 Lea Strohm1†
Lea Strohm1† Henning Ubbens
Henning Ubbens Thomas Münzel
Thomas Münzel Andreas Daiber
Andreas Daiber Steffen Daub
Steffen Daub
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 04 October 2022
Sec. Inflammation Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.995061
This article is part of the Research TopicResolution of Inflammation: Mechanisms, Mediators & Biomarkers: Volume IIView all 7 articles
Cardiovascular diseases (CVD) and cardiovascular risk factors are the leading cause of death in the world today. According to the Global Burden of Disease Study, hypertension together with ischemic heart and cerebrovascular diseases is responsible for approximately 40% of all deaths worldwide. The major pathomechanism underlying almost all CVD is atherosclerosis, an inflammatory disorder of the vascular system. Recent large-scale clinical trials demonstrated that inflammation itself is an independent cardiovascular risk factor. Specific anti-inflammatory therapy could decrease cardiovascular mortality in patients with atherosclerosis (increased markers of inflammation). Inflammation, however, can also be beneficial by conferring so-called resolution, a process that contributes to clearing damaged tissue from cell debris upon cell death and thereby represents an essential step for recovery from, e.g., ischemia/reperfusion damage. Based on these considerations, the present review highlights features of the detrimental inflammatory reactions as well as of the beneficial process of immune cell-triggered resolution. In this context, we discuss the polarization of macrophages to either M1 or M2 phenotype and critically assess the role of the CD40L-CD40-TRAF signaling cascade in atherosclerosis and its potential link to resolution. As CD40L can bind to different cellular receptors, it can initiate a broad range of inflammatory processes that may be detrimental or beneficial. Likewise, the signaling of CD40L downstream of CD40 is mainly determined by activation of TRAF1-6 pathways that again can be detrimental or beneficial. Accordingly, CD40(L)-based therapies may be Janus-faced and require sophisticated fine-tuning in order to promote cardioprotection.
Cardiovascular diseases (CVD) are classified as inflammation-related entities (Bartekova et al., 2018). Thus, it is not surprising that diseases associated with systemic inflammation, such as rheumatoid arthritis (Bergholm et al., 2002; Hänsel et al., 2003), psoriasis (Mehta et al., 2010), or type-2 diabetes (Henry et al., 2004), contribute to increased cardiovascular risk. Endothelial dysfunction occurs, for example, in severe inflammation triggered by lipopolysaccharide (LPS) (Becker et al., 2012). In 2005, the research group around Ridker could show that death caused by myocardial infarction (MI) in patients with the acute coronary syndrome can be predicted by a high CRP (C-reactive protein) level (Ridker et al., 2005). A correlation between inflammation and an increased risk of CVDs, but also between inflammation, redox reactions, and oxidative stress is evident (Karbach et al., 2014; Steven et al., 2015; Daiber et al., 2017b). However, inflammation plays a major role not only in its interaction with CVDs, as its influence can also be shown in neurodegenerative diseases such as Alzheimer´s (Prasad, 2016; Wes et al., 2016; Katsumoto et al., 2018) or Parkinson’s disease (Gorelenkova Miller et al., 2016; Troncoso-Escudero et al., 2018). Therefore, diseases associated with inflammation represent an important area of research, as they are major challenges, both for our society and our health system.
Inflammation plays a vital role in CVD and chronic diseases such as fatty liver disease, kidney disease, and neurodegenerative diseases are triggered by or via the interaction with inflammation (Steven et al., 2015; Daiber et al., 2017a; Daiber et al., 2017b). Oxidative stress is characteristic of CVD and is mainly associated with inflammation (Harrison et al., 2011; Karbach et al., 2014; Wenzel et al., 2017). This represents a risk factor in CVD (Kaptoge et al., 2014) that can be counteracted with pharmacological agents (Ridker et al., 2017). All the evidence suggests that a correlation exists between chronic inflammatory (e.g., lupus erythematosus, psoriasis, rheumatoid arthritis, autoimmune diseases) and cardiovascular diseases, as they show an increased cardiovascular risk (Hak et al., 2009; Vena et al., 2010; Soltész et al., 2011; Murdaca et al., 2012). Of particular note, psoriasis has been highlighted as an independent factor in CVDs (Mehta et al., 2010). The European Alliance of Associations for Rheumatology (EULAR) recommends rigorous screening for cardiovascular disease and an early, intensive therapeutic approach in patients who have psoriasis or rheumatoid arthritis (Peters et al., 2010). This recommendation results from the fact that endothelial dysfunction is observed in both patient groups (Balci et al., 2009; Södergren et al., 2010). In line with the immunosuppressive therapy approach, further studies have shown that targeted therapy reduced cardiovascular mortality in rheumatoid arthritis (IL-6, TNF-α, and IL-17A cascades) (Pasceri and Yeh, 1999; Choy, 2012), lupus erythematosus (IL-17A signaling) (Crispín and Tsokos, 2010), and psoriasis (IL-17/IL-23) (Di Cesare et al., 2009; Papp et al., 2012). The cytokines mentioned above (interleukins IL-6, IL-1β, and IL-17A, as well as tumor necrosis factor-α [TNF-α]) should be considered in a positive relationship with CVD (Karbach et al., 2014).
In this regard, there are clinical trials with receptor blockers or antagonists being conducted or completed in patients with CVD (van Tassell et al., 2013). Likewise, a study from the Genome-Wide Association Study (GWAS) showed that inflammatory processes play a significant role in both the development and progression of CVDs. For example, point mutations were found in or near genes involved in cell adhesion, leukocyte migration and atherosclerosis (PECAM1, rs1867624), inflammation, and coagulation (PROCR, rs867186 p. Ser219Gly) (Howson et al., 2017). In addition, further GWAS studies identified the association of several risk loci with cardiovascular inflammation (Klarin et al., 2017). Upon stress-dependent activation (e.g., inflammatory stimuli) or apoptosis of endothelial cells, they release endothelial microparticles (EMPs). These vesicles trigger oxidative stress and can lead to inflammation and eventually promote the development of atherosclerosis (Paudel et al., 2016). Immunosuppressive and anti-inflammatory properties of cardiovascular drugs such as AT1R blockers or ACE inhibitors and antidiabetic drugs such as GLP-1 receptor agonists or DPP-4 inhibitors extend beyond the drugs’ traditional therapeutic approach (Shah et al., 2011; Matsubara et al., 2012; Steven et al., 2015). Another example for the efficacy of specific anti-inflammatory therapies in CVD was shown in the CANTOS trial, in which the IL-1β antagonist canakinumab was used and improved cardiovascular outcome in a dosage-dependent manner (Iseme et al., 2017) through inhibition of the IL-1β pathway and consequent reduction in CRP levels. The use of canakinumab in patients with diabetes mellitus and additional cardiovascular disease indicate a positive outcome (Karbach et al., 2019). In summary, these findings suggest causality between CVD, and autoimmune disease via inflammation and demonstrate the possibility of an anti-inflammatory therapeutic approach to cardiovascular disease (Vena et al., 2010; Soltész et al., 2011).
The fact that oxidative stress plays a pivotal role in the development of CVD was shown by Harrison and Ohara as early as the 1990s using the experimental model of hypercholesterolemia (Ohara et al., 1993; Harrison and Ohara, 1995). Since then, it has been understood that most CVDs exhibit a mismatch between ROS (reactive oxygen species) formation and their degradation by antioxidant mechanisms (Heitzer et al., 2001; Griendling and FitzGerald, 2003). This mismatch leads to the accumulation of superoxide, peroxynitrite, hydrogen peroxide, and hypochlorous acid and puts the system into disequilibrium (Sies, 2015). Our research group has detailed the underlying molecular mechanisms of vascular dysfunction—the ROS sources involved, redox mechanism in the -NO/cGMP pathway (Daiber et al., 2014; Schulz et al., 2014; Wenzel et al., 2017). On the other hand it is also possible to initiate ROS production via crosstalk of involved activation pathways (Daiber, 2010; Dikalov, 2011; Daiber et al., 2017a). For example, this mechanism has been described for the NADPH oxidase isoform 2 (NOX-2)/mitochondrial axis (Doughan et al., 2008; Dikalova et al., 2010), xanthine dehydrogenase/oxidase conversion, and eNOS (endothelial nitric oxide synthase) uncoupling (Kröller-Schön et al., 2014; Schulz et al., 2014). The principle of redox crosstalk has also been observed in angiotensin II (AT-II)-induced hypertension: Here, activation of NOX-2 leads to Sirt3 S-glutathionylation, which contributes to acetylation of SOD2 (superoxide dismutase type 2, mitochondrial isoform) and thus decreases the enzyme’s antioxidant activity. This circumstance, in turn, increases mitochondrial superoxide and reduces nitric oxide bioavailability in the endothelium, thus aggravating hypertension (Dikalova et al., 2017; Dikalov and Dikalova, 2019). Likewise, this redox crosstalk has been associated with tobacco use and its vascular effects (Dikalov et al., 2019).
Furthermore, the uncoupling of eNOS was shown to represent a key step in the formation and progression of CVDs (Schulz et al., 2014; Münzel et al., 2005; Förstermann and Münzel, 2006). Numerous studies have shown that the interplay between oxidative stress and cardiovascular disease can be used in order to ascertain a patient’s prognosis. As an example, a positive correlation was shown between glutathione peroxidase-1 and outcomes in patients with cardiovascular disease (Blankenberg et al., 2003). Various clinical studies have shown that CVD patients have an increased blood-level of the oxidative DNA damage marker 8-hydroxy-2-deoxyguanine, which can be used as a surrogate parameter for oxidative stress (Di Minno et al., 2016). Despite these lines of evidence for an association of oxidative stress markers with CVD, large-scale clinical trials that support a beneficial impact of antioxidant therapy on the prognosis of CVD patients are lacking (Münzel et al., 2010; Schmidt et al., 2015). Finally, one can postulate that there is much evidence for the negative impact of oxidative stress on physiological and inflammatory processes (Wenzel et al., 2008; Oelze et al., 2014).
In addition to the factors influencing inflammation and its resulting diseases discussed so far, seemingly mundane factors such as unbalanced sleep (Arnett et al., 2019), poor diet, or lack of exercise are also important. They can influence molecular processes in the body and negatively affect the immune system (Frodermann et al., 2019; McAlpine et al., 2019; Tourki and Halade, 2021). Since the development of inflammation plays a vital role in CVDs, it is crucial to focus on its processes to develop strategies to treat CVDs (Murphy et al., 2020). In the disease course of CVDs, a distinction is made between mild, acute, and chronic inflammation. In patients suffering from diabetes or metabolic disorders, mild inflammation that occurs within the course of the underlying disease can become chronic if inflammatory resolution is not achieved (Fuster et al., 2016). For example, in MI, inflammation is crucially involved in plaque formation as well as plaque rupture and subsequent tissue hypoxia. The severity of the resulting ischemic heart failure (HF) depends strongly on the resolution of the inflammation, which is part of the healing process (Figure 1). This active process runs in parallel with inflammation and ensures the degradation of the resulting inflammatory products (Serhan, 2011; Serhan, 2014). Resolution-promoting lipid mediators (SPMs) are released in this process, and pro-inflammatory cells are differentiated into an immunoreactive phenotype (Halade et al., 2017; Kain et al., 2017; Sharma et al., 2020).
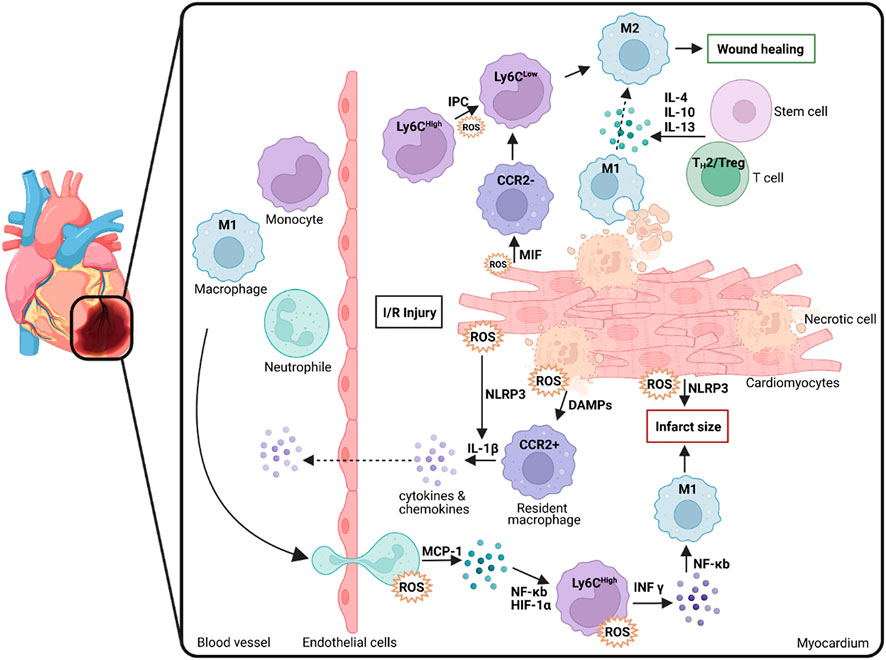
FIGURE 1. Macrophage polarization, infiltration and ROS generation following myocardial damage (e.g., through I/R injury). Macrophages are crucial in the process underlying tissue inflammation as well as wound healing and inflammatory resolution thereby determining the degree of infarct size. M1: macrophages with M1 polarization; Ly6C: Lymphocyte antigen 6 complex locus C; M2: macrophages with M2 polarization; IL: Interleukin; CCR2: C-C chemokine receptor type 2; ROS: Reactive oxygen species; IR: Ischemia/reperfusion; MIF: Macrophage migration inhibitory factor; NLRP3: Family pyrin domain containing 3; DAMP: Damage-associated molecular pattern; MCP: Monocyte Chemoattractant Protein-1 (CCL-2); NF-κB: Nuclear Factor Kappa B; HIF-1α: Hypoxia inducible factor 1 alpha; INF-γ: Interferon gamma; IPC: Ischemic preconditioning, Treg: Regulatory T cells; TH1/2: T Helper cells 1/2. Scheme was summarized from data in (Daiber et al., 2021). Created with BioRender.com.
Immediately after the onset of MI, leukocytes are dispatched to the heart to ablate damaged tissue there, ultimately effecting the inflammatory process (Halade et al., 2018). In addition, specific cytokines/chemokines, inflammatory and endothelial cells, and ROS are produced or activated (Tracy, 2006). However, the subsequent resolution or its successful progression is age-dependent, as there is increased dysregulation between pro- and anti-inflammatory molecules and subsequent pathways in older patients (Lopez et al., 2015; Halade et al., 2016). The first immune cells to respond to inflammation are neutrophils, which counteract stress-induced cell dysfunction (Kain and Halade, 2020). In inflammation and resolution, neutrophils appear according to a chronological polarization pattern in differentiated phenotypes with correspondingly divergent functions, which are subdivided as pro-inflammatory N1 and anti-inflammatory N2 neutrophils and whose role includes both tissue repair but also precisely damage to unaffected surrounding tissue (Vafadarnejad et al., 2020). At the onset of MI, N1 neutrophils are active and are subsequently replaced by N2 neutrophils, which are anti-inflammatory and enact tissue repair. In addition, monocytes belonging to the domain of leukocytes and are derived from the bone marrow circulate in the organism for up to 3 days (Duncan et al., 2020). These monocytes can differentiate into macrophages, which play an essential role in inflammation and repairing damaged tissue. Depending on phenotypic plasticity and polarization, macrophages are also divided into pro-inflammatory M1 and anti-inflammatory M2 macrophages and influence the healing process, differentiation of myofibroblasts from fibroblasts, degradation of cellular debris, tissue repair through recruitment of inflammatory cytokines/chemokines, and activation of endothelial cells (Duncan et al., 2020) (Figure 1). According to a recent study by Chiurchiù and colleagues, macrophages are a significant factor in the progression of resolution, such as regulatory T lymphocytes (Tregs) which limit pro-inflammatory responses of macrophages and T lymphocytes in their function which in turn places macrophages in an anti-inflammatory state that supports tissue repair. In addition, lymphocytes from patients with chronic heart failure have been shown to exhibit impaired immune system interference-mediated via CD8+ and CD4+ cells, indicative of impaired resolution (Chiurchiù et al., 2019).
Previously, the resolution of inflammation was thought to be a largely passive process in which inflammatory factors, enzymes involved, and signaling pathways were inhibited or blocked (Serhan, 2011). However, recent studies show a specific resolution pathway mediated by bioactive mediators (resolution-promoting mediators) (Gerlach et al., 2020). This resolution pathway is determined by several regulatory factors, such as resolution-promoting lipids (e.g., protectin, lipoxin, resolvin) and neutrophils, T cells, and macrophages, each of which is converted to its anti-inflammatory phenotype (Halade et al., 2017; Kain et al., 2017; Sharma et al., 2020). In this resolution process, the secretion of pro-inflammatory cytokines (IL-6, TNF-α) is throttled, the production of anti-inflammatory cytokines (IL-10) is stimulated, neutrophils infiltration into the tissue reduced, and phagocytic macrophages are stimulated. This resolution pathway may represent a starting point that positively influences the prognosis of CVDs. The prerequisite for a favorable prognosis after an inflammatory MI is the restoration of systemic homeostasis through the repair of the damaged tissue. In this process, macrophages play essential roles by secreting anti-inflammatory cytokines such as transforming growth factor-beta (TGF-β) or vascular endothelial growth factor (VEGF) (Ortega-Gómez et al., 2013). These cytokines are instrumental in regenerating damaged tissue, ultimately promoting homeostasis (Ortega-Gómez et al., 2013).
MI leads to ischemia of the myocardium in the medium to long term, resulting in the death of the muscle tissue. MI represents one of the most common causes of disease-related death (Sadek and Olson, 2020). As a result of the undersupply of oxygen to the tissue, after MI, necrosis occurs, triggering both systemic and local inflammation. Neutrophils and monocytes migrate into the damaged tissue and start the repair mechanism, divided into 3 phases described in detail in the preceding sections, inflammation followed by repair of the tissue, and finally, healing and resolution (Forte et al., 2018). While a plethora of macrophage phenotypes that are involved in the formation and progression of atherosclerosis have been recognized, including Mhem, Mox, and M4, the following section will focus on two extreme ends of the spectrum, namely M1 and M2 macrophages.
To limit tissue necrosis due to oxygen insufficiency and thus improve prognosis in MI, it is time-critical to reverse ischemia by reperfusion. This ischemia/reperfusion (I/R) can be achieved, for example, by pharmaceutical thrombolysis or mainly mechanical intervention, such as percutaneous coronary intervention (PCI), in which the occluded vessel can resume blood supply to the tissue using angioplasty followed by stent implantation (Vincent et al., 2017). In addition, macrophages are known to play a crucial role in the course of ischemic MI with subsequent reperfusion (Vagnozzi et al., 2020), which is related to their pro- and anti-inflammatory phenotype.
There is a broad consensus that the M1 phenotype of macrophages, especially at the onset of I/R, further increases the damage by secreting inflammatory factors and proteases and producing ROS (Formigli et al., 2001; Steffens et al., 2009). However, recent studies demonstrate that phagocytosis performed by M1 macrophages plays a vital role in subsequent tissue repair (Figure 1). M1 macrophages predominate in the myocardium and are mainly polarized by the transmembrane receptor dectin-1, leading to ROS formation but also to NF-κB activation and subsequent secretion of pro-inflammatory cytokines and chemokines (CCL5, IL-1β, IL-6, IL-12, MCP-1, TNF-α), which worsens the condition after I/R (Fan et al., 2019; Leuti et al., 2021). Diametrically, a previous study has shown that the soluble receptor for glycation end products positively affects cardiac function in the mouse in a model for MI and subsequent I/R by promoting infiltration of M1 macrophages into the damaged tissue and IFN-γ secretion (Zhang et al., 2019). This fact suggests a certain ambivalence to M1 macrophages: While they participate in tissue protection in acute ischemia, they also have the capacity to significantly damage the tissue by secreting pro-inflammatory cytokines and recruiting immune cells.
In contrast to M1 macrophages, M2 macrophages, which are polarized by type 2 T helper (TH2) cell cytokines (IL-4, IL-13), can secrete anti-inflammatory cytokines (IL-10, TGFβ) and anti-fibrogenic factors that promote a favorable prognosis through their anti-inflammatory and reparative functions (Figure 2) (Leuti et al., 2021). Another study has shown that M2 macrophages alternatively polarized by retinoic acid receptor responder protein 2 (RARRES2) can protect against damage in I/R. This study was performed in mice, and it was observed that pro-inflammatory cytokines were inhibited while the level of anti-inflammatory IL-10 was increased (Chang et al., 2015). A macrophage subtype has been in focus for some time, the regulatory M2b macrophages. Clinical studies have shown that after I/R due to MI, injecting the M2b subtype into the damaged tissue improved myocardial function and reduced cardiac fibrosis (Yue et al., 2020b). Another observation shows that this subtype is not directly involved in the healing process. However, its phenotypic expression modulates the inflammatory immune response and eventually supports a protective and tissue-building effect after I/R (Yue et al., 2020a).
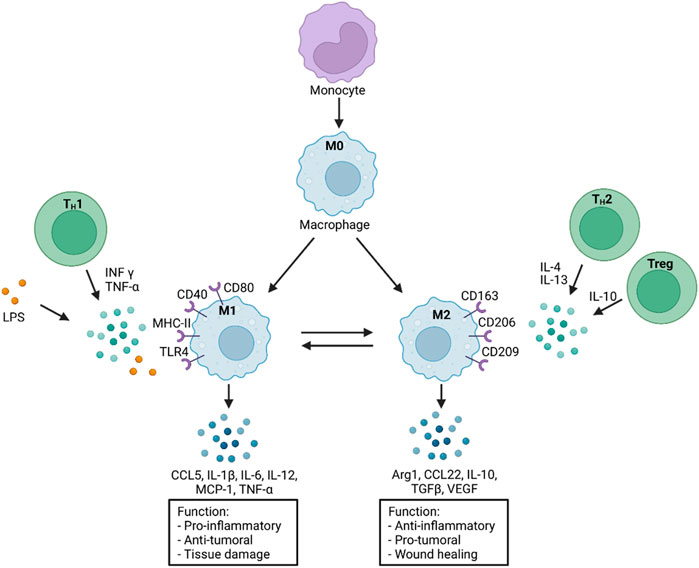
FIGURE 2. Monocyte differentiation and macrophage polarization in response to various stimuli. A selection of distinct functions is presented on the bottom. M0: macrophages without polarization; INF-γ: Interferon gamma; TNF-α: Tumor necrosis factor alpha; LPS: Lipopolysaccharide; CD: Cluster of differentiation; MHC-II: Major histocompatibility complex-II; TLR4: Toll like receptor 4; CCL5: CC-Chemokine ligand 5; MCP-1: Monocyte chemoattractant protein-1 (CCL2); IL: Interleukin; TGFβ: Transforming growth factor β; Arg1: Arginase 1; VEGF: Vascular endothelial growth factor; Treg: Regulatory T cells; TH1/2: T Helper cells 1/2. Scheme was summarized from data in (Yao et al., 2019; Daiber et al., 2021). Created with BioRender.com.
As mentioned before, macrophage recruitment as well as the differentiation of M1 and M2 macrophages is a central anchor point in the development of many chronic inflammatory diseases. For this reason, many scientists focus on macrophages to find a possible new therapeutic target to suppress inflammation. It was shown that human placental mesenchymal stem cells (pMCS) could affect macrophage function as well as the differentiation of human monocytes from M1 macrophages to anti-inflammatory M2 macrophages and are therefore potential targets for cell-based therapy. Accompanying the phenotypic shift by pMCSs, the pattern of protein expression and secretion is modulated, and typical M1 macrophage proteins (CD40), chemokines (MIP-1α), and cytokines (IL-1β and IL-12) are significantly downregulated in addition the phagocytic activity of apoptotic cells is upregulated after the differentiation towards M2 macrophages (Abumaree et al., 2013). In another approach BML-111 an analog of lipoxin4, which is an anti-inflammatory lipid mediator, was used to resolve neuroinflammation after ischemic stroke. The infarct size, as well as M1 CD40+ macrophage content, was reduced 1 week after stroke and daily injections of BML-111 in rats (Hawkins et al., 2017). Moreover, 48 h after stroke and BML-111 treatment, reduced levels of pro-inflammatory chemokines (MIP-1α and MCP-1) and cytokines (TNF-α and IFN-γ), as well as increased levels of anti-inflammatory cytokines (IL-4 and IL-10), were observed. In a third study, activated macrophages of rheumatoid arthritis patients were treated with anti-TNF-α agents, which lead to decreased production of pro-inflammatory cytokines (TNF-α, IL-6, IL-12), and decreased expression of inflammation marker (CD40), enhanced phagocytic activity as well as polarization towards M2 phenotype. The main driver of the alternative macrophage polarization after TNF-α treatment was the IL10/STAT3 signaling pathway (Degboé et al., 2019). TNF-α inhibitors are already successfully used in the treatment of Chron’s disease or rheumatoid arthritis (Gerriets et al., 2022). Another study analyzed the influence of resolvin D1 in vitro as well as in a liver I/R injury model in mice. It was shown that resolvin D1 has anti-inflammatory and pro-resolving properties and leads to enhanced M2 macrophage polarization, which was accompanied by reduced INF-γ, TNF-α, and IL-6 expression but increased IL-4 and IL-10 expression (Kang and Lee, 2016). Taken together pMCSs, cys-SPMs, BML-111 and resolvin D1 could be potential novel therapeutic targets for chronic inflammatory diseases.
In the last decades the CD40-CD40L signaling cascade became recognized as an important player in inflammatory and autoimmune diseases like atherosclerosis, diabetes type 1, rheumatoid arthritis, lupus erythematosus, and allograft rejection (Karnell et al., 2019). The signaling cascade is crucial for the development of inflammatory processes and the generation as well as stabilization of arterial thrombi (Michel et al., 2017; Daub et al., 2020).
CD40L (or CD154, 39 kDa protein) belongs to the tumor necrosis factor (TNF) family and is expressed on platelets, activated T lymphocytes, vascular smooth muscle cells, endothelial cells, monocytes, and macrophages. CD40L appears as a surface expressed protein as well as a soluble protein (sCD40L), which is mainly secreted by platelets (Foy et al., 1996; Michel et al., 2017). Four different CD40L receptor proteins are described in the literature (Figure 3): the integrins αIIbβ3, α5β1 (or VLA-5), and αMβ2 (or Mac-1), as well as CD40 (Bosmans et al., 2021). It was shown that CD40L binding to integrin receptor Mac-1 mediates monocyte adhesion, migration as well as myeloperoxidase (MPO) secretion. Inhibition of Mac-1 in vivo leads to reduced macrophage accumulation in atherosclerotic lesions, which slows down the progression of atherosclerosis (Zirlik et al., 2007). Binding of sCD40L to platelet integrin αIIbβ3 mediates stabilization of arterial thrombi (André et al., 2002). Monocytic U937 cells are activated by sCD40L-VLA-5 interaction, which results in extracellular-signaling-regulated kinase 1/2 (ERK1/2) pathway activation and gene expression (Léveillé et al., 2007). After CD40L-VLA-5 interaction increased production of pro-inflammatory chemokines and cytokines like IL-1β, IL-6, and MCP-1 were observed (Loubaki et al., 2010; Bian et al., 2018). Furthermore, platelet bound CD40L can induce multimer formation of ultra-large von Willebrand factors (UlvWFs) which are important key players in the initial phase of atherosclerotic plaque genesis. It was shown that CD40L induced UlvWF formation mediates the adhesion of platelets to endothelial cell walls as well as monocyte recruitment (Popa et al., 2018). Taken together, CD40L-integrin signaling as well as CD40L induced UlvWF formation are important mediators of thrombi initialization, thrombi stabilization, inflammation, and macrophage activation.
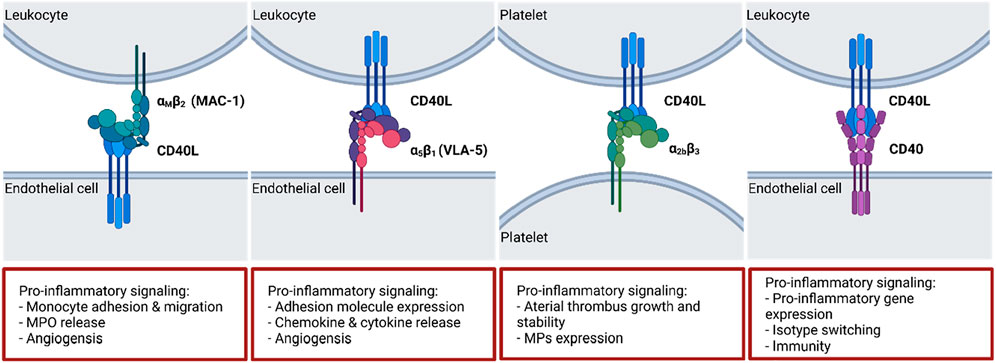
FIGURE 3. CD40(L) and its ligands and receptors. While CD40L has multiple binding partners, CD40 is largely restricted to ligation by CD40L. The dyad regulates multiple crucial biological processes including isotype switching, thrombus stabilization and cell activation. A selection of typical effects elicited by the interactions is presented on the bottom. ΑMβ2: Macrophage antigen 1 (MAC-1); α5β1: Fibronectin receptor (also: Very late antigen 5 (VLA-5)); α2bβ3: Integrin α2bβ3. Scheme was summarized from data in (Aloui et al., 2014; Michel et al., 2017). Created with BioRender.com.
CD40L with its classical binding partner CD40 plays a major role in immune responses (Figure 3). CD40 (54 kDa protein) belongs also to the TNF receptor superfamily and is expressed on thrombocytes, immune cells (B cells, dendritic cells, neutrophils, activated T cells, and macrophages) as well as on non-immune cells (endothelial cells, vascular smooth muscle cells, and fibroblasts) (Paulie et al., 1984). Mutations in CD40 or CD40L gene can disrupt its interaction capability and lead to the development of hyper IgM syndrome, which comprises defective class switch recombination, somatic hypermutation, defective T cell priming as well as macrophage and dendritic cell activation which can lead to death by infections due to the inborn immune suppression (La Morena, 2016). In general, CD40 trimerizes and binds to CD40L whereas CD40 itself has no intrinsic capabilities and needs adaptor proteins for signal transduction following its activation (Schönbeck and Libby, 2001). These adaptor proteins are TNF receptor associated factors (TRAFs), which bind to the cytosolic tail of CD40 and activate downstream pathways. CD40 contains two separate binding sites for TRAFs: one distal binding site for TRAF2/3/5 and a proximal one for TRAF6 (Bishop and Hostager, 2001; Bradley and Pober, 2001). A brief summary of the different TRAF molecules (TRAF1-7) and their role in the CD40-CD40L signaling pathway will be given in the following sections.
Heterotrimer formation of TRAF1 (46 kDa), TRAF2 (56 kDa) and CD40 leads to the activation of nuclear transcription factor (NF)-κB and pro-inflammatory p38 mitogen-activated protein kinase (MAPK) pathways (Arron et al., 2002; Wicovsky et al., 2009). TRAF1 protein levels are low in non-activated immune cells, its expression is strongly upregulated in activated lymphocytes and monocytes in an NF-κB dependent manner (Schwenzer et al., 1999; Edilova et al., 2018). It was shown that TRAF1 deficiency led to reduced pulmonary leukocyte recruitment after lipopolysaccharide inhalation presumably by impaired adhesion molecule expression (e.g., ICAM-1 and VCAM-1) as well as impaired chemokine expression (Oyoshi et al., 2007). In high fat treated mice with TRAF1 and low density lipoprotein receptor (TRAF1−/−/LDLR−/−) deficiency smaller atherosclerotic lesions, which contain less lipids and higher amounts of collagen in comparison to control mice, were observed. In detail, it was shown that the atherosclerotic lesions in TRAF1−/−/LDLR−/− mice contain less macrophages and that the adhesion of monocytes is partially inhibited by reduced adhesion molecule expression (e.g., ICAM-1 and VCAM-1) in macrophages and endothelial cells. The differentiation of macrophages was not affected in this mouse model. Furthermore, in patients suffering from acute coronary syndrome a higher TRAF1 blood mRNA expression was observed (Missiou et al., 2010). These findings indicate a pro-inflammatory role of TRAF1 signaling in atherosclerosis.
CD40 forms homodimers with TRAF2 (56 kDa) and lead to an activation of NF-κB, C-Jun N-terminal kinases (JNK) or p38 MAPK signaling (Park et al., 1999; Lalani et al., 2018). TRAF2 deficiency causes chronic inflammation and impaired T cell homeostasis, which is lethal in mice (Lin et al., 2011). It was shown, that TRAF2 protects against autoimmune inflammatory processes and TNF-α induced apoptosis via NF-κB signaling pathway (Yeh et al., 1997). TRAF3 (65 kDa) activates NF-κB signaling after CD40 binding similar to TRAF2 (Devergne et al., 1996; Duckett et al., 1997). TRAF3−/− mice show also a very limited lifespan and a deficiency of TRAF3 protein leads to permanent activation of NF-κB which leads to an multi-organ inflammation as a result of higher cytokine (e.g., IL6 and IL12) levels (Xu et al., 1996; Lalani et al., 2015). Overexpression of TRAF3 prevents CD40-induced endothelial cell activation and inhibits the expression of pro-inflammatory cytokines (Urbich et al., 2001). It seems that TRAF4 does not affect the immune cell development and is not involved in vascular inflammation and/or atherosclerosis (Gissler et al., 2022). TRAF4 is involved in more developmental processes like the formation of the respiratory upper tract (Shiels et al., 2000). TRAF5 (64 kDa) can form homodimers with TRAF3 which force the binding of TRAF5 to CD40. CD40-TRAF5 interaction also activates NF-κB and JNK signaling and as TRAF2 and TRAF3, TRAF5 seems to have also anti-inflammatory properties (Pullen et al., 1998; Leo et al., 1999; Gissler et al., 2021a). For example, TRAF5 shows protective characteristics in a myocardial ischemia/reperfusion injury (Xu et al., 2020). TRAF5 deficiency in an obese mouse model lead to enhanced adipose tissue inflammation compared to control mice which also indicate the anti-inflammatory signaling properties of TRAF5 (Gissler et al., 2021a). In addition, it was shown that TRAF2/3/5 signaling does not affect atherosclerosis or neointima formation (Donners et al., 2008; Lutgens et al., 2010).
In contrast to TRAF2/3/5, TRAF6 (63 kDa) is involved in multiple pro-inflammatory processes (Lutgens et al., 2010). NF-κB signaling is activated through CD40-TRAF6 interaction (Cao et al., 1996). TRAF6 also promotes JNK and p38 MAPK signaling cascades through binding to TRAF2 (Baud et al., 1999). The deficiency of TRAF6 in mice has a serve impact of the immune system: mice lacking TRAF6 show inflammatory cell accumulation in organs, thymic atrophy as well as lack of secondary lymphatic organs (Akiyama et al., 2005; Gissler et al., 2022). The lack of CD40-TRAF6 binding leads to reduced inflammatory cytokine production resulting in the ameliorated progression of atherosclerosis compared to CD40-TRAF2/3/5 deficient mice. Furthermore, it was shown that TRAF6 signaling leads to inflammatory gene expression and polarization of macrophages from regulatory M2 macrophages to the pro-inflammatory M1 phenotype. TRAF6 also promotes monocyte recruitment into atherosclerotic plaques (Lutgens et al., 2010).
As mentioned before, CD40-TRAF signaling can activate separate downstream pathways like NF-κB, C- JNK and/or p38 MAPK (Wajant et al., 2001) (Figure 4). Target gene classes of these pathways are cytokines/chemokines (TNF-α or IL-6), immune receptors (CD40), adhesion molecules (VCAM-1), anti-apoptotic genes (TRAF1/2), enzymes (nNOS, HO-1), stress response genes, growth factors, transcription factors or miRNAs (miR-146) (Schieven, 2005; Waetzig et al., 2005; Mussbacher et al., 2019). In summary CD40L-CD40-TRAF6 signaling seems to be the main driver in macrophage activated pro-inflammatory signaling in atherosclerosis and may therefore promote tissue damage and cell death after MI. In contrast, CD40L-CD40-TRAF2/3/5 signaling seems to have a more regulatory function in the immune system and may contribute to resolution. Regarding TRAF7 (74 kDa) and its role in inflammation or vascular disease nothing has been published so far (Gissler et al., 2022). A more detailed overview concerning the role of all TRAF proteins and their role in vascular inflammation and atherosclerosis has been published (Gissler et al., 2022).
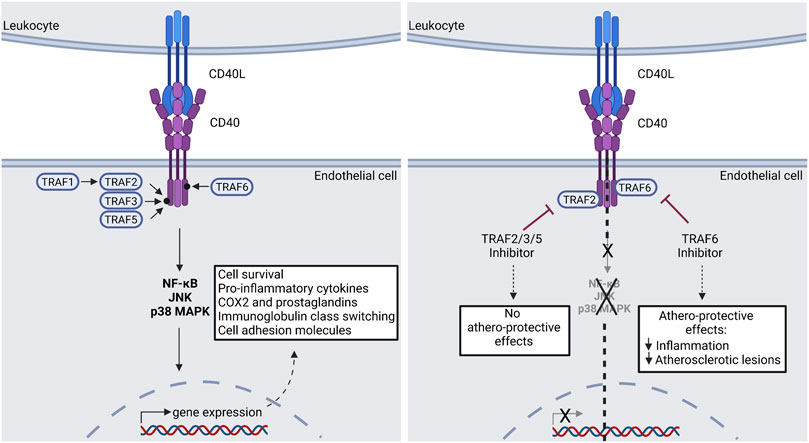
FIGURE 4. CD40(L) signal transduction via TRAF molecules. Down-stream effects of the CD40(L) dyad can be devided into two main groups: The TRAF2/3/5-dependent effects are regarded as athero-protective, while the TRAF6-dependent effects promote atherosclerosis. A potential pharmacological approach is presented in the right panel. Consequently, selective TRAF6-inhibition could be used clinically in order to treat atherosclerosis. TRAF: Tumor Necrosis Factor Associated Factor; JNK: c-Jun N-terminal kinase; p38 MAPK: p38 mitogen-activated protein kinase; NF-κB: Nuclear factor kappa B; COX2: Cyclooxygenase 2. Scheme was summarized from data in (Seijkens et al., 2018). Created with BioRender.com.
In general, CD40L deficient mice have a phenotype which is comparable to the human hyper IgM syndrome. While they are exposed to an increased risk of infection, they are viable and fertile (Renshaw et al., 1994). CD40L−/− mice in a hypertension AT-II infusion model demonstrate improved endothelial function, suppression of blood platelet monocyte interaction, improved bleeding time as well as decreased oxidative stress in aorta, heart, and blood (Hausding et al., 2013). Also, in a hyperlipidemia mouse model (16 weeks of a high fat diet) CD40L−/− has a beneficial effect of weight gain and blood cholesterol levels in contrast to control mice. In addition high fat diet associated endothelial dysfunction and vascular oxidative stress, the expression of inflammatory markers and platelet activation is improved in CD40L−/− mice (Steven et al., 2018). In contrast to these data, some studies also address the negative sites of a complete CD40L blocking. It was shown that the lack of soluble CD40L affects the stability of arterial thrombi as frequently ruptured thrombi followed by embolism were observed in CD40L−/− mice in comparison to the control mice. The interaction of CD40L and the transmembrane protein Integrin αIIbβ3 was identified as crucial for thrombi stability (André et al., 2002; Prasad et al., 2003). Monoclonal antibody treatment directed against CD40L in a kidney allograft model in monkeys also resulted in major side effects caused by thromboembolic complications (Kawai et al., 2000). A CD40L antibody (BG9855) was also used in a clinical trial with patients suffering from lupus glomerulonephritis. The trial was terminated after thromboembolic events, which occurred as side effect in study participants (Boumpas et al., 2003). A new CD40L antibody (CDP7657) that lacks a functional Fc region was developed, as the Fc region of the antibody was the supposed mediator of platelet activation, thereby inducing thrombotic events (Shock et al., 2015). A clinical phase 3 trial employing the new antibody under the name Dapirolizumab pegol is currently underway, after safety regarding thrombotic events had been demonstrated earlier (Tocoian et al., 2015; Chamberlain et al., 2017; NCT04294667). Another clinical trial with a humanized monoclonal antibody directed against CD40L (Toralizumab/IDEC-131) is ongoing. The antibody is used in patients suffering from multiple sclerosis and in Phase 1 no thromboembolic events have been reported (Fadul et al., 2021).
CD40 deficient mice are also viable and fertile but show impaired immunoglobulin class switching and germinal center formation (Kawabe et al., 1994). In comparison to CD40L−/− mice, high fat treated CD40−/− mice show no ameliorated tissue inflammation. In contrast, in these animals severe inflammation induced by an increased number of pro-inflammatory CD11+F4/80+ macrophages within adipose tissue was observed in comparison to control animals. In accordance to these results, in obese CD40−/− mice an aggravated insulin resistance was observed compared to wild-type animals (Chatzigeorgiou et al., 2014). In CD40−/− ApoE−/− mice that lack CD40 as well as the ApoE receptor, thus suffering from a severely impaired capability to handle cholesterol, reduced atherosclerosis progression was observed in comparison to control mice when subjected to a high-fat diet. In detail a reduced plaque size in aortic arches as well as a more fibrotic plaque composition was observed in these mice. In addition, the content of macrophages within the plaques was reduced and the macrophages were polarized to a more anti-inflammatory phenotype in mice lacking CD40 (Lutgens et al., 2010). The main driver of pro-inflammatory CD40 signaling seems to be leukocyte-dependent since impaired macrophage migration and decreased atherosclerosis was observed in leukocyte specific CD40-deficient mice (Lutgens et al., 2010). Several clinical trials in humans regarding CD40 blocking via antibodies are currently ongoing. For example, the monoclonal anti-CD40 antibody ASK1240 is being tested as an immunosuppressant to prevent allograft rejection. Phase 1 was completed and no thromboembolic events or cytokine release syndromes were observed, which allowed further testing in phase 2 trials (Goldwater et al., 2013; Vincenti et al., 2020). The pharmacokinetics and safety of TSK1240 antibody has subsequently been tested in patients suffering of plaque psoriasis (Anil Kumar et al., 2018). Another monoclonal CD40 antibody termed BI 655064 was already tested in phase 1 and 2a as a potential treatment agent for several autoimmune diseases like rheumatoid arthritis, systemic lupus erythematosus or lupus nephritis. So far, the antibody treatment has shown no severe side effects like thromboembolic events (Albach et al., 2018; Visvanathan et al., 2019). More clinical trials are still ongoing regarding the use of monoclonal CD40 antibodies for the treatment of autoimmune diseases like Chron´s disease (Kasran et al., 2005) or systemic lupus erythematosus (Perper et al., 2019).
Taken together it remains unclear whether the pharmacological blockade of CD40(L) as a treatment method for atherosclerosis or other inflammatory diseases will be a viable strategy for the future due to the side effects observed previously, encompassing severe immunosuppression (CD40) or thromboembolisms (CD40L).
A systemic blocking of CD40L or CD40 signaling to date does not seem promising as a long-term treatment of atherosclerosis due to the aforementioned unwanted side effects of immunosuppression or thromboembolic events. Therefore, a more targeted therapeutic strategy could lie in the development of cell type specific antibodies or targeted nanopharmaceuticals to avoid negative side effects of a complete or global CD40/CD40L blocking (Duivenvoorden et al., 2019; van de Vyver et al., 2021). Several in vivo studies regarding cell type specific deficiency of CD40/CD40L are still ongoing with promising data (Gissler et al., 2021b; Lacy et al., 2021; Bosmans et al., 2022). For example, endothelial-cell specific deletion of CD40 in ApoE−/− mice leads to a more stable plaque phenotype and to reduced leukocyte adhesion compared to control mice when subjected to a high-fat diet. No systemic parameters were affected in these mice and it was also shown that CD40 expression is upregulated in endothelial cells during atherogenesis (Gissler et al., 2021b). Also, T-cell and dendritic cell specific CD40L/CD40 deletion in ApoE−/− mice demonstrate reduced atherosclerotic plaque development as well as reduced plaque inflammation. In these mice a reduced IFN-γ expression as well as impaired T helper 1 polarization was observed, which suggests that CD40L on T cells and dendritic cells is playing an important role in T cell mediated inflammation. It was also shown that in human plasma and atherosclerotic plaques sCD40 as well as sCD40L levels correlate with circulating IFN-γ levels.
These results indicate that in human atherosclerosis, CD40-CD40L signaling is involved in the IFN-γ response which is attributed to T helper 1 cells. As the deletion of platelet-specific CD40L did not affect the size of atherosclerotic plaques or plaque phenotype, the ligand seems to be more involved in athero-thrombosis than atherosclerosis (Lacy et al., 2021). Macrophage specific deletion of CD40 in ApoE−/− mice results in a reduced activated immune profile compared to control mice. In CD40mac−/− mice, reduced atherosclerotic plaque size, reduced necrotic core area, reduced macrophage content and increased collagen production was observed all of which indicate decreased progression of atherosclerosis compared to control mice. This phenotype is probably caused by alternative macrophage activation in atherosclerotic aortas which express a more anti-inflammatory profile than CD40-containing macrophages as well as the prevention of CD40 macrophage polarization towards pro-inflammatory states. Taken together, mice with CD40-deficient macrophages show less inflammation in the atherosclerotic aorta (Bosmans et al., 2022). The cell type specific deletion of CD40 is an emerging field, which could lead to a better understanding of the involvement of CD40-CD40L signaling in inflammation and the progression of atherosclerosis. These data indicate that a cell type specific deletion of CD40(L) could be a promising tool in the future for the treatment of atherosclerosis. Certainly, more research must be done in this regard, but it can be assumed that a partial blocking of CD40 would entail fewer side effects than a global blocking.
Due to the severe side effects of a complete CD40/CD40L blockade in the first clinical trials further research was done to identify downstream targets for a more selective blockade with reduced side effects. In 2010, it was shown that CD40-TRAF6 signaling but not CD40-TRAF2/3/5 signaling is an important key player in activation of pro-inflammatory immune responses and in the development of atherosclerosis (Lutgens et al., 2010). Therefore, small TRAF stop molecules were created as a new pharmacological drug to target CD40-TRAF6 signaling (Zarzycka et al., 2015). The drug component 6877002 was first tested in an obesity mouse model. The mice were fed with a high fat diet and treated for 6 weeks with the new TRAF6 inhibitor. After treatment, improved insulin sensitivity, reduced CD11b+F480+CD11c+ (M1) macrophages and reduced hepatosteatosis were observed in comparison to the control mice. This indicates that pharmacological TRAF6 inhibition could be a target to prevent obesity related inflammation and metabolic complications (Lutgens et al., 2010; Chatzigeorgiou et al., 2014). Related to this study, pharmacological TRAF6 inhibition was tested in a diabetes mellitus type 2 mouse model for anti-inflammatory properties. The drug treatment lead to reduced body weight gain, enhanced endothelial function, reduced oxidative stress in aorta as well as to reduced inflammatory marker concentration (IL-1β, 3-NT) in blood plasma in comparison to untreated control mice (Steven et al., 2018). Also, this study demonstrates the anti-inflammatory effect of TRAF6 inhibition. An initial study regarding the potential treatment of atherosclerosis with small TRAF6 stops was already performed. There it was demonstrated that TRAF6 stop 6877002 treatment in ApoE−/− mice reduces existing atherosclerosis by decreased macrophage activation and leukocyte recruitment (Seijkens et al., 2018).
In contrast to a general CD40 inhibition, TRAF6 inhibition preserves immunity and does not affect germinal center formation or Ig isotype switching of B cells neither dendritic cell induced co-stimulation of T cells. In addition, selective TRAF6 inhibition prevents pro-inflammatory cytokine secretion of macrophages as well as immune suppressive side effects by preserving CD40-TRAF2/3/5 (anti-inflammatory) signaling and reducing thereby established atherosclerosis. For this reason, TRAF stops represent a promising treatment strategy for atherosclerosis (Seijkens et al., 2018). The TRAF stop treatment was improved by a more targeted macrophage specific TRAF6 inhibition via recombinant high density lipoprotein (rHDL) nanotherapy. For this approach TRAF stop 6877002 was incorporated into rHDL nanoparticles. After rHDL-6877002 treatment in ApoE−/− mice as well as non-human primates reduced atherosclerotic plaque size in the aortic roots as well as fewer macrophages within the plaques were observed. The advantage of this targeted nanoparticle treatment in comparison to an untargeted TRAF stop treatment is a reduced dosage application as well as reduced treatment frequency (Lameijer et al., 2018; Seijkens et al., 2018). In general, pharmacological TRAF6 inhibition with component 6877002 was not only analyzed in obesity or atherosclerotic mouse models, but was also found to be a potential therapeutic strategy in other inflammatory or autoimmune diseases like neuroinflammation/multiple sclerosis (Aarts et al., 2017), breast cancer (Bishop et al., 2020), bone cancer (Marino et al., 2022) and in heart failure (Bosch et al., 2019; Marino et al., 2022).
MiRNA expression is closely linked to many pathological processes like cardiovascular diseases, atherosclerosis and often accompanying disease like obesity or diabetes (Tang et al., 2008; Santovito et al., 2012; Iacomino and Siani, 2017; Wojciechowska et al., 2017). Especially two cytokine responsive miRNAs (miR-146 and miR-181) are described in the literature to be involved in the CD40-TRAF signaling cascade in endothelial cells (Figure 5) (Feinberg and Moore, 2016).
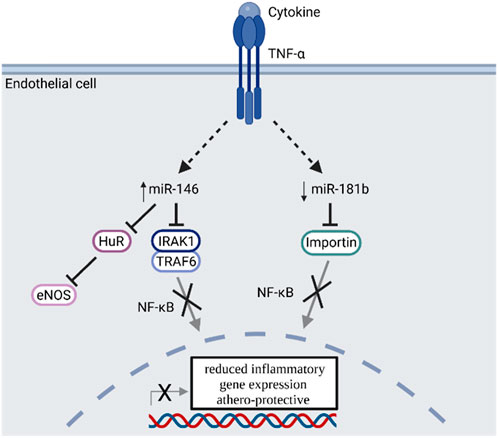
FIGURE 5. TNF-α signal transduction enhances miR-146 and reduces miR-181b expression, which inhibits NF-κB signaling and is regarded as athero-protective. TNF-α: Tumor necrosis factor alpha; miR: microRNA; HuR: Human antigen R protein; eNOS: endothelial nitric oxide synthase; IRAK1: Interleukin 1 receptor associated kinase 1; TRAF6: TNF receptor associated factor 6; NF-κB: Nuclear factor kappa B. Scheme was summarized from data in (Feinberg and Moore, 2016). Created with BioRender.com.
The expression of miR-146a in endothelial cells is induced by TNF-α or IL-1ß and effects the resolution of inflammatory gene expression by regulating NF-κB signaling via targeting TRAF6 and interleukin 1 receptor associated kinase 1 (IRAK1), as well as via the regulation of eNOS expression by targeting HuR in a NF-κB independent manner (Cheng et al., 2013). Overexpression of miR-146a results in reduced inflammatory signaling of endothelial cells. MiR-146 expression is also upregulated in atherosclerotic plaques and is involved in the cholesterol metabolism of macrophages. In oxidized LDL-stimulated macrophages, miR-146 expression leads to a reduced cytokine release and lipid uptake (Yang et al., 2011). Furthermore, miR-146 affects the balance between M1 and M2 macrophages in atherosclerotic plaques (Vergadi et al., 2014). In summary, miR-146 shows anti-inflammatory properties and plays a regulatory role in immune cells like T cells, macrophages and dendritic cells (Feinberg and Moore, 2016). Uniquely in endothelial cells but not leukocytes, miR-181b targets importin-α3 and inhibits NF-κB signaling as well as vascular inflammation (Sun et al., 2014). Furthermore miR-181a is involved in the pathogenesis of atherosclerosis as it targets c-FOS and OPN and reduces the immune-inflammatory response induced by oxidized LDL by decreasing the expression of cell surface proteins like CD40 or CD83 on dendritic cells (Wu et al., 2012). Consequently, pro-inflammatory stimuli lead to a reduced expression of miR-181 in vitro and in vivo (Sun et al., 2014). In summary miR-146 and miR-181 both regulate parts of the NF-κB signaling and have athero-protective properties. While these properties are elicited by an overexpression of miR-146, atherosclerotic plaques are protected by decreasing miR-181 expression (Figure 5).
Cardiovascular disease represents a major burden to global health, causing high mortality and reducing quality of life. There are many factors influencing CVD, ranging from rather banal yet impactful factors like lack of sleep and exercise and unhealthy nutrition, to more defined ones encompassing oxidative stress and inflammatory processes. Atherosclerosis and myocardial infarction can be regarded as driven by the latter concepts, evidenced by the degradation of damaged and regeneration of new tissue occurring in a chronologically balanced course during a cardiovascular event via an inflammatory process, termed resolution and remodeling. Crucial within this process are macrophages (M1/M2), which promote both, inflammation as well as resolution, degrading tissue and eliciting regeneration. The CD40(L) dyad is in turn intertwined in the signaling cascades influencing monocytes in their activation and immunological phenotype. A general blockade of CD40(L) signaling has of yet proven impractical regarding the treatment of CVD as the dyad is also involved in thrombus formation and stabilization. In summary, targeting the dyad on specific cells or at specific levels of the involved signaling cascades could be a promising tool in order to regain tissue-homeostasis after MI and subsequent inflammatory processes.
LS and HU wrote most parts of the article and collected the relevant references. All figures were created by LS. TM, AD, and SD critically revised the manuscript and provided funding and supervision. All authors have read and agreed with the final version of the manuscript.
SD was supported by a vascular biology research grant on “CD40L and inflammation in hypertension” of the Else-Kröner-Fresenius Foundation (2019A110). AD was supported by shared expertise grants of the DZHK (German Center for Cardiovascular Research), Partner Site Rhine-Main, Mainz, Germany. TM is PI of the DZHK (German Center for Cardiovascular Research), Partner Site Rhine-Main, Mainz, Germany. Our research was continuously supported by the European Cooperation in Science and Technology and EU-CARDIOPROTECTION COST-ACTION (CA16225), a funding scheme to enhance scientific networking in Europe.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aarts, S. A. B. M., Seijkens, T. T. P., Kusters, P. J. H., van der Pol, S. M. A., Zarzycka, B., Heijnen, P. D. A. M., et al. (2017). Inhibition of CD40-TRAF6 interactions by the small molecule inhibitor 6877002 reduces neuroinflammation. J. Neuroinflammation 14, 105. doi:10.1186/s12974-017-0875-9
Abumaree, M. H., Al Jumah, M. A., Kalionis, B., Jawdat, D., Al Khaldi, A., Abomaray, F. M., et al. (2013). Human placental mesenchymal stem cells (pMSCs) play a role as immune suppressive cells by shifting macrophage differentiation from inflammatory M1 to anti-inflammatory M2 macrophages. Stem Cell. Rev. Rep. 9, 620–641. doi:10.1007/s12015-013-9455-2
Akiyama, T., Maeda, S., Yamane, S., Ogino, K., Kasai, M., Kajiura, F., et al. (2005). Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science 308, 248–251. doi:10.1126/science.1105677
Albach, F. N., Wagner, F., Hüser, A., Igel, J., Joseph, D., Hilbert, J., et al. (2018). Safety, pharmacokinetics and pharmacodynamics of single rising doses of BI 655064, an antagonistic anti-CD40 antibody in healthy subjects: A potential novel treatment for autoimmune diseases. Eur. J. Clin. Pharmacol. 74, 161–169. doi:10.1007/s00228-017-2362-8
Aloui, C., Prigent, A., Sut, C., Tariket, S., Hamzeh-Cognasse, H., Pozzetto, B., et al. (2014). The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int. J. Mol. Sci. 15, 22342–22364. doi:10.3390/ijms151222342
André, P., Prasad, K. S. S., Denis, C. V., He, M., Papalia, J. M., Hynes, R. O., et al. (2002). CD40L stabilizes arterial thrombi by a beta3 integrin--dependent mechanism. Nat. Med. 8, 247–252. doi:10.1038/nm0302-247
Anil Kumar, M. S., Papp, K., Tainaka, R., Valluri, U., Wang, X., Zhu, T., et al. (2018). Randomized, controlled study of bleselumab (ASKP1240) pharmacokinetics and safety in patients with moderate-to-severe plaque psoriasis. Biopharm. Drug Dispos. 39, 245–255. doi:10.1002/bdd.2130
Arnett, D. K., Blumenthal, R. S., Albert, M. A., Buroker, A. B., Goldberger, Z. D., Hahn, E. J., et al. (2019). 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: A report of the American College of cardiology/American heart association task force on clinical practice guidelines. Circulation 140, e596–e646. doi:10.1161/CIR.0000000000000678
Arron, J. R., Pewzner-Jung, Y., Walsh, M. C., Kobayashi, T., and Choi, Y. (2002). Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J. Exp. Med. 196, 923–934. doi:10.1084/jem.20020774
Balci, D. D., Balci, A., Karazincir, S., Ucar, E., Iyigun, U., Yalcin, F., et al. (2009). Increased carotid artery intima-media thickness and impaired endothelial function in psoriasis. J. Eur. Acad. Dermatol. Venereol. 23, 1–6. doi:10.1111/j.1468-3083.2008.02936.x
Bartekova, M., Barancik, M., Ferenczyova, K., and Dhalla, N. S. (2018). Beneficial effects of N-acetylcysteine and N-mercaptopropionylglycine on ischemia reperfusion injury in the heart. Curr. Med. Chem. 25, 355–366. doi:10.2174/0929867324666170608111917
Baud, V., Liu, Z. G., Bennett, B., Suzuki, N., Xia, Y., and Karin, M. (1999). Signaling by proinflammatory cytokines: Oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes. Dev. 13, 1297–1308. doi:10.1101/gad.13.10.1297
Becker, L., Prado, K., Foppa, M., Martinelli, N., Aguiar, C., Furian, T., et al. (2012). Endothelial dysfunction assessed by brachial artery ultrasound in severe sepsis and septic shock. J. Crit. Care 27, 316.e9–14. doi:10.1016/j.jcrc.2011.08.002
Bergholm, R., Leirisalo-Repo, M., Vehkavaara, S., Mäkimattila, S., Taskinen, M.-R., and Yki-Järvinen, H. (2002). Impaired responsiveness to NO in newly diagnosed patients with rheumatoid arthritis. Arterioscler. Thromb. Vasc. Biol. 22, 1637–1641. doi:10.1161/01.ATV.0000033516.73864.4E
Bian, Z.-M., Field, M. G., Elner, S. G., Kahlenberg, J. M., and Elner, V. M. (2018). Distinct CD40L receptors mediate inflammasome activation and secretion of IL-1β and MCP-1 in cultured human retinal pigment epithelial cells. Exp. Eye Res. 170, 29–39. doi:10.1016/j.exer.2018.02.014
Bishop, G. A., and Hostager, B. S. (2001). Molecular mechanisms of CD40 signaling. Arch. Immunol. Ther. Exp. 49, 129–137.
Bishop, R. T., Marino, S., Carrasco, G., Li, B., Allen, R. J., Sparatore, A., et al. (2020). Combined administration of a small-molecule inhibitor of TRAF6 and Docetaxel reduces breast cancer skeletal metastasis and osteolysis. Cancer Lett. 488, 27–39. doi:10.1016/j.canlet.2020.05.021
Blankenberg, S., Rupprecht, H. J., Bickel, C., Torzewski, M., Hafner, G., Tiret, L., et al. (2003). Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N. Engl. J. Med. 349, 1605–1613. doi:10.1056/NEJMoa030535
Bosch, L., de Haan, J., Seijkens, T., van Tiel, C., Brans, M., Pasterkamp, G., et al. (2019). Small molecule-mediated inhibition of CD40-TRAF6 reduces adverse cardiac remodelling in pressure overload induced heart failure. Int. J. Cardiol. 279, 141–144. doi:10.1016/j.ijcard.2018.12.076
Bosmans, L. A., Bosch, L., Kusters, P. J. H., Lutgens, E., and Seijkens, T. T. P. (2021). The CD40-cd40l dyad as immunotherapeutic target in cardiovascular disease. J. Cardiovasc. Transl. Res. 14, 13–22. doi:10.1007/s12265-020-09994-3
Bosmans, L. A., van Tiel, C. M., Aarts, S. A. B. M., Willemsen, L., Baardman, J., van Os, B. W., et al. (2022). Myeloid CD40 deficiency reduces atherosclerosis by impairing macrophages' transition into a pro-inflammatory state. Cardiovasc. Res. Online ahead of print. doi:10.1093/cvr/cvac084
Boumpas, D. T., Furie, R., Manzi, S., Illei, G. G., Wallace, D. J., Balow, J. E., et al. (2003). A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 48, 719–727. doi:10.1002/art.10856
Bradley, J. R., and Pober, J. S. (2001). Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20, 6482–6491. doi:10.1038/sj.onc.1204788
Cao, Z., Xiong, J., Takeuchi, M., Kurama, T., and Goeddel, D. V. (1996). TRAF6 is a signal transducer for interleukin-1. Nature 383, 443–446. doi:10.1038/383443a0
Chamberlain, C., Colman, P. J., Ranger, A. M., Burkly, L. C., Johnston, G. I., Otoul, C., et al. (2017). Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann. Rheum. Dis. 76, 1837–1844. doi:10.1136/annrheumdis-2017-211388
Chang, C., Ji, Q., Wu, B., Yu, K., Zeng, Q., Xin, S., et al. (2015). Chemerin15-Ameliorated cardiac ischemia-reperfusion injury is associated with the induction of alternatively activated macrophages. Mediat. Inflamm. 2015, 563951. doi:10.1155/2015/563951
Chatzigeorgiou, A., Seijkens, T., Zarzycka, B., Engel, D., Poggi, M., van den Berg, S., et al. (2014). Blocking CD40-TRAF6 signaling is a therapeutic target in obesity-associated insulin resistance. Proc. Natl. Acad. Sci. U. S. A. 111, 2686–2691. doi:10.1073/pnas.1400419111
Cheng, H. S., Sivachandran, N., Lau, A., Boudreau, E., Zhao, J. L., Baltimore, D., et al. (2013). MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol. Med. 5, 1017–1034. doi:10.1002/emmm.201202318
Chiurchiù, V., Leuti, A., Saracini, S., Fontana, D., Finamore, P., Giua, R., et al. (2019). Resolution of inflammation is altered in chronic heart failure and entails a dysfunctional responsiveness of T lymphocytes. FASEB J. 33, 909–916. doi:10.1096/fj.201801017R
Choy, E. (2012). Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatol. Oxf. 51, v3–v11. doi:10.1093/rheumatology/kes113
Crispín, J. C., and Tsokos, G. C. (2010). IL-17 in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 943254. doi:10.1155/2010/943254
Daiber, A., Di Lisa, F., Oelze, M., Kröller-Schön, S., Steven, S., Schulz, E., et al. (2017a). Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 174, 1670–1689. doi:10.1111/bph.13403
Daiber, A., Steven, S., Weber, A., Shuvaev, V. V., Muzykantov, V. R., Laher, I., et al. (2017b). Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 174, 1591–1619. doi:10.1111/bph.13517
Daiber, A., Oelze, M., Daub, S., Steven, S., Schuff, A., Kröller-Schön, S., et al. (2014). “Vascular redox signaling, redox switches in endothelial nitric oxide synthase (eNOS uncoupling), and endothelial dysfunction,” in Systems biology of free radicals and antioxidants. Editor I. Laher (Berlin, Heidelberg: Springer), 1177–1211.
Daiber, A. (2010). Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 1797, 897–906. doi:10.1016/j.bbabio.2010.01.032
Daiber, A., Steven, S., Euler, G., and Schulz, R. (2021). Vascular and cardiac oxidative stress and inflammation as targets for cardioprotection. Curr. Pharm. Des. 27, 2112–2130. doi:10.2174/1381612827666210125155821
Daub, S., Lutgens, E., Münzel, T., and Daiber, A. (2020). CD40/CD40L and related signaling pathways in cardiovascular health and disease-the pros and cons for cardioprotection. Int. J. Mol. Sci. 21, E8533. doi:10.3390/ijms21228533
Degboé, Y., Rauwel, B., Baron, M., Boyer, J.-F., Ruyssen-Witrand, A., Constantin, A., et al. (2019). Polarization of rheumatoid macrophages by TNF targeting through an IL-10/STAT3 mechanism. Front. Immunol. 10, 3. doi:10.3389/fimmu.2019.00003
Devergne, O., Hatzivassiliou, E., Izumi, K. M., Kaye, K. M., Kleijnen, M. F., Kieff, E., et al. (1996). Association of TRAF1, TRAF2, and TRAF3 with an epstein-barr virus LMP1 domain important for B-lymphocyte transformation: Role in NF-kappaB activation. Mol. Cell. Biol. 16, 7098–7108. doi:10.1128/MCB.16.12.7098
Di Cesare, A., Di Meglio, P., and Nestle, F. O. (2009). The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J. Invest. Dermatol. 129, 1339–1350. doi:10.1038/jid.2009.59
Di Minno, A., Turnu, L., Porro, B., Squellerio, I., Cavalca, V., Tremoli, E., et al. (2016). 8-Hydroxy-2-Deoxyguanosine levels and cardiovascular disease: A systematic review and meta-analysis of the literature. Antioxid. Redox Signal. 24, 548–555. doi:10.1089/ars.2015.6508
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301. doi:10.1016/j.freeradbiomed.2011.06.033
Dikalov, S. I., and Dikalova, A. E. (2019). Crosstalk between mitochondrial hyperacetylation and oxidative stress in vascular dysfunction and hypertension. Antioxid. Redox Signal. 31, 710–721. doi:10.1089/ars.2018.7632
Dikalov, S., Itani, H., Richmond, B., Vergeade, A., Rahman, S. M. J., Boutaud, O., et al. (2019). Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am. J. Physiol. Heart Circ. Physiol. 316, H639–H646. doi:10.1152/ajpheart.00595.2018
Dikalova, A. E., Bikineyeva, A. T., Budzyn, K., Nazarewicz, R. R., McCann, L., Lewis, W., et al. (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116. doi:10.1161/CIRCRESAHA.109.214601
Dikalova, A. E., Itani, H. A., Nazarewicz, R. R., McMaster, W. G., Flynn, C. R., Uzhachenko, R., et al. (2017). Sirt3 impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension. Circ. Res. 121, 564–574. doi:10.1161/CIRCRESAHA.117.310933
Donners, M. M. P. C., Beckers, L., Lievens, D., Munnix, I., Heemskerk, J., Janssen, B. J., et al. (2008). The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood 111, 4596–4604. doi:10.1182/blood-2007-05-088906
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496. doi:10.1161/CIRCRESAHA.107.162800
Duckett, C. S., Gedrich, R. W., Gilfillan, M. C., and Thompson, C. B. (1997). Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol. Cell. Biol. 17, 1535–1542. doi:10.1128/MCB.17.3.1535
Duivenvoorden, R., Senders, M. L., van Leent, M. M. T., Pérez-Medina, C., Nahrendorf, M., Fayad, Z. A., et al. (2019). Nanoimmunotherapy to treat ischaemic heart disease. Nat. Rev. Cardiol. 16, 21–32. doi:10.1038/s41569-018-0073-1
Duncan, S. E., Gao, S., Sarhene, M., Coffie, J. W., Linhua, D., Bao, X., et al. (2020). Macrophage activities in myocardial infarction and heart failure. Cardiol. Res. Pract. 2020, 4375127. doi:10.1155/2020/4375127
Edilova, M. I., Abdul-Sater, A. A., and Watts, T. H. (2018). TRAF1 signaling in human health and disease. Front. Immunol. 9, 2969. doi:10.3389/fimmu.2018.02969
Fadul, C. E., Mao-Draayer, Y., Ryan, K. A., Noelle, R. J., Wishart, H. A., Channon, J. Y., et al. (2021). Safety and immune effects of blocking CD40 ligand in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 8, e1096. doi:10.1212/NXI.0000000000001096
Fan, Q., Tao, R., Zhang, H., Xie, H., Lu, L., Wang, T., et al. (2019). Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation 139, 663–678. doi:10.1161/CIRCULATIONAHA.118.036044
Feinberg, M. W., and Moore, K. J. (2016). MicroRNA regulation of atherosclerosis. Circ. Res. 118, 703–720. doi:10.1161/CIRCRESAHA.115.306300
Formigli, L., Manneschi, L. I., Nediani, C., Marcelli, E., Fratini, G., Zecchi Orlandini, S., et al. (2001). Are macrophages involved in early myocardial reperfusion injury? Ann. Thorac. Surg. 71, 1596–1602. doi:10.1016/S0003-4975(01)02400-6
Förstermann, U., and Münzel, T. (2006). Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 113, 1708–1714. doi:10.1161/CIRCULATIONAHA.105.602532
Forte, E., Furtado, M. B., and Rosenthal, N. (2018). The interstitium in cardiac repair: Role of the immune-stromal cell interplay. Nat. Rev. Cardiol. 15, 601–616. doi:10.1038/s41569-018-0077-x
Foy, T. M., Aruffo, A., Bajorath, J., Buhlmann, J. E., and Noelle, R. J. (1996). Immune regulation by CD40 and its ligand GP39. Annu. Rev. Immunol. 14, 591–617. doi:10.1146/annurev.immunol.14.1.591
Frodermann, V., Rohde, D., Courties, G., Severe, N., Schloss, M. J., Amatullah, H., et al. (2019). Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat. Med. 25, 1761–1771. doi:10.1038/s41591-019-0633-x
Fuster, J. J., Ouchi, N., Gokce, N., and Walsh, K. (2016). Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ. Res. 118, 1786–1807. doi:10.1161/CIRCRESAHA.115.306885
Gerlach, B. D., Marinello, M., Heinz, J., Rymut, N., Sansbury, B. E., Riley, C. O., et al. (2020). Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell. Death Differ. 27, 525–539. doi:10.1038/s41418-019-0370-1
Gerriets, V., Goyal, A., and Khaddour, K. (2022). StatPearls: Tumor necrosis factor inhibitors. Treasure Island (FL): StatPearls Publishing LLC.
Gissler, M. C., Anto-Michel, N., Pennig, J., Scherrer, P., Li, X., Marchini, T., et al. (2021a). Genetic deficiency of TRAF5 promotes adipose tissue inflammation and aggravates diet-induced obesity in mice. Arterioscler. Thromb. Vasc. Biol. 41, 2563–2574. doi:10.1161/ATVBAHA.121.316677
Gissler, M. C., Scherrer, P., Anto-Michel, N., Pennig, J., Hoppe, N., Füner, L., et al. (2021b). Deficiency of endothelial CD40 induces a stable plaque phenotype and limits inflammatory cell recruitment to atherosclerotic lesions in mice. Thromb. Haemost. 121, 1530–1540. doi:10.1055/a-1397-1858
Gissler, M. C., Stachon, P., Wolf, D., and Marchini, T. (2022). The role of tumor necrosis factor Associated factors (TRAFs) in vascular inflammation and atherosclerosis. Front. Cardiovasc. Med. 9, 826630. doi:10.3389/fcvm.2022.826630
Goldwater, R., Keirns, J., Blahunka, P., First, R., Sawamoto, T., Zhang, W., et al. (2013). A phase 1, randomized ascending single-dose study of antagonist anti-human CD40 ASKP1240 in healthy subjects. Am. J. Transpl. 13, 1040–1046. doi:10.1111/ajt.12082
Gorelenkova Miller, O., Behring, J. B., Siedlak, S. L., Jiang, S., Matsui, R., Bachschmid, M. M., et al. (2016). Upregulation of glutaredoxin-1 activates microglia and promotes neurodegeneration: Implications for Parkinson's disease. Antioxid. Redox Signal. 25, 967–982. doi:10.1089/ars.2015.6598
Griendling, K. K., and FitzGerald, G. A. (2003). Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ROS. Circulation 108, 1912–1916. doi:10.1161/01.CIR.0000093660.86242.BB
Hak, A. E., Karlson, E. W., Feskanich, D., Stampfer, M. J., and Costenbader, K. H. (2009). Systemic lupus erythematosus and the risk of cardiovascular disease: Results from the nurses' health study. Arthritis Rheum. 61, 1396–1402. doi:10.1002/art.24537
Halade, G. V., Norris, P. C., Kain, V., Serhan, C. N., and Ingle, K. A. (2018). Splenic leukocytes define the resolution of inflammation in heart failure. Sci. Signal. 11, eaao1818. doi:10.1126/scisignal.aao1818
Halade, G. V., Kain, V., Black, L. M., Prabhu, S. D., and Ingle, K. A. (2016). Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging (Albany NY) 8, 2611–2634. doi:10.18632/aging.101077
Halade, G. V., Kain, V., Ingle, K. A., and Prabhu, S. D. (2017). Interaction of 12/15-lipoxygenase with fatty acids alters the leukocyte kinetics leading to improved postmyocardial infarction healing. Am. J. Physiol. Heart Circ. Physiol. 313, H89–H102. doi:10.1152/ajpheart.00040.2017
Hänsel, S., Lässig, G., Pistrosch, F., and Passauer, J. (2003). Endothelial dysfunction in young patients with long-term rheumatoid arthritis and low disease activity. Atherosclerosis 170, 177–180. doi:10.1016/S0021-9150(03)00281-8
Harrison, D. G., Guzik, T. J., Lob, H. E., Madhur, M. S., Marvar, P. J., Thabet, S. R., et al. (2011). Inflammation, immunity, and hypertension. Hypertension 57, 132–140. doi:10.1161/HYPERTENSIONAHA.110.163576
Harrison, D. G., and Ohara, Y. (1995). Physiologic consequences of increased vascular oxidant stresses in hypercholesterolemia and atherosclerosis: Implications for impaired vasomotion. Am. J. Cardiol. 75, 75B–81B. doi:10.1016/0002-9149(95)80018-N
Hausding, M., Jurk, K., Daub, S., Kröller-Schön, S., Stein, J., Schwenk, M., et al. (2013). CD40L contributes to angiotensin II-induced pro-thrombotic state, vascular inflammation, oxidative stress and endothelial dysfunction. Basic Res. Cardiol. 108, 386. doi:10.1007/s00395-013-0386-5
Hawkins, K. E., DeMars, K. M., Alexander, J. C., Leon, L. G. de, Pacheco, S. C., Graves, C., et al. (2017). Targeting resolution of neuroinflammation after ischemic stroke with a lipoxin A4 analog: Protective mechanisms and long-term effects on neurological recovery. Brain Behav. 7, e00688. doi:10.1002/brb3.688
Heitzer, T., Schlinzig, T., Krohn, K., Meinertz, T., and Münzel, T. (2001). Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104, 2673–2678. doi:10.1161/hc4601.099485
Henry, R. M. A., Ferreira, I., Kostense, P. J., Dekker, J. M., Nijpels, G., Heine, R. J., et al. (2004). Type 2 diabetes is associated with impaired endothelium-dependent, flow-mediated dilation, but impaired glucose metabolism is not; the Hoorn Study. Atherosclerosis 174, 49–56. doi:10.1016/j.atherosclerosis.2004.01.002
Howson, J. M. M., Zhao, W., Barnes, D. R., Ho, W.-K., Young, R., Paul, D. S., et al. (2017). Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat. Genet. 49, 1113–1119. doi:10.1038/ng.3874
Iacomino, G., and Siani, A. (2017). Role of microRNAs in obesity and obesity-related diseases. Genes. Nutr. 12, 23. doi:10.1186/s12263-017-0577-z
Iseme, R. A., McEvoy, M., Kelly, B., Agnew, L., Walker, F. R., Handley, T., et al. (2017). A role for autoantibodies in atherogenesis. Cardiovasc. Res. 113, 1102–1112. doi:10.1093/cvr/cvx112
Kain, V., and Halade, G. V. (2020). Role of neutrophils in ischemic heart failure. Pharmacol. Ther. 205, 107424. doi:10.1016/j.pharmthera.2019.107424
Kain, V., Liu, F., Kozlovskaya, V., Ingle, K. A., Bolisetty, S., Agarwal, A., et al. (2017). Resolution agonist 15-epi-Lipoxin A4 programs early activation of resolving phase in post-myocardial infarction healing. Sci. Rep. 7, 9999. doi:10.1038/s41598-017-10441-8
Kang, J.-W., and Lee, S.-M. (2016). Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim. Biophys. Acta 1861, 1025–1035. doi:10.1016/j.bbalip.2016.06.002
Kaptoge, S., Seshasai, S. R. K., Gao, P., Freitag, D. F., Butterworth, A. S., Borglykke, A., et al. (2014). Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 35, 578–589. doi:10.1093/eurheartj/eht367
Karbach, S., Lagrange, J., and Wenzel, P. (2019). Thromboinflammation and vascular dysfunction. Hamostaseologie 39, 180–187. doi:10.1055/s-0038-1676130
Karbach, S., Wenzel, P., Waisman, A., Munzel, T., and Daiber, A. (2014). eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr. Pharm. Des. 20, 3579–3594. doi:10.2174/13816128113196660748
Karnell, J. L., Rieder, S. A., Ettinger, R., and Kolbeck, R. (2019). Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 141, 92–103. doi:10.1016/j.addr.2018.12.005
Kasran, A., Boon, L., Wortel, C. H., Hogezand, R. A., Schreiber, S., Goldin, E., et al. (2005). Safety and tolerability of antagonist anti-human CD40 Mab ch5D12 in patients with moderate to severe Crohn's disease. Aliment. Pharmacol. Ther. 22, 111–122. doi:10.1111/j.1365-2036.2005.02526.x
Katsumoto, A., Takeuchi, H., Takahashi, K., and Tanaka, F. (2018). Microglia in alzheimer's disease: Risk factors and inflammation. Front. Neurol. 9, 978. doi:10.3389/fneur.2018.00978
Kawabe, T., Naka, T., Yoshida, K., Tanaka, T., Fujiwara, H., Suematsu, S., et al. (1994). The immune responses in CD40-deficient mice: Impaired immunoglobulin class switching and germinal center formation. Immunity 1, 167–178. doi:10.1016/1074-7613(94)90095-7
Kawai, T., Andrews, D., Colvin, R. B., Sachs, D. H., and Cosimi, A. B. (2000). Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat. Med. 6, 114. doi:10.1038/72162
Klarin, D., Zhu, Q. M., Emdin, C. A., Chaffin, M., Horner, S., McMillan, B. J., et al. (2017). Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat. Genet. 49, 1392–1397. doi:10.1038/ng.3914
Kröller-Schön, S., Steven, S., Kossmann, S., Scholz, A., Daub, S., Oelze, M., et al. (2014). Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 20, 247–266. doi:10.1089/ars.2012.4953
La Morena, M. T. de (2016). Clinical phenotypes of hyper-IgM syndromes. J. Allergy Clin. Immunol. Pract. 4, 1023–1036. doi:10.1016/j.jaip.2016.09.013
Lacy, M., Bürger, C., Shami, A., Ahmadsei, M., Winkels, H., Nitz, K., et al. (2021). Cell-specific and divergent roles of the CD40L-CD40 axis in atherosclerotic vascular disease. Nat. Commun. 12, 3754. doi:10.1038/s41467-021-23909-z
Lalani, A. I., Moore, C. R., Luo, C., Kreider, B. Z., Liu, Y., Morse, H. C., et al. (2015). Myeloid cell TRAF3 regulates immune responses and inhibits inflammation and tumor development in mice. J. Immunol. 194, 334–348. doi:10.4049/jimmunol.1401548
Lalani, A. I., Zhu, S., Gokhale, S., Jin, J., and Xie, P. (2018). TRAF molecules in inflammation and inflammatory diseases. Curr. Pharmacol. Rep. 4, 64–90. doi:10.1007/s40495-017-0117-y
Lameijer, M., Binderup, T., van Leent, M. M. T., Senders, M. L., Fay, F., Malkus, J., et al. (2018). Efficacy and safety assessment of a TRAF6-targeted nanoimmunotherapy in atherosclerotic mice and non-human primates. Nat. Biomed. Eng. 2, 279–292. doi:10.1038/s41551-018-0221-2
Leo, E., Welsh, K., Matsuzawa, S., Zapata, J. M., Kitada, S., Mitchell, R. S., et al. (1999). Differential requirements for tumor necrosis factor receptor-associated factor family proteins in CD40-mediated induction of NF-kappaB and Jun N-terminal kinase activation. J. Biol. Chem. 274, 22414–22422. doi:10.1074/jbc.274.32.22414
Leuti, A., Talamonti, E., Gentile, A., Tiberi, M., Matteocci, A., Fresegna, D., et al. (2021). Macrophage plasticity and polarization are altered in the experimental model of multiple sclerosis. Biomolecules 11, 837. doi:10.3390/biom11060837
Léveillé, C., Bouillon, M., Guo, W., Bolduc, J., Sharif-Askari, E., El-Fakhry, Y., et al. (2007). CD40 ligand binds to alpha5beta1 integrin and triggers cell signaling. J. Biol. Chem. 282, 5143–5151. doi:10.1074/jbc.M608342200
Lin, W.-J., Su, Y.-W., Lu, Y.-C., Hao, Z., Chio, I. I. C., Chen, N.-J., et al. (2011). Crucial role for TNF receptor-associated factor 2 (TRAF2) in regulating NFκB2 signaling that contributes to autoimmunity. Proc. Natl. Acad. Sci. U. S. A. 108, 18354–18359. doi:10.1073/pnas.1109427108
Lopez, E. F., Kabarowski, J. H., Ingle, K. A., Kain, V., Barnes, S., Crossman, D. K., et al. (2015). Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 308, H269–H280. doi:10.1152/ajpheart.00604.2014
Loubaki, L., Semlali, A., Boisvert, M., Jacques, E., Plante, S., Aoudjit, F., et al. (2010). Crosstalk between T cells and bronchial fibroblasts obtained from asthmatic subjects involves CD40L/alpha 5 beta 1 interaction. Mol. Immunol. 47, 2112–2118. doi:10.1016/j.molimm.2010.03.011
Lutgens, E., Lievens, D., Beckers, L., Wijnands, E., Soehnlein, O., Zernecke, A., et al. (2010). Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J. Exp. Med. 207, 391–404. doi:10.1084/jem.20091293
Marino, S., Hannemann, N., Bishop, R. T., Zeng, F., Carrasco, G., Meurisse, S., et al. (2022). Anti-inflammatory, but not osteoprotective, effect of the TRAF6/CD40 inhibitor 6877002 in rodent models of local and systemic osteolysis. Biochem. Pharmacol. 195, 114869. doi:10.1016/j.bcp.2021.114869
Matsubara, J., Sugiyama, S., Sugamura, K., Nakamura, T., Fujiwara, Y., Akiyama, E., et al. (2012). A dipeptidyl peptidase-4 inhibitor, des-fluoro-sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. J. Am. Coll. Cardiol. 59, 265–276. doi:10.1016/j.jacc.2011.07.053
McAlpine, C. S., Kiss, M. G., Rattik, S., He, S., Vassalli, A., Valet, C., et al. (2019). Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 566, 383–387. doi:10.1038/s41586-019-0948-2
Mehta, N. N., Azfar, R. S., Shin, D. B., Neimann, A. L., Troxel, A. B., and Gelfand, J. M. (2010). Patients with severe psoriasis are at increased risk of cardiovascular mortality: Cohort study using the general practice research database. Eur. Heart J. 31, 1000–1006. doi:10.1093/eurheartj/ehp567
Michel, N. A., Zirlik, A., and Wolf, D. (2017). CD40L and its receptors in atherothrombosis-an update. Front. Cardiovasc. Med. 4, 40. doi:10.3389/fcvm.2017.00040
Missiou, A., Köstlin, N., Varo, N., Rudolf, P., Aichele, P., Ernst, S., et al. (2010). Tumor necrosis factor receptor-associated factor 1 (TRAF1) deficiency attenuates atherosclerosis in mice by impairing monocyte recruitment to the vessel wall. Circulation 121, 2033–2044. doi:10.1161/CIRCULATIONAHA.109.895037
Münzel, T., Daiber, A., Ullrich, V., and Mülsch, A. (2005). Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 25, 1551–1557. doi:10.1161/01.ATV.0000168896.64927.bb
Münzel, T., Gori, T., Bruno, R. M., and Taddei, S. (2010). Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 31, 2741–2748. doi:10.1093/eurheartj/ehq396
Murdaca, G., Colombo, B. M., Cagnati, P., Gulli, R., Spanò, F., and Puppo, F. (2012). Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 224, 309–317. doi:10.1016/j.atherosclerosis.2012.05.013
Murphy, S. P., Kakkar, R., McCarthy, C. P., and Januzzi, J. L. (2020). Inflammation in heart failure: JACC state-of-the-art review. J. Am. Coll. Cardiol. 75, 1324–1340. doi:10.1016/j.jacc.2020.01.014
Mussbacher, M., Salzmann, M., Brostjan, C., Hoesel, B., Schoergenhofer, C., Datler, H., et al. (2019). Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front. Immunol. 10, 85. doi:10.3389/fimmu.2019.00085
Oelze, M., Kröller-Schön, S., Steven, S., Lubos, E., Doppler, C., Hausding, M., et al. (2014). Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 63, 390–396. doi:10.1161/HYPERTENSIONAHA.113.01602
Ohara, Y., Peterson, T. E., and Harrison, D. G. (1993). Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Invest. 91, 2546–2551. doi:10.1172/JCI116491
Ortega-Gómez, A., Perretti, M., and Soehnlein, O. (2013). Resolution of inflammation: An integrated view. EMBO Mol. Med. 5, 661–674. doi:10.1002/emmm.201202382
Oyoshi, M. K., Barthel, R., and Tsitsikov, E. N. (2007). TRAF1 regulates recruitment of lymphocytes and, to a lesser extent, neutrophils, myeloid dendritic cells and monocytes to the lung airways following lipopolysaccharide inhalation. Immunology 120, 303–314. doi:10.1111/j.1365-2567.2006.02499.x
Papp, K. A., Leonardi, C., Menter, A., Ortonne, J.-P., Krueger, J. G., Kricorian, G., et al. (2012). Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 366, 1181–1189. doi:10.1056/NEJMoa1109017
Park, Y. C., Burkitt, V., Villa, A. R., Tong, L., and Wu, H. (1999). Structural basis for self-association and receptor recognition of human TRAF2. Nature 398, 533–538. doi:10.1038/19110
Pasceri, V., and Yeh, E. T. (1999). A tale of two diseases: Atherosclerosis and rheumatoid arthritis. Circulation 100, 2124–2126. doi:10.1161/01.CIR.100.21.2124
Paudel, K. R., Panth, N., and Kim, D.-W. (2016). Circulating endothelial microparticles: A key hallmark of atherosclerosis progression. Sci. (Cairo) 2016, 8514056. doi:10.1155/2016/8514056
Paulie, S., Koho, H., Ben-Aissa, H., Hansson, Y., Lundblad, M. L., and Perlmann, P. (1984). Monoclonal antibodies to antigens associated with transitional cell carcinoma of the human urinary bladder. II. Identification of the cellular target structures by immunoprecipitation and SDS-PAGE analysis. Cancer Immunol. Immunother. 17, 173–179. doi:10.1007/BF00205482
Perper, S. J., Westmoreland, S. V., Karman, J., Twomey, R., Seagal, J., Wang, R., et al. (2019). Treatment with a CD40 antagonist antibody reverses severe proteinuria and loss of saliva production and restores glomerular morphology in murine systemic lupus erythematosus. J. Immunol. 203, 58–75. doi:10.4049/jimmunol.1900043
Peters, M. J. L., Symmons, D. P. M., McCarey, D., Dijkmans, B. A. C., Nicola, P., Kvien, T. K., et al. (2010). EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann. Rheum. Dis. 69, 325–331. doi:10.1136/ard.2009.113696
Popa, M., Tahir, S., Elrod, J., Kim, S. H., Leuschner, F., Kessler, T., et al. (2018). Role of CD40 and ADAMTS13 in von Willebrand factor-mediated endothelial cell-platelet-monocyte interaction. Proc. Natl. Acad. Sci. U. S. A. 115, E5556–E5565. doi:10.1073/pnas.1801366115
Prasad, K. N. (2016). Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer's disease. Mech. Ageing Dev. 153, 41–47. doi:10.1016/j.mad.2016.01.002
Prasad, K. S. S., Andre, P., He, M., Bao, M., Manganello, J., and Phillips, D. R. (2003). Soluble CD40 ligand induces beta3 integrin tyrosine phosphorylation and triggers platelet activation by outside-in signaling. Proc. Natl. Acad. Sci. U. S. A. 100, 12367–12371. doi:10.1073/pnas.2032886100
Pullen, S. S., Miller, H. G., Everdeen, D. S., Dang, T. T., Crute, J. J., and Kehry, M. R. (1998). CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: Regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry 37, 11836–11845. doi:10.1021/bi981067q
Renshaw, B. R., Fanslow, W. C., Armitage, R. J., Campbell, K. A., Liggitt, D., Wright, B., et al. (1994). Humoral immune responses in CD40 ligand-deficient mice. J. Exp. Med. 180, 1889–1900. doi:10.1084/jem.180.5.1889
Ridker, P. M., Cannon, C. P., Morrow, D., Rifai, N., Rose, L. M., McCabe, C. H., et al. (2005). C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 352, 20–28. doi:10.1056/NEJMoa042378
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131. doi:10.1056/NEJMoa1707914
Sadek, H., and Olson, E. N. (2020). Toward the goal of human heart regeneration. Cell. Stem Cell. 26, 7–16. doi:10.1016/j.stem.2019.12.004
Santovito, D., Mezzetti, A., and Cipollone, F. (2012). MicroRNAs and atherosclerosis: New actors for an old movie. Nutr. Metab. Cardiovasc. Dis. 22, 937–943. doi:10.1016/j.numecd.2012.03.007
Schieven, G. L. (2005). The biology of p38 kinase: A central role in inflammation. Curr. Top. Med. Chem. 5, 921–928. doi:10.2174/1568026054985902
Schmidt, H. H. H. W., Stocker, R., Vollbracht, C., Paulsen, G., Riley, D., Daiber, A., et al. (2015). Antioxidants in translational medicine. Antioxid. Redox Signal. 23, 1130–1143. doi:10.1089/ars.2015.6393
Schönbeck, U., Libby, P., and Schonbeck, U. (2001). CD40 signaling and plaque instability. Circ. Res. 89, 1092–1103. doi:10.1161/hh2401.101272
Schulz, E., Wenzel, P., Münzel, T., and Daiber, A. (2014). Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 20, 308–324. doi:10.1089/ars.2012.4609
Schwenzer, R., Siemienski, K., Liptay, S., Schubert, G., Peters, N., Scheurich, P., et al. (1999). The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kappaB and c-Jun N-terminal kinase. J. Biol. Chem. 274, 19368–19374. doi:10.1074/jbc.274.27.19368
Seijkens, T. T. P., van Tiel, C. M., Kusters, P. J. H., Atzler, D., Soehnlein, O., Zarzycka, B., et al. (2018). Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J. Am. Coll. Cardiol. 71, 527–542. doi:10.1016/j.jacc.2017.11.055
Serhan, C. N. (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101. doi:10.1038/nature13479
Serhan, C. N. (2011). The resolution of inflammation: The devil in the flask and in the details. FASEB J. 25, 1441–1448. doi:10.1096/fj.11-0502ufm
Shah, Z., Kampfrath, T., Deiuliis, J. A., Zhong, J., Pineda, C., Ying, Z., et al. (2011). Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 124, 2338–2349. doi:10.1161/CIRCULATIONAHA.111.041418
Sharma, M., Schlegel, M. P., Afonso, M. S., Brown, E. J., Rahman, K., Weinstock, A., et al. (2020). Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ. Res. 127, 335–353. doi:10.1161/CIRCRESAHA.119.316461
Shiels, H., Li, X., Schumacker, P. T., Maltepe, E., Padrid, P. A., Sperling, A., et al. (2000). TRAF4 deficiency leads to tracheal malformation with resulting alterations in air flow to the lungs. Am. J. Pathol. 157, 679–688. doi:10.1016/S0002-9440(10)64578-6
Shock, A., Burkly, L., Wakefield, I., Peters, C., Garber, E., Ferrant, J., et al. (2015). CDP7657, an anti-cd40l antibody lacking an Fc domain, inhibits cd40l-dependent immune responses without thrombotic complications: An in vivo study. Arthritis Res. Ther. 17, 234. doi:10.1186/s13075-015-0757-4
Sies, H. (2015). Oxidative stress: A concept in redox biology and medicine. Redox Biol. 4, 180–183. doi:10.1016/j.redox.2015.01.002
Södergren, A., Karp, K., Boman, K., Eriksson, C., Lundström, E., Smedby, T., et al. (2010). Atherosclerosis in early rheumatoid arthritis: Very early endothelial activation and rapid progression of intima media thickness. Arthritis Res. Ther. 12, R158. doi:10.1186/ar3116
Soltész, P., Kerekes, G., Dér, H., Szücs, G., Szántó, S., Kiss, E., et al. (2011). Comparative assessment of vascular function in autoimmune rheumatic diseases: Considerations of prevention and treatment. Autoimmun. Rev. 10, 416–425. doi:10.1016/j.autrev.2011.01.004
Steffens, S., Montecucco, F., and Mach, F. (2009). The inflammatory response as a target to reduce myocardial ischaemia and reperfusion injury. Thromb. Haemost. 102, 240–247. doi:10.1160/TH08-12-0837
Steven, S., Dib, M., Hausding, M., Kashani, F., Oelze, M., Kröller-Schön, S., et al. (2018). CD40L controls obesity-associated vascular inflammation, oxidative stress, and endothelial dysfunction in high fat diet-treated and db/db mice. Cardiovasc. Res. 114, 312–323. doi:10.1093/cvr/cvx197
Steven, S., Münzel, T., and Daiber, A. (2015). Exploiting the pleiotropic antioxidant effects of established drugs in cardiovascular disease. Int. J. Mol. Sci. 16, 18185–18223. doi:10.3390/ijms160818185
Sun, X., He, S., Wara, A. K. M., Icli, B., Shvartz, E., Tesmenitsky, Y., et al. (2014). Systemic delivery of microRNA-181b inhibits nuclear factor-κB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ. Res. 114, 32–40. doi:10.1161/CIRCRESAHA.113.302089
Tang, X., Tang, G., and Ozcan, S. (2008). Role of microRNAs in diabetes. Biochim. Biophys. Acta 1779, 697–701. doi:10.1016/j.bbagrm.2008.06.010
Tocoian, A., Buchan, P., Kirby, H., Soranson, J., Zamacona, M., Walley, R., et al. (2015). First-in-human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti-CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus 24, 1045–1056. doi:10.1177/0961203315574558
Tourki, B., and Halade, G. V. (2021). Heart failure syndrome with preserved ejection fraction is a metabolic cluster of non-resolving inflammation in obesity. Front. Cardiovasc. Med. 8, 695952. doi:10.3389/fcvm.2021.695952
Tracy, R. P. (2006). The five cardinal signs of inflammation: Calor, dolor, rubor, tumor and penuria (apologies to aulus cornelius celsus, de medicina, c. A.D. 25). J. Gerontol. A Biol. Sci. Med. Sci. 61, 1051–1052. doi:10.1093/gerona/61.10.1051
Troncoso-Escudero, P., Parra, A., Nassif, M., and Vidal, R. L. (2018). Outside in: Unraveling the role of neuroinflammation in the progression of Parkinson's disease. Front. Neurol. 9, 860. doi:10.3389/fneur.2018.00860
Urbich, C., Mallat, Z., Tedgui, A., Clauss, M., Zeiher, A. M., and Dimmeler, S. (2001). Upregulation of TRAF-3 by shear stress blocks CD40-mediated endothelial activation. J. Clin. Invest. 108, 1451–1458. doi:10.1172/JCI13620
Vafadarnejad, E., Rizzo, G., Krampert, L., Arampatzi, P., Arias-Loza, A.-P., Nazzal, Y., et al. (2020). Dynamics of cardiac neutrophil diversity in murine myocardial infarction. Circ. Res. 127, e232–e249. doi:10.1161/CIRCRESAHA.120.317200
Vagnozzi, R. J., Maillet, M., Sargent, M. A., Khalil, H., Johansen, A. K. Z., Schwanekamp, J. A., et al. (2020). An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577, 405–409. doi:10.1038/s41586-019-1802-2
van de Vyver, A. J., Weinzierl, T., Eigenmann, M. J., Frances, N., Herter, S., Buser, R. B., et al. (2021). Predicting tumor killing and T-cell activation by T-cell bispecific antibodies as a function of target expression: Combining in vitro experiments with systems modeling. Mol. Cancer Ther. 20, 357–366. doi:10.1158/1535-7163.MCT-20-0269
van Tassell, B. W., Toldo, S., Mezzaroma, E., and Abbate, A. (2013). Targeting interleukin-1 in heart disease. Circulation 128, 1910–1923. doi:10.1161/CIRCULATIONAHA.113.003199
Vena, G. A., Vestita, M., and Cassano, N. (2010). Psoriasis and cardiovascular disease. Dermatol. Ther. 23, 144–151. doi:10.1111/j.1529-8019.2010.01308.x
Vergadi, E., Vaporidi, K., Theodorakis, E. E., Doxaki, C., Lagoudaki, E., Ieronymaki, E., et al. (2014). Akt2 deficiency protects from acute lung injury via alternative macrophage activation and miR-146a induction in mice. J. Immunol. 192, 394–406. doi:10.4049/jimmunol.1300959
Vincent, A., Covinhes, A., Barrère, C., Gallot, L., Thoumala, S., Piot, C., et al. (2017). Acute and long-term cardioprotective effects of the Traditional Chinese Medicine MLC901 against myocardial ischemia-reperfusion injury in mice. Sci. Rep. 7, 14701. doi:10.1038/s41598-017-14822-x
Vincenti, F., Klintmalm, G., Yang, H., Ram Peddi, V., Blahunka, P., Conkle, A., et al. (2020). A randomized, phase 1b study of the pharmacokinetics, pharmacodynamics, safety, and tolerability of bleselumab, a fully human, anti-CD40 monoclonal antibody, in kidney transplantation. Am. J. Transpl. 20, 172–180. doi:10.1111/ajt.15560
Visvanathan, S., Daniluk, S., Ptaszyński, R., Müller-Ladner, U., Ramanujam, M., Rosenstock, B., et al. (2019). Effects of BI 655064, an antagonistic anti-CD40 antibody, on clinical and biomarker variables in patients with active rheumatoid arthritis: A randomised, double-blind, placebo-controlled, phase IIa study. Ann. Rheum. Dis. 78, 754–760. doi:10.1136/annrheumdis-2018-214729
Waetzig, V., Czeloth, K., Hidding, U., Mielke, K., Kanzow, M., Brecht, S., et al. (2005). c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 50, 235–246. doi:10.1002/glia.20173
Wajant, H., Henkler, F., and Scheurich, P. (2001). The TNF-receptor-associated factor family: Scaffold molecules for cytokine receptors, kinases and their regulators. Cell. Signal. 13, 389–400. doi:10.1016/s0898-6568(01)00160-7
Wenzel, P., Kossmann, S., Münzel, T., and Daiber, A. (2017). Redox regulation of cardiovascular inflammation - immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 109, 48–60. doi:10.1016/j.freeradbiomed.2017.01.027
Wenzel, P., Schuhmacher, S., Kienhöfer, J., Müller, J., Hortmann, M., Oelze, M., et al. (2008). Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc. Res. 80, 280–289. doi:10.1093/cvr/cvn182
Wes, P. D., Sayed, F. A., Bard, F., and Gan, L. (2016). Targeting microglia for the treatment of Alzheimer's Disease. Glia 64, 1710–1732. doi:10.1002/glia.22988
Wicovsky, A., Henkler, F., Salzmann, S., Scheurich, P., Kneitz, C., and Wajant, H. (2009). Tumor necrosis factor receptor-associated factor-1 enhances proinflammatory TNF receptor-2 signaling and modifies TNFR1-TNFR2 cooperation. Oncogene 28, 1769–1781. doi:10.1038/onc.2009.29
Wojciechowska, A., Braniewska, A., and Kozar-Kamińska, K. (2017). MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 26, 865–874. doi:10.17219/acem/62915
Wu, C., Gong, Y., Yuan, J., Zhang, W., Zhao, G., Li, H., et al. (2012). microRNA-181a represses ox-LDL-stimulated inflammatory response in dendritic cell by targeting c-Fos. J. Lipid Res. 53, 2355–2363. doi:10.1194/jlr.M028878
Xu, W., Zhang, L., Ma, S., Zhang, Y., Cai, Z., Zhang, K., et al. (2020). TRAF5 protects against myocardial ischemia reperfusion injury via AKT signaling. Eur. J. Pharmacol. 878, 173092. doi:10.1016/j.ejphar.2020.173092
Xu, Y., Cheng, G., and Baltimore, D. (1996). Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity 5, 407–415. doi:10.1016/s1074-7613(00)80497-5
Yang, K., He, Y. S., Wang, X. Q., Lu, L., Chen, Q. J., Liu, J., et al. (2011). MiR-146a inhibits oxidized low-density lipoprotein-induced lipid accumulation and inflammatory response via targeting toll-like receptor 4. FEBS Lett. 585, 854–860. doi:10.1016/j.febslet.2011.02.009
Yao, Y., Xu, X.-H., and Jin, L. (2019). Macrophage polarization in physiological and pathological pregnancy. Front. Immunol. 10, 792. doi:10.3389/fimmu.2019.00792
Yeh, W.-C., Shahinian, A., Speiser, D., Kraunus, J., Billia, F., Wakeham, A., et al. (1997). Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7, 715–725. doi:10.1016/S1074-7613(00)80391-X
Yue, Y., Huang, S., Li, H., Li, W., Hou, J., Luo, L., et al. (2020a). M2b macrophages protect against myocardial remodeling after ischemia/reperfusion injury by regulating kinase activation of platelet-derived growth factor receptor of cardiac fibroblast. Ann. Transl. Med. 8, 1409. doi:10.21037/atm-20-2788
Yue, Y., Huang, S., Wang, L., Wu, Z., Liang, M., Li, H., et al. (2020b). M2b macrophages regulate cardiac fibroblast activation and alleviate cardiac fibrosis after reperfusion injury. Circ. J. 84, 626–635. doi:10.1253/circj.CJ-19-0959
Zarzycka, B., Seijkens, T., Nabuurs, S. B., Ritschel, T., Grommes, J., Soehnlein, O., et al. (2015). Discovery of small molecule CD40-TRAF6 inhibitors. J. Chem. Inf. Model. 55, 294–307. doi:10.1021/ci500631e
Zhang, X., Cao, X., Dang, M., Wang, H., Chen, B., Du, F., et al. (2019). Soluble receptor for advanced glycation end-products enhanced the production of IFN-γ through the NF-κB pathway in macrophages recruited by ischemia/reperfusion. Int. J. Mol. Med. 43, 2507–2515. doi:10.3892/ijmm.2019.4152
Keywords: CD40(L)-TRAF signaling, inflammation, resolution, cardiovascular disease, ischemia, reperfusion
Citation: Strohm L, Ubbens H, Münzel T, Daiber A and Daub S (2022) Role of CD40(L)-TRAF signaling in inflammation and resolution—a double-edged sword. Front. Pharmacol. 13:995061. doi: 10.3389/fphar.2022.995061
Received: 15 July 2022; Accepted: 12 September 2022;
Published: 04 October 2022.
Edited by:
Dieter Steinhilber, Goethe University Frankfurt, GermanyReviewed by:
Geoff H Werstuck, McMaster University, CanadaCopyright © 2022 Strohm, Ubbens, Münzel, Daiber and Daub. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Daiber, ZGFpYmVyQHVuaS1tYWluei5kZQ==; Steffen Daub, c3RlZmZlbi5kYXViQHVuaW1lZGl6aW4tbWFpbnouZGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.