 Lisa Buvall1
Lisa Buvall1 Robert I. Menzies1
Robert I. Menzies1 Julie Williams1
Julie Williams1 Kevin J. Woollard2
Kevin J. Woollard2 Chanchal Kumar3†Anna B. Granqvist1Maria Fritsch1Denis Feliers2Anna Reznichenko3Davide Gianni4Slavé Petrovski5Claus Bendtsen6
Chanchal Kumar3†Anna B. Granqvist1Maria Fritsch1Denis Feliers2Anna Reznichenko3Davide Gianni4Slavé Petrovski5Claus Bendtsen6 Mohammad Bohlooly-Y7
Mohammad Bohlooly-Y7 Carolina Haefliger5†Regina Fritsche Danielson1Pernille B. L. Hansen1*
Carolina Haefliger5†Regina Fritsche Danielson1Pernille B. L. Hansen1*- 1Bioscience Renal, Research and Early Development, Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
- 2Bioscience Renal, Research and Early Development, Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Cambridge, United Kingdom
- 3Translational Science and Experimental Medicine, Research and Early Development, Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
- 4Functional Genomics, Discovery Sciences, R&D, AstraZeneca, Cambridge, United Kingdom
- 5Centre for Genomics Research, Discovery Sciences, R&D, AstraZeneca, Cambridge, United Kingdom
- 6Data Sciences & Quantitative Biology, Discovery Sciences, R&D, AstraZeneca, Cambridge, United Kingdom
- 7Translational Genomics, Discovery Sciences, R&D, AstraZeneca, Gothenburg, Sweden
Kidney disease is a complex disease with several different etiologies and underlying associated pathophysiology. This is reflected by the lack of effective treatment therapies in chronic kidney disease (CKD) that stop disease progression. However, novel strategies, recent scientific breakthroughs, and technological advances have revealed new possibilities for finding novel disease drivers in CKD. This review describes some of the latest advances in the field and brings them together in a more holistic framework as applied to identification and validation of disease drivers in CKD. It uses high-resolution ‘patient-centric’ omics data sets, advanced in silico tools (systems biology, connectivity mapping, and machine learning) and ‘state-of-the-art‘ experimental systems (complex 3D systems in vitro, CRISPR gene editing, and various model biological systems in vivo). Application of such a framework is expected to increase the likelihood of successful identification of novel drug candidates based on strong human target validation and a better scientific understanding of underlying mechanisms.
1 Introduction
Chronic kidney disease (CKD) is an umbrella term for a variety of renal diseases with different etiologies, usually diagnosed on the basis of clinical and/or histopathological features, but commonly associated with longstanding diabetes and hypertension (Kidney Disease: Improving Global Outcomes, 2013). CKD is divided into five stages, 1–5, according to the estimated glomerular filtration rate (eGFR), as a measure of overall renal function, and degree of any associated albuminuria, which both correlate with increasing mortality risk (Webster et al., 2017). However, the current clinical and histological features of the different renal diseases that make up CKD do not relate to a particular pathway or factor that can define the underlying disease mechanism(s), and thereby determine a specific therapeutic intervention. This gap between the clinical and histolological classification of renal injury and the underlying drivers of disease makes use of the current CKD classification alone problematic for drug discovery (Hall and Himmelfarb, 2017). Furthermore, the structural and functional complexity of the kidney, which comprises a variety of cell types - glomerular, tubular, interstitial, and vascular—highlights the need for a deeper understanding of the underlying biology and physiology, and requirement for a broad array of tools for target validation.
Drug discovery in CKD has proved challenging. The global burden of CKD is increasing worldwide with a country-by-country prevalence ranging from 5% to 14%; CKD is ranked the fourth in a list of growing mortalities in 2020 (The Lancet Kidney Campaign, 2020). Moreover, 7.6% of all deaths from cardiovascular disease (CVD) can be attributed to CKD (G. B. D. Chronic Kidney Disease Collaboration, 2020). Despite recent progress in the treatment of CKD, such as the approval of sodium-glucose cotransporter-2 (SGLT2) inhibitors (Bakris et al., 2020; Heerspink et al., 2020), major therapeutic advances are still required to halt and reverse CKD.
The molecular drivers of renal disease are diverse and may involve over 100 biological pathways (Martini et al., 2014) and this complexity makes it difficult to recapitulate disease pathways experimentally. Two-dimensional culture of particular renal cell types has advanced our understanding of renal biology, but many components are lacking, such as cell-to-cell contact, cell-matrix interactions, and the effects of flow or pressure. Moreover, with at least 25 different cell types (Park et al., 2019), a faithful simulation of the kidney ex vivo is unfeasible at present. Therefore, efforts so far have focused on building particular capabilities that allow us to model different aspects of renal pathophysiology and anatomy in disease (Balzer et al., 2022), including omics-defined translatable primary human renal cells in vitro and better genomic characterization of animal models in vivo.
The imprecision of the current CKD classification, the complexity of the underlying renal pathophysiology, and lack of adequate translatability from preclinical findings to clinical readouts are the major challenges when identifying relevant disease drivers. Recent technological innovations and the application of multiple target identification approaches are opening up new avenues for researchers in the quest to identify and validate the right targets for CKD. Expanding collections of omics data, together with human efficacy trials are invaluable resources for ‘back-translation’ in early target identification (King et al., 2019). Advances in artificial intelligence (AI) and systems biology provide the opportunity to develop machine-learning algorithms to combine diverse sources of patient data for a more patient-centric and holistic analysis that can be applied to target identification. In addition, many studies have shown that when a target can be linked to genetic evidence in disease, or a firm understanding of the role of a target in the etiology of a disease, these are less likely to fail due to insufficient efficacy (Kamb et al., 2013; Plenge et al., 2013; Cook et al., 2014; Hurle et al., 2016; King et al., 2019). In parallel with ‘big data’ discovery, efficient mechanistic target validation requires information from a broad range of sources, including in silico, in vitro, and in vivo research. Bioinformatic analysis applied to available human CKD data and combined with readouts from complex 3D models in vitro capturing cellular cross-talk and placed in a framework based on transcriptomic readouts from the why rat animal model in vivo are now becoming essential for renal drug discovery.
The challenging nature to identify disease drivers in CKD demands a contemporary holistic approach (Figure 1). This framework is based on human target validation, starting with the generation of a list of potential targets that is derived from patient-relevant data for hypothesis testing, followed by validation using various experimental platforms. Positive validation readouts from multiple testing can facilitate data-driven selection of candidate CKD targets. In this article, we describe how combining existing knowledge of renal diseases with recent technological and scientific advances can help to address the challenges we face in identifying the optimal drug targets for treating CKD.
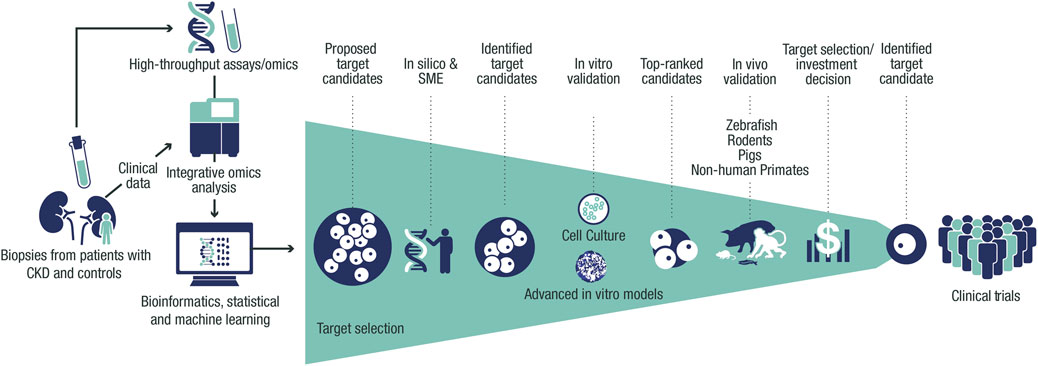
FIGURE 1. Framework for target identification and validation in CKD. Potential CKD targets identified from human data via in silico and SME approaches are prioritized using thorough validation in vitro and in vivo systems, facilitating target selection. Target identification starts with the collection of biopsies, urine, and blood from patients with CKD and controls from clinical trials and collaborations. Omics data (genetic, transcriptomic, proteomic, and metabolomic) are then generated from these samples and processed with integrative omics analyses followed by bioinformatic and statistical analyses and machine learning. This data processing results in a list of CKD targets that is assessed in silico to build further evidence of human target–disease associations. The shortlisted targets are then prioritized by applying biologically relevant in vitro validation in cultured cells and advanced in vitro models. The targets with the strongest supportive data are then validated further in vivo to build proof-of-mechanism and proof-of-principle in CKD before being presented for target selection and investment decision to enter the portfolio. CKD, chronic kidney disease; SME, subject-matter expert.
2 Identifying new disease drivers through patient-derived big data
The highly heterogeneous and progressive nature of CKD makes it amenable to using big data from large human populations to transform the way we identify potential disease drivers, better understand disease progression, and ultimately increase the success rate in drug development for this growing unmet medical need. Retrospective analyses based on data from human genetic studies, such as familial studies and genome-wide association studies (GWAS), have shown that drugs with targets supported by human genetics have an above-average chance of clinical success (Kamb et al., 2013). In CKD, the identification of rare variants, using collapsing analyses of exome sequences, can validate known disease-causing genes and identify candidate genes and modifiers (Cameron-Christie et al., 2019). Therefore, human genetics can play a key role in: i) identifying drug targets with strong human target validation for therapeutic intervention and impact, ii) validating the mechanisms of action of existing drug candidates to reduce their risk of failing, iii) implementing precision medicine strategies to identify patients with molecular diagnoses that are more likely to benefit from targeted therapies, and iv) detecting potential prohibitive drug interactions and adverse effects.
Beyond the foundation of genomics, other patient-derived large data sets that include transcriptomics, proteomics, and metabolomics, and which contain more temporal and tissue-spatial information, are critical to deciphering the downstream complexity of disease defined by the interplay between genetics and the environment. Various molecular data sets can be generated from clinical trial participants to build an understanding from gene to protein, and the metabolomic signature in patients with CKD (Eddy et al., 2020).
One of the greatest enablers for big data and multi-omic collections is increased collaboration. One example is the UK Biobank in which many pharmaceutical companies have joined a pre-competitive consortium to generate exome and genome sequence data in approximately 500,000 participants, an unparalleled clinical and genomic resource (Bycroft et al., 2018). CKD-specific collaborations have also been founded, including the Renal Pre-competitive Consortium (RPC2), in which data, resources, and expertise in molecular target identification are shared across academia and the pharmaceutical industry with the aim of accelerating novel drug development for CKD through a systems biology approach (Tomilo et al., 2018). This consortium-generated pre-competitive material (data sets and unbiased analyses) is shared equally among all partners and then with the wider scientific community, while the industry partners can conduct internal competitive research for the development of targets at their own discretion and that is fully amenable to intellectual property protection. This model has been followed by the establishment in recent years of several well-curated national CKD cohort studies for long-term patient follow-up and the collection of longitudinal clinical data and biosamples of blood and urine, and in some cases also renal tissue samples (Tomilo et al., 2018).
3 Bioinformatics-based prioritization of disease drivers
Bioinformatic approaches are used extensively to gather CKD supporting evidence for targets, including their importance as drivers of CKD. Bioinformatics continues to play a key role in the success of omics and has integrated itself seamlessly into the fabric of contemporary data-driven biology. The ability to generate multidimensional omic data sets from genomic, blood, urine, and kidney tissue sample analyses of patients with CKD has opened up new possibilities for data-driven hypothesis generation (Saez-Rodriguez et al., 2019). Various systematic knowledge-mining approaches and comprehensive functional analysis of patient-derived omic and clinical data sets can now be employed to facilitate CKD target identification, validation, and prioritization (Figure 2).
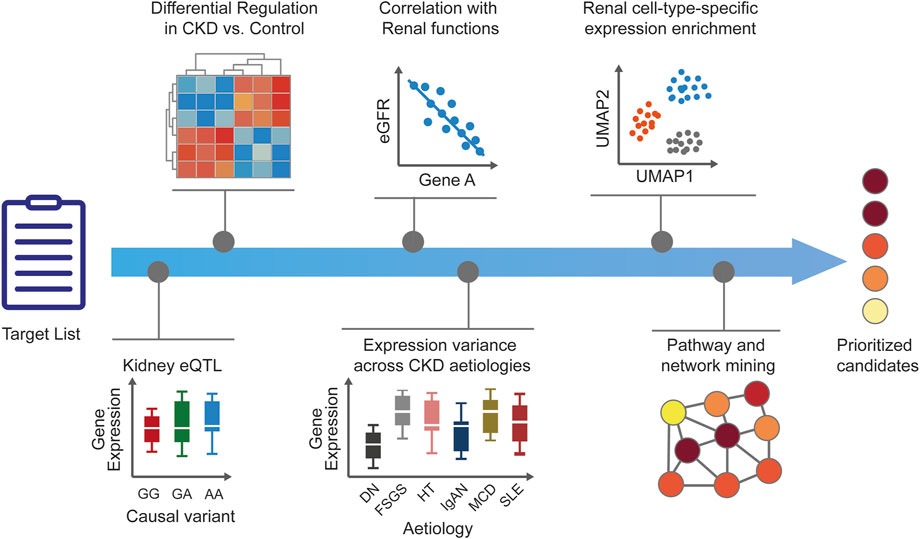
FIGURE 2. Human target validation and prioritization. After generation of CKD target lists, additional disease-relevant evidence is added to generate testable hypotheses and to prioritize the candidates. Tissue-specific expression enrichment and expression modulation in disease versus healthy states help to ascertain the role of targets in CKD. Correlation of targets with renal functional biomarkers and parameters is ascertained and target expression across CKD stages or etiologies is explored to add confidence around disease relevance. Prediction of target kidney cell type is useful for guiding downstream in vitro validation and assay selection. Pathway and network analyses can provide additional biological context for dysregulated cellular mechanisms and help infer potential mechanisms of action. The accumulated evidence supporting human target validation and mechanism of action results in a set of prioritized candidates for further experimental validation. CKD, chronic kidney disease; DN, diabetic nephropathy; eGFR, estimated glomerular filtration rate; eQTL, expression quantitative trait loci; FSGS, focal segmental glomerulosclerosis; HT, hypertensive nephropathy; IgA, immunoglobulin A nephropathy; MCD, minimal change disease; MGN, membranous glomerulonephritis; RPGN, rapid progressive glomerulonephritis; SLE, systemic lupus erythematosus.
Analysis of omic data is a multifaceted endeavor that integrates many aspects of bioinformatics, statistics, and machine learning (Hastie et al., 2009). Algorithms and workflows are employed to conduct specific aspects of data analysis, including data normalization (Kohl et al., 2014; Abbas-Aghababazadeh et al., 2018), exploratory multivariable data analysis (Gehlenborg et al., 2010), and systems-level omics integration (Mitra et al., 2013; Karczewski and Snyder, 2018). Platform-dependent omics readouts (transcriptomics, proteomics, and metabolomics) necessitate specialized data normalization to avoid technical biases obscuring biological patterns (Tyanova et al., 2016; Yamada et al., 2020), and eventually produce a data matrix that typically represents the abundance values for molecular entities (genes, proteins, and metabolites) across biological samples. Once normalized, unbiased, hypothesis-free data exploration usually begins by using various unsupervised dimensionality reduction methods (Bartenhagen et al., 2010) that permit visualization of large multidimensional data sets by summarizing thousands of variables in a few principal components that can be plotted in 2D space. Even at this initial stage, underlying patterns can be identified within the data structure by revealing the grouping of samples or their molecular features, and sometimes aided by use of hierarchical clustering.
Case-control design remains a mainstay of observational cohort studies in which omics data are generated from patients with CKD (broad or specific disease characteristics) and controls (healthy individuals or patients with non-renal disease) with the aim of making comparisons. In such a setting, differential abundance/expression analyses are used to uncover molecular features that are modulated in CKD versus control samples. Directionality of differences and changes—up- or down-regulation—sheds light on the activity state of biological processes, as well as guiding the subsequent therapeutic approach, namely development of an antagonist or agonist compound, respectively, and choosing the most appropriate modality.
Hypothesis-free data analysis methods typically yield long lists of disease-associated molecular features (such as genes), which can be ranked by, for example, statistical significance or magnitude of effect, but cannot easily be tested experimentally or have unknown biological meaning. Therefore, a substantial part of data analytics is devoted to the prioritization of candidate features to shortlist the most promising potential drug targets for subsequent validation in vitro and in vivo (Moreau and Tranchevent, 2012; Vitsios and Petrovski, 2020). The prioritization is based on annotation of those candidates with sufficient disease-related evidence to support generation of a testable hypothesis. Patient clinical characteristics are key to the interpretation of omics patterns because hypothesis-based analyses depend on a biological understanding of the disease and include testing of candidate targets for their correlations with renal function (serum creatinine, cystatin C, glomerular filtration rate [GFR], albuminuria, or proteinuria), disease progression rate, CKD stage, and histopathological diagnosis. When follow-up clinical data are available, longitudinal analyses for association with changes in renal function over time (GFR slope) and prediction of later outcomes (worsening proteinuria, cardiovascular morbidity, onset of renal failure, and mortality) are valuable in understanding the relative importance of a chosen target.
For selected candidate targets, pathway analyses and network biology approaches are used to provide additional biological context for dysregulated cellular mechanisms and to infer potential mechanisms of action (Gehlenborg et al., 2010; Choobdar et al., 2019; Reimand et al., 2019). Further hypothesis-driven analyses can inform subsequent steps for preclinical target validation. For example, prediction of the target cell type is useful in guiding the choice of cellular model in vitro, kidney enrichment of intra-renal targets can inform pharmacokinetic aspects and safety issues, and the presence of orthologous genes, pathway conservation, and consistent directionality modulation in disease models can support human-to-animal translatability and selection of animal models in vivo. Prioritized candidate targets can then enter the pipeline of preclinical testing for further target validation.
4 In vitro target validation
The appropriate use of translatable models in vitro and tools for preclinical target validation can build confidence in novel CKD targets. Preclinical target validation can include building an understanding of the mechanism of action of a particular target and an understanding of its molecular network. Confidence in a candidate target identified in silico can be gained by demonstrating pathway activity in human cell-based renal disease models in vitro.
As mentioned earlier, the complexity of kidney disease, which may involve the dysfunction of several different cell types in the glomerular, tubulo-interstitial, and vascular compartments, makes it challenging to identify translatable models in which to define the mechanism of action of potential targets in CKD. To facilitate selection of the best validation system, human omic data sources may be analyzed in combination with transcriptomic, proteomic, and metabolomic data in vitro, both in cell culture systems and in more advanced cellular models. In this way, we can identify the appropriate cell type, model system, and the right renal stressor for the target pathway, in the same way as big data and bioinformatic analyses can identify these elements in patients with CKD. CRISPR screening in renal cells can help to handle the increasing number of targets arising from big data, making it easier to triage multiple targets and select the most promising candidate for further validation.
A renal toolbox must try to mirror the renal multiplicity of cell types, the complexity of their interactions, cross-talk with other organ systems, including effects on metabolism, the microbiome, and immune system. Recently, significant progress has been made to improve the translatability of in vitro renal systems, such as the generation of podocytes derived from human induced pluripotent stem cells (iPSCs) that have gene expression signatures resembling adult human podocytes more closely than available podocyte cell lines (Yoshimura et al., 2019). In addition, primary and immortalized cell lines (Wieser et al., 2008) of several different glomerular, tubular, vasculature, and immune cell types may be used in validation assays. Co-culture systems are also advancing for studying the interplay among renal cell types and with other cells such as immune cells.
A stepwise approach may be implemented to guide the experimental design for validation of renal targets in vitro (Box 1). This process facilitates selection of the most appropriate cell models, renal stressors, and CKD readouts to define mechanism(s) of action, as well as the affected renal compartment to optimize translatability (Figure 3). By using an advanced and translatable in vitro toolbox, clear assay design, and appropriate readouts, renal validation in vitro can narrow down a list of targets and help select number of promising candidates for further validation in advanced in vitro, ex vivo, and in vivo systems.
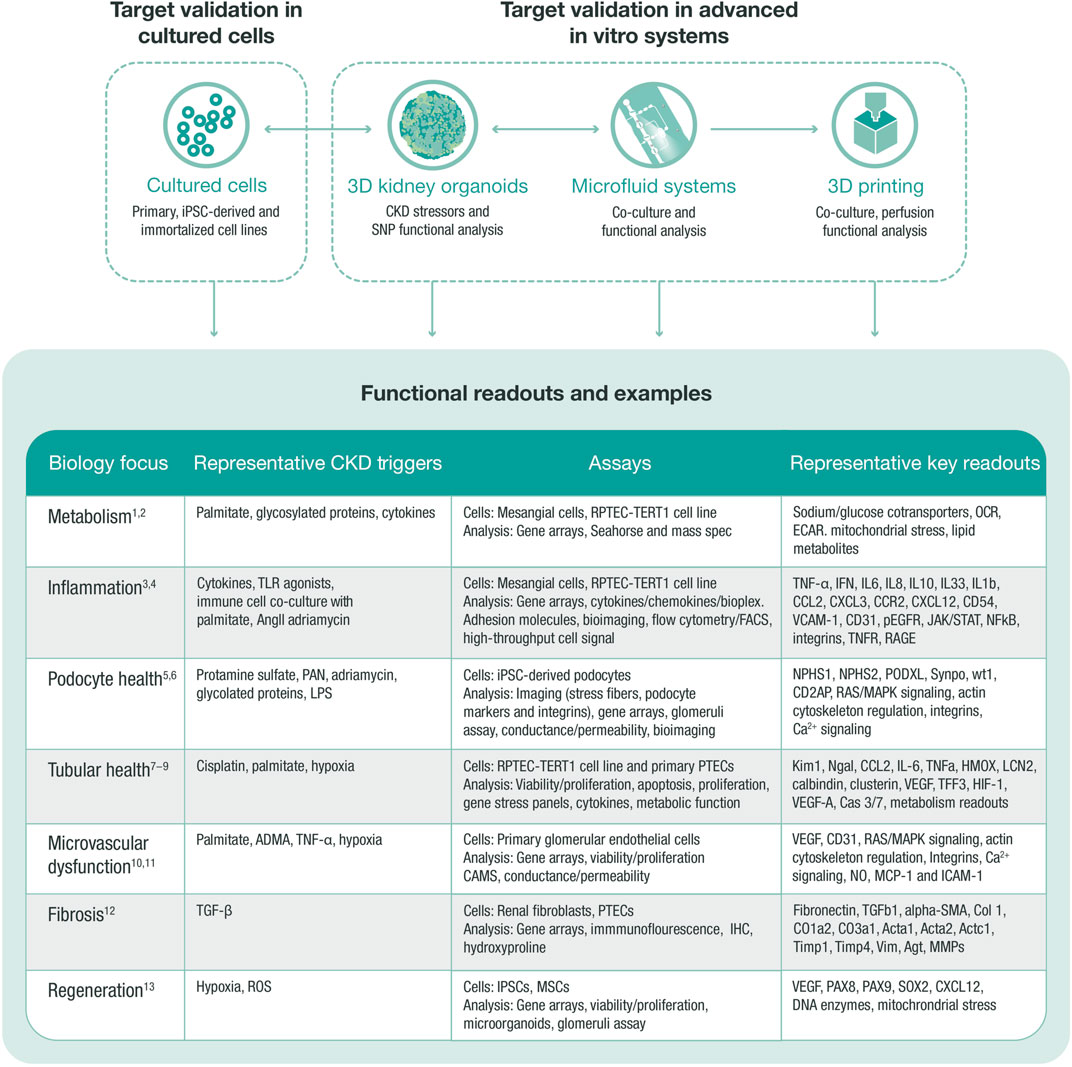
FIGURE 3. In vitro target validation process. In vitro target validation in cultured cells is used as a first approach to validate and screen several targets, aiming to triage targets with strong support for further target validation in advanced in vitro systems. Data from all systems feed into each other to select the most translatable model and define the correct stressor that regulates target and pathway. An in vitro target validation toolbox may comprise various assays, stressors, and readouts, which are chosen based on the disease biology of the target. ADMA, asymmetric dimethylarginine (arginine metabolite); CKD, chronic kidney disease; ECAR, extracellular acidification rate; HGEC, human glomerular endothelial cell; HMOX, hem oxygenase; IHC, immunohistochemistry; iPSC, induced pluripotent stem cell; MMPs, matrix metalloproteinases; MSC, mesenchymal stem cell; OCR, oxygen consumption rate; PAN, pyromycin aminonucleoside; PTEC, proximal tubular epithelial cell; RPTEC, renal proximal tubular epithelial cell; SNP, single nucleotide polymorphism; TGF, transforming growth factor; TLR, toll-like receptor; ROS, reactive oxygen species. 1. Faivre A et al. Front Med (Lausanne) 2021; 8:742072; 2. Imasawa T et al. The International Journal of Biochemistry and Cell Biology 2013; 45:2109–2118.3. Oates JC et al. American Journal of Physiology-Renal Physiology 2022; 322:F309-F321.4. Tang SCW et al. Nature Reviews Nephrology 2020; 16:206–222.5. Lee HW et al. Journal of the American Society of Nephrology 2015; 26:2741–2752.6. Perico L et al. Nature Reviews Nephrology 2016; 12:692–710.7. Prozialeck WC et al. Pharmacology and Therapeutics 2007; 114:74–93.8. Slyne J et al. Nephrology Dialysis Transplantation 2015; 30:iv60-iv67.9. Wieser M et al. American Journal of Physiology-Renal Physiology 2008; 295:F1365-F1375.10. Jourde-Chiche N et al. Nature Reviews Nephrology 2019; 15:87–108.11. Sol M et al. Front Pharmacol 2020; 11:573557.12. Liu Y. Kidney International 2006; 69:213–217.13. Yun CW et al. International Journal of Molecular Sciences 2019; 20:1619.
All model systems have their limitations and for the different cell culture models you need to be aware that targets, pathways, and functions can be changed or even lost when cells are removed from their natural environment. The interactions with neighboring cells, changed physiological conditions, and loss of 3D structure can affect the readouts from a simplified 2D culture system and have, for example, been shown to impact gene regulation in proximal tubular cells. The 2D models in vitro should therefore not stand alone in a target validation package but be combined with other models described below. 2D models are useful systems before setting up more complex, low throughput, 3D systems in vitro system, and models in vivo. Each system can play a role in adding to knowledge of mechanisms and the pros and cons are all summarized in Figure 4 and how and when they are used are based on the biology question being asked.
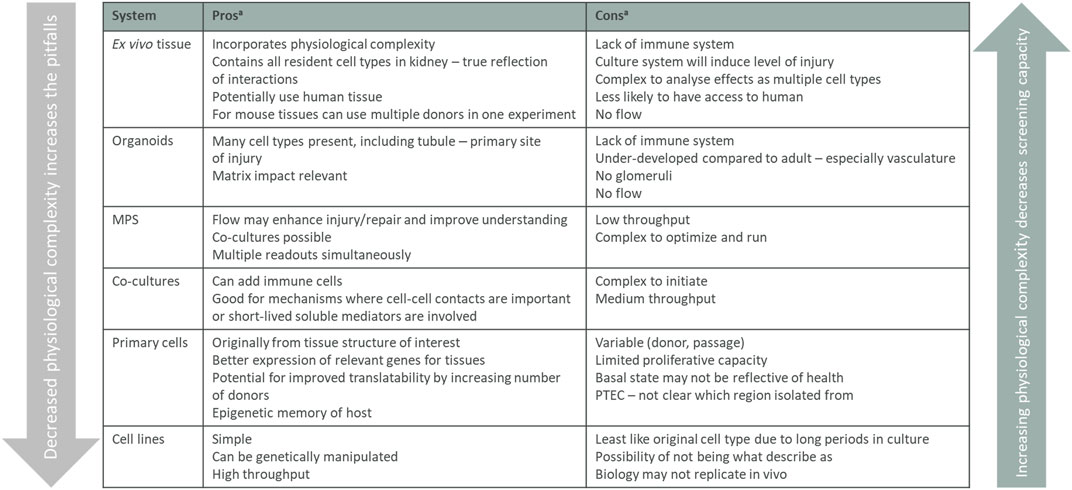
FIGURE 4. Summary of in vitro validation models. Listed in vitro models arranged from the assay containing the most tissue complexity down to single cells, listing the pros and cons for the different models. The physiological complexity of the different models impacts both the screening capacity of the assay and what biology that translates. MPS, micro-physiological systems. aconsiderations are listed in a step-wise manner and main considerations only are stated.
4.1 Functional genomics
The discovery of CRISPR technology and its applications to large-scale genetic loss-of-function screens, also known as functional genomics, holds great promise for the identification of novel validated drug targets, including in CKD. Functional genomics screens allow the systematic perturbation of large numbers of genes or proteins, revealing cellular phenotypes that allow for an inference of gene function. The success of functional genomic platforms in driving the identification of the most relevant targets in renal disease will be determined by advances based on three strategic pillars that are the foundation of functional genomics: (i) development of more advanced in vitro systems, (ii) creation of validated screening libraries and technologies to alter gene and protein function, and (iii) establishment of ‘end-to-end’ computational pipelines that can facilitate the quantitative analysis of cellular phenotypes resulting from genetic or other perturbations.
According to these three strategic pillars the choice and development of the appropriate in vitro system will be critical for the identification of translatable targets in CKD. The value generated by functional genomic efforts will be directly proportional to the translatability of the cellular models employed in screening campaigns in which the targets were originally discovered. Consequently, there is a strong drive among researchers in the field to use cellular systems in vitro that can closely recapitulate disease-relevant phenotypes, rather than use easier to screen 2D cell lines with limited their physiological relevance. A successful example is the use of CRISPR methodology with iPSC-derived podocytes and primary renal cells, such as implementation of the ObLiGaRe doxycycline inducible (ODIn) Cas9 system in iPSCs (Lundin et al., 2020).
5 Advanced in vitro systems
Advanced in vitro systems aim to bridge the gap between in vitro and in vivo by facilitating the exploration of mechanisms that rely on factors such as cross-talk between cells, cell-matrix interactions or changes in pressure or flow that are not present in 2D cultures. The improved maturity of cells in these advanced systems means that basal expression levels of many genes are reduced, providing a cleaner background on which to detect pathophysiological changes in gene expression. In vivo, the presence of multiple cell types can either protect the target cell from injury or cascade damage throughout its structure. These nuances in advanced models allow us to answer some intricate questions without the use of in vivo models, thereby achieving a reduction in the use of experimental animals. In addition, the same cellular manipulations applied to 2D systems can still be used.
The toolbox available for renal drug discovery is growing with the advent of microphysiological systems that include both glomerular and tubular structures, as well as 3D bio-printed tubules, and more complex renal organoids. From these systems investigation of the target choice can progress to ex vivo models, including isolated glomeruli, blood vessels, and tubules, through to kidney slices. Each model has its own advantages and increases our understanding of a target’s impact on disease biology—knowledge that would not be gained from simpler systems.
5.1 Glomerular and tubular surrogates
Several different approaches have been taken to tackle the problem of mimicking glomerular structure and function, including co-culture membrane-based chips (Zhou et al., 2016; Musah et al., 2017), in-gel self-aggregation methods (Waters et al., 2017), and isolated glomeruli on chips (Wang et al., 2017). The addition of flow and a more physiological matrix has given rise to structures that show a greater resemblance to their intact homolog (Musah et al., 2017). The replacement of thick artificial membranes with structures having a more natural morphology has also improved functionality (Slater et al., 2011). The main readouts from such systems are imaging, transcriptomics, and permeability. A further step forward has been allowing the cells to generate their own structures (Waters et al., 2017), which can give rise to tubular structures surrounded by a supporting matrix of cells and interstitium; however, the ability to perfuse these structures is so far limited. Disease phenotypes have been modeled in many of these systems, including diabetic (Wang et al., 2017), hypertensive (Zhou et al., 2016) and fibrotic kidney disease (Waters et al., 2017). However, while these are of interest for drug development, they frequently lack complexity or appropriate controls, such as adjusting for the osmotic stress of high glucose. Despite these limitations, interesting findings can be made from co-culture systems, such as the ability of podocytes to reduce endothelial inflammation (Kuravi et al., 2014).
The renal tubule may seem to be a less difficult structure to replicate than the glomerulus. However, it too comprises several different cell types with segment-specific properties, mainly to do with fluid, electrolyte and solute transport, and is surrounded by the ‘black box’ of the interstitium, which is ill-defined and even harder to reproduce. Therefore, a reductionist approach has been taken and the majority of systems have chosen to model the proximal tubule. These range from monocultures on membranes under flow (Jang et al., 2013) to more complex 3D bio-printed tubules with associated vascular structures (Homan et al., 2016). Characterization has highlighted the difficulties of fully replicating the in vivo characteristics of tubular cells and with no system to date expressing the full gamut of receptors/transporters or with their correct localization. Introducing flow has certainly improved this, as has the addition of matrix, as well as endothelial cells, which all improve the phenotype of tubular epithelial cells (Aydin et al., 2008; Miya et al., 2011). However, the majority of these model tubular systems have been used to screen for drug nephrotoxicity and changes in drug metabolism and secretion (DesRochers et al., 2013; Soo et al., 2018). Disease model development has not been pursued so far; however, the system can replicate epithelial–mesenchymal transition (Zhou et al., 2014).
5.2 Organoids
Pluripotent stem cells can be differentiated to form 3D kidney organoids representative of a first trimester developing fetal kidney. These complex self-aggregating structures usually contain at least 11 different cell types (Harder et al., 2019), representing the major nephron segments, including cells of the glomerulus (podocytes), tubule (proximal, distal, and connecting segments), vasculature (although often scant), and interstitial stroma. 3D bioprinting technology offers the opportunity to improve morphology and throughput (Higgins et al., 2018). The addition of flow has been shown to improve vascularization (Homan et al., 2019).
Kidney organoids are a robust model in which to study human kidney development (Little et al., 2016), a potential source of human kidney cells for bioengineering and regeneration (personalized medicine), a system for toxicological assessment of pharmaceuticals (Czerniecki et al., 2018), and a tool for drug development. Numerous readouts can be used in these systems. CRISPR-Cas9 gene editing in iPSCs has produced several fluorescence reporter cell lines that allow easy visualization and lineage tracking of individual cell populations (Borestrom et al., 2018), as well as the inducible ODIn Cas9 system (Lundin et al., 2020) for speedy target validation in iPSC-derived organoids. Furthermore, high-throughput screening platforms and automated multidimensional phenotyping analysis permit measurement of multiple parameters with multiple stimuli (Ramm et al., 2016). Organoids can also be used to model human renal diseases, either by genetic manipulation of the progenitor cells or by isolating cells from patients (Freedman et al., 2015; Hale et al., 2018; Shamshirgaran et al., 2021). This opens the possibility of testing the reversal of genetic conditions using CRISPR-Cas9 technology (Forbes et al., 2018). Alternatively, the use of patient-derived organoids facilitates a true personalized medicine approach, including patient-specific drug validation (Low et al., 2019; van den Berg et al., 2019). Finally, external agents can be used to injure organoids, either to assess potential nephrotoxicity or to induce a renal disease phenotype. Employing omics techniques then allows investigation of new pathophysiological pathways for the identification of human translatable biomarkers (Harder et al., 2019). Some caveats do exist concerning the use of kidney organoids, such as their immaturity (although advances are being made (Garreta et al., 2019)), the presence of ‘non-renal’ cells (although strategies are evolving to circumvent this (Wu et al., 2018)), the lack of all cell types (due to their differentiation via a single pathway (Nishinakamura, 2019)), and the level of reproducibility among different iPSCs (Subramanian et al., 2019).
Advanced cell culture systems offer an opportunity to investigate disease mechanisms in a simpler manner than in vivo, yet provide some of the complexity of the kidney. Challenges remain in reconstituting the full gamut of renal physiology in vitro (Ashammakhi et al., 2018). Although throughput is necessarily much lower than 2D culture systems, capacities are ramping up with improved technologies such as the Draper system (Vedula et al., 2017; Fu et al., 2019). Differences seen in the responses to various stressors and drugs in these systems highlight the importance of adding layers of complexity to our toolbox. Further advances will come with the integration of multi-organ mimics to generate the so-called ‘human on a chip’ (Vernetti et al., 2017; Ramme et al., 2019). Adoption of such systems has been slow because of practical considerations (Ewart and Roth, 2020), but their potential for transforming translation from ‘bench to bedside’ cannot be over-emphasized. Furthermore, for ethical reasons the ability to forgo animal research would be priceless.
6 Ex vivo systems
Ex vivo approaches allow screening of compound efficacy and the assessment of target engagement and proof of mechanism, which inform the design of experiments in vivo. This is a crucial intermediate step between in vitro and in vivo work.
Isolated vascular and tubular structures can be used to understand underlying renal physiology and pathophysiology. The use of micro-dissected nephron segments (Helbert et al., 1997) and vascular segments has progressed from reverse transcription-polymerase chain reaction-based analyses determining changes in a few individual genes (Jensen et al., 2001), to large omics and functional studies using isolated perfused kidney tissue segments to gain a more refined physiological understanding (Giesecke et al., 2019). Expression data at the transcriptomic level (Lee et al., 2015) and more recently at the protein level (Limbutara et al., 2020) are a valuable resource that can facilitate investigations of segment-specific gene and protein expression patterns for novel targets. Isolated tubules and glomeruli have been used for mechanistic and functional studies, and experiments using perfused afferent and efferent arterioles have increased our understanding of the mechanisms regulating renal blood flow and GFR (Poulsen et al., 2011; Hansen, 2013; de Bruijn et al., 2015). The method is excellent for providing an in-depth understanding, although more difficult to use for high-throughput studies. Isolated renal vascular and microvascular segments ex vivo also afford investigation of renal endothelial and vascular smooth muscle cell function (Stulak et al., 2001) In addition, isolated rodent or human kidneys can be used to bridge from in vitro to in vivo, as well as confirming the translatability of a certain target (Taft, 2004; Weissenbacher et al., 2019).
6.1 Isolated glomeruli
Glomerular isolation ex vivo is driven by the need to study the glomerular compartment without tubular interference and the need to understand cell-cell and cell-matrix interactions. The advantage of this technique is the ability to isolate a complete glomerulus with all its original 3D structure, extracellular matrix, and complex renal cell composition. The drawback is that it is hard to recapitulate some important physiological features such as flow and pressure, thus limiting its relevance when studying glomerular endothelial function. However, there is additional benefit from isolating these structures directly from tissue compared with reconstituting them using cells and artificial devices, and include retaining the complex matrix structures that are important in the physiology of the overall structure. Isolated glomeruli are viable for up to 7–10 days for molecular analysis, pathway profiling, and omics characterization, and can be obtained from several sources: rodents, pigs, non-human primates, human biopsies, or human kidneys unsuitable for transplantation (Desideri et al., 2018; Rush et al., 2018; Wang et al., 2019). It is possible to apply both well-established molecular biology techniques and new approaches, such as high-content imaging and machine learning, to isolated glomeruli. Structures can be subjected to common stress factors such as cytotoxic chemical and various biological stressors, including cytokines, hypoxia, and ischemia-reperfusion, as well as mechanical/physical stressors (for example, flow and stretching), immune complexes, and immune cell co-culture methods. Isolated glomeruli can also be used for target identification, target validation, compound selection, phenotypic screening, proof-of-mechanism studies, molecular pathway characterization, and investigation of glomerular disease mechanisms.
6.2 Precision-cut kidney slices
Further to studying isolated glomeruli and tubules, precision-cut kidney slices (PCKS) allow the study of renal tissue in all its complexity while still in a defined culture environment, establishing a bridge between in vitro and in vivo studies (Stribos et al., 2016). Historically, PCKS have been used to study renal fibrosis, a very complex process that cannot be elucidated using in vitro systems. Two strategies can be used: slices can be taken from injured kidneys, for example, after unilateral ureteral obstruction (Genovese et al., 2016) or slices can be taken from healthy kidneys and treated with pro-fibrotic stimuli such as TGF-β (Stribos et al., 2017). An advantage of PCKS is that they can be cultured for 5–7 days, making them suitable for longitudinal studies and amenable to the types of analyses that are performed on kidneys isolated from mice. Histology and immunochemistry can be performed, and the kidney slices can be used for transcriptomic analyses (Bigaeva et al., 2019). It is worth noting that preparation of the kidneys for slicing induces stress, transiently increasing the expression of kidney injury markers, although these return to baseline after approximately 24 h. In addition to the well-established pro-fibrotic model, PCKS may be used to study the effect of inflammatory and hypoxic stimuli on kidney structure and injury.
Kidney slices can also be used as a predictive tool to test the efficacy of candidate drug compounds (Bigaeva et al., 2020) as part of initial screens before performing experiments in vivo. This can eliminate compounds are expected to show some efficacy in this model, but do not, and to determine an effect size of a compound and calculate group sizes for the experiments in vivo. The use of PCKS as a screening tool can reduce the number of animals needed to validate a test compound. As with brain slices, PCKS are also suitable for some imaging studies and can be superfused, for example, to examine tubule segment-specific changes in mitochondrial function (Stulak et al., 2001; Hall et al., 2009). PCKS represent a versatile tool to study aspects of kidney function and injury in an intact environment.
6.3 Zebrafish screen
Zebrafish are an efficient and high-throughput target validation system because they are genetically tractable and have a basic renal anatomy, with glomerular and tubular filtration processing (Wingert and Davidson, 2008). The pronephros in the zebrafish larvae consists of a single glomerulus connected to two tubular structures. The tubules express several of the important proximal and distal transporters, but lack the loop of Henle, because, as freshwater fish, they do not need to concentrate their urine. CRISPR-Cas9 is used to create insertions and deletions in the zebrafish genome to create loss-of-function models of targeted genes. Approximately 70% of human genes have at least one zebrafish ortholog, allowing validation of the majority of potential targets (Howe et al., 2017). The role of candidate genes in renal function is assessed by the proportion of larvae displaying edema and/or renal cysts, followed by detailed analysis of filtration and morphology (using electron microscopy and immunohistochemistry) (Hanke et al., 2015). This model will also capture the effects of altered gene expression in other organs, as well as any impact on embryogenesis, development, and survival (Gehrig et al., 2018). Zebrafish larvae may be used as an intermediate screening tool to triage the gene hit list, between cell culture/ex vivo systems and in vivo models in higher species, permitting quicker selection of potential targets. In addition, the adult zebrafish can be used as an efficacy model for studying both acute kidney injury and regeneration (McKee and Wingert, 2015) by using either nephrotoxic substances, laser ablation or genome editing to drive the injury (Morales and Wingert, 2017). Zebrafish are also commonly used as a safety model when screening for drug-induced kidney injury (Kato et al., 2020). However, cautious interpretation is warranted, as zebrafish physiology is far remote from human physiology.
7 Validating mechanism of action in vivo
In parallel with data generation from in vitro and ex vivo models, the target validation process to define the most translatable in vivo model to provide proof of mechanism and proof of principle in CKD takes place. Similar to the workflows for in vitro and ex vivo models, several disease models with different mechanistic drivers may be used to capture the complexity of CKD (Figure 5). To facilitate selection of the most relevant preclinical model in vivo for a certain target, RNA sequencing can be performed on renal tissue from a panel of disease models to track the regulation of specific genes and signaling pathways. This information is also critical for an understanding the human translatability of the models in the context of disease and treatment mechanisms. The preclinical model is also selected based on the target biology, available biomarkers, and knowledge of the pathology of the disease models.
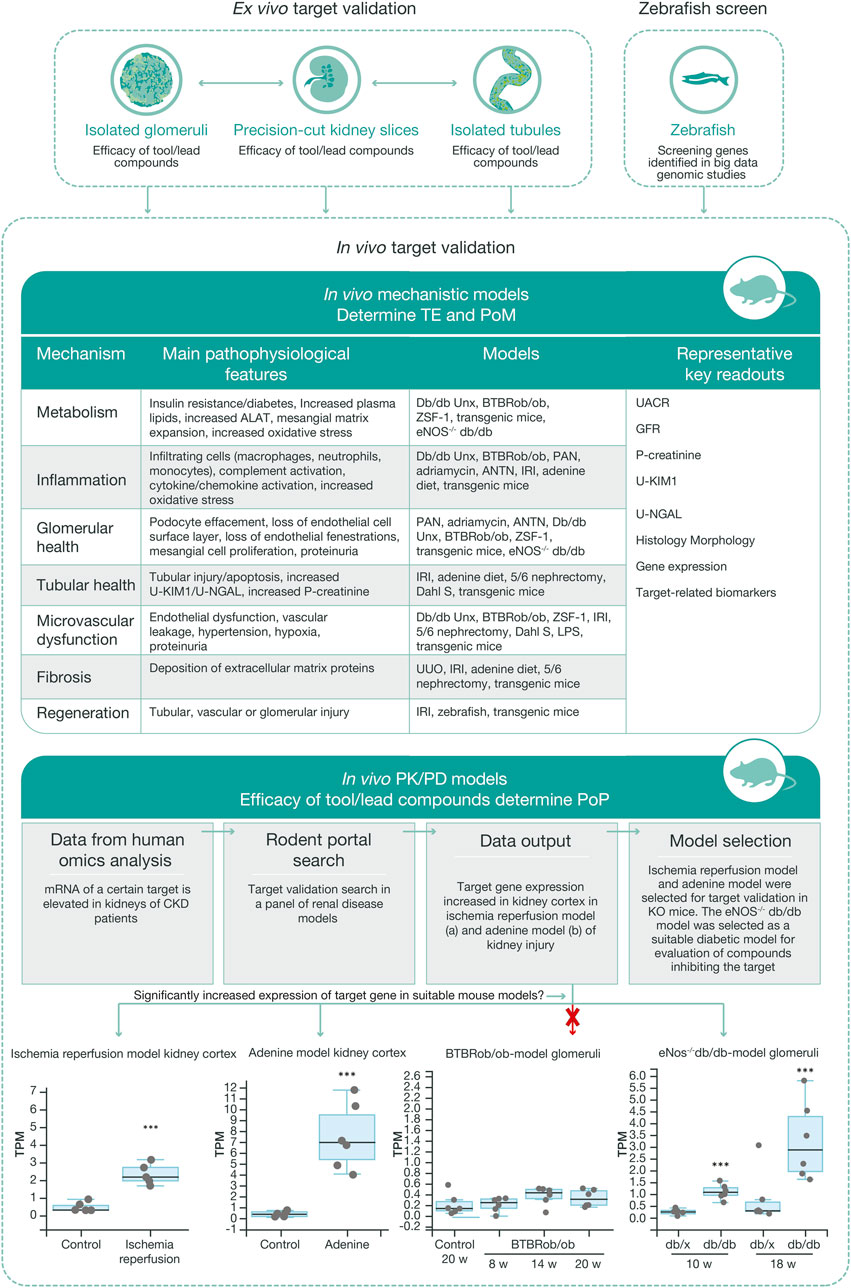
FIGURE 5. Workflow for in vivo target validation and compound testing. Ex vivo models are chosen to validate genes of interest and assess the efficacy of compounds targeting these gene products, based on their cellular expression and target engagement, respectively. Data from the ex vivo experiments guide the design of mechanistic and PKPD in vivo studies that generate data regarding target engagement and proof of mechanism, and on exposure and proof of principle, respectively. Various mechanistic models are used to validate targets and measure efficacy of test compounds. Selection of the most relevant in vivo PKPD model for a specific target or signaling pathway is based on ex vivo experimental data, human omics data, and RNA sequencing data from our ‘rodent portal’, generated from a panel of rodent renal disease models. The bottom panel illustrates differential upregulation of a target gene in different disease models, which in turn allows selection of the most appropriate model for the tested protocol. ALAT, alanine aminotransferase; ANTN, accelerated/non-accelerated nephrotoxic nephritis; BTBR, black and tan brachyuric; eNOS, endothelial nitric oxide synthase; IRI, ischemic reperfusion injury; KIM1, kidney injury molecule one; LPS, lipopolysaccharide; NGAL, neutrophil gelatinase; PAN, pyromycin aminonucleoside; PKPD, pharmacokinetic–pharmacodynamic; POM, proof of mechanism; TE, target engagement; UUO, unilateral ureter obstruction.
BOX 1 Experimental design process for in vitro renal target validation
1. Confirm that the target is the most effective and accessible target in the disease pathway2. Select a cell culture model in which the target is present3. Confirm a renal stressor that regulates the target in the same way as observed in patients with CKD more generally or a specific renal disease4. Confirm whether genetic modification (CRISPR knockout/overexpression) of the regulated target in the defined renal cell type results in a CKD-like phenotype5. Confirm that tool compounds or genetic modification can produce a treatment effect in a relevant translatable cell model6. Confirm whether genetic modification or tool compounds that agonize/antagonize a target in advanced models have an effect on clinical measures of renal function7. Continue target validation in advanced in vitro/ex vivo systems and establish a mode of action in translatable models in vivo
When designing a preclinical study, the aim is to use a mechanistic model in which target engagement can be easily captured, followed by a relevant proof-of-mechanism disease model in which an efficacy readout can be captured. The experiment in the mechanistic model is usually short and confirms that test molecule engages its target by measuring biomarkers directly linked to the target’s biology (activation of specific signaling pathways, altered gene or protein expression) in the plasma, urine, or in the kidney itself. During these studies, identification of additional markers of target engagement could influence the design of future clinical trials. For example, a viable target engagement model measuring the impact of a mineralocorticoid antagonist would measure plasma aldosterone and/or urinary electrolytes (Bamberg et al., 2018). This model is also used as a screening tool in the compound development process to ensure sufficient exposure in a subsequent pharmacokinetic–pharmacodynamic (PKPD) model. Once a compound (either an experimental molecule or a tool) has been shown to modify the disease pathway of interest at a relevant level of plasma or tissue exposure, a proof-of-mechanism study is designed. The model in this study should be relevant to the intended patient population and allow measurement of the physiological outcomes that will also be assessed in a clinical trial, such as urinary albumin excretion or a change in GFR, as potential human efficacy endpoint (Greene et al., 2019). Results from the proof-of-mechanism/PKPD model will affect the future design of clinical trials by guiding the modelling of the compound’s pharmacokinetics and calculation of receptor occupancy.
Translatability of models in vivo to human disease is a key aspect of target validation in vivo (Silver and Gerarduzzi, 2019). However, in most cases more than one disease model is needed to address mechanistic actions and functional readouts. A growing number of renal models in vivo have been developed (Betz and Conway, 2016; Mullins et al., 2016), representing damage to the different renal cell types that reflect key pathological changes seen in human disease. A good understanding of injury mechanisms in each model allows for the selection of a specific model to study a given target, based on its renal localization and mechanism of action. Therefore, selection of the most relevant disease model is crucial. For some candidate drugs the same model can address both the mechanism of action and functional efficacy; however, multiple models are usually necessary to build confidence. It is also important to recognize that any model of human kidney disease will capture only a ‘snapshot’ of the disease process (functional and/or structural), since human kidney disease occurs over decades.
Another approach to evaluating the relevance of a target is to use genetically modified animals for target validation and as safety tools to understand a super-physiological antagonization or agonization of gene function, as well as in vivo tool for ‘on/off’ target effects of candidate drugs. However, for several rodent preclinical models, inter-strain variability needs to be taken into account when selecting the disease model. For example, C57/Bl6 mice, one of the most commonly used background strains for generating knockout mice, are known to be less susceptible to renal injury than many other mouse strains (Breyer et al., 2005). In addition, issues like absence of human-like comorbidities or oftentimes impractical treatment regimen (e.g., preceding injury) may erode the predictive validity of a research model.
In vivo target validation together with the in silico and in vitro methodologies discussed earlier, is crucial to build a sound scientific understanding of the disease mechanisms in play.
8 Conclusion
CKD has steadily climbed the ranking of leading causes of death in recent years as a result of an aging population and increasing prevalence of risk factors such as diabetes and hypertension (Couser et al., 2011). Until now, research into finding therapies that be used to treat all forms CKD has focused on improving cardiovascular outcomes and in slowing disease progression, mainly with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, which also help to lower blood pressure, a driver of CKD progression, and more recent use of SGLT2 inhibitors. However, little progress has been made in halting or reversing disease progression.
A new patient-centric approach is key for future target identification and validation. Starting the target discovery based on patient data and a patient-centric approach using translatable models is essential to the identification and validation of targets that can result in successful CKD drug discovery programs and increase our understanding of mechanistic disease drivers. Scientific expertise in multiple areas of renal biology, pathophysiology and clinical aspects of renal disease is crucial, together with computational biology, systems-wide integration of omics with clinical data sets, and functional validation. Selection of the most appropriate in vitro, ex vivo, and in vivo systems for target validation and aiming to maximize translatability to patient disease can be guided by cutting-edge in silico and omics-based analyses.
The technological revolution continues to deliver patient omics data sets and AI systems for analysis of big data which allow researchers to use the patient as the starting point. Increased access to big data makes it impossible for one researcher to oversee all data. Therefore, the development of tools like knowledge graphs transform the way researchers work with big data. In recent years, much progress has also been made generating omics data from advanced in vitro models such as micro-physiological systems which facilitate studies of genes and pathways not possible in simple 2D cell culture systems. These advanced systems enable understanding of the complexity of the kidney by capturing the effect of 3D structure and cell-cell interactions. Despite the progress that has been made with advanced systems, there is still a need to capture adult renal cell biology, rather than the fetal signature that organoids present with today. In addition, more advanced models including multi-organ systems are needed to capture the full gamut of systemic responses and improve the validation tools for the future.
In parallel, the renal in vivo research community continue to build on the understanding of disease mechanisms and translatability by using metabolomics, proteomics, bulk and single cell NGS data in animal models. The omics data sets are key for understanding the translatability of animal models to patients. Owing to the complexity of chronic kidney disease with cellular crosstalk and multiorgan involvement, we still need to secure reliable in vivo models to support pharmacology read-out.
The increasing number of clinical trials for renal disease during the last decade have resulted in a better understanding of relevant endpoints, biomarkers and disease drivers. Clinical backtranslation beyond patient omics data will be a key factor and continue to improve therapeutic target identification and validation.
The toolbox of state-of-the-art models now available to us to simulate the complexity of CKD can facilitate obtaining readouts that are translatable from preclinical to clinical studies, which until now has been a major challenge for drug discovery in renal medicine. This toolbox needs to be continuously enriched by infusion of novel raw data acquired by researchers across the disciplines, as well as by development of powerful tools to mine and interpret those data. The recent years technological success will continue to deliver improved tools in the future. Despite the increased use of complex micro-physiological systems, there is still a need for testing in vivo for systemic pharmacological responses. We foresee that with the current progress in humanized in vitro models with increased complicity, the need for animal testing will decrease in the future. Using micro-physiological systems is expected to increase translatability to patients and reduce the number of animals used in pharmacological testing, a key ethical parameter in therapeutic target validation.
In conclusion, the novel patient-centric approach building on the combination of in silico analysis of human data, together with extensive in vitro complex humanized models and in vivo validation in CKD research improves the probability to identify disease drivers that could be successful as potential drug targets (Figure 6).
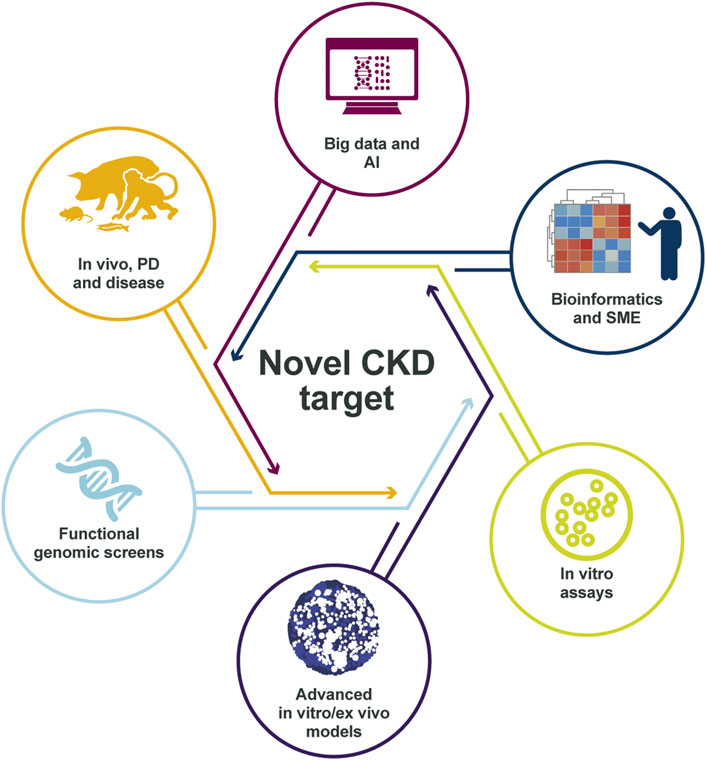
FIGURE 6. Identification of the right CKD target. The target identification and validation framework relies on multiple data sources and validation models, integrating many disease-relevant data sets, to create a holistic scientific understanding of the mechanisms that link the target to disease biology. The in-depth scientific understanding of pathophysiology and target link to disease is, in our view, essential for delivering successful medicines to patients with CKD in the future. AI, artificial intelligence; CKD, chronic kidney disease; PD, pharmacodynamics; SME, subject-matter expert.
Author contributions
LB and PH planned the manuscript outline. LB, RM, JW, KW, AG, MF, DF, DG, CK, and PH wrote the manuscript. All authors reviewed the manuscript
Funding
All authors are employees and stockholders of AstraZeneca and the study is founded by AstraZeneca. The funder had the following involvement with the study: study design, collection, analysis, and interpretation of data, the writing of this article and the decision to submit for publication. Editorial support on an earlier draft was provided by Richard Claes, of Oxford PharmaGenesis, funded by AstraZeneca.
Acknowledgments
We thank Meera Kodukulla, Maria Ryaboshapkina, Robert Unwin, Anna Jonebring, Linn Kullberg, Ryan Hicks, Ulrike Kunzel, Graham Belfield, Maryam Clausen, Paola Castaldi, Sonja Hess, Santhosh Vankayala Ramaiah, Maria Strömstedt, Alex Zhou, Hanna Wallentin, Linda Cederblad, Birgitta Rosengren, Simone Romoli, Marcello Maresca, John Weisman, Krister Bamberg, Sophia Cameron Christie, Rob Howes, Sam Barichievy, and Sara Engberg of AstraZeneca for their contributions to the development of this manuscript.
Conflict of interest
All authors LB, RB, JW, KW, CK, AG, MF, DF, AR, DG, SP, CB, MB, CH, RD, and PH are employees and stockholders of AstraZeneca and the study is founded by AstraZeneca.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbas-Aghababazadeh, F., Li, Q., and Fridley, B. L. (2018). Comparison of normalization approaches for gene expression studies completed with high-throughput sequencing. PLoS One 13 (10), e0206312. doi:10.1371/journal.pone.0206312
Ashammakhi, N., Wesseling-Perry, K., Hasan, A., Elkhammas, E., and Zhang, Y. S. (2018). Kidney-on-a-chip: Untapped opportunities. Kidney Int. 94 (6), 1073–1086. doi:10.1016/j.kint.2018.06.034
Aydin, S., Signorelli, S., Lechleitner, T., Joannidis, M., Pleban, C., Perco, P., et al. (2008). Influence of microvascular endothelial cells on transcriptional regulation of proximal tubular epithelial cells. Am. J. Physiol. Cell Physiol. 294 (2), C543–C554. doi:10.1152/ajpcell.00307.2007
Bakris, G., Oshima, M., Mahaffey, K. W., Agarwal, R., Cannon, C. P., Capuano, G., et al. (2020). Effects of canagliflozin in patients with baseline eGFR <30 ml/min per 1.73 m(2): Subgroup Analysis of the randomized CREDENCE trial. Clin. J. Am. Soc. Nephrol. 15 (12), 1705–1714. doi:10.2215/CJN.10140620
Balzer, M. S., Doke, T., Yang, Y.-W., Aldridge, D. L., Hu, H., Mai, H., et al. (2022). Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat. Commun. 13 (1), 4018. doi:10.1038/s41467-022-31772-9
Bamberg, K., Johansson, U., Edman, K., William-Olsson, L., Myhre, S., Gunnarsson, A., et al. (2018). Preclinical pharmacology of AZD9977: A novel mineralocorticoid receptor modulator separating organ protection from effects on electrolyte excretion. PLoS One 13 (2), e0193380. doi:10.1371/journal.pone.0193380
Bartenhagen, C., Klein, H. U., Ruckert, C., Jiang, X., and Dugas, M. (2010). Comparative study of unsupervised dimension reduction techniques for the visualization of microarray gene expression data. BMC Bioinforma. 11, 567. doi:10.1186/1471-2105-11-567
Betz, B., and Conway, B. R. (2016). An update on the use of animal models in diabetic nephropathy research. Curr. Diab. Rep. 16 (2), 18. doi:10.1007/s11892-015-0706-2
Bigaeva, E., Gore, E., Simon, E., Zwick, M., Oldenburger, A., de Jong, K. P., et al. (2019). Transcriptomic characterization of culture-associated changes in murine and human precision-cut tissue slices. Arch. Toxicol. 93 (12), 3549–3583. doi:10.1007/s00204-019-02611-6
Bigaeva, E., Puerta Cavanzo, N., Stribos, E. G. D., de Jong, A. J., Biel, C., Mutsaers, H. A. M., et al. (2020). Predictive value of precision-cut kidney slices as an ex vivo screening platform for therapeutics in human renal fibrosis. Pharmaceutics 12 (5), E459. doi:10.3390/pharmaceutics12050459
Borestrom, C., Jonebring, A., Guo, J., Palmgren, H., Cederblad, L., Forslow, A., et al. (2018). A CRISP(e)R view on kidney organoids allows generation of an induced pluripotent stem cell-derived kidney model for drug discovery. Kidney Int. 94 (6), 1099–1110. doi:10.1016/j.kint.2018.05.003
Breyer, M. D., Bottinger, E., Brosius, F. C., Coffman, T. M., Harris, R. C., Heilig, C. W., et al. (2005). Mouse models of diabetic nephropathy. J. Am. Soc. Nephrol. 16 (1), 27–45. doi:10.1681/ASN.2004080648
Bycroft, C., Freeman, C., Petkova, D., Band, G., Elliott, L. T., Sharp, K., et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562 (7726), 203–209. doi:10.1038/s41586-018-0579-z
Cameron-Christie, S., Wolock, C. J., Groopman, E., Petrovski, S., Kamalakaran, S., Povysil, G., et al. (2019). Exome-based rare-variant analyses in CKD. J. Am. Soc. Nephrol. 30 (6), 1109–1122. doi:10.1681/ASN.2018090909
Choobdar, S., Ahsen, M. E., Crawford, J., Tomasoni, M., Fang, T., Lamparter, D., et al. (2019). Assessment of network module identification across complex diseases. Nat. Methods 16 (9), 843–852. doi:10.1038/s41592-019-0509-5
Chronic Kidney Disease Collaboration, G. B. D. (2020). Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the global burden of disease study 2017. Lancet 395 (10225), 709–733. doi:10.1016/S0140-6736(20)30045-3
Cook, D., Brown, D., Alexander, R., March, R., Morgan, P., Satterthwaite, G., et al. (2014). Lessons learned from the fate of AstraZeneca's drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 13 (6), 419–431. doi:10.1038/nrd4309
Couser, W. G., Remuzzi, G., Mendis, S., and Tonelli, M. (2011). The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 80 (12), 1258–1270. doi:10.1038/ki.2011.368
Czerniecki, S. M., Cruz, N. M., Harder, J. L., Menon, R., Annis, J., Otto, E. A., et al. (2018). High-throughput screening enhances kidney organoid differentiation from human pluripotent stem cells and enables automated multidimensional phenotyping. Cell Stem Cell 22 (6), 929–940. doi:10.1016/j.stem.2018.04.022
de Bruijn, P. I., Bleich, M., Praetorius, H. A., and Leipziger, J. (2015). P2X receptors trigger intracellular alkalization in isolated perfused mouse medullary thick ascending limb. Acta Physiol. 213 (1), 277–284. doi:10.1111/apha.12417
Desideri, S., Onions, K. L., Qiu, Y., Ramnath, R. D., Butler, M. J., Neal, C. R., et al. (2018). A novel assay provides sensitive measurement of physiologically relevant changes in albumin permeability in isolated human and rodent glomeruli. Kidney Int. 93 (5), 1086–1097. doi:10.1016/j.kint.2017.12.003
DesRochers, T. M., Suter, L., Roth, A., and Kaplan, D. L. (2013). Bioengineered 3D human kidney tissue, a platform for the determination of nephrotoxicity. PLoS One 8 (3), e59219. doi:10.1371/journal.pone.0059219
Eddy, S., Mariani, L. H., and Kretzler, M. (2020). Integrated multi-omics approaches to improve classification of chronic kidney disease. Nat. Rev. Nephrol. 16, 657–668. doi:10.1038/s41581-020-0286-5
Ewart, L., and Roth, A. (2020). Opportunities and challenges with microphysiological systems: A pharma end-user perspective. Nat. Rev. Drug Discov. 20, 327–328. doi:10.1038/d41573-020-00030-2
Forbes, T. A., Howden, S. E., Lawlor, K., Phipson, B., Maksimovic, J., Hale, L., et al. (2018). Patient-iPSC-derived kidney organoids show functional validation of a ciliopathic renal phenotype and reveal underlying pathogenetic mechanisms. Am. J. Hum. Genet. 102 (5), 816–831. doi:10.1016/j.ajhg.2018.03.014
Freedman, B. S., Brooks, C. R., Lam, A. Q., Fu, H., Morizane, R., Agrawal, V., et al. (2015). Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 6, 8715. doi:10.1038/ncomms9715
Fu, J., Akat, K. M., Sun, Z., Zhang, W., Schlondorff, D., Liu, Z., et al. (2019). Single-cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic kidney disease. J. Am. Soc. Nephrol. 30 (4), 533–545. doi:10.1681/ASN.2018090896
Garreta, E., Prado, P., Tarantino, C., Oria, R., Fanlo, L., Marti, E., et al. (2019). Fine tuning the extracellular environment accelerates the derivation of kidney organoids from human pluripotent stem cells. Nat. Mat. 18 (4), 397–405. doi:10.1038/s41563-019-0287-6
Gehlenborg, N., O'Donoghue, S. I., Baliga, N. S., Goesmann, A., Hibbs, M. A., Kitano, H., et al. (2010). Visualization of omics data for systems biology. Nat. Methods 7 (3), S56–S68. doi:10.1038/nmeth.1436
Gehrig, J., Pandey, G., and Westhoff, J. H. (2018). Zebrafish as a model for drug screening in genetic kidney diseases. Front. Pediatr. 6, 183. doi:10.3389/fped.2018.00183
Genovese, F., Karpati, Z. S., Nielsen, S. H., and Karsdal, M. A. (2016). Precision-cut kidney slices as a tool to understand the dynamics of extracellular matrix remodeling in renal fibrosis. Biomark. Insights 11, 77–84. doi:10.4137/BMI.S38439
Giesecke, T., Himmerkus, N., Leipziger, J., Bleich, M., Koshimizu, T. A., Fahling, M., et al. (2019). Vasopressin increases urinary acidification via V1a receptors in collecting duct intercalated cells. J. Am. Soc. Nephrol. 30 (6), 946–961. doi:10.1681/ASN.2018080816
Greene, T., Ying, J., Vonesh, E. F., Tighiouart, H., Levey, A. S., Coresh, J., et al. (2019). Performance of GFR slope as a surrogate end point for kidney disease progression in clinical trials: A statistical simulation. J. Am. Soc. Nephrol. 30 (9), 1756–1769. doi:10.1681/ASN.2019010009
Hale, L. J., Howden, S. E., Phipson, B., Lonsdale, A., Er, P. X., Ghobrial, I., et al. (2018). 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat. Commun. 9 (1), 5167. doi:10.1038/s41467-018-07594-z
Hall, A. M., Unwin, R. J., Parker, N., and Duchen, M. R. (2009). Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J. Am. Soc. Nephrol. 20 (6), 1293–1302. doi:10.1681/ASN.2008070759
Hall, Y. N., and Himmelfarb, J. (2017). The CKD classification system in the precision medicine era. Clin. J. Am. Soc. Nephrol. 12 (2), 346–348. doi:10.2215/CJN.09310916
Hanke, N., King, B. L., Vaske, B., Haller, H., and Schiffer, M. (2015). A fluorescence-based assay for proteinuria screening in larval zebrafish (Danio rerio). Zebrafish 12 (5), 372–376. doi:10.1089/zeb.2015.1093
Hansen, P. B. (2013). Functional and pharmacological consequences of the distribution of voltage-gated calcium channels in the renal blood vessels. Acta Physiol. 207 (4), 690–699. doi:10.1111/apha.12070
Harder, J. L., Menon, R., Otto, E. A., Zhou, J., Eddy, S., Wys, N. L., et al. (2019). Organoid single cell profiling identifies a transcriptional signature of glomerular disease. JCI Insight 4 (1), 122697. doi:10.1172/jci.insight.122697
Hastie, T., Tibshirani, R., and Friedman, J. (2009). The elements of statistical learning: Data mining, inference and prediction. New York: Springer.
Heerspink, H. J. L., Stefansson, B. V., Correa-Rotter, R., Chertow, G. M., Greene, T., Hou, F. F., et al. (2020). Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 383 (15), 1436–1446. doi:10.1056/NEJMoa2024816
Helbert, M. J., Dauwe, S. E., Van der Biest, I., Nouwen, E. J., and De Broe, M. E. (1997). Immunodissection of the human proximal nephron: Flow sorting of S1S2S3, S1S2 and S3 proximal tubular cells. Kidney Int. 52 (2), 414–428. doi:10.1038/ki.1997.348
Higgins, J. W., Chambon, A., Bishard, K., Hartung, A., Arndt, D., Brugnano, J., et al. (2018). Bioprinted pluripotent stem cell-derived kidney organoids provide opportunities for high content screening. bioRxiv, 505396. doi:10.1101/505396
Homan, K. A., Gupta, N., Kroll, K. T., Kolesky, D. B., Skylar-Scott, M., Miyoshi, T., et al. (2019). Flow-enhanced vascularization and maturation of kidney organoids in vitro. Nat. Methods 16 (3), 255–262. doi:10.1038/s41592-019-0325-y
Homan, K. A., Kolesky, D. B., Skylar-Scott, M. A., Herrmann, J., Obuobi, H., Moisan, A., et al. (2016). Bioprinting of 3D convoluted renal proximal tubules on perfusable chips. Sci. Rep. 6, 34845. doi:10.1038/srep34845
Howe, D. G., Bradford, Y. M., Eagle, A., Fashena, D., Frazer, K., Kalita, P., et al. (2017). The zebrafish model organism database: New support for human disease models, mutation details, gene expression phenotypes and searching. Nucleic Acids Res. 45 (D1), D758–D768. doi:10.1093/nar/gkw1116
Hurle, M. R., Nelson, M. R., Agarwal, P., and Cardon, L. R. (2016). Impact of genetically supported target selection on R&D productivity. Nat. Rev. Drug Discov. 15 (9), 596–597. doi:10.1038/nrd.2016.187
Jang, K. J., Mehr, A. P., Hamilton, G. A., McPartlin, L. A., Chung, S., Suh, K. Y., et al. (2013). Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 5 (9), 1119–1129. doi:10.1039/c3ib40049b
Jensen, B. L., Stubbe, J., Hansen, P. B., Andreasen, D., and Skott, O. (2001). Localization of prostaglandin E(2) EP2 and EP4 receptors in the rat kidney. Am. J. Physiol. Ren. Physiol. 280 (6), F1001–F1009. doi:10.1152/ajprenal.2001.280.6.F1001
Kamb, A., Harper, S., and Stefansson, K. (2013). Human genetics as a foundation for innovative drug development. Nat. Biotechnol. 31 (11), 975–978. doi:10.1038/nbt.2732
Karczewski, K. J., and Snyder, M. P. (2018). Integrative omics for health and disease. Nat. Rev. Genet. 19 (5), 299–310. doi:10.1038/nrg.2018.4
Kato, Y., Tonomura, Y., Hanafusa, H., Nishimura, K., Fukushima, T., and Ueno, M. (2020). Adult zebrafish model for screening drug-induced kidney injury. Toxicol. Sci. 174 (2), 241–253. doi:10.1093/toxsci/kfaa009
Kidney Disease: Improving Global Outcomes (KDIGO) (2013). 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. 3, 1–150.
King, E. A., Davis, J. W., and Degner, J. F. (2019). Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 15 (12), e1008489. doi:10.1371/journal.pgen.1008489
Kohl, M., Megger, D. A., Trippler, M., Meckel, H., Ahrens, M., Bracht, T., et al. (2014). A practical data processing workflow for multi-OMICS projects. Biochim. Biophys. Acta 1844 (1), 52–62. doi:10.1016/j.bbapap.2013.02.029
Kuravi, S. J., McGettrick, H. M., Satchell, S. C., Saleem, M. A., Harper, L., Williams, J. M., et al. (2014). Podocytes regulate neutrophil recruitment by glomerular endothelial cells via IL-6-mediated crosstalk. J. Immunol. 193 (1), 234–243. doi:10.4049/jimmunol.1300229
Lee, J. W., Chou, C. L., and Knepper, M. A. (2015). Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J. Am. Soc. Nephrol. 26 (11), 2669–2677. doi:10.1681/ASN.2014111067
Limbutara, K., Chou, C. L., and Knepper, M. A. (2020). Quantitative proteomics of all 14 renal tubule segments in rat. J. Am. Soc. Nephrol. 31 (6), 1255–1266. doi:10.1681/ASN.2020010071
Little, M. H., Combes, A. N., and Takasato, M. (2016). Understanding kidney morphogenesis to guide renal tissue regeneration. Nat. Rev. Nephrol. 12 (10), 624–635. doi:10.1038/nrneph.2016.126
Low, J. H., Li, P., Chew, E. G. Y., Zhou, B., Suzuki, K., Zhang, T., et al. (2019). Generation of human PSC-derived kidney organoids with patterned nephron segments and a de novo vascular network. Cell Stem Cell 25 (3), 373–387. doi:10.1016/j.stem.2019.06.009
Lundin, A., Porritt, M. J., Jaiswal, H., Seeliger, F., Johansson, C., Bidar, A. W., et al. (2020). Development of an ObLiGaRe Doxycycline Inducible Cas9 system for pre-clinical cancer drug discovery. Nat. Commun. 11 (1), 4903. doi:10.1038/s41467-020-18548-9
Martini, S., Nair, V., Keller, B. J., Eichinger, F., Hawkins, J. J., Randolph, A., et al. (2014). Integrative biology identifies shared transcriptional networks in CKD. J. Am. Soc. Nephrol. 25 (11), 2559–2572. doi:10.1681/ASN.2013080906
McKee, R. A., and Wingert, R. A. (2015). Zebrafish renal pathology: Emerging models of acute kidney injury. Curr. Pathobiol. Rep. 3 (2), 171–181. doi:10.1007/s40139-015-0082-2
Mitra, K., Carvunis, A. R., Ramesh, S. K., and Ideker, T. (2013). Integrative approaches for finding modular structure in biological networks. Nat. Rev. Genet. 14 (10), 719–732. doi:10.1038/nrg3552
Miya, M., Maeshima, A., Mishima, K., Sakurai, N., Ikeuchi, H., Kuroiwa, T., et al. (2011). Enhancement of in vitro human tubulogenesis by endothelial cell-derived factors: Implications for in vivo tubular regeneration after injury. Am. J. Physiol. Ren. Physiol. 301 (2), F387–F395. doi:10.1152/ajprenal.00619.2010
Morales, E. E., and Wingert, R. A. (2017). Zebrafish as a model of kidney disease. Results Probl. Cell Differ. 60, 55–75. doi:10.1007/978-3-319-51436-9_3
Moreau, Y., and Tranchevent, L. C. (2012). Computational tools for prioritizing candidate genes: Boosting disease gene discovery. Nat. Rev. Genet. 13 (8), 523–536. doi:10.1038/nrg3253
Mullins, L. J., Conway, B. R., Menzies, R. I., Denby, L., and Mullins, J. J. (2016). Renal disease pathophysiology and treatment: contributions from the rat. Dis. Model Mech. 9 (12), 1419–1433. doi:10.1242/dmm.027276
Musah, S., Mammoto, A., Ferrante, T. C., Jeanty, S. S. F., Hirano-Kobayashi, M., Mammoto, T., et al. (2017). Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat. Biomed. Eng. 1, 0069. doi:10.1038/s41551-017-0069
Nishinakamura, R. (2019). Human kidney organoids: Progress and remaining challenges. Nat. Rev. Nephrol. 15 (10), 613–624. doi:10.1038/s41581-019-0176-x
Park, J., Liu, C. L., Kim, J., and Susztak, K. (2019). Understanding the kidney one cell at a time. Kidney Int. 96 (4), 862–870. doi:10.1016/j.kint.2019.03.035
Plenge, R. M., Scolnick, E. M., and Altshuler, D. (2013). Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 12 (8), 581–594. doi:10.1038/nrd4051
Poulsen, C. B., Al-Mashhadi, R. H., Cribbs, L. L., Skott, O., and Hansen, P. B. (2011). T-type voltage-gated calcium channels regulate the tone of mouse efferent arterioles. Kidney Int. 79 (4), 443–451. doi:10.1038/ki.2010.429
Ramm, S., Adler, M., and Vaidya, V. S. (2016). A high-throughput screening assay to identify kidney toxic compounds. Curr. Protoc. Toxicol. 69, 1–9. doi:10.1002/cptx.12
Ramme, A. P., Koenig, L., Hasenberg, T., Schwenk, C., Magauer, C., Faust, D., et al. (2019). Autologous induced pluripotent stem cell-derived four-organ-chip. Future Sci. OA 5 (8), FSO413. doi:10.2144/fsoa-2019-0065
Reimand, J., Isserlin, R., Voisin, V., Kucera, M., Tannus-Lopes, C., Rostamianfar, A., et al. (2019). Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 14 (2), 482–517. doi:10.1038/s41596-018-0103-9
Rush, B. M., Small, S. A., Stolz, D. B., and Tan, R. J. (2018). An efficient sieving method to isolate intact glomeruli from adult rat kidney. J. Vis. Exp. 141. doi:10.3791/58162
Saez-Rodriguez, J., Rinschen, M. M., Floege, J., and Kramann, R. (2019). Big science and big data in nephrology. Kidney Int. 95 (6), 1326–1337. doi:10.1016/j.kint.2018.11.048
Shamshirgaran, Y., Jonebring, A., Svensson, A., Leefa, I., Bohlooly, Y. M., Firth, M., et al. (2021). Rapid target validation in a Cas9-inducible hiPSC derived kidney model. Sci. Rep. 11 (1), 16532. doi:10.1038/s41598-021-95986-5
Silver, S. A., and Gerarduzzi, C. (2019). Found in translation: Reasons for optimism in the pursuit to prevent chronic kidney disease after acute kidney injury. Can. J. Kidney Health Dis. 6, 2054358119868740. doi:10.1177/2054358119868740
Slater, S. C., Beachley, V., Hayes, T., Zhang, D., Welsh, G. I., Saleem, M. A., et al. (2011). An in vitro model of the glomerular capillary wall using electrospun collagen nanofibres in a bioartificial composite basement membrane. PLoS One 6 (6), e20802. doi:10.1371/journal.pone.0020802
Soo, J. Y., Jansen, J., Masereeuw, R., and Little, M. H. (2018). Advances in predictive in vitro models of drug-induced nephrotoxicity. Nat. Rev. Nephrol. 14 (6), 378–393. doi:10.1038/s41581-018-0003-9
Stribos, E. G. D., Seelen, M. A., van Goor, H., Olinga, P., and Mutsaers, H. A. M. (2017). Murine precision-cut kidney slices as an ex vivo model to evaluate the role of transforming growth factor-β1 signaling in the onset of renal fibrosis. Front. Physiol. 8, 1026. doi:10.3389/fphys.2017.01026
Stribos, E. G., Hillebrands, J. L., Olinga, P., and Mutsaers, H. A. (2016). Renal fibrosis in precision-cut kidney slices. Eur. J. Pharmacol. 790, 57–61. doi:10.1016/j.ejphar.2016.06.057
Stulak, J. M., Lerman, A., Porcel, M. R., Caccitolo, J. A., Romero, J. C., Schaff, H. V., et al. (2001). Renal vascular function in hypercholesterolemia is preserved by chronic antioxidant supplementation. J. Am. Soc. Nephrol. 12 (9), 1882–1891. doi:10.1681/ASN.V1291882
Subramanian, A., Sidhom, E. H., Emani, M., Vernon, K., Sahakian, N., Zhou, Y., et al. (2019). Single cell census of human kidney organoids shows reproducibility and diminished off-target cells after transplantation. Nat. Commun. 10 (1), 5462. doi:10.1038/s41467-019-13382-0
Taft, D. R. (2004). The isolated perfused rat kidney model: A useful tool for drug discovery and development. Curr. Drug Discov. Technol. 1 (1), 97–111. doi:10.2174/1570163043484824
The Lancet Kidney Campaign (2020). Chronic kidney disease (CKD): Effect on global mortality. The Lancet kidney campaign. Available from: https://els-jbs-prod-cdn.jbs.elsevierhealth.com/pb/assets/raw/pb/assets/raw/lancet/campaigns/kidney/chronic-kidney-disease-facts.jpg (Accessed Aug 23, 2022).
{kind=link}
Tomilo, M., Ascani, H., Mirel, B., Magnone, M. C., Quinn, C. M., Karihaloo, A., et al. (2018). Renal pre-competitive consortium (RPC2): Discovering therapeutic targets together. Drug Discov. Today 23 (10), 1695–1699. doi:10.1016/j.drudis.2018.05.021
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13 (9), 731–740. doi:10.1038/nmeth.3901
van den Berg, A., Mummery, C. L., Passier, R., and van der Meer, A. D. (2019). Personalised organs-on-chips: Functional testing for precision medicine. Lab. Chip 19 (2), 198–205. doi:10.1039/c8lc00827b
Vedula, E. M., Alonso, J. L., Arnaout, M. A., and Charest, J. L. (2017). A microfluidic renal proximal tubule with active reabsorptive function. PLoS One 12 (10), e0184330. doi:10.1371/journal.pone.0184330
Vernetti, L., Gough, A., Baetz, N., Blutt, S., Broughman, J. R., Brown, J. A., et al. (2017). Functional coupling of human microphysiology systems: Intestine, liver, kidney proximal tubule, blood-brain barrier and skeletal muscle. Sci. Rep. 7, 42296. doi:10.1038/srep42296
Vitsios, D., and Petrovski, S. (2020). Mantis-ml: Disease-agnostic gene prioritization from high-throughput genomic screens by stochastic semi-supervised learning. Am. J. Hum. Genet. 106 (5), 659–678. doi:10.1016/j.ajhg.2020.03.012
Wang, H., Sheng, J., He, H., Chen, X., Li, J., Tan, R., et al. (2019). A simple and highly purified method for isolation of glomeruli from the mouse kidney. Am. J. Physiol. Ren. Physiol. 317 (5), F1217–F1223. doi:10.1152/ajprenal.00293.2019
Wang, L., Tao, T., Su, W., Yu, H., Yu, Y., and Qin, J. (2017). A disease model of diabetic nephropathy in a glomerulus-on-a-chip microdevice. Lab. Chip 17 (10), 1749–1760. doi:10.1039/c7lc00134g
Waters, J. P., Richards, Y. C., Skepper, J. N., Southwood, M., Upton, P. D., Morrell, N. W., et al. (2017). A 3D tri-culture system reveals that activin receptor-like kinase 5 and connective tissue growth factor drive human glomerulosclerosis. J. Pathol. 243 (3), 390–400. doi:10.1002/path.4960
Webster, A. C., Nagler, E. V., Morton, R. L., and Masson, P. (2017). Chronic kidney disease. Lancet 389 (10075), 1238–1252. doi:10.1016/S0140-6736(16)32064-5
Weissenbacher, A., Lo Faro, L., Boubriak, O., Soares, M. F., Roberts, I. S., Hunter, J. P., et al. (2019). Twenty-four-hour normothermic perfusion of discarded human kidneys with urine recirculation. Am. J. Transpl. 19 (1), 178–192. doi:10.1111/ajt.14932
Wieser, M., Stadler, G., Jennings, P., Streubel, B., Pfaller, W., Ambros, P., et al. (2008). hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am. J. Physiol. Ren. Physiol. 295 (5), F1365–F1375. doi:10.1152/ajprenal.90405.2008
Wingert, R. A., and Davidson, A. J. (2008). The zebrafish pronephros: A model to study nephron segmentation. Kidney Int. 73 (10), 1120–1127. doi:10.1038/ki.2008.37
Wu, H., Uchimura, K., Donnelly, E. L., Kirita, Y., Morris, S. A., and Humphreys, B. D. (2018). Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell 23 (6), 869–881. doi:10.1016/j.stem.2018.10.010
Yamada, R., Okada, D., Wang, J., Basak, T., and Koyama, S. (2020). Interpretation of omics data analyses. J. Hum. Genet. 10, 93–102. doi:10.1038/s10038-020-0763-5
Yoshimura, Y., Taguchi, A., Tanigawa, S., Yatsuda, J., Kamba, T., Takahashi, S., et al. (2019). Manipulation of nephron-patterning signals enables selective induction of podocytes from human pluripotent stem cells. J. Am. Soc. Nephrol. 30 (2), 304–321. doi:10.1681/ASN.2018070747
Zhou, M., Ma, H., Lin, H., and Qin, J. (2014). Induction of epithelial-to-mesenchymal transition in proximal tubular epithelial cells on microfluidic devices. Biomaterials 35 (5), 1390–1401. doi:10.1016/j.biomaterials.2013.10.070
Keywords: chronic kidney disease, validation, systems biology, omics, machine learning, drug discovery, artificial intelligence
Citation: Buvall L, Menzies RI, Williams J, Woollard KJ, Kumar C, Granqvist AB, Fritsch M, Feliers D, Reznichenko A, Gianni D, Petrovski S, Bendtsen C, Bohlooly-Y M, Haefliger C, Danielson RF and Hansen PBL (2022) Selecting the right therapeutic target for kidney disease. Front. Pharmacol. 13:971065. doi: 10.3389/fphar.2022.971065
Received: 16 June 2022; Accepted: 17 October 2022;
Published: 02 November 2022.
Edited by:
Paul Cockwell, University Hospitals Birmingham NHS Foundation Trust, United KingdomReviewed by:
Silvia Mihaila, Utrecht University, NetherlandsShuwang Ge, Huazhong University of Science and Technology, China
Ying Luo, East China Normal University, China
Santosh Varughese, Christian Medical College and Hospital, India
Copyright © 2022 Buvall, Menzies, Williams, Woollard, Kumar, Granqvist, Fritsch, Feliers, Reznichenko, Gianni, Petrovski, Bendtsen, Bohlooly-Y, Haefliger, Danielson and Hansen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pernille B. L. Hansen, cGVybmlsbGUubGFlcmtlZ2FhcmRoYW5zZW5AYXN0cmF6ZW5lY2EuY29t
†Present address: Carolina Haefliger, Debiopharm International, Lausanne, SwitzerlandChanchal Kumar, Translational and Disease Understanding, Research and Development, Grünenthal Ltd., Stokenchurch, United Kingdom