
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 16 September 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.989575
This article is part of the Research Topic New Insights into the Histone Lysine Specific Demethylase (LSD1) Inhibitors for Cancer Treatment View all 4 articles
 Guan-Jun Yang1,2,3
Guan-Jun Yang1,2,3 Yan-Jun Liu2Li-Jian Ding4
Yan-Jun Liu2Li-Jian Ding4 Fan Tao2Ming-Hui Zhu2Zhen-Yuan Shi2Juan-Ming Wen2Meng-Yao Niu2
Fan Tao2Ming-Hui Zhu2Zhen-Yuan Shi2Juan-Ming Wen2Meng-Yao Niu2 Xiang Li2Zhan-Song Xu2Wan-Jia Qin2
Xiang Li2Zhan-Song Xu2Wan-Jia Qin2 Chen-Jie Fei1,2,3
Chen-Jie Fei1,2,3 Jiong Chen1,2,3*
Jiong Chen1,2,3*Breast cancer (BC) is a kind of malignant cancer in women, and it has become the most diagnosed cancer worldwide since 2020. Histone methylation is a common biological epigenetic modification mediating varieties of physiological and pathological processes. Lysine-specific demethylase 1 (LSD1), a first identified histone demethylase, mediates the removal of methyl groups from histones H3K4me1/2 and H3K9me1/2 and plays a crucial role in varieties of cancer progression. It is also specifically amplified in breast cancer and contributes to BC tumorigenesis and drug resistance via both demethylase and non-demethylase manners. This review will provide insight into the overview structure of LSD1, summarize its action mechanisms in BC, describe the therapeutic potential of LSD1 inhibitors in BC, and prospect the current opportunities and challenges of targeting LSD1 for BC therapy.
Breast cancer (BC) is a kind of malignant tumour in women occurring in the breast glandular epithelial tissues, and it has become the most diagnosed cancer worldwide since 2020 (Siegel et al., 2021; Siegel et al., 2022). The incidence of BC is increasing year by year and patients are getting younger and younger, posing a serious threat to women’s health (Siegel et al., 2022). Although advances in early diagnosis and treatment of BC have partially alleviated the crisis of some BC patients, there are still a large number of patients suffering from BC due to their complex pathogenesis, insensitivity to existing drugs, and easy-to-develop drug resistance (Yang et al., 2019; Yang et al., 2021b). Therefore, there is an urgent need to find new and effective targeted therapies for this type of BC.
Lysine-specific demethylase 1 (LSD1) is a flavin-dependent lysine-specific histone demethylase first identified in 2004 and mediated transcriptional activation or repression via erasing methyl groups from H3K9me2/1 and H3K4me2/1, respectively (Figure 1) (Shi et al., 2004; Fang et al., 2020; Kim et al., 2021; Malagraba et al., 2022; Song et al., 2022). Recently studies found that LSD1 could also remove the methyl groups from several non-histone proteins such as ERα, MTA1, HIF-1α, AGO2, HSP90, MEFD2, and STAT3 and be involved in many cancer cell events (Majello et al., 2019). LSD1 exhibits its catalytic mechanism via consuming oxidation of FAD and O2 and yielding HCHO and H2O2 in cellulo (Yang et al., 2018a). Apart from demethylase activity, LSD1 exhibits non-demethylase activity via interacting with its client proteins and is involved in physiological and pathological processes (Gu et al., 2020). LSD1 is also overexpressed in varieties of cancers and mediates their progression (Yang et al., 2018a; Fang et al., 2019; Fang et al., 2020). LSD1 is aberrantly expressed in BC and promotes proliferation and metastasis of BC cells (Liu et al., 2020a; Zhou et al., 2021a). Moreover, LSD1 also is involved in regulating resistance of chemotherapy and immunotherapy in BC (Kim et al., 2013; Boulding et al., 2018; Yang et al., 2018d; Qin et al., 2019; Verigos et al., 2019; Tu et al., 2020; Sobczak et al., 2022). Given the multifaceted functions of LSD1 in BC progression, new therapeutic strategies targeting LSD1 are constantly being developed, such as the discovery of novel LSD1 inhibitors, the development of dual-target inhibitors, and the combination therapies with chemical agents or immunomodulators (Kim et al., 2013; Boulding et al., 2018; Yang et al., 2018d; Qin et al., 2019; Verigos et al., 2019; Tu et al., 2020; Sobczak et al., 2022). Therefore, LSD1 is a potential target for BC therapy.
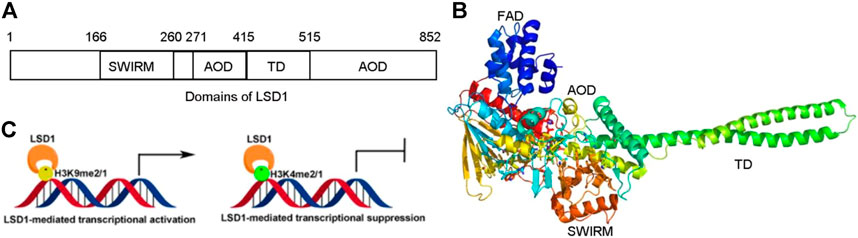
FIGURE 1. Structure and function of LSD1. (A) Distribution of domains of LSD1. (B) LSD1-mediated transcriptional modulation. (C) Overview structure of LSD1.
Herein, the structures, functions and the regulatory roles of LSD1 in tumorigenesis were introduced, the reported LSD1 inhibitors and their therapeutic mechanisms for BC treatment were summarized, and the current challenges and the potential opportunities of LSD1 inhibitors for BC therapy were prospected.
LSD1 is a FAD-dependent demethylase encoding a peptide with 852 amino acid residues. LSD1 consists of highly conserved three distinct domains: a SWI3/Rac8/Moira (SWIRM) domain, a tower domain (TD), and a catalytic amine oxidase domain (AOD) (Figures 1A,B) (Forneris et al., 2008; Yang et al., 2018a). The SWIRM domain is an indispensable domain for LSD1-mediated histone modification and chromatin remodeling (Metzger et al., 2005). The TD is a special domain with two antiparallel helices, and it can bind to RCOR1 and form the CoREST complex (Pilotto et al., 2015; Marabelli et al., 2016). The AOD domain is the catalytic domain of LSD1, and it consists of two well-defined motifs: the substrate-recognition motif and the FAD-binding motif (Yang et al., 2018a). The latter is highly conserved and responsible for binding sites of some reported LSD1 inhibitors. The two motifs assemble into a big cavity containing the interface of enzyme activity centre (Forneris et al., 2008). In the active state of LSD1, the second lobe of the AOD domain could form a hydrophobic binding pocket SWIRM domain (Yoneyama et al., 2007). This binding pocket mediates LSD1 binding to histone H3 and is also chosen as the binding pocket for developing LSD1 inhibitors (Zhou et al., 2015).
LSD1 plays heterogeneous roles via transcriptionally modulating its downstream genes in demethylase-dependent or -independent modes in varieties of cancers (Song et al., 2022; Yang et al., 2022). It acts as an oncogene in some cancers, while functioning as a cancer-suppressor gene in the other cancers (Yang et al., 2018a; Hu et al., 2019). In addition, LSD1 is also regulated by multiple epigenetic regulators in BC.
Upregulating of H3K4me2/1 often contributes to the transcriptional activation (Figure 1C). LSD1 removes methyl groups from active Histone 3 via assembling into different co-repressor complexes with several distinct proteins and shaping chromatin into a repressive conformation. For example, LSD1 was found to form a co-repressor complex with SIN3A/HDAC and maintain sensitivity to chemotherapy via reducing inhibiting several genes such as TGFB2, CASP7, TERT, MDM2, and HIF1α in BC (Yang et al., 2018d). It also could assemble into LSD1/CoREST/BRMS1 and inhibit the metastasis of BC cells via reduced levels of Vimentin, COL5A2, INSIG2, MRPL33, SLC1A1, KLK11, and OLFML3 (Qiu et al., 2018).
LSD1 also works as a transcriptional co-activator via demethylating H3K9me2/1 (Figure 1C). It can promote estrogen transcription in breast cancer cells through interacting with estrogen receptor (ER) (Bennesch et al., 2016). Additionally, LSD1 also regulates chromatin events such as DNA replication, heterochromatin, and imprinting propagation (Zhu et al., 2012; Yang et al., 2018a).
The function of LSD1 has also been found to be regulated by many epigenetic components. For example, miR-708 inhibits BC proliferation and invasion via directly binding to the 3′ UTR of LSD1 and reducing its level (Ma et al., 2016). In addition, epigenetic modifications such as phosphorylation (Peng et al., 2015; Feng et al., 2016; Zhou et al., 2016), acetylation (Luo et al., 2016), methylation (Liu et al., 2020a), and ubiquitination (Wu et al., 2013; Yi et al., 2016; Zhou et al., 2016; Gong et al., 2021) also contribute to the function of LSD1 (Figure 2). PKCα can phosphorylate LSD1 at S112, activating its demethylase activity and enhancing the occupancy on E-cadherin promoter, and finally promote EMT and metastasis in BC (Feng et al., 2016). Phosphorylated modification is also found to regulate LSD1-medatied DNA damage (Peng et al., 2015). To be specific, LSD1 was di-phosphorylated at S131 and S137 by CK2 and Wip1, respectively, which promoted its interaction with RNF168 and RNF168-dependent 53BP1 ubiquitination and subsequent recruitment to the DNA damage sites. CK1α was also found to phosphorylate LSD1 at S687 and then induce S683 phosphorylation of LSD1 by nuclear GSK3β (Zhou et al., 2016). The di-phosphorylated LSD1 would be deubiquitylated by USP22 and induced stabilization itself. MOF, a lysine acetyltransferase first found acetylated histone H4 at K16 residue, also could catalyze the acetylation of LSD1 at K432, K433, and K436 and suppress LSD1-medaited EMT in epithelial cells (Luo et al., 2016). Arginine methyltransferase 4 (PRMT4) was also found to mediate deubiquitination and stabilization of LSD1 via dimethylating it at R838 and promoting its deubiquitination by USP7 (Liu et al., 2020a). Apart from USP7 and USP22, USP28 also could remove ubiquitin modifications from LSD1, maintain its stability, and thus confer stemness to BC cells (Wu et al., 2013). Recently, OTUD7B was also found to mediate deubiquitination of LSD1 at K226/277 residues, which stabilized LSD1 and promoted its assembly into co-repressor complex (Gong et al., 2021).
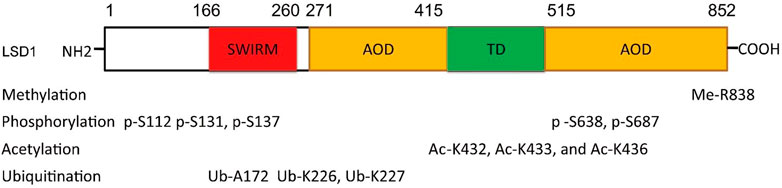
FIGURE 2. Residue sites were modified by varieties of posttranslational modifications in LSD1.
LSD1 is overexpressed in several subtypes of BC and functions as an oncogene mediating proliferation, differentiation, invasion, and metastasis of BC cells (Figure 3; Tables 1–3) (Ma et al., 2016; Feng et al., 2017; Yang et al., 2018a; Hu et al., 2019; Zhou et al., 2021a; Ji et al., 2021). When normal human mammary epithelial cells are exposed to carcinogens, their LSD1 levels would be upregulated and promote the carcinogenesis via reducing p57kip2 level (Bradley et al., 2007). LSD1 exhibits its oncogene functions via interacting with distinct ligands in different BC subtypes. As a major biomarker of ER-positive BC, ERα and its transcriptional activity are regulated by LSD1 via assembling into complex with different ligands to mediate BC proliferation (Lim et al., 2010; Pollock et al., 2012; Zhu et al., 2012; Andresen et al., 2017). For example, CAC1 antagonized LSD1-mediated ERα activation and suppressed the proliferation of BC cells (Kim et al., 2013), while ASXL2 promoted proliferation of BC cells via forming a complex ASXL2/LSD1/UTX/MLL to activate ERα activity (Park et al., 2016). In addition, LSD1 reduces tumor suppressor gene Lefty1 via interacting with β-catenin in BC cells (Yakulov et al., 2013), and suppresses BC cell growth through binding to histone deacetylases (HDACs) (Huang et al., 2012; Vasilatos et al., 2013). It also sensitizes BC cells to chemotherapy via assembling into a complex with SIN3A/HDAC and inhibits BC proliferation and metastasis via interacting with HDAC5 (Cao et al., 2017; Cao et al., 2018; Yang et al., 2018d). Further studies found that LSD1 promotes BC metastasis via H3K4me2 demethylase occupying the gene promoters of Snail and Slug and reducing their levels (Lin et al., 2010; Wu et al., 2012; Lin et al., 2014; Phillips and Kuperwasser, 2014; Bai et al., 2017). Interestingly, androgen receptor (AR) is also involved in BC metastasis via interacting with LSD1 to reduce E-cadherin and upregulate Vimentin (Feng et al., 2017).
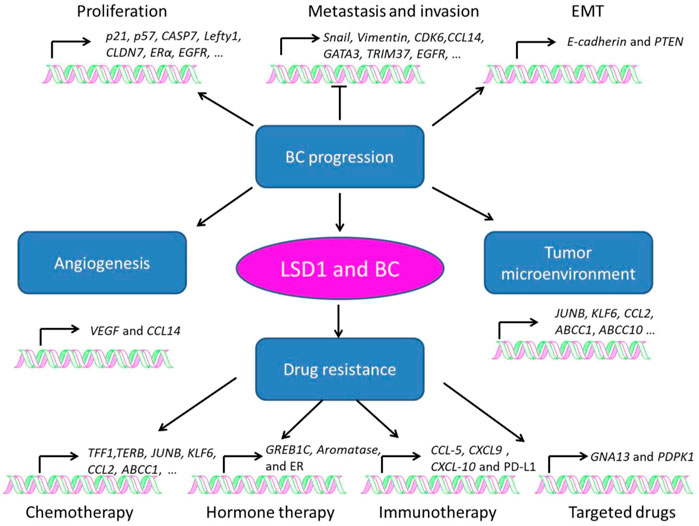
FIGURE 3. Role of LSD1 in breast cancer progression, angiogenesis, tumor microenvironment, and drug resistance.
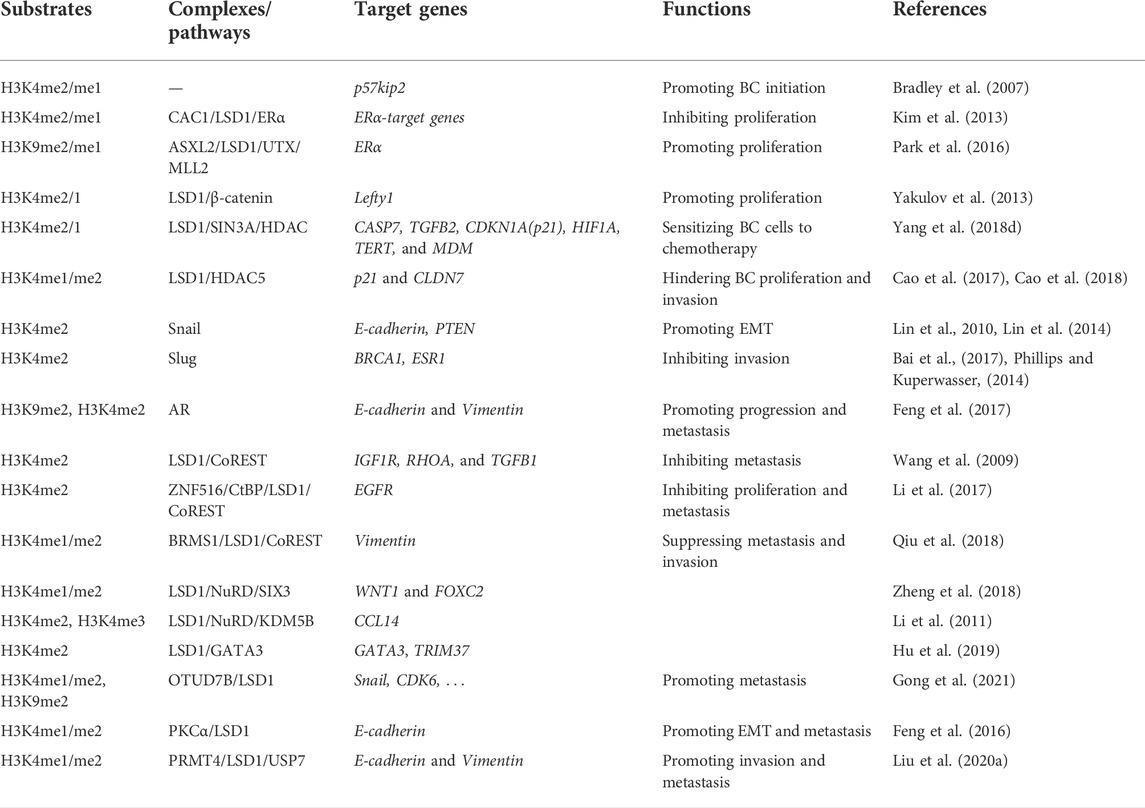
TABLE 1. Roles of LSD1 in BC progression.

TABLE 2. Regulatory roles of LSD1 in BC angiogenesis and microenvironment.

TABLE 3. Roles of LSD1 in drug resistance of BC.
Interestingly, LSD1 also exert its function as a tumor suppressor gene via forming different complexes with distinct ligand proteins (Li et al., 2017). LSD1 was also found to inhibit proliferation, invasion, and metastasis in vitro and in vivo via assembling into LSD1/NuRD complex (Wang et al., 2009; Li et al., 2017). This complex exhibits its heterogeneous tumor suppressor functions dependent of the different subunits in varieties of BC cells (Wang et al., 2009; Li et al., 2017). In MCF-7 cells, zinc-finger protein 516 (ZNF516) inhibited EGFR transcription, and thus reduced the proliferation and invasion of BC in vitro and in vivo via targeting CtBP/LSD1/CoREST complex (Li et al., 2017). Breast carcinoma metastasis suppressor 1 (BRMS1) is another gene coordinating with LSD1/NuRD complex to inhibit the metastasis of MCF7 cells (Qiu et al., 2018). In MDA-MB-231 cells, LSD1/NuRD complex suppressed BC tumorigenesis and metastasis via recruiting the homeotic protein SIX3 (Zheng et al., 2018). CC chemokine ligand 14 (CCL14) is a chemokine promoting angiogenesis in viral infection and tumor progression (Mortier et al., 2008; Li et al., 2011). KDM5B reduce CCL14 transcription to impede metastasis via targeting LSD1/NuRD complex (Li et al., 2011). In addition, in luminal BC, LSD1 suppressed invasion, migration, and metastasis BC cells via raising GATA3 and repressing TRIM37 (Hu et al., 2019).
Apart from as a subunit of many complexes mediating BC progression, the function of LSD1 was also regulated by several epigenetic enzymes in BC (Feng et al., 2016; Liu et al., 2020a; Gong et al., 2021). Feng et al. (2016) highlighted the PKCα-mediated phosphorylation of the S112 residue of LSD1 which was crucial for epithelial-mesenchymal transition (EMT) and metastasis of BC cells. Liu et al. (2020a) found that PRMT4 methylated and stabilized LSD1 via promoting it and it binding to deubiquitinase USP7 in BC cells. Recently, Gong et al. (2021) revealed that OTUD7B could remove the Poly-Ub Chains of LSD1 at K226/277 residues, maintain the integrity of LSD1/CoREST/HDACs co-repressor complexes, and inhibit BC metastasis.
Angiogenesis is a pivotal process for BC growth and metastasis (Ayoub et al., 2022). LSD1 is also involved in this process via modulating several pathways (Table 2) (Li et al., 2011; Lee et al., 2017). Li et al. (2011) found that LSD1 worked as a co-repressor and suppressed angiogenesis and metastasis of BC cells via assembling into a complex with KDM5B and NuRD and reducing the transcription of CCL4. Hypoxia-inducible factor alpha (HIF1α), a transcription factor promoting breast cancer angiogenesis, is also found to be regulated by LSD1/NuRD complex. To be specific, this complex demethylated HIF1α to stabilize it, and then stabilized HIF1α would upregulate the vascular endothelial growth factor (VEGF) via cooperating with CBP and metastasis-associated antigen 1, and induce angiogenesis in BC (Lee et al., 2017).
Tumor microenvironment refers to the internal and external environment of tumor cells where they survive, grow, and metastasize (Xiao and Yu, 2021). Tumor stroma consists of several heterogeneous cells such as cancer-associated fibroblasts (CAFs) and macrophages (Mф), which promote tumorigenesis via the secretion of varieties of chemokines, cytokines, and growth factors (Disis, 2010). While LSD1 increases CAF burden and reducing innate M1 Mф infiltration at the primary tumor site in BC (Boulding et al., 2018). In TNBC, LSD1 also mediated CD8+ lymphocyte trafficking to the tumor microenvironment and reducing Mф polarization toward M1-like phenotype (Qin et al., 2019; Tan et al., 2019; Shen et al., 2022).
Drug resistance is one of the major causes that leads to distant metastasis, poor prognosis, and death of BC (Wen et al., 2020). LSD1 is widely involved in the resistance to chemotherapy (Kim et al., 2013; Boulding et al., 2018; Verigos et al., 2019; Sobczak et al., 2022), hormone therapy (Bennani-Baiti, 2012; Cortez et al., 2012; Benedetti et al., 2019; Sukocheva et al., 2020), immunotherapy (Qin et al., 2019; Tu et al., 2020), and targeted therapy (Strachowska et al., 2021; Liu et al., 2022) of BC (Table 3). Briefly, LSD1 promotes chemoresistance via functioning as a co-activator through interacting with different ligand proteins (Kim et al., 2013; Boulding et al., 2018; Verigos et al., 2019; Strachowska et al., 2021). It mediates resistance to hormone therapy via activating ER transcriptional activity (Cortez et al., 2012; Benedetti et al., 2019). In addition, LSD1 is also involved in resistance to PD-1 antibody treatment and BRD4 inhibitors via its transcriptional inhibitory activity against multiple oncogenes (Qin et al., 2019; Tu et al., 2020; Liu et al., 2022).
Considering the crucial roles of LSD1 in BC progression, it has the potential as a therapeutic target for BC treatment. Currently, tens of LSD1 inhibitors have been documented with anti-BC activity with some of these having entered clinical trials. Herein, they were classified into seven subcategories based on their structural characteristics: PCPA-based LSD1 inhibitors, polyamine analogues, natural products, propargylamine derivatives, benzohydrazide derivatives, phenyl oxazole derivatives, and dual-target inhibitors.
Since LSD1 and monoamine oxidases (MAOs) share high similarity in their catalytic domains, several LSD1 inhibitors have been discovered based on reported MAO inhibitors via in silicon screening and chemical structure optimizations (Yang et al., 2007; Yang et al., 2018a). Tranylcypromine hydrochloride (2-PCPA, 1), an irreversible monoamine oxidase (MAO) inhibitor with half maximal inhibitory concentration (IC50) of 11.5 and 7.0 μM for MAO A and MAO B in vitro, respectively, has also exhibited inhibitory activity against LSD1 (IC50 = 22.3 μM) (Figure 4) via covalently binding to its FAD-binding motif (Ji et al., 2017; Cao et al., 2018). Further study verified that compound 1 also inhibited migration, invasion, and metastasis of TNBC cell lines BT-549 and MDA-MB-231 cells and tumor bone metastasis in vivo. In the mechanism, 1 blocked the interaction between LSD1 and slug, and thus upregulated suppressor E-cadherin and reduced epithelial markers (Ferrari-Amorotti et al., 2014). To improve the potency, and selectivity of 2-PCPA, some 2-PCPA derivatives have been designed and synthesized based on multiple optimization strategies. GlaxoSmithKline lnc. has designed 2 PCPA-based LSD1 inhibitors 2 and 3 with IC50 of 1.7 μM, and 0.016 μM, respectively, (Figure 4) (Tu et al., 2020; Zhou et al., 2021b). N-alkylated 2-PCPA derivative 2, a selective and orally bioavailable for LSD1 inhibitor induced IFN-γ/TNF-α-expressing CD8 T cell infiltration into the tumors of 4T1 immunotherapy-resistant mice and sensitized to immunotherapy via a LSD1-EOMES switch. Interestingly, 2 showed much potent anti-BC activity than PD-L1 antibody (Tu et al., 2020). Compound 3 promoted the antigen presentation and enhanced the tumor-killing activity of tumor-specific cytotoxic T-cells in 4T1 mouse model (Zhou et al., 2021b). Compounds 4 and 5 (Figure 4) are two PCPA-4-hydroxytamoxifen conjugates, which released 4-hydroxytamoxifen catalyzing by LSD1 in vitro and in cellulo and exhibited anti-proliferative activity against MCF-7 cells at concentrations as low as 0.1 μM. In addition, both of the two conjugates have better in cellulo anti-proliferative activity than their parent compounds (Ota et al., 2016). NCD38 (6), a selective LSD1 inactivator optimized from PCPA with IC50 of 0.59 μM (Sugino et al., 2017), could reduce the stemness of TNBC cells and tumor growth in vitro (Zhou et al., 2021a). ORY-1001 (7), a PCPA derivative in phase II clinical trial for acute myelocytic leukemia, could inhibit TNBC cells and HER2-positive BC cells in distinct mechanisms (Figure 4) (Cuyàs et al., 2020; Wang et al., 2022). Compound 7 inhibited HER2-positive BC via reducing SOX2-driven breast cancer stem cells. In the mechanism, compound 7 disturbed the assembly between LSD1 and co-repressor RCOR1/CoREST via blocking the binding between LSD1 and FAD cofactor, and thus enhanced transcriptional repression of SOX2 (Cuyàs et al., 2020). In TNBC, compound 7 suppressed the proliferation of TNBC cells via devitalizing androgen receptor (Wang et al., 2022).
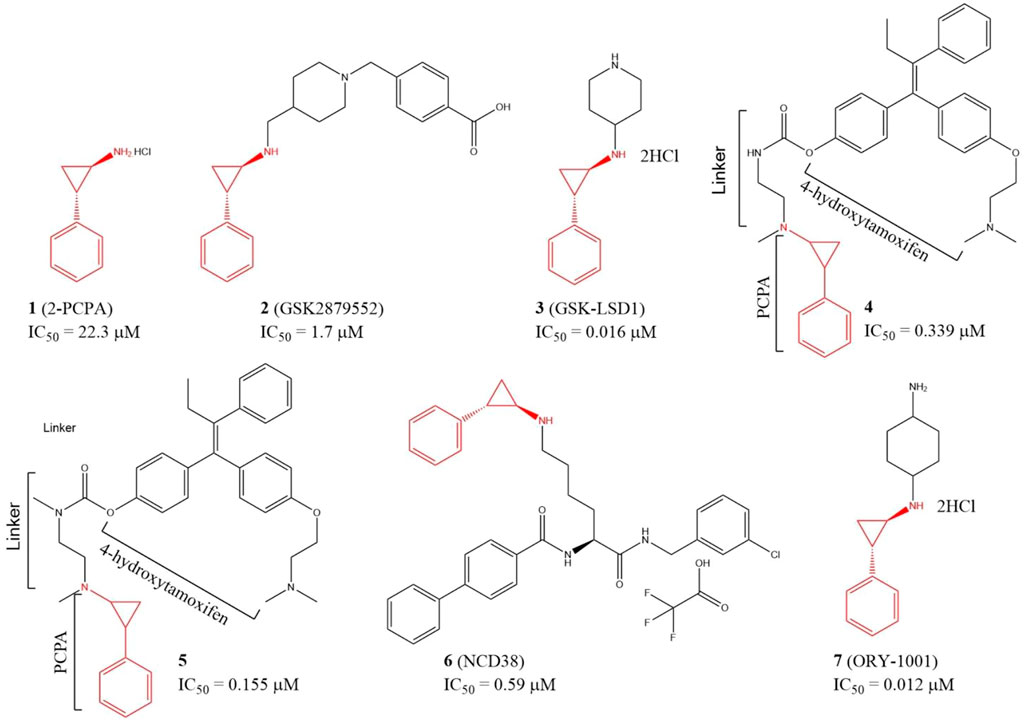
FIGURE 4. Chemical structures of PCPA-based LSD1 inhibitors in BC therapy.
Polyamine analogues, previously identified as the FAD-dependent spermine oxidase inhibitors, were also found to inhibit LSD1 demethylase activity in 2007 (Huang et al., 2007). Bisguanidine 8 and biguanide 9 exhibited demethylase inhibitory activity by over 50% at 1 μM but both of them lack selectivity among MAOs (Nowotarski et al., 2015). Four (bis)-thioureidopropyldiamine compounds (10–13) showed improved selectivity compared with their lead compounds 8 and 9 (Figure 5). Among them, compound 13 exhibited the best selectivity and potency with IC50 values of 5.0 and 4.8 μM in vitro, respectively. In fact, 13 also showed much more potent anticancer activity against MCF7 cells than 2-PCPA (Nowotarski et al., 2015). Further study showed that compounds 10–13 suppressed the proliferation of MCF-7 cells via increasing H3K4me2 levels, which significantly upregulated tumor suppressor genes p16, GATA4, HCAD, and SFRP2. The docking assay indicated that 11 could form three hydrogen bonds with residues N535 and A539, and FAD within the LSD catalytic pocket. In addition, the hydrophobic interactions between hydrophobic residues in lining the LSD1 pocket and 11 also contributed to their binding ability.
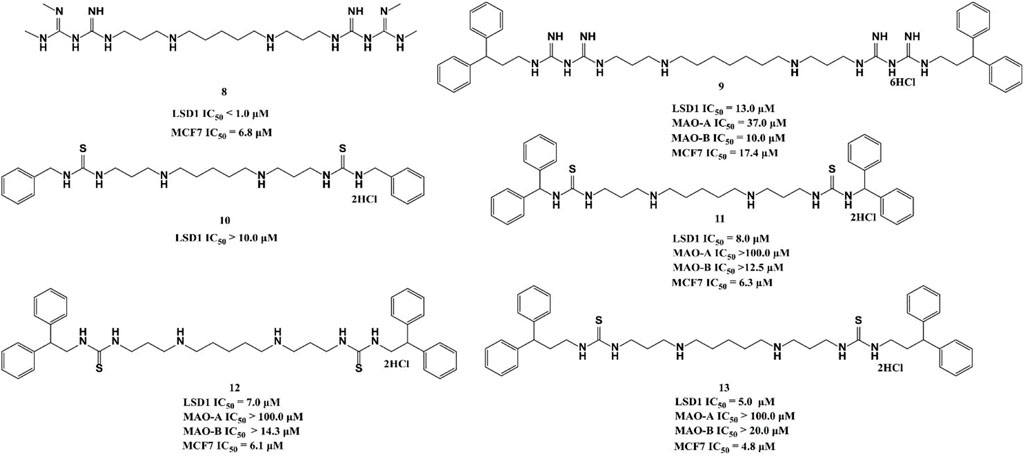
FIGURE 5. Chemical structures of polyamine analogue-based LSD1 inhibitors.
Natural products are one of the major sources in drug discovery due to their diverse chemical scaffolds and activity profiles (Fang et al., 2020; Yang et al., 2020; Yang et al., 2021a; Cheng et al., 2022b; Song et al., 2022). Many natural products and their derivatives have been found with in vitro inhibitory activity against LSD1.
Kong’s group identified six flavonoid compounds (Figure 6, 14–19) with inhibitory activity against LSD1 from Scutellaria baicalensis Georgi using countercurrent chromatography (CCC) (Han et al., 2018). Among them, compound 19 is the best LSD1 inhibitor with the in vitro IC50 of 2.98 µM and in cellulo IC50 of 17.94 µM against MDA-MB-231 cells. Isoquercitrin (Figure 6, 20), a flavonoid compound with anti-BC activity extracted from Bidens bipinnata L, is an LSD1 inhibitor that inhibited proliferation of TNBC cell line MDA-MB-231 via activating mitochondrial-mediated apoptosis (Xu et al., 2019). Biochanin A (21), a dietary flavonoid from Cicer arietinum L, could inhibit the proliferation and metastasis of BC in cellulo and in vivo (Moon et al., 2008; Sehdev et al., 2009; Ren et al., 2018). Compound 21 was found to be effective and reversible with IC50 of 2.95 μM and it preferably suppressed LSD1 over MAO-A/B (>32 μM) (Wang et al., 2020). In gastric MGC-803 cells, Biochanin A induced the accumulation of H3K4me1/2 and inhibited cell growth moderately (IC50 = 6.77 µM) (Wang et al., 2020). Oleacein (Figure 5, 22), a dihydroxy-phenol found in extra virgin olive oil, is a FAD competitive LSD1 inhibitor with IC50 of 2.5 μM in vitro (Cuyàs et al., 2019). Compound 22 reduced the stemness of BC stem cells via blocking the interaction between LSD1 and the methylated histone H3, disintegrating the assembled co-repressor complex LSD1/RCOR1/CoREST, disturbing the occupation of LSD1 to the SOX2 promoter and finally reducing the SOX2 level. Capsaicin (Figure 6, 23), a bioactive compound from chili peppers with the broad spectrum of anticancer activity in various subtype of BC cells (Chou et al., 2009; Thoennissen et al., 2010; Wu et al., 2020; Chen et al., 2021), was found to act as reversible LSD1 inhibitor with an IC50 value of 0.6 μM (Jia et al., 2020). Compound 23 competitively occupied with FAD-binding sites within the catalytic pocket of LSD1, raised H3K4me1/2 levels and suppressed the proliferation and migration of BC cells.
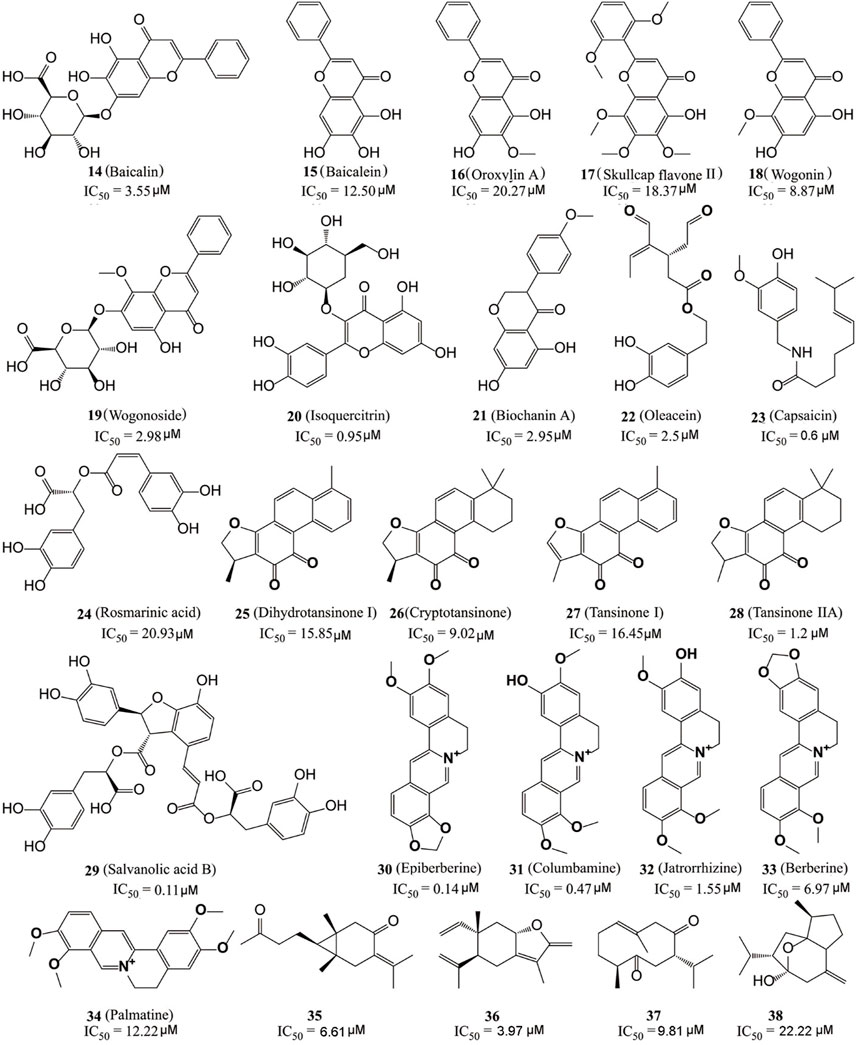
FIGURE 6. Structure of natural LSD1 inhibitors.
The dried root of Salvia miltiorrhiza is a traditional Chinese medicine used for over 1,000 years to treat cardiovascular and cerebrovascular diseases, gynecological diseases, diabetes, and insomnia (Su et al., 2015; Jia et al., 2019; Shi et al., 2019). S. miltiorrhiza has also been reported to improve the survival rate of BC patients and several bioactive components (Figure 6, 24–29) mediated its pharmacological actions via multiple anticancer pathways (Jin et al., 2021; Mahmoud et al., 2021). For example, rosmarinic acid (24) could suppress proliferation, metastasis and angiogenesis, and sensitize BC cells to paclitaxel via NF-κB-p53 pathways (Mahmoud et al., 2021). Dihydroisotanshinone I (25) inhibited the proliferation of BC cells via inducing their ferroptosis and apoptosis (Lin et al., 2019). Cryptotanshinone (26) inhibited migration through inactivating PKM2/β-Catenin signaling, and mediated drug resistance via reducing the oligomer formation of breast cancer resistance protein on the cell membrane, and thus blocking its efflux function (Zhou et al., 2020; Ni et al., 2021). Tanshinone I (27) inhibited the proliferation of MDA-MB-231 cells via activating the AMP-activated protein kinase mediating autophagic signaling (Zheng et al., 2020). Tanshinone IIA (28) sensitized BC cells to adriamycin via attenuates the stemness of BC cells by targeting the miR-125b/STARD13 signaling (Li et al., 2022). Recently, the six extracted monomeric compounds from roots of S. miltiorrhiza have been identified as LSD1 inhibitors with their IC50 values within the range 0.11–20.93 μM (Lin et al., 2020). Among them, salvianolic acid B (29) showed the best inhibitory activity against LSD1 (IC50 = 0.11 μM) and it demethylase-dependently inhibited the proliferation and migration of MDA-MB-231 cells with an IC50 value of 54.98 μM at 24 h against MDA-MB-231 cells.
Coptis chinensis, another traditional Chinese medicine widely used over 2,000 years for treatment of atherosclerosis, diabetes, and inflammation (Alami et al., 2020) has also been used to treat a varieties of cancers including BC (Wu et al., 2019), and isoquinoline alkaloids (Figure 6, 30–34) have been identified as the main anticancer active components of C. chinensis with inhibitory activity against LSD1 (Yi et al., 2016). Epierberine (30) inhibited the proliferation and metastasis of BC cells via induced cell cycle arrest and induced apoptosis by regulating Wnt/β-catenin pathway (Dian et al., 2022). Jatrorrhizine (32) exhibited anti-proliferative activity via attenuating TNIK/Wnt/β-catenin signaling in BC cells with IC50 values of 11.08, 17.11, and 22.14 μM against MCF-7, MDA-MB-231, and 4T1 cells, respectively (Sun et al., 2019). Berberine (27) sensitized the BC cells to chemotherapeutic agents via reducing XRCC1-mediated base excision repair (Gao et al., 2019). Palmatine (34) reduced the lung metastasis of TNBC via downregulating metastasis-associated protein 1 (MTA1) and increasing p53 level (Ativui et al., 2022). Recently, Li Z. R. et al. (2020) found that five protoberberine alkaloids (30–34) also showed the inhibitory activities against LSD1. All the IC50 values of them were as low as micromoles and highly selective to LSD1 over MAOs.
Recently, Ren et al. (2021) also identified four sesquiterpene-based LSD1 inhibitors (compounds 35–38) with their IC50 values within the range 3.97–22.22 μM from zedoary turmeric oil using CCC strategy. Compound 36 had the best inhibitory activity (IC50 = 3.95 μM) of them and also exhibited the anti-metastasis activity against MDA-MB-231 cells.
Due to the similar structure between MAOs and LSD1, two MAOs inhibitors (Figure 7, 39 and 40) with active propargylamine group also showed a weak inhibitory activity against LSD1 in the millimolar range (Lee et al., 2006; Matos et al., 2009). To improve the potency of LSD1 inhibitor, a covalent and irreversible LSD1 inhibitor (41) was designed through combining the active propargylamine group with N-terminal 21 amino acids of LSD1 substrate H3, compound 41 selectively inhibited LSD1 demethylase activity with a Ki value of 0.107 μM (Culhane et al., 2006). Based on the structure of 41, Schmitt et al. (2013) designed and synthesized a set of with propargyl warhead (Figure 7, 42–45). Among them, compound 44 was the best LSD1 irreversible inhibitor with IC50 of 44.0 μM in vitro, while 43 has the best anti-proliferative activity against MCF7 cells (IC50 = 91.5 μM) for 24 h (Schmitt et al., 2013). The docking analysis of the binding mode between 42 and LSD1 showed that residues Y761 and D555 formed hydrogen bonds with the amine of N-propargylamine warhead and the amide nitrogen atom of the benzamide moiety, respectively. The aromatic substituents extensively form hydrophobic and T-shaped aromatic interactions with F558, F560, Y807, and H812.
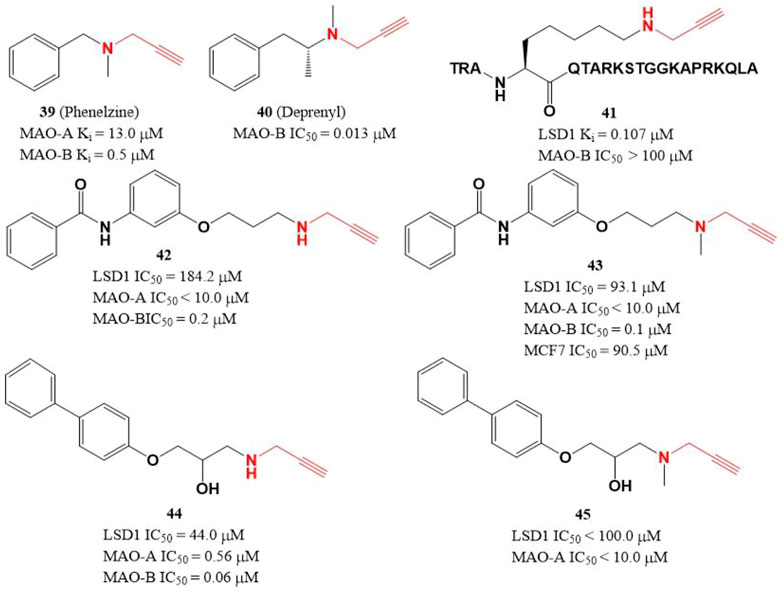
FIGURE 7. Structure of propargylamine-based LSD1 inhibitors.
Sorna et al. (2013) identified six benzohydrazides (Figure 8, 46–51) IC50 values within 0.19–0.333 μM range based on virtual screening from a compound library containing 2,000,000 compounds and biochemical assay. Then, three compounds 53–55, with IC50 of 0.128, 0.013, and 0.014 μM, respectively, were gotten though structure-based optimization. Compound 55, the best potent LSD1 inhibitor of them, is reversible and specific for LSD1 and inhibits the proliferation and survival of seven BC cell lines with IC50 from 0.468 to 2.730 μM range, which is more potent than a known MAO inhibitor 2-PCPA. Further study showed that 55 suppressed the proliferation of BC cells via reducing Sox2 expression, promoting G1 cell cycle arrest, and inducing the expression of differentiation-related genes in demethylase-dependent manner (Zhang et al., 2013). The binding mode analysis using ICM software showed that 55 could form three H-bonding interactions with the residues G314, R350, and Y510 via its benzohydrazide scaffold.
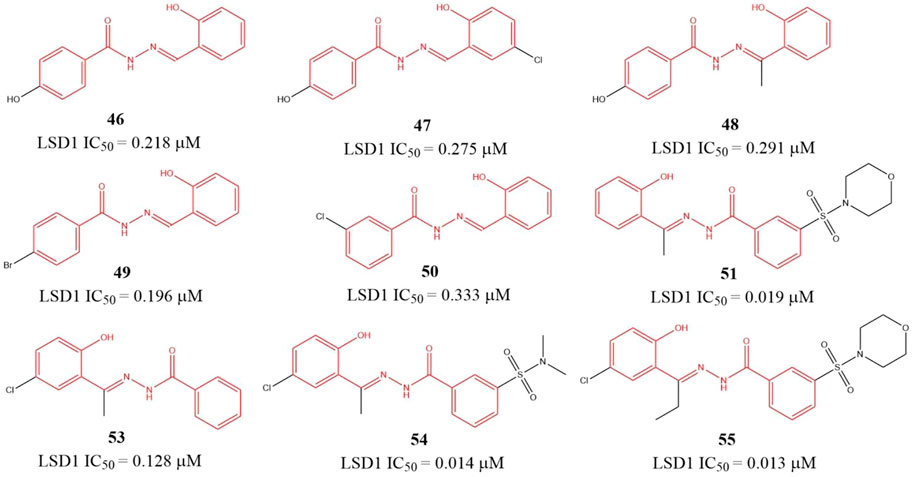
FIGURE 8. Structure of benzohydrazide-based LSD1 inhibitors.
The polyamine/guanidine and methionine were often key pharmacophores for designing MAOs inhibitors. Dulla et al. designed a series of compounds (Figure 9, 56–59) with related pharmacophores using oxazole as a linker (Dulla et al., 2013). The results showed that compounds 56–58 exhibited good in vitro inhibitory activity against LSD1 with IC50 of 16.1, 10.1, and 9.5 μM, respectively. Interestingly, all the synthesized compounds including compound 59 showed an excellent cytotoxicity activity against MDA-MB-231 cells (IC50 = 1.035–1.328 nM) in cellulo, which suggested these compounds have other targets contributing their anticancer activity. Docking analysis showed that the free -NH2 and the oxazole nitrogen of compound 56 formed H-bonding with residues T624 and R316 of LSD1, respectively, and its oxazole ring is involved in a π-cation interaction with residue R316. In case of 57, its free -NH2 formed a H-bond with S760, the sulfur atom formed H-bond with M332 and V333, and the phenyl ring interacted with W751 and Y761 residues via π-π stacking. In case of 57, its replaced guanidine group could form two H-bonds with residues E308 and R310 whereas its phenyl ring interacted with R316 via π-cation interaction.
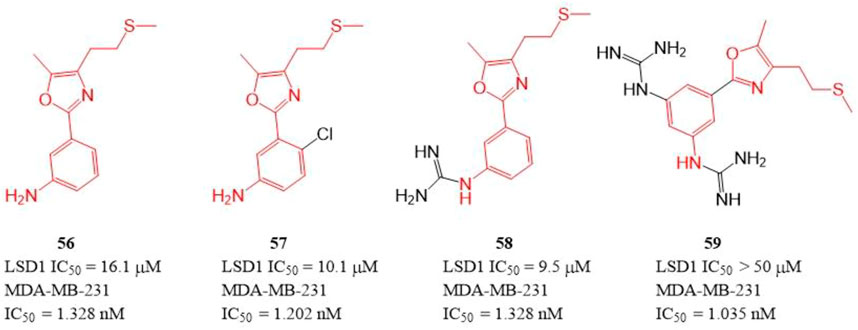
FIGURE 9. Structure of phenyl oxazole-based LSD1 inhibitors.
In BC, LSD1 and KDM6A have been found to co-express and co-localize with ER, and regulate hormone receptor signaling (Benedetti et al., 2019), suggesting that developing dual-targeting agents against the two proteins are potent strategy to improve the potency of LSD1 inhibitors. Based on this hypothesis, Altucci’s group designed four dual-KDM inhibitors (Figure 10, 60–63) targeting LSD1 and KDM6A. Among them, compound 61 exhibited the best anti-BC activity with no significant toxicity and good oral potency. In the mechanism, 61 induced cell arrest and apoptosis of hormone-positive BC in cellulo and in vivo and exhibited lower toxicity against non-cancerous cells (HaCaT) compared with clinical HDAC inhibitor Vorinostat. Additionally, it also reduced the resistance to endocrine therapies, suggesting dual-target is a feasible and potent strategy to overcome drug resistance for BC therapy.
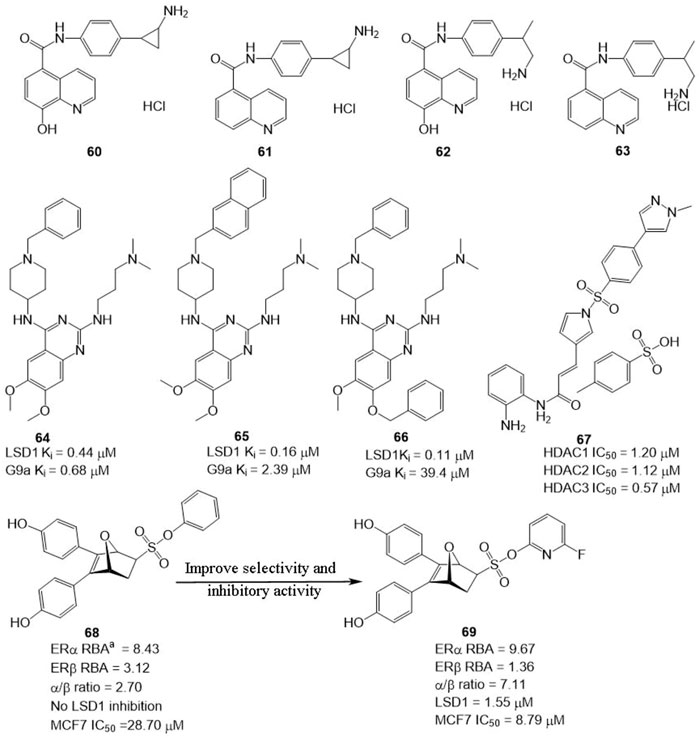
FIGURE 10. Structure of reported dual-target inhibitors. a RBA values =
Compound 64 was identified as a non-covalent dual LSD1/G9a inhibitor with anti-leukemia activity (Speranzini et al., 2016; Menna et al., 2022). Mai’s group optimized the structure of 64 via modifying its quinazoline core and got two non-covalent, and more potent LSD1/G9a inhibitors (Figure 10, 65 and 66). Compared to lead compound 63, compounds 65 and 66 exhibited a better inhibitory activity against LSD1 but reduced the inhibitory activity against G9a. Further study showed that 65 and 66 showed better anticancer activity against MDA-MBA-231, THP-1, and MV4-11 cells without significant toxicity to non-cancer AHH-1 cells compared with epigenetic drug UNC0638, which suggested that the LSD1 antagonistic activity of LSD1/G9a inhibitors endorsed with anticancer activity (Menna et al., 2022).
LSD1 has been found to promote BC proliferation via interacting with histone deacetylases (HDACs) (Cao et al., 2017). Huang’s group found that HDAC inhibitor sulforaphane suppressed the proliferation of BC cells via blocking activity of upstream transcription factor 1 (USF1) and promoting ubiquitination degradation of LSD1, and LSD1 inhibitor significantly sensitized sulforaphane to BC in cellulo and in vivo (Cao et al., 2018), which inhibited that the combined therapy or discovery of LSD1/HDAC5 inhibitors is a potential strategy for BC treatment. Zylla et al. (2022) study found that HDAC/LSD1 inhibitor 67 (Figure 10) exhibited anti-proliferative and anti-metastatic activity against TNBC in cellulo and in vivo.
Currently, hormonal drugs and chemotherapy have been widely combined in the utilization for BC therapy in clinic. But combined strategies also showed undeniable disadvantages during clinic use. To overcome this phenomenon, Zhou’s group developed a set of dual-target inhibitors based compound 68. Among these conjugators, compound 69 has the best in vitro and in cellulo activity with IC50 values of 9.67, 1.36, 1.55, and 8.79 μM against ERα, ERβ, LSD1, and MCF7, respectively (He et al., 2020). Most notably, 69 exhibited better anti-BC activity in MCF-7 cells than clinical agent 4-hydroxytamoxifen.
Mounting evidence supports that LSD1 is overexpressed in many subtypes of BC and promotes their proliferation (Pollock et al., 2012; Yang et al., 2018d; Xu et al., 2019; Wang et al., 2022), differentiation (Wu et al., 2013; Zhang et al., 2013; Ji et al., 2021), metastasis (Li et al., 2011; Qiu et al., 2018; Zheng et al., 2018; Hu et al., 2019; Gong et al., 2021), and drug resistance (Bennani-Baiti, 2012; Verigos et al., 2019; Zhou et al., 2021a; Liu et al., 2022), which makes LSD1 become a promising target for BC therapy. But the detailed mechanisms of the LSD1 in BC progression are unclear and more potential anti-tumor pathways or downstream genes are yet to clarify due to the heterogeneity of varieties of BC subtypes. For example, metastasis and drug resistance are two main factors responsible for BC-caused death in clinic (Cheng et al., 2021; Cheng et al., 2022a). Although there are several reported researches on the roles of LSD1 in BC metastasis and drug resistance, the specific functions of LSD1 in these two cancer cell events are yet to be investigated. In addition, many studies showed that LSD1 functioned as an oncogene or suppressor gene in BC progression dependent on its transcriptional regulatory activity via assembling into different complexes with its client proteins. Therefore, mapping the protein-protein interactome of LSD1 is a potential strategy to further clarify its function in BC development (Yatim et al., 2012; Yang et al., 2022). Meanwhile, the exploration on the functions of LSD1 in homeostasis is also imperative to avoid the potential health risks during the development and advancement of clinical trials of LSD1 inhibitors. Moreover, most of the current studies about LSD1 functions mainly focus on its histone demethylase activity, and few works are available about the roles of non-histone substrates, epigenetic modifications, and non-enzyme activity of LSD1 in BC progression (Feng et al., 2016; Liu et al., 2020a; Gong et al., 2021). Thus, it is imperative to further carry out research in these areas.
Currently, some LSD1 inhibitors have entered clinical trials to combat small lung cancer cells and acute myeloid leukemia and several of them showed encouraging results (Fang et al., 2019). But there is no LSD1 inhibitor in clinical trials for BC. Many factors contribute to this phenomenon apart from the complex etiological factor of BC. First, most of the reported LSD1 inhibitors have poor selectivity, which adds to the uncertainty of drug therapy. Second, some LSD1 inhibitors such as compounds 56–59 exhibited intracellular activity inconsistent with their inhibitory activity against demethylase activity (Dulla et al., 2013), which suggested that there are potential off-target effects and unpredictable risks of some identified LSD1 inhibitors, when they were advanced into clinical trials for BC therapy. Third, like other enzyme inhibitors (Yang et al., 2018c; Wu et al., 2018; Yang et al., 2019; Yang et al., 2021c; Yang et al., 2021d), most of the reported LSD1 inhibitors were only detected in anti-BC activity in vitro or in cellulo assays with a dearth of the studies about in vivo toxicology, pharmacokinetics, and effectiveness in animals. Finally, due to the existence of several alternative signaling and isoenzymes in BC cells, LSD1 inhibitor used alone may be not sufficient to achieve the desired therapeutic effect sometimes. To solve these dilemmas, pharmaceutical chemists and pharmacologists have proposed several strategies. Given allosteric regulation is a common characteristic for metabolic enzymes (Kremer and Lyssiotis, 2022), identifying allosteric pockets or sites and developing corresponding inhibitors is a feasible strategy for the discovery of LSD1 inhibitors. Targeting the protein–protein interaction (PPI) is also an effective method to improve the selectivity of enzymes (Yang et al., 2018a; Cheng et al., 2020; Yang et al., 2021a; Yang et al., 2021c; Yang et al., 2021d; Yang et al., 2022), blocking the interaction between LSD1 and its client proteins may be also a useful strategy to develop selective LSD1 inhibitors. Metal complexes have showed promising in vivo activity for BC treatment (Yang et al., 2018b; Cheng et al., 2022b), and Yang et al. (2017) have also identified a rhodium-based LSD1 inhibitor with in cellulo anticancer activity against prostate cancer, which suggested this kind of compound is a unique source for the discovery of potent and selective LSD1 inhibitors against BC treatment. Several drug design strategy such as computer aided drug optimization and proteolysis targeting chimera (PROTAC)-strategy have been introduced to discover lead compounds against LSD1 with better biocompatibility and in vivo potency (Liu et al., 2020b; Martín-Acosta and Xiao, 2021). Moreover, combined therapy and developing dual-target inhibitors have also been used to improve the anticancer potency and overcome drug resistance of LSD1 inhibitors (Ota et al., 2016; Cao et al., 2018; Benedetti et al., 2019; Qin et al., 2019; Verigos et al., 2019; Liu et al., 2022). Drug delivery by nanocarriers is a potent strategy to increase drug utilization rate, improve potency, and reduce toxicity and drug resistance of single agent or combined therapy (Singh and Mitragotri, 2020; Vallet-Regí et al., 2022). LSD1 has been found to exhibit its activity by promoting gastric cancer cell stemness via delivered by endogenous small extracellular vesicles in vivo (Zhao et al., 2021), and the drug delivery of LSD1 siRNA using nanocarriers has exhibited potent anticancer activities in several studies (Suzuki, 2019; Li B. et al., 2020; Jiang et al., 2021).
In a word, LSD1 plays a crucial role in BC progression and drug resistance, and its inhibition is a potential anti-BC strategy due to its efficacy in current preclinical studies. Therefore, the discovery of more specific LSD1 inhibitors is imperative to deepen our understanding of the role of LSD1 in BC tumorigenesis and verify the feasibility of targeting LSD1 as an anti-BC therapy in clinic.
G-JY and JC: supervision, funding acquisition, and writing—original draft preparation. Y-JL, L-JD, FT, M-HZ, and Z-YS: preparation of figures and tables. J-MW, M-YN, XL, Z-SL, W-JQ, and C-JF: investigation and validation, writing, reviewing, and editing. G-JY and JC: conception, writing, reviewing ,and editing. The authors contributed to the data preparation and drafted and revised the manuscript.
This work was supported by the National Natural Science Foundation of China (31972821), the General Scientific Research Project of Education of Zhejiang Province (Y202147351), the Starting Research Fund of Ningbo University (421912073), and the State Key Laboratory for Managing Biotic and Chemical Threats to the Quality and Safety of Agro-products (2010DS700124-ZZ2008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alami, M. M., Xue, J., Ma, Y., Zhu, D., Abbas, A., Gong, Z., et al. (2020). Structure, function, diversity, and composition of fungal communities in rhizospheric soil of Coptis chinensis Franch under a successive cropping system. Plants 9 (2), 244. doi:10.3390/plants9020244
Andresen, M. S., Ali, H. O., Myklebust, C. F., Sandset, P. M., Stavik, B., Iversen, N., et al. (2017). Estrogen induced expression of tissue factor pathway inhibitor-2 in MCF7 cells involves lysine-specific demethylase 1. Mol. Cell. Endocrinol. 443, 80–88. doi:10.1016/j.mce.2017.01.016
Ativui, S., Danquah, C. A., Ossei, P. P. S., and Ofori, M. (2022). Palmatine attenuates metastatic lung colonization of triple negative breast cancer cells. Front. Pharmacol. 13, 853230. doi:10.3389/fphar.2022.853230
Ayoub, N. M., Jaradat, S. K., Al-Shami, K. M., and Alkhalifa, A. E. (2022). Targeting angiogenesis in breast cancer: Current evidence and future perspectives of novel anti-angiogenic approaches. Front. Pharmacol. 13, 838133. doi:10.3389/fphar.2022.838133
Bai, J. W., Chen, M. N., Wei, X. L., Li, Y. C., Lin, H. Y., Chen, M., et al. (2017). The zinc-finger transcriptional factor Slug transcriptionally downregulates ERα by recruiting lysine-specific demethylase 1 in human breast cancer. Oncogenesis 6 (5), e330. doi:10.1038/oncsis.2017.38
Benedetti, R., Dell'Aversana, C., De Marchi, T., Rotili, D., Liu, N. Q., Novakovic, B., et al. (2019). Inhibition of histone demethylases LSD1 and UTX regulates ERα signaling in breast cancer. Cancers 11 (12), 2027. doi:10.3390/cancers11122027
Bennani-Baiti, I. M. (2012). Integration of ERα-PELP1-HER2 signaling by LSD1 (KDM1A/AOF2) offers combinatorial therapeutic opportunities to circumventing hormone resistance in breast cancer. Breast Cancer Res. 14 (5), 112. doi:10.1186/bcr3249
Bennesch, M. A., Segala, G., Wider, D., and Picard, D. (2016). LSD1 engages a corepressor complex for the activation of the estrogen receptor α by estrogen and cAMP. Nucleic Acids Res. 44 (18), 8655–8670. doi:10.1093/nar/gkw522
Boulding, T., McCuaig, R., Tan, A., Hardy, K., Wu, F., Dunn, J., et al. (2018). LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 8 (1), 73. doi:10.1038/s41598-017-17913-x
Bradley, C., van der Meer, R., Roodi, N., Yan, H. P., Chandrasekharan, M., Sun, Z. W., et al. (2007). Carcinogen-induced histone alteration in normal human mammary epithelial cells. Carcinogenesis 28 (10), 2184–2192. doi:10.1093/carcin/bgm100
Cao, C. Y., Vasilatos, S. N., Bhargava, R., Fine, J. L., Oesterreich, S., Davidson, N. E., et al. (2017). Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 36 (1), 133–145. doi:10.1038/onc.2016.186
Cao, C. Y., Wu, H., Vasilatos, S. N., Chandran, U., Qin, Y., Wan, Y., et al. (2018). HDAC5-LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 143 (6), 1388–1401. doi:10.1002/ijc.31419
Chen, M. J., Xiao, C. C., Jiang, W., Yang, W. P., Qin, Q. H., Tan, Q., et al. (2021). Capsaicin inhibits proliferation and inducesapoptosis in breast cancer by down-regulating FBI-1-mediated NF-κB pathway. Drug Des. devel. Ther. 15, 125–140. doi:10.2147/DDDT.S269901
Cheng, S. S., Qu, Y. Q., Wu, J., Yang, G. J., Liu, H., Wang, W. H., et al. (2022a). Inhibition of the CDK9–cyclin T1 protein–protein interaction as a new approach against triple-negative breast cancer. Acta Pharm. Sin. B 12 (3), 1390–1405. doi:10.1016/j.apsb.2021.10.024
Cheng, S. S., Yang, G. J., Wang, W. H., Leung, C. H., and Ma, D. L. (2020). The design and development of covalent protein-protein interaction inhibitors for cancer treatment. J. Hematol. Oncol. 13, 26. doi:10.1186/s13045-020-00850-0
Cheng, S. S., Yang, G. J., Wang, W. H., Ma, D. L., and Leung, C. H. (2021). Discovery of a tetrahydroisoquinoline-based CDK9-cyclin T1 protein–protein interaction inhibitor as an anti-proliferative and anti-migration agent against triple-negative breast cancer cells. Genes Dis.. doi:10.1016/j.gendis.2021.06.005
Cheng, S. S., Yang, G. J., Wang, W. H., Song, Y. Q., Ko, C. N., Han, Q. B., et al. (2022b). Identification of a cytisine-based EED-EZH2 protein-protein interaction inhibitor preventing metastasis in triple-negative breast cancer cells. Acta Mat. Med. 1 (2), 197–211. doi:10.15212/AMM-2022-0006
Chou, C. C., Wu, Y. C., Wang, Y. F., Chou, M. J., Kuo, S. J., and Chen, D. R. (2009). Capsaicin-induced apoptosis in human breast cancer MCF-7 cells through caspase-independent pathway. Oncol. Rep. 21 (3), 665–671. PMID: 19212624. doi:10.3892/or_00000269
Cortez, V., Mann, M., Tekmal, S., Suzuki, T., Miyata, N., Rodriguez-Aguayo, C., et al. (2012). Targeting the PELP1-KDM1 axis as a potential therapeutic strategy for breast cancer. Breast Cancer Res. 14 (4), R108. doi:10.1186/bcr3229
Culhane, J. C., Szewczuk, L. M., Liu, X., Da, G., Marmorstein, R., and Cole, P. A. (2006). A mechanism-based inactivator for histone demethylase LSD1. J. Am. Chem. Soc. 128 (14), 4536–4537. doi:10.1021/ja0602748
Cuyàs, E., Gumuzio, J., Lozano-Sánchez, J., Carreras, D., Verdura, S., Llorach-Parés, L., et al. (2019). Extra virgin olive oil contains a phenolic inhibitor of the histone demethylase LSD1/KDM1A. Nutrients 11 (7), 1656. doi:10.3390/nu11071656
Cuyàs, E., Gumuzio, J., Verdura, S., Brunet, J., Bosch-Barrera, J., Martin-Castillo, B., et al. (2020). The LSD1 inhibitor iadademstat (ORY-1001) targets SOX2-driven breast cancer stem cells: A potential epigenetic therapy in luminal-B and HER2-positive breast cancer subtypes. Aging 12 (6), 4794–4814. doi:10.18632/aging.102887
Dian, L. L., Xu, Z. Z., Sun, Y. F., Li, J. H., Lu, H. F., Zheng, M., et al. (2022). Berberine alkaloids inhibit the proliferation and metastasis of breast carcinoma cells involving Wnt/β-catenin signaling and EMT. Phytochemistry 200, 113217. doi:10.1016/j.phytochem.2022.113217
Disis, M. (2010). Immune regulation of cancer. J. Clin. Oncol. 28, 4531–4538. doi:10.1200/JCO.2009.27.2146
Dulla, B., Kirla, K. T., Rathore, V., Deora, G. S., Kavela, S., Maddika, S., et al. (2013). Synthesis and evaluation of 3-amino/guanidine substituted phenyl oxazoles as a novel class of LSD1 inhibitors with anti-proliferative properties. Org. Biomol. Chem. 11 (19), 3103–3107. doi:10.1039/c3ob40217g
Fang, Y., Liao, G. C., and Yu, B. (2019). LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 12 (1), 129. doi:10.1186/s13045-019-0811-9
Fang, Y., Yang, C., Yu, Z. Q., Li, X., Mu, Q. C., Liao, G. C., et al. (2020). Natural products as LSD1 inhibitors for cancer therapy. Acta Pharm. Sin. B 11 (3), 621–631. doi:10.1016/j.apsb.2020.06.007
Feng, J., Li, L., Zhang, N., Liu, J., Zhang, L., Gao, H., et al. (2017). Androgen and AR contribute to breast cancer development and metastasis: An insight of mechanisms. Oncogene 36 (20), 2775–2790. doi:10.1038/onc.2016.432
Feng, J. X., Xu, G. Y., Liu, J. W., Zhang, N., Li, L. L., Ji, J. F., et al. (2016). Phosphorylation of LSD1 at Ser112 is crucial for its function in induction of EMT and metastasis in breast cancer. Breast Cancer Res. Treat. 159 (3), 443–456. doi:10.1007/s10549-016-3959-9
Ferrari-Amorotti, G., Chiodoni, C., Shen, F., Cattelani, S., Soliera, A., Manzotti, G., et al. (2014). Suppression of invasion and metastasis of triple-negative breast cancer lines by pharmacological or genetic inhibition of slug activity. Neoplasia 16 (12), 1047–1058. doi:10.1016/j.neo.2014.10.006
Forneris, F., Binda, C., Battaglioli, E., and Mattevi, A. (2008). LSD1: Oxidative chemistry for multifaceted functions in chromatin regulation. Trends biochem. Sci. 33 (4), 181–189. doi:10.1016/j.tibs.2008.01.003
Gao, X. J., Wang, J., Li, M. Q., Wang, J., Lv, J., Zhang, L., et al. (2019). Berberine attenuates XRCC1-mediated base excision repair and sensitizes breast cancer cells to the chemotherapeutic drugs. J. Cell. Mol. Med. 23 (10), 6797–6804. doi:10.1111/jcmm.14560
Gong, Z., Li, A., Ding, J., Li, Q., Zhang, L., Li, Y., et al. (2021). OTUD7B deubiquitinates LSD1 to govern its binding partner specificity, homeostasis, and breast cancer metastasis. Adv. Sci. 8 (15), e2004504. doi:10.1002/advs.202004504
Gu, F. Y., Lin, Y. X., Wang, Z., Wu, X. X., Ye, Z. Y., Wang, Y. Z., et al. (2020). Biological roles of LSD1 beyond its demethylase activity. Cell. Mol. Life Sci. 77 (17), 3341–3350. doi:10.1007/s00018-020-03489-9
Han, C., Wang, S. S., Li, Z. R., Chen, C., Hou, J. Q., Xu, D. Q., et al. (2018). Bioactivity-guided cut countercurrent chromatography for isolation of lysine-specific demethylase 1 inhibitors from Scutellaria baicalensis Georgi. Anal. Chim. Acta 1016, 59–68. doi:10.1016/j.aca.2018.01.014
He, M., Ning, W. T., Hu, Z. Y., Huang, J., Dong, C. N., and Zhou, H. B. (2020). Design, synthesis and biological evaluation of novel dual-acting modulators targeting both estrogen receptor α (ERα) and lysine-specific demethylase 1 (LSD1) for treatment of breast cancer. Eur. J. Med. Chem. 195, 112281. doi:10.1016/j.ejmech.2020.112281
Hu, X., Xiang, D. X., Xie, Y., Tao, L. W., Zhang, Y., Jin, Y., et al. (2019). LSD1 suppresses invasion, migration and metastasis of luminal breast cancer cells via activation of GATA3 and repression of TRIM37 expression. Oncogene 38 (44), 7017–7034. doi:10.1038/s41388-019-0923-2
Huang, Y., Greene, E., Murray Stewart, T., Goodwin, A. C., Baylin, S. B., Woster, P. M., et al. (2007). Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc. Natl. Acad. Sci. U. S. A. 104, 8023–8028. doi:10.1073/pnas.0700720104
Huang, Y., Vasilatos, S. N., Boric, L., Shaw, P. G., and Davidson, N. E. (2012). Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res. Treat. 131 (3), 777–789. doi:10.1007/s10549-011-1480-8
Ji, X. Y., Guo, D. X., Ma, J., Yin, M., Yu, Y., Liu, C., et al. (2021). Epigenetic remodeling hydrogel patches for multidrug-resistant triple-negative breast cancer. Adv. Mat. 33 (18), e2100949. doi:10.1002/adma.202100949
Ji, Y. Y., Lin, S. D., Wang, Y. J., Su, M., Zhang, W., Gunosewoyo, H., et al. (2017). Tying up tranylcypromine: Novel selective histone lysine specific demethylase 1 (LSD1) inhibitors. Eur. J. Med. Chem. 141, 101–112. doi:10.1016/j.ejmech.2017.09.073
Jia, G., Cang, S. D., Ma, P. Z., and Song, Z. Y. (2020). Capsaicin: A “hot” KDM1A/LSD1 inhibitor from peppers. Bioorg. Chem. 103, 104161. doi:10.1016/j.bioorg.2020.104161
Jia, Q. Q., Zhu, R. Y., Tian, Y. M., Chen, B. B., Li, R., Li, L., et al. (2019). Salvia miltiorrhiza in diabetes: A review of its pharmacology, phytochemistry, and safety. Phytomedicine 58, 152871. doi:10.1016/j.phymed.2019.152871
Jiang, L. X., Gong, X. M., Liao, W. D., Lv, N. H., and Yan, R. W. (2021). Molecular targeted treatment and drug delivery system for gastric cancer. J. Cancer Res. Clin. Oncol. 147, 973–986. doi:10.1007/s00432-021-03520-x
Jin, Z., Yu, C. H., and Peng, C. (2021). Anticancer effect of tanshinones on female breast cancer and gynecological cancer. Front. Pharmacol. 12, 824531. doi:10.3389/fphar.2021.824531
Kim, D. H., Kim, K., and Baek, S. H. (2021). Roles of lysine-specific demethylase 1 (LSD1) in homeostasis and diseases. J. Biomed. Sci. 28 (1), 41. doi:10.1186/s12929-021-00737-3
Kim, J., Park, U. H., Moon, M., Um, S. J., and Kim, E. J. (2013). Negative regulation of ERα by a novel protein CAC1 through association with histone demethylase LSD1. FEBS Lett. 587 (1), 17–22. doi:10.1016/j.febslet.2012.10.054
Kremer, D. M., and Lyssiotis, C. A. (2022). Targeting allosteric regulation of cancer metabolism. Nat. Chem. Biol. 18 (5), 441–450. doi:10.1038/s41589-022-00997-6
Lee, J. Y., Park, J. H., Choi, H. J., Won, H. Y., Joo, H. D., Shin, D. H., et al. (2017). LSD1 demethylates HIF1α to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 36 (39), 5512–5521. doi:10.1038/onc.2017.158
Lee, M. G., Wynder, C., Schmidt, D. M., McCafferty, D. G., and Shiekhattar, R. (2006). Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 13 (6), 563–567. doi:10.1016/j.chembiol.2006.05.004
Li, B. L., Li, R., Zou, H. L., Ariga, K., Li, N. B., and Leong, D. T. (2020). Engineered functionalized 2D nanoarchitectures for stimuli-responsive drug delivery. Mat. Horiz. 7 (2), 455–469. doi:10.1039/C9MH01300H
Li, L., Liu, X. H., He, L., Yang, J. G., Pei, F., Li, W. M., et al. (2017). ZNF516 suppresses EGFR by targeting the CtBP/LSD1/CoREST complex to chromatin. Nat. Commun. 8 (1), 691. doi:10.1038/s41467-017-00702-5
Li, Q., Shi, L., Gui, B., Yu, W. H., Wang, J. M., Zhang, D., et al. (2011). Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 71 (21), 6899–6908. doi:10.1158/0008-5472.Can-11-1523
Li, X. M., Jia, Q., Zhou, Y. Y., Jiang, X., Song, L., Wu, Y. Y., et al. (2022). Tanshinone IIA attenuates the stemness of breast cancer cells via targeting the miR-125b/STARD13 axis. Exp. Hematol. Oncol. 11 (1), 2. doi:10.1186/s40164-022-00255-4
Li, Z. R., Suo, F. Z., Guo, Y. J., Cheng, H. F., Niu, S. H., Shen, D. D., et al. (2020). Natural protoberberine alkaloids, identified as potent selective LSD1 inhibitors, induce AML cell differentiation. Bioorg. Chem. 97, 103648. doi:10.1016/j.bioorg.2020.103648
Lim, S., Janzer, A., Becker, A., Zimmer, A., Schüle, R., Buettner, R., et al. (2010). Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31 (3), 512–520. doi:10.1093/carcin/bgp324
Lin, T., Ponn, A., Hu, X., Law, B. K., and Lu, J. (2010). Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 29 (35), 4896–4904. doi:10.1038/onc.2010.234
Lin, Y. L., Han, C., Xu, Q. Q., Wang, W. L., Li, L. N., Zhu, D. R., et al. (2020). Integrative countercurrent chromatography for the target isolation of lysine-specific demethylase 1 inhibitors from the roots of Salvia miltiorrhiza. Talanta 206, 120195. doi:10.1016/j.talanta.2019.120195
Lin, Y. S., Shen, Y. C., Wu, C. Y., Tsai, Y. Y., Yang, Y. H., Lin, Y. Y., et al. (2019). Danshen improves survival of patients with breast cancer and dihydroisotanshinone I induces ferroptosis and apoptosis of breast cancer cells. Front. Pharmacol. 10, 1226. doi:10.3389/fphar.2019.01226
Lin, Y. W., Kang, T. B., and Zhou, B. P. (2014). Doxorubicin enhances Snail/LSD1-mediated PTEN suppression in a PARP1-dependent manner. Cell Cycle 13 (11), 1708–1716. doi:10.4161/cc.28619
Liu, B. B., Liu, X. H., Han, L. L., Chen, X., Wu, X. D., Wu, J. J., et al. (2022). BRD4-directed super-enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. Proc. Natl. Acad. Sci. U. S. A. 119 (6), e2109133119. doi:10.1073/pnas.2109133119
Liu, J., Ma, J., Liu, Y., Xia, J., Li, Y. Y., Wang, Z. P., et al. (2020b). PROTACs: A novel strategy for cancer therapy. Semin. Cancer Biol. 67, 171–179. doi:10.1016/j.semcancer.2020.02.006
Liu, J. W., Feng, J. X., Li, L. L., Lin, L. Y., Ji, J. F., Lin, C., et al. (2020a). Arginine methylation-dependent LSD1 stability promotes invasion and metastasis of breast cancer. EMBO Rep. 21 (2), e48597. doi:10.15252/embr.201948597
Luo, H. C., Shenoy, A. K., Li, X. H., Jin, Y., Jin, L. H., Cai, Q. S., et al. (2016). MOF acetylates the histone demethylase LSD1 to suppress epithelial-to-mesenchymal transition. Cell Rep. 15 (12), 2665–2678. doi:10.1016/j.celrep.2016.05.050
Ma, L., Ma, S., Zhao, G. M., Yang, L. Q., Zhang, P., Yi, Q. T., et al. (2016). miR-708/LSD1 axis regulates the proliferation and invasion of breast cancer cells. Cancer Med. 5 (4), 684–692. doi:10.1002/cam4.623
Mahmoud, M. A., Okda, T. M., Omran, G. A., and Abd-Alhaseeb, M. M. (2021). Rosmarinic acid suppresses inflammation, angiogenesis, and improves paclitaxel induced apoptosis in a breast cancer model via NF3 κB-p53-caspase-3 pathways modulation. J. Appl. Biomed. 19 (4), 202–209. doi:10.32725/jab.2021.024
Majello, B., Gorini, F., Saccà, C. D., and Amente, S. (2019). Expanding the role of the histone lysine-specific demethylase LSD1 in cancer. Cancers 11 (3), 324. doi:10.3390/cancers11030324
Malagraba, G., Yarmohammadi, M., Javed, A., Barceló, C., and Rubio-Tomás, T. (2022). The role of LSD1 and LSD2 in cancers of the gastrointestinal system: An update. Biomolecules 12 (3), 462. doi:10.3390/biom12030462
Marabelli, C., Marrocco, B., and Mattevi, A. (2016). The growing structural and functional complexity of the LSD1/KDM1A histone demethylase. Curr. Opin. Struct. Biol. 41, 135–144. doi:10.1016/j.sbi.2016.07.011
Martín-Acosta, P., and Xiao, X. S. (2021). PROTACs to address the challenges facing small molecule inhibitors. Eur. J. Med. Chem. 210, 112993. doi:10.1016/j.ejmech.2020.112993
Matos, M. J., Viña, D., Quezada, E., Picciau, C., Delogu, G., Orallo, F., et al. (2009). A new series of 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 19 (12), 3268–3270. doi:10.1016/j.bmcl.2009.04.085
Menna, M., Fiorentino, F., Marrocco, B., Lucidi, A., Tomassi, S., Cilli, D., et al. (2022). Novel non-covalent LSD1 inhibitors endowed with anticancer effects in leukemia and solid tumor cellular models. Eur. J. Med. Chem. 237, 114410. doi:10.1016/j.ejmech.2022.114410
Metzger, E., Wissmann, M., Yin, N., Müller, J. M., Schneider, R., Peters, A. H. F. M., et al. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437 (7057), 436–439. doi:10.1038/nature04020
Moon, Y. J., Shin, B. S., An, G., and Morris, M. E. (2008). Biochanin A inhibits breast cancer tumor growth in a murine xenograft model. Pharm. Res. 25, 2158–2163. doi:10.1007/s11095-008-9583-6
Mortier, A., Van Damme, J., and Proost, P. (2008). Regulation of chemokine activity by posttranslational modification. Pharmacol. Ther. 120 (2), 197–217. doi:10.1016/j.pharmthera.2008.08.006
Ni, W., Fan, H., Zheng, X., Xu, F., Wu, Y., Li, X., et al. (2021). Cryptotanshinone inhibits ERα-dependent and-independent BCRP oligomer formation to reverse multidrug resistance in breast cancer. Front. Oncol. 11, 624811. doi:10.3389/fonc.2021.624811
Nowotarski, S. L., Pachaiyappan, B., Holshouser, S. L., Kutz, C., Li, Y., Huang, Y., et al. (2015). Structure-activity study for (bis)ureidopropyl- and (bis)thioureidopropyldiamine LSD1 inhibitors with 3-5-3 and 3-6-3 carbon backbone architectures. Bioorg. Med. Chem. 23 (7), 1601–1612. doi:10.1016/j.bmc.2015.01.049
Ota, Y., Itoh, Y., Kaise, A., Ohta, K., Endo, Y., Masuda, M., et al. (2016). Targeting cancer with PCPA-drug conjugates: LSD1 inhibition-triggered release of 4-hydroxytamoxifen. Angew. Chem. Int. Ed. Engl. 55 (52), 16115–16118. doi:10.1002/anie.201608711
Park, U. H., Kang, M. R., Kim, E. J., Kwon, Y. S., Hur, W., Yoon, S. K., et al. (2016). ASXL2 promotes proliferation of breast cancer cells by linking ERα to histone methylation. Oncogene 35 (28), 3742–3752. doi:10.1038/onc.2015.443
Peng, B., Wang, J., Hu, Y., Zhao, H. L., Hou, W. Y., Zhao, H. L., et al. (2015). Modulation of LSD1 phosphorylation by CK2/WIP1 regulates RNF168-dependent 53BP1 recruitment in response to DNA damage. Nucleic Acids Res. 43 (12), 5936–5947. doi:10.1093/nar/gkv528
Phillips, S., and Kuperwasser, C. (2014). Slug: Critical regulator of epithelial cell identity in breast development and cancer. Cell adh. Migr. 8 (6), 578–587. doi:10.4161/19336918.2014.972740
Pilotto, S., Speranzini, V., Tortorici, M., Durand, D., Fish, A., Valente, S., et al. (2015). Interplay among nucleosomal DNA, histone tails, and corepressor CoREST underlies LSD1-mediated H3 demethylation. Proc. Natl. Acad. Sci. U. S. A. 112 (9), 2752–2757. doi:10.1073/pnas.1419468112
Pollock, J. A., Larrea, M. D., Jasper, J. S., McDonnell, D. P., and McCafferty, D. G. (2012). Lysine-specific histone demethylase 1 inhibitors control breast cancer proliferation in ERα-dependent and -independent manners. ACS Chem. Biol. 7 (7), 1221–1231. doi:10.1021/cb300108c
Qin, Y., Vasilatos, S. N., Chen, L., Wu, H., Cao, Z. S., Fu, Y. M., et al. (2019). Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 38 (3), 390–405. doi:10.1038/s41388-018-0451-5
Qiu, R. F., Shi, H., Wang, S., Leng, S., Liu, R. Q., Zheng, Y., et al. (2018). BRMS1 coordinates with LSD1 and suppresses breast cancer cell metastasis. Am. J. Cancer Res. 8 (10), 2030–2045. PMCID: PMC6220148.
Ren, C. L., Lin, Y. L., Liu, X. Q., Yan, D., Xu, X., Zhu, D. R., et al. (2021). Target separation and antitumor metastasis activity of sesquiterpene-based lysine-specific demethylase 1 inhibitors from zedoary turmeric oil. Bioorg. Chem. 108, 104666. doi:10.1016/j.bioorg.2021.104666
Ren, G. X., Shi, Z. X., Teng, C., and Yao, Y. (2018). Antiproliferative activity of combined Biochanin A and ginsenoside Rh₂ on MDA-MB-231 and MCF-7 human breast cancer cells.. Molecules 23 (11), 2908. doi:10.3390/molecules23112908
Schmitt, M. L., Hauser, A. T., Carlino, L., Pippel, M., Schulz-Fincke, J., Metzger, E., et al. (2013). Nonpeptidic propargylamines as inhibitors of lysine specific demethylase 1 (LSD1) with cellular activity. J. Med. Chem. 56 (18), 7334–7342. doi:10.1021/jm400792m
Sehdev, V., Lai, J. C., and Bhushan, A. (2009). Biochanin A modulates cell viability, invasion, and growth promoting signaling pathways in HER-2-positive breast cancer cells. J. Oncol. 2009, 121458. doi:10.1155/2009/121458
Shen, D. D., Pang, J. R., Bi, Y. P., Zhao, L. F., Li, Y. R., Zhao, L. J., et al. (2022). LSD1 deletion decreases exosomal PD-L1 and restores T-cell response in gastric cancer. Mol. Cancer 21 (1), 75. doi:10.1186/s12943-022-01557-1
Shi, M., Huang, F. F., Deng, C. P., Wang, Y., and Kai, G. Y. (2019). Bioactivities, biosynthesis and biotechnological production of phenolic acids in Salvia miltiorrhiza. Crit. Rev. Food Sci. Nutr. 59 (6), 953–964. doi:10.1080/10408398.2018.1474170
Shi, Y. J., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119 (7), 941–953. doi:10.1016/j.cell.2004.12.012
Siegel, R. L., Miller, K. D., Fuchs, H. E., and Jemal, A. (2021). Cancer statistics, 2021. Ca. Cancer J. Clin. 72 (1), 7–33. doi:10.3322/caac.21654
Siegel, R. L., Miller, K. D., Fuchs, H. E., and Jemal, A. (2022). Cancer statistics, 2022. Ca. Cancer J. Clin. 72, 7–33. doi:10.3322/caac.21708
Singh, B., and Mitragotri, S. (2020). Harnessing cells to deliver nanoparticle drugs to treat cancer. Biotechnol. Adv. 42, 107339. doi:10.1016/j.biotechadv.2019.01.006
Sobczak, M., Strachowska, M., Gronkowska, K., and Robaszkiewicz, A. (2022). Activation of ABCC genes by Cisplatin depends on the CoREST occurrence at their promoters in A549 and MDA-MB-231 cell lines. Cancers 14 (4), 894. doi:10.3390/cancers14040894
Song, Y. H., Zhang, H. Q., Yang, X. K., Shi, Y. T., and Yu, B. (2022). Annual review of lysine-specific demethylase 1 (LSD1/KDM1A) inhibitors in 2021. Eur. J. Med. Chem. 228, 114042. doi:10.1016/j.ejmech.2021.114042
Sorna, V., Theisen, E. R., Stephens, B., Warner, S. L., Bearss, D. J., Vankayalapati, H., et al. (2013). High-throughput virtual screening identifies novel N'-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 56 (23), 9496–9508. doi:10.1021/jm400870h
Speranzini, V., Rotili, D., Ciossani, G., Pilotto, S., Marrocco, B., Forgione, M., et al. (2016). Polymyxins and quinazolines are LSD1/KDM1A inhibitors with unusual structural features. Sci. Adv. 2 (9), e1601017. doi:10.1126/sciadv.1601017
Strachowska, M., Gronkowska, K., Michlewska, S., and Robaszkiewicz, A. (2021). CBP/p300 bromodomain inhibitor-I-CBP112 declines transcription of the key ABC transporters and sensitizes cancer cells to chemotherapy drugs. Cancers 13 (18), 4614. doi:10.3390/cancers13184614
Su, C. Y., Ming, Q. L., Rahman, K., Han, T., and Qin, L. P. (2015). Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 13 (3), 163–182. doi:10.1016/S1875-5364(15)30002-9
Sugino, N., Kawahara, M., Tatsumi, G., Kanai, A., Matsui, H., Yamamoto, R., et al. (2017). A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia 31 (11), 2303–2314. doi:10.1038/leu.2017.59
Sukocheva, O. A., Lukina, E., Friedemann, M., Menschikowski, M., Hagelgans, A., and Aliev, G. (2020). The crucial role of epigenetic regulation in breast cancer anti-estrogen resistance: Current findings and future perspectives. Semin. Cancer Biol. 82, 35–59. doi:10.1016/j.semcancer.2020.12.004
Sun, Y. F., Gao, X. Y., Wu, P. P., Wink, M., Li, J. H., Dian, L. L., et al. (2019). Jatrorrhizine inhibits mammary carcinoma cells by targeting TNIK mediated Wnt/β-catenin signalling and epithelial-mesenchymal transition (EMT). Phytomedicine 63, 153015. doi:10.1016/j.phymed.2019.153015
Suzuki, T. (2019). Lysine-specific histone demethylases 1/2 (LSD1/2) and their inhibitors. Chem. Epigenetics 2019, 197–219. doi:10.1007/7355_2019_74
Tan, A. H. W., Tu, W. J., McCuaig, R., Hardy, K., Donovan, T., Tsimbalyuk, S., et al. (2019). Lysine-specific histone demethylase 1A regulates macrophage polarization and checkpoint molecules in the tumor microenvironment of triple-negative breast cancer. Front. Immunol. 10, 1351. doi:10.3389/fimmu.2019.01351
Thoennissen, N. H., O'kelly, J., Lu, D., Iwanski, G. B., La, D. T., Abbassi, S., et al. (2010). Capsaicin causes cell-cycle arrest and apoptosis in ER-positive and-negative breast cancer cells by modulating the EGFR/HER-2 pathway. Oncogene 29 (2), 285–296. doi:10.1038/onc.2009.335
Tu, W. J., McCuaig, R. D., Tan, A. H. Y., Hardy, K., Seddiki, N., Ali, S., et al. (2020). Targeting nuclear LSD1 to reprogram cancer cells and reinvigorate exhausted T cells via a novel LSD1-EOMES switch. Front. Immunol. 11, 1228. doi:10.3389/fimmu.2020.01228
Vallet-Regí, M., Schüth, F., Lozano, D., Colilla, M., and Manzano, M. (2022). Engineering mesoporous silica nanoparticles for drug delivery: Where are we after two decades? Chem. Soc. Rev. 51 (13), 5365–5451. doi:10.1039/d1cs00659b
Vasilatos, S. N., Katz, T. A., Oesterreich, S., Wan, Y., Davidson, N. E., and Huang, Y. (2013). Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis 34 (6), 1196–1207. doi:10.1093/carcin/bgt033
Verigos, J., Karakaidos, P., Kordias, D., Papoudou-Bai, A., Evangelou, Z., Harissis, H. V., et al. (2019). The histone demethylase LSD1/ΚDM1A mediates chemoresistance in breast cancer via regulation of a stem cell program. Cancers 11 (10), 1585. doi:10.3390/cancers11101585
Wang, L., Li, L. Z., Han, Q. X., Wang, X. F., Zhao, D., and Liu, J. Q. (2020). Identification and biological evaluation of natural product Biochanin A. Bioorg. Chem. 97, 103674. doi:10.1016/j.bioorg.2020.103674
Wang, T., Zhang, F. L., and Sun, F. L. (2022). ORY-1001, a KDM1A inhibitor, inhibits proliferation, and promotes apoptosis of triple negative breast cancer cells by inactivating androgen receptor. Drug Dev. Res. 83 (1), 208–216. doi:10.1002/ddr.21860
Wang, Y., Zhang, H., Chen, Y. P., Sun, Y. M., Yang, F., Yu, W. H., et al. (2009). LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138 (4), 660–672. doi:10.1016/j.cell.2009.05.050
Wen, N., Lv, Q., and Du, Z. G. (2020). MicroRNAs involved in drug resistance of breast cancer by regulating autophagy. J. Zhejiang Univ. Sci. B 21 (9), 690–702. doi:10.1631/jzus.B2000076
Wu, D., Jia, H. Y., Zhang, Z. R., and Li, S. J. (2020). Capsaicin suppresses breast cancer cell viability by regulating the CDK8/PI3K/Akt/Wnt/β-catenin signaling pathway. Mol. Med. Rep. 22 (6), 4868–4876. doi:10.3892/mmr.2020.11585
Wu, J. S., Luo, Y., Deng, D. H., Su, S. Y., Li, S., Xiang, L., et al. (2019). Coptisine from Coptis chinensis exerts diverse beneficial properties: A concise review. J. Cell. Mol. Med. 23 (12), 7946–7960. doi:10.1111/jcmm.14725
Wu, K. J., Zhong, H. J., Yang, G. J., Wu, C., Huang, J. M., Li, G. D., et al. (2018). Small molecule Pin1 inhibitor blocking NF-κB signaling in prostate cancer cells. Chem. Asian J. 13 (3), 275–279. doi:10.1002/asia.201701216
Wu, Y. D., Wang, Y. F., Yang, X. W. H., Kang, T. B., Zhao, Y. X., Wang, C., et al. (2013). The deubiquitinase USP28 stabilizes LSD1 and confers stem-cell-like traits to breast cancer cells. Cell Rep. 5 (1), 224–236. doi:10.1016/j.celrep.2013.08.030
Wu, Z. Q., Li, X. Y., Hu, C. Y., Ford, M., Kleer, C. G., and Weiss, S. J. (2012). Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. U. S. A. 109 (41), 16654–16659. doi:10.1073/pnas.1205822109
Xiao, Y., and Yu, D. H. (2021). Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 221, 107753. doi:10.1016/j.pharmthera.2020.107753
Xu, X., Peng, W. H., Liu, C. Y., Li, S. X., Lei, J. L., Wang, Z., et al. (2019). Flavone-based natural product agents as new lysine-specific demethylase 1 inhibitors exhibiting cytotoxicity against breast cancer cells in vitro. Bioorg. Med. Chem. 27 (2), 370–374. doi:10.1016/j.bmc.2018.12.013
Yakulov, T., Raggioli, A., Franz, H., and Kemler, R. (2013). Wnt3a-dependent and -independent protein interaction networks of chromatin-bound β-catenin in mouse embryonic stem cells. Mol. Cell. Proteomics 12 (7), 1980–1994. doi:10.1074/mcp.M112.026914
Yang, C., Li, D., Zang, S. H., Zhang, L., Zhong, Z. F., and Zhou, Y. T. (2022). Mechanisms of carcinogenic activity triggered by lysine-specific demethylase 1A. Front. Pharmacol. 2804, 955218. doi:10.3389/fphar.2022.955218
Yang, C., Wang, W. H., Liang, J. X., Li, G. D., Vellaisamy, K., Wong, C. Y., et al. (2017). A rhodium (III)-based inhibitor of lysine-specific histone demethylase 1 as an epigenetic modulator in prostate cancer cells. J. Med. Chem. 60 (6), 2597–2603. doi:10.1021/acs.jmedchem.7b00133
Yang, G. J., Wang, W. H., Mok, S. W. F., Wu, C., Law, B. Y. K., Miao, X. M., et al. (2018b). Selective Inhibition of lysine-specific demethylase 5A (KDM5A) using a rhodium(III) complex for triple-negative breast cancer therapy. Angew. Chem. Int. Ed. Engl. 57 (40), 13091–13095. doi:10.1002/anie.201807305
Yang, G. J., Wu, J., Leung, C. H., Ma, D. L., and Chen, J. (2021c). A review on the emerging roles of pyruvate kinase M2 in anti-leukemia therapy. Int. J. Biol. Macromol. 193, 1499–1506. doi:10.1016/j.ijbiomac.2021.10.213
Yang, G. J., Ko, C. N., Zhong, H. J., Leung, C. H., and Ma, D. L. (2019). Structure-based discovery of a Selective KDM5A inhibitor that exhibits anti-cancer activity via inducing cell cycle arrest and senescence in breast cancer cell lines. Cancers 11 (1), 92. doi:10.3390/cancers11010092
Yang, G. J., Lei, P. M., Wong, S. Y., Ma, D. L., and Leung, C. H. (2018a). Pharmacological inhibition of LSD1 for cancer treatment. Molecules 23 (12), 3194. doi:10.3390/molecules23123194
Yang, G. J., Song, Y. Q., Wang, W. H., Han, Q. B., Ma, D. L., and Leung, C. H. (2021b). An optimized BRD4 inhibitor effectively eliminates NF-κB-driven triple-negative breast cancer cells. Bioorg. Chem. 114, 105158. doi:10.1016/j.bioorg.2021.105158
Yang, G. J., Wang, W. H., Lei, P. M., Leung, C. H., and Ma, D. L. (2020). A 7-methoxybicoumarin derivative selectively inhibits BRD4 BD2 for anti-melanoma therapy. Int. J. Biol. Macromol. 164, 3204–3220. doi:10.1016/j.ijbiomac.2020.08.194
Yang, G. J., Wu, J., Miao, L., Zhu, M. H., Zhou, Q. J., Lu, X. J., et al. (2021d). Pharmacological inhibition of KDM5A for cancer treatment. Eur. J. Med. Chem. 226, 113855. doi:10.1016/j.ejmech.2021.113855
Yang, G. J., Zhong, H. J., Ko, C. N., Wong, S. Y., Vellaisamy, K., Ye, M., et al. (2018c). Identification of a rhodium(iii) complex as a Wee1 inhibitor against TP53-mutated triple-negative breast cancer cells. Chem. Commun. 54 (20), 2463–2466. doi:10.1039/c7cc09384e
Yang, G. J., Zhu, M. H., Lu, X. J., Liu, Y. J., Lu, J. F., Leung, C. H., et al. (2021a). The emerging role of KDM5A in human cancer. J. Hematol. Oncol. 14 (1), 30. doi:10.1186/s13045-021-01041-1
Yang, M. J., Culhane, J. C., Szewczuk, L. M., Jalili, P., Ball, H. L., Machius, M., et al. (2007). Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 46, 8058–8065. doi:10.1021/bi700664y
Yang, Y., Huang, W., Qiu, R. F., Liu, R. Q., Zeng, Y., Gao, J., et al. (2018d). LSD1 coordinates with the SIN3A/HDAC complex and maintains sensitivity to chemotherapy in breast cancer. J. Mol. Cell Biol. 10 (4), 285–301. doi:10.1093/jmcb/mjy021
Yatim, A., Benne, C., Sobhian, B., Laurent-Chabalier, S., Deas, O., Judde, J. D., et al. (2012). NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol. Cell 48 (3), 445–458. doi:10.1016/j.molcel.2012.08.022
Yi, L., Cui, Y., Xu, Q. F., and Jiang, Y. J. (2016). Stabilization of LSD1 by deubiquitinating enzyme USP7 promotes glioblastoma cell tumorigenesis and metastasis through suppression of the p53 signaling pathway. Oncol. Rep. 36 (5), 2935–2945. doi:10.3892/or.2016.5099
Yoneyama, M., Tochio, N., Umehara, T., Koshiba, S., Inoue, M., Yabuki, T., et al. (2007). St, ructural and functional differences of SWIRM domain subtypes. J. Mol. Biol. 369 (1), 222–238. doi:10.1016/j.jmb.2007.03.027
Zhang, X. M., Lu, F., Wang, J., Yin, F., Xu, Z. S., Qi, D. H., et al. (2013). Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep. 5 (2), 445–457. doi:10.1016/j.celrep.2013.09.018
Zhao, L. J., Li, Y. Y., Zhang, Y. T., Fan, Q. Q., Ren, H. M., Zhang, C., et al. (2021). Lysine demethylase LSD1 delivered via small extracellular vesicles promotes gastric cancer cell stemness. EMBO Rep. 22, e50922. doi:10.15252/embr.202050922
Zheng, L. H., Zhang, Y., Liu, G. J., Cheng, S., Zhang, G., An, C., et al. (2020). Tanshinone I regulates autophagic signaling via the activation of AMP-activated protein kinase in cancer cells. Anticancer. Drugs 31 (6), 601–608. doi:10.1097/CAD.0000000000000908
Zheng, Y., Zeng, Y., Qiu, R. F., Liu, R. Q., Huang, W., Hou, Y. Q., et al. (2018). The homeotic protein SIX3 suppresses carcinogenesis and metastasis through recruiting the LSD1/NuRD(MTA3) complex. Theranostics 8 (4), 972–989. doi:10.7150/thno.22328
Zhou, A. D., Lin, K. Y., Zhang, S. C., Chen, Y. H., Zhang, N., Xue, J. F., et al. (2016). Nuclear GSK3β promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat. Cell Biol. 18 (9), 954–966. doi:10.1038/ncb3396
Zhou, C., Kang, D., Xu, Y. G., Zhang, L. Y., and Zha, X. M. (2015). Identification of novel selective lysine-specific demethylase 1 (LSD1) inhibitors using a pharmacophore-based virtual screening combined with docking. Chem. Biol. Drug Des. 85 (6), 659–671. doi:10.1111/cbdd.12461
Zhou, J., Su, C. M., Chen, H. A., Du, S., Li, C. W., Wu, H., et al. (2020). Cryptanshinone inhibits the glycolysis and inhibits cell migration through PKM2/β-catenin axis in breast cancer. Onco. Targets. Ther. 13, 8629–8639. doi:10.2147/OTT.S239134
Zhou, M., Venkata, P. P., Viswanadhapalli, S., Palacios, B., Alejo, S., Chen, Y. H., et al. (2021a). KDM1A inhibition is effective in reducing stemness and treating triple negative breast cancer. Breast Cancer Res. Treat. 185 (2), 343–357. doi:10.1007/s10549-020-05963-1
Zhou, Z. L., Van der Jeught, K., Fang, Y. Z., Yu, T., Li, Y. J., Ao, Z., et al. (2021b). An organoid-based screen for epigenetic inhibitors that stimulate antigen presentation and potentiate T-cell-mediated cytotoxicity. Nat. Biomed. Eng. 5 (11), 1320–1335. doi:10.1038/s41551-021-00805-x
Zhu, Q. S., Huang, Y., Marton, L. J., Woster, P. M., Davidson, N. E., and Jr Casero, R. A. (2012). Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids 42, 887–898. doi:10.1007/s00726-011-1004-1
Keywords: LSD1, histone demethylase, breast cancer, inhibitors, H3K4me1/2, H3K9me1/2
Citation: Yang G-J, Liu Y-J, Ding L-J, Tao F, Zhu M-H, Shi Z-Y, Wen J-M, Niu M-Y, Li X, Xu Z-S, Qin W-J, Fei C-J and Chen J (2022) A state-of-the-art review on LSD1 and its inhibitors in breast cancer: Molecular mechanisms and therapeutic significance. Front. Pharmacol. 13:989575. doi: 10.3389/fphar.2022.989575
Received: 08 July 2022; Accepted: 15 August 2022;
Published: 16 September 2022.
Edited by:
Chao Han, China Pharmaceutical University, ChinaCopyright © 2022 Yang, Liu, Ding, Tao, Zhu, Shi, Wen, Niu, Li, Xu, Qin, Fei and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiong Chen, amNoZW4xOTc1QDE2My5jb20=, Y2hlbmppb25nQG5idS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.