 Xuan Jiang1,2
Xuan Jiang1,2 Yongquan Chen
Yongquan Chen Shenglong Zhu
Shenglong Zhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 23 November 2022
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.942061
This article is part of the Research Topic Bioactive Substances-Mediated Targeted Therapy of Cardio-Cerebrovascular Diseases View all 12 articles
Background: The rising prevalence of obesity and its complications is a big challenge for the global public health. Obesity is accompanied by biological dysfunction of skeletal muscle and the development of muscle atrophy. The deep knowledge of key molecular mechanisms underlying myogenic differentiation is crucial for discovering novel targets for the treatment of obesity and obesity-related muscle atrophy. However, no effective target is currently known for obesity-induced skeletal muscle atrophy.
Methods: Transcriptomic analyses were performed to identify genes associated with the regulation of myogenic differentiation and their potential mechanisms of action. C2C12 cells were used to assess the myogenic effect of Apol9a through immunocytochemistry, western blotting, quantitative polymerase chain reaction, RNA interference or overexpression, and lipidomics.
Results: RNA-seq of differentiated and undifferentiated C2C12 cells revealed that Apol9a expression significantly increased following myogenic differentiation and decreased during obesity-induced muscle atrophy. Apol9a silencing in these C2C12 cells suppressed the expression of myogenesis-related genes and reduced the accumulation of intracellular triglycerides. Furthermore, RNA-seq and western blot results suggest that Apol9a regulates myogenic differentiation through the activation of extracellular signal-regulated kinase 1/2 (ERK1/2). This assumption was subsequently confirmed by intervention with PD98059.
Conclusion: In this study, we found that Apol9a regulates myogenic differentiation via the ERK1/2 pathway. These results broaden the putative function of Apol9a during myogenic differentiation and provide a promising therapeutic target for intervention in obesity and obesity-induced muscle atrophy.
Obesity is widely reported to be a potential risk factor for type 2 diabetes mellitus (T2DM), a metabolic disorder typified by chronic hyperinsulinemia, hyperglycemia, and insulin resistance (Astrup and Finer, 2000; Lingvay et al., 2021). Diabetes is reportedly stimulated during obesity because of a failure in appropriate glucose utilization by skeletal muscle, which is the primary target site of insulin-stimulated glucose uptake (DeFronzo and Tripathy, 2009; Lingvay et al., 2021; Mengeste et al., 2021). Obesity-related ectopic fat deposition induces biological dysfunction in skeletal muscle, such as insulin resistance (IR), mitochondrial dysfunction, and inflammation (Wu and Ballantyne, 2017). These processes further exacerbate skeletal muscle loss and physical dysfunction. Maintaining skeletal muscle health is fundamental for general health.
Myogenesis, which includes satellite cell activation, myoblast differentiation, and myotube formation, is responsible for the maintenance of skeletal muscle mass and integrity in general (Schiaffino et al., 2013; Sousa-Victor et al., 2022). Myogenesis dysregulation causes muscle wasting diseases such as sarcopenia and cachexia, increasing the risk of frailty, morbidity, and lethality (Nishikawa et al., 2021a; Wiedmer et al., 2021). Numerous studies have discovered that muscle wasting is caused by various factors that inhibit myogenesis, such as oxidative stress, mitochondrial malfunction, and aging (Bonaldo and Sandri, 2013; McCarthy and Berg, 2021). Obesity has been shown to aggravate the negative effects of muscle loss, leading to sarcopenic obesity (Roh and Choi, 2020; Nishikawa et al., 2021b). Additionally, obesity reduces the capacity of skeletal muscle differentiation and may impact muscle plasticity and function (Brown et al., 2015). Furthermore, another study showed that the low level of inflammation induced by obesity downregulates myogenesis (Du et al., 2010). A deep understanding of myogenesis is crucial for comprehending the mechanisms regulating skeletal muscle mass under pathological disorders. The myogenic differentiation process comprises multiple pathways, and numerous potential targets in myogenesis regulation remain unknown.
To discover the potential targets in the process of myogenic differentiation, we applied RNA sequencing to identify unknown genes that regulate myogenesis in a classical mouse cell model. As a result, we identified apolipoprotein L9a (Apol9a) as a key moderator regulating myogenic differentiation. Apol9a is a member of the murine apolipoprotein L gene family, and interferon-inducible mouse Apol9a is secreted by macrophages to promote epithelial cell proliferation (Kreit et al., 2015; Sun et al., 2015). However, the biological function of Apol9a in skeletal muscles remains unclear. Experiments have indicated that Apol9a knockdown impairs skeletal muscle differentiation, principally by activating the ERK1/2 pathway. Activated ERK1/2 signaling promotes skeletal muscle cell proliferation but negatively regulates myogenic differentiation (Jones et al., 2001) and modulates nuclear factor of activated T cells c1 (NFATc1) (Chen et al., 2017). Studies have indicated that ERK1/2 activation is elevated in atrophic and damaged skeletal muscles (Penna et al., 2010). Our results highlight the potential importance of Apol9a in myogenic differentiation, suggesting that modulation of Apol9a-ERK activity may help reduce the risk of obesity-related muscle atrophy.
Six-week-old male C57BL/6 mice were purchased from Jicui Yaokang Biological Technology Co., (Nanjing, China). All mice were maintained under standard conditions of 22°C ± 2°C, 50%–60% relative humidity, and alternate dark/light cycles. Mice were fed a high-fat diet (HFD) (60 kcal%, D12492, Research Diets) for 10 weeks to induce obesity. Control mice were fed a basal diet (10 kcal%, D12450B, Research Diets). Both groups of mice were sacrificed at 16 weeks of age, and their gastrocnemius muscles were dissected for experimental analyses. Skeletal muscle triglyceride (TG) contents were measured using a Triglyceride Quantification Kit (Nanjing Jiancheng, China). All experiments in this study were authorized by the Ethics Committee of Jiangnan University [NO: JN. No20211030c0700625 (426)].
C2C12 mouse myoblast cells were purchased from the ATCC (CRL-1772, United States). The C2C12 cell line was cultured in growth medium (GM) supplemented with basic DMEM (11995–065, Gibco, United States), 10% FBS (1009–141, Gibco, United States), and 1% penicillin plus 1% streptomycin (SV30010, Hyclone, United States) at 37°C and 5% CO2. GM was replaced with differentiation medium (DM) containing DMEM with 2% horse serum (HS, 16050–130, Gibco, United States) and incubated for 4 days in order to induce C2C12 cell differentiation when C2C12 cells reached 90% confluency.
To explore the effect of Apol9a on C2C12 cells, small interfering RNA (siRNA, GenePharma, Shanghai, China) was used to knockdown intracellular Apol9a, and the overexpression plasmid (GENEWIZ) was used to overexpress Apol9a. C2C12 cells were seeded into 6-well plates and cultured for 8 h (equivalent to a cell density of 30%–40%). Next, siRNA and the overexpression plasmid were transfected into cells at a concentration of 50 nM. The jetPRIME® transfection reagent (114–15, Polyplus transfection) was used to transfer siRNAs, the overexpression plasmid, and the empty control plasmid. At 12 h after transfection, the transfection medium was replaced with DM to induce differentiation for 4 days. Specific knockdown of Apol9a was validated using two different siRNAs. The two siRNA sequences for mouse Apol9a was 5′-UUGUAUCCAAGGCCAAGUUGUTT-3′and 5′-AGCCCUUGAGCAGCACAUGAATT-3′. A random siRNA sequence (A06001, GenePharma, shanghai, China) was applied as control. To inhibit the ERK1/2 signaling pathway, C2C12 myoblasts were treated with 50 μm PD98059 (HY-12028, Med Chem Express) in DM for 4 d (after 8 h transfection). Dimethyl sulfoxide (DMSO, D8418, Sigma-Aldrich) was used as a solvent control.
Myotube diameter measurements were obtained using ImageJ software. The diameter was measured at the widest region of each myotube, and the mean diameter size was compared between conditions.
Total RNA was isolated from both C2C12 myoblasts and skeletal muscle tissue by using the RNA extraction kit (K101, JN. BIOTOOLS, Wuxi, China). cDNA was synthesized using the BTS I 1st Strand cDNA Synthesis Kit (K102, JN. BIOTOOLS, Wuxi, China), and 1 μg of total RNA to be used for quantitative real-time PCR (qPCR) was reverse transcribed. qPCR was performed using the Power SYBR Green Master Mix kit (4367659, Invitrogen, United States) in a CFX 96TMRealTime PCR Detection System (Bio-Rad, United States). The amplification conditions for qPCR were as follows: 94°C for 5 min, followed by 45 cycles of 94°C for 15 s and 45 s at 59°C. The fold change of gene expression was analyzed according to the 2−ΔΔCT method. β-actin was utilized as the internal control for normalization. The sequences of all primers used in this research are presented in Supplementary Table S1.
Total proteins were extracted from the cell lysate and determined using the Quick BCA Protein Assay Kit (SYW3-1, Solarbio, China). The lysate proteins were resolved through SDS-PAGE with a 10% gel, transferred to PVDF membranes (IPVH00005, Merck Millipore), blocked, and incubated with primary antibodies against Apol9a (AC-15–1076, Ango Biotechnology, 1:1000); myosin heavy chain (MyHC) (MF-20, 1:1000, Developmental Studies Hybridoma Bank); myogenin (MyoG) (ab 1835, Abcam, 1:2000); myoblast determination protein (MyoD) (18943-1-AP, 1:1000, Proteintech); Myf5 (BD-PT2930, 1:1000, Biodragon); phospho-Erk1/2 (4370, 1:1000, CST); Erk1/2 (4695, 1:1000, CST); phospho-JNK (4668, 1:1000, CST); JNK (9252, 1:1000, CST); phospho-p38 (4511, 1:1000, CST); p38 (9212, 1:1000, CST); and β-actin (ab8227, 1:2000, Abcam). After all membranes were washed, they were treated with specific secondary antibodies (AS014, AS003, Abclonal, China). Specific protein bands were detected using an ECL kit (WBKLS0100, Merck Millipore). The images were observed using the Bio-Rad ChemiDoc MP Imaging System, and then, image bands were quantified through densitometry by using ImageJ plus software. The expression level of each target protein was normalized to that of β-actin, which was used as an internal control.
C2C12 myoblasts were fixed with paraformaldehyde (4%) for 20 min and treated with 1% Triton X-100 for 5 min. The fixed cell samples were incubated with blocking buffer (A8010, Solarbio, China) in phosphate-buffered saline (PBS) and then incubated with anti-MyHC (MF20, 1:50, DSHB) antibody at 4°C overnight. After washing the samples with PBS, the cell samples were treated with a 1:100 dilution of secondary antibody, which was derived from goat anti-mouse IgG (AS001, Abclonal) and conjugated with FITC, for 2 h. Cell nuclei were counterstained with DAPI staining solution (C1002, Beyotime, China). Finally, the stained cells were viewed using a fluorescence microscope (Leica DM2500, Leica Microsystems), and the images were captured. The fusion index was calculated as the percentage of nuclei in fused myotubes out of the total nuclei. The number of nuclei in each image was evaluated using the ImageJ plus software.
Fatty acid extraction and methyl-esterification were performed as previously described (Zhu et al., 2022a). Briefly, the recovered fatty acid methyl esters were examined through GC-MS (QP2010 ultra mass spectrometer, GC 2010 plus, Thermo Scientific). The electron energy was fixed at 70 eV, and the temperature programming profile was as follows: kept for 5 min at 60°C, up to 120°C in increments of 10°C/min, maintained for 5 min at 120°C, up to 190°C in increments of 5°C/min, kept for 7 min at 190°C, up to 230°C in increments of 2°C/min, and kept for 10 min at 230°C. A 5-MS column (Restek, United States) was used to resolve all samples. The detector and ion source were run at 240°C and 230°C, respectively. The peaks obtained were identified by comparing their retention times with those of known standards (Sigma). Pentadecanoic acid (C15:0) was used as the internal standard, and each sample was normalized to total cellular protein concentration, as measured using the Quick BCA protein assay kit (SYW3-1, Solarbio, China).
Cell samples were produced in the manner previously described (Zhu et al., 2022b). Lipidomic analysis was conducted using LC-MS (QExactive Plus Orbitrap mass spectrometer, Thermo Scientific). Acetonitrile:MilliQ water (6:4 v/v) and isopropanol:acetonitrile (9:1 v/v) were used as solvents A and B, respectively; both solvents contained 10 mm ammonium acetate. Column chromatography was performed using the Waters ACQUITY UPLC CSHTM C18 column. The gradient profile was as follows: 32%–100% solvent B over 24 min, back to 32% solvent B and 6 min before the next injection, and equilibrate the column. LipidSearch program v4.1.16 (Thermo Scientific) was used to identify the type of lipids. A pool of all lipid extracts was prepared and used for quality control. SIMCA-P (Sweden) was used to import the raw MS data, and an orthogonal projection was performed for latent structures-discriminant analysis (OPLS-DA). The resulting heatmap plots were drawn using R software.
RNA sequencing (RNA-seq) was conducted as previously described (Zhang et al., 2021; Zhu et al., 2022c). Briefly, total RNA of C2C12 myoblasts transfected with si-NC or si-Apol9a was isolated using the RNA extraction kit (K101, JN. BIOTOOLS, China). Library construction and paired-end sequencing were performed by GENEWIZ Biotech (Suzhou, China). Raw data files were matched to the mouse reference genome by using STAR software (http://www.code.google.com/p/rna-star/). For each sample, fold change was estimated using the fragments per kilobase per million reads values, and differential expression analysis was performed using the DESeq2 package. The standard of fold change ≥1.5 and p < 0.05 were defined to screen differentially expressed genes (DEGs). Gene ontology (GO) analysis was conducted to perform the functional enrichment analysis of specific DEGs using Metascape (metascape.org). All RNA-seq data files have been deposited in the SRA database (Accession: PRJNA845924).
Statistical analysis was performed using SPSS software version 22.0 and GraphPad Prism 8.0. The results are shown as mean ± SEM from at least three independent experiments. Differences between groups were evaluated using an unpaired Student’s t-test (between two groups) or one-way ANOVA (between multiple groups). *p < 0.05, **p < 0.01, and ***p < 0.001 were considered statistically significant.
To evaluate the effects of obesity on skeletal muscle differentiation, we used the HFD-induced obesity mouse model (Feraco et al., 2021). Body weight and TG content in skeletal muscle tissue were considerably higher in HFD mice than in normal diet (ND) mice (Figures 1A,C). Meanwhile, the HFD mice exhibited a lower gastrocnemius muscle mass (Figure 1B) than the ND mice. Furthermore, a significant decrease in the mRNA expression levels of myogenesis-related genes (including Myf5, MyoD, MyoG, and MyHC) was observed in the gastrocnemius muscle from HFD mice (Figure 1D). Based on Pearson’s correlation analysis, we investigated the correlations between body weight, gastrocnemius muscle mass, expression levels of myogenesis-related genes, and TG levels in HFD mice (Figure 1E). A notable negative correlation was observed between TG content and gastrocnemius muscle mass. Together, these results demonstrate that muscle mass and myogenesis capacity were decreased in obese mice.
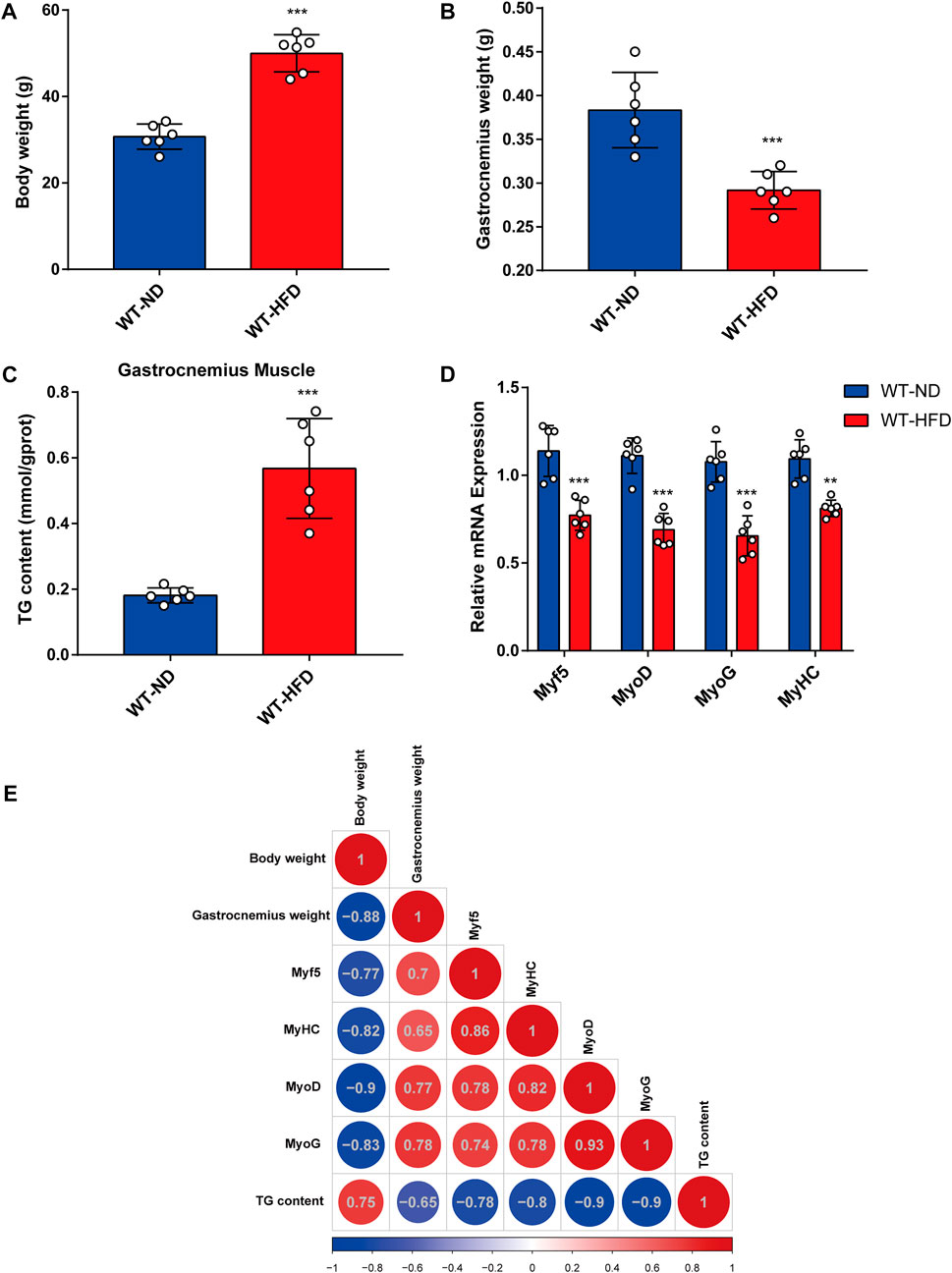
FIGURE 1. Correlation between obesity and myogenic differentiation. Six-week-old male mice were divided into two groups, each fed with normal chow and high-fat diet for 8 weeks. (A) Body weight comparison. (B) Gastrocnemius muscle weight (GW) comparison. (C) Comparison of triglyceride (TG) levels in gastrocnemius muscles from the ND-fed and HFD-fed mice as assessed using the Triglyceride Quantification Kit. (D) The mRNA expression levels of myogenesis-related genes (Myf5, MyoD, MyoG, and MyHC) in the ND-fed and HFD-fed mice as measured through quantitative real-time PCR (qPCR). (E) Correlation heatmap (Pearson correlation) of body weight, TG level, GW, and mRNA levels of myogenesis-related genes. Positive and negative correlations are shown by red and blue colors, respectively. Date are shown as mean ± SEM (n = 6 for each group). *p < 0.05, **p < 0.01, and ***p < 0.001 vs. WT-ND.
To determine the critical genes involved in myogenic differentiation, we compared the transcriptome of differentiated C2C12 cells (an established myoblast cell model) with that of undifferentiated C2C12 cells (Yaffe and Saxel, 1977). After myogenic differentiation, C2C12 cells became fused and the myotube diameter increased (Figure 2A). As demonstrated in the volcano plot (Figure 2B), 1353 genes were upregulated following myogenic differentiation and 2731 genes were downregulated (fold change ≥1.5). The upregulated genes were subjected to GO analysis, and the top 10 enriched GO terms were all related to muscle differentiation (Figure 2C). Among the upregulated gene sets, we observed that many apolipoprotein family genes were significantly upregulated following myogenic differentiation (Figure 2D). The most significant change involved Apol9a, an understudied cytoplasmic, interferon-inducible gene with antiviral activity (Kreit et al., 2015). To our knowledge, a role for Apol9a in myogenic differentiation has not been previously reported. Using qPCR, we confirmed that Apol9a mRNA expression levels increased following myogenesis (Figure 2E). However, Apol9a mRNA expression levels decreased in obese mice (Figure 2F). Together, these results indicate that Apol9a may play a role in myogenic differentiation.
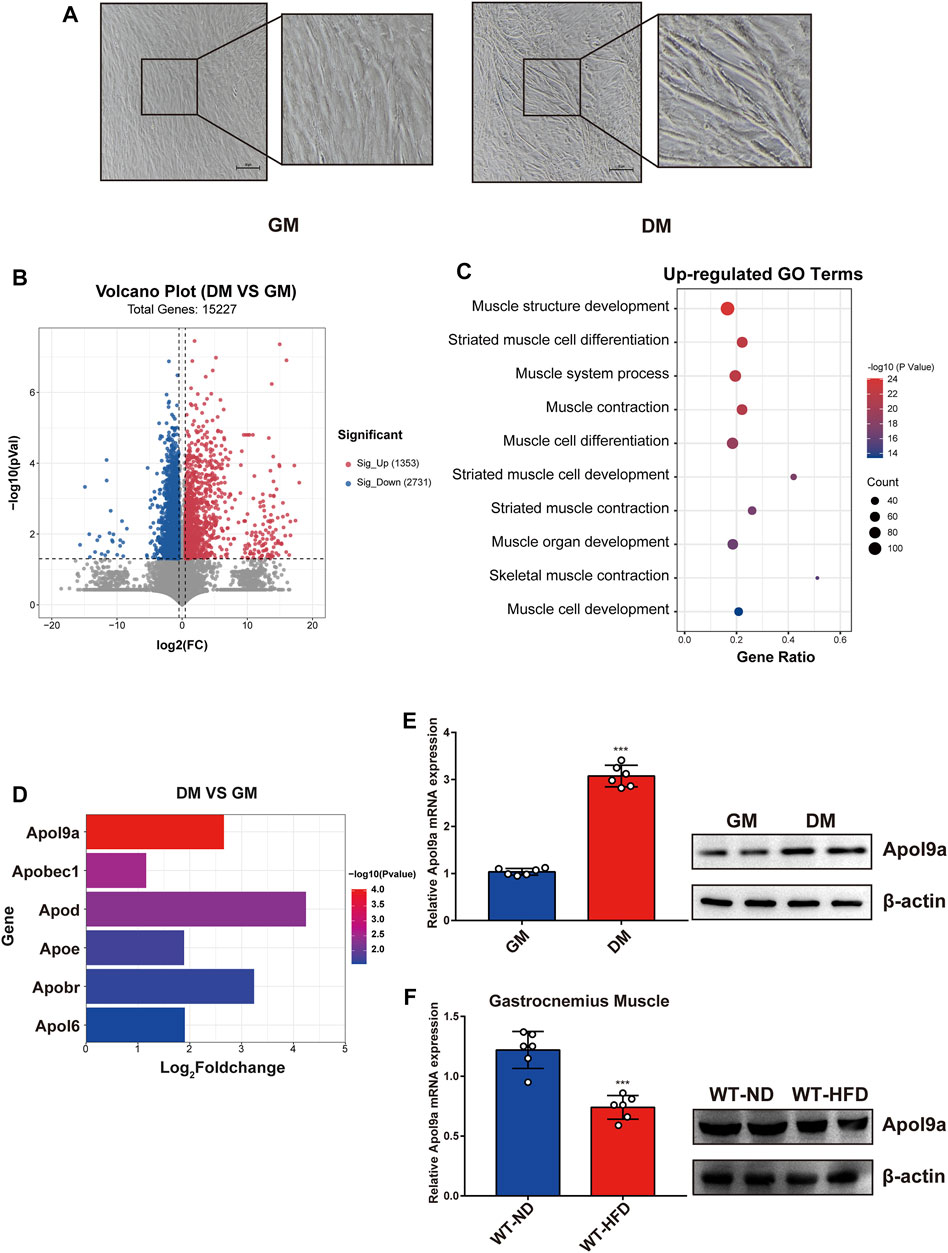
FIGURE 2. Apol9a is increased in myogenic differentiation and decreased in obese mice. RNA sequencing (RNA-seq) data of C2C12 cells with or without myogenic induction were collected. (A) Representative images from C2C12 cells in growth medium (GM) and differentiation medium (DM). Scale bar, 100 μm. (B) Volcano plot showing differentially expressed genes (DEGs) in differentiated or undifferentiated C2C12 cells. (C) Ten most upregulated GO terms during C2C12 differentiation according to the RNA-seq data. (D) Expression profiles of apolipoprotein family genes during C2C12 differentiation. (E) The relative mRNA expression (left) and protein levels (right) of Apol9a during C2C12 differentiation were measured using qPCR and western blotting. (F) The relative mRNA expression (left) and protein levels (right) of Apol9a in obese skeletal muscle were measured through qPCR and western blotting. The data are presented as mean ± SEM from at least three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
To further investigate the function of Apol9a in the progress of myogenesis, siRNA was used to interfere with Apol9a expression. C2C12 cells were transfected with Apol9a siRNA or NC siRNA, and then, the GM was replaced with DM for 4 days. Apol9a mRNA expression levels were reduced approximately 60% in Apol9a siRNA-transfected cells compared with the si-NC control group (Figure 3A). Myogenic differentiation is known to start with the induction of a specific set of transcription genes known as myogenic regulatory factors (MRFs), which include Myf5, MyoD, and MyoG (Chargé and Rudnicki, 2004; Ciciliot and Schiaffino, 2010). Comparison with the si-NC control group, the protein and mRNA expression levels of several myogenic differentiation markers, including Myf5, MyoD, MyoG, and MyHC, were remarkably decreased in differentiated C2C12 cells after si-Apol9a transfection (Figures 3B–D). To validate the effect of Apol9a deficiency on myotube formation, we evaluated the expression of MyHC protein through immunofluorescence in differentiated C2C12 cells and a quantitative analysis of the fusion index. As shown in Figure 3E, Apol9a knockdown cells displayed a different morphology and appeared to have shorter myofibers compared with the control group. Moreover, the fusion index was significantly decreased after Apol9a silencing (Figure 3E). Furthermore, we explored the effect of Apol9a overexpression on myogenic differentiation. Compared with the empty vector group, the Apol9a mRNA expression level was markedly increased in pcDNA3.1-Apol9a-transfected cells. Compared with the empty vector group, the Apol9a mRNA expression level was significantly increased in pcDNA3.1-Apol9a-transfected cells (Figure 4A), and the myotube diameter was significantly longer after Apol9a overexpression (Figure 4B). Western blot analysis showed that Apol9a overexpression significantly upregulated MyoG and MyHC protein levels (Figures 4C,D). Together, these results demonstrate that Apol9a knockdown inhibited myogenesis and Apol9a overexpression promoted myogenic differentiation.
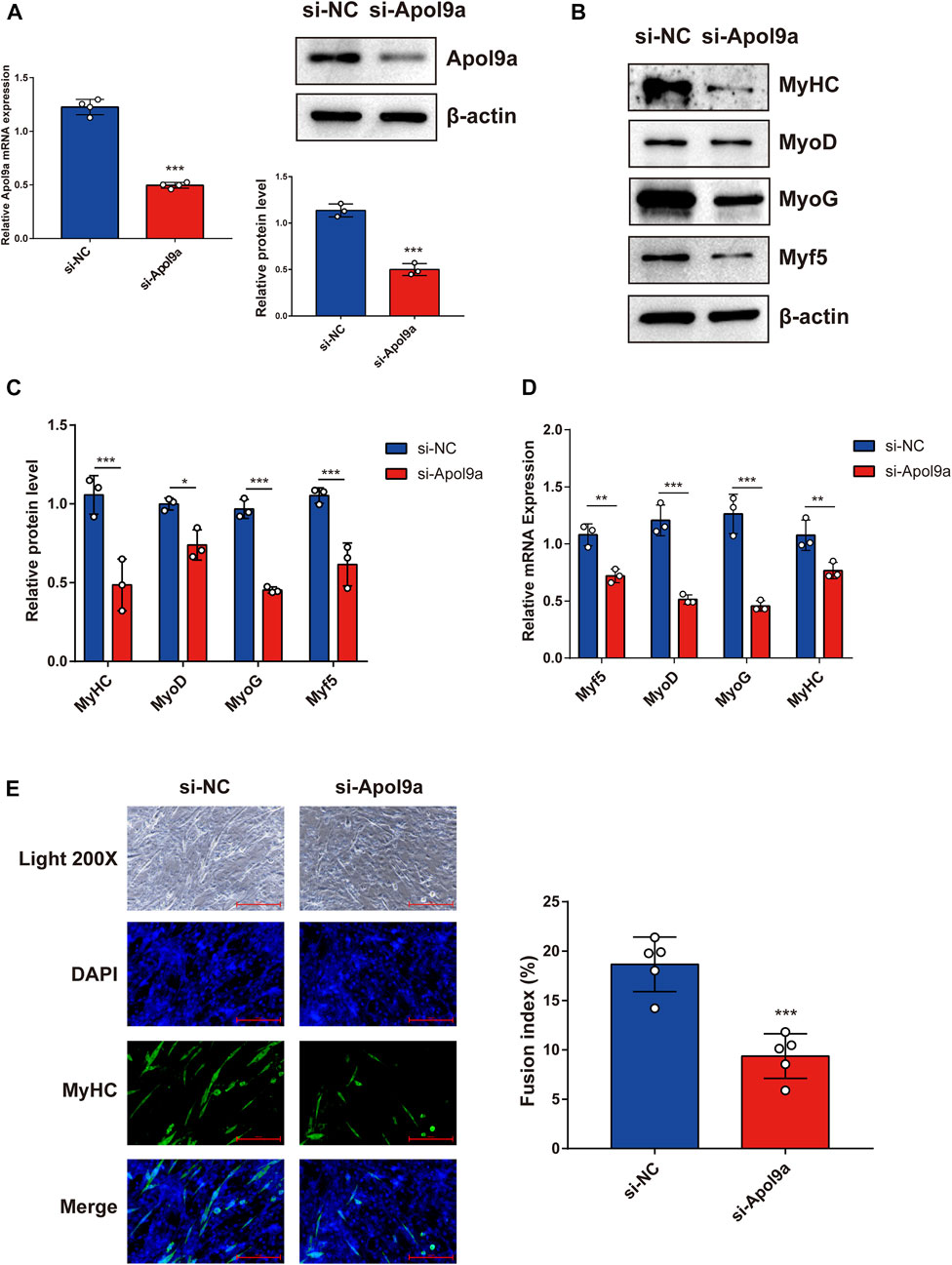
FIGURE 3. Apol9a knockdown inhibits C2C12 myogenesis. C2C12 cells were induced to differentiate after si-NC or si-Apol9a transfection for 4 days. (A) qPCR and western blot validation of the efficiency of Apol9a knockdown in C2C12 cells. (B) Protein expression levels of MyHC, MyoD, Myf5, and MyoG were estimated through western blot analysis after si-NC and si-Apol9a transfection. (C) Gray scale analysis of western blot results of (B) determined using ImageJ software. (D) qPCR was performed to assess the mRNA expression levels of several myogenic differentiation genes (Myf5, MyoD, MyoG, and MyHC). (E) Myotube formation can be observed in fluorescence images of DAPI (blue) and MyHC antibody-stained (green) myotubes of C2C12 cells transfected si-NC or si-Apol9a. Scale bar, 100 μm. The fusion index (%) was quantified and is shown on the right. The data are expressed as mean ± SEM of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs. si-NC control.
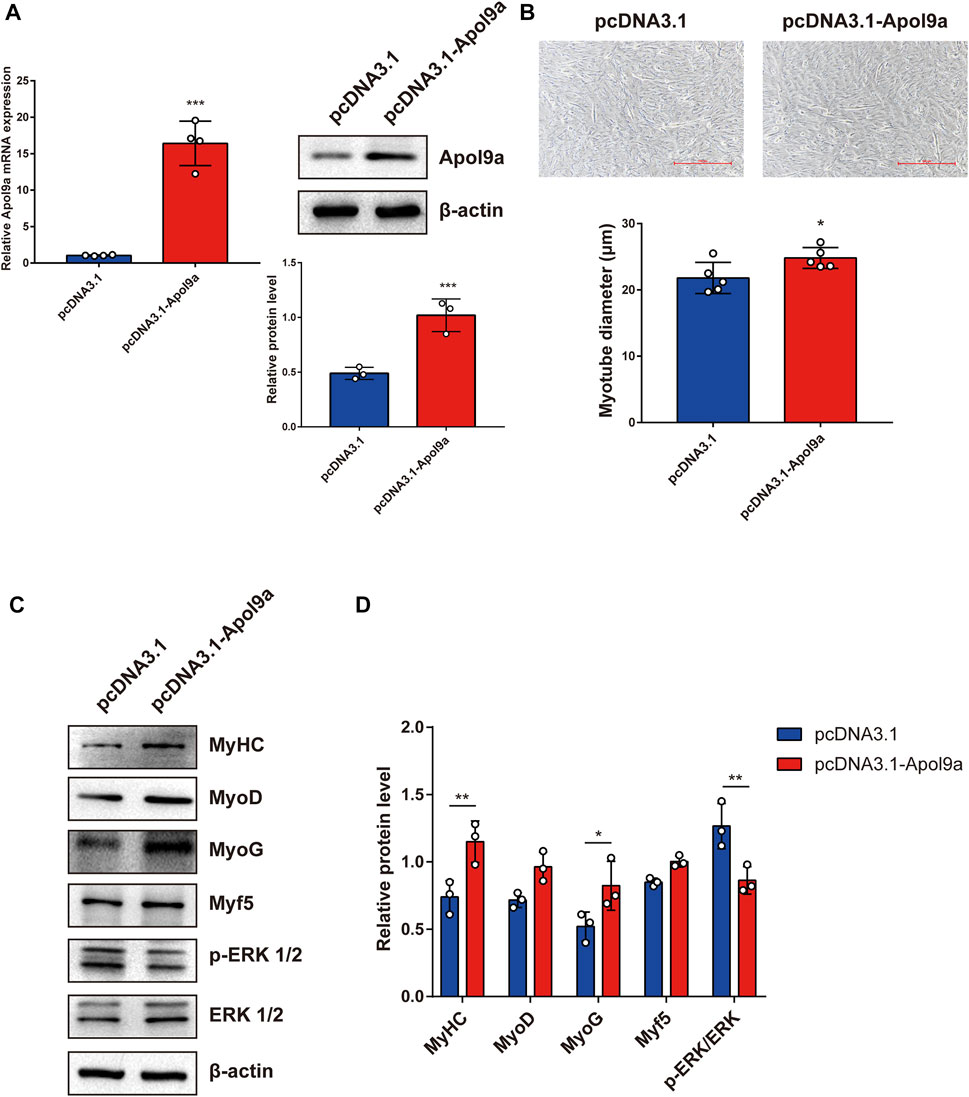
FIGURE 4. Apol9a overexpression promotes myogenic differentiation. C2C12 cells were induced to differentiate after pcDNA3.1 or pcDNA3.1-Apol9a transfection for 4 days. (A) qPCR and western blot validation of the efficiency of Apol9a overexpression in C2C12 cells. (B) Morphology of C2C12 cells treated with pcDNA3.1 or pcDNA3.1-Apol9a. The results of quantification of average myotube diameter are shown in the right. (C) Protein expression levels of MyHC, MyoD, Myf5, MyoG, p-ERK 1/2, ERK 1/2, and β-actin were estimated through western blot analysis after transfection. (D) Gray scale analysis of western blot results of (C) determined using ImageJ software. The data are expressed as mean ± SEM from at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs. pcDNA3.1 group.
Apol9a is an apolipoprotein family member, and previous research has suggested that Apol9a has a role in lipid transport (Arvind and Rangarajan, 2016). To identify changes in the types of fatty acids and changes in lipid composition induced by Apol9a knockdown after cell differentiation, the types and contents of fatty acid in C2C12 cells with or without Apol9a knockdown after myogenic differentiation were determined through GC-MS and LC-MS, respectively. After Apol9a knockdown, the levels of various fatty acids (C16:0, C18:0, C18:2, and C22:6) significantly decreased (in comparison with the si-NC transfected cells) (Figure 5A). To further analyze changes in lipid composition, we applied LC-MS-based lipidomics to identify the specific types of lipids whose levels decreased in Apol9a knockdown cells. Moreover, orthogonal partial least squares-discriminant analysis (OPLS-DA) indicated different clustering of lipids from NC and Apol9a-silenced groups (Figure 5B). While the intensity levels of C16:0 fatty acid were mainly decreased in phosphatidylcholine (PC) and phosphatidylethanolamine (PE), those of C18:0 fatty acid were reduced in TG and PE (Figure 5D). In addition, a significant decrease was observed in the intensity levels of C18:2 fatty acid, especially in PC and TG (Figures 5C,E). Elevated skeletal muscle TG is reported to be related to insulin resistance in obesity and impaired skeletal muscle differentiation (Wu and Ballantyne, 2017), (Eum et al., 2020). Furthermore, our heatmap analysis revealed that many TG species (including C18:0 and C18:2 fatty acids) demonstrate a remarkable decrease (VIP >1 and p < 0.05) in C2C12 cells after Apol9a knockdown and myogenic differentiation (Figure 5F). Together, these results provide evidence that inhibition of myogenesis following Apol9a knockdown may be related to changes in lipid composition, especially changes in TG species.
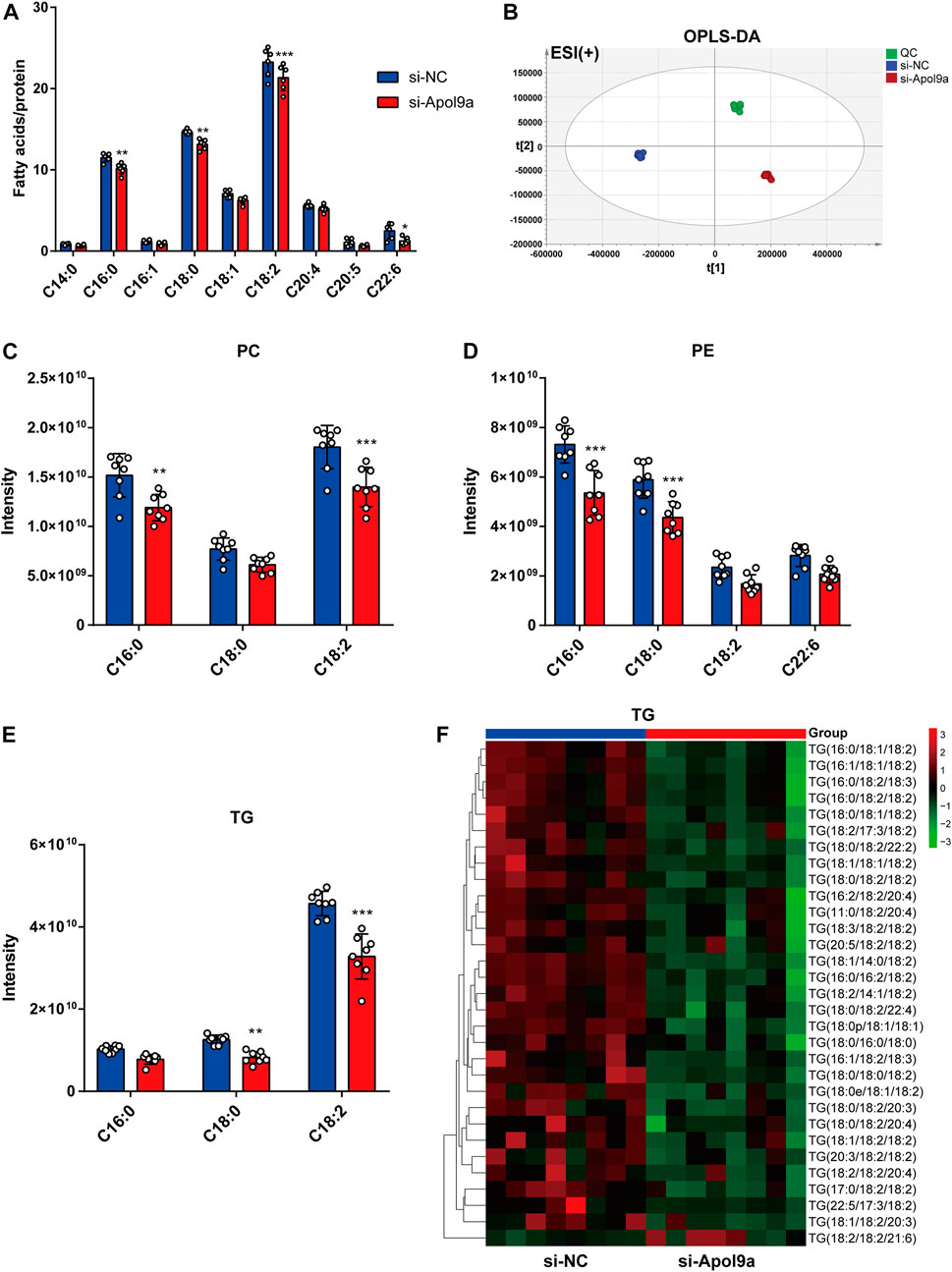
FIGURE 5. Fatty acid methyl ester analysis and lipidomic analysis. After myogenic differentiation for 4 days, C2C12 cells transfected with si-NC or si-Apol9a were collected, and cellular fatty acid and lipidomic analyses were performed. (A) Cellular fatty acid profiles were detected through GC-MS following C2C12 differentiation. (B) orthogonal partial least squares discriminant analysis (OPLS-DA) of lipid profiles in positive ion modes from C2C12 cells with or without Apol9a knockdown. (C) The composition of phosphatidylcholine (PC) (C16:0, C18:0, C18:2) among all lipids. (D) The composition of phosphatidylethanolamine (PE) (C16:0, C18:0, C18:2, C22:6) among all lipids. (E) The composition of triacylglycerol (TG) (C16:0, C18:0, C18:2) among all lipids. (F) Heatmap showing the differential lipid composition of TGs (C18:0, C18:2). The data are presented as mean ± SEM (n = 8, each group). *p < 0.05, **p < 0.01, ***p < 0.001 vs. si-NC group.
To elucidate the underlying molecular mechanisms of Apol9a regulation of myogenesis, RNA sequencing was used to evaluate gene expression differences in C2C12 cells with or without Apol9a knockdown. Principal component analysis (PCA) was then performed to discriminate changes in expression between experimental groups. The PCA results revealed that the two groups could be completely separated based on their transcriptomes (Figure 6A). Based on the criteria of fold change ≥1.5 and p < 0.05, 580 differentially expressed genes (DEGs) between the two groups were identified, including 282 downregulated genes and 298 upregulated genes (Figure 6B). Then, GO analysis was applied to find the potential functions of the upregulated and downregulated genes. As shown in Figure 6C, significantly enriched functional terms, including “tissue homeostasis,” “response to lipoprotein particle,” “negative regulation of cell differentiation,” and “MAPK cascade,” were identified using the upregulated gene set. However, no specifically enriched biological processes were identified using the downregulated gene set (data not shown). The MAPK cascade pathway is essential for the regulation of myogenic differentiation (Jones et al., 2001; Keren et al., 2006; Xie et al., 2018; Boyer et al., 2019). We hypothesized that the ERK1/2 signaling pathway may play a role in the inhibition of myogenic differentiation induced by Apol9a knockdown. This hypothesis was subsequently confirmed by western blotting analyses of the ERK1/2 phosphorylation status and protein levels in C2C12 cells. Thus, while Apol9a knockdown markedly increased ERK1/2 phosphorylation during myogenesis, other MAPKs (JNK and p38) remained unchanged (Figure 6D). Moreover, phosphorylated ERK1/2 protein levels were significantly reduced after C2C12 differentiation (Figure 6E). In summary, our data demonstrate that Apol9a knockdown activates the ERK1/2 signaling pathway during myogenic differentiation.
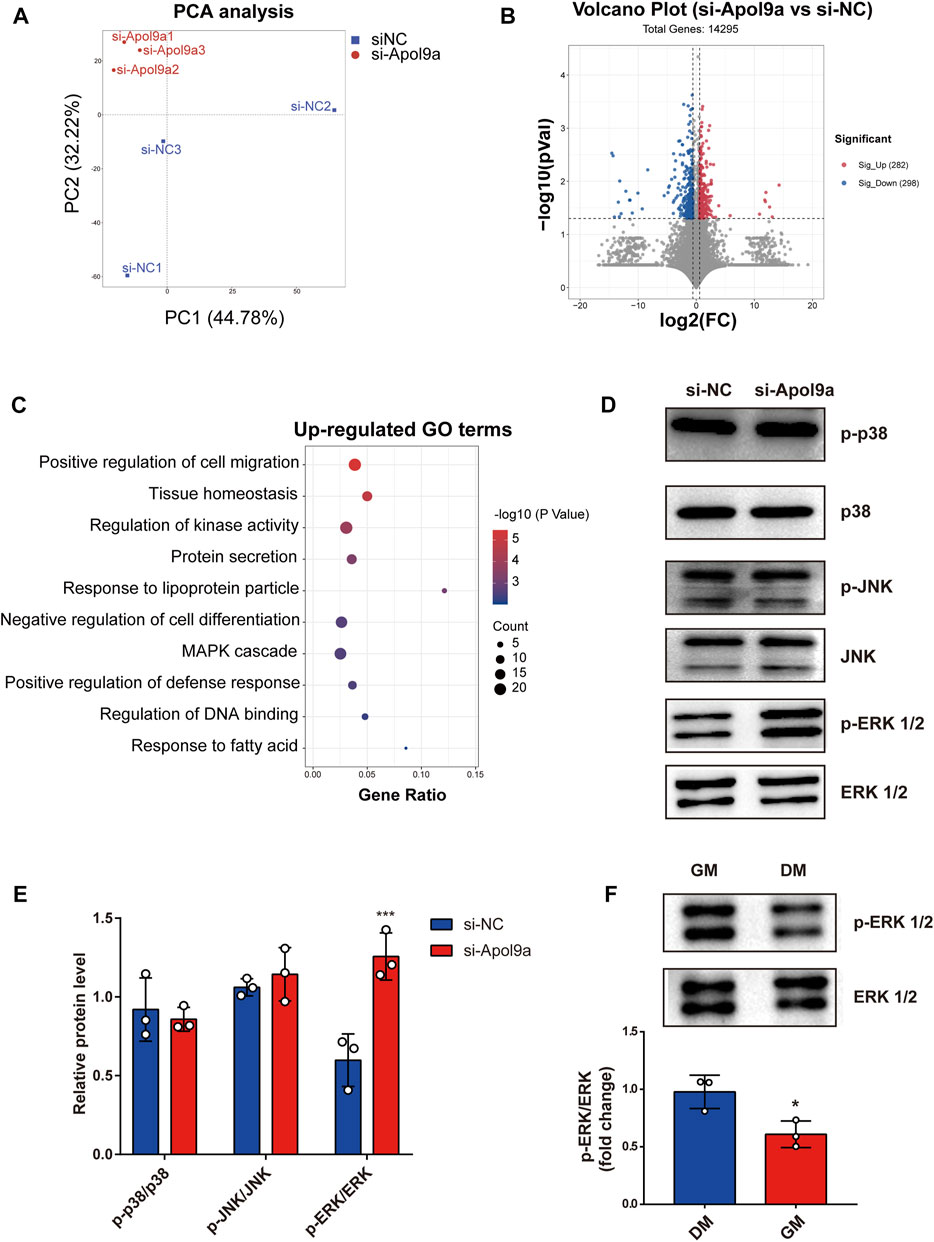
FIGURE 6. Transcriptomic data. C2C12 cells with or without si-Apol9a treatment was harvested after myogenic differentiation, and then transcriptomic analysis was performed. (A) Principal component analysis of RNA-seq data from C2C12 cells with (si-Apol9a) or without (si-NC) Apol9a knockdown after myogenic differentiation (n = 3 per group). (B) Volcano plot showing the DEGs in C2C12 cells with (si-Apol9a) or without (si-NC) Apol9a knockdown after myogenic differentiation. (C) Gene ontology analysis of upregulated DEGs. (D) Western blot analysis showed the protein levels of p-P38, P38, p-JNK, JNK, p-ERK1/2, and ERK1/2. (E) Gray scale analysis of western blot results determined using ImageJ software. (F) Western blot analysis for p-ERK1/2 and ERK1/2. C2C12 cells were cultured in GM or DM for 4 days. The data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. si-NC group.
To test the hypothesis that Apol9a regulates myogenic differentiation via the ERK1/2 signaling-dependent pathway, we applied PD98059, a specific inhibitor of ERK1/2, to the C2C12 cells. In agreement with our hypothesis, Apol9a knockdown-induced inhibition of myogenesis was almost completely reversed by PD98059 (Figure 7A). Furthermore, western blot analysis and immunofluorescence experiments demonstrated that ERK inhibition blocked the downregulation of myogenic genes (MyHC, MyoD, and MyoG) otherwise observed during Apol9a knockdown (Figures 7B,C). Together, these results provide evidence that Apol9a regulates myogenic differentiation via the ERK pathway.
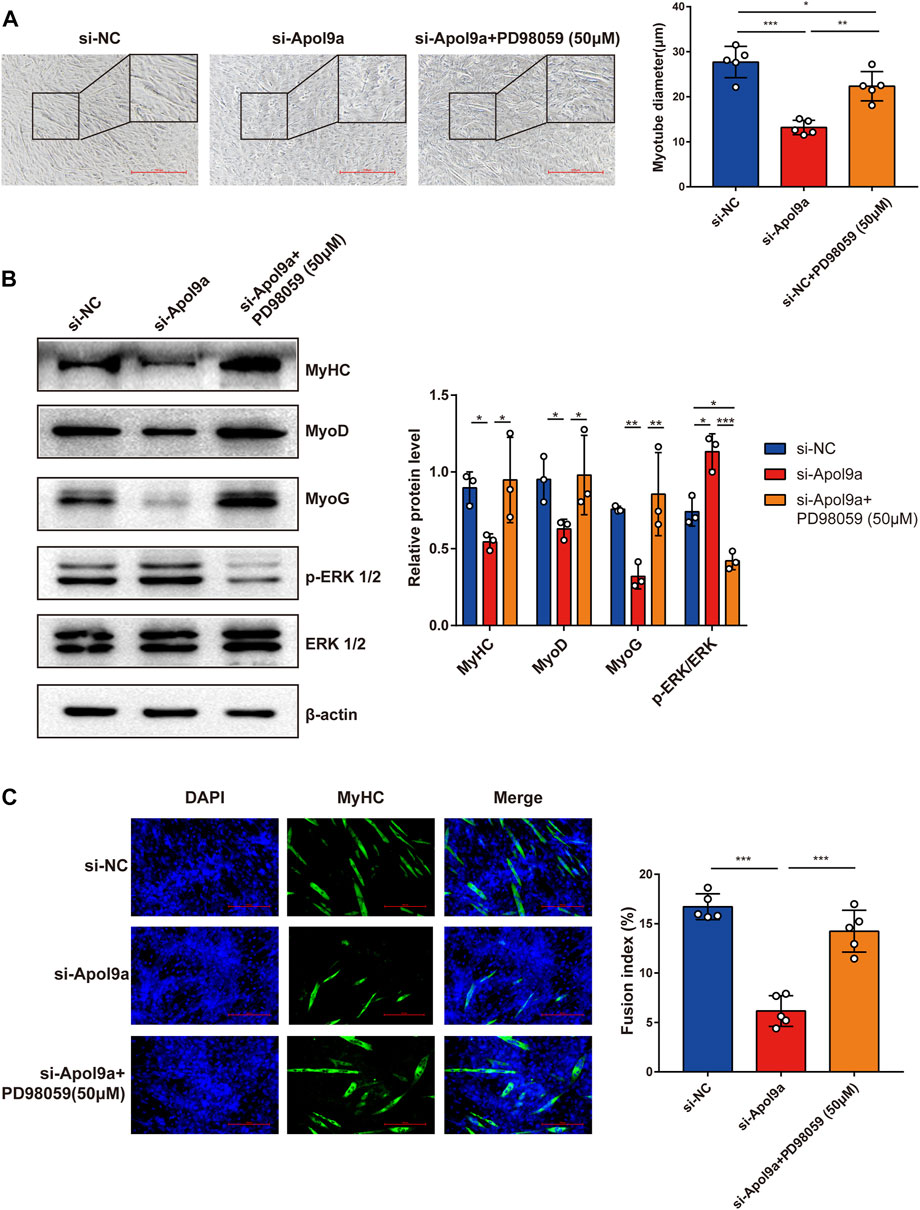
FIGURE 7. ERK inhibitor reverses Apol9a knockdown-induced inhibition of myogenesis. C2C12 cells were transfected with NC or Apol9a siRNA. After 12 h, the GM was replaced with the DM to induce differentiation or DM containing PD98059 (50 μm). (A) Morphology of C2C12 cells treated with si-NC, si-Apol9a, and si-Apol9a plus PD98059 (50 μm). The results of quantification of average myotube diameter are shown in the right. (B) Protein expression levels of MyHC, MyoD, MyoG, p-ERK1/2, and ERK1/2 as detected through western blot analysis. Gray intensity analysis is shown on the right. (C) Representative images of immunofluorescence for MyHC (green) are shown. The fusion index (%) was quantified and is shown on the right. Scale bar, 100 μm. Each experiment in this study was repeated at least three independent times. *p < 0.05, **p < 0.01, ***p < 0.001.
Skeletal muscle health is very crucial for human life at all stages. Moreover, skeletal muscle mass and myogenesis capacity are impaired in many pathological conditions, including obesity (Fu et al., 2016; Geiger et al., 2020). We confirmed that skeletal muscle mass and myogenesis capacity are decreased in obesity condition through the HFD-induced obesity mouse model. Although considerable attention has been paid to the investigation of therapeutic pharmacological supplements for improving muscle mass and function, progress is limited. Skeletal muscle plasticity is compromised due to impaired myogenesis (Suetta, 2017). By furthering our understanding of the molecular events regulating myogenesis, we may be able to develop an effective strategy for maintaining skeletal muscle integrity and plasticity.
In this study, Apol9a was identified as a new target gene critically involved in myogenesis. To our knowledge, this is the first study to report the underlying mechanisms and importance of Apol9a in skeletal muscle function. Apol9a belongs to the murine apolipoprotein L family and was previously reported to be an interferon-stimulated protein (Smith and Malik, 2009; Kreit et al., 2014). However, not much more is known about the function of Apol9a. In general, apolipoproteins play a role in lipid transportation (Su and Peng, 2020). For example, mouse apolipoprotein L9 is reported to be a PE-binding protein (Thekkinghat et al., 2019). In agreement with its proposed role in lipid transportation, our lipidomic data indicates that Apol9a knockdown decreased the PE level. Moreover, the TG and PC contents were reduced after Apol9a knockdown. Obesity leads to ectopic lipid deposition and abnormal lipid metabolism in skeletal muscle (Girousse et al., 2019). Our study confirmed that Apol9a is involved in multiple lipid transport activities, and its related functions should be further studied.
To explore the molecular mechanisms of Apol9a regulation of myogenic differentiation, RNA-seq was used to reconstruct de novo transcriptomes of Apol9a silenced cells. The results provide evidence that ERK signaling plays a crucial role in myogenesis. ERK1/2 signaling pathways are known to regulate numerous cellular processes, including cell proliferation, differentiation, and apoptosis (Cagnol and Chambard, 2010). While activated, ERK1/2 signaling plays a positive role in skeletal muscle cell proliferation, it negatively regulates myogenic differentiation (Jones et al., 2001). Moreover, studies have reported that ERK1/2 activation is elevated in atrophied and damaged skeletal muscles (Weston et al., 2003; Barreto et al., 2016; Oishi et al., 2019). Skeletal muscle differentiation is known to be mediated by several muscle regulatory factors, including MyoD, MyoG, and MyHC (Chal and Pourquié, 2017; Xu et al., 2017; Engquist and Zammit, 2021). Apol9a silencing significantly increased ERK1/2 phosphorylation and markedly suppressed the expression of muscle regulatory marker genes, such as Myf5, MyoD, MyoG, and MyHC. The experiments of Apol9a overexpression proved that Apol9a had a promoting effect on myogenic differentiation and significantly inhibited the ERK 1/2 pathway. In addition, MyoG and MyHC are the most significantly altered proteins corresponding to Apo9a overexpression. Application of an ERK inhibitor (PD98059) significantly reversed the aforementioned effects, indicating that Apol9a-ERK may be an upstream regulator of myogenic differentiation. Together, our results provide evidence that Apol9a-ERK may be a key therapeutic target for the management of skeletal muscle integrity and plasticity.
In summary, we indicate that Apol9a is a novel regulator of myogenic differentiation. Apol9a mRNA expression levels were significantly increased during myogenesis and decreased during obesity-induced muscle atrophy. Apol9a knockdown inhibited myogenic differentiation, possibly via the ERK1/2 signaling pathway. This study broadens our knowledge of the myogenic differentiation process and identifies a promising therapeutic target for intervention in obesity-related muscle atrophy.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA845924.
The animal study was reviewed and approved by the Ethics Committee of Jiangnan University.
SZ and XJ designed the concept and study. XJ, RW, SC, and SZ performed the experiments. XJ and SJ conducted the bioinformatics analysis. SZ, XJ, and WW collected the data. SZ, XJ, and YC prepared, wrote and edited the manuscript. All authors reviewed and gave their approval of the final manuscript to be published.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82000808, 82000685), the Major Special Fund for Translational Medicine (Grant Nos. 2020ZHZD03), the Fundamental Research Funds for the Central Universities (Grant Nos. JUSRP12048) and the Innovation and Application Project of Medical and Public Health Technology of Wuxi Science and Technology (Grant Nos. N20202005). This current study was mainly sponsored by the Fund of Wuxi Healthcare Commission and the National Natural Science Foundation of China.
We would like to thank Jingwei Zhang for cell culture and technical assistance, Haiyong Zhao for RNA-seq analysis support, Yizhou Zhou for GC-MS and LC-MS analysis, Zhe Jing for qPCR, University of Jiangnan Department of Experimental Medicine animal care takers, and Lengyun Wei for discussions. We are particularly grateful to YC for his valuable suggestions and all members in his lab.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.942061/full#supplementary-material
Apol9a, apolipoprotein L 9a; MyHC, myosin heavy chain; MyoD, myoblast determination protein; MyoG, myogenin; HFD, high-fat diet; qPCR, Real-time quantitative polymerase chain reaction; TG, triacylglycerol; PCA, principal component analysis; FAME, fatty acid methyl ester; OPLS-DA, orthogonal partial least squares-discriminant analysis; PE, phosphatidylethanolamine; PC, phosphatidylcholine; VIP, Variable Importance in Projection.
Arvind, T. A., and Rangarajan, P. N. (2016). Mouse Apolipoprotein L9 is a phosphatidylethanolamine-binding protein. Biochem. Biophys. Res. Commun. 479 (4), 636–642. doi:10.1016/j.bbrc.2016.09.161
Astrup, A., and Finer, N. (2000). Redefining type 2 diabetes: 'diabesity' or 'obesity dependent diabetes mellitus. Obes. Rev. 1 (2), 57–59. doi:10.1046/j.1467-789x.2000.00013.x
Barreto, R., Waning, D. L., Gao, H., Liu, Y., Zimmers, T. A., and Bonetto, A. (2016). Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 7 (28), 43442–43460. doi:10.18632/oncotarget.9779
Bonaldo, P., and Sandri, M. (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 6 (1), 25–39. doi:10.1242/dmm.010389
Boyer, J. G., Prasad, V., Song, T., Lee, D., Fu, X., Grimes, K. M., et al. (2019). ERK1/2 signaling induces skeletal muscle slow fiber-type switching and reduces muscular dystrophy disease severity. JCI Insight 5 (10), 127356. doi:10.1172/jci.insight.127356
Brown, L. A., Lee, D. E., Patton, J. F., Perry, R. A., Brown, J. L., Baum, J. I., et al. (2015). Diet-induced obesity alters anabolic signalling in mice at the onset of skeletal muscle regeneration. Acta Physiol. (Oxf). 215 (1), 46–57. doi:10.1111/apha.12537
Cagnol, S., and Chambard, J. C. (2010). ERK and cell death: Mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. Febs J. 277 (1), 2–21. doi:10.1111/j.1742-4658.2009.07366.x
Chal, J., and Pourquié, O. (2017). Making muscle: Skeletal myogenesis in vivo and in vitro. Development 144 (12), 2104–2122. doi:10.1242/dev.151035
Chargé, S. B., and Rudnicki, M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84 (1), 209–238. doi:10.1152/physrev.00019.2003
Chen, X., Luo, Y., Huang, Z., Jia, G., Liu, G., and Zhao, H. (2017). Akirin2 regulates proliferation and differentiation of porcine skeletal muscle satellite cells via ERK1/2 and NFATc1 signaling pathways. Sci. Rep. 7, 45156. doi:10.1038/srep45156
Ciciliot, S., and Schiaffino, S. (2010). Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des. 16 (8), 906–914. doi:10.2174/138161210790883453
DeFronzo, R. A., and Tripathy, D. (2009). Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32, S157–S163. doi:10.2337/dc09-S302
Du, M., Yan, X., Tong, J. F., Zhao, J., and Zhu, M. J. (2010). Maternal obesity, inflammation, and fetal skeletal muscle development. Biol. Reprod. 82 (1), 4–12. doi:10.1095/biolreprod.109.077099
Engquist, E. N., and Zammit, P. S. (2021). The satellite cell at 60: The foundation years. J. Neuromuscul. Dis. 8 (2), S183–s203. doi:10.3233/JND-210705
Eum, J. Y., Lee, G. B., Yi, S. S., Kim, I. Y., Seong, J. K., and Moon, M. H. (2020). Lipid alterations in the skeletal muscle tissues of mice after weight regain by feeding a high-fat diet using nanoflow ultrahigh performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1141, 122022. doi:10.1016/j.jchromb.2020.122022
Feraco, A., Gorini, S., Armani, A., Camajani, E., Rizzo, M., and Caprio, M. (2021). Exploring the role of skeletal muscle in insulin resistance: Lessons from cultured cells to animal models. Int. J. Mol. Sci. 22 (17), 9327. doi:10.3390/ijms22179327
Fu, X., Zhu, M., Zhang, S., Foretz, M., Viollet, B., and Du, M. (2016). Obesity impairs skeletal muscle regeneration through inhibition of AMPK. Diabetes 65 (1), 188–200. doi:10.2337/db15-0647
Geiger, A. E., Daughtry, M. R., Yen, C. N., Kirkpatrick, L. T., Shi, H., and Gerrard, D. E. (2020). Dual effects of obesity on satellite cells and muscle regeneration. Physiol. Rep. 8 (15), e14511. doi:10.14814/phy2.14511
Girousse, A., Gil-Ortega, M., Bourlier, V., Bergeaud, C., Sastourné-Arrey, Q., Moro, C., et al. (2019). The release of adipose stromal cells from subcutaneous adipose tissue regulates ectopic intramuscular adipocyte deposition. Cell Rep. 27 (2), 323–333. doi:10.1016/j.celrep.2019.03.038
Jones, N. C., Fedorov, Y. V., Rosenthal, R. S., and Olwin, B. B. (2001). ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J. Cell. Physiol. 186 (1), 104–115. doi:10.1002/1097-4652(200101)186:1<104::AID-JCP1015>3.0.CO;2-0
Keren, A., Tamir, Y., and Bengal, E. (2006). The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 252 (1-2), 224–230. doi:10.1016/j.mce.2006.03.017
Kreit, M., Paul, S., Knoops, L., De Cock, A., Sorgeloos, F., and Michiels, T. (2014). Inefficient type I interferon-mediated antiviral protection of primary mouse neurons is associated with the lack of apolipoprotein l9 expression. J. Virol. 88 (7), 3874–3884. doi:10.1128/JVI.03018-13
Kreit, M., Vertommen, D., Gillet, L., and Michiels, T. (2015). The interferon-inducible mouse apolipoprotein L9 and prohibitins cooperate to restrict theiler's virus replication. PLoS One 10 (7), e0133190. doi:10.1371/journal.pone.0133190
Lingvay, I., Sumithran, P., Cohen, R. V., and le Roux, C. W. (2021). Obesity management as a primary treatment goal for type 2 diabetes: Time to reframe the conversation. Lancet 399, 394–405. doi:10.1016/S0140-6736(21)01919-X
McCarthy, D., and Berg, A. (2021). Weight loss strategies and the risk of skeletal muscle mass loss. Nutrients 13 (7), 2473. doi:10.3390/nu13072473
Mengeste, A. M., Rustan, A. C., and Lund, J. (2021). Skeletal muscle energy metabolism in obesity. Obesity 29 (10), 1582–1595. doi:10.1002/oby.23227
Nishikawa, H., Asai, A., Fukunishi, S., Nishiguchi, S., and Higuchi, K. (2021). Metabolic syndrome and sarcopenia. Nutrients 13 (10), 3519. doi:10.3390/nu13103519
Nishikawa, H., Goto, M., Fukunishi, S., Asai, A., Nishiguchi, S., and Higuchi, K. (2021). Cancer cachexia: Its mechanism and clinical significance. Int. J. Mol. Sci. 22 (16), 8491. doi:10.3390/ijms22168491
Oishi, Y., Ogata, T., Ohira, Y., and Roy, R. R. (2019). Phosphorylated ERK1/2 protein levels are closely associated with the fast fiber phenotypes in rat hindlimb skeletal muscles. Pflugers Arch. 471 (7), 971–982. doi:10.1007/s00424-019-02278-z
Penna, F., Costamagna, D., Fanzani, A., Bonelli, G., Baccino, F. M., and Costelli, P. (2010). Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One 5 (10), e13604. doi:10.1371/journal.pone.0013604
Roh, E., and Choi, K. M. (2020). Health consequences of sarcopenic obesity: A narrative review. Front. Endocrinol. 11, 332. doi:10.3389/fendo.2020.00332
Schiaffino, S., Dyar, K. A., Ciciliot, S., Blaauw, B., and Sandri, M. (2013). Mechanisms regulating skeletal muscle growth and atrophy. Febs J. 280 (17), 4294–4314. doi:10.1111/febs.12253
Smith, E. E., and Malik, H. S. (2009). The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 19 (5), 850–858. doi:10.1101/gr.085647.108
Sousa-Victor, P., García-Prat, L., and Muñoz-Cánoves, P. (2022). Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat. Rev. Mol. Cell Biol. 23 (3), 204–226. doi:10.1038/s41580-021-00421-2
Su, X., and Peng, D. (2020). The exchangeable apolipoproteins in lipid metabolism and obesity. Clin. Chim. Acta. 503, 128–135. doi:10.1016/j.cca.2020.01.015
Suetta, C. (2017). Plasticity and function of human skeletal muscle in relation to disuse and rehabilitation: Influence of ageing and surgery. Dan. Med. J. 64 (8), B5377.
Sun, L., Miyoshi, H., Origanti, S., Nice, T. J., Barger, A. C., Manieri, N. A., et al. (2015). Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe 17 (1), 85–97. doi:10.1016/j.chom.2014.11.004
Thekkinghat, A. A., Yadav, K. K., and Rangarajan, P. N. (2019). Apolipoprotein L9 interacts with LC3/GABARAP and is a microtubule-associated protein with a widespread subcellular distribution. Biol. Open 8 (9), bio045930. doi:10.1242/bio.045930
Weston, A. D., Sampaio, A. V., Ridgeway, A. G., and Underhill, T. M. (2003). Inhibition of p38 MAPK signaling promotes late stages of myogenesis. J. Cell Sci. 116 (14), 2885–2893. doi:10.1242/jcs.00525
Wiedmer, P., Jung, T., Castro, J. P., Pomatto, L. C. D., Sun, P. Y., Davies, K. J. A., et al. (2021). Sarcopenia - molecular mechanisms and open questions. Ageing Res. Rev. 65, 101200. doi:10.1016/j.arr.2020.101200
Wu, H., and Ballantyne, C. M. (2017). Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Invest. 127 (1), 43–54. doi:10.1172/JCI88880
Xie, S. J., Li, J. H., Chen, H. F., Tan, Y. Y., Liu, S. R., Zhang, Y., et al. (2018). Inhibition of the JNK/MAPK signaling pathway by myogenesis-associated miRNAs is required for skeletal muscle development. Cell Death Differ. 25 (9), 1581–1597. doi:10.1038/s41418-018-0063-1
Xu, X., Ji, S., Li, W., Yi, B., Li, H., Zhang, H., et al. (2017). LncRNA H19 promotes the differentiation of bovine skeletal muscle satellite cells by suppressing Sirt1/FoxO1. Cell. Mol. Biol. Lett. 22, 10. doi:10.1186/s11658-017-0040-6
Yaffe, D., and Saxel, O. (1977). Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270 (5639), 725–727. doi:10.1038/270725a0
Zhang, J., Wang, W., Feng, N., Jiang, X., Zhu, S., and Chen, Y. Q. (2021). Ndufa6 regulates adipogenic differentiation via Scd1. Adipocyte 10 (1), 646–657. doi:10.1080/21623945.2021.2007590
Zhu, S., Wang, W., Zhang, J., Ji, S., Jing, Z., and Chen, Y. Q. (2022). Slc25a5 regulates adipogenesis by modulating ERK signaling in OP9 cells. Cell. Mol. Biol. Lett. 27 (1), 11. doi:10.1186/s11658-022-00314-y
Zhu, S., Zhang, J., Jiang, X., Wang, W., and Chen, Y. Q. (2022). Free fatty acid receptor 4 deletion attenuates colitis by modulating Treg Cells via ZBED6-IL33 pathway. EBioMedicine 80, 104060. doi:10.1016/j.ebiom.2022.104060
Keywords: obesity, myogenesis, Apol9a, ERK1/2, C2C12
Citation: Jiang X, Ji S, Cui S, Wang R, Wang W, Chen Y and Zhu S (2022) Apol9a regulates myogenic differentiation via the ERK1/2 pathway in C2C12 cells. Front. Pharmacol. 13:942061. doi: 10.3389/fphar.2022.942061
Received: 12 May 2022; Accepted: 10 November 2022;
Published: 23 November 2022.
Edited by:
Guixue Wang, Chongqing University, ChinaReviewed by:
Kai Qiu, Chinese Academy of Agricultural Sciences (CAAS), ChinaCopyright © 2022 Jiang, Ji, Cui, Wang, Wang, Chen and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shenglong Zhu, c2hlbmdsb25nemh1QGppYW5nbmFuLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.