
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 20 September 2022
Sec. Inflammation Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.965661
 Yijun Hu1,2
Yijun Hu1,2 Hong Zhou1Huanxin Zhang1Yunlong Sui1
Hong Zhou1Huanxin Zhang1Yunlong Sui1 Zhen Zhang1Yuntao Zou1Kunquan Li1Yunyi Zhao1Jiangbo Xie1Lunzhong Zhang1*
Zhen Zhang1Yuntao Zou1Kunquan Li1Yunyi Zhao1Jiangbo Xie1Lunzhong Zhang1*Dexmedetomidine (DEX) is a highly selective α2 receptor agonist that is routinely used in the clinic for sedation and anesthesia. Recently, an increasing number of studies have shown that DEX has a protective effect against brain injury caused by traumatic brain injury (TBI), subarachnoid hemorrhage (SAH), cerebral ischemia and ischemia–reperfusion (I/R), suggesting its potential as a neuroprotective agent. Here, we summarized the neuroprotective effects of DEX in several models of neurological damage and examined its mechanism based on the current literature. Ultimately, we found that the neuroprotective effect of DEX mainly involved inhibition of inflammatory reactions, reduction of apoptosis and autophagy, and protection of the blood–brain barrier and enhancement of stable cell structures in five way. Therefore, DEX can provide a crucial advantage in neurological recovery for patients with brain injury. The purpose of this study was to further clarify the neuroprotective mechanisms of DEX therefore suggesting its potential in the clinical management of the neurological injuries.
Dexmedetomidine (DEX) exerts its sedative effect by reducing sympathetic tone through α2 adrenergic receptors, and this agent is more selective than its predecessor, the α2 adrenergic receptor agonist clonidine; their α2:α1 ratios are 1620:1 and 220:1, respectively (Virtanen et al., 1988). Unlike most sedatives, DEX has a reversible sedative effect, similar to the unconscious state of natural sleep, and patients can be easily awakened. Therefore, DEX is often used in awake craniotomy or awake sedation. In addition, this drug has anxiolytic and analgesic potential (Lobo et al., 2016; Barends et al., 2017). DEX causes less respiratory depression than traditional sedatives, but there may be an increased risk of hypotension and bradycardia. Notably, the liver is involved in the pharmacokinetics of DEX, so this drug should be administered with caution in patients with liver impairment (Riker et al., 2009; Jakob et al., 2012; Weerink et al., 2017).
An increasing number of studies have shown that DEX has neuroprotective effects and can reduce the inflammatory response and oxidative stress, inhibit apoptosis, protect the blood–brain barrier (BBB), maintain the balance of the coagulation-anticoagulant system and prevent vasospasm (Wang et al., 2016; Liu et al., 2022). A meta-analysis of 879 patients confirmed the neuroprotective effects of DEX in inhibiting inflammatory responses, reducing neuroendocrine hormone release, and maintaining intracranial homeostasis (Jiang et al., 2017). This literature discusses different neuroprotective mechanisms of DEX. Figure 1 shows the overall framework of the literature.
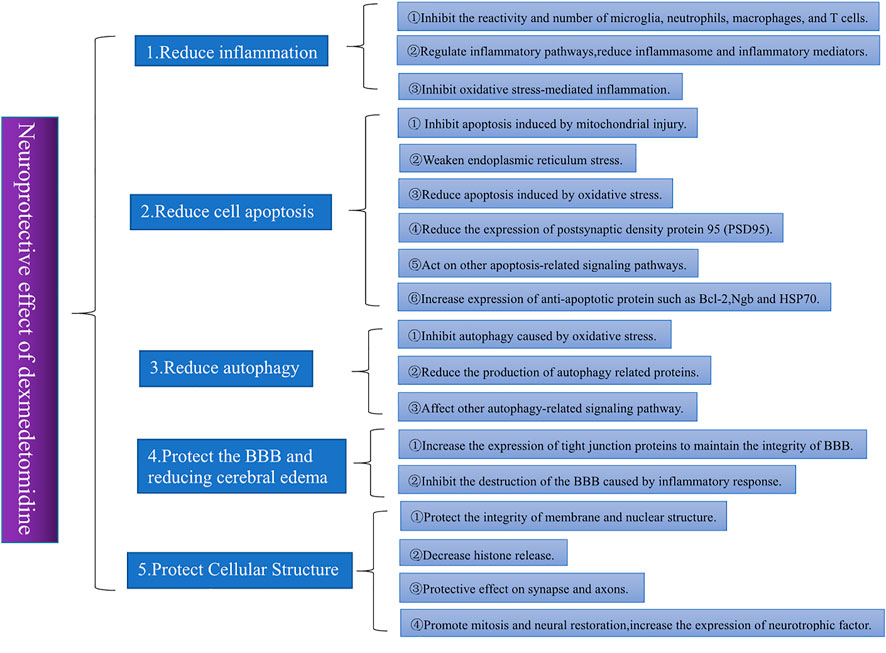
FIGURE 1. Different trials often offer different explanations for the neuroprotective effects of DEX. By sorting and summarizing relevant studies in recent years, we found that the neuroprotective effects of DEX are mainly reflected in five aspects: 1) reducing inflammation, 2) reducing apoptosis, 3) reducing autophagy, 4) protecting the BBB and reducing cerebral edema, and 5) protecting the cellular structure. Among these aspects, the reduction of the inflammatory response is the most important. Almost all studies on the neuroprotective effect of DEX involve the improvement of nervous system inflammation.
Recent studies have shown that excessive inflammation in the nervous system is a key factor in the secondary brain damage caused by traumatic brain injury (TBI), subarachnoid hemorrhage (SAH), cerebral ischemia and ischemia–reperfusion (I/R), which mainly manifest as the activation and migration of immune cells, the activation of inflammation-related pathways and the release of inflammatory mediators (Zhang et al., 2013; Miao and Liao, 2014; Van Lieshout et al., 2018).
DEX treatment significantly reduced the number of neutrophils and microglia in damaged nerve tissue (Wang et al., 2018a; Yin et al., 2018; Karakaya et al., 2022). In addition, Wang et al. showed through morphological observation and showed that the transformation of microglia from round to large was slowed, indicating that DEX inhibited microglial reactivity and that microglial reactivity led to the release of inflammatory mediators such as interleukin-1 beta (IL-1β) and activated peripheral migrating macrophages and T cells, leading to inflammatory infiltration of these cells (Mantovani et al., 2014; McKee and Lukens, 2016; Wang et al., 2018a; Yin et al., 2018; Zheng et al., 2018). Activated macrophages can secrete chemokines that promote the accumulation of immune cells such as neutrophils, further exacerbating the inflammatory response (Russo and McGavern, 2016). Tumor necrosis factor receptor superfamily member 5 (CD40) and T-lymphocyte activation antigen CD86 (CD86) are activation markers on the surface of macrophages. Ding et al. showed that DEX treatment could reduce the expression of CD40 and CD86, suggesting its inhibitory effects on macrophage activation and infiltration (Yin et al., 2018; Ding et al., 2019). After several days of TBI, T cells accumulate at the damaged site and are activated by microglia, which may further cause secondary injury. Therefore, T cells play an important role in neuroinflammation (Krämer et al., 2019). Karakaya et al. found that a high concentration of DEX (200 μg/kg) could reduce the amount of T-cell migration occurring approximately 3 days after TBI and reduce T-cell motility (Karakaya et al., 2022).
Immune cell activation leads to enhanced activation of inflammation-related pathways and promotes the release of a variety of inflammatory mediators. DEX can act on different processes associated with inflammation-related pathways, ultimately reducing the release of inflammatory mediators and alleviating tissue damage. Toll-like receptor 4 (TLR4) on the surface of microglia plays an important role in regulating the inflammatory response caused by cerebral infarction, cerebral hemorrhage and TBI. Myeloid differentiation primary response 88 (MyD88) is an important component of the TLR4 signaling pathway. When TLR4 is activated, MyD88 activates downstream nuclear factor-kappaB (NF-κB) to produce a series of inflammatory factors that may cause nervous system damage, such as IL-1β, tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and interleukin-18 (IL-18) (Li et al., 2011; Fang et al., 2013; Zhang et al., 2016). Zhu et al. showed that microglia and macrophages play important roles in acute neuroinflammation through the TLR4/MyD88/NF-κB pathway (Zhu et al., 2014). An increase in these inflammatory mediators impairs the BBB, increases neuroinflammation, and induces apoptosis (Zheng and Wong, 2017). IL-1β is the most important inflammatory mediator in the posttraumatic inflammatory response because it peaks within hours of brain tissue injury and promotes the release of other cytokines, prompting nearby microglia to transition from a surveillant to a reactive state and leading to the accumulation of other inflammatory cells (McKee and Lukens, 2016). Many recent studies have shown that DEX can reduce the release of inflammatory mediators, such as IL-1β, TNF-α, IL-6 and monocyte chemotactic protein 1 (MCP-1) by inhibiting TLR4-or NF-κB-related pathways, thus reducing inflammatory damage in the nervous system. Table 1 shows changes in various inflammatory mediators and cells (Wang et al., 2018a; Yin et al., 2018; Li et al., 2019; Feng et al., 2021b; Huang and Hao, 2021).
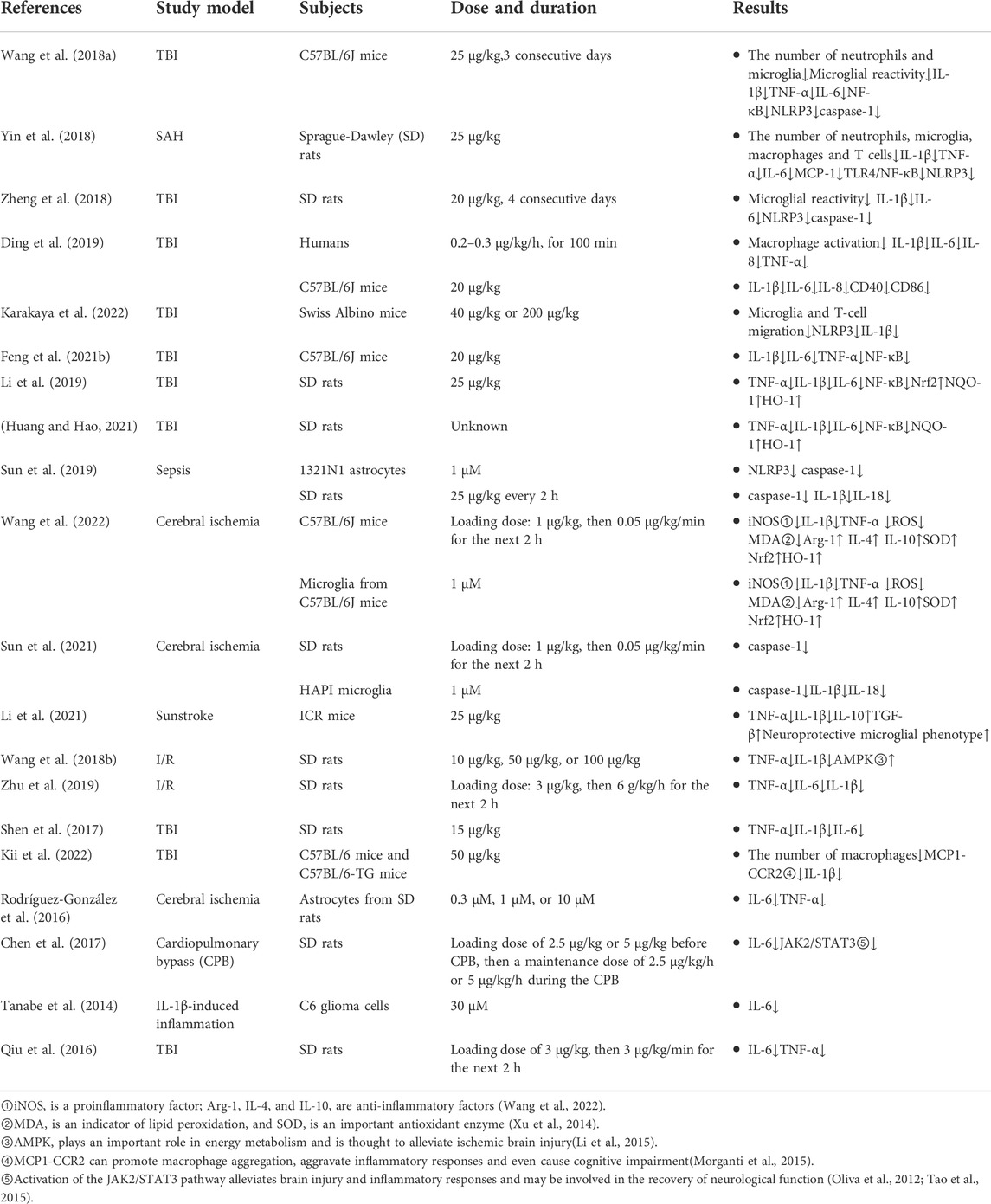
TABLE 1. Changes in various inflammatory mediators and cells.
The NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome plays a key role in nervous system inflammation by inducing the expression of immune- and inflammation-related genes. NLRP3 activation requires NF-κB activation, in addition to pathogens or injury-related molecules that initiate the assembly of NLRP3 (Zhou et al., 2016; Yang et al., 2018). Activated NLRP3 further activates caspase-1 and promotes the processing and release of the inflammatory cytokine IL-18, while activated caspase-1 cleaves inactive pro-IL-1β to produce IL-1β, which promotes the inflammatory response (Liu et al., 2013; McKee and Lukens, 2016; Mamik and Power, 2017). Many studies have proven that DEX can reduce the effect of caspase-1 on the precursors of inflammatory mediators by inhibiting the activation of the NLRP3 inflammasome, thus reducing the release of inflammatory mediators and ultimately alleviating neuroinflammation (Wang et al., 2018a; Yin et al., 2018; Zheng et al., 2018; Sun et al., 2019; Karakaya et al., 2022).
Oxidative stress plays an important role in the inflammatory response. Reactive oxygen species (ROS) are one of the most important inflammatory mediators. Nuclear factor erythroid 2-related factor 2 (Nrf2) is an important transcription factor that controls the antioxidant activities of cells, inhibits the increase in intracellular ROS during the inflammatory response and reduces the transformation of macrophages into the proinflammatory M1 phenotype (Wu et al., 2018b; Liu et al., 2018). Nicotinamide adenine dinucleotide phosphate quinone oxidoreductase-1 (NQO-1) and heme oxygenase-1 (HO-1) are important antioxidant enzymes that can alleviate cell damage caused by oxidative stress by promoting the removal of ROS. Li et al. showed that DEX could increase the expression of Nrf2 and its downstream proteins NQO-1 and HO-1 in TBI rat brain tissue (Li et al., 2019). Moreover, HO-1 can also reduce the infiltration of inflammatory cells, while NQO-1 has a neuroprotective effect (Gueron et al., 2009; Sekine et al., 2016; Qu et al., 2020). Additionally, the Nrf2/NLRP3 pathway may play an important role in inhibiting the inflammatory response. Shan et al. examined the mechanism by which DEX alleviates neuropathic pain and accidentally discovered that DEX inhibited the expression of NLRP3 by activating Nrf2, thus reducing the levels of IL-1β and other inflammatory mediators (Shan et al., 2021). In-vivo and in-vitro experiments by Wang et al. showed that DEX could transform microglial cells from the proinflammatory phenotype to the neuroprotective phenotype by promoting Nrf2 translocation from the cytoplasm to the nucleus and upregulating HO-1 expression. In addition, DEX can reduce the release of inflammatory mediators through the Nrf2/HO-1/NLRP3 pathway (Wang et al., 2022). Mechanistically, heme is broken down by HO-1 into biliverdin and carbon monoxide, which can inhibit NLRP3 inflammasome activation (Gomperts et al., 2017).
In various nervous system injury models, a large number of apoptotic cells were found at the injury site, and DEX successfully reduced the number of apoptotic nerve cells and affected the expression of apoptosis-related proteins (Kose et al., 2013; Yin et al., 2018; Gao et al., 2019; Huang and Jiang, 2019; Sun et al., 2019; Feng et al., 2021b).
The B-cell lymphoma-2 (Bcl-2) protein family plays an important role in the mitochondrial apoptosis pathway, and the proapoptotic protein B-cell lymphoma-2 associated X (Bax) and antiapoptotic protein Bcl-2 are important components of this protein family. The balance between these two proteins maintains the normal process of apoptosis in the physiological state (Youle and Strasser, 2008). Bax promotes apoptosis through three pathways by ①activating other proapoptotic factors; ②inactivating the antiapoptotic protein Bcl-2; and ③triggering the release of corresponding cytokines into the cytoplasm to activate the apoptosis-related protein caspase-3 (Shamas-Din et al., 2011; Zhao et al., 2017). Changes in the levels of caspase-3, which mediates cell apoptosis, can reflect the degree of apoptosis (Wu et al., 2017). A series of studies have shown that DEX can reverse the increase in Bax and the decrease in Bcl-2 after nerve injury and inhibit the level of caspase-3, thus alleviating apoptosis after nerve injury. Zhang et al. showed that DEX can not only affect the expression of these apoptosis-related proteins but also increase the protein expression of heat shock protein 70 (HSP70). HSP70 is a molecular chaperone that can reduce cell damage and has an antiapoptotic effect. Table 2 shows changes in various apoptosis-related proteins (Feder and Hofmann, 1999; Chen et al., 2017; Huang et al., 2018a; Zhang et al., 2018b; Yin et al., 2018; Gao et al., 2019; Li et al., 2019; Feng et al., 2021b; Feng et al., 2021a; Huang and Hao, 2021).
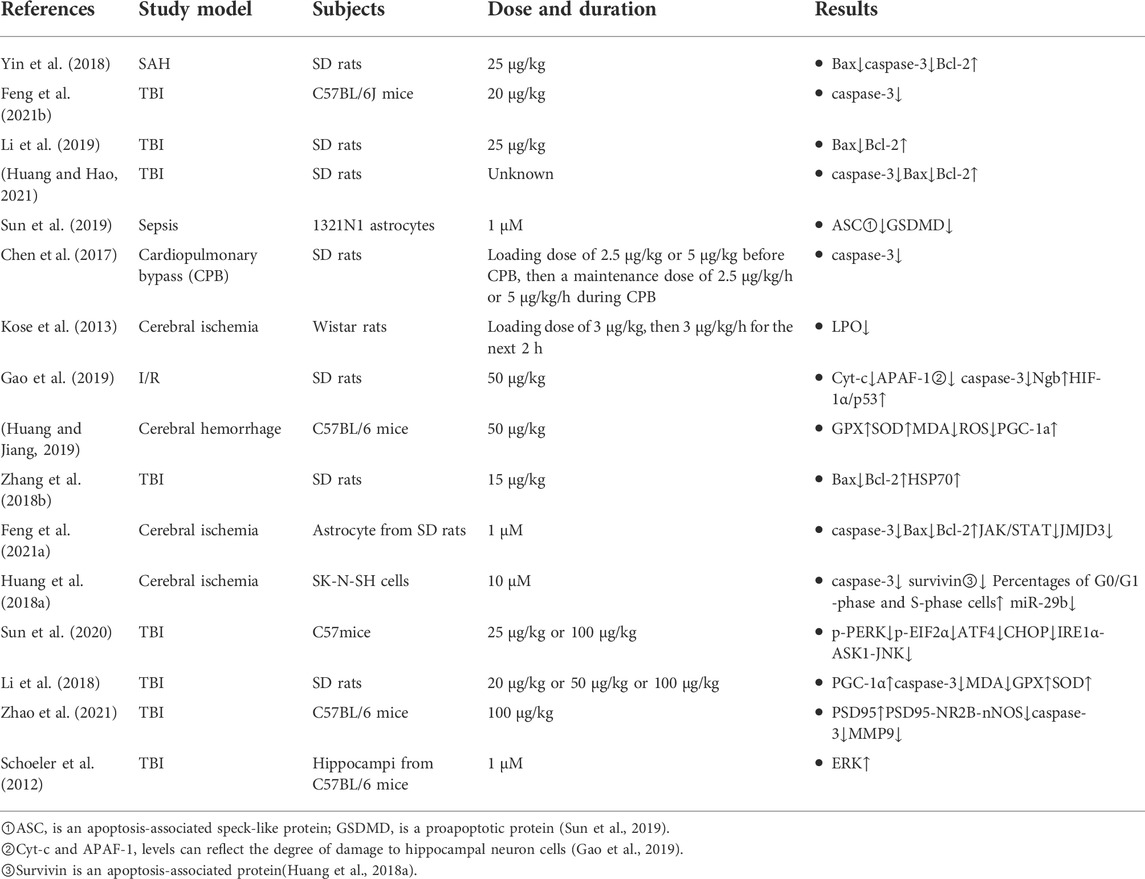
TABLE 2. Changes in various apoptosis-related proteins.
When nerve cells are damaged, ischemia, hypoxia or oxidative stress will change the internal environment of the endoplasmic reticulum, causing Ca2+ dysregulation in the endoplasmic reticulum, and the increased number of unfolded and misfolded proteins, which triggers endoplasmic reticulum stress, leads to the unfolded protein response (UPR) and ultimately induces apoptosis (Sano and Reed, 2013; Dash et al., 2015). Sun et al. showed that the expression levels of the endoplasmic reticulum stress markers phosphorylated protein kinase RNA-like ER kinase (p-PERK), phosphorylated eukaryotic initiation factor 2α (p-EIF2α), activating transcription factor 4 (ATF4) and C/EBP-homologous protein (CHOP) were significantly increased in TBI mice. CHOP also plays an important role in endoplasmic reticulum pathway-mediated apoptosis, but DEX decreased the levels of these endoplasmic reticulum stress marker proteins. This study suggested that inhibiting the apoptosis-related signaling pathway IRE1α/ASK1/JNK may reduce apoptosis and alleviate nervous system injury (Oyadomari and Mori, 2004; Sun et al., 2020).
An increasing number of studies have proven that apoptosis induced by oxidative stress plays an important role in secondary damage in the nervous system (Kose et al., 2013; Li et al., 2018; Huang and Jiang, 2019). Oxidative stress can produce excessive ROS, which can cause damage and impair cell membrane permeability by oxidizing DNA, proteins and membrane lipids and further lead to a decrease in Na+-K+-ATPase activity on the membrane, which will cause an imbalance in the K+ and Mg2+ concentrations in the cell. These two ions are important for protein synthesis (White et al., 1993). Kose et al. showed that DEX could reduce lipid peroxidation, and the lipid peroxidation index, protect the cell membrane and membrane receptors, and reduce the damage to the cell structure caused by oxidative stress (Kose et al., 2013). Malondialdehyde (MDA) is an indicator of lipid peroxidation, while superoxide dismutase (SOD) and glutathione peroxidase (GPX) are important antioxidant enzymes. These antioxidant enzymes can convert hydrogen peroxide and lipid peroxides into nontoxic compounds. Huang and Lia et al. suggested that DEX could reduce the apoptosis of nerve cells in patients with brain injury caused by oxidative stress through the proliferator-activated receptor-gamma coactivator 1α (PGC-1α) pathway, and DEX decreased MDA levels while increasing SOD and GPX levels (Xu et al., 2014; Li et al., 2018; Huang and Jiang, 2019).
Zhao et al. (2021) showed that the expression of postsynaptic density protein 95 (PSD95) was decreased in TBI mouse brain tissues, while the levels of the PSD95-N- methyl-D-aspartic acid (PSD95-NMDA) complex were significantly increased. Excessive levels of the PSD95-NMDA complex can lead to the massive release of NO and activate matrix metallopeptidase 9 (MMP9), thus inducing apoptosis, and DEX can inhibit the formation of the PSD95-NMDA complex. In addition, after TBI, a large amount of glutamate is released into the synaptic cleft to activate NMDA receptors and anchor PSD95. This procedure greatly promotes Ca2+ influx to produce a large amount of reactive oxygen and active nitrogen, leading to oxidative stress and apoptosis. DEX inhibits the interaction between NMDA and PSD95, thus reducing the oxidative stress response (Gu et al., 2002; Luo et al., 2011).
Schoeler et al. (2012) examined an in vitro TBI model of hippocampal cells and showed that DEX may protect neurons by activating the extracellular regulated kinase (ERK) signaling pathway, which has been shown to protect neuronal cells from mechanical injury-induced apoptosis in previous studies (Ma et al., 2011).
Studies have proven that excessive Jumonji domain-containing protein 3 demethylase (JMJD3) expression can increase the levels of the proapoptotic proteins Bax and caspase-3, leading to cell apoptosis. Feng et al. stimulated astrocytes with oxygen and glucose deprivation (OGD) in vitro to simulate ischemic hypoxia after cerebral infarction and demonstrated that DEX could regulate the expression of apoptosis-related proteins by inhibiting the JAK/STAT pathway (which is involved in many crucial biological processes, including cell proliferation, differentiation, apoptosis, and immune regulation) and reducing the expression of JMJD3 (Zhang et al., 2018a; Feng et al., 2021a). Huang et al. used an OGD-induced in vitro cerebral ischemia model and showed that DEX could reverse the OGD-induced decreases in the percentages of G0/G1 and S phase cells, increase cell proliferation, reduce cell apoptosis, and inhibit the expression of microRNA-29b (miR-29b) (Huang et al., 2018a). Moreover, the authors demonstrated in another trial that miR-29b could promote neuronal apoptosis by targeting MCL-1 (Huang et al., 2018b).
Neuroglobin (Ngb) is an endogenous anti-injury factor that has a significant protective effect on nerve cells and is thought to play an important role in inhibiting mitochondrial apoptosis: this factor is called the “hemoglobin of the nervous system” (Mammen et al., 2002; Fiocchetti et al., 2021). Gao et al. found that DEX could increase the expression of Ngb and inhibit mitochondrial damage-mediated apoptosis in a rat I/R injury model, possibly by activating the hypoxia-inducible factor 1α (HIF-1α)/p53 signaling pathway. HIF-1α is a regulatory factor that promotes the expression of a series of genes that enable cells to adapt to a hypoxic environment (Demidenko and Blagosklonny, 2011; Gao et al., 2019).
Feng et al. (2021b) showed that autophagy was overactivated in the brains of TBI mice, and the autophagy markers Beclin-1 and light chain 3I/II (LC3I/II) were significantly increased, leading to the destruction of the BBB, brain edema, and cell apoptosis. However, DEX significantly reduced Beclin-1 and LC3I/II levels, alleviating autophagy and damage. In mice that were pretreated with the autophagy activator rapamycin, the neuroprotective effect of DEX on reducing autophagy was reversed. In this study, DEX inhibited TBI-induced autophagy by regulating the Nrf2/ROS signaling pathway.
Circlrp1b (a circRNA that regulates autophagy) and DNA-damage regulated autophagy modulator 2 (Dram2) play synergistic roles in autophagy, inflammation and impaired nerve function after TBI, while miR-27a-3p plays the opposite role in these processes. Li et al. showed that the circlrp1b/miR-27a-3p/Dram2 pathway played an important role in autophagy after TBI. DEX decreased the levels of the autophagy-related proteins Dram2, autophagy protein 5 (ATG5), Beclin-1 and LC3I/II, reversing the TBI-induced increased levels of circlrp1b and Dram2 and decreased levels of mir-27a-3p, thus alleviating the autophagic response (Li et al., 2020).
Shen et al. and Zhu et al. showed that DEX could reduce the expression of Beclin-1 and LC3I/II, which are autophagy-related proteins induced by brain injury in rat TBI and I/R models, respectively, and also reduced the degree of edema, vacuolation and autophagosomes, as shown by electron microscopy. The PI3K/Akt pathway plays an important role in cell growth, metabolism and survival, while mechanistic target of rapamycin (mTOR) is the core protein that regulates autophagy. Shen et al. suggested that DEX activated the PI3K/Akt/mTOR signaling pathway to alleviate autophagy (Li et al., 2016; Li et al., 2017; Shen et al., 2017; Zhu et al., 2019). Zhu et al. showed that the reduction in autophagy was due to the inhibition of c-Jun N-terminal kinase (JNK) pathway activation, which was previously thought to be widely involved in stress, cell division, apoptosis and other processes. In the study, Zhu et al. showed that this pathway may also be involved in autophagy (Zhong et al., 2017; Zhu et al., 2019). Table 3 shows changes in various autophagic proteins.

TABLE 3. Changes in various autophagic proteins.
BBB injury is a common primary brain injury after TBI. Unavoidable mechanical damage caused by external forces applied to the brain during the acute stage, which can lead to brain edema, further increasing intracranial pressure and worsening patient prognosis (Zhao et al., 2016).
Tight junction proteins, including occludin, zona occludens-1 (ZO-1) and claudin-5, are important components of the BBB, and damage to the BBB is accompanied by the degradation of tight junction proteins. Wang et al. and Shen et al. showed that DEX treatment could increase the expression of tight junction proteins to maintain the integrity of the BBB (Shen et al., 2017; Wang et al., 2018a). As the expression of tight junction proteins increased, the permeability of the BBB was restored. Experimentally, BBB permeability and damage are often assessed by observing the extravasation of Evans blue dye. A series of studies have shown that DEX can significantly reduce the leakage of Evans blue in the cerebral cortex. Reversing the increase in BBB permeability caused by TBI reduces brain edema (Shen et al., 2017; Wang et al., 2018a; Sun et al., 2020; Feng et al., 2021b). In addition, Yin et al. used a model of SAH and showed that DEX alleviated brain edema by increasing tight junction protein expression and reducing the extravasation of Evans blue dye to indicate a protective effect on the BBB (Yin et al., 2018). Table 4 shows changes in various tight junction proteins.
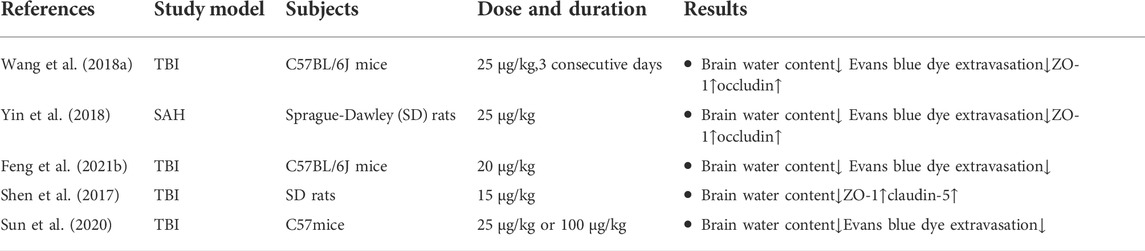
TABLE 4. Changes in brain water content and various tight junction proteins.
In addition to acute mechanical injury, the inflammatory response after TBI also plays an important role in BBB disruption. A growing body of research suggests that an excessive inflammatory response after brain injury can lead to BBB breakdown and nerve damage (Abdul-Muneer et al., 2015; Gao et al., 2015). Peripheral neutrophils, macrophages and other inflammatory cells can migrate to the brain through the damaged BBB, further exacerbating the inflammatory response (Xu et al., 2017). DEX can reduce the expression of the inflammatory mediators IL-1β, TNF-α, IL-6 and NLRP3 inflammasome in the brain after TBI (Wang et al., 2018a; Feng et al., 2021b; Li et al., 2021). Shen et al. suggested that DEX could not only reduce the inflammatory response but also alleviate BBB injury after TBI by activating the PI3K/Akt/mTOR signaling pathway, which may increase the expression of the tight junction proteins occludin, claudin-1 and ZO-1 (Shen et al., 2017).
In addition, DEX has been shown to play an important role in protecting cellular structures after nerve injury. Sun et al. found that DEX could protect the integrity of the astrocyte membrane and nuclear structure, and this type of cell provides support for neurons through glial transmission, synaptic remodeling, gap connections, energy metabolism, information transmission and other processes (Rama Rao and Kielian, 2015; Sun et al., 2019). In this study, abnormally elevated nuclear histones were detected in the cytoplasm during cell injury, and the cytoskeletal structure was destroyed. However, DEX significantly improved these outcomes, and the number of histones released from the nucleus was significantly reduced. The overall cytoskeletal structure of the cells was relatively intact, maintaining normal cellular morphology. In addition, these findings showed that histones released from the nucleus can cause neuronal damage (Sun et al., 2019).
Synaptic damage often leads to neuronal dysfunction and even apoptosis after TBI. PSD95 plays a supporting role in synaptic development, promotes improvements in synaptic function, and plays a role in synaptic integration and functional recovery when nerve cells are damaged (Keith and El-Husseini, 2008; Merlo et al., 2014; Mo et al., 2016). Zhao et al. showed that the expression of PSD95 was significantly decreased after TBI, while DEX significantly reversed this outcome. Mechanistically, DEX reduces the production of PSD95-NMDA compounds, which has a negative effect on the repair of damaged synapses and recovery from cognitive impairment (Zhao et al., 2021). Wu et al. used an anti-synaptophysin antibody to immunostain samples and found that the intensity and number of synaptophysin-positive cells after TBI were lower than those in the control group, indicating synaptic degeneration, and DEX treatment protected synapses by increasing the intensity of synaptophysin staining (Wu et al., 2018a). In addition, this study showed a protective effect of DEX on axons. β-amyloid precursor protein (β-APP) is an important marker of axonal injury because β-APP can be rapidly transported in normal axons. When axons are damaged, β-APP transport is impaired, resulting in the accumulation of β-APP (Kaur et al., 1999). Therefore, axonal injury can be assessed by β-APP immunostaining. Compared with the control treatment, DEX treatment significantly weakened β-APP immunoreactivity, suggesting that DEX could alleviate the axonal injury caused by TBI (Wu et al., 2018a). Table 5 shows the protective effect of DEX on the cell structure.

TABLE 5. Protective effect of DEX on the cell structure.
Extracellular regulated kinase1/2 (ERK1/2) is an indispensable mediator of mitosis, and cyclic adenosine monophosphate response element binding protein (CREB) maintains the survival of damaged neurons and promotes the subsequent regeneration and repair process. Teng et al. and Shi et al. showed that DEX could increase the expression of ERK1/2 and CREB in damaged nerve cells (Cao et al., 2012; Shi et al., 2018; Teng et al., 2019). Yang et al. compared mRNA and miRNA expression in TBI model rats in the control group and DEX treatment group by RNA sequencing and conducted bioinformatics analysis: the results showed that Lyn and Cdk1 may be central genes involved in DEX-mediated neuroprotection (Yang et al., 2021). Cdk1 is critical in mitotic substrate processing, and Lyn regulates B-cell signaling pathways to maintain tolerance to autoantigens. In addition, activation of the Lyn-ERK1/2-CREB pathway increases the expression of brain-derived neurotrophic factor (BDNF) (Zhang et al., 2010; Malumbres, 2014; Brodie et al., 2018). Rodríguez et al. and Degos et al. showed that DEX could increase the expression of BDNF in astrocytes to play a neuroprotective role (Degos et al., 2013; Rodríguez-González et al., 2016).
The effects of DEX on inflammation after TBI are summarized in Figure 2. At the cellular level, DEX reduces the density of microglial cells, inhibits microglial reactivity, and reduces inflammatory cell infiltration by inhibiting the reactivity of microglia and the accumulation of macrophages and T cells (Mantovani et al., 2004; Wang et al., 2018a; Zheng et al., 2018; Ding et al., 2019; Karakaya et al., 2022; Kii et al., 2022). At the molecular level, DEX inhibits the microglial TLR4/MyD88/NF-κB pathway and reduces the expression of various inflammatory mediators and the NLRP3 inflammasome (Wang et al., 2018a; Zheng et al., 2018; Li et al., 2019; Feng et al., 2021b; Huang and Hao, 2021; Karakaya et al., 2022). The oxidative stress process mediates inflammation through ROS and promotes the infiltration of macrophages (Liu et al., 2018; Kii et al., 2022). Therefore, we believe that the effect of DEX on microglia plays an important role in alleviating inflammatory injury in TBI, possibly by inhibiting the TLR4/MyD88/NF-κB/NLRP3 pathway. Based on these studies, we found that DEX could affect most of the inflammatory processes shown in Figure 2, but DEX-mediated inhibition of microglia and the promotion of Nrf2 expression may be the initial links in its neuroprotective effect (Wang et al., 2018a; Zheng et al., 2018; Li et al., 2019; Feng et al., 2021b; Huang and Hao, 2021; Karakaya et al., 2022). Recent studies have shown that these two processes can drive all other processes after being initiated by DEX. However, the direct effect of DEX on other processes cannot be ruled out. Notably, in models of ischemic brain injury and neuralgia, DEX acts on oxidative stress processes to reduce the number of inflammatory microglia and NLRP3 inflammasome formation. This result suggests that the oxidative stress process may be directly linked to the inflammatory response through microglia and the NLRP3 inflammasome (Shan et al., 2021; Wang et al., 2022). However, it is undeniable that there may be some heterogeneity in the mechanism of inflammation due to different experimental models.
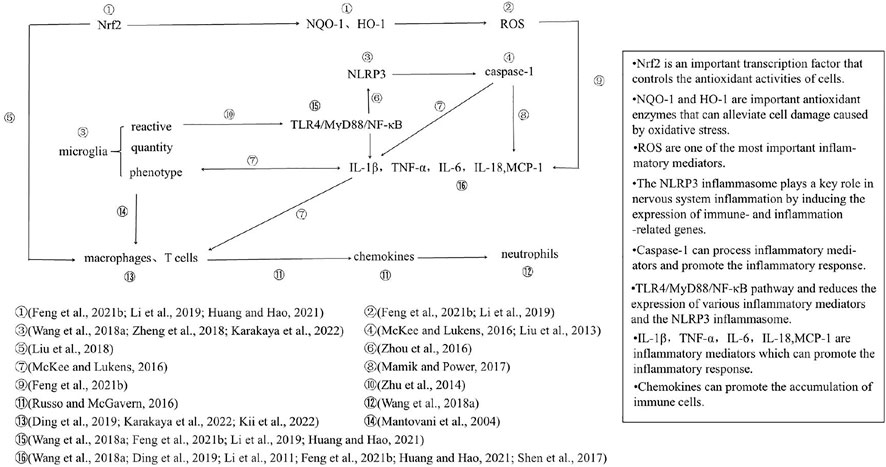
FIGURE 2. The TBI model plays a leading role in research on the neuroprotective effect of DEX. Therefore, trials with TBI as the model were screened and summarized to explore the neuroprotective mechanism of DEX under the same injury conditions. In the TBI model, the action of DEX involves two main processes, the inflammatory response and oxidative stress, which act on multiple levels of cells, signaling pathways and molecules; oxidative stress process ultimately promotes the release of inflammatory mediators and infiltration of inflammatory cells. Once again, it was confirmed that DEX plays an important role in alleviating inflammation in patients with neurological injury.
DEX decreases apoptosis mainly by inhibiting the mitochondrial pathway of apoptosis; mitigating endoplasmic reticulum stress and oxidative stress; and decreasing the expression of proapoptotic proteins. According to the literature, these three apoptosis pathways are relatively independent, and there is no evidence to verify a connection between them. The mechanism by which DEX inhibits mitochondrial apoptosis may involve reducing the expression of JMJD3 by inhibiting the JAK/STAT pathway, reducing the production of the proapoptotic proteins Bax and caspase-3, and activating the HIF-1α/p53 signaling pathway to enhance the adaptation of nerve cells to adverse environments (Gao et al., 2019; Feng et al., 2021a). The DEX-mediated inhibition of endoplasmic reticulum stress pathway-mediated apoptosis may be related to inhibiting the IRE1α-ASK1-JNK pathway (Sun et al., 2020). DEX may inhibit oxidative stress through the PGC-1α pathway and the interaction between NMDA and PSD95 (Li et al., 2018; Zhao et al., 2021).
Based on recent studies, we expounded on the role and possible mechanism by which DEX alleviates autophagy, protects the BBB and maintains normal cellular structure. In addition, we found that the various protective effects of DEX on the nervous system were not independent. For example, inhibiting oxidative stress can reduce autophagy and apoptosis, while reducing inflammation can prevent the destruction of the BBB (Wang et al., 2018a; Feng et al., 2021b). DEX activates the PI3K/Akt/mTOR signaling pathway and plays an active role in alleviating autophagy and protecting the BBB (Shen et al., 2017). The neuroprotective effect of dexmedetomidine and its relations in various aspects are shown in Figure 3. In addition, the protective effect of DEX in patients with brain injury is not limited to neuroprotection. For example, studies have shown that DEX can reduce delirium and agitation in patients after TBI (Roberson et al., 2021; Soltani et al., 2021), and DEX can also reduce paroxysmal sympathetic hyperactivity (PSH) caused by nervous system damage (Tang et al., 2017; Branstetter et al., 2020).
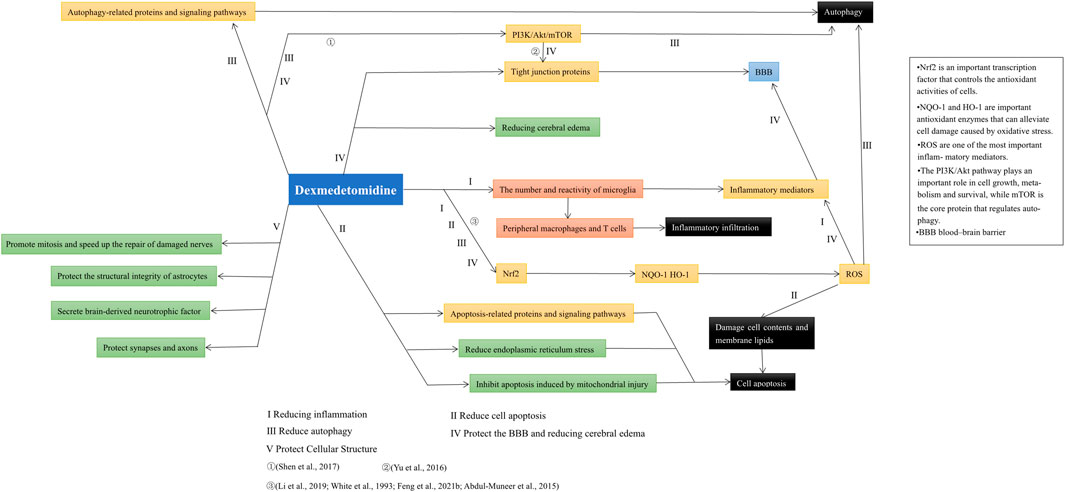
FIGURE 3. After summarizing the literature included in this paper, it was found that the neuroprotective effects of dexmedetomidine are not completely independent. Its effect on Nrf2 signaling pathway can reduce inflammation, apoptosis, autophagy, and protect BBB by inhibiting ROS generation. Additionally, the PI3K/Akt/mTOR pathway plays an important role in alleviating autophagy and protecting the BBB.
This review indicates that DEX can play a neuroprotective role in different aspects of brain injury caused by a variety of factors and that DEX can significantly improve patient prognosis. However, this study has some limitations. First, most of the current experiments on the neuroprotective effects of DEX are animal experiments or in vitro cell experiments, and clinical data are lacking. In addition, priority is given to TBI and cerebral ischemia models; few other models of neurological damage are available for review. Therefore, in this paper, the goal was not to differentiate damage types but to examine neuroprotective effects; different types of nerve damage have certain similarities. Further understanding of the neuroprotective effect of DEX on different injury types will be important in the future.
YH, LZ, and HZ drafted the manuscript and critically revised it for intellectual content. HXZ, YS, and ZZ collected the relevant literature. YZ and KL drew the figures and summarized their content. YYZ and JX designed the tables and summarized their content. YH finalized and submitted the manuscript. All authors read and approved the final version of the manuscript before submission.
This work was supported by a grant from the Shandong Traditional Chinese Medicine Science and Technology Project Task Book (year 2020, No. 2020M143).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdul-Muneer, P. M., Chandra, N., and Haorah, J. (2015). Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 51 (3), 966–979. doi:10.1007/s12035-014-8752-3
Barends, C. R., Absalom, A., van Minnen, B., Vissink, A., and Visser, A. (2017). Dexmedetomidine versus midazolam in procedural sedation. A systematic review of efficacy and safety. PLoS One 12 (1), e0169525. doi:10.1371/journal.pone.0169525
Branstetter, J. W., Ohman, K. L., Johnson, D. W., and Gilbert, B. W. (2020). Management of paroxysmal sympathetic hyperactivity with dexmedetomidine and propranolol following traumatic brain injury in a pediatric patient. J. Pediatr. Intensive Care 9 (1), 64–69. doi:10.1055/s-0039-1698758
Brodie, E. J., Infantino, S., Low, M. S. Y., and Tarlinton, D. M. (2018). Lyn, lupus, and (B) lymphocytes, a lesson on the critical balance of kinase signaling in immunity. Front. Immunol. 9, 401. doi:10.3389/fimmu.2018.00401
Cao, H., Ren, W. H., Zhu, M. Y., Zhao, Z. Q., and Zhang, Y. Q. (2012). Activation of glycine site and GluN2B subunit of NMDA receptors is necessary for ERK/CREB signaling cascade in rostral anterior cingulate cortex in rats: Implications for affective pain. Neurosci. Bull. 28 (1), 77–87. doi:10.1007/s12264-012-1060-x
Chen, Y., Zhang, X., Zhang, B., He, G., Zhou, L., and Xie, Y. (2017). Dexmedetomidine reduces the neuronal apoptosis related to cardiopulmonary bypass by inhibiting activation of the JAK2-STAT3 pathway. Drug Des. devel. Ther. 11, 2787–2799. doi:10.2147/dddt.S140644
Dash, P. K., Hylin, M. J., Hood, K. N., Orsi, S. A., Zhao, J., Redell, J. B., et al. (2015). Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J. Neurotrauma 32 (20), 1608–1620. doi:10.1089/neu.2014.3772
Degos, V., Charpentier, T. L., Chhor, V., Brissaud, O., Lebon, S., Schwendimann, L., et al. (2013). Neuroprotective effects of dexmedetomidine against glutamate agonist-induced neuronal cell death are related to increased astrocyte brain-derived neurotrophic factor expression. Anesthesiology 118 (5), 1123–1132. doi:10.1097/ALN.0b013e318286cf36
Demidenko, Z. N., and Blagosklonny, M. V. (2011). The purpose of the HIF-1/PHD feedback loop: To limit mTOR-induced HIF-1α. Cell Cycle 10 (10), 1557–1562. doi:10.4161/cc.10.10.15789
Ding, M., Chen, Y., Luan, H., Zhang, X., Zhao, Z., and Wu, Y. (2019). Dexmedetomidine reduces inflammation in traumatic brain injury by regulating the inflammatory responses of macrophages and splenocytes. Exp. Ther. Med. 18 (3), 2323–2331. doi:10.3892/etm.2019.7790
Fang, H., Wang, P. F., Zhou, Y., Wang, Y. C., and Yang, Q. W. (2013). Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J. Neuroinflammation 10, 27. doi:10.1186/1742-2094-10-27
Feder, M. E., and Hofmann, G. E. (1999). Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 61, 243–282. doi:10.1146/annurev.physiol.61.1.243
Feng, P., Zhang, A., Su, M., Cai, H., Wang, X., and Zhang, Y. (2021a). Dexmedetomidine inhibits apoptosis of astrocytes induced by oxygen-glucose deprivation via targeting JAK/STAT3 signal pathway. Brain Res. 1750, 147141. doi:10.1016/j.brainres.2020.147141
Feng, X., Ma, W., Zhu, J., Jiao, W., and Wang, Y. (2021b). Dexmedetomidine alleviates early brain injury following traumatic brain injury by inhibiting autophagy and neuroinflammation through the ROS/Nrf2 signaling pathway. Mol. Med. Rep. 24 (3), 661. doi:10.3892/mmr.2021.12300
Fiocchetti, M., Cracco, P., Montalesi, E., Solar Fernandez, V., Stuart, J. A., and Marino, M. (2021). Neuroglobin and mitochondria: The impact on neurodegenerative diseases. Arch. Biochem. Biophys. 701, 108823. doi:10.1016/j.abb.2021.108823
Gao, W., Zhao, Z., Yu, G., Zhou, Z., Zhou, Y., Hu, T., et al. (2015). VEGI attenuates the inflammatory injury and disruption of blood-brain barrier partly by suppressing the TLR4/NF-κB signaling pathway in experimental traumatic brain injury. Brain Res. 1622, 230–239. doi:10.1016/j.brainres.2015.04.035
Gao, Y., Yin, H., Zhang, Y., Dong, Y., Yang, F., Wu, X., et al. (2019). Dexmedetomidine protects hippocampal neurons against hypoxia/reoxygenation-induced apoptosis through activation HIF-1α/p53 signaling. Life Sci. 232, 116611. doi:10.1016/j.lfs.2019.116611
Gomperts, E., Belcher, J. D., Otterbein, L. E., Coates, T. D., Wood, J., Skolnick, B. E., et al. (2017). The role of carbon monoxide and heme oxygenase in the prevention of sickle cell disease vaso-occlusive crises. Am. J. Hematol. 92 (6), 569–582. doi:10.1002/ajh.24750
Gu, Z., Kaul, M., Yan, B., Kridel, S. J., Cui, J., Strongin, A., et al. (2002). S-Nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 297 (5584), 1186–1190. doi:10.1126/science.1073634
Gueron, G., De Siervi, A., Ferrando, M., Salierno, M., De Luca, P., Elguero, B., et al. (2009). Critical role of endogenous heme oxygenase 1 as a tuner of the invasive potential of prostate cancer cells. Mol. Cancer Res. 7 (11), 1745–1755. doi:10.1158/1541-7786.Mcr-08-0325
Huang, G. R., and Hao, F. G. (2021). Dexmedetomidine inhibits inflammation to alleviate early neuronal injury via TLR4/NF-κB pathway in rats with traumatic brain injury. Crit. Rev. Eukaryot. Gene Expr. 31 (1), 41–47. doi:10.1615/CritRevEukaryotGeneExpr.2021037390
Huang, J., and Jiang, Q. (2019). Dexmedetomidine protects against neurological dysfunction in a mouse intracerebral hemorrhage model by inhibiting mitochondrial dysfunction-derived oxidative stress. J. Stroke Cerebrovasc. Dis. 28 (5), 1281–1289. doi:10.1016/j.jstrokecerebrovasdis.2019.01.016
Huang, Z., Liu, G., Zeng, Q., Gao, R., Zhang, S., Wang, L., et al. (2018a). MiR-29b expression is associated with a dexmedetomidine-mediated protective effect against oxygen-glucose deprivation-induced injury to SK-N-SH cells in vitro. Cell Biol. Int. 42 (3), 344–352. doi:10.1002/cbin.10906
Huang, Z., Lu, L., Jiang, T., Zhang, S., Shen, Y., Zheng, Z., et al. (2018b). miR-29b affects neurocyte apoptosis by targeting MCL-1 during cerebral ischemia/reperfusion injury. Exp. Ther. Med. 16 (4), 3399–3404. doi:10.3892/etm.2018.6622
Jakob, S. M., Ruokonen, E., Grounds, R. M., Sarapohja, T., Garratt, C., Pocock, S. J., et al. (2012). Dexmedetomidine vs midazolam or propofol for sedation during prolonged mechanical ventilation: Two randomized controlled trials. Jama 307 (11), 1151–1160. doi:10.1001/jama.2012.304
Jiang, L., Hu, M., Lu, Y., Cao, Y., Chang, Y., and Dai, Z. (2017). The protective effects of dexmedetomidine on ischemic brain injury: A meta-analysis. J. Clin. Anesth. 40, 25–32. doi:10.1016/j.jclinane.2017.04.003
Karakaya, D., Cakir-Aktas, C., Uzun, S., Soylemezoglu, F., and Mut, M. (2022). Tailored therapeutic doses of dexmedetomidine in evolving neuroinflammation after traumatic brain injury. Neurocrit. Care 36 (3), 802–814. doi:10.1007/s12028-021-01381-3
Kaur, B., Rutty, G. N., and Timperley, W. R. (1999). The possible role of hypoxia in the formation of axonal bulbs. J. Clin. Pathol. 52 (3), 203–209. doi:10.1136/jcp.52.3.203
Keith, D., and El-Husseini, A. (2008). Excitation control: Balancing PSD-95 function at the synapse. Front. Mol. Neurosci. 1, 4. doi:10.3389/neuro.02.004.2008
Kii, N., Sawada, A., Yoshikawa, Y., Tachibana, S., and Yamakage, M. (2022). Dexmedetomidine ameliorates perioperative neurocognitive disorders by suppressing monocyte-derived macrophages in mice with preexisting traumatic brain injury. Anesth. Analg. 134 (4), 869–880. doi:10.1213/ane.0000000000005699
Kose, E. A., Bakar, B., Kasimcan, O., Atilla, P., Kilinc, K., Muftuoglu, S., et al. (2013). Effects of intracisternal and intravenous dexmedetomidine on ischemia-induced brain injury in rat: A comparative study. Turk. Neurosurg. 23 (2), 208–217. doi:10.5137/1019-5149.Jtn.6757-12.0
Krämer, T. J., Hack, N., Brühl, T. J., Menzel, L., Hummel, R., Griemert, E. V., et al. (2019). Depletion of regulatory T cells increases T cell brain infiltration, reactive astrogliosis, and interferon-γ gene expression in acute experimental traumatic brain injury. J. Neuroinflammation 16 (1), 163. doi:10.1186/s12974-019-1550-0
Li, F., Wang, X., Deng, Z., Zhang, X., Gao, P., and Liu, H. (2018). Dexmedetomidine reduces oxidative stress and provides neuroprotection in a model of traumatic brain injury via the PGC-1α signaling pathway. Neuropeptides 72, 58–64. doi:10.1016/j.npep.2018.10.004
Li, F., Wang, X., Zhang, Z., Zhang, X., and Gao, P. (2019). Dexmedetomidine attenuates neuroinflammatory-induced apoptosis after traumatic brain injury via Nrf2 signaling pathway. Ann. Clin. Transl. Neurol. 6 (9), 1825–1835. doi:10.1002/acn3.50878
Li, G. Z., Zhang, Y., Zhao, J. B., Wu, G. J., Su, X. F., and Hang, C. H. (2011). Expression of myeloid differentiation primary response protein 88 (Myd88) in the cerebral cortex after experimental traumatic brain injury in rats. Brain Res. 1396, 96–104. doi:10.1016/j.brainres.2011.04.014
Li, H., Lu, C., Yao, W., Xu, L., Zhou, J., and Zheng, B. (2020). Dexmedetomidine inhibits inflammatory response and autophagy through the circLrp1b/miR-27a-3p/Dram2 pathway in a rat model of traumatic brain injury. Aging (Albany NY) 12 (21), 21687–21705. doi:10.18632/aging.103975
Li, P., Shen, T., Luo, X., Yang, J., Luo, Z., Tan, Y., et al. (2021). Modulation of microglial phenotypes by dexmedetomidine through TREM2 reduces neuroinflammation in heatstroke. Sci. Rep. 11 (1), 13345. doi:10.1038/s41598-021-92906-5
Li, X., Li, J., Zhang, Y., and Zhou, Y. (2017). Di-n-butyl phthalate induced hypospadias relates to autophagy in genital tubercle via the PI3K/Akt/mTOR pathway. J. Occup. Health 59 (1), 8–16. doi:10.1539/joh.16-0089-OA
Li, X., Wang, L., Zhou, X. E., Ke, J., de Waal, P. W., Gu, X., et al. (2015). Erratum: Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 25 (3), 398. doi:10.1038/cr.2015.27
Li, Y., Yang, W., Quinones-Hinojosa, A., Wang, B., Xu, S., Zhu, W., et al. (2016). Interference with protease-activated receptor 1 alleviates neuronal cell death induced by lipopolysaccharide-stimulated microglial cells through the PI3K/akt pathway. Sci. Rep. 6, 38247. doi:10.1038/srep38247
Liu, H., Busl, K. M., and Doré, S. (2022). Role of dexmedetomidine in aneurysmal subarachnoid hemorrhage: A comprehensive scoping review. J. Neurosurg. Anesthesiol. 34 (2), 176–182. doi:10.1097/ana.0000000000000728
Liu, H. D., Li, W., Chen, Z. R., Hu, Y. C., Zhang, D. D., Shen, W., et al. (2013). Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem. Res. 38 (10), 2072–2083. doi:10.1007/s11064-013-1115-z
Liu, Z. M., Chen, Q. X., Chen, Z. B., Tian, D. F., Li, M. C., Wang, J. M., et al. (2018). RIP3 deficiency protects against traumatic brain injury (TBI) through suppressing oxidative stress, inflammation and apoptosis: Dependent on AMPK pathway. Biochem. Biophys. Res. Commun. 499 (2), 112–119. doi:10.1016/j.bbrc.2018.02.150
Lobo, F. A., Wagemakers, M., and Absalom, A. R. (2016). Anaesthesia for awake craniotomy. Br. J. Anaesth. 116 (6), 740–744. doi:10.1093/bja/aew113
Luo, P., Fei, F., Zhang, L., Qu, Y., and Fei, Z. (2011). The role of glutamate receptors in traumatic brain injury: Implications for postsynaptic density in pathophysiology. Brain Res. Bull. 85 (6), 313–320. doi:10.1016/j.brainresbull.2011.05.004
Luo, X., Zheng, X., and Huang, H. (2016). Protective effects of dexmedetomidine on brain function of glioma patients undergoing craniotomy resection and its underlying mechanism. Clin. Neurol. Neurosurg. 146, 105–108. doi:10.1016/j.clineuro.2016.05.004
Ma, Y., Liu, W., Wang, Y., Chao, X., Qu, Y., Wang, K., et al. (2011). VEGF protects rat cortical neurons from mechanical trauma injury induced apoptosis via the MEK/ERK pathway. Brain Res. Bull. 86 (5-6), 441–446. doi:10.1016/j.brainresbull.2011.07.007
Mamik, M. K., and Power, C. (2017). Inflammasomes in neurological diseases: Emerging pathogenic and therapeutic concepts. Brain 140 (9), 2273–2285. doi:10.1093/brain/awx133
Mammen, P. P., Shelton, J. M., Goetsch, S. C., Williams, S. C., Richardson, J. A., Garry, M. G., et al. (2002). Neuroglobin, a novel member of the globin family, is expressed in focal regions of the brain. J. Histochem. Cytochem. 50 (12), 1591–1598. doi:10.1177/002215540205001203
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., and Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25 (12), 677–686. doi:10.1016/j.it.2004.09.015
McKee, C. A., and Lukens, J. R. (2016). Emerging roles for the immune system in traumatic brain injury. Front. Immunol. 7, 556. doi:10.3389/fimmu.2016.00556
Merlo, L., Cimino, F., Angileri, F. F., La Torre, D., Conti, A., Cardali, S. M., et al. (2014). Alteration in synaptic junction proteins following traumatic brain injury. J. Neurotrauma 31 (16), 1375–1385. doi:10.1089/neu.2014.3385
Miao, Y., and Liao, J. K. (2014). Potential serum biomarkers in the pathophysiological processes of stroke. Expert Rev. Neurother. 14 (2), 173–185. doi:10.1586/14737175.2014.875471
Mo, S. F., Liao, G. Y., Yang, J., Wang, M. Y., Hu, Y., Lian, G. N., et al. (2016). Protection of neuronal cells from excitotoxicity by disrupting nNOS-PSD95 interaction with a small molecule SCR-4026. Brain Res. 1648 (1), 250–256. doi:10.1016/j.brainres.2016.07.012
Morganti, J. M., Jopson, T. D., Liu, S., Riparip, L. K., Guandique, C. K., Gupta, N., et al. (2015). CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci. 35 (2), 748–760. doi:10.1523/jneurosci.2405-14.2015
Neumann, H., and Takahashi, K. (2007). Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 184 (1-2), 92–99. doi:10.1016/j.jneuroim.2006.11.032
Oliva, A. A., Kang, Y., Sanchez-Molano, J., Furones, C., and Atkins, C. M. (2012). STAT3 signaling after traumatic brain injury. J. Neurochem. 120 (5), 710–720. doi:10.1111/j.1471-4159.2011.07610.x
Oyadomari, S., and Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11 (4), 381–389. doi:10.1038/sj.cdd.4401373
Qiu, Y. S., Jia, Y. P., and Xu, Q. (2016). [The protective effect of alpha-2 adrenergic agonist on cranial nerve in rats with brain injury]. Zhonghua Yi Xue Za Zhi 96 (28), 2246–2250. doi:10.3760/cma.j.issn.0376-2491.2016.28.010
Qu, Z., Chen, Y., Luo, Z. H., Shen, X. L., and Hu, Y. J. (2020). 7-methoxyflavanone alleviates neuroinflammation in lipopolysaccharide-stimulated microglial cells by inhibiting TLR4/MyD88/MAPK signalling and activating the Nrf2/NQO-1 pathway. J. Pharm. Pharmacol. 72 (3), 385–395. doi:10.1111/jphp.13219
Rama Rao, K. V., and Kielian, T. (2015). Neuron-astrocyte interactions in neurodegenerative diseases: Role of neuroinflammation. Clin. Exp. Neuroimmunol. 6 (3), 245–263. doi:10.1111/cen3.12237
Riker, R. R., Shehabi, Y., Bokesch, P. M., Ceraso, D., Wisemandle, W., Koura, F., et al. (2009). Dexmedetomidine vs midazolam for sedation of critically ill patients: A randomized trial. Jama 301 (5), 489–499. doi:10.1001/jama.2009.56
Roberson, S. W., Patel, M. B., Dabrowski, W., Ely, E. W., Pakulski, C., and Kotfis, K. (2021). Challenges of delirium management in patients with traumatic brain injury: From pathophysiology to clinical practice. Curr. Neuropharmacol. 19 (9), 1519–1544. doi:10.2174/1570159x19666210119153839
Rodríguez-González, R., Sobrino, T., Veiga, S., López, P., Rodríguez-García, J., del Río, S. V., et al. (2016). Neuroprotective effects of dexmedetomidine conditioning strategies: Evidences from an in vitro model of cerebral ischemia. Life Sci. 144, 162–169. doi:10.1016/j.lfs.2015.12.007
Russo, M. V., and McGavern, D. B. (2016). Inflammatory neuroprotection following traumatic brain injury. Science 353 (6301), 783–785. doi:10.1126/science.aaf6260
Sano, R., and Reed, J. C. (2013). ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833 (12), 3460–3470. doi:10.1016/j.bbamcr.2013.06.028
Schoeler, M., Loetscher, P. D., Rossaint, R., Fahlenkamp, A. V., Eberhardt, G., Rex, S., et al. (2012). Dexmedetomidine is neuroprotective in an in vitro model for traumatic brain injury. BMC Neurol. 12, 20. doi:10.1186/1471-2377-12-20
Sekine, H., Okazaki, K., Ota, N., Shima, H., Katoh, Y., Suzuki, N., et al. (2016). The mediator subunit MED16 transduces NRF2-activating signals into antioxidant gene expression. Mol. Cell. Biol. 36 (3), 407–420. doi:10.1128/mcb.00785-15
Shamas-Din, A., Brahmbhatt, H., Leber, B., and Andrews, D. W. (2011). BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta 1813 (4), 508–520. doi:10.1016/j.bbamcr.2010.11.024
Shan, W., Liao, X., Tang, Y., and Liu, J. (2021). Dexmedetomidine alleviates inflammation in neuropathic pain by suppressing NLRP3 via Nrf2 activation. Exp. Ther. Med. 22 (4), 1046. doi:10.3892/etm.2021.10479
Shen, M., Wang, S., Wen, X., Han, X. R., Wang, Y. J., Zhou, X. M., et al. (2017). Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed. Pharmacother. 95, 885–893. doi:10.1016/j.biopha.2017.08.125
Shi, Y., Peng, X. H., Li, X., Luo, G. P., and Wu, M. F. (2018). Neuroprotective role of dexmedetomidine pretreatment in cerebral ischemia injury via ADRA2A-mediated phosphorylation of ERK1/2 in adult rats. Exp. Ther. Med. 16 (6), 5201–5209. doi:10.3892/etm.2018.6878
Soltani, F., Tabatabaei, S., Jannatmakan, F., Nasajian, N., Amiri, F., Darkhor, R., et al. (2021). Comparison of the effects of haloperidol and dexmedetomidine on delirium and agitation in patients with a traumatic brain injury admitted to the intensive care unit. Anesth. Pain Med. 11 (3), e113802. doi:10.5812/aapm.113802
Sun, D., Wang, J., Liu, X., Fan, Y., Yang, M., and Zhang, J. (2020). Dexmedetomidine attenuates endoplasmic reticulum stress-induced apoptosis and improves neuronal function after traumatic brain injury in mice. Brain Res. 1732, 146682. doi:10.1016/j.brainres.2020.146682
Sun, K., Zhang, J., Yang, Q., Zhu, J., Zhang, X., Wu, K., et al. (2021). Dexmedetomidine exerts a protective effect on ischemic brain injury by inhibiting the P2X7R/NLRP3/Caspase-1 signaling pathway. Brain Res. Bull. 174, 11–21. doi:10.1016/j.brainresbull.2021.05.006
Sun, Y. B., Zhao, H., Mu, D. L., Zhang, W., Cui, J., Wu, L., et al. (2019). Dexmedetomidine inhibits astrocyte pyroptosis and subsequently protects the brain in in vitro and in vivo models of sepsis. Cell Death Dis. 10 (3), 167. doi:10.1038/s41419-019-1416-5
Tanabe, K., Matsushima-Nishiwaki, R., Kozawa, O., and Iida, H. (2014). Dexmedetomidine suppresses interleukin-1β-induced interleukin-6 synthesis in rat glial cells. Int. J. Mol. Med. 34 (4), 1032–1038. doi:10.3892/ijmm.2014.1863
Tang, Q., Wu, X., Weng, W., Li, H., Feng, J., Mao, Q., et al. (2017). The preventive effect of dexmedetomidine on paroxysmal sympathetic hyperactivity in severe traumatic brain injury patients who have undergone surgery: A retrospective study. PeerJ 5, e2986. doi:10.7717/peerj.2986
Tao, Z., Cheng, M., Wang, S. C., Lv, W., Hu, H. Q., Li, C. F., et al. (2015). JAK2/STAT3 pathway mediating inflammatory responses in heatstroke-induced rats. Int. J. Clin. Exp. Pathol. 8 (6), 6732–6739.
Teng, L., Chen, W., Yin, C., Zhang, H., and Zhao, Q. (2019). Dexmedetomidine improves cerebral ischemia-reperfusion injury in rats via extracellular signal-regulated kinase/cyclic adenosine monophosphate response element binding protein signaling pathway. World Neurosurg. 127, e624–e630. doi:10.1016/j.wneu.2019.03.232
Van Lieshout, J. H., Dibué-Adjei, M., Cornelius, J. F., Slotty, P. J., Schneider, T., Restin, T., et al. (2018). An introduction to the pathophysiology of aneurysmal subarachnoid hemorrhage. Neurosurg. Rev. 41 (4), 917–930. doi:10.1007/s10143-017-0827-y
Virtanen, R., Savola, J. M., Saano, V., and Nyman, L. (1988). Characterization of the selectivity, specificity and potency of medetomidine as an alpha 2-adrenoceptor agonist. Eur. J. Pharmacol. 150 (1-2), 9–14. doi:10.1016/0014-2999(88)90744-3
Wang, D., Xu, X., Wu, Y. G., Lyu, L., Zhou, Z. W., and Zhang, J. N. (2018a). Dexmedetomidine attenuates traumatic brain injury: Action pathway and mechanisms. Neural Regen. Res. 13 (5), 819–826. doi:10.4103/1673-5374.232529
Wang, N., Nie, H., Zhang, Y., Han, H., Wang, S., Liu, W., et al. (2022). Dexmedetomidine exerts cerebral protective effects against cerebral ischemic injury by promoting the polarization of M2 microglia via the Nrf2/HO-1/NLRP3 pathway. Inflamm. Res. 71 (1), 93–106. doi:10.1007/s00011-021-01515-5
Wang, Y., Han, R., and Zuo, Z. (2016). Dexmedetomidine-induced neuroprotection: Is it translational? Transl. Perioper. Pain Med. 1 (4), 15–19.
Wang, Z., Zhou, W., Dong, H., Ma, X., and He, Z. (2018b). Dexmedetomidine pretreatment inhibits cerebral ischemia/reperfusion-induced neuroinflammation via activation of AMPK. Mol. Med. Rep. 18 (4), 3957–3964. doi:10.3892/mmr.2018.9349
Weerink, M. A. S., Struys, M., Hannivoort, L. N., Barends, C. R. M., Absalom, A. R., and Colin, P. (2017). Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin. Pharmacokinet. 56 (8), 893–913. doi:10.1007/s40262-017-0507-7
White, B. C., Grossman, L. I., and Krause, G. S. (1993). Brain injury by global ischemia and reperfusion: A theoretical perspective on membrane damage and repair. Neurology 43 (9), 1656–1665. doi:10.1212/wnl.43.9.1656
Wu, G. J., Chen, J. T., Tsai, H. C., Chen, T. L., Liu, S. H., and Chen, R. M. (2017). Protection of dexmedetomidine against ischemia/reperfusion-induced apoptotic insults to neuronal cells occurs via an intrinsic mitochondria-dependent pathway. J. Cell. Biochem. 118 (9), 2635–2644. doi:10.1002/jcb.25847
Wu, J., Vogel, T., Gao, X., Lin, B., Kulwin, C., and Chen, J. (2018a). Neuroprotective effect of dexmedetomidine in a murine model of traumatic brain injury. Sci. Rep. 8 (1), 4935. doi:10.1038/s41598-018-23003-3
Wu, P. S., Ding, H. Y., Yen, J. H., Chen, S. F., Lee, K. H., and Wu, M. J. (2018b). Anti-inflammatory activity of 8-hydroxydaidzein in LPS-stimulated BV2 microglial cells via activation of nrf2-antioxidant and attenuation of akt/NF-κB-Inflammatory signaling pathways, as well As inhibition of COX-2 activity. J. Agric. Food Chem. 66 (23), 5790–5801. doi:10.1021/acs.jafc.8b00437
Xu, J., Wang, H., Ding, K., Zhang, L., Wang, C., Li, T., et al. (2014). Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic. Biol. Med. 71, 186–195. doi:10.1016/j.freeradbiomed.2014.03.009
Xu, X., Gao, W., Cheng, S., Yin, D., Li, F., Wu, Y., et al. (2017). Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J. Neuroinflammation 14 (1), 167. doi:10.1186/s12974-017-0934-2
Yang, L., Wu, H., Yang, F., Li, P., Huang, Y., Zhang, X., et al. (2021). Identification of candidate genes and pathways in dexmedetomidine-induced neuroprotection in rats using RNA sequencing and bioinformatics analysis. Ann. Palliat. Med. 10 (1), 372–384. doi:10.21037/apm-20-2346
Yang, S. J., Shao, G. F., Chen, J. L., and Gong, J. (2018). The NLRP3 inflammasome: An important driver of neuroinflammation in hemorrhagic stroke. Cell. Mol. Neurobiol. 38 (3), 595–603. doi:10.1007/s10571-017-0526-9
Yin, D., Zhou, S., Xu, X., Gao, W., Li, F., Ma, Y., et al. (2018). Dexmedetomidine attenuated early brain injury in rats with subarachnoid haemorrhage by suppressing the inflammatory response: The TLR4/NF-κB pathway and the NLRP3 inflammasome may be involved in the mechanism. Brain Res. 1698, 1–10. doi:10.1016/j.brainres.2018.05.040
Youle, R. J., and Strasser, A. (2008). The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9 (1), 47–59. doi:10.1038/nrm2308
Yu, C., Jia, G., Deng, Q., Zhao, H., Chen, X., Liu, G., et al. (2016). The effects of Glucagon-like peptide-2 on the tight junction and barrier function in IPEC-J2 cells through phosphatidylinositol 3-kinase-Protein kinase B-mammalian target of rapamycin signaling pathway. Asian-Australas. J. Anim. Sci. 29 (5), 731–738. doi:10.5713/ajas.15.0415
Zhang, H., Wang, J., Huang, J., Shi, T., Ma, X., Luo, X., et al. (2018a). Inhibiting Jumoji domain containing protein 3 (JMJD3) prevent neuronal apoptosis from stroke. Exp. Neurol. 308, 132–142. doi:10.1016/j.expneurol.2018.07.007
Zhang, M. H., Zhou, X. M., Cui, J. Z., Wang, K. J., Feng, Y., and Zhang, H. A. (2018b). Neuroprotective effects of dexmedetomidine on traumatic brain injury: Involvement of neuronal apoptosis and HSP70 expression. Mol. Med. Rep. 17 (6), 8079–8086. doi:10.3892/mmr.2018.8898
Zhang, Q. G., Han, D., Hu, S. Q., Li, C., Yu, C. Z., Wang, R., et al. (2010). Positive modulation of AMPA receptors prevents downregulation of GluR2 expression and activates the Lyn-ERK1/2-CREB signaling in rat brain ischemia. Hippocampus 20 (1), 65–77. doi:10.1002/hipo.20593
Zhang, R., Liu, Y., Yan, K., Chen, L., Chen, X. R., Li, P., et al. (2013). Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J. Neuroinflammation 10, 106. doi:10.1186/1742-2094-10-106
Zhang, X. S., Li, W., Wu, Q., Wu, L. Y., Ye, Z. N., Liu, J. P., et al. (2016). Resveratrol attenuates acute inflammatory injury in experimental subarachnoid hemorrhage in rats via inhibition of TLR4 pathway. Int. J. Mol. Sci. 17 (8), E1331. doi:10.3390/ijms17081331
Zhao, W. Y., Chen, S. B., Wang, J. J., Xu, C., Zhao, M. L., Dong, H. J., et al. (2017). Establishment of an ideal time window model in hypothermic-targeted temperature management after traumatic brain injury in rats. Brain Res. 1669, 141–149. doi:10.1016/j.brainres.2017.06.006
Zhao, Z., Ren, Y., Jiang, H., and Huang, Y. (2021). Dexmedetomidine inhibits the PSD95-NMDA receptor interaction to promote functional recovery following traumatic brain injury. Exp. Ther. Med. 21 (1), 4. doi:10.3892/etm.2020.9436
Zhao, Z., Wang, D., Jia, Y., Tian, Y., Wang, Y., Wei, Y., et al. (2016). Analysis of the association of fluid balance and short-term outcome in traumatic brain injury. J. Neurol. Sci. 364, 12–18. doi:10.1016/j.jns.2016.03.007
Zheng, B., Zhang, S., Ying, Y., Guo, X., Li, H., Xu, L., et al. (2018). Administration of Dexmedetomidine inhibited NLRP3 inflammasome and microglial cell activities in hippocampus of traumatic brain injury rats. Biosci. Rep. 38 (5), BSR20180892. doi:10.1042/bsr20180892
Zheng, V. Z., and Wong, G. K. C. (2017). Neuroinflammation responses after subarachnoid hemorrhage: A review. J. Clin. Neurosci. 42, 7–11. doi:10.1016/j.jocn.2017.02.001
Zhong, L., Zhang, Z. L., Li, X., Liao, C., Mou, P., Wang, T., et al. (2017). TREM2/DAP12 complex regulates inflammatory responses in microglia via the JNK signaling pathway. Front. Aging Neurosci. 9, 204. doi:10.3389/fnagi.2017.00204
Zhou, K., Shi, L., Wang, Y., Chen, S., and Zhang, J. (2016). Recent advances of the NLRP3 inflammasome in central nervous system disorders. J. Immunol. Res. 2016, 9238290. doi:10.1155/2016/9238290
Zhu, H. T., Bian, C., Yuan, J. C., Chu, W. H., Xiang, X., Chen, F., et al. (2014). Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-κB signaling pathway in experimental traumatic brain injury. J. Neuroinflammation 11, 59. doi:10.1186/1742-2094-11-59
Zhu, Y., Li, S., Liu, J., Wen, Q., Yu, J., Yu, L., et al. (2019). Role of JNK signaling pathway in dexmedetomidine post-conditioning-induced reduction of the inflammatory response and autophagy effect of focal cerebral ischemia reperfusion injury in rats. Inflammation 42 (6), 2181–2191. doi:10.1007/s10753-019-01082-2
Akt V-akt murine thymoma viral oncogene homolog
AMPK AMP-activated protein kinase
APAF-1 apoptotic protease activating factor 1
Arg-1 arginase-1
ASC apoptosis-associated speck-like protein containing a CARD
ASK1 apoptosis signal-regulating kinase 1
ATF4 activating transcription factor 4
ATG5 autophagy protein 5
Bax B-cell lymphoma-2 associated X
BBB blood–brain barrier
Bcl-2 B-cell lymphoma-2
BDNF brain-derived neurotrophic factor
CCR2* C-C chemokine receptor type 2
CD40* tumor necrosis factor receptor superfamily member 5
CD86* T-lymphocyte activation antigen CD86
Cdk1 cyclin-dependent kinase 1
CHOP C/EBP-homologous protein
CREB cyclic adenosine monophosphate response element binding protein
Cyt-c cytochrome c
DEX dexmedetomidine
Dram2 DNA-damage regulated autophagy modulator 2
ERK extracellular regulated kinase
GPX glutathione peroxidase
GSDMD gasdermin D
HIF-1α hypoxia-inducible factor-1α
HO-1 heme oxygenase-1
HSP70 heat shock protein 70
I/R ischemia–reperfusion
IL-1β interleukin-1beta
IL-4 interleukin-4
IL-6 interleukin-6
IL-8 interleukin-8
IL-10 interleukin-10
IL-18 interleukin-18
iNOS inducible nitric oxide synthase
IRE1α inositol-requiring enzyme 1α
JAK2 Janus kinase 2
JMJD3 Jumonji domain-containing protein 3 demethylase
JNK c-Jun N-terminal kinase
LC3I/II light chain 3I/II
LPO lipid peroxidation
MCL-1 myeloid cell leukemia-1
MCP-1 monocyte chemotactic protein 1
MDA malondialdehyde
miR-29b microRNA-29b
MMP9 matrix metallopeptidase 9
mTOR mechanistic target of rapamycin
MyD88 myeloid differentiation primary response 88
NF-κB nuclear factor-kappaB
Ngb neuroglobin
NLRP3* NACHT, LRR and PYD domains-containing protein 3
NMDA N-methyl-D-aspartate
nNOS neuronal nitric oxide synthase
NQO-1 nicotinamide adenine dinucleotide phosphate quinone oxidoreductase-1
NR2B N-methyl-D-aspartic acid receptor 2B
Nrf2* nuclear factor erythroid 2-related factor 2
OGD oxygen and glucose deprivation
p-EIF2α phosphorylated eukaryotic initiation factor 2α
PGC-1α proliferator-activated receptor-gamma coactivator 1α
PI3K phosphoinositide 3-kinase
p-PERK phosphorylated protein kinase RNA-like ER kinase
PSD95 postsynaptic density protein 95
PSH paroxysmal sympathetic hyperactivity
ROS reactive oxygen species
SAH subarachnoid hemorrhage
SOD superoxide dismutase
STAT3 signal transducer and activator of transcription 3
TBI traumatic brain injury
TGF-β transforming growth factor-β
TLR4 Toll-like receptor 4
TNF-α tumor necrosis factor alpha
UPR unfolded protein response
ZO-1 zona occludens-1
β-APP β-amyloid precursor protein
Keywords: dexmedetomidine, neuroprotective, inflammatory response, cell apoptosis, blood-brain barrier, cell structure protection, autophagy
Citation: Hu Y, Zhou H, Zhang H, Sui Y, Zhang Z, Zou Y, Li K, Zhao Y, Xie J and Zhang L (2022) The neuroprotective effect of dexmedetomidine and its mechanism. Front. Pharmacol. 13:965661. doi: 10.3389/fphar.2022.965661
Received: 13 June 2022; Accepted: 16 August 2022;
Published: 20 September 2022.
Edited by:
Olumayokun Olajide, University of Huddersfield, United KingdomReviewed by:
Jinn-Rung Kuo, Department of Neurology, Chi Mei Medical Center, TaiwanCopyright © 2022 Hu, Zhou, Zhang, Sui, Zhang, Zou, Li, Zhao, Xie and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lunzhong Zhang, emhhbmdsdW56aG9uZ0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.