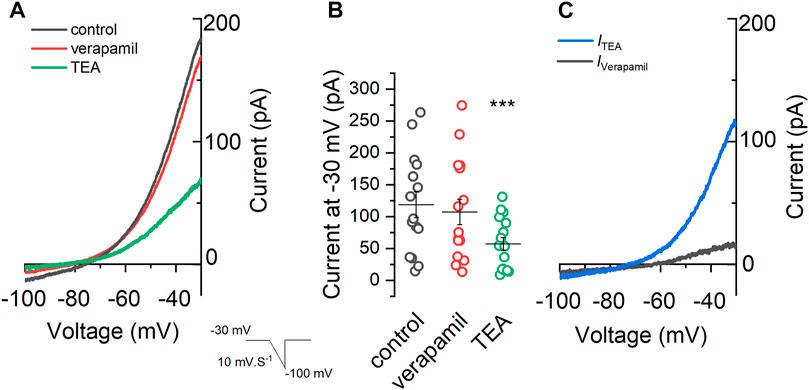
FIGURE 3. Effect of verapamil on the M-current. (A) A voltage-ramp from −30 mV to −100 mV (7 s) was applied in the presence of 0.5 μM TTX, 100 μM CdCl2 and 1 mM CsCl (control condition, black) in order to activate the M-current. After that 50 μM verapamil (red) and 15 mM TEA (green) were sequentially added . (B) Summary of the current values at −30 mV under different conditions. We found a clear difference among the currents observed in the presence of TEA, in control conditions and in the presence of verapamil (n = 14, One-way repeated measures ANOVA, Bonferroni test, p = 0.00004 and p = 0.00003, respectively), while no difference was found between current in control conditions versus verapamil (p = 1). (C) The TEA-sensitive (ITEA) current (blue) is obtained by subtracting the control (A, black) current from the current in the presence of TEA (A, green). Showing a clear rectification at values below −60 mV and almost no current at value below −60 mv, as expected for the M-current. Contrarily, the verapamil-sensitive current (IVerapamil), obtained by subtracting the control current and the current in the presence of verapamil, showed a linear leak-like kinetic.
The voltage-ramps shown in Figure 3C suggest that verapamil might be inhibiting a voltage-independent potassium leak current and we know that SCG neurons do express a good amount of leak TREK2 channels (Cadaveira-Mosquera et al., 2012). To check this hypothesis, we tested the effect of verapamil in the presence of several channel blockers and antagonists using the current-clamp bridge-mode technique (Table 2). Initially, we tested whether a combination of drugs, that we know does not affect the TREK channels (Cadaveira-Mosquera et al., 2011), could prevent the depolarization normally induced by verapamil. This mixture (called Cocktail A) contained blockers of sodium (TTX), potassium (TEA), calcium (cadmium) and cationic h (cesium) currents. We found that the depolarization evoked by verapamil in the presence of the cocktail A (Figure 4B; Table 2) is similar to that found in control conditions (Figures 4A,E), supporting the hypothesis that such effect is not due to voltage-dependent ion channels.

TABLE 2. Verapamil depolarizing effect in both current- and voltage-clamp. Significance was determined using a One-way ANOVA test followed by a Bonferroni post-hoc test (**p = 0.01 and *p = 0.04, respectively).
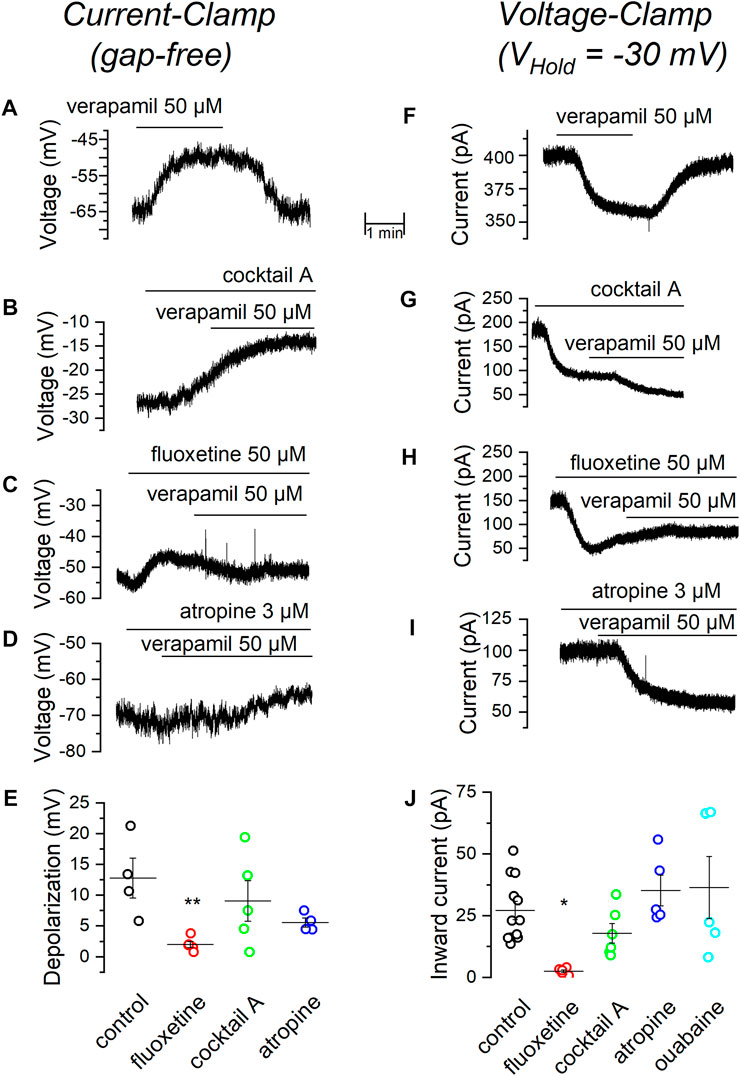
FIGURE 4. Effect of verapamil in the presence of different blockers. (A) Depolarization induced by 50 µM verapamil in current-clamp (gap free) conditions. In the presence of the standard cocktail of blockers. (B), fluoxetine (C) and atropine (D). A summary of results comparing the change in membrane potential elicited by 50 µM verapamil under different conditions is shown in. (E,F) In voltage-clamp conditions (VHold = −30 mV), current induced by 50 µM verapamil alone. (F), in the presence of the standard cocktail of blockers (G), fluoxetine (H) and atropine (I). A summary of results comparing the inward current induced by 50 µM verapamil under different conditions is shown in (J). Values are summarized in Table 2, **p = 0.01, *p = 0.04.
Because a major component of the currents stabilizing the resting membrane potential in mSCG neurons is driven by leak channels of the TREK subfamily (Cadaveira-Mosquera et al., 2011; Rivas-Ramírez et al., 2020), we decided to investigate whether these channels are responsible for the verapamil-induced membrane depolarization seen in our previous experiments. For this purpose, we applied verapamil in the presence of the TREK channel blocker fluoxetine (Kennard et al., 2005). The presence of 50 µM fluoxetine strongly prevented verapamil from inducing membrane depolarization (Figures 4C,E), suggesting again that the inhibition of TREK channels by verapamil could account for its depolarizing effect.
It has been reported that muscarinic agonists inhibit TREK currents in SCG neurons by reducing PIP2 (Lamas et al., 2012; Rivas-Ramirez et al., 2015). We wondered whether the effect of verapamil could be somehow related to the interaction of this drug with the muscarinic cascade. For this purpose, we added verapamil in the presence of atropine, a general muscarinic antagonist. Atropine failed to affect the membrane depolarization induced by verapamil (Figure 4D). In summary, only the presence of the TREK blocker fluoxetine was able to reduce the depolarization produced by the addition of verapamil (Figure 4E).
As a next step, we focused into the current responsible for the depolarization induced by verapamil and therefore performed a series of experiments in the voltage-clamp configuration. With the membrane potential clamped at -30 mV (Cadaveira-Mosquera et al., 2011), the application of 50 µM verapamil induced an inward current (Figure 4F; Table 2) which, as in CC conditions, was insensitive to the application of the cocktail A (Figure 4G). This verapamil-evoked current was however completely suppressed in the presence of the TREK channel blocker fluoxetine (Figure 4H). As in CC mode, 3 µM atropine had no effect on the current-induced by verapamil (Figure 4I). To discard the possibility that the verapamil-induced inward current could be produced by the activation of the Na+/K+ pump, in an additional set of experiments we tested 500 µM ouabain (Lamas et al., 2002; Mahmmoud et al., 2014), which also failed to affect the verapamil-induced inward current. In agreement with the result obtained in current-clamp mode, in voltage-clamp mode only the presence of the TREK inhibitor was able to block the effect of verapamil (Figure 4J).
Our previous experiments suggest that the inhibition of TREK channels could account for the depolarization induced by verapamil in mSCG neurons. If this were true, verapamil should increase membrane resistance. In order to confirm this hypothesis, we clamped the membrane potential at -30 mV and analyzed its conductance in the presence and absence of the drug (see Methods). Figure 5A shows how the application of verapamil reduces the size of the small currents evoked by the hyperpolarizing pulses used to measure membrane conductance from 3.49 ± 0.69 nS in control conditions to 2,14 ± 0.37 nS in the presence of 50 µM verapamil (Figure 5B). This indicates that the closure of outward currents, presumably through TREK channels, and not the activation of inward currents, is the responsible for the effect of verapamil in our cells.
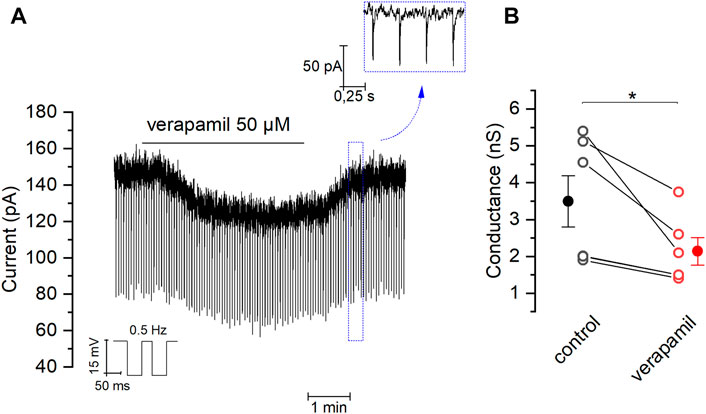
FIGURE 5. Effect of verapamil on the membrane conductance (A) Brief negative pulses (−15 mV at 0.5 Hz) were applied from a holding potential of −30 mV in order to measure the membrane conductance. We chose 20 random measurements of conductance (see material and methods) in absence and presence of verapamil, comparing the average for each cell (B) Effect of verapamil in conductance (n = 6, paired sample t-Test, *p = 0.03). Filled dot and error bars represents mean ± SEM.
We previously showed that TREK-2 and TRESK are the most abundant members of K2P channels expressed in the mSCG (Cadaveira-Mosquera et al., 2012). Therefore, we decided to test if the effect of verapamil is specifically related to the activation of TREK-2 and not TRESK channels. For this purpose, we applied riluzole, which in the presence of cocktail A, induces the activation of TREK-2 channels (Duprat et al., 2000; Cadaveira-Mosquera et al., 2011), and at the same time inhibits TRESK channels (Fernández-Fernández et al., 2018). With the membrane clamped at -30 mV, the application of riluzole in standard solution evoked an outward current (IRIL) of 216.7 ± 39.7 pA (n = 6) (Figure 6A). As we expected, IRIL was unaffected by the presence of cocktail A (see Methods) (173.8 ± 16.5 pA, n = 12) (Figure 6B) or in the presence of a more complete cocktail (cocktail B) aiming to additionally block calcium-dependent potassium channels (SK and BK) and TRP channels (156.5 ± 35.3 pA, n = 5) (Figure 6C). Taken together, these data confirm that, in mSCG cells, IRIL is indeed driven by TREK-2 channels, not finding significant differences between the conditions (Figure 6D).
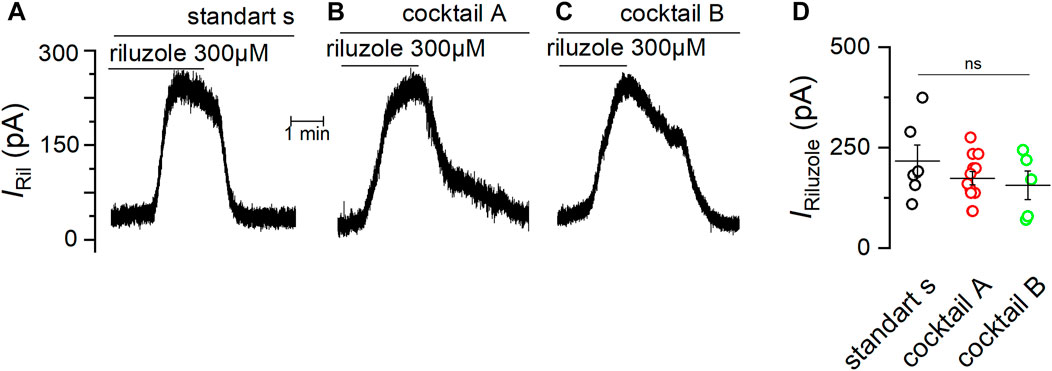
FIGURE 6. The current induced by riluzole is unaffected by different channel blockers. Experiments in VC mode with membrane potential fixed at −30 mV showing the outward current induced by 300 µM riluzole in standard solution (A), in the presence of cocktail (A,B) and in the presence of the cocktail (B,C) are shown (D) Summary of the current induced by riluzole (IRIL) under different conditions. Significance was determined using a one-way ANOVA test (p = 0.37).
After verifying that the IRIL was driven by TREK channels in mSCG, we investigated whether the riluzole-activated current was modified by verapamil. Figure 7A shows how the presence of verapamil strongly reduced IRil obtained in cocktail B. The effect was dose-dependent (Figure 7B) and inhibition values reached by 1000, 300, 100, 30, 10 and 3 μM verapamil on IRIL were: 82.7 ± 0.2% (n = 4), 75.15 ± 0.5% (n = 5), 35 ± 1% (n = 5), 14.8 ± 0.7% (n = 4), 16.9 ± 1% (n = 4) and 13.3 ± 1.6% (n = 6) respectively. Data points were fit using the Hill equation with an estimated IC50 of 96.09 μM and a Hill coefficient of 1.3. It has been suggested that the binding site of verapamil on other potassium channels located intracellularly (Cohen et al., 1987; Rauer and Grissmer, 1999) and that their adhesion causes a collapse of the pore (Baba et al., 2015). However, it is also described that verapamil might exert its blockade by interacting with the extracellular side of these channels (Keith et al., 1994). Our dose-responses curves of verapamil with both the membrane depolarization and IRIL report a Hill coefficient that suggest that this effect happens through a similar mechanism which might require at least two interaction sites acting cooperatively in the channel (Monod et al., 1965; Weiss, 1997).
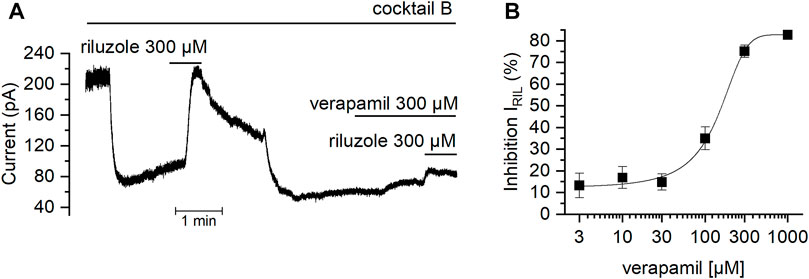
FIGURE 7. Effect of verapamil on the current activated by riluzole (A) With the membrane potential clamped at −30 mV in the presence of cocktail B (see Methods), the TREK activator riluzole was added in the absence and in the presence of verapamil. Note that when verapamil is present, the current activated by riluzole is strongly reduced. (B) Dose-dependent effect of verapamil over the current activated by riluzole.
In the present study, we aimed to investigate the specific effects that the antiarrhythmic verapamil exerts on different potassium conductances evoked in isolated neurons from the mSCG, which is an important regulator of the heart within the sympathetic branch of the autonomic nervous system (Kawashima, 2005). For the first time, we show that verapamil causes a dose-dependent depolarization of the resting membrane potential of mSCG neurons. In the intracardiac ganglion of the parasympathetic system, verapamil has been reported not to affect the RMP, yet it causes a decrease in the firing rate of isolated neurons from this ganglion (Hogg et al., 1999). The depolarization of the RMP seen in our experiments does not lead to an increase in the number of action potentials fired in response to depolarizing current steps. This apparent inconsistency might be explained by the different repertory of ion channels present in both types of neurons, which in this way could account for the overall opposite effect that verapamil exerts in both sympathetic and parasympathetic autonomic nervous systems (Hogg et al., 1999; Sun et al., 2000). An important issue to take into account and that could constitute a limitation to this conclusion is the fact that all the recorded cells showed a phasic firing pattern (Figures 1C,D). Although this pattern has been shown to be preponderant in rodent SCG cells (Malin and Nerbonne, 2000; Martinez-Pinna et al., 2018), the fact that SCG cells with a tonic firing pattern could see their frequency affected cannot be ignored. Indeed, in our study we report a significant yet discrete inhibition of the potassium delayed rectifier channel current (IKv) by 50 µM verapamil as shown before both in heterologous and native systems (Chouabe et al., 1998; Baba et al., 2015; Diesch and Grissmer, 2017; Orvos et al., 2019). However, in the intracardiac ganglion, 10 µM verapamil is already enough to inhibit more than half of IKv currents, claimed by the authors to be the underlying mechanism causing a decrease in AP firing upon verapamil application (Hogg et al., 1999). Additionally, we demonstrate for the first time that, in contrast to the sinoatrial and atrioventricular nodes cardiac action potential where verapamil exerts an increase in its refractory period (Nademanee and Singh, 1988), in mSCG cells this drug produces a decrease of both peak and acceleration of the TAU of IA. In this respect, although we have not found an increase in firing rate, it is possible that this is due to the low concentration used or the counteracting effect caused by the inhibition of IKv currents. On the other hand, a lower strength in and a faster inactivation, in other words, a reduction in IA, could lead to a reduction in the firing frequency that could be evident in cells with a tonic firing pattern. In fact, as shown in Figure 2D, there is a reduction in the number of action potentials, although this reduction was not significant. In agreement with previous reports, we also show here that bath application of verapamil does not affect the M-current (Chouabe et al., 1998; Hogg et al., 1999).
Our results show that the depolarization caused by verapamil in the mSCG neurons is caused by the blockade of an outward current, associated with a decrease in the membrane conductance and therefore a closure of channels. We have tested a combination of channel blockers in order to block voltage-dependent potassium, cationic, calcium and sodium currents, but this had no effect on the depolarization or the inward current induced by verapamil. The inability of TEA to prevent the depolarizing effect of verapamil would emphasize our finding that the M-current is insensitive to the drug. In the same way, the presence of atropine both in CC mode and in VC mode, and ouabain in VC mode, would rule out the participation of metabotropic receptors and Na+/K+ pump respectively as mediators of the effect of verapamil.
We have previously demonstrated that under our experimental conditions, a major component of the outward current seen under resting conditions is driven by K2P channels of the TREK subfamily (Cadaveira-Mosquera et al., 2011). In fact, of all the blockers used here only the application of fluoxetine, an inhibitor of TREK channels (Kennard et al., 2005; Heurteaux et al., 2006), was able to fully abolish the effect of verapamil in both voltage- and current-clamp conditions. Altogether, these results support our thesis that verapamil blocks TREK channels in mSCG neurons. Our dose-responses curves of verapamil reported a Hill coefficient that suggest that this effect happens through at least two interaction sites acting cooperatively in the channel. The polysite pharmacology of TREK channels is well known (Pope and Minor, 2021). It has been shown how TREK channels have several binding sites for small molecules found on the extracellular side of the channel, comprising at least four binding sites for these molecules including the keystone inhibitor site, the K2P modulator pocket, the fenestration site, and the modulatory lipid site. Each one offering a different and very rich structural environment for the control of the channels by the active molecules (Dong et al., 2015; Lolicato et al., 2017; Schewe et al., 2019). Therefore, it is not surprising that, like fluoxetine, verapamil shows affinity for more than one binding site in the structure of TREK channels.
Blocking the TREK channels was further confirmed by the fact that the neuroprotective agent riluzole, an activator of TREK subfamily channels (Duprat et al., 2000), evokes an outward current that was largely inhibited by the application of verapamil in a dose-dependent manner. Importantly, this inhibition takes place even in the presence of our standard cocktail of drugs also supplemented with apamin, paxilline, 4-aminopyridine and clemizole. This indicates that calcium, potassium and sodium voltage-gated currents, the h current, calcium dependent potassium channels (SK and BK) and TRP channels, are all not mediating the inhibitory effect that verapamil exerts on the current activated by riluzole, strongly suggesting that indeed TREK channels are those inhibited by verapamil. Among them, TREK-2 is most likely the main channel blocked by verapamil under these conditions, as it has been shown to be the most abundant TREK channel present in mSCG neurons (Cadaveira-Mosquera et al., 2011; Cadaveira-Mosquera et al., 2012). Alternatively, very recently the presence of TREK-2/TRESK heterodimers has been described in trigeminal primary sensory neurons (Lengyel et al., 2020). Although we have no news saying that chimera is expressed in the SCG, it cannot be ruled out. Supporting our hypothesis, the inhibition produced by verapamil in the presence of fluoxetine would strongly indicate that the effect of the drug would be driven mainly by TREK-2.
The sympathetic system exerts a positive control over the sinoatrial node and the cardiomyocytes. In particular, the SCG functions as a relay station, from where postganglionar neurons synapse with the cardiac tissue (Hernandez-Ochoa et al., 2009; Peng-Sheng Chen et al., 2014). From the SCG, motor pathways that reach the heart set the activity of the sinoatrial and atrioventricular nodes, and downstream, the cardiomyocytes (Pather et al., 2003; Hernandez-Ochoa et al., 2009; Klabunde, 2011). Therefore, any alteration of this system could be harmful for the normal functioning of the heart. In fact, the SCG has been implicated in various cardiovascular conditions (Kong et al., 2013; Na et al., 2014; Murakami et al., 2015; Cheng et al., 2018). In the present study, we have shown that verapamil causes a clear depolarization of mSCG neurons by blocking TREK-2 channels, an effect which would contribute to the increase of sympathetic activity. Additionally, another explanation could be given. Although the maximum depolarization (∼10 mV induced by 300 μM) produced by verapamil would place the RMP at a value (∼ to -55 mV) in which the voltage-gated sodium channels would still be available (Vandael et al., 2015), it could be that a slower repolarization induced by a greater inactivation of the IA together with an eventual lower availability of voltage-dependent sodium channels leads to an inactivation of the sympathetic system which could act synergistically with the reduction of the activity of the L-type calcium channels, promoting greater sympathetic-parasympathetic dysregulation. Although during atrial fibrillation (AF) verapamil causes a reduction in the nodal rhythm (Stern et al., 1982), the increase in excitation of peripheral sympathetic neurons by verapamil, described in the present study, could also lead to various pathological situations such as AF (Nguyen et al., 2009), the dysregulation of the sympathetic-parasympathetic balance (Zhang et al., 2009; Vaseghi et al., 2014), exercise-induced tachycardia (Ozdemir et al., 2003) and dysfunctions of the sinus rhythm (Elvan et al., 1996). It should be taken into account that the therapeutic concentration of verapamil in an adult of normal complexion can be in the nM range (Megarbane et al., 2011), so the effects shown in vitro (μM range) would have to be weighed and analyzed in a broader context. Anyway, considering the above, the therapeutic use of verapamil should take into consideration possible collateral effects of this drug on peripheral sympathetic relay stations such as the SCG.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Spanish Research Council and the University of Vigo Scientific Committee, under Spanish and European directives for the protection of experimental animals (RD 05/03/2013; EU 06/03/2010).
Conceptualization, SH-P and JL; validation, JL; investigation, SH-P, AC-R, LR-R, DF-F, and JL; resources, JL; writing of original draft, SH-P; re-view and editing of manuscript, SH-P, DF-F, and JL; supervision, JL; funding acquisition, JL.
This research was funded by the Spanish government M.I.C.I.U. PID2019-109425GB-I00. All the funding was awarded to JL. SH-P personal funding, Gobierno de Castilla La Mancha mediante convocatoria de Ayudas Regionales a la Investigación en Biomedicina y Ciencias de la Salud, No de expediente II-202_15.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Baba, A., Tachi, M., Maruyama, Y., and Kazama, I. (2015). Suppressive effects of diltiazem and verapamil on delayed rectifier K(+)-channel currents in murine thymocytes. Pharmacol. Rep. 67 (5), 959–964. doi:10.1016/j.pharep.2015.01.009
Bergson, P., Lipkind, G., Lee, S. P., Duban, M. E., and Hanck, D. A. (2011). Verapamil block of T-type calcium channels. Mol. Pharmacol. 79 (3), 411–419. doi:10.1124/mol.110.069492
Bodnar, M., Schlichthorl, G., and Daut, J. (2015). The potassium current carried by TREK-1 channels in rat cardiac ventricular muscle. Pflugers Arch. 467 (5), 1069–1079. doi:10.1007/s00424-014-1678-9
Bond, R. C., Choisy, S. C., Bryant, S. M., Hancox, J. C., and James, A. F. (2014). Inhibition of a TREK-like K+ channel current by noradrenaline requires both β1- and β2-adrenoceptors in rat atrial myocytes. Cardiovasc. Res. 104 (1), 206–215. doi:10.1093/cvr/cvu192
Cadaveira-Mosquera, A., Perez, M., Reboreda, A., Rivas-Ramirez, P., Fernandez-Fernandez, D., and Lamas, J. A. (2012). Expression of K2P channels in sensory and motor neurons of the autonomic nervous system. J. Mol. Neurosci. 48 (1), 86–96. doi:10.1007/s12031-012-9780-y
Cadaveira-Mosquera, A., Ribeiro, S. J., Reboreda, A., Perez, M., and Lamas, J. A. (2011). Activation of TREK currents by the neuroprotective agent riluzole in mouse sympathetic neurons. J. Neurosci. 31 (4), 1375–1385. doi:10.1523/JNEUROSCI.2791-10.2011
Cheng, L. J., Wang, X. H., Liu, T., Tse, G., Fu, H. Y., and Li, G. P. (2018). Modulation of ion channels in the superior cervical ganglion neurons by myocardial ischemia and fluvastatin treatment. Front. Physiol. 9, 1157. doi:10.3389/fphys.2018.01157
Chouabe, C., Drici, M. D., Romey, G., Barhanin, J., and Lazdunski, M. (1998). HERG and KvLQT1/IsK, the cardiac K+ channels involved in long QT syndromes, are targets for calcium channel blockers. Mol. Pharmacol. 54 (4), 695–703.
Cohen, L., Vereault, D., Wasserstrom, J. A., Retzinger, G. S., and Kezdy, F. J. (1987). Evidence that uncharged verapamil inhibits myocardial-contractility. J. Pharmacol. Exp. Ther. 242 (2), 721–725.
De Gama, B. Z., Lazarus, L., Partab, P., and Satyapal, K. S. (2012). The sympathetic and parasympathetic contributions to the cardiac plexus: A fetal study. Int. J. Morphol. 30 (4), 1569–1576. doi:10.4067/s0717-95022012000400048
Decher, N., Kiper, A. K., and Rinne, S. (2017). Stretch-activated potassium currents in the heart: Focus on TREK-1 and arrhythmias. Prog. Biophys. Mol. Biol. 130, 223–232. doi:10.1016/j.pbiomolbio.2017.05.005
Devenyi, R. A., Ortega, F. A., Groenendaal, W., Krogh-Madsen, T., Christini, D. J., and Sobie, E. A. (2017). Differential roles of two delayed rectifier potassium currents in regulation of ventricular action potential duration and arrhythmia susceptibility. J. Physiol. 595 (7), 2301–2317. doi:10.1113/JP273191
Diesch, A. K., and Grissmer, S. (2017). Kinetic aspects of verapamil binding (On-Rate) on wild-type and six hK(v)1.3 mutant channels. Cell. Physiol. biochem. 44 (1), 172–184. doi:10.1159/000484625
Doan, T. N., and Kunze, D. L. (1999). Contribution of the hyperpolarization-activated current to the resting membrane potential of rat nodose sensory neurons. J. Physiol. 514 (1), 125–138. doi:10.1111/j.1469-7793.1999.125af.x
Dong, Y. Y., Pike, A. C., Mackenzie, A., McClenaghan, C., Aryal, P., Dong, L., et al. (2015). K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science 347 (6227), 1256–1259. doi:10.1126/science.1261512
Duprat, F., Lesage, F., Patel, A. J., Fink, M., Romey, G., and Lazdunski, M. (2000). The neuroprotective agent riluzole activates the two P domain K+ channels TREK-1 and TRAAK. Mol. Pharmacol. 57 (5), 906–912.
Elvan, A., Wylie, K., and Zipes, D. P. (1996). Pacing-induced chronic atrial fibrillation impairs sinus node function in dogs: Electrophysiological remodeling. Circulation 94 (11), 2953–2960. doi:10.1161/01.cir.94.11.2953
Fernández-Fernández, D., Cadaveira-Mosquera, A., Rueda-Ruzafa, L., Herrera-Pérez, S., Veale, E. L., Reboreda, A., et al. (2018). Activation of TREK currents by riluzole in three subgroups of cultured mouse nodose ganglion neurons. Plos One 13 (6), e0199282. doi:10.1371/journal.pone.0199282
Freeze, B. S., McNulty, M. M., and Hanck, D. A. (2006). State-dependent verapamil block of the cloned human Ca(v)3.1 T-type Ca2+ channel. Mol. Pharmacol. 70 (2), 718–726. doi:10.1124/mol.106.023473
Grandi, E., Sanguinetti, M. C., Bartos, D. C., Bers, D. M., Chen-Izu, Y., Chiamvimonvat, N., et al. (2017). Potassium channels in the heart: Structure, function and regulation. J. Physiol. 595 (7), 2209–2228. doi:10.1113/JP272864
Hadley, J. K., Passmore, G. M., Tatulian, L., Al-Qatari, M., Ye, F., Wickenden, A. D., et al. (2003). Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J. Neurosci. 23 (12), 5012–5019. doi:10.1523/jneurosci.23-12-05012.2003
Hernandez-Ochoa, E. O., Prosser, B. L., Wright, N. T., Contreras, M., Weber, D. J., and Schneider, M. F. (2009). Augmentation of Ca(v)1 channel current and action potential duration after uptake of S100A1 in sympathetic ganglion neurons. Am. J. Physiol. Cell Physiol. 297 (4), C955–C970. doi:10.1152/ajpcell.00140.2009
Heurteaux, C., Lucas, G., Guy, N., El Yacoubi, M., Thummler, S., Peng, X. D., et al. (2006). Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat. Neurosci. 9 (9), 1134–1141. doi:10.1038/nn1749
Hogg, R. C., Trequattrini, C., Catacuzzeno, L., Petris, A., Franciolini, F., and Adams, D. J. (1999). Mechanisms of verapamil inhibition of action potential firing in rat intracardiac ganglion neurons. J. Pharmacol. Exp. Ther. 289 (3), 1502–1508.
Ito, H., Takikawa, R., Kurachi, Y., and Sugimoto, T. (1989). Anti-cholinergic effect of verapamil on the muscarinic acetylcholine receptor-gated K+ channel in isolated Guinea-pig atrial myocytes. Naunyn. Schmiedeb. Arch. Pharmacol. 339 (1-2), 244–246. doi:10.1007/BF00165150
Kato, M., Dote, K., Sasaki, S., Takemoto, H., Habara, S., and Hasegawa, D. (2004). Intracoronary verapamil rapidly terminates reperfusion tachyarrhythmias in acute myocardial infarction. Chest 126 (3), 702–708. doi:10.1378/chest.126.3.702
Kawashima, T. (2005). The autonomic nervous system of the human heart with special reference to its origin, course, and peripheral distribution. Anat. Embryol. 209 (6), 425–438. doi:10.1007/s00429-005-0462-1
Keith, R. A., Mangano, T. J., Defeo, P. A., Ernst, G. E., and Warawa, E. J. (1994). Differential inhibition of neuronal calcium entry and [3H]-D-aspartate release by the quaternary derivatives of verapamil and emopamil. Br. J. Pharmacol. 113 (2), 379–384. doi:10.1111/j.1476-5381.1994.tb16999.x
Kennard, L. E., Chumbley, J. R., Ranatunga, K. M., Armstrong, S. J., Veale, E. L., and Mathie, A. (2005). Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br. J. Pharmacol. 144 (6), 821–829. doi:10.1038/sj.bjp.0706068
Klabunde, R. (2011). “6 - neurohumoral control of the heart and circulation,” in Cardiovascular physiology concepts. Second ed (United States: Lippincott Williams & Wilkins), 124–147.
Kong, F. J., Liu, S. M., Xu, C. S., Liu, J., Li, G. D., Li, G. L., et al. (2013). Electrophysiological studies of upregulated P2X(7) receptors in rat superior cervical ganglia after myocardial ischemic injury. Neurochem. Int. 63 (3), 230–237. doi:10.1016/j.neuint.2013.06.003
Lamas, J. A. (2012). “Mechanosensitive K2P channels, TREKking through the autonomic nervous system,” in Mechanically gated channels and their regulation. Editors A. Kamkin, and I. Lozinsky (Dordrecht: Springer Science+Business Media), 635–668. Mechanosensitivity in cells and tissues.
Lamas, J. A., Reboreda, A., and Codesido, V. (2002). Ionic basis of the resting membrane potential in cultured rat sympathetic neurons. Neuroreport 13 (5), 585–591. doi:10.1097/00001756-200204160-00010
Lamas, J. A., Selyanko, A. A., and Brown, D. A. (1997). Effects of a cognition-enhancer, linopirdine (DuP 996), on M-type potassium currents (I-K(M)) and some other voltage- and ligand-gated membrane currents in rat sympathetic neurons. Eur. J. Neurosci. 9 (3), 605–616. doi:10.1111/j.1460-9568.1997.tb01637.x
Lengyel, M., Czirjak, G., Jacobson, D. A., and Enyedi, P. (2020). TRESK and TREK-2 two-pore-domain potassium channel subunits form functional heterodimers in primary somatosensory neurons. J. Biol. Chem. 295 (35), 12408–12425. doi:10.1074/jbc.RA120.014125
Limberg, S. H., Netter, M. F., Rolfes, C., Rinne, S., Schlichthorl, G., Zuzarte, M., et al. (2011). TASK-1 channels may modulate action potential duration of human atrial cardiomyocytes. Cell. Physiol. biochem. 28 (4), 613–624. doi:10.1159/000335757
Liu, J., Kim, K. H., London, B., Morales, M. J., and Backx, P. H. (2011). Dissection of the voltage-activated potassium outward currents in adult mouse ventricular myocytes: I(to,f), I(to,s), I(K,slow1), I(K,slow2), and I(ss). Basic Res. Cardiol. 106 (2), 189–204. doi:10.1007/s00395-010-0134-z
Lolicato, M., Arrigoni, C., Mori, T., Sekioka, Y., Bryant, C., Clark, K. A., et al. (2017). K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature 547 (7663), 364–368. doi:10.1038/nature22988
Mahmmoud, Y. A., Shattock, M., Cornelius, F., and Pavlovic, D. (2014). Inhibition of K+ transport through Na+, K+-ATPase by capsazepine: Role of membrane span 10 of the alpha-subunit in the modulation of ion gating. Plos One 9 (5), e96909. doi:10.1371/journal.pone.0096909
Malin, S. A., and Nerbonne, J. M. (2000). Elimination of the fast transient in superior cervical ganglion neurons with expression of KV4.2W362F: Molecular dissection of IA. J. Neurosci. 20 (14), 5191–5199. doi:10.1523/jneurosci.20-14-05191.2000
Martinez-Pinna, J., Soriano, S., Tuduri, E., Nadal, A., and de Castro, F. (2018). A calcium-dependent chloride current increases repetitive firing in mouse sympathetic neurons. Front. Physiol. 9, 508. doi:10.3389/fphys.2018.00508
Megarbane, B., Karyo, S., Abidi, K., Delhotal-Landes, B., Aout, M., Sauder, P., et al. (2011). Predictors of mortality in verapamil overdose: Usefulness of serum verapamil concentrations. Basic Clin. Pharmacol. Toxicol. 108 (6), 385–389. doi:10.1111/j.1742-7843.2010.00666.x
Miranda, P., Cadaveira-Mosquera, A., Gonzalez-Montelongo, R., Villarroel, A., Gonzalez-Hernandez, T., Lamas, J. A., et al. (2013). The neuronal serum- and glucocorticoid-regulated kinase 1.1 reduces neuronal excitability and protects against seizures through upregulation of the M-current. J. Neurosci. 33 (6), 2684–2696. doi:10.1523/JNEUROSCI.3442-12.2013
Monod, J., Wyman, J., and Changeux, J. P. (1965). On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12, 88–118. doi:10.1016/s0022-2836(65)80285-6
Murakami, M., Yoshikawa, T., Nakamura, T., Ohba, T., Matsuzaki, Y., Sawamura, D., et al. (2015). Involvement of the histamine H1 receptor in the regulation of sympathetic nerve activity. Biochem. Biophys. Res. Commun. 458 (3), 584–589. doi:10.1016/j.bbrc.2015.02.009
Na, S., Kim, O. S., Ryoo, S., Kweon, T. D., Choi, Y. S., Shim, H. S., et al. (2014). Cervical ganglion block Attenuates the progression of pulmonary hypertension via nitric oxide and arginase pathways. Hypertension 63 (2), 309–315. doi:10.1161/HYPERTENSIONAHA.113.01979
Nademanee, K., and Singh, B. N. (1988). Control of cardiac-arrhythmias by calcium antagonism. Ann. N. Y. Acad. Sci. 522, 536–552. doi:10.1111/j.1749-6632.1988.tb33397.x
Nguyen, B. L., Fishbein, M. C., Chen, L. S., Chen, P. S., and Masroor, S. (2009). Histopathological substrate for chronic atrial fibrillation in humans. Heart rhythm. 6 (4), 454–460. doi:10.1016/j.hrthm.2009.01.010
Orvos, P., Kohajda, Z., Szlovak, J., Gazdag, P., Arpadffy-Lovas, T., Toth, D., et al. (2019). Evaluation of possible proarrhythmic potency: Comparison of the effect of dofetilide, cisapride, sotalol, terfenadine, and verapamil on hERG and native I-Kr currents and on cardiac action potential. Toxicol. Sci. 168 (2), 365–380. doi:10.1093/toxsci/kfy299
Ozdemir, O., Soylu, M., Demir, A. D., Topaloglu, S., Alyan, O., Geyik, B., et al. (2003). Increased sympathetic nervous system activity as cause of exercise-induced ventricular tachycardia in patients with normal coronary arteries. Tex. Heart Inst. J. 30 (2), 100–104.
Park, H., Kim, E. J., Ryu, J. H., Lee, D. K., Hong, S. G., Han, J., et al. (2018). Verapamil inhibits TRESK (K(2P)18.1) current in trigeminal ganglion neurons independently of the blockade of Ca2+ influx. Int. J. Mol. Sci. 19 (7), E1961. doi:10.3390/ijms19071961
Pather, N., Partab, P., Singh, B., and Satyapal, K. S. (2003). The sympathetic contributions to the cardiac plexus. Surg. Radiol. Anat. 25 (3-4), 210–215. doi:10.1007/s00276-003-0113-2
Peng-Sheng Chen, L. S. C., and Lin, S.-F. (2014). “Neural activity and atrial tachyarrhythmias,” in Cardiac electrophysiology: From cell to bedside. Editor P. J. J. Douglas Zipes. Sixth ed., 399–407.
Pope, L., and Minor, D. L. (2021). The polysite pharmacology of TREK K2P channels. Adv. Exp. Med. Biol. 1349, 51–65. doi:10.1007/978-981-16-4254-8_4
Rauer, H., and Grissmer, S. (1999). The effect of deep pore mutations on the action of phenylalkylamines on the Kv1.3 potassium channel. Br. J. Pharmacol. 127 (5), 1065–1074. doi:10.1038/sj.bjp.0702599
Rivas-Ramirez, P., Cadaveira-Mosquera, A., Lamas, J. A., and Reboreda, A. (2015). Muscarinic modulation of TREK currents in mouse sympathetic superior cervical ganglion neurons. Eur. J. Neurosci. 42 (2), 1797–1807. doi:10.1111/ejn.12930
Rivas-Ramírez, P., Reboreda, A., Rueda-Ruzafa, L., Herrera-Pérez, S., and Lamas, J. A. (2020). Contribution of KCNQ and TREK channels to the resting membrane potential in sympathetic neurons at physiological temperature. Int. J. Mol. Sci. 21 (16), E5796. doi:10.3390/ijms21165796
Romero, M., Reboreda, A., Sanchez, E., and Lamas, J. A. (2004). Newly developed blockers of the M-current do not reduce spike frequency adaptation in cultured mouse sympathetic neurons. Eur. J. Neurosci. 19 (10), 2693–2702. doi:10.1111/j.1460-9568.2004.03363.x
Schewe, M., Sun, H., Mert, U., Mackenzie, A., Pike, A. C. W., Schulz, F., et al. (2019). A pharmacological master key mechanism that unlocks the selectivity filter gate in K(+) channels. Science 363 (6429), 875–880. doi:10.1126/science.aav0569
Schmidt, C., Wiedmann, F., Schweizer, P. A., Becker, R., Katus, H. A., and Thomas, D. (2012). Novel electrophysiological properties of dronedarone: Inhibition of human cardiac two-pore-domain potassium (K2P) channels. Naunyn. Schmiedeb. Arch. Pharmacol. 385 (10), 1003–1016. doi:10.1007/s00210-012-0780-9
Staudacher, I., Illg, C., Chai, S., Deschenes, I., Seehausen, S., Gramlich, D., et al. (2018). Cardiovascular pharmacology of K2P17.1 (TASK-4, TALK-2) two-pore-domain K(+) channels. Naunyn. Schmiedeb. Arch. Pharmacol. 391 (10), 1119–1131. doi:10.1007/s00210-018-1535-z
Stern, E. H., Pitchon, R., King, B. D., Guerrero, J., Schneider, R. R., and Wiener, I. (1982). Clinical use of oral verapamil in chronic and paroxysmal atrial-fibrillation. Chest 81 (3), 308–311. doi:10.1378/chest.81.3.308
Sun, N., Hong, T., Zhang, R., and Yang, X. (2000). The effects of verapamil SR and bisoprolol on reducing the sympathetic nervous system's activity. Hypertens. Res. 23 (5), 537–540. doi:10.1291/hypres.23.537
Unudurthi, S. D., Wu, X. Q., Qian, L., Amari, F., Onal, B., Li, N., et al. (2016). Two-pore K+ channel TREK-1 regulates sinoatrial node membrane excitability. J. Am. Heart Assoc. 5 (4), e002865. doi:10.1161/JAHA.115.002865
Vandael, D. H., Ottaviani, M. M., Legros, C., Lefort, C., Guerineau, N. C., Allio, A., et al. (2015). Reduced availability of voltage-gated sodium channels by depolarization or blockade by tetrodotoxin boosts burst firing and catecholamine release in mouse chromaffin cells. J. Physiol. 593 (4), 905–927. doi:10.1113/jphysiol.2014.283374
Vaseghi, M., Ajijola, O. A., Mahajan, A., and Shivkumar, K. (2014). “Sympathetic innervation, denervation, and cardiac arrhythmias,” in Cardiac electrophysiology: From cell to bedside. Amsterdam: Elsiever, 409–417.
Weiss, J. N. (1997). The Hill equation revisited: Uses and misuses. FASEB J. 11 (11), 835–841. doi:10.1096/fasebj.11.11.9285481
Wiedmann, F., Schmidt, C., Lugenbiel, P., Staudacher, I., Rahm, A. K., Seyler, C., et al. (2016). Therapeutic targeting of two-pore-domain potassium (K(2P)) channels in the cardiovascular system. Clin. Sci. 130 (9), 643–650. doi:10.1042/CS20150533
Zhang, Y. H., Popovic, Z. B., Bibevski, S., Fakhry, I., Sica, D. A., Van Wagoner, D. R., et al. (2009). Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ. Heart Fail. 2 (6), 692–699. doi:10.1161/CIRCHEARTFAILURE.109.873968
Keywords: verapamil, TREK, superior cervical ganglion, riluzole, TREK-2
Citation: Herrera-Pérez S, Rueda-Ruzafa L, Campos-Ríos A, Fernández-Fernández D and Lamas JA (2022) Antiarrhythmic calcium channel blocker verapamil inhibits trek currents in sympathetic neurons. Front. Pharmacol. 13:997188. doi: 10.3389/fphar.2022.997188
Received: 18 July 2022; Accepted: 31 August 2022;
Published: 15 September 2022.
Edited by:
Oscar Casis, Universidad del País Vasco UPV/EHU, SpainReviewed by:
Marcos Matamoros, Washington University in St. Louis, United StatesCopyright © 2022 Herrera-Pérez, Rueda-Ruzafa, Campos-Ríos, Fernández-Fernández and Lamas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: S. Herrera-Pérez, c3NhbHZhNEBtZS5jb20=; J. A. Lamas, YW50b25pb2xhbWFzQHV2aWdvLmVz
†Present Address: D. Fernández-Fernández, Department of Biomedicine, University of Basel, Basel, Switzerland
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.