 Xinbo Yang
Xinbo Yang Xianrong Xing2
Xianrong Xing2- 1School of Information Science and Engineering, Shandong Normal University, Jinan, China
- 2Department of Pharmacy, Shandong Medical College, Jinan, China
- 3School of Clinical Medicine, Harbin Medical University, Daqing, China
The novel coronavirus disease (COVID-19) caused by SARS-CoV-2 virus spreads rapidly to become a global pandemic. Researchers have been working to develop specific drugs to treat COVID-19. The main protease (Mpro) of SARS-CoV-2 virus plays a pivotal role in mediating viral replication and transcription, which makes it a potential therapeutic drug target against COVID-19. In this study, a virtual drug screening method based on the Mpro structure (Protein Data Bank ID: 6LU7) was proposed, and 8,820 compounds collected from the DrugBank database were used for molecular docking and virtual screening. A data set containing 1,545 drug molecules, derived from compounds with a low binding free energy score in the docking experiment, was established. N-1H-Indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine, ergotamine, antrafenine, dihydroergotamine, and phthalocyanine outperformed the other compounds in binding conformation and binding free energy over the N3 inhibitor in the crystal structure. The bioactivity and ADMET properties of these five compounds were further investigated. These experimental results for five compounds suggested that they were potential therapeutics to be developed for clinical trials. To further verify the results of molecular docking, we also carried out molecular dynamics (MD) simulations on the complexes formed by the five compounds and Mpro. The five complexes showed stable affinity in terms of root mean square distance (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and hydrogen bond. It was further confirmed that the five compounds had potential inhibitory effects on SARS-CoV-2 Mpro.
1 Introduction
From December 2019, the world witnessed an outbreak of an acute respiratory disease (Han et al., 2020; Rothan and Byrareddy, 2020). In the early stages of the disease outbreak, Zhou et al. (2020) obtained the full-length genomic sequences of the virus collected from five patients. These sequences exhibited 79.6% homology with SARS-CoV. In addition, the newly found virus exhibited 96% identity to bat coronavirus at the whole-genome level. The International Committee of Taxonomy of Viruses named the virus “severe acute respiratory syndrome coronavirus 2” (SARS-CoV-2), and the World Health Organization (WHO) announced this new disease as a novel coronavirus disease 2019 (COVID-19) (Anand et al., 2020; Wang et al., 2020). According to data from the WHO, over 526 million confirmed cases and over six million deaths have been recorded by 29 May 2022.
Similar to SARS-CoV, SARS-CoV-2 also belongs to the β-coronavirus class but is more contagious and mutable (Tang B. et al., 2020; Shereen et al., 2020). Vaccination has been widely promoted as an important preventive measure against COVID-19. As on 9 September 2022, the WHO reported that there were 371 COVID-19 vaccine candidates in development, of which 172 have entered clinical trials (COVID-19 vaccine tracker and landscape, 2022, https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines). Among the vaccines in clinical development, the number of types ranked was protein subunit vaccines (32%), RNA vaccines (23%), viral vector (non-replicating) vaccines (13%), inactivated virus vaccines (13%), DNA vaccines (9%), and other types of vaccines. As research on protein subunit vaccines was relatively mature and was the priority vaccine development method, the number of protein subunit vaccines was the largest among COVID-19 vaccines. However, persistent mutations of the virus can affect the vaccine’s preventive effect, especially Omicron, which largely evaded the antibodies elicited by the vaccine (Planas et al., 2022). SARS-CoV-2 comprises a single-stranded positive-sense RNA genome that encodes both structural and non-structural proteins. The non-structural proteins include RNA-dependent RNA polymerase, coronavirus main protease (Mpro, also known as 3C-like protease, 3CLpro), and papain-like protease (PLpro). When the viral genome enters the host cell, the host cell protein translation mechanism translates it into a viral polyprotein, which is then cleaved into effector proteins by the viral proteases Mpro and PLpro (Tang X. et al., 2020; Liu et al., 2020; Zhang et al., 2020). Since Mpro can cleave polyproteins at no less than 11 conserved sites, it plays a vital role in the replication of viral particles (ul Qamar et al., 2020). Therefore, it is an attractive target for the screening of antiviral inhibitors. The high-resolution crystal structure of SARS-CoV-2 Mpro was presented by the Zihe Rao and Haitao Yang’s research team. They also provided a basis for drug screening and design based on the structure of the Mpro (Jin et al., 2020).
The research and development of a new drug is a time-consuming process that requires huge financial investment. In the current global crisis, the repositioning of existing drugs seems to be a potentially useful tool in searching for new therapeutic options (Serafin et al., 2020). Computer-assisted virtual screening provides an inexpensive and rapid alternative to high-throughput screening for drug discovery. Furthermore, virtual screening technology can optimize the selection of potential drugs (de Carvalho Gallo et al., 2018). In the past few decades, virtual screening has played an important role in the discovery of small molecule inhibitors of therapeutic targets. Various ligands and structure-based virtual screening methods have been used to identify small-molecule ligands for proteins of interest (Bharatham et al., 2017; Singh and Jana, 2017; Li et al., 2020). Virtual screening technology has revealed several compound molecules that can inhibit SARS-CoV activity (Wei et al., 2006; Niu et al., 2008; Wang et al., 2017).
In this study, we investigated potential Mpro inhibitors using a docking-based virtual screening approach. We used a variety of screening strategies, such as molecular docking, molecular dynamics (MD) simulations, biological activity, and ADMET prediction. The AutoDock Tools were used to prepare the Mpro receptor model of SARS-CoV-2. A Vina-based molecular docking program was encoded, and Mpro and compounds (from DrugBank, with the 3D structure) were docked. The compounds were sorted based on the combined free energy score. The potential drug compounds with inhibitory effects on Mpro were determined by analyzing the binding mode between the compounds with better scoring results and Mpro. The bioactivity and ADMET properties of the five selected compounds were further explored. Simultaneously, we performed MD simulation experiments on the complexes of five compounds and Mpro. The purpose of this study was to identify potential drug compounds from DrugBank by molecular docking and MD simulations. This method can rapidly predict whether a compound has inhibitory effect on the activity of Mpro based on the physicochemical properties of the compound and the stability of the protein–ligand complex.
2 Materials and methods
2.1 Receptor (SARS-CoV-2 Mpro protein) preparation
SARS-CoV-2 Mpro is a key CoV enzyme, which plays a pivotal role in mediating viral replication and transcription, making it an attractive drug target for treating COVID-19 (Anand et al., 2002; Yang et al., 2003; Jin et al., 2020).
The complex crystal structure of Mpro and the N3 inhibitor (PDB ID: 6LU7) (Jin et al., 2020) was downloaded from the Protein Data Bank (http://www.rcsb.org). Mpro was isolated from the complex crystal structure using PyMOL. The separation process of Mpro and N3 inhibitor is shown in Figure 1. Figure 1A shows the complex crystal structure of Mpro protein with N3, and (c) shows the 3D structure of Mpro.
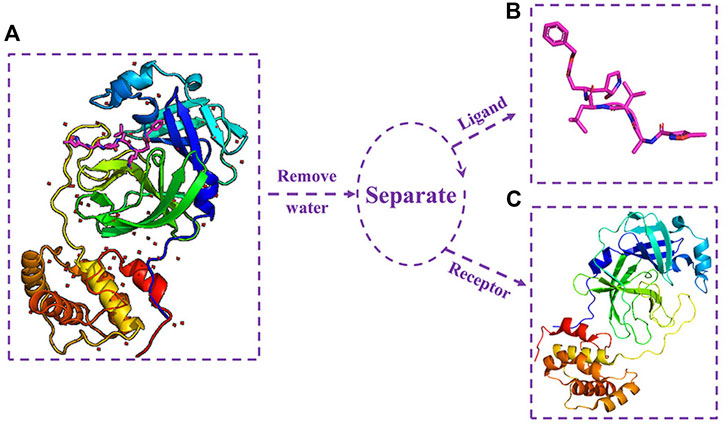
FIGURE 1. (A) Complex crystal structure of Mpro protein with N3; (B) three-dimensional structure of N3 inhibitor; and (C) three-dimensional structure of Mpro protein.
2.2 Ligand data set preparation
For the docking experimental ligand (which is composed of a drug molecule data set and N3 inhibitor), the N3 inhibitor was isolated from the SARS-CoV-2 main protease crystal complex. Figure 1B shows the 3D structure of N3 inhibitor.
The drug molecule data set contained 8,820 molecules with their 3D structures. They were obtained from DrugBank (https://www.drugbank.ca/) in the SDF format (Wishart et al., 2018).
2.3 Pre-processing of receptor and ligands
The docking program requires files stored in the Protein Data Bank, especially in the Partial Charge and Atom Type (PDBQT) format. Mpro standardization involved Gasteiger charges and the addition of polar hydrogen atoms. The conversion of the file format from the Protein Data Bank (PDB) format to PDBQT format was implemented using AutoDock Tools.
Data standardization was performed as a part of the pre-processing. The drug molecules of the ligand data set were first added to polar hydrogen atoms using Open Babel software (O’Boyle et al., 2011). Subsequently, Gasteiger charges were added using Raccoon (Forli et al., 2016) and broken down into 8,820 small molecule files in a PDBQT format.For the N3 inhibitor, Open Babel software was used to add polar hydrogen atoms and Gasteiger charges, followed by converting the format from PDB to PDBQT.
2.4 Molecular docking and screening
Molecular docking was performed using AutoDock Vina and the standardized docking data. In this study, the center of grid box was set to (–10.807, 12.541, 68.917) Å for (center_x, center_y, center_z). Meanwhile, the size of the grid box was defined as (30, 30, 30) Å for (size_x, size_y, size_z). Figure 2A shows the setting information of the grid box, and (b) shows the 3D structure of the grid box for Mpro. To generate as many different binding modes as possible, the num-modes was set to 20 (maximum number of binding modes to generate), and the energy range was set to 6 kcal/mol (maximum energy difference between the best binding mode and the worst one displayed [kcal/mol]). The number of CPUs was set to 20 (CPU = 20), and the explicit random seed was set to 200.
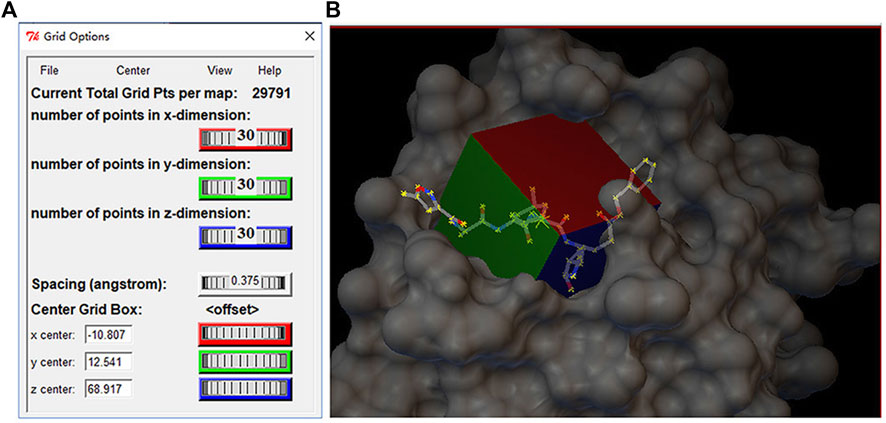
FIGURE 2. (A) Setting information of the grid box and(B) three-dimensional structure of gird box in the Mpro protein.
We encoded a bash script file to implement the docking process. This script file encapsulated the Vina program and the parameters required for the Vina program, including the parameters set in the previous paragraph and the input and output parameters. It could automatically execute the Vina program and perform docking experiments with each ligand molecule and the receptor and finally showed the score of each ligand molecule. It was used to calculate the free energy score of Mpro with different conformations of each ligand.
Screening for the potential drug molecule was achieved by implementing a specific Python script program. The optimal docking score of Mpro with the N3 inhibitor was used as a reference standard for the analysis and screening to establish a candidate drug molecule data set. Figure 3 shows the experimental process of molecular docking and virtual screening.
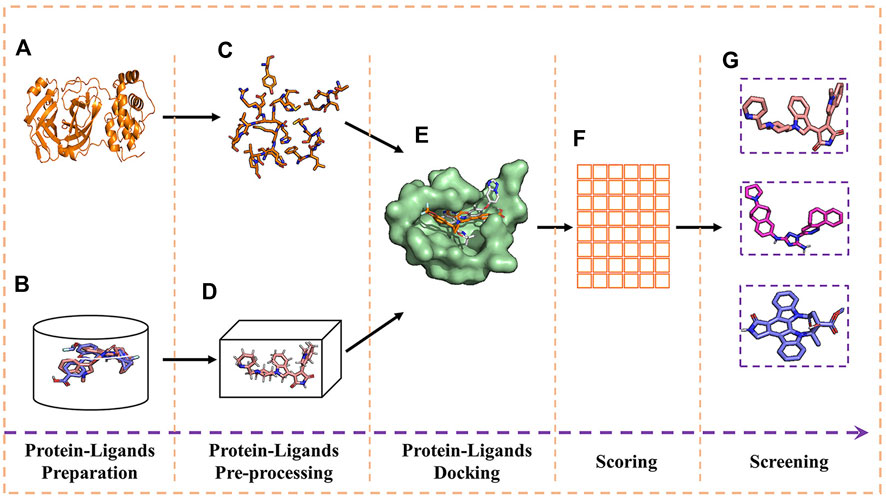
FIGURE 3. Experimental process of molecular docking and virtual screening. (A) Receptor; (B) ligands; (C) receptor pre-processing; (D) ligand pre-processing; (E) docking of receptor and ligands; (F) table of free energy score; and (G) virtual screening for the docking result.
Discovery Studio Visualizer was used to analyze the interactions and types of interactions between compounds and Mpro (docking complexes).
2.5 Molecular dynamics simulation
Molecular dynamics (MD) simulation is an effective method to predict the stability of protein–ligand complexes (Bharadwaj et al., 2021). In this study, we used GROMACS (version: 2022.2, https://www.gromacs.org/) for molecular dynamics simulation. The topology file for Mpro protease was generated using the gmx pdb2gmx command, with the addition of the gromos53a6 force field. The topologies of the five drug molecules were generated using the PRODRG (http://davapc1.bioch.dundee.ac.uk/) server, and their respective topology files with parameters set to chirality: Yes; charge: Full; and EM: NO was also generated.
The simulation system adopted a rectangular solvated box and used gmx grompp for the energy detection and minimization processes. The maximum number of minimization steps to perform was set to 50,000, the energy step size was set to 0.01, and the energy minimization algorithm adopted the steepest descent minimization. At the same time, the system was stabilized by 100 ps NVT and NPT balance. The V-rescale thermal bath coupling algorithm was used in the NVT ensemble, and the Parrinello–Rahman pressure coupling method was used in the NPT ensemble. Finally, we performed 100 ns MD simulations of the equilibrium system at a temperature of 300 K and pressure of 1 bar. The RMSD, RMSF, Rg, number of hydrogen bonds, and protein–ligand interactions of the MD simulation results were recorded and analyzed for further validation of our virtual screening results. The results of molecular dynamics simulations were visualized using qtgrace (version: V26) software.
2.6 Bioactivity and ADMET property prediction
As an alternative to clinical experiments, computer technology was a fast and efficient method to predict the pharmacodynamic properties of compounds (Zaki et al., 2022). We combined the DrugBank database and used Molinspiration Cheminformatics (https://www.molinspiration.com) and admetSAR web service (Yang et al., 2019) to predict the bioactivity and ADMET properties of the five screened compounds, respectively.
3 Results
3.1 Molecular docking and screening
In this study, AutoDock Vina was used to perform the docking of the screened molecules with modeled Mpro. Each ligand generated 20 conformations. These conformations were further subjected to virtual screening evaluation. From the docking search, the conformation with the lowest docked energy was selected as the best conformation. The molecular docking results for AutoDock Vina are presented in Supplementary Table S1.
The binding energy of the interaction of N3 with Mpro was -7.8 kcal/mol. N3 is mainly stabilized by interacting with Mpro through the formation of covalent and hydrogen bonds. The S atom of Cys145 of Mpro forms a covalent bond with C20 of N3. As shown in Figure 4, N3 forms seven hydrogen bonds with Gly143, His164, Glu166, Thr190, Gln189, His163, and Phe140 of Mpro (Tang B. et al., 2020). These results show the active pocket position of Mpro. As a reference, 1,545 compounds, with energy values lower than -7.8 kcal/mol, were obtained. The groups of these compounds in DrugBank are listed in Table 1. According to the order of energy value, the top 30 compounds from the molecular docking analysis are listed in Table 2.
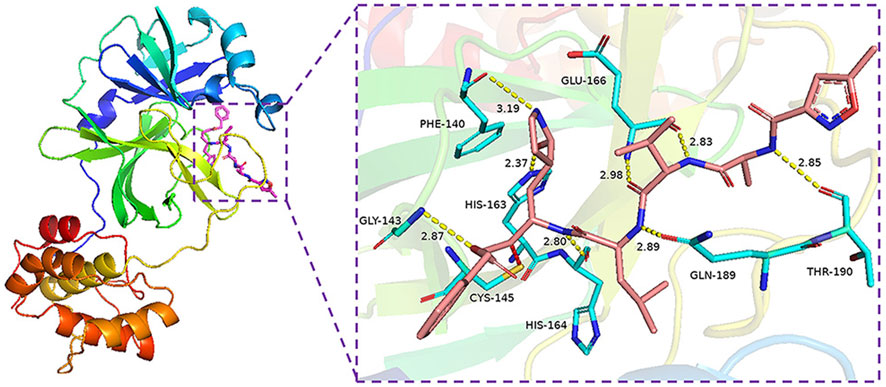
FIGURE 4. Interaction of covalent bonding and hydrogen bonding between Mpro protein and N3.

TABLE 1. Groups of 1,545 compounds in DrugBank.
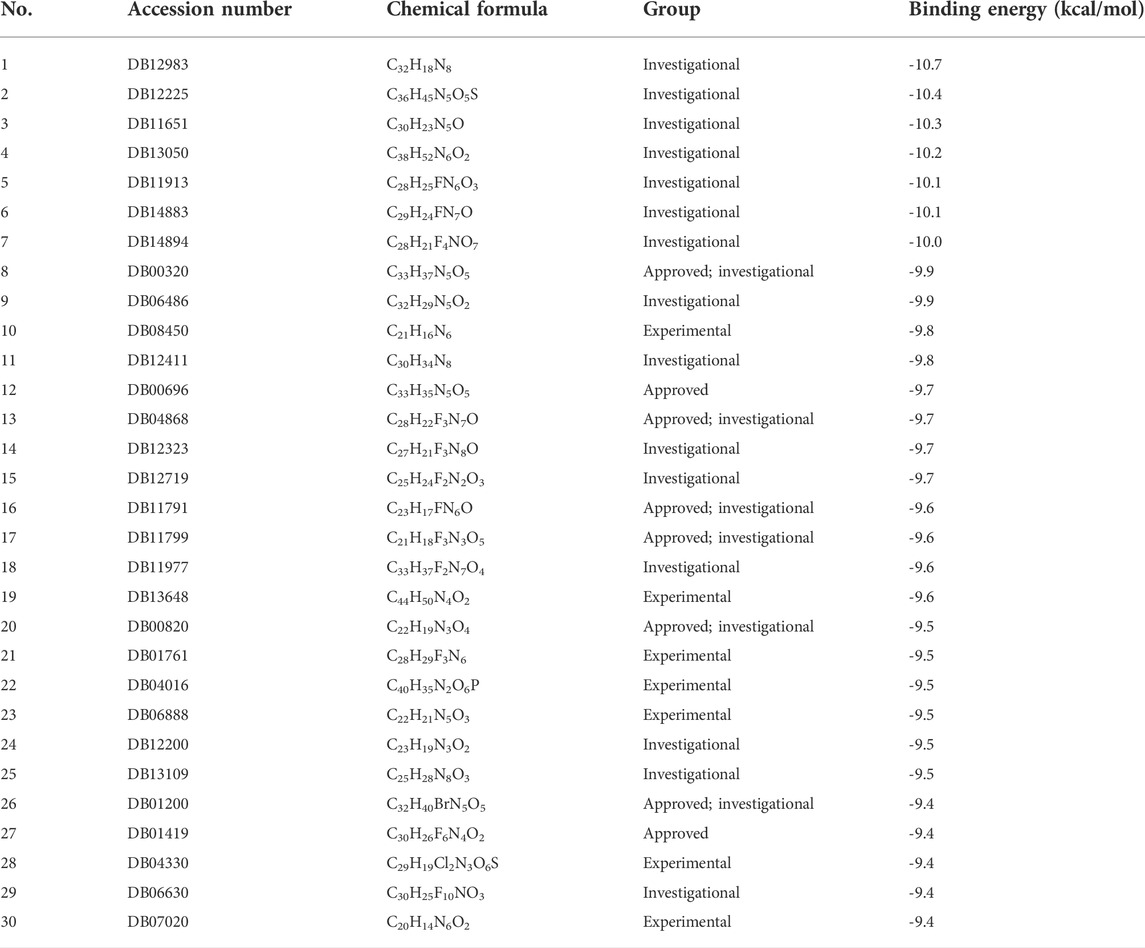
TABLE 2. Top 30 compounds from the docking results.
The top 30 compounds were distributed among four different groups of compounds. Among these, we selected the compounds with the best docking energy. For the “approved” type, we selected two compounds. Next, we analyzed the interactions of the compounds C33H35N5O5 (DB00696, generic name: ergotamine), C30H26F6N4O2 (DB01419, generic name: antrafenine), C33H37N5O5 (DB00320, generic name: dihydroergotamine), C21H16N6 (DB08450, generic name: N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl) quinazolin-4-amine), and C32H18N8 (DB12983, generic name: phthalocyanine) with Mpro.
The conformation diagrams of these compounds are displayed in Figure 5. The interactions of the Mpro protease with each of the five molecules are shown in Figures 6–10. Hydrophobic interactions were visualized using LIGPLOT (Laskowski and Swindells, 2011) Figure 7. Other interactions, including conventional hydrogen bond, carbon–hydrogen bond, pi–donor hydrogen bond, alkyl, pi–alkyl, halogen (fluorine), and pi–pi t-shaped, were visualized using Discovery Studio Visualizer. The interactions of residues with their respective ligands are shown in Supplementary Table S2.
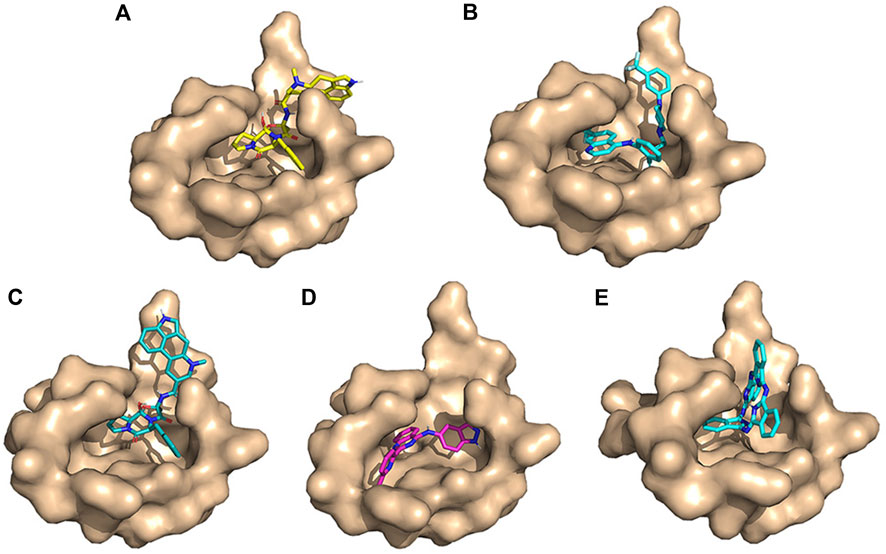
FIGURE 5. Conformation diagrams of these compounds. (A) Ergotamine; (B) antrafenine; (C) dihydroergotamine; (D) N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine; and (E) phthalocyanine.
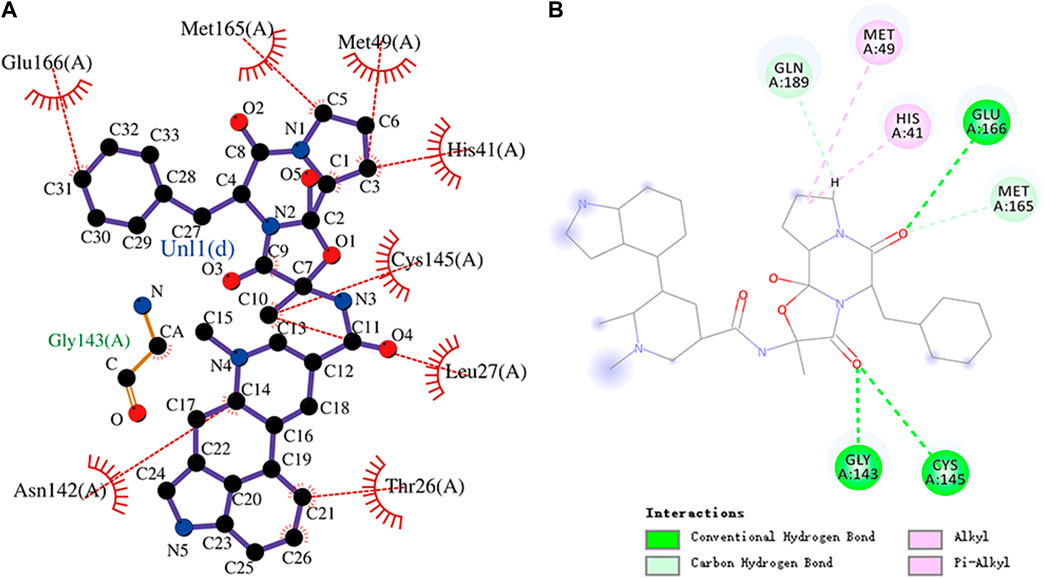
FIGURE 6. (A) Hydrophobic interaction between Mpro and ergotamine and (B) two-dimensional plot of ergotamine interaction with the amino acid residues.
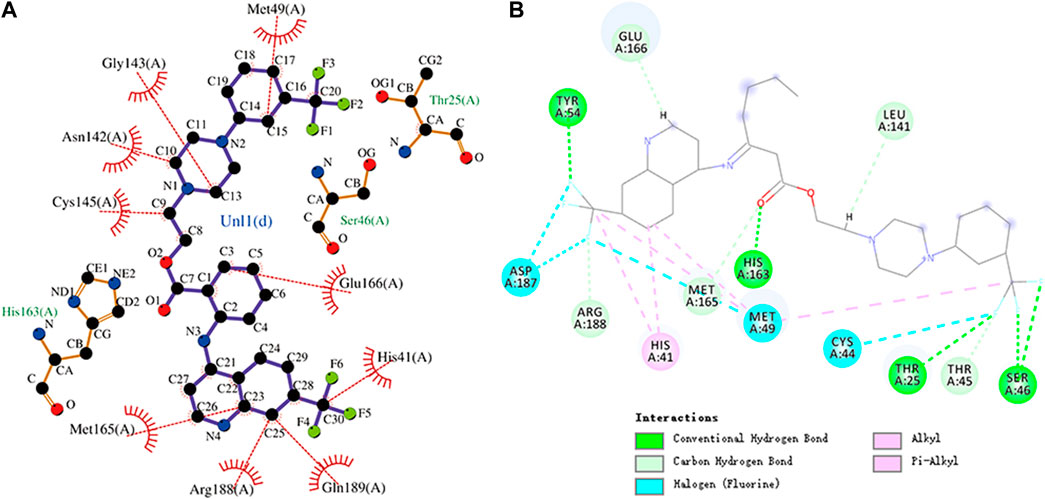
FIGURE 7. (A) Hydrophobic interaction between Mpro and antrafenine and (B) two-dimensional plot of antrafenine interaction with the amino acid residues.
3.2 Molecular dynamics simulation
We performed 100 ns MD simulations for each of the five compounds and N3 inhibitors in complex with Mpro. As shown in Figure 8, complex N3–Mpro trajectory stabilized around 20 ns, N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine–Mpro stabilized around 25 ns, phthalocyanine–Mpro stabilized around 35 ns, antrafenine–Mpro stabilized around 25 ns, ergotamine–Mpro stabilized around 25 ns, and dihydroergotamine–Mpro stabilized around 25 ns The results showed that the RMSD of the five complexes in the MD trajectory interval (35–50 ns) fluctuated from 2.47 to 3.59 Å for dihydroergotamine Figure 9, 2.71–3.71 Å for ergotamine, 2.43–4.75 Å for phthalocyanine, 2.99–4.56 Å for antrafenine, 2.25–3.40 Å for N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine, and 1.91–3.33 Å for the N3 inhibitor and Mpro complex. The smaller the RMSD value, the smaller the fluctuation of the complex structure. Compared with N3 inhibitors, the RMSD of the five molecule–Mpro complexes have little difference Figure 10.
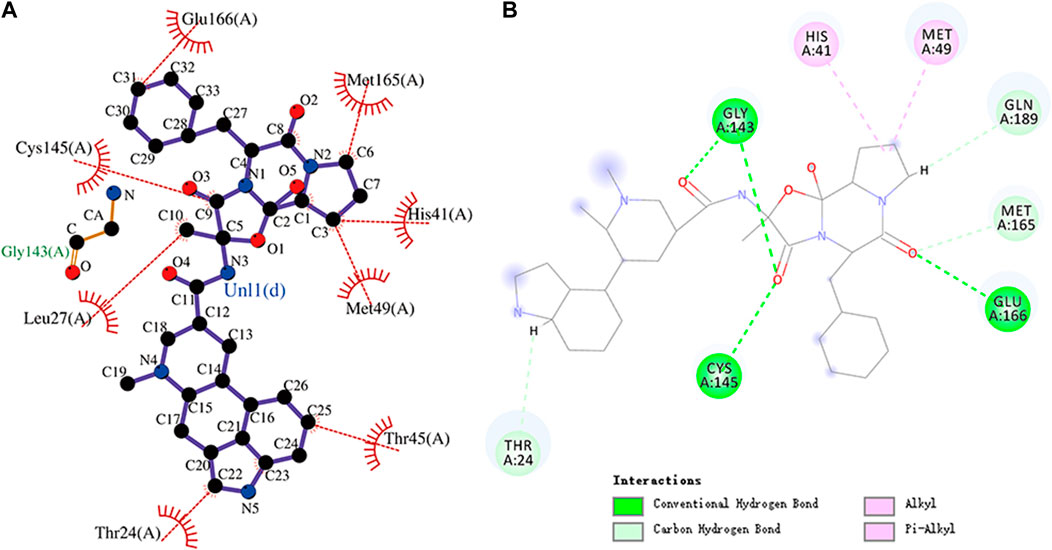
FIGURE 8. (A) Hydrophobic interaction between Mpro and dihydroergotamine and (B) two-dimensional plot of dihydroergotamine interaction with the amino acid residues.
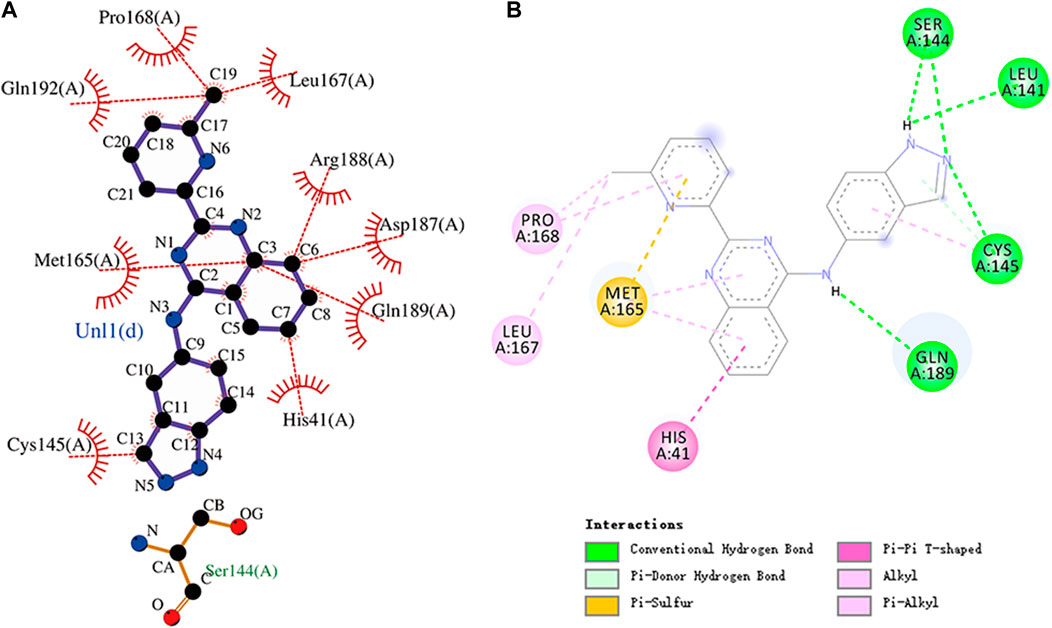
FIGURE 9. (A) Hydrophobic interaction between Mpro and N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine and (B) two-dimensional plot of N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine interaction with the amino acid residues.
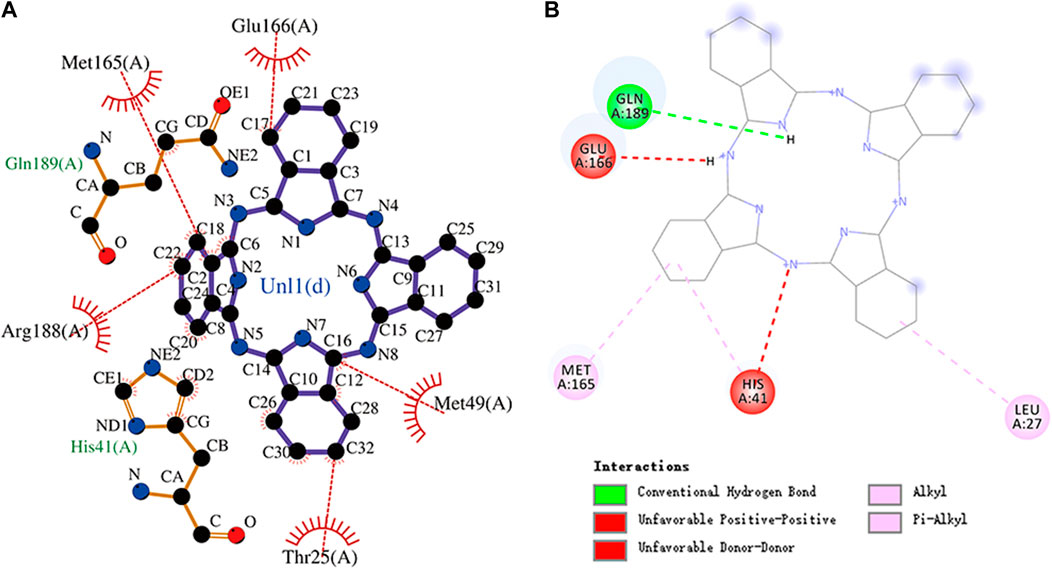
FIGURE 10. (A) Hydrophobic interaction between Mpro and phthalocyanine and (B) two-dimensional plot of phthalocyanine interaction with the amino acid residues.
Rg is an important indicator for evaluating the compactness of the docking architecture. The smaller the cyclotron radius, the better the compactness Figure 11, and hence, the more stable the protein structure. The Rg results of the five molecules and N3 inhibitors in the complex with Mpro are shown in Figure 12. The average Rg of the N3–Mpro complex was about 22.5 Å. The average Rg of the five complexes showed little difference and was lower than that of the N3–Mpro complex except for ergotamine–Mpro complex. The average Rg of the drug dihydroergotamine–Mpro, phthalocyanine–Mpro, N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl) quinazolin-4-amine–Mpro, and antrafenine–Mpro complexes were all about 22 Å. The average Rg of the ergotamine–Mpro complex was about the highest (22.8 Å). This shows that the results of the Rg analysis are consistent with the results of the RMSD trajectory analysis.
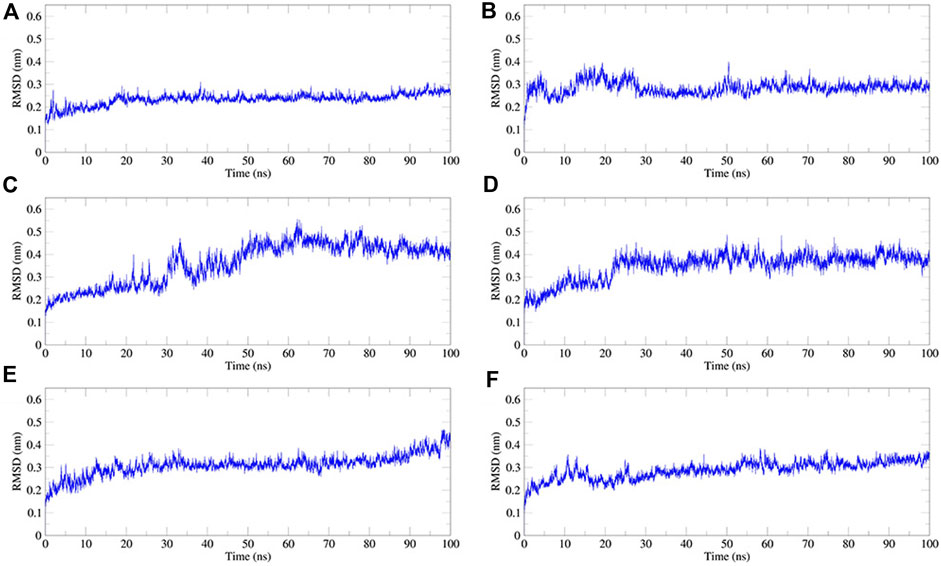
FIGURE 11. Plot of root mean square deviation (RMSD) values, during 100 ns MD simulation of compound–Mpro complexes. (A) N3–Mpro RMSD; (B) N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine–Mpro RMSD; (C) phthalocyanine–Mpro RMSD; (D) antrafenine–-Mpro RMSD (E) ergotamine–Mpro RMSD; and (F) dihydroergotamine–Mpro RMSD.
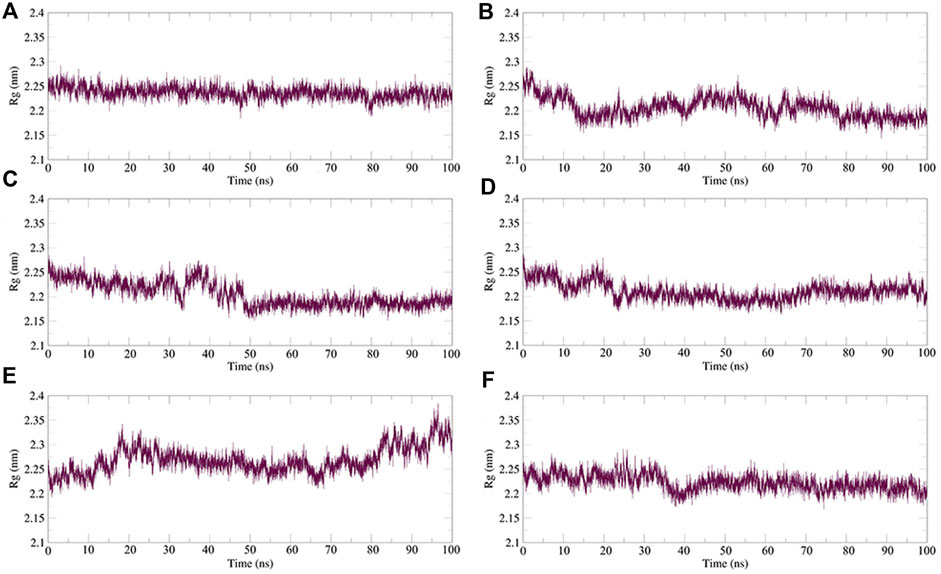
FIGURE 12. Plot of radius of gyration (Rg) values, during 100 ns MD simulation of compound–Mpro complexes. (A) N3–Mpro Rg; (B) N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine–Mpro Rg; (C) phthalocyanine–Mpro Rg; (D) antrafenine–Mpro Rg; (E) ergotamine–Mpro Rg; and (F) dihydroergotamine–Mpro Rg.
The RMSF can be used to observe how individual amino acids fluctuate during the simulation. It is possible to compare the effects of different small-molecule ligands on the spatial structural fluctuations of proteins by calculating the RMSF value. The smaller the value of RMSF, the smaller the disturbance of the small-molecule ligand to the protein, and therefore the stronger the stability of the complex. We calculated the RMSF value for each of the five small molecules and N3 inhibitors bound to Mpro. The calculated values are shown in Figure 13. The RMSF results showed that the average RMSF value of Mpro bound to dihydroergotamine was 1.75 Å, which indicates less fluctuation in the complex structure. However, the residues Met49 (7.07 Å), Tyr54 (3.67 Å), Arg188 (2.30 Å), and Thr24 (2.30 Å) showed a slight fluctuation in the dihydroergotamine–Mpro complex during the simulation. Thus, from the perspective of RMSF, the stability order of the complex formed with the main protease is dihydroergotamine, antrafenine, ergotamine, N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl) quinazolin-4-amine, and phthalocyanine.
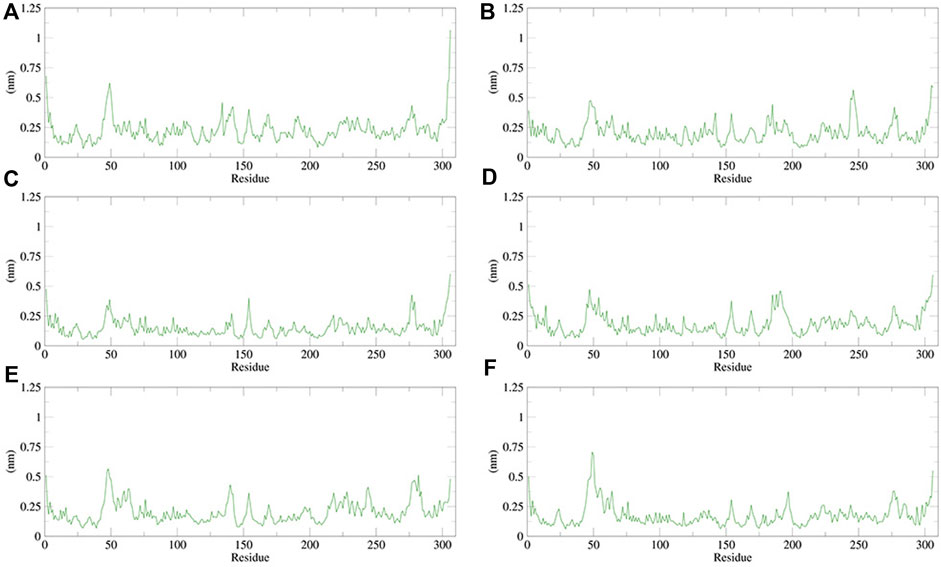
FIGURE 13. Plot of root mean square fluctuations (RMSF) values, during 100 ns MD simulation of compound–Mpro complexes. (A) N3–Mpro RMSF; (B) N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine–Mpro RMSF; (C) phthalocyanine–Mpro RMSF; (D) antrafenine–Mpro RMSF; (E) ergotamine–Mpro RMSF; and (F) dihydroergotamine–Mpro RMSF.
Hydrogen bonds between the ligand and key residues of the main protease were investigated using 100 ns MD simulations as shown in Figure 14. During the 100 ns simulation, there were multiple hydrogen bonds between the five compounds and Mpro. The results confirmed that the five compounds in the MD system had a strong inhibitory effect on Mpro, and there was a good binding effect between the compounds and Mpro in the pocket of Mpro.
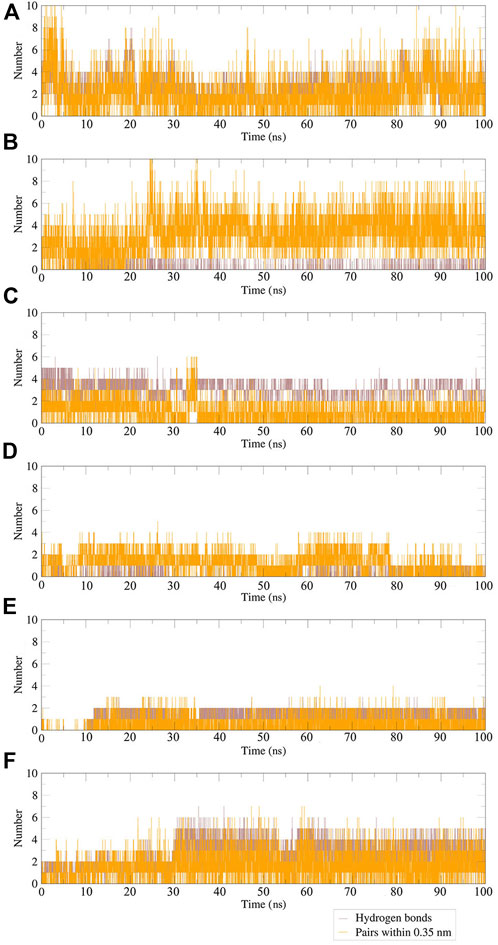
FIGURE 14. Plot of hydrogen bonds in compound–Mpro complexes during 100 ns MD simulation. (A) N3–Mpro; (B) N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine–Mpro; (C) antrafenine–Mpro; (D) ergotamine–Mpro; (E) dihydroergotamine–Mpro; (F) phthalocyanine–Mpro.
3.3 Pharmacodynamic properties
The results of bioactivity and ADMET are shown in Supplementary Tables S3, S4, respectively.
Molinspiration Cheminformatics can predict the GPCR ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor, and enzyme inhibitor values of compounds to evaluate their biological activities. According to reports, a bioactivity score of -5.0–0.0 is considered moderately active, and a score of ≥0 is considered active (Mokhnache et al., 2019; Rahman et al., 2021). From the predicted results, it can be concluded that N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine (score 0.38) is an active enzyme inhibitor, and the other four compounds can be approximately considered active enzyme inhibitors.
The admetSAR can predict ADMET for pharmacodynamic studies of five compounds in the host. In Supplementary Table S4, parameters such as molecular weight, water solubility (logS), human intestinal absorption, blood–brain barrier, Caco-2 permeable, human oral bioavailability, and toxicity of the compounds were listed. The results showed that the solubility value of antrafenine was slightly lower than -4, while the values of other compounds were higher than -4. This indicated that the solubility of the five compounds was suitable (Rahman et al., 2021). None of the five compounds were carcinogenic. But it should be noted that phthalocyanine and N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine were shown to have AMES toxicity. Regarding drug-likeliness, only N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine met the requirements of the five rules, and other compounds were larger than the ideal molecular weight of 500. By comparison, it was concluded that the drug candidate order of the five compounds was ergotamine, dihydroergotamine, antrafenine, N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine, and phthalocyanine.
4 Discussion
The docking of small-molecule compounds to receptor binding sites and the estimation of the binding affinity of the complex are important components of the structure-based drug design process. AutoDock Vina is an open-source program for drug discovery, molecular docking, and virtual screening, which significantly improves the average accuracy of the binding mode predictions (Herowati and Widodo, 2014; Xiang et al., 2015).
Ergotamine is an α-1 selective adrenergic agonist that is commonly used in the treatment of migraine disorders. The binding energy of ergotamine and Mpro was -9.7 kcal/mol. Ergotamine forms hydrogen bonds with residues Gly143, Cys145, and Glu166, respectively. Glu166, Met165, Met49, His41, Cys145, Leu27, Thr26, and Asn142 residues and the hydrophobic groups of ergotamine can engage through hydrophobic interactions. Ergotamine has an alkyl interaction with residue Met49 and a pi–alkyl interaction with residue His41. The molecular docking representation of ergotamine with Mpro is shown in Figure 6.
The interaction energy between antrafenine and Mpro was -9.4 kcal/mol. Antrafenine forms hydrogen bonds with residues Thr25, Ser46, Tyr54, and His163, respectively. Met49, Glu166, His41, Gln189, Arg188, Met165, Cys145, Asn142, and Gly143 residues and antrafenine can engage through hydrophobic interactions. There were also alkyl, pi–alkyl, and halogen (fluorine) interactions between antrafenine and residues. The molecular docking representation of antrafenine with Mpro is shown in. Moreover, antrafenine is a piperazine derivative drug, which exhibits analgesic and anti-inflammatory effects similar to naproxen.
The interaction energy between dihydroergotamine and Mpro was -9.9 kcal/mol. Dihydroergotamine forms hydrogen bonds with residues Gly143, Cys145, and Glu166, respectively. Moreover, dihydroergotamine is stabilized by the interaction with Mpro through hydrophobic interactions, involving Glu166, Leu27, Cys145, Thr24, Thr45, Met49, His41, and Met165 residues. There were also carbon–hydrogen bond, alkyl, and pi–alkyl interactions between dihydroergotamine and residues. The molecular docking representation of dihydroergotamine with Mpro is shown in .
N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine is an experimental drug molecule. The binding energy of N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine and Mpro was -9.8 kcal/mol. N-1H-indazol-5-yl-2-(6-methylpyridin -2-yl)quinazolin-4-amine forms hydrogen bonds with residues Leu141, Ser144, Cys145, and Gln189, respectively. Gln192, Pro168, and Leu167 residues and the methyl of N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine can engage through hydrophobic interactions. Furthermore, Arg188, Asp187, Met165, Gln189, His41, and Cys145 residues can form hydrophobic interactions with N-1H-indazol-5-yl-2-(6-methylpyridin -2-yl)quinazolin-4-amine. There were also pi–donor hydrogen bond, alkyl, pi–alkyl, halogen (fluorine), and pi–pi T-shaped interactions between N-1H-indazol-5-yl-2-(6 -methylpyridin-2-yl)quinazolin-4-amine and residues. The molecular docking representation of N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl)quinazolin-4-amine with Mpro is shown in.
Phthalocyanine is an 18-electron large conjugated system compound comprising four isoindole units. The center of the conjugated ring structure has a large cavity that can accommodate metal ions (such as iron, cobalt, and nickel). Phthalocyanine has the lowest binding energy value of -10.7 kcal/mol. Glu166, Met165, Arg188, His41, Met49, and Thr25 residues and the isoindole ring of phthalocyanine can engage through hydrophobic interactions. The carbonyl group of Gln189 (hydrogen bond acceptor) forms a hydrogen bond with the NH group of acting phthalocyanine (hydrogen bond donor). Phthalocyanine also has alkyl and pi–alkyl interactions with residues. However, it should be noted that phthalocyanine has unfavorable interactions with residues His41 and Glu166, respectively. The molecular docking representation of phthalocyanine with Mpro is shown in.
5 Conclusion
Molecular docking and molecular dynamics simulations have been widely used in drug screening and drug design. In this study, we present several exciting findings about SARS-CoV-2 Mpro. The compounds analyzed in this study can be used as potential inhibitors of SARS-CoV-2 Mpro: Ergotamine is an approved medication for the treatment of migraine disorders, and antrafenine is used as an anti-inflammatory and analgesic agent for the relief of mild-to-moderate pain. Furthermore, we have uncovered dihydroergotamine, N-1H-indazol-5-yl-2-(6-methylpyridin-2-yl) quinazolin-4-amine, and phthalocyanine, which may be developed as potential treatments against SARS-CoV-2 infections. Structural optimization and clinical trials are needed for these compounds to become strong drug candidates. At present, no biological experiments have been carried out in this study. However, through high-throughput molecular docking and molecular dynamics simulations, it was confirmed that these five compounds can form stable conformational structures with Mpro and have potential inhibitory effects on SARS-CoV-2. At the same time, this study provides research ideas and helps for drug designing and drug reusing for the treatment of SARS-CoV-2.
Data availability statement
Publicly available datasets were analyzed in this study. These data can be found at: the complex crystal structure of Mpro protein with an N3 inhibitor (PDB ID: 6lu7) was downloaded from the Protein Data Bank (http://www.rcsb.org). The drug molecule data set contains 8,820 molecules, with 3D structures, which is in the SDF format obtained from DrugBank (https://www.drugbank.ca/).
Author contributions
XY and YZ contributed to the conception and design of the study. YL and XY contributed to the collection and collation of data. XY and XX performed the statistical analysis. XY and XX wrote the first draft of the manuscript. All authors contributed to manuscript revision and read and approved the submitted version.
Funding
This work was supported in part by the National Natural Science Foundation of China (No. 81871508) and the Major Program of Shandong Province Natural Science Foundation (ZR2019ZD04).
Acknowledgments
The authors kindly thank all participants for their contributions to this study. The authors also thank the Protein Data Bank and DrugBank for their data support services.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.962863/full#supplementary-material
References
Anand, K., Karade, S., Sen, S., and Gupta, R. (2020). Sars-cov-2: camazotz’s curse. Med. J. Armed Forces India 76, 136–141. doi:10.1016/j.mjafi.2020.04.008
Anand, K., Palm, G. J., Mesters, J. R., Siddell, S. G., Ziebuhr, J., and Hilgenfeld, R. (2002). Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 21, 3213–3224. doi:10.1093/emboj/cdf327
Bharadwaj, K. K., Sarkar, T., Ghosh, A., Baishya, D., Rabha, B., Panda, M. K., et al. (2021). Macrolactin a as a novel inhibitory agent for sars-cov-2 mpro: Bioinformatics approach. Appl. Biochem. Biotechnol. 193, 3371–3394. doi:10.1007/s12010-021-03608-7
Bharatham, N., Finch, K. E., Min, J., Mayasundari, A., Dyer, M. A., Guy, R. K., et al. (2017). Performance of a docking/molecular dynamics protocol for virtual screening of nutlin-class inhibitors of mdmx. J. Mol. Graph. Model. 74, 54–60. doi:10.1016/j.jmgm.2017.02.014
de Carvalho Gallo, J. C., de Mattos Oliveira, L., Araújo, J. S. C., Santana, I. B., and dos Santos Junior, M. C. (2018). Virtual screening to identify leishmania braziliensis n-myristoyltransferase inhibitors: Pharmacophore models, docking, and molecular dynamics. J. Mol. Model. 24, 260. doi:10.1007/s00894-018-3791-8
Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., and Olson, A. J. (2016). Computational protein–ligand docking and virtual drug screening with the autodock suite. Nat. Protoc. 11, 905–919. doi:10.1038/nprot.2016.051
Han, Q., Lin, Q., Jin, S., and You, L. (2020). Coronavirus 2019-ncov: A brief perspective from the front line. J. Infect. 80, 373–377. doi:10.1016/j.jinf.2020.02.010
Herowati, R., and Widodo, G. P. (2014). Molecular docking studies of chemical constituents of tinospora cordifolia on glycogen phosphorylase. Procedia Chem. 13, 63–68. doi:10.1016/j.proche.2014.12.007
Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., et al. (2020). Structure of mpro from sars-cov-2 and discovery of its inhibitors. Nature 582, 289–293. doi:10.1038/s41586-020-2223-y
Laskowski, R. A., and Swindells, M. B. (2011). Ligplot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model 51 (10), 2778–2786. doi:10.1021/ci200227u
Li, T., Tan, X., Yang, R., Miao, Y., Zhang, M., Xi, Y., et al. (2020). Discovery of novel glyceraldehyde-3-phosphate dehydrogenase inhibitor via docking-based virtual screening. Bioorg. Chem. 96, 103620. doi:10.1016/j.bioorg.2020.103620
Liu, C., Zhou, Q., Li, Y., Garner, L. V., Watkins, S. P., Carter, L. J., et al. (2020). Research and development on therapeutic agents and vaccines for Covid-19 and related human coronavirus diseases. ACS Cent. Sci. 6, 315–331. doi:10.1021/acscentsci.0c00272
Mokhnache, K., Madoui, S., Khither, H., and Charef, N. (2019). Drug-likeness and pharmacokinetics of a bis-phenolic ligand: Evaluations by computational methods. Sch. J. App Med. Sci. 1, 167–173.
Niu, C., Yin, J., Zhang, J., Vederas, J. C., and James, M. N. (2008). Molecular docking identifies the binding of 3-chloropyridine moieties specifically to the s1 pocket of sars-cov mpro. Bioorg. Med. Chem. 16, 293–302. doi:10.1016/j.bmc.2007.09.034
O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011). Open babel: An open chemical toolbox. J. Cheminform. 3, 33–14. doi:10.1186/1758-2946-3-33
Planas, D., Saunders, N., Maes, P., Guivel-Benhassine, F., Planchais, C., Buchrieser, J., et al. (2022). Considerable escape of sars-cov-2 omicron to antibody neutralization. Nature 602, 671–675. doi:10.1038/s41586-021-04389-z
Rahman, F., Tabrez, S., Ali, R., Alqahtani, A. S., Ahmed, M. Z., and Rub, A. (2021). Molecular docking analysis of rutin reveals possible inhibition of sars-cov-2 vital proteins. J. Tradit. Complement. Med. 11, 173–179. doi:10.1016/j.jtcme.2021.01.006
Rothan, H. A., and Byrareddy, S. N. (2020). The epidemiology and pathogenesis of coronavirus disease (Covid-19) outbreak. J. Autoimmun. 109, 102433. doi:10.1016/j.jaut.2020.102433
Serafin, M. B., Bottega, A., Foletto, V. S., da Rosa, T. F., Hörner, A., and Hörner, R. (2020). Drug repositioning is an alternative for the treatment of coronavirus Covid-19. Int. J. Antimicrob. Agents 55, 105969. doi:10.1016/j.ijantimicag.2020.105969
Shereen, M. A., Khan, S., Kazmi, A., Bashir, N., and Siddique, R. (2020). COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 24, 91–98. doi:10.1016/j.jare.2020.03.005
Singh, A., and Jana, N. K. (2017). Discovery of potential zika virus rna polymerase inhibitors by docking-based virtual screening. Comput. Biol. Chem. 71, 144–151. doi:10.1016/j.compbiolchem.2017.10.007
Tang, B., Bragazzi, N. L., Li, Q., Tang, S., Xiao, Y., and Wu, J. (2020a). An updated estimation of the risk of transmission of the novel coronavirus (2019-ncov). Infect. Dis. Model. 5, 248–255. doi:10.1016/j.idm.2020.02.001
Tang, X., Wu, C., Li, X., Song, Y., Yao, X., Wu, X., et al. (2020b). On the origin and continuing evolution of sars-cov-2. Natl. Sci. Rev. 7, 1012–1023. doi:10.1093/nsr/nwaa036
ul Qamar, M. T., Alqahtani, S. M., Alamri, M. A., and Chen, L.-L. (2020). Structural basis of sars-cov-2 3clpro and anti-Covid-19 drug discovery from medicinal plants. J. Pharm. Anal. 10, 313–319. doi:10.1016/j.jpha.2020.03.009
Wang, L., Bao, B.-B., Song, G.-Q., Chen, C., Zhang, X.-M., Lu, W., et al. (2017). Discovery of unsymmetrical aromatic disulfides as novel inhibitors of sars-cov main protease: Chemical synthesis, biological evaluation, molecular docking and 3d-qsar study. Eur. J. Med. Chem. 137, 450–461. doi:10.1016/j.ejmech.2017.05.045
Wang, L., Wang, Y., Ye, D., and Liu, Q. (2020). Review of the 2019 novel coronavirus (sars-cov-2) based on current evidence. Int. J. Antimicrob. Agents 55, 105948. doi:10.1016/j.ijantimicag.2020.105948
Wei, D.-Q., Zhang, R., Du, Q.-S., Gao, W.-N., Li, Y., Gao, H., et al. (2006). Anti-sars drug screening by molecular docking. Amino Acids 31, 73–80. doi:10.1007/s00726-006-0361-7
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). Drugbank 5.0: A major update to the drugbank database for 2018. Nucleic Acids Res. 46, D1074–D1082. doi:10.1093/nar/gkx1037
Xiang, L., Xu, Y., Zhang, Y., Meng, X., and Wang, P. (2015). Virtual screening studies of Chinese medicine coptidis rhizoma as alpha7 nicotinic acetylcholine receptor agonists for treatment of alzheimer’s disease. J. Mol. Struct. 1086, 207–215. doi:10.1016/j.molstruc.2015.01.021
Yang, H., Lou, C., Sun, L., Li, J., Cai, Y., Wang, Z., et al. (2019). Admetsar 2.0: web-service for prediction and optimization of chemical admet properties. Bioinformatics 35, 1067–1069. doi:10.1093/bioinformatics/bty707
Yang, H., Yang, M., Ding, Y., Liu, Y., Lou, Z., Zhou, Z., et al. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 100, 13190–13195. doi:10.1073/pnas.1835675100
Zaki, A. A., Ashour, A., Elhady, S. S., Darwish, K. M., and Al-Karmalawy, A. A. (2022). Calendulaglycoside a showing potential activity against sars-cov-2 main protease: Molecular docking, molecular dynamics, and sar studies. J. Tradit. Complement. Med. 12, 16–34. doi:10.1016/j.jtcme.2021.05.001
Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., et al. (2020). Crystal structure of sars-cov-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412. doi:10.1126/science.abb3405
Keywords: SARS-CoV-2, main protease, molecular docking, virtual screening, drug repurposing, molecular dynamics
Citation: Yang X, Xing X, Liu Y and Zheng Y (2022) Screening of potential inhibitors targeting the main protease structure of SARS-CoV-2 via molecular docking. Front. Pharmacol. 13:962863. doi: 10.3389/fphar.2022.962863
Received: 06 June 2022; Accepted: 12 September 2022;
Published: 05 October 2022.
Edited by:
Mithun Rudrapal, Rasiklal M. Dhariwal Institute of Pharmaceutical Education and Research, IndiaReviewed by:
Ismail Celik, Erciyes University, TurkeyVishma Pratap Sur, Institute of Biotechnology (ASCR), Czechia
Copyright © 2022 Yang, Xing, Liu and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinbo Yang, dWJlaWhhbmdAMTI2LmNvbQ==; Yuanjie Zheng, eWp6aGVuZ0BzZG51LmVkdS5jbg==